
Abstract
Bile acids (BAs) are a large family of molecules that have a steroidal structure and are synthesized from cholesterol in the liver. BAs are physiological detergents important for the emulsification of dietary fats, drugs, and lipid-soluble vitamins in the intestine, and their subsequent absorption and transport to the liver for metabolism is followed by distribution to other tissues and organs. BAs also act as signalling molecules and are important for the regulation of their own synthesis, uptake and secretion as well as the control of cholesterol synthesis and the regulation of lipid and glucose metabolism. These processes are accomplished via the direct activation of the nuclear receptor farnesoid X receptor (FXR), TGR5, the pregnane X receptor (PXR) and the vitamin D receptor (VDR). In addition, other nuclear receptors, such as the constitutive androstane receptor (CAR) and the liver X receptor (LXR), can be indirectly influenced by BA, and these receptors, in turn, influence BA synthesis via feedback mechanisms and have a considerable influence on the metabolic processes of the entire organism. This chapter will focus on BA homeostasis, which is affected by BA synthesis, metabolism and disposition in the liver and intestine. Furthermore, the roles of BAs as signalling molecules and therapeutic drugs to treat several diseases and metabolic imbalances will be discussed. Since there are cross-species differences in the synthesis and metabolism of BAs, the chapter will focus on humans and mice and will point out differences between these two species.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Introduction
Bile acids (BAs) are a large family of molecules that have a steroidal structure and are synthesized from cholesterol in the liver. BAs are physiological detergents important for the emulsification of dietary fats, drugs, and lipid-soluble vitamins in the intestine, and their subsequent absorption and transport to the liver for metabolism is followed by distribution to other tissues and organs. BAs also act as signalling molecules and are important for the regulation of their own synthesis, uptake, and secretion as well as the control of cholesterol synthesis and the regulation of lipid and glucose metabolism. These processes are accomplished via the direct activation of the nuclear receptor farnesoid X receptor (FXR), TGR5, the pregnane X receptor (PXR), and the vitamin D receptor (VDR) [1]. In addition, other nuclear receptors, such as the constitutive androstane receptor (CAR) and the liver X receptor (LXR), can be indirectly influenced by BA, and these receptors, in turn, influence BA synthesis via feedback mechanisms and have a considerable influence on the metabolic processes of the entire organism. This chapter will focus on BA homeostasis, which is affected by BA synthesis, metabolism, and disposition in the liver and intestine. Furthermore, the roles of BAs as signalling molecules and therapeutic drugs to treat several diseases and metabolic imbalances will be discussed. Since there are cross-species differences in the synthesis and metabolism of BAs, the chapter will focus on humans and mice and will point out differences between these two species [2, 3].
Bile Acid Homeostasis
Bile Acid Synthesis in the Liver
The synthesis of BAs in the liver is performed by hepatocytes and involves 17 distinct enzymes located in the cytosol, endoplasmic reticulum, mitochondria, and peroxisomes [4]. These enzymes catalyze the oxidation and modifications of cholesterol at its steroid ring and facilitate the oxidative cleavage of three carbons from the cholesterol side chain to form C24 BAs [5]. These modifications can be classified into two distinct pathways—the neutral and the alternative BA synthesis pathways. The neutral pathway (also known as the classic pathway) is the major BA synthesis pathway in the liver. Via the neutral pathway, cholesterol is converted to 7α-hydroxycholesterol (7α-HOC) by the rate-limiting enzyme cholesterol 7α-hydroxylase (CYP7A1), which is located in the endoplasmic reticulum. The sterol 12α-hydroxylase (CYP8B1) converts the intermediate 7α-hydroxy-4 cholesten-3-one (C4) to 7α, 12α-dihydroxy-4-cholesten-3-one, resulting in the synthesis of cholic acid (CA). Without 12αhydroxylation by CYP8B1, C4 is converted to chenodeoxycholic acid (CDCA). The mitochondrial sterol 27-hydroxylase (CYP27A1) catalyzes steroid side chain oxidation in both CA and CDCA synthesis.
In the alternative pathway, CYP27A1 converts cholesterol to 3β-hydroxy-5-cholestenoic acid, which is then hydroxylated by oxysterol 7α-hydroxylase (CYP7B1) to form 3β,7α-dihydroxy-5-cholestenoic acid. Finally, CDCA is generated from 27-hydroxycholesterol (27-HOC). In the mouse liver, most CDCA is converted to α- and β-muricholic acid (MCA). MCA is only found in trace amounts in humans [6, 7]. In rodents, the alternative pathway can account for up to 25% of the total BA synthesis, whereas in humans, this route contributes less than 10% of the total amount of BAs. However, recent studies have shown that the alternative pathway is significantly more active in childhood, while in adults, the classical pathway makes a more significant contribution to the composition of BA pool [8] (Fig. 1).

