
Abstract
The Locally Self-consistent Multiple Scattering (LSMS) code solves the first principles Density Functional theory Kohn-Sham equation for a wide range of materials with a special focus on metals, alloys and metallic nano-structures. It has traditionally exhibited near perfect scalability on massively parallel high performance computer architectures. We present our efforts to exploit GPUs to accelerate the LSMS code to enable first principles calculations of O(100,000) atoms and statistical physics sampling of finite temperature properties. Using the Cray XK7 system Titan at the Oak Ridge Leadership Computing Facility we achieve a sustained performance of 14.5PFlop/s and a speedup of 8.6 compared to the CPU only code.
This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Curie Temperature
- Multiple Scattering Theory
- Intermediate Block
- High Performance Computer Architecture
- Global Ground State
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Multiple Scattering Theory
Density Functional Theory [4], especially in the Kohn-Sham formulation [6] represents a major, well established, methodology for investigating materials from first principles. Most computational approaches to solving the Kohn-Sham equation for electrons in materials attempt to solve the eigenvalue problem for periodic systems directly. The solution of the eigenvalue problem for dense matrices results in cubic scaling in the system size. Additionally these spectral methods require approximations such as the use of pseudopotentials or linearized basis sets for all electron methods to make the size of the basis set manageable.
In this paper we utilize a different approach to solving the Kohn-Sham equation using multiple scattering theory in real space. The basis of this method is formed by the Kohn-Korringa-Rostocker (KKR) method [5, 7], that allows for the solution of the all electron DFT equations without the need for linearization.
1.1 The LSMS Algorithm
For the energy evaluation, we employ the first principles framework of density functional theory (DFT) in the local density approximation (LDA). To solve the Kohn-Sham equations arising in this context, we use a real space implementation of the multiple scattering formalism. The details of this method for calculating the Green function and the total ground state energy \(E[n(\varvec{r}),\varvec{m}(\varvec{r})]\) are described elsewhere [3, 14]. Linear scaling is achieved by limiting the scattering distance of electrons in the solution of the multiple scattering problem. Additionally the LSMS code allows the constraint of the magnetic moment directions [11] which enables the sampling of the excited magnetic states in the Wang-Landau procedure described below.
For the present discussion it is important to note that the computationally most intensive part is the calculation of the scattering path matrix \(\tau \) for each atom in the system by inverting the multiple scattering matrix.
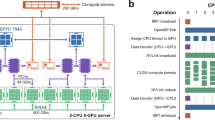
While the rank of the scattering path matrix \(\tau \) is proportional to the number of sites in the local interaction zone and to \((l_{max}+1)^2\) (typically a few thousand, e.g. for \(l_{max}=3\) and 113 atoms in the local interaction zone, the rank is 3616.), the only part of \(\tau \) that will be required in the subsequent calculations of site diagonal observables (i.e. magnetic moments, charge densities, and total energy) is a small (typically \(32 \times 32\)) diagonal block of this matrix. This will allow us to employ the algorithm described in the next section for maximum utilization of the on node floating point compute capabilities (Fig. 1).

Parallelization scheme of the LSMS method [14].
From benchmarking a typical calculation of 1024 iron atoms with a local interaction zone radius of \(12.5 a_0\) on the AMD processors on Titan, we find that the majority of time (\(95\,\%\)) and floating point operations are spent inside the inversion of the multiple scattering matrix to obtain the \(\tau \)-matrix. About half of the remaining time is used to construct this matrix. Thus our approach to accelerate the LSMS code for GPUs concentrated on these two routines that will be presented in detail in the following two sections.
1.2 Scattering Matrix Construction
To calculate the \(\tau \)-matrix (Eq. 1) the first step involves constructing the scattering matrix
to be inverted. The m matrix is constructed from blocks that are associated with the sites i and j in the local interaction zone.
Each of these block can in principle be evaluated in parallel. The size of these individual blocks is given by the cut-off in the l expansion of the scattering expansion. For a spin-canted calculation the size of a block is \(2(l_{max}+1)^2\), i.e. for a typical \(l_{max}=3\) each of the \(m_{ij}\) blocks has rank 32. The indices inside each block label the angular momentum l, m. For a typical number of O(100) atoms in the local interaction zone, there are O(10, 000) blocks of the m matrix that need to be calculated, thus providing significant parallelism that can be exploited on accelerators. On the accelerator the m matrix is first initialized to a unit matrix to account for the \(I\delta _{ij}\) part. The single site scattering matrices are currently calculated on the CPU, as this involves only the numerical evaluation of ordinary differential equations for initial values determined by the energy and the angular momentum l and are communicated as needed to remote nodes and transferred to the GPU memory.
