
Abstract
Cholesterol-dependent cytolysins (CDCs) constitute a family of pore forming toxins secreted by Gram-positive bacteria. These toxins form transmembrane pores by inserting a large β-barrel into cholesterol-containing membrane bilayers. Binding of water-soluble CDCs to the membrane triggers the formation of oligomers containing 35–50 monomers. The coordinated insertion of more than seventy β-hairpins into the membrane requires multiple structural conformational changes. Perfringolysin O (PFO), secreted by Clostridium perfringens, has become the prototype for the CDCs. In this chapter, we will describe current knowledge on the mechanism of PFO cytolysis, with special focus on cholesterol recognition, oligomerization, and the conformational changes involved in pore formation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- β-barrel
- Cholesterol
- Cholesterol-dependent cytolysins
- Lysteriolysin O
- Membrane
- Perfringolysin O
- Pneumolysin
- Pore formation
- Streptolysin O
- Toxin
Introduction
Perfringolysin O (PFO) is the prototypical example of a growing family of bacterial pore-forming toxins known as the Cholesterol Dependent Cytolysins (CDCs, [19, 25, 81]). CDCs are secreted by Gram-positive bacteria including Bacillus, Listeria, Lysinibacillus, Paenibacillus, Brevibacillus, Streptococcus, Clostridium, Gardnerella, Arcanobacterium, and Lactobacillus (see [25, 35, 64]). There are 30 members of the CDC family reported for Gram-positive bacteria and, surprisingly, two CDC-coding DNA sequences have been found in the Gram-negative Desulfobulbus propionicus and Enterobacter lignolyticus. However, in contrast with the Gram-positive bacteria that produce CDCs, the Gram-negative ones have not been shown to inhabit humans or indeed animals of any kind [29]. Despite their extremely diverse lineage, the majority of CDCs show an amino acid sequence identity greater than 39 % when compared to PFO [25]. The C-terminus (domain 4 or D4) of PFO is responsible for membrane binding and is the domain with the highest percentage of amino acid identity when sequences are compared with other CDC members.
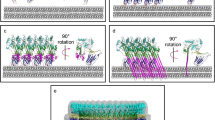
Most CDCs possess a cleavable signal sequence which targets the toxins for secretion to the extracellular medium. The secreted water-soluble toxins diffuse until encountering their target, a cholesterol-containing mammalian cell membrane (Fig. 5.1, step I). An exception to the cholesterol requirement for targeting was found for intermedilysin which uses the human receptor CD59 for membrane targeting [17]. However this toxin still requires cholesterol to insert into the membrane and form a transmembrane pore [16]. After binding, CDC monomers diffuse across the surface of the membrane and interact reversibly with other monomers until formation of a stable dimer (Fig. 5.1, step II, [30, 54]. These initial dimers grow by the incorporation of additional monomers into a large ring shaped complex (known as the pre-pore complex (Fig. 5.1, step III, [75]. Each of these complexes contains 35–50 monomers, and upon insertion into the membrane, they form large β-barrel pores (up to 250–300 Å in diameter, Fig. 5.1, step IV, [8, 73, 74]).

Cartoon representation of the different steps/intermediates identified for the PFO mechanism of pore formation. A water-soluble monomer is secreted by the bacterium and binds to the target membrane via D4 (step I). Membrane-bound monomers diffuse across the membrane surface interacting transiently until they form a stable dimer (step II). The initial dimer starts growing with the addition of other monomers until completion of a circular ring or pre-pore complex (step III). In the last step, each monomer inserts two amphipathic transmembrane hairpins into the bilayer aided by the vertical collapse of D2 forming a large β-barrel pore (step IV). Domains are numbered and color coded as follows: D1 (green), D2 (yellow), D3 (red), and D4 (blue). Only a few PFO monomers are shown in the side view at the bottom to simplify the figure. On the top is a schematic top view for each step of the pore formation mechanism shown below. The membrane bilayer is depicted by a gray rectangle
In this chapter, we will discuss CDCs through the lens of one of the most studied and well understood CDCs, PFO [19, 25, 81]. We will focus on the targeting of PFO to cholesterol-containing membranes and on the multiple conformational changes the protein undergoes in order to spontaneously transition from a water-soluble monomer to a large multimeric transmembrane complex. We will also comment on the most recent findings about the PFO cytolytic mechanism.
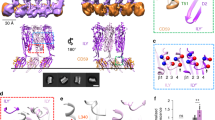
PFO is secreted by Clostridium perfringens as a 52.6 kDa protein, and the crystal structure of the water-soluble monomer revealed four distinct domains (Fig. 5.2a, [68]). The overall three dimensional structure observed for PFO is conserved for all other CDCs whose high resolution structures have been solved [5, 57, 82]. Domain 1 (D1) consists of the top portion of the elongated molecule. D1 is the only domain that does not undergo large structural rearrangements during pore formation. Domain 2 (D2) adopts mostly a β-strand secondary structure that collapses vertically during pore-formation to allow the insertion of the β-hairpins that form the transmembrane β-barrel [7, 8, 63, 80]. Domain 3 (D3) contains both the β-sheet involved in the oligomerization of the toxin and the six short α-helixes that unfurl into two amphipathic β-hairpins to form the β-barrel [62, 73, 74]. Domain 4 (D4) consists of a β-sandwich and contains a conserved Trp rich loop as well as three other conserved loops at the distal tip (Fig. 5.2b and c). D4 is responsible for cholesterol recognition and the initial binding of the toxin to the membrane [22, 61].

