
Abstract
Tumor microenvironment (TME) is composed of tumor cells, immune cells, cytokines, extracellular matrix, etc. The immune system and the metabolisms of glucose, lipids, amino acids, and nucleotides are integrated in the tumorigenesis and development. Cancer cells and immune cells show metabolic reprogramming in the TME, which intimately links immune cell functions and edits tumor immunology. Recent findings in immune cell metabolism hold the promising possibilities toward clinical therapeutics for treating cancer. This chapter introduces the updated understandings of metabolic reprogramming of immune cells in the TME and suggests new directions in manipulation of immune responses for cancer diagnosis and therapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
The tumor microenvironment (TME), first proposed by Lord in 1979, is a complex integrated system for tumor cell growth [1]. This environment is composed of tumor cells, endothelial cells, immune cells, fibroblasts, and extracellular matrix. The immune cells in the TME including macrophages, neutrophils, and lymphocytes play important roles in tumor immune escape, tolerance, and suppression. Traditional theory states that the incidence of cancer is due to failure of immune surveillance [2]. However, this theory was unable to fully explain the interplay between immunity and tumorigenesis. The immunoediting theory, proposed by Schreiber and Dunn, elucidated the role of the immune system in cancer development from a new perspective [3]. Depending on the cellular and environmental context, the immune cells may kill tumor cells but also promote tumor development. This theory divides the carcinogenesis into three stages: immune clearance, immune balance, and immune escape. Immune clearance is similar to the immune surveillance. Immune balance is a stalemate stage that the tumor is not completely removed by the immune system, in which the tumor characteristics are reshaped by the immune system. Immune escape refers to the stage that the tumor escapes immune surveillance after tumor cells are edited by the immune system and progress to clinical tumor stage [4].
During the development of cancer, tumor cells and immune cells interact in a dynamic microenvironment that determines the outcome of tumorigenesis [5]. In the immune clearance phase, the immune system removes tumor cells through antigen-specific and nonspecific mechanisms, wherein lymphocytes are the main effector cells. If the immune surveillance stage functions effectively and the tumor cells are cleared, the immunoediting stage will not ensue. However, the immune system is not always effective in removing every tumor cells, especially the ones with low immunogenicity. These tumor cells often escape temporarily from the immune cell-mediated destruction. The immune balance stage then follows [6]. At this stage, the immune system constantly kills high immunogenic tumor cells, whereas it is “blind” to the tumor cells with low immunogenicity, resulting in the gradual emergence of the tumors with low immunogenic and high malignant phenotypes. The immune system continues to impose pressure to select for the tumor cells with accommodating immunological phenotypes. This Darwin’s natural selection-like process is referred to as immune remodeling. The surviving tumor cells with low immunogenicity repeatedly stimulate the immune system and eventually induce the immune tolerance [5,6,7]. Tumor cells remodeled by the immune system can aberrantly produce inhibitory cytokines including interleukin-10 (IL-10), transforming growth factor-β (TGFβ), and vascular endothelial growth factor (VEGF) and induce immunosuppressive cells including regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), fostering an immunosuppressive TME. In the TME, T cells are tolerant to tumor-associated antigen and functionally suppressive; the professional antigen presentation cells (APCs) are functionally defective, leading to hindered antitumor immune response and the systemic immunosuppression [8]. Consequently, establishing the immune-tolerant TME not only reduces the ability of the immune system to reject tumors but also promotes the immune escape of tumors that should have been rejected.
The metabolomics is an emerging direction of immune and cancer research [9, 10]. The metabolism of carbohydrates, lipids, and amino acids is the material basis of all biological processes. For the development of malignancy, the tumor cells must face two challenges: first, obtaining the nutrients needed for the rapid growth; second, evading the surveillance and attack from the host immune system. Tumor cell’s unique metabolic program can be used to meet these challenges. Glycolysis is the major metabolic process used by malignant tumors, even when oxygen supply is adequate, which is termed as “Warburg effect” [11]. Glycolysis decreases the pH value of the TME; therefore, tumor cells can inhibit the activities of antigen-presenting cells (APCs) and cytotoxic T lymphocytes (CTLs) by controlling the acidity of the microenvironment, eventually leading to tumor cell immune escape [12].
Recent studies indicate that tumor-associated immune cells show altered metabolism to affect their differentiation, survival, and function [10]. Since the TME heavily influences carcinogenesis, the metabolic programming and reprogramming of immune cells in the TME and how immune metabolism affects the tumor initiation, development, and metastasis are of great interest and significance [10, 12]. In this chapter, we review the current knowledge on the metabolism of the cell types in the TME, discuss the emerging concept of the metabolic reprogramming in tumor-associated immune cells, and propose the impact of immune metabolism on carcinogenesis and clinical applications.
5.2 Metabolism of Cancer Cells and Cancer Stem Cells in TME
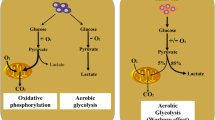
Aberrant proliferation of cancer cells is fueled by altered metabolism (Fig. 5.1). Oncogenic mutations trigger a switch from oxidative phosphorylation (OXPHOS) to glycolysis in tumor cells, and hypoxia further enhances this reprogramming [13, 14]. OXPHOS generates 36 mol ATP from 1 mol glucose, while glycolysis produces only 4 mol ATP [15]. This seeming inefficient metabolic feature relies on abnormal upregulation of the glucose transporters such as GLUT1 [16]. Moreover, mutations in tricarboxylic acid (TCA) cycle enzymes such as succinate dehydrogenase (SDH) or fumarate hydratase (FH) also promote glycolysis and inhibit OXPHOS [14, 17, 18]. Lactic acid is the end product of glycolysis. Lactic acid production can be used as a biomarker of tumor metastasis and overall survival [14, 15]. The secretion of lactic acid by monocarboxylate transporter 4 (MCT4) by cancer cells depends on the intracellular and extracellular concentrations of lactic acid [19, 20]. The extracellular lactate effects include restricting monocyte conversion to dendritic cells (DCs), suppressing cytokine release from DC and CTL, and inhibiting monocyte migration and CTL function. In addition, the release of lactic acid by tumor cells to the extracellular space can block the lactic acid secretion from immune cells to trigger cell death due to excessive intracellular lactic acid [21, 22].

Overview of cancer cell metabolism. This diagram depicts the cellular metabolic pathways in cancer cells. Abbreviations: 3DG 3-deoxyglucosone, 3PG 3-phosphoglycerate, 6PGD 6-phosphogluconate dehydrogenase, AGE advanced glycation end product, AR aldose reductase, ARG arginase, ATP adenosine triphosphate, CPT carnitine palmitoyltransferase, DHAP dihydroxyacetone phosphate, eNOS endothelial nitric oxide synthase, ETC electron transport chain, F6P fructose 6-phosphate, F1,6P2 fructose 1,6-bisphosphate, F2,6P2 fructose 2,6 bisphosphate, FA fatty acid, G6P glucose 6-phosphate, G6PD glucose 6-phosphate dehydrogenase, GAPDH glyceraldehyde 3-phosphate dehydrogenase, GFAT glutamine-6-phosphate amidotransferase, GlucN6P glucosamine-6-phosphate, GLS glutaminase, GLUT glucose transporter, GS glutamine synthetase, GSH glutathione, hCYS homocysteine, HMG-CoA hydroxymethylglutaryl coenzyme A, IDH isocitrate dehydrogenase, LDH lactate dehydrogenase, MCT monocarboxylate transporter, ME malic enzyme, MET methionine, meTHF 5.10-methylene-tetrahydrofolate, mTHF 5-methyltetrahydrofolate, MS methionine synthetase, NAD nicotinamide adenine dinucleotide, NADPH nicotinamide adenine dinucleotide phosphate, NO nitric oxide, ODC ornithine decarboxylase, PFK1 phosphofructokinase-1, PFKFB3 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3, PGK phosphoglycerate kinase, ROS reactive oxygen species, RPI ribose-5-phosphate isomerase, SAH S-adenosylhomocysteine, SAM S-adenosylmethionine, TCA cycle tricarboxylic acid cycle, THF tetrahydrofolate, TKT transketolase, UDP-GlcNAc uridine diphosphate N-acetylglucosamine
Cancer cells also obtain energy from high levels of glutamine to support the proliferation [23]. Glutaminolysis is the main metabolic pathway regulated in mitochondria, through which glutamate is catabolized into alpha-ketoglutarate (α-KG) and glutamate. α-KG is then converted to pyruvate via TCA cycle and then to lactate [24]. Although combined activation of c-Myc and HIF-1 can induce lactate dehydrogenase A (LDHA) and pyruvate dehydrogenase kinase 1 (PDK1) that contribute to glycolysis [25, 26], mTOR-SIRT4-glutamate dehydrogenase (GDH) axis and c-Myc orchestrate glutaminolysis. Moreover, c-Myc can induce glutamine transporters (SLC5A1) and glutaminase 1 (GLS1) [27, 28]. Glutaminolysis enables cancer cells to reduce NADP+ to NADPH which is an electron donor for reductive steps in lipid synthesis and nucleotide metabolism and the maintenance of reduced glutathione (GSH); thus, glutaminolysis is essential for cancer cells to regulate redox state [29].
In the presence of extracellular nutrients and enough oxygen, cancer cells synthesize fatty acids. However, under metabolic stress, cancer cells scavenge extracellular lipids to maintain viability and growth. Mechanistically, hypoxia, oncogenic RAS, and mTORC1 stimulate cancer cells to uptake lysophospholipids and desaturated fatty acids [30,31,32]. Also, fatty acid oxidation is enhanced in cancer cells to enable survival and proliferation [33]. Cancer cells express high levels of monoacylglycerolipase (MAGL), an enzyme that hydrolyze endocannabinoid 2-arachidonoylglycerol (2-AG) and convert monoacylglycerols to free fatty acid and glycerol that are essential for supplying energy for cancer cells [34].
Cancer stem cells (CSCs) are a small group of tumor cells with stem cell characteristics that initiate and maintain tumor growth and correlate with tumor metastasis. CSCs possess unlimited self-renewal, capacity to propagate tumors through asymmetric cell division, and therapeutic resistance [35, 36]. Tumor cells compete with CSCs for the space and energy generating macromolecules in the TME to limit CSC division and to drive tumor dormancy. The death of tumor cells therefore will relieve the CSCs from such competition and lead to the self-renewal and proliferation of CSCs [37]. Accumulating evidence indicates that the metabolic reprogramming is essential for CSCs to maintain stemness [38, 39]. Activated mitochondrial metabolism by genotoxic stress or hypoxia can lead to increased ROS and prostaglandin E2 that awaken dominant G0-phased CSCs to proliferative state [40]. The monocarboxylate transporter 1 (MCT1) is highly expressed in cancer stem-like cells to promote lactate uptake for the self-renewal and invasion [19, 20]. A recent study showed that CSCs express CD36, a scavenger receptor, which uptake oxidative low-density lipoproteins (ox-LDL) to maintain stemness [41]. Redox stress is a hallmark of cancer tissues that mediates robust metabolism in adjacent MCT1-positive proliferating CSCs which utilize lactate derived from glycolytic cancer cells to fuel mitochondrial metabolism. According to the distinct metabolic changes, cancer cells and CSCs dynamically regulate tumor progression.