The structure constants \(G^{ij}_{0,LL'}(E)\) are geometry dependent and the atomic distances \(R_{ij}\) and directions \(\hat{R}_{ij}\) are transferred to the GPU memory at the beginning of the program and remain there unchanged. The structure constants are given by the expression
where L are the combined (l, m) indices and \(C^L_{L'L''}\) are the Gaunt coefficients. The factor \(D^{ij}_{L''}(E)\) is given by the following equation.
Here \(h_l(x)\) are the spherical Hankel functions and \(Y_L(\hat{r})\) are the spherical harmonics. These structure constants are evaluated inside a CUDA kernel that is allows parallelization in \(L, L'\) and executed in multiple streams for i, j. The final product \(t_i G_{ij}\) is evaluated using batched cuBLAS double complex matrix multiplications.
1.3 Matrix Inversion
The most computationally intensive part of the LSMS calculation is the matrix inversion to obtain the multiple scattering matrix \(\tau \). (Eq. 1) The amount of computational effort can be reduced by utilizing the fact that for each local interaction zone only the left upper block (\(\tau _{00}\)) of the scattering path matrix \(\tau \) is required. LSMS uses an algorithm that reduces the amount of work needed while providing excellent performance due to its reliance on dense matrix-matrix multiplications that are available in highly optimized form in vendor or third party provided implementations (i.e. ZGEMM in the BLAS library).
The method employed in LSMS to calculate the required block of the inverse relies on the well known expression for writing the inverse of a matrix in term of inverses and products of subblocks:
where
and similar expressions for V, W, and Y. This method can be applied multiple times to the subblock U until the desired block \(\tau _{00}\) of the scattering path matrix is obtained.
The operations needed to obtain the \(\tau _{00}\) thus are matrix multiplications and the inversion of the diagonal subblocks. For the matrix multiplication on the GPUs we can exploit the optimized version of these routines that are readily available in the cuBLAS library. The size of the intermediate block sizes used in the matrix inversion serves as a tuning parameter to optimize the performance of our block inversion algorithm. As the block size becomes larger, the resulting matrices entering the matrix multiplication usually result in significant improvements of the matrix multiplication performance at the cost of performing more floating-point operations then are strictly needed to obtain the final \(\tau _{00}\) block. Thus there exists a optimum intermediate block size that minimizes the runtime of the block inversion. For the CPU only code this is achieve at a rank of the intermediate blocks of approximately 1000. For the GPU version we employ an optimized matrix inversion algorithm written in CUDA that executes the whole inversion in a single kernel in GPU memory, thus avoiding costly memory transfers and kernel launches. The maximal rank of double complex matrices that can be handled by this algorithm is 175, thus providing the size limit for the intermediate blocks and the block size for which we observe the maximum performance of the GPU version reported in this paper.
2 Wang-Landau Monte-Carlo Sampling
The LSMS method allows the calculation of energies for a set of parameters or constraints \(\{\xi _i\}\) that specify a state of the system that is not the global ground state. Examples of this include arbitrary orientations of the magnetic moments or chemical occupations of the lattice sites. Thus we can calculate the energy \(E(\{\xi _i\})\) associated with these sets of parameters. Evaluating the partition function
where \(\beta = 1/k_B T\) is the inverse temperature and the sum is over all possible configurations \(\{\xi _i\}\), allows the investigation of the statistical physics of the system and the evaluation of its finite temperature properties. In all but the smallest most simple systems (e.g. for a few Ising spins), it is computational intractable to perform this summation directly. Monte-Carlo methods have been used successfully to evaluate these very high dimensional sums or integrals using statistical importance sampling. The most widely used method is the Metropolis method [8], which generates samples in phase space with a probability that is given by the Boltzmann factor \(e^{-\beta E(\{\xi _i\})}\).
For our work we have chosen to employ the Wang-Landau Monte-Carlo method [12, 13], which is a method to calculate the density of states g(E) of the system for the phase space spanned by the set of classical parameters that describe the system.
We have parallelized the Wang-Landau procedure by employing multiple, parallel walkers that update the same histogram and density of states [2] as illustrated in Fig. 2.

Parallelization strategy of the combined Wang-Landau/LSMS algorithm. The Wang-Landau process generates random spin configurations for M walkers and updates a single density of states g(E). The energies for these N atom systems are calculated by independent LSMS processes. This results in two levels of communication, between the Wang-Landau driver and the LSMS instances, and the internal communication inside the individual LSMS instances spanning N processes each.