Three dimensional structure of PFO showing the location of important elements that modulate cholesterol interaction. a ribbon representation of the water-soluble PFO monomer with domains colored as indicated in Fig. 5.1. Also in color are three key residues that influence cholesterol interaction T490, L491, R468 (Red), and the Trp rich loop (TRL, orange). b A view of the tip of D4 from the bottom showing the exposed surface of the Trp rich loop residues (orange), the three small loops (green), and the residues indicated in A (red). c The ribbon rendering of the same bottom view of D4 shown in B. PFO (1PFO) structure representation was rendered using PyMol (DeLano Scientific LLC)
Membrane Recognition and Binding
One of the unique features of the mammalian cell membrane is the presence of cholesterol. C. perfringens and other pathogens have exploited this property of mammalian membranes to target their CDCs without compromising the integrity of their own membranes. It has long been known that binding of PFO and other CDCs requires high levels of cholesterol in model membranes prepared with phosphatidylcholine [1, 47, 67]. Based on the requirement of high cholesterol levels, targeting of PFO to cholesterol rich domains or “lipid rafts” has been suggested [49]. However, it has become clear that exposure of cholesterol at the membrane surface is a key factor to trigger PFO binding, and “lipid rafts” may not be necessary for toxin binding [15, 26, 44, 46, 52, 76]. Moreover, the localization of PFO oligomers on the membrane surface may change from the original binding site after insertion of the β-barrel [39, 45].
It has also been shown that the binding of PFO to cholesterol containing membranes is modulated by amino acids located in the loops that connect the β-strands at the bottom of D4 (Fig. 5.2c, [11, 13, 34, 44, 78, 79]), however the precise molecular mechanism of CDC-cholesterol interaction remains poorly understood.
Cholesterol Recognition
The first step in the binding of a water-soluble CDC to the membrane involves the formation of a non-specific collisional complex between a monomer and the lipid bilayer. This step is diffusional and electrostatic interactions may play an important role (e.g. introduction or elimination of negative charges alters binding, [34, 79]). While on the membrane surface, insertion of non-polar and aromatic amino acids and/or specific interactions with membrane lipids may anchor the protein to the membrane [6]. However, non-polar amino acids are rarely exposed on the surface of water-soluble proteins, and therefore conformational changes are often required to expose these residues to the hydrophobic core of the membrane bilayer. As a result, multiple conformational changes are triggered during the transition of PFO from a water-soluble monomer to a membrane-inserted oligomer.
In model membranes prepared exclusively with phosphatidylcholine >30 mol% cholesterol is required to trigger binding of PFO [22, 47], streptolysin O [67], lysteriolysin O [3], or tetanolysin [1] but the amount of cholesterol needed does vary depending on membrane phospholipid composition. The “cholesterol threshold” can be reduced by the presence of double bonds in the acyl chains of the phospholipids or by the presence of phospholipids with smaller head groups [14, 15, 46]. Therefore, it has become clear that modifications to the phospholipids that form the membrane can alter the ability of PFO to detect cholesterol at the membrane surface [44]. Despite their influence on membrane binding the presence of phospholipids is not required, since cholesterol alone (in the absence of any other lipid) is sufficient to trigger PFO oligomerization and formation of ring-like complexes ([26] and references therein). Accessibility of cholesterol at the membrane surface seems to be the key to trigger the binding of PFO to membranes [15, 44, 52, 76].
What is Cholesterol Accessibility?
It has long been recognized that cholesterol modulates important membrane properties including permeability, fluidity, thickness, and domain formation, among others. The cholesterol-dependent association of certain proteins and peptides with membranes has often been associated with the effect of cholesterol on one or more of these membrane physical properties. More recently, studies with molecules that directly interact with cholesterol, like cyclic sugar polymers (e.g. cyclodextrins, [60]), enzymes (e.g. cholesterol-oxidase, [38]), and bacterial toxins (e.g. PFO, [15, 44, 46, 76]) have shown that the accessibility of cholesterol at the membrane surface also plays a critical role in cell biology.
Cholesterol is insoluble in aqueous solutions, but it is readily soluble in phospholipid bilayers. The solubility limit of cholesterol in lipid bilayers is dictated by the nature of the phospholipids (acyl chain length and saturation, and head group size, [50]). If the concentration of cholesterol in a bilayer increases to levels above its solubility limit, cholesterol aggregates would form crystals and precipitate out into the aqueous solution [2, 40, 83].
Given its hydrophobic nature, in a lipid bilayer cholesterol orients parallel to the acyl chains of the phospholipids with the only polar group (an OH) facing the surface of the membrane, in close proximity to the phospholipid head groups (Fig. 5.3). At low concentrations, the interaction of cholesterol with other membrane components (lipids, proteins, etc.) reduces the ability of cholesterol to interact with water-soluble molecules at the membrane surface. In other words, when present in low amounts, cholesterol is not accessible to interact with molecules like PFO or cyclodextrins. As the concentration of cholesterol increases, its accessibility remains low until a saturation point is reached. The concentration of cholesterol at the saturation point will depend on the phospholipid or phospholipid mixture present in the membrane (Fig. 5.3a). At this point, a small increase in the sterol concentration causes a sharp increase in the ability of water-soluble molecules to interact with cholesterol [22, 38, 60]. Different models have been proposed to explain changes on cholesterol accessibility at the membrane surface: the cholesterol:phospholipid complex model and the umbrella model [32, 41]. Despite their thermodynamics or steric basis, the models are not mutually exclusive [37, 42]. Recent molecular dynamics simulations of simple membrane models [52] suggested that cholesterol accessibility is related to the overall cholesterol depth within the membrane bilayer and not to the appearance of a new pool of cholesterol molecules (sometimes referred as free cholesterol or active cholesterol). In favor of clarity in this chapter we will refer to the effect that cause an increase in the interaction of cholesterol with water-soluble molecules, as an increase in cholesterol accessibility at the membrane surface (Fig. 5.3).

