
Abstract
A series of vanadium-incorporated tungstophosphoric acid catalysts supported on titania were prepared and characterised by FT-infrared, X-ray diffraction, laser Raman spectroscopy, X-ray photoelectron spectroscopy and temperature-programmed reduction techniques. The characterisation data reveals the incorporation of vanadium into Keggin unit of TPA and its subsequent dispersion on titania. The catalytic activity of these catalysts was evaluated for oxidative cleavage of olefins to corresponding carbonyl compounds using H2O2 as oxidant at room temperature. Influence of vanadium presence in primary as well as secondary structure of tungstophosphoric acid on catalytic activity was also studied. Different parameters such as H2O2 to substrate mole ratio, solvent effect and temperature on activity and selectivity were also evaluated. The preparation of the catalyst is simple, easily recoverable and reusable many times without loss of activity and selectivity. The catalyst is also active even for oxidative cleavage of aliphatic olefins.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Oxidation of organic substrates to oxygen-rich compounds is one of the paramount reactions developed in synthetic chemistry. The oxidative functionalisation and cleavage of olefins to corresponding carbonyl compounds are significant transformations in organic synthesis [1]. These compounds are having high commercial value as they have been used as intermediates for many chemical feedstocks, agrochemicals, fragrances, pharmaceuticals and polymers. The classical method for the scission of alkenes is ozonolysis followed by appropriate workup [2–5]. However, its utility is often limited as per safety concerns, and serious accidents have been reported [6, 7]. Alternatively, olefins can also be cleaved by the use of homogeneous catalysts such as Pd(OAc)2 [8], AuCl in combination of TBHP oxidant [9] and RuCl3 [10]. From the economical and environmental point of view, these homogeneous catalysts are not preferable. Therefore, there is a scope to develop heterogeneous catalyst for the oxidative cleavage of carbon–carbon double bonds under more eco-friendly conditions. In this regard, recently, supported Keggin type of heteropoly acid catalysts [11–13], vanadium complexes [14] and strontium ferrite spinel [15] have been reported for the oxidation of styrene. Although these processes are efficient, they have limitations: it takes longer reaction time and requires high reaction temperature, and utility of the catalyst for different olefin substrates are not reported.
From the past decade polyoxometalates especially Keggin type of heteropoly acids (HPAs) were extensively used for the acid and oxidation catalysts for several industrial applications [16–18]. HPAs possess incredibly strong Bronsted acidity and on the other hand appropriate redox properties. Both the acid and redox properties of HPAs can be tuned by varying its chemical composition at molecular levels. An essential drawback of these catalysts is their low thermal stability, low surface area and high solubility in polar solvents. Efforts are being made to overcome these difficulties by supporting the HPAs on various acidic supports or by simply exchange of protons with different metal ions. We have been working on modified Keggin-type heteropoly acids for various oxidation reactions [19–21].
Herein, we demonstrate vanadium-substituted tungstophosphoric acid supported on titania as a heterogeneous catalyst for the selective oxidative cleavage of olefins to carbonyl compounds at room temperature. The scope of the catalyst was also studied for different olefins. The effect off V content and its location in Keggin ion of TPA on the oxidative cleavage of olefins is also one of the aims of the present study.
2 Experimental Section
2.1 Preparation of Catalysts
Vanadium-substituted TPA (H4PW11V1O40; TPAV1) was prepared according to the similar method reported in the literature [22] using NaVO3, Na3PO4 and Na2WO4·2H2O. Sodium metavanadate (NaVO3, 7.33 mmol) was dissolved in 50 mL of deionised water at 80 °C and mixed with disodium hydrogen phosphate (Na2HPO4, 7.3 mmol), which was previously dissolved in 20 mL of water. The mixture was cooled to room temperature. Concentrated H2SO4 (5 mL) was then added to give a red solution. Sodium tungstate dihydrate (Na2WO4·2H2O, 80.63 mmol) was dissolved in 50 mL of distilled water separately and added to the above red solution drop-wise with vigorous stirring followed by slow addition of concentrated H2SO4. TPAV1 was obtained with ether extraction followed by evaporation. The yellow solid obtained was dissolved in water and concentrated until crystals appeared. Similarly, TPAV2 and TPAV3 catalysts were also prepared by varying the amounts of starting materials.
2.2 Preparation of VOTPA Catalyst
The catalyst with vanadium exchanged with the protons of TPA (denoted as VOTPA) was prepared by the exchange of the protons of H3PW12O40 (TPA) with (VO)+2 ions [23]. Calculated amount of V2O5 was dissolved in oxalic acid at 100 °C followed by cooling the solution to room temperature. This solution was added to the aqueous solution of TPA with constant stirring. The excess water was removed on a water bath, and the sample was further dried at 120 °C for 12 h.
2.3 Preparation of Titania-Supported TPAV1 Catalyst
A series of catalysts with 10–25 wt% of TPAV1 supported on TiO2 were prepared by impregnation method. Calculated amount of TPAV1 was dissolved in deionised water and added to TiO2. The excess water was removed on a water bath followed by drying at 120 °C for 12 h and finally calcined at 300 °C for 2 h.