The “classic pathway” and the “alternative pathway” to synthesize cholic acid (CA) and chenodeoxycholic acid (CDCA). In the “classic pathway” CYP7A1 converted cholesterol into 7-hydroxycholesterol in the endoplasmic reticulum. Via several steps, involving the enzymes Cyp8B1 and HSD3B7, the two primary bile acids CA and CDCA are synthesized. In the “alternative pathway” the enzymes Cyp27A1 and Cyp7B1 are used to synthesize CDCA from cholesterol
Newly synthesized BAs are conjugated to the amino acids glycine and taurine, secreted through the apical membrane of hepatocytes, and stored in the gallbladder. The conjugation of BAs by only two amino acids is a result of the substrate specificity of pancreatic carboxypeptidases that cleave all other conjugating moieties [9]. Only conjugation with taurine or glycine makes BAs indigestible and unabsorbable in the proximal small intestine where most lipid absorption occurs. Such conjugated BAs are generally less hydrophobic than unconjugated bile acid whereby the hydrophobicity depends on the type of conjugation. In healthy individuals, nearly all BAs are present in their conjugated form [10].
Humans have two primary BAs (CA, CDCA), whereas five primary BAs (CA, CDCA, the muricholic acids α MCA and β MCA, and UDCA) are synthesized in mice [11] (Table 1).
The regulation of BA synthesis is closely linked to the whole-body BA pool, which must be relatively constant. To maintain homeostasis, fine-tuned feedback mechanisms exist that are mediated via cholesterol intake and several nuclear receptors, which act as both BA and biological sensors [12].
Excess cholesterol consumption negatively regulates cholesterol uptake and synthesis by proteolysis of the sterol regulatory element-binding proteins (SREBPs) which in turn are able to coordinate the synthesis of the two major components of membranes: fatty acids and cholesterol [13]. Since cholesterol is the source for BA synthesis, this mechanism not only reduces the amount of cholesterol, but also reduces the synthesis of BAs.
Another method of regulating the BA level in an organism is by downregulating bile acid biosynthesis via suppression of the key enzyme Cyp7A1 by several BA receptors [14], which will be discussed in detail in the section “Bile Acids and Their Interaction with Nuclear Receptors.”
Bile Acid Metabolism
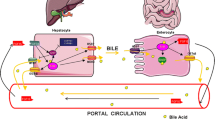
Hepatocytes secrete conjugated BAs into the bile, which is stored in the gall bladder and then reabsorbed by passive diffusion before being transported from the intestinal epithelial cells back to the liver following enterohepatic circulation (EHC) (Fig. 2). The process of BA secretion into the bile is accomplished by the polarity of hepatocytes, the mechanisms of which are complex and include intracellular and membrane trafficking systems and components, the cytoskeleton, tight junctions (TJs), and intracellular molecules [15]. In response to the peptide hormone cholecystokinin, the gallbladder releases bile into the duodenum to enable fat solubilization and absorption. The majority of the BAs that reach the distal intestine are reabsorbed during EHC.

The enterohepatic circulation of bile acids and its associated nuclear receptors. Cholesterol is degraded in the liver by CYP17A1 and CYP8B1 to the primary BA’s cholic acid (CA) and chenodeoxycholic acid (CDCA). Through the two transporters bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2) in the canalicular membranes of the hepatocytes, CA and CDCA can be transported into the bile. In the gut, CA and CDCA are metabolized to the secondary bile acids deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA). Most bile acids (primary and secondary) are reabsorbed in the intestine and returned to the liver via the enterohepatic circulation (EHC). The transporters apical sodium-dependent bile acid transporter (ASBT) and organic solute transporter alpha and beta (OSTα/β), which are expressed in the intestine are responsible for the absorption of BAs into the portal blood. At the basolateral membrane of hepatocytes, the transporters sodium/taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptide (OATP) reabsorb the BAs. The nuclear receptors FXR, PXR, and CAR are mainly metabolizing enzymes in the liver for induction Phase I and II to eliminate the excess BAs in the form of bile and urine
In detail, the EHC can be described as the movement of BA molecules from the hepatocytes into canalicular bile, through the biliary tract and into the duodenum. As a result of propulsive small intestinal motility, BAs then flow in the intestines where they are actively absorbed from the distal ileum and passively from the large intestine. In the next step, BAs are transported to the liver via the portal venous blood and are then efficiently taken up by the hepatocytes. To complete the EHC, the BAs enter the hepatocytes and are actively secreted into canalicular bile again [16]. This BA uptake by hepatocytes, like BA synthesis, is a process that does not occur in all hepatocytes in the liver lobe in the same manner. Hydrophilic BAs cross the cell membrane of hepatocytes by diffusion and show no zonal differences in transport velocity. In contrast, hydrophobic BAs transverse the cytosol bound to BA transporters or are distributed to organelles. Such transport systems like vesicles, glutathione S-transferase, and 3-α-hydroxysteroid dehydrogenase show a strong periportal distribution which leads to a faster transport in periportal hepatocytes compared to pericentral hepatocytes [17].
In addition to the enterohepatic circulation of BAs, a small proportion of conjugated BAs escapes this active transport and are deconjugated and further modified by the luminal bacteria into secondary BAs. Thereby, bacterial 7-dehydroxylase removes a hydroxyl group from C-7 and converts CA to deoxycholic acid (DCA) and CDCA to lithocholic acid (LCA) [18]. CYP3A1 and epimerases also convert CDCA to secondary BAs, including hyocholic acid (HCA), murideoxycholic acid (MDCA), ω-muricholic acid (ω-MCA), hyodeoxycholic acid (HDCA), and ursodeoxycholic acid (UDCA). Most LCA and ω-MCA are excreted into feces [19] (Fig. 2) (Table 1).