5.3 Metabolism of Immune Cells in TME
The immune cells infiltrated in TME include cell subsets belong to both innate and adaptive immune systems. The current findings suggest that metabolic reprogramming is a common feature of both cancer cells and immune cells in the TME. The adjustment of the metabolic program regulates the differentiation and functions of tumor-associated immune cells and thus the progression of tumors [9, 10, 12]. Understanding how the immune metabolism changes and how the altered metabolism regulates immune cells in TME is of great interest and vital for developing effective therapies to treat cancer.
5.3.1 Macrophages
Macrophages have both anti- and pro-tumor functions by regulating tumor growth, angiogenesis, invasion, and metastasis [42, 43]. Based on the phenotypes, tumor-associated macrophages were divided arguably into two types, M1 and M2. M1 cells can be induced by interferon (IFN) γ and lipopolysaccharide (LPS), secrete pro-inflammatory cytokines including TNFα and IL-12, and express high levels of MHC molecules and nitric oxide synthase (NOS). M1 cells play crucial roles in pathogen clearance and tumor antigen presentation. M2 cells can be induced by IL-4 and IL-10, express moderate levels of MHC molecules and IL-12, but produce abundant anti-inflammatory cytokine such as IL-10, mannose receptor, and arginases to promote immunosuppression, tumor cell extravasations, and metastasis [44]. Most tumor-promoting tumor-associating macrophages (TAMs) are M2 type, while both M1 and M2 cells do coexist in the TME. The function of TAMs appears plastic as they have been found to display an inflammatory phenotype in the early phase of tumor initiation but exhibit immunosuppressive characteristics during tumor progression and metastasis [42]. These observations are consistent with the immunoediting theory that TME is a dynamically changing system.
Distinct polarization leads to different metabolic modes of macrophages (Fig. 5.2). M1 macrophages undergo glycolysis, while M2 macrophages exhibit increased oxygen consumption rate and show increased OXPHOS and decreased lactate release [45, 46]. TME is heterogeneous and dynamic during carcinogenesis. The glucose metabolism of TAMs varies in distinct phases. Indeed, at the early inflammatory stage of cancer initiation, TAMs favor glycolysis; in contrast, TAMs show OXPHOS at the later stage of tumor progression [47, 48]. This shift is mediated by cytokines and lactic acid. The lactate released from tumor cells promotes hypoxia and induces TAMs by enhancing the expression of arginase 1 (ARG1) to catalyze the metabolism of arginine to ornithine and polymines to promote collagen synthesis and tumor growth [49]. The activation of PI3K-Akt pathway may also contribute to the glucose uptake and glycolysis since it upregulates the expression of glucose transporters (e.g., GLUT1) and key enzymes (e.g., hexokinase and phosphofructokinase-1); promotes acetyl-CoA synthesis to link the metabolism of fatty acids, glucose, cholesterol, and amino acids; and facilitates the diversion of citrate from TCA cycle to acetyl-CoA by phosphorylating and activating ATP citrate lyase [46, 50, 51]. The switch to glycolysis in TAMs is controlled by Akt-mTOR-HIF-1 axis, resulting in the abundant TCA cycle intermediates and succinate accumulation [52, 53]. Factors in TME such as HMGB1 and DAMPs can stimulate Toll-like receptors (TLRs) that activate PI3K-Akt in myeloid cells like TAMs resulting in glycolysis and enhanced inflammation in TME [54]. PI3K-Akt activation in myeloid cells is also involved in resistance to anti-angiogenic therapy [55]. The activation of mTOR, a downstream molecule of PI3K-Akt, counterintuitively promotes M2 cells [52]. Of interest, this activation feedback negatively regulates PI3K [56]. The activation of c-Myc skews macrophage to M2 polarization and promotes tumor-promoting function of TAMs by increasing CCL18, TGFβ, VEGF, and MMPs [57]. However, whether and how c-Myc controls the metabolic reprogramming of TAMs remains unclear.

Metabolic reprogramming of TAMs. An overview of the key metabolisms in M1 and M2 macrophages and their roles in tumor initiation and progression. During tumor initiation, macrophages are in M1 phenotype and metabolize through a glycolytic shift, HIF-1 activation, and impaired OXPHOS to mediate the expression of NO, ROI, IL-1β, and TNF, to support genetic instability and cancer-related inflammation that leads to tumorigenesis. HIF-1 also enhances angiogenic molecule VEGF-A. In TAMs (M2 macrophages) in the tumor progression stage, AMPK is activated via nutrient deprivation, Th2-derived IL-4, lactate accumulation, and activated PKM2 suppresses glycolysis while upregulating OXPHOS. This induces immunosuppressive macrophages that promote tumor growth. Amino acid, iron, and fat metabolism that contribute to this process are also shown
Moreover, inflammatory TAMs also express PKM2 which in its inactive dimeric form binds to HIF-1 to promote glycolysis in M1 macrophages, while in its active tetrameric form switches macrophages into M2 phenotype [58]. TAMs enhance cancer-related inflammation via HIF-1 which transcribes several key enzymes in glucose metabolism, such as GLUT1, HK2, and PGK1 [59, 60]. HIF-1 can also induce ROS and RNI production which contributes to the genetic mutation and transformation [61]. Moreover, hypoxic TME-increased glycolysis leads to lactic acid accumulation in the TME and skews TAMs to an M2 phenotype with high expression of ARG1, VEGF-A, Tie-2, and IL-10. These immunosuppressive and pro-angiogenic factors promote TAM-related tumorigenesis [49, 62, 63]. The distinct modes of arginine metabolism in TAMs lead to different functions, with M1 macrophages producing NO showing antitumor effect, while polyamine-producing M2 macrophages induce cancer cell proliferation, remodeling, and growth [64,65,66]. TAMs that express ARG1 also contribute to T cell immunosuppression via inducing T cell apoptosis [67].
The lipid metabolism in TME macrophages is altered in response to a variety of stimuli. LPS and IFN-γ, the M1 macrophage inducers, suppress fatty acid intake and oxidation, while M2 macrophages are prone to increase fatty acid oxidation (FAO) [68]. The uptake of lipids, especially triacylglycerol (TAG), is also critical for FAO and M2 activation [69]. The underlying mechanism involves peroxisome proliferator-activated receptors (PPARs), liver X receptors (LXRs), and signal transducer and activator of transcription (STAT) [68, 70,71,72]. PPARs and LXRs are nuclear receptors activated by lipids, such as free fatty acids, eicosanoids, and cholesterol metabolites. PPARγ mediates M2 macrophage polarization to promote tumor progression and metastasis. PPARδ activation in macrophages is triggered by the clearance of apoptotic cells. The fatty acid synthase and PPAR activation are induced in TAMs to contribute to tumor growth. The phagocytosis of apoptotic tumor cells containing oxysterols activates LXRs in macrophages, leading to an M2 immunosuppressive phenotype.
Arachidonic acid metabolism also mediates the switch of macrophage phenotypes. For example, M1 stimulation leads to increased prostaglandin E2, a cyclooxygenase (COX)-derived eicosanoid, while IL-4 induces the upregulation of 15-lipoxygenase (15-LOX) in macrophages [73, 74]. The alveolar macrophages express high levels of COX-1 and 5-LOX, but TAMs express high levels of COX-2 [72]. Anti-inflammatory factors including IL-10, IL-4, and TGFβ induce AMPK activation which drive TAMs to an immunosuppressive M2 phenotype and induce OXPHOS [75]. These findings indicate distinct metabolic modes in TAMs mediate both anti- and pro-tumor responses.
Glutamine metabolism is another important pathway for the differentiation and functions of TAMs, and macrophages express high levels of glutaminase. Glutamine is required for macrophage phagocytosis and antigen presentation. Also, key enzymes in glutamine metabolism, such as AKG, GPT2, GLUL, and GATM, are enhanced in M2 macrophages [49, 76]. However, the mechanisms by which glutamine metabolism regulates TAMs remain unknown. In addition, M2 macrophages can generate indoleamine-2, 3-dioxygenase (IDO), an enzyme degrading tryptophan [77]. Since IDO upregulates regulatory T cells via tryptophan catabolite, TAMs promotes Treg cell generation to inhibit T cell function and establish an immune-tolerant microenvironment [78].
Polarized macrophages show altered iron metabolism. M1 macrophages express high level of H-ferritin, a protein for iron storage, but M2 macrophages express increased ferroportin, the iron exporter. Thus, M1 macrophages favor iron sequestration and inhibit tumor growth, while M2 macrophages exhibit enhanced iron release which promotes tumor progression [79, 80]. Hemeoxygenase-1 (HO-1), an iron-releasing enzyme metabolizing heme to carbon monoxide (CO), biliverdin, and ferrous iron, is inhibited in M2 but not in M1 macrophages [81]. Importantly, iron is also involved in regulating HIF-1 stability by activating prolyl hydroxylases (PHDs) in TAMs [82, 83]. The evidence suggests the iron metabolism is also crucial for TAM-mediated regulation of carcinogenesis.
5.3.2 Neutrophils
Neutrophils, also known as polymorphonuclear cells (PMNs), account for 50–60 % of the peripheral blood leukocytes, with a potent phagocytic function. Neutrophil to lymphocyte ratio is a risk predictive index for tumor recurrence [84]. In the TME, neutrophils also have two biological phenotypes: antitumor N1 exerts tumor cytotoxicity to reject tumor by enhancing antitumor immune memory; pro-tumor N2 plays the opposing roles, i.e., enhancing tumor growth, invasion, and metastasis, promoting tumor angiogenesis, mediating immunosuppression, and producing enzymes to damage normal tissue cells to facilitate tumor growth, invasion, and metastasis. Such dichotomy of neutrophils is similar to that of macrophages [85, 86]. Tumor-derived TGFβ can switch N1 neutrophils to N2 which blunts CD8+ T cell responses to promote tumor growth [87]. Similar to macrophages, tumor-associated neutrophils (TANs) can produce several factors such as ARG1, ROS, MMPs, IL-6, and IL-1β to promote cancer progression, angiogenesis, and metastasis [85, 86].