Cholesterol accessibility changes at the membrane surface as a function of the lipid composition. a when interactions with other membranes components saturate the accessibility of cholesterol increases at the membrane surface. b At constant cholesterol concentration, an increase in the number of double bonds on the acyl chains of the phospholipids increases cholesterol accessibility. c At constant cholesterol concentration, an increase in the concentration of phospholipids with smaller head groups increases cholesterol accessibility. The red lines depict a hypothetical increase on cholesterol accessibility. The actual change on cholesterol accessibility for each schematic graph may differ from a simple linear response. Some cholesterol molecules are colored red to visualize the increase on accessibility but they are indistinguishable from other cholesterol molecules in the membrane
Domain 4 and the Conserved Loops
PFO D4 consists of two four-stranded β-sheets located at the C-terminus of the protein (Fig. 5.2). There are four loops that interconnect the eight β-strands at the distal tip of the toxin, three short loops (L1, L2, and L3) and a longer Trp rich loop (also known as the conserved undecapeptide). These loops insert into the membrane upon binding and are presumably responsible for the interaction of the toxin with cholesterol [13, 61, 79]. Two of these loops (L2 and L3, Fig. 5.2c) connect β-strands from opposite β-sheets, while L1 and the Trp rich loop connect β-strands from the same β-sheet. L1 and the Trp rich loop are parallel to each other and abutted perpendicularly by L2 forming a pocket in the bottom of the protein. The loops that form the pocket are the most conserved segments in D4, and modifications to any of these loops affects the cholesterol binding properties of PFO ([13, 34, 44, 58], see below). The remaining L3 is far less conserved and distant from the pocket formed by the other three loops.
The Trp rich loop is the longest of the D4 loops, containing 11 residues (E C T G L A W E W W R). It is a signature feature of the CDCs and is highly conserved among species. The three-dimensional structure of this loop seems to be more variable [5, 57, 68, 82], but this may simply reflect its flexibility [57]. Initially, the Trp rich loop was thought to be responsible for cholesterol recognition and binding, and this idea was supported by several studies showing that modifications in it greatly decreased the pore-forming activity of the protein [4, 36, 43, 55, 58, 71, 72]. However, recent studies showed that the other loops in D4 are also responsible for cholesterol recognition [13]. The Trp rich loop has now been suggested to play a role in both the pre-pore to pore transition [79] and the coupling of monomer binding with initiation of the pre-pore assembly [11]. Dowd and colleagues recently showed that modification of a charged amino acid in the Trp rich loop (Arg468, Fig. 5.2b) resulted in complete elimination of the pore-forming activity of PFO and had a significant effect on the membrane binding of the toxin [11, 58]. The R468A PFO derivative was not able to oligomerize after membrane binding, suggesting that this modification disrupts the previously reported allosteric coupling between D4 and D3 [22]. Despite the novel functions assigned to the Trp rich loop, its role in binding cannot be neglected since many modifications to this segment have been shown to have a significant effect in toxin-membrane interaction [58].
Unlike the flexible Trp rich loop, the three-dimensional structure of the other three short loops is more conserved. The L3 is located on the far edge of D4, away from a pocket formed by the Trp rich loop, L1, and L2 (Fig. 5.2c). Modifications introduced into L3 have been shown to have either a negligible effect on cholesterol interaction, or to decrease the amount of cholesterol required for binding [13, 34]. For example, the elimination of the charge of D434 in L3 reduced the amount of cholesterol required to trigger binding [34]. These results suggest that L3 plays a limited role in cholesterol recognition, and its effect on binding may be related to nonspecific interactions with the membrane that stabilize the bound monomer at lower cholesterol levels.
Proposed Cholesterol Recognition Motif
It has been proposed that PFO contains a cholesterol recognition motif composed of only two adjacent amino acids in L1, Thr490 and Leu491 [13]. These amino acids are completely conserved throughout all reported CDCs, and modifications to them greatly affect the binding of the protein to both cell and model membranes [13]. These data suggest a prominent role for these two amino acids in cholesterol recognition, however other well conserved amino acids in that region have not been analyzed yet (e.g. H398, Y402 and A404). Moreover, no direct interaction between cholesterol and these two residues has been shown so far. The fact that both amino acids must be mutated to eliminate binding in a motif containing only two amino acids, coupled with the fact that there are many additional conserved amino acids in the vicinity, suggest that other amino acids may also play a role in cholesterol recognition and form part of the cholesterol binding site. Further studies are required in this area.
The Effect of Cholesterol Accessibility on Perfringolysin O Binding
While cholesterol accessibility is necessary for PFO binding, the analysis of PFO derivatives with modifications on D4 revealed that sterol accessibility is not sufficient to trigger stable PFO-membrane association [34]. As mentioned above, native PFO readily binds to model membranes containing 40 mol% cholesterol (and an equimolar mixture of other phospholipids, see [34]), revealing that cholesterol is accessible at the membrane surface. However, the PFOC459A−L491S derivative was not able to bind to the same membranes, clearly indicating that the cholesterol molecules were not sufficiently accessible to trigger toxin binding. Binding of the PFOC459A−L491S derivative was recovered when the cholesterol concentration was increased to 50 mol%, suggesting that the affinity of this derivative for cholesterol is lower than that of native PFO, and more cholesterol was required at the membrane surface to trigger stable binding (note that L491 is one of the two amino acids proposed to be essential for cholesterol recognition). It is not clear how cholesterol accessibility varies with increasing amount of cholesterol in the membranes. For simplicity we have represented this variation as a linear function of cholesterol concentration (Fig. 5.3) however cholesterol accessibility may have a non-linear dependence in these systems. Further investigations are required in this area to establish the precise mechanism of PFO-cholesterol interaction as a function of cholesterol accessibility.
Mutations in Domain 4 Affect the Cholesterol Threshold Required to Trigger Binding
The effect that a particular amino acid modification has on PFO activity is often characterized by alterations to the hemolytic properties of the toxin (i.e. pore formation). The EC50 or effective toxin concentration required for 50 % lysis is a good indicator of these effects. Another method frequently used to characterize the effect of modifications in PFO derivatives is the percentage of hemolysis as compared with the one obtained for the native toxin under the same experimental conditions. However, it is worth noticing that the latter method is highly influenced by the toxin/red blood cell ratio used in the assay (see Fig. S2 in [34]). Similarly, when the characterization of PFO derivatives is done using model membranes, the protein/lipid ratio should be carefully taken into account.