Bile Acids as Signalling Molecules in Metabolic Regulation
In addition to the immensely important function of BAs as physiological detergents for the absorption and transport of nutrients, fats, and vitamins from food, BAs also act as signalling molecules. This property enables BAs to regulate their own synthesis, the uptake, secretion, and synthesis of cholesterol, and the metabolism of lipids, glucose, and energy. This can be realized via the direct activation of the nuclear receptor farnesoid X receptor (FXR), the pregnane X receptor (PXR), the vitamin D receptor (VDR), and the Gs protein-coupled receptor TGR5 [1]. In addition, other nuclear receptors, such as the constitutive androstane receptor (CAR) and the Liver X receptor (LXR), can be indirectly influenced by BAs, which in turn influence BA synthesis via feedback mechanisms and have a considerable influence on global metabolism (Fig. 2).
Bile Acids and Their Interaction with Nuclear Receptors
BAs have the ability to directly and indirectly activate different nuclear receptors such as FXR, PXR, VDR as well as CAR to specifically influence their own biosynthesis and metabolic regulatory processes.
For FXR, two distinct genes (FXRα, NR1H4 and FXRβ, NR1H5) are known that are evolutionarily conserved between humans and rodents, but in contrast to the mouse, FXRβ is a pseudogene in humans [20]. The activation of FXR can only be achieved via binding of CDCA and its conjugates as well as LCA and DCA whereas the other BAs are not able to activate FXR [21] (Fig. 1). The binding of these BAs induces the formation of a heterodimer between FXR and retinoid X receptor (RXRFXR) which is able to bind to the inverted repeats of AGGTCA-like sequences with one nucleotide spacing (IR1) located in the promoters of the FXR target genes. The RXRFXR heterodimer then induces the expression of the negative nuclear receptor SHP, which inhibits the nuclear receptors liver-related homolog-1 and hepatocyte nuclear factor 4α (HNF4α). This inhibition results in inhibition of the transcription of CYP7A1, the driver enzyme of BA synthesis [6, 22]. In the intestine, activation of FXR by BAs induces the expression of the intestinal hormone fibroblast growth factor 15 (FGF15; or FGF19 in humans), which activates hepatic FGF receptor 4 (FGFR4) signalling to inhibit BA synthesis. In addition, activation of FXR also leads to downregulation of the intestinal BA transporter ASBT and the hepatic uptake transporters NTCP and OATP, which also reduces the BA concentration. This effect of FXR is further enhanced by a simultaneous upregulation of the hepatic efflux transporters BA export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2) [23, 24]. However, FXR inhibits not only CYP7A1 but also CYP8B1 and CYP27A1 transcription, as alterations to FXR activation are controlled by complicated mechanisms [25]. In addition to the modulation of BA synthesis, FXR activation also results in an overall decrease in triglyceride levels and modulation of glucose metabolism [26].
Also highly expressed in the liver and the intestine is the pregnane X receptor, PXR, which is also able to regulate CYP7A1 indirectly by inhibiting HNF4α and PGC-1α transactivation of Cyp7a1 gene expression [27]. Only secondary BAs are able to bind to the receptor and induce BA catabolism in this way, whereby the most potent ligand of PXR is LCA [28]. It is unclear whether BA can activate the receptor under physiological conditions when plasma levels are lower than 100 nmol/L, but it is probable that after rupture of the intrahepatic bile duct in cholestasis, PXR increases BAs clearance by CYP3A induction and decreases its biosynthesis by suppressing CYP7A1 expression [29]. After BA binding, PXR is translocated into the nucleus to form a heterodimer with RXR and activates target gene transcription. The range of regulated genes is wide, including many phase I and II enzymes and the uptake and efflux transporters.
Additionally, the vitamin D receptor has a dramatic influence on BA homeostasis. The alteration of BA synthesis via VDR is associated with the HNF4α Cyp7A1 axis, whereby the expression of VRD in the liver is restricted to nonparenchymal liver cells such as Kupffer cells or sinusoidal endothelial cells [30]. Like PXR, VDR can be activated by LCA and its metabolite 3-ketoLCA, while most Bas, including CDCA, CA, DCA, or muricholic acid, do not activate VDR [31]. The function of VDR in BA homeostasis is similar to that of PXR, namely, it exerts a protective role against BA toxicity and protects against bile duct infections. The latter is achieved through the stimulation of VDR by both vitamin D and LCA, which induces the production of the antimicrobial peptide cathelicidin in the bile duct epithelial cells [32]. In addition, LCA binding also increases the expression of CYP3A4, leading to elevated BA clearance, while the expression of CYP7A1 can be reduced through VDR activation [33]. In addition to influencing BA homeostasis, the VDR also has other functions such as regulating mineral homeostasis and metabolism, and a broader range of biological functions are connected to its expression such as cell growth, differentiation, antiproliferation, apoptosis, adaptive and innate immune responses [34]. Being widely expressed in various tissues, VDR represents an important therapeutic target in the treatment of diverse disorders that will be discussed later in detail.
Since the regulation of BA homeostasis by targeting nuclear receptors is very similar, it is assumed that FXR, PXR, and VDR coordinately regulate BAs, lipoproteins, drugs, glucose, and energy metabolism (Fig. 2).