3 Scaling and Performance
The WL-LSMS code has been known for its performance on scalability, as has been shown on CPU only architectures, such as the previous Jaguar system at the Oak Ridge Leadership Computing Facility [2]. The acceleration of significant portions of the code for GPUs, combined with major restructuring of the high level structure of the LSMS code was able to maintain the excellent scalability of the code. In Fig. 3 we show the near perfect weak scaling of the LSMS code in the number of atom, while maintaining the number of atom per compute node over five orders of magnitude from 16 iron atoms to 65, 536 atoms, while achieving a speedup factor of 8.6 for the largest systems compared to using the CPUs only on the Titan system at Oak Ridge. This performance puts calculations of million atom size systems within reach for the next generation of supercomputers such as the planned Summit system at the Oak Ridge Leadership Computing Facility.

Weak scaling of LSMS on Titan utilizing the GPU accelerators for a bulk iron calculation. 16 atoms on 4 nodes require 67.343 s per iteration step and 65536 atoms on 16384 nodes require 69.988 s, resulting in a parallel scaling efficiency of 96 % across Titan.
For the statistical sampling with the Wang-Landau method as described above, we tested the scaling of the code in the number of walkers. The performance tests were done for 1024 iron atoms and the energies were self-consistently calculated for a LIZ radius of \(12.5a_0\) allowing us to achieve a sampling rate of nearly one Monte-Carlo sample per second on Titan. The scaling of the energy samples per wall-time is shown in Fig. 4.

Scaling of Wang Landau LSMS on Titan for 1024 Fe calculations, using 128 nodes per Monte-Carlo walker. The code shows good scaling in the number of states sampled with increasing number of walkers. With two walkers on 257 nodes the code generates 0.0208 samples/second and with 96 walkers on 12289 nodes 0.9128 samples/second are generated, thus achieving a parallel efficiency of 92 %.
To assess the improvements in the computational and power efficiency that resulted from the porting of the significant portions of the code to GPU accelerators, we have run an identical WL-LSMS calculation for 1024 iron atoms with 290 walkers on 18561 nodes on Titan at the Oak Ridge Leadership Computing Facility for 20 Monte-Carlo steps per walker. We measured the instantaneous power consumption at the power supply to the compute cabinets which includes the power for compute, memory and communication as well as line losses and the secondary cooling system inside the cabinets, but excludes the power consumption of the file system and the chilled water supply. The measurements were performed both for a CPU only run as well a for a computation utilizing the GPUs. The results are shown in Fig. 5. The difference in power consumption between the compute intensive LSMS calculations, that take most of the time, and Monte-Carlo steps that are marked by a significant drop in the power consumption is obvious and this allows a clear comparison of the two runs.

Power consumption traces for identical WL-LSMS runs with 1024 Fe atoms on 18,561 Titan nodes (99 % of Titan). 14.5 PF sustained vs 1.86 PF CPU only. Runtime is 8.6X faster for the accelerated code, Energy consumed is 7.3X less. GPU accelerated code consumed 3,500 kW-hr, CPU only code consumed 25,700 kW-hr.
4 Applications
In this section we review results that we have obtained using the method described in this paper to calculating the Curie temperatures of various materials. In particular we have applied this method to iron and cementite [1] and to \(\mathrm {Ni}_2\mathrm {MnGa}\) [9]. For the underlying LSMS calculations the atoms are placed on lattices with lattice parameters corresponding to the experimental room temperature values. The self-consistently converged potentials for the ferromagnetic or ferrimagnetic ground states were used for all the individual frozen-potential energy calculations in the combined Wang-Landau/LSMS algorithm. The calculations were performed by randomly choosing a site in the supercell and randomly picking a new moment direction. In the case of Fe and \(\text {Fe}_3\text {C}\) the convergence criterion for the Wang-Landau density of states was chosen to be the convergence of Curie temperature. The density of state thus obtained was used to calculate the specific heat. The peak in the specific heat allows us to identify the Curie temperature to be 980 K for iron, in good agreement with the experimental value of 1050 K. The Curie temperature obtained for \(\text {Fe}_3\text {C}\) is 425 K which again is in good agreement with the experimental value of 480 K. [1] For \(\mathrm {Ni}_2\mathrm {MnGa}\) the Curie temperature reported is 185 K, well below the experimental value of 351 K. [9] The small cell used in these calculation (144 atoms) will have resulted in a significant finite size error. Additionally, it is known that the localized moment picture that underpins our WL-LSMS calculations does miss important contributions to the fluctuations that determine the finite temperature magnetism in nickel, which will contribute to the reduction of the calculated Curie temperature from the experimental value. This was already observed by Staunton et al. [10] in disordered local moment calculations that underestimate the Curie temperature of pure Ni and they find 450 K as opposed to the experimental value of 631 K. We propose to include fluctuations in the magnitude of local moments. Preliminary calculations with a Heisenberg model that is extended with the inclusion of the magnitude of the local moment as a variable indicates that this can result in an increase in the Curie temperature.