The metabolic reprogramming, aerobic glycolysis and pentose phosphate pathway (PPP), controls the functions of neutrophils [88, 89]. Very few mitochondria are in neutrophils; OXPHOS and ATP production are ineffective in these cells. Neutrophils rely on PPP to produce NADPH that is essential for maintaining redox balance and cell survival [90]. The chemotaxis, calcium mobilization, and oxidative burst are driven by glycolysis since G6P deficiency blunts these functions of neutrophils [91]. Glycolysis and PPP are also involved in the formation of neutrophil extracellular traps (NETs), a mixture of DNA, histones, and antimicrobial peptides that traps and kills bacteria [90, 92]. NETs segregate circulating tumor cells and accumulate in the vasculature to promote the inflammatory adherence, contributing cancer-induced organ failure and metastasis [93]. Moreover, neutrophil-derived leukotrienes contribute to the colonization of distant tissues via selectively expanding the sub-pool of cancer cells to retain high tumorigenic potential. Knocking down of 5-lipoxygenase (5-LOX), the key enzyme for leukotriene synthesis, blunts the pro-metastatic activity of neutrophils [94]. Together, these observations suggest that metabolic change regulates the TAN functions and tumor development. Whether and how the lipid and amino acid metabolism are reprogrammed and how these metabolic changes regulate neutrophil functions await further investigation.
5.3.3 Basophils and Eosinophils
Basophils, a cell population derived from myeloid cells, respond to IgE-dependent and IgE-independent stimuli and crosstalk with other immune cells such as lymphocytes, macrophages, and DCs [95]. These cells are involved in Th2 responses through producing cytokines including IL-4, IL-13, and IL-25 and also contribute to immunoglobulin synthesis, tumor angiogenesis, and hematopoiesis by secreting IL-6, GM-CSF, and VEGF and arguably present antigens to T cells. Hence, basophils regulate both innate and adaptive immunity. However, the metabolic reprogramming in tumor-associated basophils remains unknown.
Eosinophils are granulocytic leukocytes derived from hematopoietic progenitors. They interact with both innate and adaptive immune cells. IL-3, IL-5, and GM-CSF are crucial for eosinophil development, while CCL11, CCL24, and CCL26 contribute to eosinophil chemotaxis [96, 97]. Eosinophils express MHC-II and costimulatory molecules such as CD40 and CD80/86 to promote T cell activation and proliferation [98]. Eosinophils also produce IDO and TGFβ to mediate Treg and Th2 polarization [99]. Interestingly, the infiltration of eosinophils in TME associates with improved prognosis in various types of solid tumors [96] but with poor outcome in Hodgkin lymphoma [100]. The antitumor activity of eosinophils is through their degranulation in the tumor. Consistently, eosinophils with CCL11 deficiency exhibit impaired antitumor potential [101]. Also, the necrosis and chemokines in cancer tissues induce the differentiation and migration of eosinophils [102]. Systemic IL-2 and IL-25 therapy promotes eosinophil degranulation [103, 104]. The recruitment of eosinophils in TME is mainly mediated by high-mobility group box 1 (HMGB1), a factor in damage-associated molecular patterns, which elicits eosinophil degranulation by binding to the receptor for advanced glycation end products (RAGE) [105]. Some receptors normally expressed in NK cells such as NKG2D and 2B4 are also expressed in eosinophils to mediate tumor cytotoxicity [106, 107]. These observations suggest that reprogramming the metabolism in eosinophils may trigger the degranulation and improve antitumor immunity.
5.3.4 Mast Cells
Mast cells are derived from bone marrow hematopoietic stem cell-differentiated precursor cells. These precursor cells enter the cavity or mucosa from blood and then mature. As an important class of innate immune cells, mast cells play a key role in allergic diseases such as asthma. After activation, mast cells not only produce and release a variety of cytokines and chemokines, including histamine, serotonin, interleukins, leukotrienes, prostaglandins, and proteases to promote inflammation, but regulate the functional activity of DC, T cells, CD4+ CD25+ regulatory T cells, B cells, and other immune cells in TME [108]. Similar to that in TAMs, PI3K-Akt activation in mast cells correlates with enhanced glycolysis [109]. The metabolic reprogramming of the tumor-associated mast cells is yet unknown.
5.3.5 CAFs
Fibroblasts are a large proportion of cells in the TME. These cells synthesize collagen, laminin, fibronectin, and other matrix components to establish a structural framework in the matrix. Fibroblasts can be activated during wound healing, inflammation, and stress. The TME-activated fibroblasts called cancer-associated fibroblasts (CAF) or myofibroblasts specifically express α-smooth muscle actin (αSMA) [110]. Apart from the local fibroblasts, CAFs can be derived from vascular smooth muscle cells, pericytes, marrow-derived mesenchymal cells, and through epithelial-mesenchymal transformation (EMT) [111]. CAF can reshape the extracellular matrix by secreting matrix-degrading enzymes, particularly metalloproteinases and derivatives to contribute to tumor drug resistance [112]. CAFs promote tumor growth, angiogenesis, and metastasis by secreting several factors, including insulin-like growth factor, hepatocyte growth factor, basic fibroblast growth factor, Wnt ligands and MMPs, as well as cytokines and chemokines such as CCL7, CXCL12, and VEGF-A [110, 111].
It has been observed that CAFs engage in aerobic glycolysis. Mechanistically, IDH3α reduced by TGFβ or platelet-derived growth factor (PDGF) breaks the equilibrium between α-KG and fumarate/succinate that are allosteric regulators of PHDs, thereby in turn increasing HIF-1 transactivation and enhancing glycolysis [113]. CAFs also display increased glutamine metabolism and decreased OXPHOS [114, 115]. They provide lactic acid, amino acids, and ketone bodies to cancer cells, while cancer cells produce ROS to activate HIF-1 in CAFs to maintain the glycolysis [110, 111]. The metabolic change of CAFs impacts the secretion of cytokines and chemokines. For example, α-KG and fumarate/succinate are allosteric regulators of lysine-specific demethylase, PHDs, and methylcytosine demethylase that are epigenetic regulators [116]. Also, glycolysis and glutamine metabolism in CAFs are regulated by p62-mTORC1-c-Myc pathway that promotes ROS and IL-6 production and enhance tumor progression [114, 117].
Distinct from cancer cells, although CAFs favor glycolysis, the proliferation is much slower when compared with normal fibroblasts, suggesting that the biosynthesis of CAFs is not dependent on glycolysis [118]. However, cancer cells uptake the CAF-secreted lactate for tumor anabolic metabolism, growth, and metastasis [117]. GLUT4 is overexpressed on CAFs to release lactic acid, while GLUT1 is upregulated in cancer cells to import glucose and metabolites. The lactate released from CAFs further acidifies the TME to facilitate tumor progression and drug resistance [119]. Hence, CAFs not only secrete growth factors but also fuel cancer cells by providing lactate and other glucose metabolites. The cooperation of metabolites shuttling between CAFs and cancer cells aggregates the TME that facilitate tumor development.
5.3.6 NK and NKT Cells
Natural killer cells (NK cells) are a subtype of lymphocytes in innate immune system. They express Ly49, NCR, and CD16 and play antitumor action by secreting IFN-γ and cytotoxic molecules such as perforin and granzyme or by antibody-dependent cell-mediated cytotoxicity (ADCC) and T cell activation [120, 121]. Under resting state and short-term activation, they favor OXPHOS, while upon prolonged activation by high-dose IL-15, NK cells switch to glycolysis [122]. In the TME, both IL-15 and hypoxia can lead to enhanced glycolysis in NK cells [123]. However, the mechanism by which TME affects the metabolic reprogramming and how the altered metabolism modulates the activity of NK cells remain to be elucidated.
NKT cells are a heterogeneous group of T cells that share properties of both NK cells and T cells. They recognize non-polymorphic CD1d (an APC molecule) that binds self and foreign lipids and glycolipids [124]. Upon activation, these cells produce abundant IL-2, IL-4, IFN-γ, GM-CSF, and IL-21 so that they play antitumor functions in TME [125]. However, the metabolic reprogramming in tumor-associated NKT cells is yet unknown.
5.3.7 Endothelial Cells
In the TME, vasculatures deliver nutrients and oxygen to the tumor, which is the basis of tumor survival and development. Tumor cells release pro-angiogenic signals to drive the metabolic reprogramming of endothelial cells (ECs) [126]. Inhibition of VEGF signaling is a clinically approved strategy, although the benefits are limited since tumors acquire drug resistance within months after treatment [127].
It has been found that the structure and function of vessels in TME and in normal tissues are significantly different. They are dilated, tortuous, and hyperpermeable. The ECs are poorly connected and lack a regular pattern. The basement membrane has nonuniform thickness and composition [128]. These cause deprived oxygen and nutrients. Hypoxia switches cancer cell metabolism away from OXPHOS to glycolysis, from glucose to glutamine as the major substrate for fatty acid synthesis (FAS). Tumor-associated ECs resemble cancer cells to undergo a shifting from quiescence to rapid growth during vessel sprouting. These ECs are highly plastic [126, 129]. They require a baseline glycolysis flux to function as an endothelium and maintain vascular barrier homeostasis. ECs preserve high concentration of oxygen in the blood. Also, ECs protect themselves from oxidative stress using glycolysis. They can also move from normoxic to hypoxic areas [130]. Glycolysis can produce ATP faster than OXPHOS. Hence, similar to cancer cells, glycolysis contributes to vascular sprouting and the survival and proliferation of ECs (Table 5.1) [131, 132]. Lactate dehydrogenase B, GLUT1, and glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) are increased in tumor-associated ECs. PFKFB3 promotes the synthesis of fructose-2,6-bisphosphate(F2,6P2) and the activation of 6-phosphofructo-1-kinase (PFK1) which converts fructose-6-phosphate (F6P) to fructose-1,6-bisphosphate (F1,6P2) [133]. Indeed, interfering with glycolysis by inhibiting PFKFB3 (by 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO)) or other key enzymes blunts angiogenesis-associated tumor growth [134]. However, the systemic complete and permanent inhibition of glycolysis may also induce undesired effects. Fortunately, Partial and transient reduction of glycolysis renders ECs more quiescent without overt detrimental side effects [134]. Glycolytic metabolites such as lactate can be uptaken by ECs through MCT1. Instead of being metabolized, lactate induces HIF-1 activation and competes with α-KG to bind to prolyl hydroxylase 2 (PHD2), thereby enhancing the expression of angiogenesis-related genes [135, 136]. Therefore, the partial and transient reduction of glycolysis may be sufficient to inhibit pathological angiogenesis in the TME. These results indicate that targeting glycolysis in ECs inhibits angiogenesis, but the viability should be concerned.
The decreased supply of glucose in ECs can also be compensated by glycogenolytic production of glucose-1-phosphate (G1P), which can be converted into glucose-6-phosphate (G6P). The glycogenolysis-derived G6P only minimally contributes to energy production in normal or low glucose conditions. G6P catabolism might be important in the oxidative PPP (oxPPP) process to generate energy [137, 138]. PPP also produce ribose-5-phosphate (Rb-5-P) which can feedback into glycolysis through the biogenesis of F6P [139, 140]. Moreover, NADPH produced by PPP protects ECs against ROS, enhances NO synthesis, and contributes to ATP production [141]. Therefore, PPP may promote angiogenesis and regulates redox homeostasis.