Like PXR, the nuclear receptor CAR plays important roles in the regulation of BA metabolism and detoxication by inducing genes involved in BA conjugation and transport in order to maintain homeostasis [35]. Since BAs cannot directly bind to or activate CAR, the regulation occurs through well-known CAR activators such as TCPOBOP and Phenobarbital (PB). Both drugs are able to decrease the total BA level in the mouse liver, mainly by decreasing the amount of Taurine-conjugated CA (T-CA) to inhibit Cyp8b1. In mice, Cyp8b1 activation results in an increase in the proportion of muricholic acid (MCA). Furthermore, TCPOBOP is also able to increase Cyp7a1 expression in the liver to return the BAs to physiological concentrations. CAR activation also increases bile flow by increasing BA-independent flow, but the biliary excretion of BAs is not altered. In addition, CAR is also able to regulate BA homeostasis via the induction of LCA sulfation [35]. However, whether modulations of CAR are promising tools to regulate BA homeostasis in humans is questionable because of the differences in BA regulation between mice and humans. For example, in contrast to mice, humans have very low levels of MCA in the BA pool. Furthermore, glycine conjugates of BAs are the predominant amino acid conjugate in humans, while the taurine conjugates are predominant in mice.
Bile Acids and Their Interaction with TGR5
TGR5 (Takeda G protein-coupled receptor 5), also known as Gpbar-1 is a GS protein-coupled receptor that is responsive to various unconjugated and conjugated Bas. Taurine-conjugated lithocholic acid and taurodeoxycholic acid TDCA are the most potent endogenous BA agonists for TGR5 [36, 37]. In addition, secondary BAs, like LCA and DCA, are produced in the intestine by gut bacteria and are able to bind to the receptor [6]. The receptor is located on cholangiocytes, the epithelial surface of the gallbladder and intestinal cells, the basolateral surface of smooth muscle, neural cells, brown adipose tissue, immune cells including dendritic cells and macrophages, and enteroendocrine cells that produce glucagon-like peptide 1 (GLP-1). However, hepatocytes do not express TGR5 [38]. In the liver, BAs activate the phosphorylation of endothelial nitric oxide synthase (NOS) through TGR5 activation and cAMP release, leading to nitric oxide (NO) synthesis in sinusoidal endothelial cells [39]. In gallbladder smooth muscle, TGR5 activation lowers intracellular calcium levels, decreasing the rhythmic discharge of intracellular Ca2+ necessary to induce contraction [40]. Another mechanism for BA regulation via TGR5 is by the alteration of the alternative BA synthesis pathway via CYP7B1expression. These results come from recent studies using Tgr5-deficient mice that show a reduced Cyp7b1 expression and a dramatic change in the BA composition in the gallbladder.
In addition to the regulation of BA synthesis, modulation of TGR5 also leads to changes in the hepatic fatty acid uptake and oxidation rate, making TGR5 targeting a potent drug candidate for BA-associated liver diseases [41]. In addition, TGR5 binding is also a liver-specific process that promotes liver regeneration. TGR5 expressed in Kupffer cells (KC), biliary epithelium, and sinusoidal endothelial cells constitutes a permeable barrier between hepatocytes and blood. Recently, it was shown that TGR5 takes control over bile hydrophobicity and cytokine secretion after partial hepatectomy to prevent liver injury in mice [42].
Bile Acids and Diseases
Since BAs have multiple important functions in the body, it is easily imaginable that pathological changes in BA synthesis, secretion, or transformation can lead to many diseases which affect the entire organism.
The main problem caused by an increase in BAs is due to their detergent activity against cell membranes, which can cause cytotoxic effects that lead to mitochondrial and endoplasmic reticulum apoptosis, cell necrosis, and ultimately cancer [3, 43]. Thereby, BA toxicity highly correlates with hydrophobicity, which is ranked: LCA/DCA > CA, UDCA, MCA, and HCA [2]. The consequences of high concentrations of BAs in intra- and extrahepatic systems are manifold and affect various functions of almost all organs.
For example, anomalies in intestinal BAs can induce systemic intestinal infections by disrupting the barrier function of the small intestine and promoting the translocation of bacteria [44]. A strong increase in hydrophobic BAs upregulates the amount of pro-inflammatory cytokines and NF-κB, changes the composition of intestinal microbiota, increases endotoxin levels, aggravates the inflammatory response caused by glucose tolerance and insulin resistance, and augments intestinal permeability [45]. Other bowel diseases can result from high concentrations of intestinal DCA, which enhance the excretion of chloride ions, increase intestinal permeability, and inhibit mucosal healing [46].
In the liver, the most common diseases caused by alterations in BA homeostasis are cholestatic liver diseases. Cholestasis is mainly caused by a disruption of bile flow, which leads to a lack of bile in the intestine and an accumulation of toxic BAs in the liver. In addition, in cholestatic patients, a dramatically increased BA concentration can be found in systemic circulation (up to 100-fold in humans) [31, 47]. In addition to disruption of bile flow, impediment of the bile ducts by tumor or stones, mutations in genes that encode BA transporters, and dysregulation of the bile transport system by drugs, pregnancy, and pathophysiological conditions can cause disease [31, 48]. Cholestasis can be classified into primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC), which are the two most common chronic cholestatic liver diseases in adults. The development of PBC is an immune-mediated injury of intrahepatic biliary epithelial cells leading to cholestasis, fibrosis, and biliary cirrhosis [49].