The effect of a particular amino acid modification is dependent on how much cholesterol is accessible at the membrane surface ([34], and see below). As mentioned above, the PFOC459A−L491S derivative showed negligible binding to liposomes containing 40 mol% cholesterol, but the binding of PFOC459A−L491S was indistinguishable from that of native PFO when membranes containing 50 mol% cholesterol were used. The deleterious effect of many mutations to the toxin can be overcome by an increase in the cholesterol content in the membrane [34, 44]. Interestingly, while most modifications to the D4 loops do not affect the sharp sigmoidal shape of the binding isotherm, the amount of cholesterol required for 50 % binding (or “cholesterol threshold”) may change significantly for different PFO derivatives. Therefore, when comparing PFO derivatives using model membranes it is more accurate to quantify the effect of a particular modification as the relative change in the “cholesterol threshold” compared to one obtained for native PFO using the same batch of membranes [34].
We have shown recently that modifications to the binding domain of PFO were able to increase or decrease the “cholesterol threshold” of a PFO derivative [34]. These derivatives were successfully used to detect changes in the cholesterol content of cells and model membranes. While PFO has long been put forth as a probe for cholesterol-rich membranes, the advent of new PFO derivatives with varied “cholesterol thresholds” adds a layer of selectivity to the cholesterol sensing measurements.
Oligomerization on the Membranes Surface
Upon binding to a cholesterol containing membrane, PFO diffuses across the surface of the lipid bilayer and oligomerizes into a large ring shaped complex (Fig. 5.1). This complex contains 35–50 individual PFO monomers (~250–300 Å inner diameter) and it is referred to as the pre-pore complex [8, 51, 75]. Transition of the pre-pore complex to the final membrane-inserted complex occurs by the insertion of numerous β-hairpins (two per monomer) that perforate the membrane forming a large transmembrane β-barrel [73]. The conformation of the individual PFO monomers in the pre-pore complex is not vastly changed from that of their water-soluble form. There are subtle structural changes triggered by membrane binding and oligomerization of the protein that allow for proper alignment of the monomers and the overall geometry of the pore [62]. Formation of complete rings at the membrane surface seems to be regulated by the relatively slow formation of an initial CDC dimer [30, 54].
Nucleation of the Pre-pore Complex
Oligomerization of the CDCs is triggered by membrane binding and interaction with cholesterol (or exceptionally by interaction with a protein receptor for intermedilysin). Cholesterol binding is sufficient to trigger the conformational changes that unblock the hidden oligomerization interface in the water-soluble monomer [26, 62]. Blockage of the oligomerization interface in the monomer prevents premature oligomerization of the toxin in solution. This regulatory mechanism can be overridden if the monomers are present at high concentration in solution (e.g. for pneumolysin, [20, 77]), but oligomerization is rare at physiological concentrations (i.e. nM range or lower).
The most significant of the conformational changes that follows membrane binding involves the exposure of the core β-sheet that comprises a large part of D3. A short β-strand (β5) separates from the core β-sheet in D3 and exposes β4 for its interaction with the always-exposed β1 strand of another PFO molecule, promoting oligomerization [31, 62]. This conformational change is thought to be facilitated by a pair of Gly residues, Gly324 and Gly325, located in the loop between β4 and β5. These Gly residues are highly conserved, and act as a hinge between the two β-strands [62]. In addition to the separation of β5 from β4, it has been suggested that there is a disruption of the D2 and D3 interface. This disruption is thought to be caused by the rotation of D4 which breaks the weak interactions between D2 and D3. These conformational changes cause the rotation of D3 away from D1 and ultimately the unfurling of the transmembrane hairpins [30, 69].
Hotze et al. [30] have recently suggested that the initial interaction between two membrane-bound PFO monomers is weak and transient and rarely of sufficient length to allow for the transition to a stable dimer with β1 and β4 strands properly aligned. However, if the transition occurs, addition of further PFO monomers to the complex becomes favorable and oligomerization ensues. Therefore, formation of a stable initial dimer constitutes the rate limiting step in oligomerization that diminishes the formation of uncompleted rings on the membrane surface (Fig. 5.1, step II, [30]). While it has been originally proposed that the separation of β5 from β4 happens upon membrane binding [62], it is still unclear whether these structural changes are caused by toxin binding or as a consequence of monomer-monomer oligomerization.
Alignment of Core β-Sheets
Addition of monomers to the growing oligomer requires the proper alignment of the core β-strands of the newly added PFO monomer with a β strand at the edge of the oligomer. Formation of hydrogen bonds between adjacent β-strands is energetically favorable but non-specific in nature. If the alignment is not correct, proper growing of the oligomer would not be possible. Thus, it is critical to regulate the alignment of neighbor β-strands to prevent the formation of truncated pre-pore complexes. It has been suggested that the correct alignment of adjacent β-strands among individual PFO monomers is dictated by π-stacking interactions between aromatic residues located in β1 (Tyr181) and β4 (Phe318) [62]. Modifications on either of these residues have proven to be extremely deleterious to the ability of PFO to form pores [34, 62]. Interestingly, despite being a critical interaction, it appears that only Tyr181 is completely conserved among the CDCs. A few CDC family members do not contain an aromatic residue in the corresponding location of Phe318 in PFO, suggesting that proper alignment of adjacent β-strands may follow another regulatory mechanism for these members (i.e. lectinolysin, intermedilysin, vaginolysin, pneumolysin, mitilysin, pseudoneumolysin, and the two newly identified members, see [25, 35, 64]).
Mechanism of Pore Formation
The last step in the cytolytic mechanism of PFO is the formation of the transmembrane pore. The pre-pore complex transitions into a membrane-inserted complex forming a large transmembrane β-barrel (Fig. 5.1, step IV). This transition involves the unfurling of six short α-helixes located in D3 down to two amphipathic β-hairpins, and the collapse of D2 to bring down the β-hairpins so they can span the hydrophobic core of the membrane. Large secondary and tertiary structural changes are required to coordinate the insertion of more than 140 individual β-strands and the removal of thousands of lipid molecules to form a β-barrel pore. The use of two β-hairpins per monomer to create a transmembrane β-barrel was first described for PFO [27, 73], and it is likely that this mechanism is also employed by other important pore-forming proteins like the Membrane Attack Complex/Perforin (MACPF) proteins [12, 21, 66].