ECs have a high level of glutaminolysis to support ATP synthesis and fuel cell proliferation in the conditions of decreased glucose supply (Table 5.1) [142]. ECs can uptake glutamine from extracellular milieu and also produce glutamine from glutamate. Inhibition of glutaminolysis induces EC senescence [143]. Glutamine metabolism also promotes ornithine synthesis, a precursor of mitogenic polyamines [144]. In addition, glutamine metabolite glucosamine can inhibit oxPPP and NO production by reducing NADPH [145]. How glutaminolysis mediate angiogenesis has not been described.
Most ECs contain mitochondria that compose less than 5 % of the cell volume. The mitochondria-derived ROS activates HIFs by inhibiting PHDs, which in turn enhances glycolytic metabolism and angiogenesis [146]. Glycolysis-derived ATP also in part contributes to maintaining EC mitochondrial network. However, the role of mitochondria in tumorigenesis is yet unknown. FAO is induced in ECs upon glucose deprivation, in which process AMPK is activated [147]. Whether and how FAO influences angiogenesis is also unclear.
5.3.8 Dendritic Cells
DCs are divided into immature (imDCs), semi-mature (smDCs), and mature (mDCs). imDCs show low expression of MHC class I molecules, lack of B7 costimulatory molecules, etc. These cells induce immune tolerance since they cannot effectively activate T cells [148]. Recent studies showed that semi-mature and mDCs can also induce immune tolerance. smDCs have a unique feature which can be induced by granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-4, and TNF with bone marrow cells in vitro. smDCs obtain a significantly different molecular phenotype, as compared with mDCs and imDCs, showing high expression of MHC class I molecules and moderate expression of costimulatory molecules, but they do not secrete inflammatory cytokines such as IL-1, IL-6, and IL-12 [149, 150]. These cells produce inflammation-inhibiting factors including IL-10 and can enhance the activation of CD4+CD25+ Tregs [151].
As a major population of professional antigen-presenting cells, DCs are activated and mature by sensing pathogen-associated or damage-related stimuli, displaying upregulation of MHC molecules and costimulatory molecules (e.g., CD80/86, CD40), cytokines (e.g., IL-12), and chemokine receptors (e.g., CCR7). Then DCs migrate to lymphoid organs to present antigens and activate T cells. However, tumor-associated DCs are immunosuppressive [152,153,154]. Moreover, increased imDC numbers and decreased mDC and DC numbers with impaired functions were observed in cancer microenvironment [149, 152].
The differentiation and function of DCs also rely on the metabolic reprogramming. Under resting state, DCs favor OXPHOS, while they switch to glycolysis after activation [155]. Also, glycolysis is essential for upregulated costimulatory molecules (CD80/86, CD40) and cytokines (IL-12) and DC survival [50]. Indeed, the activated DCs show increased NO production, enhanced PI3K-Akt activation, and impaired OXPHOS [50, 155].
In the hypoxic TME, HIF-1 drives the transcription of mTOR and glycolysis-associated genes, thereby promoting DC glycolysis, maturation, and activation [156]. However, hypoxia also inhibits the recruitment of monocyte-derived DCs from peripheral blood [157]. Hypoxia upregulates adenosine receptor (A2b), and adenosine can bind this receptor to blunt DC differentiation and switch them to a Th2-promoting profile, i.e., express IL-10, TGFβ, and COX-2 that lead to abnormal differentiation of myeloid cells thereby causing Treg activation and DC defection [158]. Mechanistically, adenosine can induce AMP-activated protein kinase (AMPK) that promotes OXPHOS and inhibits glycolysis [50]. The lactic acid also contributes to inhibit DCs by inhibiting glycolysis [159]. These impaired DCs also secrete VEGF-A to promote angiogenesis and tumor growth [160].
PI3K-Akt activation also contributes to enhanced expression of GLUT1, the glucose transporter key for glycolysis, and disabled-2 adaptor (DAB2) on DCs which suppresses T cell response against tumors [161]. The mTOR inhibition by rapamycin in BMDCs promotes their antitumor activity, while mTOR inhibition in human monocyte-derived and plasmacytoid DCs blunts the immunostimulatory actions and T cell response [155].
Similar to macrophages, tumor-associated DCs (TADCs) can express enhanced ARG1, IDO, and iNOS that deplete arginine and tryptophan in the TME so that suppression of CD8+ T cell function and survival is mediated [162]. Moreover, the retinoic acid metabolism in TADCs can promote Treg-mediated immunosuppression [163]. These amino acid metabolism pathways suggest potential targets for cancer immunotherapy. Recently, it was found that vaccines can trigger GCN2, a nutrient sensor, in DCs that in turn activate the antigen-presenting function of DCs and CTL response [164].
TADCs are also regulated by lipid metabolism. During DC activation, the FAS can promote the antigen presentation by increasing ER and Golgi expansion [165, 166]. The scavenging receptors such as CD36 and MSR1 upregulated in TADCs contribute to the uptake of lipids [167]. The lipid accumulation subsequently attenuates DC function and T cell activation. Hence, it has been proposed that the switch of glucose metabolism from glycolysis to OXPHOS in the TADCs may turn on the FAS and lipid uptake, therefore impairing DCs in TME and inducing an immunotolerogenic condition [155, 165, 168]. Furthermore, the limited nutrients in TME can induce ER stress that will induce excessive lipid accumulation via TCA cycle in TADCs and further inhibit CTL priming [169].
Abovementioned finding suggests that the altered metabolic programming in TADCs impairs their activation and antitumor function. Rectifying the abnormal metabolism may restore DCs’ ability to reject tumor in the TME.
5.3.9 Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs, originally discovered in the 1970s, are a group of myeloid immature cells in tumor-bearing mice [170, 171]. The morphology of these cells is similar to the granulocyte-monocyte progenitor cells that are yet to differentiate into macrophages, DCs, or granulocytes. These cells are CD11b+Gr1+ and can be subcategorized into monocytic (Ly6ChighLy6G−) and granulocytic (Ly6ClowLy6G+) based on the expression of Ly6C and Ly6G. In humans, MDSCs also express hematopoietic stem cell surface markers CD34 and immature marker CD31, hardly expressing MHC class I molecules and maturation markers [172, 173]. MDSCs can secrete MMP9 and undergo endothelialization to induce angiogenesis and promote tumor survival and invasion [174]. With the development and progression of tumor, large amounts of cytokines, such as VEGF, IL-10, and TGFβ, can be released in the TME. These cytokines not only recruit MDSCs to tumor but also promote their differentiation into immunosuppressive cells [175]. On the other hand, MDSCs are immunosuppressive through multiple means: MDSCs can suppress the maturation and antigen presentation function of DCs directly or indirectly [176]. By direct interaction, MDSCs suppress IFN-γ production by CD8 T cells [177]. MDSCs secrete TGFβ, arginine enzymes, and reactive oxygen and nitrogen species to inhibit T cell activation and proliferation [177] and to promote Treg cell generation [178].
Glycolysis, glutaminolysis, and TCA cycle metabolism and arginine metabolism are upregulated during MDSC maturation. Fatty acid uptake and FAO mediate the immunosuppressive function of MDSCs in TME [179]. Amino acid metabolism and oxidative stress also mediate MDSC immunosuppressive functions. By expressing IDO, MDSCs metabolize tryptophan to kynurenine, induce Treg expansion, and inhibit T cell functions [180]. L-arginine and L-cysteine can be depleted by MDSCs, leading to the downregulation of CD3ζ and the inhibition of T cell activation [181, 182]. MDSCs express high levels of ARG and iNOS. ROS such as peroxynitrites are produced under the conditions of limited L-arginine availability. ROS induces T cell apoptosis by nitrotyrosylating and preventing tyrosine phosphorylation of key signaling proteins for T cell activation [183]. Peroxynitrites also nitrate TCR, IL-2R, and CD8 molecules, leading to T cell signaling disruption [184, 185]. Recent studies indicated that a lipid mediator derived from arachidonic acid via cyclooxygenase 2 (COX-2), i.e., prostaglandin E2 (PGE2), can be secreted by tumor cells and MDSCs [186, 187]. PGE2 enhances MDSC development by inducing ARG, iNOS, and IDO and promotes MDSC recruitment to the TME by inducing CXCL12 [188]. Taken together, the metabolic reprogramming of MDSCs provided a potential target for regulating the immunosuppressive network in the TME.
5.3.10 T Cells
The infiltration and activation of T cells in TME control tumor progression. CD8+ T cells are the major effector cells in tumor immunity. Upon activation by APCs, CD8+ T cells migrate to the tumor tissue to kill target cells through perforin (to damage cell membranes), granzymes (to enter target cells and degrade DNA), and FasL. In addition, CD8 T cells secrete cytokines such as IFN-γ and TNFα to promote antitumor immune response [189]. CD4+ T cells differentiate into distinct subtypes to promote or repress tumorigenesis. CD4+ Th1 T cells produce IFN-γ to promote tumor immune rejection. Th1 cell infiltration in TME is associated with good clinical prognosis. In contrast, Th2 cells and Tregs temper tumor rejection and facilitate tumor immune escape. Th17 cells may both promote and inhibit tumor progression in a tumor-type and stage-dependent manner [190].
For activated T cells to proliferate and release cytotoxic factors and cytokines, they switch the metabolism to aerobic glycolysis to increase the uptake of glucose and glutamine [191, 192]. However, the activation of PD-1 can inhibit the uptake and utilization of these nutrients and promote FAO, thereby inducing T cell anergy, exhaustion, and autophagy [193,194,195]. Since H+ secretion increases when tumor cells release lactic acid, TME becomes acidic, leading to reduced T cell function [196].
In the TME, tumor cells compete for nutrients to hinder antitumor functions of T cells and lead to the metabolic reprogramming of T cells (Table 5.2). The efficiency in tumor cell uptake of glucose is ten times of that in activated T cells. Because glucose is the sole source of the energy required for effector T cells [192]. The lack of glucose severely affects IFN-γ production and the cytolytic activity of CTLs. Moreover, the accumulation of metabolic wastes in TME, such as lactate and kynurenine, can also inhibit T cell function [197]. Acidification and hypoxia of TME also impairs the proliferation and function of CTL [192, 196]. De novo FAS is critical for the development of effector T cells, while the generation and survival of memory T cells need FAO and OXPHOS [198].