In addition, a high concentration of BAs accelerates the senescence of hepatic secretory cells, facilitates the generation of tumor-promoting factors, and induces the progression of nonalcoholic steatohepatitis (NASH) and liver cancer [50]. Additionally, the hepatic control of xenobiotic and drug metabolism is closely associated with the regulatory network of BA homeostasis.
The fact that the characteristic changes of BAs in blood and tissues can indirectly indicate disease states has led to BAs being used as biomarkers for many diseases [50].
Bile Acids as Therapeutic Agents
BAs have an immense potential as therapeutic agents to produce beneficial effects in cases of primary biliary cirrhosis (PBC), primary sclerosing cholangitis, gallstones, digestive tract diseases, cystic fibrosis, and cancer [8]. Because BAs also influence metabolic-associated diseases such as nonalcoholic liver disease and diabetes, clinical studies have started to investigate these possibilities.
Bile Acids as Therapeutic Agents for Liver Diseases
BAs are being used as therapeutics, especially in patients with cholestatic liver diseases such as PCB. Thereby the administration of UDCA is the accepted therapy to treat PCB. This treatment inhibits intestinal BA absorption, resulting in an increase in BA secretion rich in bicarbonate to eliminate toxic substances from hepatocytes. As result, the entire BA pool is enriched with less toxic, hydrophilic BAs, which relieves parenchymal necrosis and apoptosis [51]. Unfortunately, up to 20% of PBC patients are UDCA nonresponders and have a reduced prognosis compared to healthy individuals [52]. For those patients, obeticholic acid (OCA, also known as INT-747) has been recently registered as a second-line therapy after demonstrating beneficial effects on liver biochemistry in approximately 50% of patients with an inadequate response to UDCA [49].
The treatment of PSC is much more complicated compared to PCB since several clinical studies showed no survival benefit from treatment with UDCA and other drugs. Therefore, liver transplantation is the only intervention shown to prolong survival of the patients [49].
BAs are also useful in the treatment (dissolution) of gallstones by increasing the concentration of bile acid and lowering cholesterol levels in the bile (resulting in less saturated bile).
Since BAs also have a significant influence on energy metabolism, targeted changes in BA receptors and the BA pool are used to increase glycemic control in diabetes. The main focus of the investigations is on nuclear transcription factors such as FXR in the liver and intestines, and the G protein-coupled receptor TGR5 in enteroendocrine cells and pancreatic β cells, as these interact directly with BAs [53]. In general, due to the many organs in which the signature of BAs is mediated by TGR5 and FXR, the mode of action of such inhibitors is highly complex which will have to be investigated further in the future (Fig. 3).

The role of bile acids in the entire organism
FXR Agonists for the Treatment of Metabolic Liver Diseases
One once-promising FXR agonist, obeticholic acid (OCA, also known as INT-747) is a semisynthetic derivative of chenodeoxycholic acid and was approved in the United States for the treatment of PBC after meeting the primary endpoint of reduced alkaline phosphatase level in a 2016 phase III clinical trial [54, 55]. This drug has also shown promising results in the treatment of NASH in a phase II trial, [56] demonstrating an improvement in the histological features of NASH, including hepatic steatosis, inflammation, hepatocyte ballooning, and liver fibrosis, and is currently in a phase III clinical trial in NASH patients [57]. However, recently, the FDA announced a warning about an increased risk of serious liver injury and death associated with OCA in patients with moderate to severe decreases in liver function. In addition, treatment with OCA in humans can lead to an increase in low-density lipoprotein cholesterol and a reduction of high-density lipoprotein cholesterol as well as increased pruritus [54].
In addition to OCA, other FXR agonists such as GS-9674 and Tropifexor (also known as LJN-452) have promising potential as therapeutic agents for cholestatic liver diseases and are currently undergoing phase II trials for PBC [58]. The advantages of these two compounds are that they are non-BA formulations and are thus expected to cause less pruritus and hyperlipidemia compared to OCA.
Recently, the non-bile acid FXR agonist, EDP-305, demonstrated a promising safety and tolerability profile in a phase I study including healthy individuals and patients with presumed NAFLD [59]. The drug is now under evaluation for patients with PBC [23].
TGR5 Agonists for the Treatment of Metabolic Liver Diseases
Targeting the BA receptor TGR5 is also a useful treatment for several metabolic diseases. The activation of TGR5 decreases body weight and forces the secretion of the hormone GLP-1, which promotes insulin release from β cells of the pancreas [60] (Fig. 3).
Furthermore, since TGR5 is highly expressed in monocytes and macrophages, where it modulates immune responses [61], targeting TGR5 lowers the levels of pro-inflammatory cytokines in monocytes. This finding has led to new insights into the modulatory role of BAs in pathology, where inflammatory processes play a central role, including colitis and atheroma development.
A new semisynthetic derivative of cholic acid, 6α-ethyl-23(S)-methyl-3α,7α,12α-trihydroxy-5β-cholan-24-oic acid (INT-777), is a selective TGR5 agonist that has a protective effect on many inflammatory diseases, such as sepsis, atherosclerosis, diabetic nephropathy, and hepatic steatosis [62].