5 Conclusions
We have shown that for some classes of calculations, it is possible to make efficient use of GPU accelerators with a reasonable amount of code modification work. The acceleration of the code additionally results in significant energy savings while maintaining its scalability. Consequently the code and work presented in this paper enables the first principles investigation of materials at scales that were previously hard to access and pushes the possibilities for first principles statistical physics. Ongoing work involves extending the capabilities of LSMS presented in this paper to non spherical atomic potentials and to solving the Dirac equation for the electrons in solids, which will allow the first principles investigation of the coupling of magnetic and atomic degrees of freedom and other effects involving atomic displacements. These additions to the code will require additional work to accelerate the single site solvers for GPUs as in these cases a significant amount of compute resources will be needed to solve the single site equation for non spherical scatterers.
References
Eisenbach, M., Nicholson, D.M., Rusanu, A., Brown, G.: First principles calculation of finite temperature magnetism in Fe and Fe3C. J. Appl. Phys. 109(7), 07E138 (2011)
Eisenbach, M., Zhou, C.G., Nicholson, D.M., Brown, G., Larkin, J., Schulthess, T.C.: A scalable method for ab initio computation of free energies in nanoscale systems. In: Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis, SC 2009, pp. 64:1–64:8. ACM, New York (2009)
Eisenbach, M., Györffy, B.L., Stocks, G.M., Újfalussy, B.: Magnetic anisotropy of monoatomic iron chains embedded in copper. Phys. Rev. B 65, 144424 (2002)
Hohenberg, P., Kohn, W.: Inhomogeneous electron gas. Phys. Rev. 136, B864–B871 (1964)
Kohn, W., Rostoker, N.: Solution of the Schrödinger equation in periodic lattices with an application to metallic Lithium. Phys. Rev. 94, 1111–1120 (1954). http://link.aps.org/doi/10.1103/PhysRev.94.1111
Kohn, W., Sham, L.J.: Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138 (1965)
Korringa, J.: On the calculation of the energy of a Bloch wave in a metal. Physica 13, 392–400 (1947)
Metropolis, N., Rosenbluth, A.W., Rosenbluth, M.N., Teller, A.H., Teller, E.: Equation of state calculations by fast computing machines. J. Chem. Phys. 21, 1087 (1953)
Nicholson, D.M., Odbadrakh, K., Rusanu, A., Eisenbach, M., Brown, G., Evans III, B.M.: First principles approach to the magneto caloric effect: application to \({\rm Ni}_{2}\)MnGa. J. Appl. Phys. 109(7), 07A942 (2011)
Staunton, J., Gyorffy, B.: Onsager cavity fields in itinerant-electron paramagnets. Phys. Rev. Lett. 69, 371–374 (1992)
Stocks, G.M., Eisenbach, M., Újfalussy, B., Lazarovits, B., Szunyogh, L., Weinberger, P.: On calculating the magnetic state of nanostructures. Prog. Mater. Sci. 52(2–3), 371–387 (2007)
Wang, F., Landau, D.P.: Determining the density of states for classical statistical models: a random walk algorithm to produce a flat histogram. Phys. Rev. E 64, 056101 (2001)
Wang, F., Landau, D.P.: Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 86(10), 2050–2053 (2001)
Wang, Y., Stocks, G.M., Shelton, W.A., Nicholson, D.M.C., Temmerman, W.M., Szotek, Z.: Order-N multiple scattering approach to electronic structure calculations. Phys. Rev. Lett. 75, 2867 (1995)
Acknowledgements
This work has been sponsored by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Material Sciences and Engineering Division (basic theory and applications) and by the Office of Advanced Scientific Computing (software optimization and performance measurements). This research used resources of the Oak Ridge Leadership Computing Facility, which is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC05-00OR22725.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Singapore
About this paper
Cite this paper
Eisenbach, M., Larkin, J., Lutjens, J., Rennich, S., Rogers, J.H. (2016). GPU Acceleration of the Locally Selfconsistent Multiple Scattering Code for First Principles Calculation of the Ground State and Statistical Physics of Materials. In: Chen, W., et al. Big Data Technology and Applications. BDTA 2015. Communications in Computer and Information Science, vol 590. Springer, Singapore. https://doi.org/10.1007/978-981-10-0457-5_24
Download citation
DOI: https://doi.org/10.1007/978-981-10-0457-5_24
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-0456-8
Online ISBN: 978-981-10-0457-5
eBook Packages: Computer ScienceComputer Science (R0)