Naive T cells get energy from OXPHOS, fatty acid oxidation (FAO), and low levels of glutaminolysis, whereas these cells need much more nutrient for activation [191, 192, 198]. Therefore, activated T cells show enhanced glycolysis, PPP, and glutaminolysis and decreased FAO (Table 5.2). Glycolysis is required for the functions of effector T cells, while PPP and glutamine metabolism are involved in biosynthesis. Upon activation, signaling pathways involving PI3K-Akt, mTOR, HIF-1, and c-Myc are triggered in CD8+, Th1, Th2, and Th17 cells to promote the expression of key factors in nutrient metabolism, such as GLUT1, PDK1, and HK2 [191,192,193, 198]. This in turn leads to further enhanced glycolysis and glutaminolysis. In addition, the activated T cells exhibit a noncanonical Myc-dependent transcriptome coupling glycolysis and glutaminolysis to polyamine biosynthesis to maintain T cell proliferation [198, 199]. The mTOR activation regulates the balance between effector and memory T cells by modulating T-bet, a key transcription factor for Th1 cell differentiation [200]. The energy sensor AMPK, activated by increased ratio of AMP and ATP, nutrient deprivation, and anti-inflammatory cytokines (e.g., IL-4, IL-10, and TGFβ), suppresses IFN-γ and granzyme B production and induces OXPHOS and glutamine-dependent mitochondrial metabolism in T cells to suppress T cell-mediated antitumor response [201, 202]. Tumor-derived oxysterol induces LXR activation to inhibit neutrophil recruitment and DC migration, switches on M2 polarization, and suppresses T cell response to aid tumor immune tolerance [70].
The metabolism of amino acids, such as arginine, tryptophan, glutamine, and cysteine, is important for TIL functions (Table 5.2). For example, deficiency of arginine impairs protein synthesis in TILs, leading to reduced TIL activation [203, 204]. L-arginine metabolism is dependent on the activities of NOS and ARG. NOS converts arginine to NO and citrulline, and ARG hydrolyzes arginine into urea and ornithine. Administration of ARG and NOS-specific inhibitors can activate TILs [205]. Mechanistically, NO can react with ROS to produce reactive nitrogen species (RNS) such as peroxynitrite which induce lymphocyte anergy and apoptosis by nitration of tyrosine residues or the mitochondrial permeability transition pore (voltage-dependent anion channel) [184, 206]. Moreover, RNS modifies chemokines such as CCL2, by nitration or nitrosylation, to inhibit T cell infiltration into the tumor. Indeed, drugs targeting nitration in TME induce T cell infiltration [207]. Effector CD8+ T cells can also impair the CAF-mediated chemoresistance by inhibiting cysteine and glutathione metabolism in fibroblasts [208], suggesting a novel intersection for combined chemotherapy and immunotherapy in cancer treatment.
As mentioned earlier, tryptophan deprivation in TME contributes to tumor progression. IDO, the rate-limiting enzyme in tryptophan metabolism, inhibits the proliferation of effector T cells by depleting tryptophan in the TME [209, 210]. IDO is mainly expressed in mesenchymal cells, such as ECs, macrophages, and DCs. Tumor cells also express IDO upon IFN stimulation [211, 212]. By metabolizing tryptophan, IDO leads to the release of kynurenine. The reduction of tryptophan and the increase of kynurenine synergistically inhibit the activation and proliferation of antitumor T cells [209]. Therefore, IDO upregulation in cancer patients correlates with impaired T cell accumulation, proliferation, and function and poor prognosis. Agents inhibiting IDO, such as INCB024360 and 1MT, can promote the antitumor T cell function [209, 213].
A recent study revealed a new mechanism that cholesterol metabolism regulates the antitumor responses of CD8+ T cells [214]. Cholesterol is abundant in the plasma membrane, which is key for the TCR clustering and immunological synapse formation. The deficiency of ACAT1, a key cholesterol esterification enzyme, led to potentiated effector function and proliferation of CD8+ T cells. However, ACAT1 knockout in CD8+ T cells could not affect the glycolysis, OXPHOS, and FAO levels. Inhibition of PD-1 did not alter the expressions of ACAT1 and other cholesterol esterification genes. Combination of anti-PD-1 antibody and ACAT1 inhibition synergistically blunted the tumor development.
Tregs are generally divided into natural regulatory T cells (nTregs, CD4+CD25+) and induced regulatory T cells (iTregs, CD4+CD25−), both of them express Foxp3. They express IL-10, TGFβ, and IL-2 receptor α-chain (CD25), but do not produce IL-2. Tregs suppress immune response and T cell activation through cell-cell interaction and cytokines [215, 216]. Tregs are present in various tumors and play an immunosuppressive role. AhR-enhanced IDO1 and kynurenine mediate Treg generation [217], while mTOR activation inhibits Treg development [218]. Thus the elimination of Tregs in the TME, to some extent and excluding in colorectal cancer, inhibits tumor growth. The function of regulatory T cells (Tregs) is not affected by lactic acid and acidic environment. Of interest, Tregs favor OXPHOS, FAO, and activated AMPK for the nutrient metabolism (Table 5.2) [218,219,220]. Hence they survive well in the nutrient-deprived TME. Moreover, the HIF-1 upregulation in the hypoxic TME promotes Treg expansion [221]. Interestingly, HIF-1 shows an important metabolic checkpoint for the differentiation of Tregs or Th17 cells [222].
Th17 cells are a newly discovered class of T helper cell subsets. Naive CD4+ T cells preferentially differentiate to Th17 under the stimulation of TGFβ and IL-6. In addition, IL-23 is a key factor for the maintenance and expansion of Th17 cells [223]. Recent studies found that the presence of Th17 in the TME antagonizes the IFN-γ-producing Th1 cells to favor tumor growth [224]. Th17 cells also rely on glycolysis and FAS for differentiation and activation [225] (Table 5.2).
5.3.11 B Cells
By producing antibodies and immune complexes, B cells can regulate the functions of myeloid cells to promote tumor growth. It has been reported that c-Myc, but not HIF-1, mediates LPS and antigen-stimulated activation of B cells and triggers the glycolysis and mitochondrial metabolic activity. Tumor cells express BAFF which can also induce the glycolysis and antibody production of B cells by a GLUT1-dependent manner [226]. Therefore, metabolic reprogramming of B cells will be also of interest to be investigated to better understand the immunoediting in tumor progression.
5.4 Clinical Diagnostic and Therapeutic Applications
5.4.1 PET/CT and PET/MRI
Cancer cells favor glycolysis to metabolize glucose, regardless of oxygen tension, which is termed as Warburg effect. Anaerobic glycolysis (fermentation) is more rapid but less efficient than OXPHOS to generate ATP. This process produces lactate and contributes to immunosuppression. At present, Warburg effect has offered an opportunity to diagnose and monitor therapy response in many clinical cancers. Position emission tomography-computed tomography/magnetic resonance imaging (PET/CT or PET/MRI) is a clinical imaging technique combining PET and CT/MRI [227,228,229,230]. PET imaging shows the spatial distribution of metabolic activity (especially the Warburg effect), while CT/MRI precisely aligns the anatomic imaging. PET with distinct radiotracers can evaluate altered metabolisms of glucose, fatty acids, amino acids, and other cancer markers. Hence, molecular imaging with PET is very precise for detection and directing therapy and has been applied widely in several cancer types.
5.4.2 PD-L1, PD-1, CTLA-4, and IDO
Recent breakthrough in cancer immunotherapy based on the clinical application of monoclonal antibodies targeting T cell immune checkpoints, including PD-1 and CTLA-4, clearly demonstrates the significance of effector T cell activation in antitumor response [231]. PD-L1:PD-1 and CTLA-4 signaling dampens antitumor responses. The expression of PD-L1, a key immune checkpoint, can be induced by hypoxia in tumor-associated DCs. PD-L1 is a target gene of HIF-1 and NF-κB, two central transcriptional factors in hypoxic responses. PD-L1/PD-1 pathway inhibits glycolysis and promotes FAO and lipolysis to mediate T cell metabolic reprogramming [232,233,234]. Of interest, PD-1 is also expressed in cancer cells such as melanoma cells. Activation of melanoma-PD-1 promotes tumor progression by mTOR pathway [235]. Preclinical data suggested that inhibition of PD-1 and prostaglandin E synthases synergistically promotes tumor eradication [236]. Therefore, it has been proposed to use PD-1 as a radiolabeled PET imaging tracer to efficiently distinguish PD-L1-positive and PD-L1-negative tumors.
CTLA-4 is a target gene of Foxp3 and has a major role in enhancing Treg activity and suppressing T helper cells [237]. IDO activity can be induced by CTLA-4 in plasmacytoid DCs via reversing CD80 signaling [238]. The increase of IDO, PD-L1, and CTLA-4 in the peripheral blood of cancer patients correlates with advanced disease and poor outcome, independent of the stages of cancer [239, 240]. Therefore, combination treatments targeting several of these markers to modulate metabolisms in immune cells may have a synergistic effect.
5.4.3 CAR-T
Cancer immunotherapy based on the adoptive transfer of autologous T cells has shown promising efficacies. Chimeric antigen receptor (CAR)-T cells have been used to exert potent antitumor effect [241, 242]. CARs consist of cytoplasmic domain of the Fc receptor γ chain or CD3ζ modules and that of costimulatory cytoplasmic domains such as CD28, 4-1BB, and ICOS [243]. Little is known about the metabolic reprogramming of CAR-T cells. As described in the above section, naïve and memory T cells rely on fatty acid oxidation, while activated effector T cells shift to glycolysis and enhanced OXPHOS. A recent report showed that CD28 or 4-1BB CD3ζ CAR-T cells exhibited increased survival and proliferation, promoting central memory T cells. CD28-CD3ζ CAR-T cells favored aerobic glycolysis, while 4-1BB-CD3ζ CAR-T cells preferred FAO [243]. The choice of CAR impacted the T cell metabolic reprogramming and differentiation, suggesting that modulating the metabolism in T cells may also be important to enhance the antitumor effects of CAR-T cells.
5.4.4 Drug Repositioning
Drug repositioning, also known as drug repurposing, re-profiling, re-tasking, or therapeutic switching, is the application of known drugs and compounds to new indications. It is an emerging and important application in drug development for cancer therapy [244]. The computational approaches can enhance the efficiency and success rates, particularly in terms of high-throughput shotgun repurposing. For example, proton pump inhibitor (PPI) is an acid-activated drug that inhibits H/K-ATPase to treat gastric cancer. PPI can synergistically modulate the acidic TME with chemotherapy and improve chemoresistance [245, 246]. Terfenadine is a histamine receptor H1 antagonist, which can prevent VEGF secretion from mast cells in hypoxic microenvironment and induce ROS-mediated apoptosis and autophagy of melanoma cells [247]. Simvastatin specifically inhibits HMG-CoA reductase so that it restrains p53 mutation from activating mevalonate pathway for cholesterol synthesis in breast and ovarian cancer cells [248]. System xc− cystine/glutamate antiporter, a heterodimer composed of the 4F2 heavy chain (SLC3A2) and the light chain xCT, is a membrane amino acid transporter that mediates the exchange of extracellular cystine and intracellular glutamate [249]. Sulfasalazine is a specific inhibitor of xCT cystine transporter. It blocks the reduced GSH synthesis, leading to oxidative stress in cancer cells, resulting in the suppression of NSCLC and gastric tumor progression and breast cancer metastasis [249,250,251]. Metformin is an oral drug used to treat type 2 diabetes mellitus by suppressing glucose production by the liver [252]. Recently, metformin was reported to inhibit tumor progression and ameliorate the prognosis [253,254,255]. By inhibiting ATP-binding cassette subfamily G member 1 (ABCG2) and ectonucleotide pyrophosphatase/phosphodiesterase family member 1 (ENPP1), metformin suppresses cancer cell chemoresistance to drugs [256]. These drugs are being applied in clinical trials and show promising results.