Conclusion
BAs are extremely interesting compounds whose manifold functions are not yet fully understood. On the one hand, they serve to dissolve food components and are simultaneously secreted and received as signal molecules by various organs (Fig. 3). Since it is known that alterations in BA homeostasis are essentially responsible for various diseases, great efforts have been made to develop pharmaceutical concepts to restore this balance. Unfortunately, it is precisely the regulation of this equilibrium, which involves a wide variety of tissues, that causes the greatest difficulties in the treatment with these drugs. For example, the effects of FXR agonists not only reduce cholesterol metabolism in the liver but also might promote reverse cholesterol transport out of tissues. Therefore, in patients who are treated with such drugs, cholesterol changes need prospective monitoring and analysis in future studies of these therapies for liver disease [56].
References
Keitel V, Häussinger D. Perspective: TGR5 (Gpbar-1) in liver physiology and disease. Clin Res Hepatol Gastroenterol. 2012;36:412–9. https://doi.org/10.1016/j.clinre.2012.03.008.
Thakare R, Alamoudi JA, Gautam N, Rodrigues AD, Alnouti Y. Species differences in bile acids I. Plasma and urine bile acid composition. J Appl Toxicol. 2018;38:1323–35. https://doi.org/10.1002/jat.3644.
Thakare R, Alamoudi JA, Gautam N, Rodrigues AD, Alnouti Y. Species differences in bile acids II. Bile acid metabolism. J Appl Toxicol. 2018;38:1336–52. https://doi.org/10.1002/jat.3645.
Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56:1085–99. https://doi.org/10.1194/jlr.R054114.
Chiang JY. Recent advances in understanding bile acid homeostasis. F1000Res. 2017;6:2029. https://doi.org/10.12688/f1000research.12449.1.
Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017;1:3–9. https://doi.org/10.1016/j.livres.2017.05.001.
Ellis E, Goodwin B, Abrahamsson A, Liddle C, Mode A, Rudling M, Bjorkhem I, Einarsson C. Bile acid synthesis in primary cultures of rat and human hepatocytes. Hepatology. 1998;27:615–20. https://doi.org/10.1002/hep.510270241.
Šarenac TM, Mikov M. Bile acid synthesis: from nature to the chemical modification and synthesis and their applications as drugs and nutrients. Front Pharmacol. 2018;9:939. https://doi.org/10.3389/fphar.2018.00939.
Huijghebaert SM, Hofmann AF. Pancreatic carboxypeptidase hydrolysis of bile acid-amino conjugates: selective resistance of glycine and taurine amidates. Gastroenterology. 1986;90:306–15.
Hofmann AF. The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci (Landmark Ed). 2009;14:2584–98.
Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall H-U, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–35. https://doi.org/10.1016/j.cmet.2013.01.003.
Tu H, Okamoto AY, Shan B. FXR, a bile acid receptor and biological sensor. Trends Cardiovasc Med. 2000;10:30–5.
Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. https://doi.org/10.1016/S0092-8674(00)80213-5.
Santamaría E, Rodríguez-Ortigosa CM, Uriarte I, Latasa MU, Urtasun R, Alvarez-Sola G, Bárcena-Varela M, Colyn L, Arcelus S, Jiménez M, Deutschmann K, Peleteiro-Vigil A, Gómez-Cambronero J, Milkiewicz M, Milkiewicz P, Sangro B, Keitel V, Monte MJ, Marin JJ, Fernández-Barrena MG, Ávila MA, Berasain C. The epidermal growth factor receptor ligand amphiregulin protects from cholestatic liver injury and regulates bile acids synthesis. Hepatology. 2018;69:1632. https://doi.org/10.1002/hep.30348.
Hanley J, Dhar DK, Mazzacuva F, Fiadeiro R, Burden JJ, Lyne A-M, Smith H, Straatman-Iwanowska A, Banushi B, Virasami A, Mills K, Lemaigre FP, Knisely AS, Howe S, Sebire N, Waddington SN, Paulusma CC, Clayton P, Gissen P. Vps33b is crucial for structural and functional hepatocyte polarity. J Hepatol. 2017;66:1001–11. https://doi.org/10.1016/j.jhep.2017.01.001.
Hofmann AF, Hagey LR. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci. 2008;65:2461–83. https://doi.org/10.1007/s00018-008-7568-6.
Baier PK, Hempel S, Waldvogel B, Baumgartner U. Zonation of hepatic bile salt transporters. Dig Dis Sci. 2006;51:587–93. https://doi.org/10.1007/s10620-006-3174-3.
Li T, Chiang JYL. Bile acids as metabolic regulators. Curr Opin Gastroenterol. 2015;31:159–65. https://doi.org/10.1097/MOG.0000000000000156.
Ashby K, Navarro Almario EE, Tong W, Borlak J, Mehta R, Chen M. Review article: therapeutic bile acids and the risks for hepatotoxicity. Aliment Pharmacol Ther. 2018;47:1623–38. https://doi.org/10.1111/apt.14678.