A key step in the pore formation mechanism of the CDCs is the unfurling of six short α-helixes in D3 to form two extended amphipathic β-hairpins [26, 70, 73]. These conformational changes are necessary to minimize the exposure of hydrophobic residues in the water-soluble form of the PFO monomer [24, 73]. After insertion, the hydrophobic side of the amphipathic hairpin faces the non-polar lipid core, and the hydrophilic side faces the aqueous pore (Fig. 5.4, [73, 74]). The exact molecular mechanism for the pre-pore to pore conversion remains unknown, but thermal energy plays a key factor since at low temperatures (e.g. 4 °C) the PFO oligomer remains locked at the pre-pore complex state [28, 75].

A schematic view that depicts the position and orientation of the transmembrane hairpins (TMH1 and TMH2) of PFO in the membrane-inserted complex as determined by Sato et al. [70]. The tilted membrane and the rectangle representing the rest of the PFO molecule are depicted in gray and blue, respectively. The amino acids that compose the D3 β–sheet and the transmembrane hairpins are depicted by their single letter code and color-coded according to conservation in the CDC family. Amino acids conserved in more than 90 % of the 28 CDC members are shown in red, in orange if conservation was higher than 70 % but lower than 90 %, and in black if not highly conserved. Highlighted in green are the aromatic amino acids that are thought to be involved in π-stacking interaction that stabilize PFO pre-pore confirmation and help to align individual PFO monomers for pore formation (see text for details)
Sato et al. [70] have recently shown that in the pre-pore complex the β-strands that form the transmembrane pore are flexible and mobile. These transmembrane β-hairpins are located high above the membrane in the pre-pore complex [63, 80] and are able to extend and test hydrogen bonding arrangements, but they do not fully form a β-barrel structure [28, 70]. This partially unfolded state of the β-hairpins is thought to represent an intermediate step in pre-pore to membrane-inserted complex transition for PFO [70]. The partial alignment of the β-hairpins in the pre-pore complex may constitute a kinetic barrier that deters the insertion of incomplete rings favoring the formation of complete pre-pore complexes.
The unfurling of the two α-helical bundles into two β-hairpins is favored by the formation of multiple hydrogen bonds, both between hairpins within a PFO monomer and between hairpins on adjacent monomers (Fig. 5.4). Crosslinking experiments revealed that the β-hairpins in the inserted β-barrel adopt a ~20° angle to the plane of the membrane, and the adjacent inter-monomer strands align themselves with a shift of two amino acids (Fig. 5.4, [70]). As mentioned above, PFO oligomerization is aided by the proper alignment of β-strands from adjacent monomers via π-stacking interaction between the completely conserved Tyr181 and the highly conserved Phe318. Inspection of the extended hairpins in the β-barrel conformation (Fig. 5.4) revealed another potential π-stacking interaction that may act to stabilize the hairpins in their extended conformation. These are the completely conserved Phe211 in the transmembrane hairpin 1 (TMH1) and highly conserved Phe294 (present in all but vaginolysin, lectinolysin, and intermedilysin of the 30 members) in the transmembrane hairpin 2 (TMH2). Interestingly, the F211C modification decreased the hemolytic activity of PFO [74] and the PFO derivative containing the F294C modification could not be stably produced [73].
The vertical collapse of D2 to bring D3 closer to the membrane surface is another important step in pore formation [7, 63]. In the pre-pore complex, PFO is positioned perpendicular to the membrane leaving D3 about 40 Å above the membrane surface [63, 80]. In this position, the β-strands that form the pore would barely reach the membrane surface and could not penetrate the membrane. The required vertical collapse of D2 would drop D3 to the membrane surface and allow the β-hairpins to punch through the membrane and form a β-barrel. Unfortunately, little is known about the mechanism of the transmembrane β-barrel insertion.
Formation of a pre-pore complex and formation of hydrogen-bonds between adjacent β-strands helps the toxin to overcome the energetic barrier of inserting non-hydrogen bonded β-hairpins [25]. The insertion of incomplete rings may also occur, especially when free monomers are no longer available to complete the circular complex. Trapped metastable arc-like structures may form a pore by themselves, but the formation of a lipid edge at one side of the pore is not energetically favored, and the arcs would have a tendency to associate with other arcs or any proximal complete rings [18, 53, 59].
One of the most intriguing aspects of the CDCs cytolytic mechanism is what happens to the lipids that are displaced to form the pore. The insertion of the β-barrel requires the displacement of more than 1,000 lipid molecules from the membrane [27]. It is not clear how such a large amount of molecules are removed from the center of the pre-pore complex, but the hydrophilic nature of the inner portion of PFO the β-barrel could aid in this process.
Conclusions and Future Perspectives
Despite the lack of high resolution structures for intermediates or for the final membrane-inserted complex, the pore formation mechanism of the CDCs is becoming one of the better understood mechanisms among pore-forming toxins. The elucidation of the three-dimensional structure of the water-soluble PFO monomer [68] in combination with the use of site-directed mutagenesis and fluorescence spectroscopy techniques played a critical role in these advances [23, 33].
More recently a lot of attention has been focused on the study of PFO-cholesterol interaction. These studies revealed that not only changes in cholesterol levels may increase or decrease the ability of the toxin to bind to membranes [22, 47], but also changes in the phospholipid composition of the lipid bilayer [14, 15, 44, 46, 47, 76].
Accessibility of cholesterol at the membrane surface seems to be a key factor to trigger PFO binding. However, simply having accessible sterol molecules is not enough to stabilize PFO monomers at the membrane surface. Different “grades” of cholesterol accessibility are required to trigger binding of PFO derivatives with modifications at the conserved loops of D4 [34, 44]. More studies are required to elucidate the molecular details of how cholesterol accessibility modulates PFO binding.