5.4.5 Metabolism-Based Antiangiogenic Therapy
The current paradigm for anti-angiogenic therapy is to block VEGF and VEGFRs [257]. However, the tumor cells can rely on other signaling pathways for pro-angiogenesis. The hypoxic conditions caused by treatment often lead to the outgrowth of resistant tumor clones [258]. The EC metabolism requirement potentially provides novel anti-angiogenesis therapeutic opportunities. Silencing PFKFB3 by 3PO is capable to reduce EC glycolysis and vessel sprouting, without switching to aerobic respiration [134]. More importantly, the effect of 3PO on glycolysis is reversible, as normal sprouting was recovered after administration for 6 h in vivo The moderate reduction in glycolysis by 3PO is sufficient for increasing the fraction of quiescent ECs and reducing EC proliferation and migration [132, 133]. Combination of 3PO and VEGFR tyrosine kinase inhibitor SU5416 significantly impaired angiogenesis, as compared with the optimal doses of any one of these inhibitors alone [134]. In addition, 3PO is a chemotherapeutic agent to block tumor proliferation [259]. Together these findings indicate that targeting tumor-associated EC metabolism is a potential therapeutic strategy.
5.5 Concluding Remarks
The metabolism of cancer cells and immune cells in TME is instrumental for tumor initiation, progression, and metastasis. The deprivation of nutrients from the environment suppresses antitumor immune cells, such as CD8+ T cells, M1 macrophages, and N1 neutrophils, promoting the differentiation and activation of pro-tumor immune cells, including MDSCs, M2 macrophages, and Tregs [260, 261]. The extent to which metabolism pathways represent true vulnerabilities for tumor development remains unclear. Targeting glycolysis, glutaminolysis, and FAO has provided clinical benefits; the strategies integrating redox homeostasis and PPP may also generate new opportunities [9, 10, 119].
The most serious challenge in reshaping the immune profiles in TME is to understand the metabolic heterogeneity which is extremely complex depending not only on tumor and immune cell types but also on tumor stages and etiology (Fig. 5.3). Activation of signaling pathways including PI3K-Akt, mTOR, HIF-1, c-Myc, etc. in tumor-associated immune cells regulates their metabolism for survival, differentiation, and pro- or antitumor functions [10]. For example, HIF-1 activation in cancer cells and immune cells may upregulate glycolytic metabolism and enhance cancer-related inflammation during the initiation. In the tumor progression stage, HIF-1 elevation in TAMs, TADCs, MDSCs, and Tregs contributes to immunosuppression and angiogenesis by PD-L1 expression, lactate release, and adenosine-adenosine receptor interaction that facilitate tumor growth [32, 46, 58, 60, 136, 221, 232, 233]. Other factors and molecules, such as noncoding RNAs, complements, and coagulation-related factors, also regulate immune cell survival, differentiation, and functions in TME [262,263,264]. However, whether and how these molecules regulate metabolic reprogramming in tumor-associated immune cells will be of interest to be investigated.

Metabolic crosstalk between cancer cells and TME-immune cells
The metabolite exchange adds an essential dimension of heterogeneity in the TME to contribute to tumor growth, metastasis and clinical resistance [12, 260, 261, 265]. Nonetheless, we posit that targeting the immune cell metabolism in TME in addition to the traditional cancer therapies will lead to more precise and efficient diagnosis and treatment of these fatal diseases.
References
Lord EM, Penney DP, Sutherland RM, Cooper RA Jr (1979) Morphological and functional characteristics of cells infiltrating and destroying tumor multicellular spheroids in vivo. Virchows Arch B Cell Pathol Incl Mol Pathol 31:103–116
Shearer WT, Fink MP (1977) Immune surveillance system: its failure and activation. Prog Hematol 10:247–310
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360
Teng MW, Galon J, Fridman WH, Smyth MJ (2015) From mice to humans: developments in cancer immunoediting. J Clin Invest 125:3338–3346
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331:1565–1570
Galon J, Angell HK, Bedognetti D, Marincola FM (2013) The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity 39:11–26
Desrichard A, Snyder A, Chan TA (2016) Cancer neoantigens and applications for immunotherapy. Clin Cancer Res 22:807–812
Inman BA, Frigola X, Dong H, Kwon ED (2007) Costimulation, coinhibition and cancer. Curr Cancer Drug Targets 7:15–30
Boroughs LK, DeBerardinis RJ (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 17:351–359
Biswas S, Metabolic K (2015) Reprogramming of immune cells in cancer progression. Immunity 43:435–449
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Chiarugi P, Cirri P (2015) Metabolic exchanges within tumor microenvironment. Cancer Lett 380:272–280
Krebs H, The A (1972) Pasteur effect and the relations between respiration and fermentation. Essays Biochem 8:1–34
Cairns RA, Harris IS, Mak TW (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11:85–95
Garcia-Heredia JM, Carnero A (2015) Decoding Warburg’s hypothesis: tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 6:41582–41599
Chan DA et al (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med 3:94ra70
Lussey-Lepoutre C et al (2015) Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun 6:8784
Clark GR et al (2014) Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab 99:E2046–E2050
Sonveaux P et al (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest 118:3930–3942
Semenza GL (2008) Tumor metabolism: cancer cells give and take lactate. J Clin Invest 118:3835–3837
Fischer K et al (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109:3812–3819
Gottfried E et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 107:2013–2021
De Vitto H, Perez-Valencia J, Radosevich JA (2016) Glutamine at focus: versatile roles in cancer. Tumour Biol 37:1541–1558
McKeehan WL (1982) Glycolysis, glutaminolysis and cell proliferation. Cell Biol Int Rep 6:635–650
Mushtaq M, Darekar S, Klein G, Kashuba E (2015) Different mechanisms of regulation of the Warburg effect in lymphoblastoid and Burkitt lymphoma cells. PLoS One 10:e0136142
Dang CV, Kim JW, Gao P, Yustein J (2008) The interplay between MYC and HIF in cancer.Nat Rev Cancer 8:51–56
Gao P et al (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458:762–765
Wise DR et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105:18782–18787
DeBerardinis RJ et al (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104:19345–19350
Haissaguerre M, Saucisse N, Cota D (2014) Influence of mTOR in energy and metabolic homeostasis. Mol Cell Endocrinol 397:67–77
Salloum D, Mukhopadhyay S, Tung K, Polonetskaya A, Foster DA (2014) Mutant ras elevates dependence on serum lipids and creates a synthetic lethality for rapamycin. Mol Cancer Ther 13:733–741
Bensaad K et al (2014) Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep 9:349–365
Carracedo A, Cantley LC, Pandolfi PP (2013) Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 13:227–232
Nomura DK et al (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140:49–61
Sugiarto S et al (2011) Asymmetry-defective oligodendrocyte progenitors are glioma precursors. Cancer Cell 20:328–340
Borah A, Raveendran S, Rochani A, Maekawa T, Kumar DS (2015) Targeting self-renewal pathways in cancer stem cells: clinical implications for cancer therapy. Oncogenesis 4:e177
Takeishi S, Nakayama KI (2016) To wake up cancer stem cells, or to let them sleep, that is the question. Cancer Sci 107:875–881
Viale A et al (2014) Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514:628–632
Kurtova AV et al (2015) Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517:209–213
Zhuang ZH et al (2008) Early upregulation of cyclooxygenase-2 in human papillomavirus type 16 and telomerase-induced immortalization of human esophageal epithelial cells. J Gastroenterol Hepatol 23:1613–1620
Hale JS et al (2014) Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 32:1746–1758
Franklin RA et al (2014) The cellular and molecular origin of tumor-associated macrophages. Science 344:921–925
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266
Mantovani A et al (2004) The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686
Yang K et al (2013) T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity 39:1043–1056
Cheng SC et al (2014) mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345:1250684
Baardman J, Licht I, de Winther MP, Van den Bossche J (2015) Metabolic-epigenetic crosstalk in macrophage activation. Epigenomics 7:1155–1164
Bosca L et al (2015) Metabolic signatures linked to macrophage polarization: from glucose metabolism to oxidative phosphorylation. Biochem Soc Trans 43:740–744
Colegio OR et al (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513:559–563
Krawczyk CM et al (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115:4742–4749
Chang M, Hamilton JA, Scholz GM, Elsegood CL (2009) Glycolytic control of adjuvant-induced macrophage survival: role of PI3K, MEK1/2, and Bcl-2. J Leukoc Biol 85:947–956
Byles V et al (2013) The TSC-mTOR pathway regulates macrophage polarization. Nat Commun 4:2834
Howell JJ, Manning BD (2011) mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab 22:94–102
Schmid MC et al (2011) Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19:715–727
Rivera LB et al (2015) Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep 11:577–591
Rozengurt E, Soares HP, Sinnet-Smith J (2014) Suppression of feedback loops mediated by PI3K/mTOR induces multiple overactivation of compensatory pathways: an unintended consequence leading to drug resistance. Mol Cancer Ther 13:2477–2488
Pello OM et al (2012) Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 119:411–421
Palsson-McDermott EM et al (2015) Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21:65–80
Semenza GL, Roth PH, Fang HM, Wang GL (1994) Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem 269:23757–23763
Semenza G, Targeting L (2003) HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Biswas SK, Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11:889–896
Lewis CE, De Palma M, Naldini L (2007) Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res 67:8429–8432
Lemke G, Lu Q (2003) Macrophage regulation by Tyro 3 family receptors. Curr Opin Immunol 15:31–36
Chang CI, Liao JC, Kuo L (2001) Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res 61:1100–1106
Ho VW, Sly LM (2009) Derivation and characterization of murine alternatively activated (M2) macrophages. Methods Mol Biol 531:173–185
Stuehr DJ, Nathan CF (1989) Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med 169:1543–1555
Ellyard JI, Quah BJ, Simson L, Parish CR (2010) Alternatively activated macrophage possess antitumor cytotoxicity that is induced by IL-4 and mediated by arginase-1. J Immunother 33:443–452
Odegaard JI, Chawla A (2011) Alternative macrophage activation and metabolism. Annu Rev Pathol 6:275–297
Huang SC et al (2014) Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15:846–855
Traversari C, Sozzani S, Steffensen KR, Russo V (2014) LXR-dependent and -independent effects of oxysterols on immunity and tumor growth. Eur J Immunol 44:1896–1903
Li Y et al (2011) Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 30:3887–3899
Park H, Lee J, Park T, Lee S, Yi W (2015) Enhancement of photo-current conversion efficiency in a CdS/CdSe quantum-dot-sensitized solar cell incorporated with single-walled carbon nanotubes. J Nanosci Nanotechnol 15:1614–1617
Martinez FO, Gordon S, Locati M, Mantovani A (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177:7303–7311