Garcia M, Thirouard L, Sedès L, Monrose M, Holota H, Caira F, Volle DH, Beaudoin C. Nuclear receptor metabolism of bile acids and xenobiotics: a coordinated detoxification system with impact on health and diseases. Int J Mol Sci. 2018;19:E3630. https://doi.org/10.3390/ijms19113630.
Keitel V, Häussinger D. Role of TGR5 (GPBAR1) in liver disease. Semin Liver Dis. 2018;38:333–9. https://doi.org/10.1055/s-0038-1669940.
Malerød L, Sporstøl M, Juvet LK, Mousavi SA, Gjøen T, Berg T, Roos N, Eskild W. Bile acids reduce SR-BI expression in hepatocytes by a pathway involving FXR/RXR, SHP, and LRH-1. Biochem Biophys Res Commun. 2005;336:1096–105. https://doi.org/10.1016/j.bbrc.2005.08.237.
Goldstein J, Levy C. Novel and emerging therapies for cholestatic liver diseases. Liver Int. 2018;38:1520–35. https://doi.org/10.1111/liv.13880.
Trauner M, Fuchs CD, Halilbasic E, Paumgartner G. New therapeutic concepts in bile acid transport and signaling for management of cholestasis. Hepatology. 2017;65:1393–404. https://doi.org/10.1002/hep.28991.
Chiang JYL. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–66. https://doi.org/10.1194/jlr.R900010-JLR200.
Kalaany NY, Mangelsdorf DJ. LXRS and FXR: the yin and yang of cholesterol and fat metabolism. Annu Rev Physiol. 2006;68:159–91. https://doi.org/10.1146/annurev.physiol.68.033104.152158.
Bhalla S, Ozalp C, Fang S, Xiang L, Kemper JK. Ligand-activated pregnane X receptor interferes with HNF-4 signaling by targeting a common coactivator PGC-1alpha. Functional implications in hepatic cholesterol and glucose metabolism. J Biol Chem. 2004;279:45139–47. https://doi.org/10.1074/jbc.M405423200.
Juřica J, Dovrtělová G, Nosková K, Zendulka O. Bile acids, nuclear receptors and cytochrome P450. Physiol Res. 2016;65:S427–40.
Copple BL, Li T. Pharmacology of bile acid receptors: Evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res. 2016;104:9–21. https://doi.org/10.1016/j.phrs.2015.12.007.
Gascon-Barré M, Demers C, Mirshahi A, Néron S, Zalzal S, Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology. 2003;37:1034–42. https://doi.org/10.1053/jhep.2003.50176.
Li T, Chiang JYL. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–83. https://doi.org/10.1124/pr.113.008201.
D’Aldebert E, Biyeyeme Bi Mve M-J, Mergey M, Wendum D, Firrincieli D, Coilly A, Fouassier L, Corpechot C, Poupon R, Housset C, Chignard N. Bile salts control the antimicrobial peptide cathelicidin through nuclear receptors in the human biliary epithelium. Gastroenterology. 2009;136:1435–43. https://doi.org/10.1053/j.gastro.2008.12.040.
Han S, Li T, Ellis E, Strom S, Chiang JYL. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol Endocrinol. 2010;24:1151–64. https://doi.org/10.1210/me.2009-0482.
Belorusova AY, Rochel N. Structural studies of vitamin D nuclear receptor ligand-binding properties. Vitam Horm. 2016;100:83–116. https://doi.org/10.1016/bs.vh.2015.10.003.
Li T, Chiang JYL. Nuclear receptors in bile acid metabolism. Drug Metab Rev. 2013;45:145–55. https://doi.org/10.3109/03602532.2012.740048.
Duboc H, Taché Y, Hofmann AF. The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis. 2014;46:302–12. https://doi.org/10.1016/j.dld.2013.10.021.
Reich M, Klindt C, Deutschmann K, Spomer L, Häussinger D, Keitel V. Role of the G protein-coupled bile acid receptor TGR5 in liver damage. Dig Dis. 2017;35:235–40. https://doi.org/10.1159/000450917.
Malhi H, Camilleri M. Modulating bile acid pathways and TGR5 receptors for treating liver and GI diseases. Curr Opin Pharmacol. 2017;37:80–6. https://doi.org/10.1016/j.coph.2017.09.008.
Keitel V, Reinehr R, Gatsios P, Rupprecht C, Görg B, Selbach O, Häussinger D, Kubitz R. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. https://doi.org/10.1002/hep.21458.
Lavoie B, Balemba OB, Godfrey C, Watson CA, Vassileva G, Corvera CU, Nelson MT, Mawe GM. Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of KATP channels. J Physiol (Lond). 2010;588:3295–305. https://doi.org/10.1113/jphysiol.2010.192146.
Donepudi AC, Boehme S, Li F, Chiang JYL. G protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis. Hepatology. 2016;65:813–27. https://doi.org/10.1002/hep.28707.
Li G, Guo L. Farnesoid X receptor, the bile acid sensing nuclear receptor, in liver regeneration. Acta Pharm Sin B. 2015;5:93–8. https://doi.org/10.1016/j.apsb.2015.01.005.
Palmeira CM, Rolo AP. Mitochondrially-mediated toxicity of bile acids. Toxicology. 2004;203:1–15. https://doi.org/10.1016/j.tox.2004.06.001.
Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56:1283–92. https://doi.org/10.1016/j.jhep.2012.01.019.
Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol. 2011;178:175–86. https://doi.org/10.1016/j.ajpath.2010.11.026.
Raimondi F, Santoro P, Barone MV, Pappacoda S, Barretta ML, Nanayakkara M, Apicella C, Capasso L, Paludetto R. Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G906–13. https://doi.org/10.1152/ajpgi.00043.2007.
Mertens KL, Kalsbeek A, Soeters MR, Eggink HM. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front Neurosci. 2017;11:617. https://doi.org/10.3389/fnins.2017.00617.
Heubi JE, Setchell KDR, Bove KE. Inborn errors of bile acid metabolism. Clin Liver Dis. 2018;22:671–87. https://doi.org/10.1016/j.cld.2018.06.006.
Santiago P, Scheinberg AR, Levy C. Cholestatic liver diseases: new targets, new therapies. Therap Adv Gastroenterol. 2018;11:1756284818787400. https://doi.org/10.1177/1756284818787400.
Liu Y, Rong Z, Xiang D, Zhang C, Liu D. Detection technologies and metabolic profiling of bile acids: a comprehensive review. Lipids Health Dis. 2018;17:121. https://doi.org/10.1186/s12944-018-0774-9.
Yang H, Duan Z. Bile acids and the potential role in primary biliary cirrhosis. Digestion. 2016;94:145–53. https://doi.org/10.1159/000452300.
Ronca V, Carbone M, Bernuzzi F, Malinverno F, Mousa HS, Gershwin ME, Invernizzi P. From pathogenesis to novel therapies in the treatment of primary biliary cholangitis. Expert Rev Clin Immunol. 2017;13:1121–31. https://doi.org/10.1080/1744666X.2017.1391093.
Rajani C, Jia W. Bile acids and their effects on diabetes. Front Med. 2018;12:608. https://doi.org/10.1007/s11684-018-0644-x.
Erstad DJ, Farrar CT, Ghoshal S, Masia R, Ferreira DS, Chen Y-CI, Choi J-K, Wei L, Waghorn PA, Rotile NJ, Tu C, Graham-O’Regan KA, Sojoodi M, Li S, Li Y, Wang G, Corey KE, Or YS, Jiang L, Tanabe KK, Caravan P, Fuchs BC. Molecular magnetic resonance imaging accurately measures the antifibrotic effect of EDP-305, a novel farnesoid X receptor agonist. Hepatol Commun. 2018;2:821–35. https://doi.org/10.1002/hep4.1193.
Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, Drenth JPH, Pockros PJ, Regula J, Beuers U, Trauner M, Jones DE, Floreani A, Hohenester S, Luketic V, Shiffman M, van Erpecum KJ, Vargas V, Vincent C, Hirschfield GM, Shah H, Hansen B, Lindor KD, Marschall H-U, Kowdley KV, Hooshmand-Rad R, Marmon T, Sheeron S, Pencek R, MacConell L, Pruzanski M, Shapiro D. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–43. https://doi.org/10.1056/NEJMoa1509840.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–65. https://doi.org/10.1016/S0140-6736(14)61933-4.
Iracheta-Vellve A, Calenda CD, Petrasek J, Ambade A, Kodys K, Adorini L, Szabo G. FXR and TGR5 agonists ameliorate liver injury, steatosis, and inflammation after binge or prolonged alcohol feeding in mice. Hepatol Commun. 2018;2:1379–91. https://doi.org/10.1002/hep4.1256.
Liles JT, Karnik S, Hambruch E, Kremoser C, Birkel M, Watkins WJ, Tumas D, Breckenridge D, French D. Fxr agonism by Gs-9674 decreases steatosis and fibrosis in a murine model of nash. J Hepatol. 2016;64:S169. https://doi.org/10.1016/S0168-8278(16)01682-2.
Silveira MG, Lindor KD. Investigational drugs in phase II clinical trials for primary biliary cholangitis. Expert Opin Investig Drugs. 2017;26:1115–21. https://doi.org/10.1080/13543784.2017.1371135.
Roda A, Pellicciari R, Gioiello A, Neri F, Camborata C, Passeri D, de FF, Spinozzi S, Colliva C, Adorini L, Montagnani M, Aldini R. Semisynthetic bile acid FXR and TGR5 agonists: physicochemical properties, pharmacokinetics, and metabolism in the rat. J Pharmacol Exp Ther. 2014;350:56–68. https://doi.org/10.1124/jpet.114.214650.
Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–40. https://doi.org/10.1074/jbc.M209706200.
Li B, Yang N, Li C, Li C, Gao K, Xie X, Dong X, Yang J, Yang Q, Tong Z, Lu G, Li W. INT-777, a bile acid receptor agonist, extenuates pancreatic acinar cells necrosis in a mouse model of acute pancreatitis. Biochem Biophys Res Commun. 2018;503:38–44. https://doi.org/10.1016/j.bbrc.2018.05.120.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Matz-Soja, M. (2020). Bile Acids as Regulatory Signalling Molecules. In: Rozman, D., Gebhardt, R. (eds) Mammalian Sterols . Springer, Cham. https://doi.org/10.1007/978-3-030-39684-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-39684-8_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-39683-1
Online ISBN: 978-3-030-39684-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)