A detailed understanding of the cytolytic mechanism combined with the ability to modify the toxin and produce novel PFO derivatives, have now opened the door for the development of tools to study cell biology. Non-lytic PFO derivatives have been developed to study cholesterol accessibility at the surface of cell membranes [34, 48]. Other lytic PFO derivatives have been used to specifically permeabilize the plasma membrane of cells to study the biochemistry of intact organelles in their native environment (e.g. mitochondria [10]). Moreover, the striking on-and-off binding properties of PFO have been exploited to study the role of cholesterol in cell physiology and the intracellular traffic of cholesterol [9, 56]. It is clear that the understanding of the molecular mechanism of these fascinating proteins secreted by pathogens has had, and will have a great impact on the studies of biochemistry and physiology in whole cells [10], as well as on the studies of cholesterol-dependent mechanisms in cell biology [9, 56, 65].
Abbreviations
- CDCs:
-
Cholesterol-dependent cytolysins
- D1, D2, D3, and D4:
-
Domain 1, domain 2, domain 3, and domain 4
- L1, L2, and L3:
-
Loop 1, loop 2, and loop 3
- PFO:
-
Perfringolysin O
- TMH1 and TMH2:
-
Transmembrane hairpin 1 and transmembrane hairpin 2
References
Alving CR, Habig WH, Urban KA, Hardegree MC (1979) Cholesterol-dependent tetanolysin damage to liposomes. Biochim Biophys Acta 551:224–228
Bach D, Wachtel E (2003) Phospholipid/cholesterol model membranes: formation of cholesterol crystallites. Biochim Biophys Acta 1610:187–197
Bavdek A, Gekara NO, Priselac D, Gutierrez Aguirre I, Darji A, Chakraborty T, Macek P, Lakey JH, Weiss S, Anderluh G (2007) Sterol and pH interdependence in the binding, oligomerization, and pore formation of listeriolysin O. Biochemistry 46:4425–4437
Billington SJ, Songer JG, Jost BH (2002) The variant undecapeptide sequence of the Arcanobacterium pyogenes haemolysin, pyolysin, is required for full cytolytic activity. Microbiology 148:3947–3954
Bourdeau RW, Malito E, Chenal A, Bishop BL, Musch MW, Villereal ML, Chang EB, Mosser EM, Rest RF, Tang W-J (2009) Cellular functions and x-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J Biol Chem 284:14645–14656
Cho W, Stahelin RV (2005) Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct 34:119–151
Czajkowsky DM, Hotze EM, Shao Z, Tweten RK (2004) Vertical collapse of a cytolysin prepore moves its transmembrane beta-hairpins to the membrane. EMBO J 23:3206–3215
Dang TX, Hotze EM, Rouiller I, Tweten RK, Wilson-Kubalek EM (2005) Prepore to pore transition of a cholesterol-dependent cytolysin visualized by electron microscopy. J Struct Biol 150:100–108
Das A, Goldstein JL, Anderson DD, Brown MS, Radhakrishnan A (2013) Use of mutant 125I-Perfringolysin O to probe transport and organization of cholesterol in membranes of animal cells. Proc Natl Acad Sci USA 110:10580–10585
Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, Henry RR, McDonald WG, Colca JR, Simon MI, Ciaraldi TP, Murphy AN (2013) Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proc Natl Acad Sci USA 110:5422–5427
Dowd KJ, Tweten RK (2012) The cholesterol-dependent Cytolysin Signature Motif: a critical element in the Allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog 8:e1002787
Dunstone MA, Tweten RK (2012) Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr Opin Struct Biol 22:342–349
Farrand AJ, LaChapelle S, Hotze EM, Johnson AE, Tweten RK (2010) Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc Natl Acad Sci USA 107:4341–4346
Flanagan JJ, Heuck AP, Johnson AE (2002) Cholesterol-Phospholipid interactions play an important role in Perfringolysin O binding to membrane. FASEB J 16:A929
Flanagan JJ, Tweten RK, Johnson AE, Heuck AP (2009) Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48:3977–3987
Giddings KS, Johnson AE, Tweten RK (2003) Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc Natl Acad Sci USA 100:11315–11320
Giddings KS, Zhao J, Sims PJ, Tweten RK (2004) Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat Struct Mol Biol 11:1173–1178
Gilbert RJ (2005) Inactivation and activity of cholesterol-dependent cytolysins: what structural studies tell us. Structure 13:1097–1106
Gilbert RJ (2010) Cholesterol-dependent cytolysins. Adv Exp Med Biol 677:56–66
Gilbert RJC, Rossjohn J, Parker MW, Tweten RK, Morgan PJ, Mitchell TJ, Errington N, Rowe AJ, Andrew PW, Byron O (1998) Self-interaction of pneumolysin, the pore-forming protein toxin of Streptococcus pneumoniae. J Mol Biol 284:1223–1237
Hadders MA, Beringer DX, Gros P (2007) Structure of C8 a-MACPF reveals mechanism of membrane attack in complement immune defense. Science 317:1552–1554
Heuck AP, Hotze EM, Tweten RK, Johnson AE (2000) Mechanism of membrane insertion of a multimeric β-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol Cell 6:1233–1242
Heuck AP, Johnson AE (2002) Pore-forming protein structure analysis in membranes using multiple independent fluorescence techniques. Cell Biochem Biophys 36:89–101
Heuck AP, Johnson AE (2005) Membrane recognition and pore formation by bacterial pore-forming Toxins. In: Tamm LK (ed) Protein-lipid interactions. From membrane domains to cellular networks. Wiley-VCH, Weinheim, pp 163–186
Heuck AP, Moe PC, Johnson BB (2010) The cholesterol-dependent cytolysins family of Gram-positive bacterial toxins. In: Harris JR (ed) Cholesterol binding proteins and cholesterol transport, Subcellular biochemistry, vol 51. Springer, The Netherlands pp 551–577