Biswas SK, Mantovani A (2012) Orchestration of metabolism by macrophages. Cell Metab 15:432–437
Zhou D et al (2014) Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal 26:192–197
Jha AK et al (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42:419–430
Zhao Q et al (2012) Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol 188:1117–1124
Frumento G, Piazza T, Di Carlo E, Ferrini S (2006) Targeting tumor-related immunosuppression for cancer immunotherapy. Endocr Metab Immune Disord Drug Targets 6:233–237
Cairo G, Recalcati S, Mantovani A, Locati M (2011) Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol 32:241–247
Recalcati S et al (2010) Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol 40:824–835
Deng R et al (2013) Inhibition of tumor growth and alteration of associated macrophage cell type by an HO-1 inhibitor in breast carcinoma-bearing mice. Oncol Res 20:473–482
Wilkinson N, Pantopoulos K (2013) IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2alpha mRNA translation. Blood 122:1658–1668
Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S (2013) The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med 65:1174–1194
Kayadibi H, Sertoglu E, Uyanik M, Tapan S (2014) Neutrophil-lymphocyte ratio is useful for the prognosis of patients with hepatocellular carcinoma. World J Gastroenterol 20:9631–9632
Galdiero MR et al (2013) Tumor associated macrophages and neutrophils in cancer. Immunobiology 218:1402–1410
Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A (2013) Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 228:1404–1412
Fridlender ZG et al (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16:183–194
Federzoni EA et al (2012) PU.1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood 119:4963–4970
Ahmed N, Weidemann MJ (1994) Purine metabolism in promyelocytic HL60 and dimethylsulphoxide-differentiated HL60 cells. Leuk Res 18:441–451
Azevedo EP et al (2015) A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced Neutrophil Extracellular Trap (NET) formation. J Biol Chem 290:22174–22183
Suzuki H, Kakinuma K (1983) Evidence that NADPH is the actual substrate of the oxidase responsible for the “respiratory burst” of phagocytosing polymorphonuclear leukocytes. J Biochem 93:709–715
Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ (2015) Metabolic requirements for neutrophil extracellular traps formation. Immunology 145:213–224
Cools-Lartigue J et al (2013) Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 123:3446–3458
Wculek SK, Malanchi I (2015) Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528:413–417
Karasuyama H, Yamanishi Y (2014) Basophils have emerged as a key player in immunity. Curr Opin Immunol 31:1–7
Davis BP, Rothenberg ME (2014) Eosinophils and cancer. Cancer Immunol Res 2:1–8
Berek C (2016) Eosinophils: important players in humoral immunity. Clin Exp Immunol 183:57–64
Shi HZ (2004) Eosinophils function as antigen-presenting cells. J Leukoc Biol 76:520–527
Cho KS et al (2014) Adipose-derived stem cells ameliorate allergic airway inflammation by inducing regulatory T cells in a mouse model of asthma. Mediat Inflamm 2014:436476
Pinto A et al (1997) The role of eosinophils in the pathobiology of Hodgkin’s disease. Ann Oncol 8(Suppl 2):89–96
Nolen BM, Lokshin AE (2010) Targeting CCL11 in the treatment of ovarian cancer. Expert Opin Ther Targets 14:157–167
Vendramini-Costa DB, Carvalho JE (2012) Molecular link mechanisms between inflammation and cancer. Curr Pharm Des 18:3831–3852
Simon HU et al (2003) Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells’ syndrome. Eur J Immunol 33:834–839
Nadif R, Zerimech F, Bouzigon E, Matran R (2013) The role of eosinophils and basophils in allergic diseases considering genetic findings. Curr Opin Allergy Clin Immunol 13:507–513
Lotfi R et al (2009) Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J Immunol 183:5023–5031
Elishmereni M, Bachelet I, Nissim Ben-Efraim AH, Mankuta D, Levi-Schaffer F (2013) Interacting mast cells and eosinophils acquire an enhanced activation state in vitro. Allergy 68:171–179
Kataoka S, Konishi Y, Nishio Y, Fujikawa-Adachi K, Tominaga A (2004) Antitumor activity of eosinophils activated by IL-5 and eotaxin against hepatocellular carcinoma. DNA Cell Biol 23:549–560
Krystel-Whittemore M, Dileepan KN, Wood JG (2015) Mast cell: a multi-functional master cell. Front Immunol 6:620
Sekar Y, Moon TC, Slupsky CM, Befus AD (2010) Protein tyrosine nitration of aldolase in mast cells: a plausible pathway in nitric oxide-mediated regulation of mast cell function. J Immunol 185:578–587
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Shiga K et al (2015) Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel) 7:2443–2458
Johansson AC et al (2012) Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol Cancer Res 10:1158–1168
Zhang D et al (2015) Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep 10:1335–1348
Guido C et al (2012) Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 3:798–810
Lisanti MP, Martinez-Outschoorn UE, Sotgia F (2013) Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle 12:2723–2732
Janzer A et al (2012) The H3K4me3 histone demethylase Fbxl10 is a regulator of chemokine expression, cellular morphology, and the metabolome of fibroblasts. J Biol Chem 287:30984–30992
Martinez-Outschoorn UE, Lisanti MP, Sotgia F (2014) Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol 25:47–60
Shi H et al (2015) Overexpression of monocarboxylate anion transporter 1 and 4 in T24-induced cancer-associated fibroblasts regulates the progression of bladder cancer cells in a 3D microfluidic device. Cell Cycle 14:3058–3065
Xing Y, Zhao S, Zhou BP, Mi J (2015) Metabolic reprogramming of the tumour microenvironment. FEBS J 282:3892–3898
Orr MT, Lanier LL (2010) Natural killer cell education and tolerance. Cell 142:847–856
Vivier E et al (2011) Innate or adaptive immunity? The example of natural killer cells. Science 331:44–49
Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA (2015) Activation-specific metabolic requirements for NK Cell IFN-gamma production. J Immunol 194:1954–1962
Velasquez SY et al (2016) Short-term hypoxia synergizes with interleukin 15 priming in driving glycolytic gene transcription and supports human natural killer cell activities. J Biol Chem 291:12960–12977
Le Nours J et al (2016) Atypical natural killer T-cell receptor recognition of CD1d-lipid antigens. Nat Commun 7:10570
Dahlberg CI, Sarhan D, Chrobok M, Duru AD, Alici E (2015) Natural killer cell-based therapies targeting cancer: possible strategies to gain and sustain anti-tumor activity. Front Immunol 6:605
Hida K, Maishi N, Torii C, Hida Y (2016) Tumor angiogenesis-characteristics of tumor endothelial cells. Int J Clin Oncol 21:206–212
Ribatti D (2016) Tumor refractoriness to anti-VEGF therapy. Oncotarget 7:46668–46677
Goel S et al (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91:1071–1121
Dudley AC (2012) Tumor endothelial cells. Cold Spring Harb Perspect Med 2:a006536
Lee SL, Fanburg BL (1987) Glycolytic activity and enhancement of serotonin uptake by endothelial cells exposed to hypoxia/anoxia. Circ Res 60:653–658
Jakobsson L et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12:943–953
De Bock K, Georgiadou M, Carmeliet P (2013) Role of endothelial cell metabolism in vessel sprouting. Cell Metab 18:634–647
De Bock K et al (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154:651–663
Schoors S et al (2014) Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab 19:37–48
Vegran F, Boidot R, Michiels C, Sonveaux P, Feron O (2011) Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res 71:2550–2560
Sonveaux P et al (2012) Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS One 7:e33418
Leopold JA, Zhang YY, Scribner AW, Stanton RC, Loscalzo J (2003) Glucose-6-phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bioavailable nitric oxide. Arterioscler Thromb Vasc Biol 23:411–417
Tian WN et al (1998) Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem 273:10609–10617
Locasale JW et al (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43:869–874
Jiang P et al (2011) p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 13:310–316
Anastasiou D et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334:1278–1283
Spolarics Z, Lang CH, Bagby GJ, Spitzer JJ (1991) Glutamine and fatty acid oxidation are the main sources of energy for Kupffer and endothelial cells. Am J Phys 261:G185–G190
Unterluggauer H et al (2008) Premature senescence of human endothelial cells induced by inhibition of glutaminase. Biogerontology 9:247–259
Li H et al (2001) Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am J Physiol Endocrinol Metab 280:E75–E82
Wu G, Haynes TE, Li H, Yan W, Meininger CJ (2001) Glutamine metabolism to glucosamine is necessary for glutamine inhibition of endothelial nitric oxide synthesis. Biochem J 353:245–252
Kim YW, Byzova TV (2014) Oxidative stress in angiogenesis and vascular disease. Blood 123:625–631
Dagher Z, Ruderman N, Tornheim K, Ido Y (2001) Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ Res 88:1276–1282
Wang Q et al (2006) Induction of allospecific tolerance by immature dendritic cells genetically modified to express soluble TNF receptor. J Immunol 177:2175–2185
Dudek AM, Martin S, Garg AD, Agostinis P (2013) Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol 4:438
Perrot I et al (2007) Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol 178:2763–2769
Cahill EF, Tobin LM, Carty F, Mahon BP, English K (2015) Jagged-1 is required for the expansion of CD4+ CD25+ FoxP3+ regulatory T cells and tolerogenic dendritic cells by murine mesenchymal stromal cells. Stem Cell Res Ther 6:19
Melief CJ (2008) Cancer immunotherapy by dendritic cells. Immunity 29:372–383
Steinman RM, Banchereau J (2007) Taking dendritic cells into medicine. Nature 449:419–426
Apetoh L, Locher C, Ghiringhelli F, Kroemer G, Zitvogel L (2011) Harnessing dendritic cells in cancer. Semin Immunol 23:42–49
Dong H, Bullock TN (2014) Metabolic influences that regulate dendritic cell function in tumors. Front Immunol 5:24
Kumar V, Gabrilovich DI (2014) Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 143:512–519
Mancino A et al (2008) Divergent effects of hypoxia on dendritic cell functions. Blood 112:3723–3734
Yang M et al (2010) HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol Cell Biol 88:165–171
Nasi A, Rethi B (2013) Disarmed by density: a glycolytic break for immunostimulatory dendritic cells? Oncoimmunology 2:e26744
Novitskiy SV et al (2008) Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112:1822–1831
Ahmed MS et al (2015) Dab2, a negative regulator of DC immunogenicity, is an attractive molecular target for DC-based immunotherapy. Oncoimmunology 4:e984550
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL (2015) Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol 194:2985–2991
Galvin KC et al (2013) Blocking retinoic acid receptor-alpha enhances the efficacy of a dendritic cell vaccine against tumours by suppressing the induction of regulatory T cells. Cancer Immunol Immunother 62:1273–1282
Ravindran R et al (2014) Vaccine activation of the nutrient sensor GCN2 in dendritic cells enhances antigen presentation. Science 343:313–317
Everts B et al (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15:323–332
Rehman A et al (2013) Role of fatty-acid synthesis in dendritic cell generation and function. J Immunol 190:4640–4649
Harshyne LA, Zimmer MI, Watkins SC, Barratt-Boyes SM (2003) A role for class A scavenger receptor in dendritic cell nibbling from live cells. J Immunol 170:2302–2309
Malinarich F et al (2015) High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol 194:5174–5186