Heuck AP, Savva CG, Holzenburg A, Johnson AE (2007) Conformational changes that effect Oligomerization and initiate pore formation are triggered throughout Perfringolysin O upon binding to cholesterol. J Biol Chem 282:22629–22637
Heuck AP, Tweten RK, Johnson AE (2001) Beta-barrel pore-forming toxins: intriguing dimorphic proteins. Biochemistry 40:9065–9073
Heuck AP, Tweten RK, Johnson AE (2003) Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J Biol Chem 278:31218–31225
Hotze EM, Le HM, Sieber JR, Bruxvoort C, McInerney MJ, Tweten RK (2013) Identification and characterization of the first cholesterol-dependent cytolysins from Gram-negative bacteria. Infect Immun 81:216–225
Hotze EM, Wilson-Kubalek E, Farrand AJ, Bentsen L, Parker MW, Johnson AE, Tweten RK (2012) Monomer-Monomer interactions propagate structural transitions necessary for pore formation by the cholesterol-dependent cytolysins. J Biol Chem 287:24534–24543
Hotze EM, Wilson-Kubalek EM, Rossjohn J, Parker MW, Johnson AE, Tweten RK (2001) Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane beta-sheet from a prepore intermediate. J Biol Chem 276:8261–8268
Huang J, Feigenson GW (1999) A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys J 76:2142–2157
Johnson AE (2005) Fluorescence approaches for determining protein conformations, interactions and mechanisms at membranes. Traffic 6:1078–1092
Johnson BB, Moe PC, Wang D, Rossi K, Trigatti BL, Heuck AP (2012) Modifications in Perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry 51:3373–3382
Jost BH, Lucas E, Billington S, Ratner A, McGee D (2011) Arcanolysin is a cholesterol-dependent cytolysin of the human pathogen Arcanobacterium haemolyticum. BMC Microbiol 11:239
Korchev YE, Bashford CL, Pederzolli C, Pasternak CA, Morgan PJ, Andrew PW, Mitchell TJ (1998) A conserved tryptophan in pneumolysin is a determinant of the characteristics of channels formed pneumolysin in cells and planar lipid bilayers. Biochem J 329:571–577
Lange Y, Steck TL (2008) Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog Lipid Res 47:319–332
Lange Y, Ye J, Steck TL (2005) Activation of membrane cholesterol by displacement from phospholipids. J Biol Chem 280:36126–36131
Lin Q, London E (2013) Altering hydrophobic sequence lengths shows that hydrophobic mismatch controls affinity for ordered lipid domains (rafts) in the multitransmembrane strand protein Perfringolysin O. J Biol Chem 288:1340–1352
Mason PR, Tulenko TN, Jacob RF (2003) Direct evidence for cholesterol crystalline domains in biological membranes: role in human pathobiology. Biochim Biophys Acta 1610:198–207
McConnell HM, Radhakrishnan A (2003) Condensed complexes of cholesterol and phospholipids. Biochim Biophys Acta 1610:159–173
Mesmin B, Maxfield FR (2009) Intracellular sterol dynamics. Biochim Biophys Acta 1791:636–645
Michel E, Reich KA, Favier R, Berche P, Cossart P (1990) Attenuated mutants of the intracellular bacterium Listeria monocytogenes obtained by single amino acid substitutions in listeriolysin O. Mol Microbiol 4:2167–2178
Moe PC, Heuck AP (2010) Phospholipid hydrolysis caused by Clostridium perfringens α-toxin facilitates the targeting of perfringolysin O to membrane bilayers. Biochemistry 49:9498–9507
Nelson LD, Chiantia S, London E (2010) Perfringolysin O association with ordered lipid domains: implications for transmembrane protein raft affinity. Biophys J 99:3255–3263
Nelson LD, Johnson AE, London E (2008) How interaction of Perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of Perfringolysin O-lipid raft interaction. J Biol Chem 283:4632–4642
Ohno-Iwashita Y, Iwamoto M, Ando S, Iwashita S (1992) Effect of lipidic factors on membrane cholesterol topology—mode of binding of θ-toxin to cholesterol in liposomes. Biochim Biophys Acta 1109:81–90
Ohno-Iwashita Y, Shimada Y, Hayashi M, Iwamoto M, Iwashita S, Inomata M (2010) Cholesterol-binding toxins and anti-cholesterol antibodies as structural probes for cholesterol localization In: Harris JR (ed) Cholesterol binding and cholesterol transport proteins. Subcellular Biochemistry. vol 51, Springer, The Netherlands pp 597–621
Ohno-Iwashita Y, Shimada Y, Waheed A, Hayashi M, Inomata M, Nakamura M, Maruya M, Iwashita M (2004) Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe 10:125–134
Ohvo-Rekilä H, Ramstedt B, Leppimäki P, Slotte JP (2002) Cholesterol interactions with phospholipids in membranes. Prog Lipid Res 41:66–97
Olofsson A, Hebert H, Thelestam M (1993) The projection structure of Perfringolysin O (Clostridium perfringens θ-toxin). FEBS Lett 319:125–127
Olsen BN, Bielska AA, Lee T, Daily MD, Covey DF, Schlesinger PH, Baker NA, Ory DS (2013) The structural basis of cholesterol accessibility in membranes. Biophys J 105:1838–1847
Palmer M, Harris R, Freytag C, Kehoe M, Tranum-Jensen J, Bhakdi S (1998) Assembly mechanism of the oligomeric streptolysin O pore: the early membrane lesion is lined by a free edge of the lipid membrane and is extended gradually during oligomerization. EMBO J 17:1598–1605
Palmer M, Valeva A, Kehoe M, Bhakdi S (1995) Kinetics of Streptolysin O self-assembly. Eur J Biochem 231:388–395
Pinkney M, Beachey E, Kehoe M (1989) The thiol-activated toxin streptolysin O does not require a thiol group for cytolytic activity. Infect Immun 57:2553–2558
Pocognoni CA, De Blas GA, Heuck AP, Belmonte SA, Mayorga LS (2013) Perfringolysin O as a useful tool to study human sperm physiology. Fertil Steril 99(99–106):e102
Polekhina G, Feil SC, Tang J, Rossjohn J, Giddings KS, Tweten RK, Parker MW (2006) Comparative three-dimensional structure of cholesterol-dependent cytolysins. In: Alouf JE, Popoff MR (eds) The comprehensive sourcebook of bacterial protein toxins, 3rd edn. Academic Press, Oxford, England, pp 659–670
Polekhina G, Giddings KS, Tweten RK, Parker MW (2005) Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. Proc Natl Acad Sci USA 102:600–605
Praper T, Sonnen A, Viero G, Kladnik A, Froelich CJ, Anderluh G, Dalla Serra M, Gilbert RJC (2010) Human perforin employs different avenues to damage membranes. J Biol Chem 286:2946–2955
Radhakrishnan A, McConnell HM (2000) Chemical activity of cholesterol in membranes. Biochemistry 39:8119–8124
Ramachandran R, Heuck AP, Tweten RK, Johnson AE (2002) Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat Struct Mol Biol 9:823–827
Ramachandran R, Tweten RK, Johnson AE (2004) Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit beta-strand alignment. Nat Struct Mol Biol 11:697–705
Ramachandran R, Tweten RK, Johnson AE (2005) The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc Natl Acad Sci USA 102:7139–7144
Rampersaud R, Planet PJ, Randis TM, Kulkarni R, Aguilar JL, Lehrer RI, Ratner AJ (2011) Inerolysin, a cholesterol-dependent cytolysin produced by Lactobacillus iners. J Bacteriol 193:1034–1041
Reid PC, Sakashita N, Sugii S, Ohno-Iwashita Y, Shimada Y, Hickey WF, Chang T-Y (2004) A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res 45:582–591
Rosado CJ, Buckle AM, Law RHP, Butcher RE, Kan W-T, Bird CH, Ung K, Browne KA, Baran K, Bashtannyk-Puhalovich TA, Faux NG, Wong W, Porter CJ, Pike RN, Ellisdon AM, Pearce MC, Bottomley SP, Emsley J, Smith AI, Rossjohn J, Hartland EL, Voskoboinik I, Trapani JA, Bird PI, Dunstone MA, Whisstock JC (2007) A common fold mediates vertebrate defense and bacterial attack. Science 317:1548–1551
Rosenqvist E, Michaelsen TE, Vistnes AI (1980) Effect of streptolysin O and digitonin on egg lecithin/cholesterol vesicles. Biochim Biophys Acta 600:91–102
Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW (1997) Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 89:685–692
Rossjohn J, Polekhina G, Feil SC, Morton CJ, Tweten RK, Parker MW (2007) Structures of Perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J Mol Biol 367:1227–1236
Sato TK, Tweten RK, Johnson AE (2013) Disulfide-bond scanning reveals assembly state and beta-strand tilt angle of the PFO beta-barrel. Nat Chem Biol 9:383–389
Saunders FK, Mitchell TJ, Walker JA, Andrew PW, Boulnois GJ (1989) Pneumolysin, the thiol-activated toxin of Streptococcus pneumoniae, does not require a thiol group for in vitro activity. Infect Immun 57:2547–2552
Sekino-Suzuki N, Nakamura M, Mitsui K-I, Ohno-Iwashita Y (1996) Contribution of individual tryptophan residues to the structure and activity of θ-toxin (perfringolysin O), a cholesterol-binding cytolysin. Eur J Biochem 241:941–947
Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK (1999) The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99:293–299
Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK (1998) Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37:14563–14574
Shepard LA, Shatursky O, Johnson AE, Tweten RK (2000) The mechanism of pore assembly for a cholesterol-dependent cytolysin: formation of a large prepore complex precedes the insertion of the transmembrane beta-hairpins. Biochemistry 39:10284–10293
Sokolov A, Radhakrishnan A (2010) Accessibility of cholesterol in endoplasmic reticulum membranes and activation of SREBP-2 switch abruptly at a common cholesterol threshold. J Biol Chem 285:29480–29490
Solovyova AS, Nollmann M, Mitchell TJ, Byron O (2004) The solution structure and oligomerization behavior of two bacterial toxins: pneumolysin and perfringolysin O. Biophys J 87:540–552
Soltani CE, Hotze EM, Johnson AE, Tweten RK (2007) Specific protein-membrane contacts are required for prepore and pore assembly by a cholesterol-dependent cytolysin. J Biol Chem 282:15709–15716
Soltani CE, Hotze EM, Johnson AE, Tweten RK (2007) Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc Natl Acad Sci USA 104:20226–20231
Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR (2005) Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 121:247–256
Tweten RK (2005) Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun 73:6199–6209
Xu L, Huang B, Du H, Zhang X, Xu J, Li X, Rao Z (2010) Crystal structure of cytotoxin protein suilysin from Streptococcus suis. Protein Cell 1:96–105
Ziblat R, Leiserowitz L, Addadi L (2010) Crystalline domain structure and cholesterol crystal nucleation in single hydrated DPPC:cholesterol:POPC Bilayers. J Am Chem Soc 132:9920–9927
Acknowledgments
Work in the author’s laboratory was supported by Grant Number GM 097414 from the National Institute of Health (A.P.H). B.B.J. was partially supported by the National Science Foundation, Integrative Graduate Education and Research Traineeship (IGERT), Institute for Cellular Engineering (DGE-0654128).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Johnson, B.B., Heuck, A.P. (2014). Perfringolysin O Structure and Mechanism of Pore Formation as a Paradigm for Cholesterol-Dependent Cytolysins. In: Anderluh, G., Gilbert, R. (eds) MACPF/CDC Proteins - Agents of Defence, Attack and Invasion. Subcellular Biochemistry, vol 80. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8881-6_5
Download citation
DOI: https://doi.org/10.1007/978-94-017-8881-6_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-8880-9
Online ISBN: 978-94-017-8881-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)