Cubillos-Ruiz JR et al (2015) ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161:1527–1538
Li S, Champlin R, Fitchen JH, Gale RP (1985) Abnormalities of myeloid progenitor cells after “successful” bone marrow transplantation. J Clin Invest 75:234–241
Strober S (1984) Natural suppressor (NS) cells, neonatal tolerance, and total lymphoid irradiation: exploring obscure relationships. Annu Rev Immunol 2:219–237
Young MR, Kolesiak K, Wright MA, Gabrilovich DI (1999) Chemoattraction of femoral CD34+ progenitor cells by tumor-derived vascular endothelial cell growth factor. Clin Exp Metastasis 17:881–888
Bronte V et al (2000) Identification of a CD11b(+)/Gr-1(+)/CD31(+) myeloid progenitor capable of activating or suppressing CD8(+) T cells. Blood 96:3838–3846
Tartour E et al (2011) Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev 30:83–95
Morse MA, Hall JR, Plate JM (2009) Countering tumor-induced immunosuppression during immunotherapy for pancreatic cancer. Expert Opin Biol Ther 9:331–339
Maeda A, Kawamura T, Ueno T, Usui N, Miyagawa S (2014) Monocytic suppressor cells derived from human peripheral blood suppress xenogenic immune reactions. Xenotransplantation 21:46–56
Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P (2011) Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 118:5498–5505
Draghiciu O, Lubbers J, Nijman HW, Daemen T (2015) Myeloid derived suppressor cells-an overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 4:e954829
Hossain F et al (2015) Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res 3:1236–1247
Zoso A et al (2014) Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur J Immunol 44:3307–3319
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70:68–77
Nagaraj S, Schrum AG, Cho HI, Celis E, Gabrilovich DI (2010) Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol 184:3106–3116
Lu T et al (2011) Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest 121:4015–4029
Brito C et al (1999) Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol 162:3356–3366
Kasic T et al (2011) Modulation of human T-cell functions by reactive nitrogen species. Eur J Immunol 41:1843–1849
Wu AA, Drake V, Huang HS, Chiu S, Zheng L (2015) Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 4:e1016700
Crook KR et al (2015) Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol 97:573–582
Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P (2011) PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res 71:7463–7470
Zhang N, Bevan MJ (2011) CD8(+) T cells: foot soldiers of the immune system. Immunity 35:161–168
Liao W, Lin JX, Leonard WJ (2013) Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 38:13–25
Siska PJ, Rathmell JC (2015) T cell metabolic fitness in antitumor immunity. Trends Immunol 36:257–264
MacIver NJ, Michalek RD, Rathmell JC (2013) Metabolic regulation of T lymphocytes. Annu Rev Immunol 31:259–283
Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD (2009) Anergic T cells are metabolically anergic. J Immunol 183:6095–6101
Patsoukis N et al (2015) PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 6:6692
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348
Calcinotto A et al (2012) Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res 72:2746–2756
Chang CH et al (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153:1239–1251
Maciolek JA, Pasternak JA, Wilson HL (2014) Metabolism of activated T lymphocytes. Curr Opin Immunol 27:60–74
Wang Q, Liang B, Shirwany NA, Zou MH (2011) 2-Deoxy-D-glucose treatment of endothelial cells induces autophagy by reactive oxygen species-mediated activation of the AMP-activated protein kinase. PLoS One 6:e17234
Zeng H, Chi H (2014) mTOR signaling and transcriptional regulation in T lymphocytes. Transcription 5:e28263
Fernandez-Ramos AA, Poindessous V, Marchetti-Laurent C, Pallet N, Loriot MA (2016) The effect of immunosuppressive molecules on T-cell metabolic reprogramming. Biochimie 127:23–36
Blagih J et al (2015) The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42:41–54
Bronte V, Zanovello P (2005) Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol 5:641–654
Rodriguez PC et al (2004) Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64:5839–5849
Bronte V et al (2005) Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med 201:1257–1268
Aulak KS et al (2001) Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci U S A 98:12056–12061
Molon B et al (2011) Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med 208:1949–1962
Wang W et al (2016) Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell 165:1092–1105
Liu X et al (2010) Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 115:3520–3530
Uyttenhove C et al (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 9:1269–1274
Carbotti G et al (2015) IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 6:43267–43280
Leung BS, Stout LE, Shaskan EG, Thompson RM (1992) Differential induction of indoleamine-2,3-dioxygenase (IDO) by interferon-gamma in human gynecologic cancer cells. Cancer Lett 66:77–81
Sorensen RB et al (2011) Spontaneous cytotoxic T-cell reactivity against indoleamine 2,3-dioxygenase-2. Cancer Res 71:2038–2044
Yang W et al (2016) Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531:651–655
Mucida D et al (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317:256–260
Liu Z et al (2015) Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528:225–230
Vacchelli E et al (2014) Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 3:e957994
Michalek RD et al (2011) Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol 186:3299–3303
Lochner M, Berod L, Sparwasser T (2015) Fatty acid metabolism in the regulation of T cell function. Trends Immunol 36:81–91
Beier UH et al (2015) Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J 29:2315–2326
Bollinger T et al (2014) HIF-1alpha- and hypoxia-dependent immune responses in human CD4+CD25high T cells and T helper 17 cells. J Leukoc Biol 96:305–312
Shi LZ et al (2011) HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208:1367–1376
McGeachy MJ, Cua DJ (2008) Th17 cell differentiation: the long and winding road. Immunity 28:445–453
Leung S et al (2010) The cytokine milieu in the interplay of pathogenic Th1/Th17 cells and regulatory T cells in autoimmune disease. Cell Mol Immunol 7:182–189
Berod L et al (2014) De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20:1327–1333
Caro-Maldonado A et al (2014) Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol 192:3626–3636
Bashir U et al (2015) PET/MRI in oncological imaging: state of the art. Diagnostics (Basel) 5:333–357
Flechsig P, Mehndiratta A, Haberkorn U, Kratochwil C, Giesel FL (2015) PET/MRI and PET/CT in lung lesions and thoracic malignancies. Semin Nucl Med 45:268–281
Mehanna H et al (2016) PET-CT Surveillance versus neck dissection in advanced head and neck cancer. N Engl J Med 374:1444–1454
Radford J et al (2015) Results of a trial of PET-directed therapy for early-stage Hodgkin’s lymphoma. N Engl J Med 372:1598–1607
McGranahan N et al (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351:1463–1469
Palazon A et al (2012) The HIF-1alpha hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov 2:608–623
Noman MZ et al (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211:781–790
Gowrishankar K et al (2015) Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One 10:e0123410
Kleffel S et al (2015) Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell 162:1242–1256
Zelenay S et al (2015) Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162:1257–1270
Chen C, Rowell EA, Thomas RM, Hancock WW, Wells AD (2006) Transcriptional regulation by Foxp3 is associated with direct promoter occupancy and modulation of histone acetylation. J Biol Chem 281:36828–36834
Onodera T et al (2009) Constitutive expression of IDO by dendritic cells of mesenteric lymph nodes: functional involvement of the CTLA-4/B7 and CCL22/CCR4 interactions. J Immunol 183:5608–5614
Chevolet I et al (2015) Characterization of the immune network of IDO, tryptophan metabolism, PD-L1, and in circulating immune cells in melanoma. Oncoimmunology 4:e982382
Hellmann MD, Friedman CF, Wolchok JD (2016) Combinatorial cancer immunotherapies. Adv Immunol 130:251–277
Dai H, Wang Y, Lu X, Han W (2016) Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst 108:djv439
Jackson HJ, Rafiq S, Brentjens RJ (2016) Driving CAR T-cells forward. Nat Rev Clin Oncol 13:370–383
Kawalekar OU et al (2016) Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44:380–390
Wurth R et al (2016) Drug-repositioning opportunities for cancer therapy: novel molecular targets for known compounds. Drug Discov Today 21:190–199
Wang BY et al (2015) Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J Exp Clin Cancer Res 34:85
Luciani F et al (2004) Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst 96:1702–1713
Nicolau-Galmes F et al (2011) Terfenadine induces apoptosis and autophagy in melanoma cells through ROS-dependent and -independent mechanisms. Apoptosis 16:1253–1267
Strandberg TE et al (2004) Mortality and incidence of cancer during 10-year follow-up of the Scandinavian Simvastatin Survival Study (4S). Lancet 364:771–777
Ishimoto T et al (2011) CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 19:387–400
Yae T et al (2012) Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3:883
Yoshikawa M et al (2013) xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res 73:1855–1866
Ferrannini E (2014) The target of metformin in type 2 diabetes. N Engl J Med 371:1547–1548
Psutka SP et al (2015) The association between metformin use and oncologic outcomes among surgically treated diabetic patients with localized renal cell carcinoma. Urol Oncol 33(67):e15–e23
Zhang Y et al (2014) Involvement of metformin and AMPK in the radioresponse and prognosis of luminal versus basal-like breast cancer treated with radiotherapy. Oncotarget 5:12936–12949
Chen H et al (2015) Synergistic effects of metformin in combination with EGFR-TKI in the treatment of patients with advanced non-small cell lung cancer and type 2 diabetes. Cancer Lett 369:97–102
Takahashi RU et al (2015) Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat Commun 6:7318
Singh M, Ferrara N (2012) Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat Biotechnol 30:648–657
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8:592–603
Clem B et al (2008) Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther 7:110–120
Whiteside TL (2006) The role of immune cells in the tumor microenvironment. Cancer Treat Res 130:103–124
Gajewski TF, Schreiber H, Fu YX (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14:1014–1022
Mauge L, Terme M, Tartour E, Helley D (2014) Control of the adaptive immune response by tumor vasculature. Front Oncol 4:61
Kohlhapp FJ, Mitra AK, Lengyel E, Peter ME (2015) MicroRNAs as mediators and communicators between cancer cells and the tumor microenvironment. Oncogene 34:5857–5868
Pio R, Ajona D, Lambris JD (2013) Complement inhibition in cancer therapy. Semin Immunol 25:54–64
Yuneva MO et al (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15:157–170
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Li, Y., Wan, Y.Y., Zhu, B. (2017). Immune Cell Metabolism in Tumor Microenvironment. In: Li, B., Pan, F. (eds) Immune Metabolism in Health and Tumor. Advances in Experimental Medicine and Biology, vol 1011. Springer, Dordrecht. https://doi.org/10.1007/978-94-024-1170-6_5
Download citation
DOI: https://doi.org/10.1007/978-94-024-1170-6_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-024-1168-3
Online ISBN: 978-94-024-1170-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)