3 Characterisation of Catalysts
FT-IR spectra of catalysts were taken on a DIGILAB (USA) IR spectrometer by the KBr disc method. XRD patterns were measured on a RIGAKU MINI FLEX diffractometer using CuKα radiation (λ = 1.54 Å). The 2-theta angels were scanned from 2° to 60° at a rate of 2° min−1.
The Raman spectra of the samples were collected with a Horiba-Jobin Yvon LabRAM-HR spectrometer equipped with a confocal microscope, 2,400/900 grooves/mm gratings and a notch filter. The visible laser excitation at 532 nm (visible/green) was supplied by a Yag doubled diode pumped laser (20 mW). The scattered photons were directed and focused onto a single-stage monochromator and measured with a UV-sensitive LN2-cooled CCD detector.
Temperature-programmed reduction (TPR) of the catalysts was carried out in a flow of 10 % H2/Ar mixture gas at a flow rate of 30 mL/min with a temperature ramp of 10 °C/min. Before the H2–TPR run, the catalysts were pretreated with argon gas at 250 °C for 2 h. The hydrogen consumption was monitored using a thermal conductivity detector.
XPS measurements were conducted on a KRATOS AXIS 165 with a dual-anode (Mg and Al) apparatus using Mg KR anode. The nonmonochromatised Al KR X-ray source (1486.6 eV) was operated at 12.5 kV and 16 mA. Before acquisition of the data, the sample was outgassed for about 3 h at 100 °C under a vacuum of 1.0 × 10−7 Torr to minimise surface contamination. The XPS instrument was calibrated using Au as standard. For energy calibration, the carbon 1s photoelectron line was used. The carbon 1s binding energy was taken as 285 eV. A charge neutralisation of 2 eV was used to balance the charge up of the sample. The spectra were deconvoluted using a Sun Solaris-based Vision-2 curve resolver. The location and the full width at half-maximum (fwhm) value for the species were first determined using the spectrum of the pure sample. Symmetric Gaussian shapes were used in all cases.
4 General Reaction Procedure
In a typical experiment, a 25 mL round bottom flask equipped with magnetic stirrer was charged with styrene (1 mmol), acetonitrile (3 mL), 30 % H2O2 (3 mmol) and 20 % TPAV1/TiO2 (50 mg) catalyst. The resulting mixture was stirred at room temperature for 10 h. After completion of the reaction, the catalyst was separated by filtration and dried over anhydrous sodium sulphate. The products were identified by GC–MS (SHIMADZU-2010) analysis by separating them on a DB-5 column.
5 Results and Discussion
Initially, styrene was taken as model substrate for the oxidative cleavage of olefins and conducted the reaction at room temperature under atmospheric pressure using vanadium containing TPA catalyst. The results are shown in Table 4.1. The main oxidation product is benzaldehyde and the others are styrene epoxide, 1-phenylethane-1,2-diol, benzoic acid and formaldehyde (Scheme 4.1). Oxidation of styrene without using catalyst did not yield any product.

Oxidative cleavage of styrene
TPA itself is not active at room temperature. TPA showed about 50 % conversion with 79 % selectivity towards benzaldehyde when the reaction temperature increased to 80 °C. Monovanadium-substituted TPA (TPAV1) catalyst gave up to 70 % conversion of styrene with 80 % selectivity to benzaldehyde at room temperature. TPA is known for its high acidic character and substitution of tungsten by vanadium in its primary structure results in generation of redox properties [23]. As the more number of tungsten atoms are replaced by vanadium atoms (TPAV2, TPAV3), there was an increase in conversion of styrene, but conversely decrease in selectivity to benzaldehyde was observed.
Location of vanadium in the heteropoly tungstate also has a significant effect on the oxidation ability of the catalyst [23]. Location of vanadium also played a crucial role in the oxidation of styrene. When the vanadium was present in the secondary structure of TPA (VOTPA), the activity of the catalyst is low compared (Table 4.1, entry 6) with the catalysts where the vanadium is present in the primary structure of TPA. It is observed that TPAV1 was efficient catalyst for the oxidation of styrene at room temperature. However, it suffers from homogeneity.
In order to make TPAV1 heterogeneous catalyst, it was supported on titania. The activity of TPAV1/TiO2 catalysts in the oxidation of styrene as function of TPAV1 loading at room temperature is shown in Table 4.2. TiO2 itself is not found to be active and its role is to disperse the active component on its surface. The conversion of styrene increased with increase in the content of TPAV1 and attained maximum conversion (97) at a loading of 20 wt%. Further increasing the loading, a marginal decrease in conversion is observed.
In search of the reason for the observed variation in activity with change in the content of TPAV1 on titania, these catalysts are characterised to know their surface and structural characteristics.
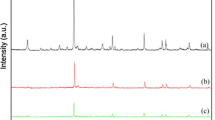
The XRD patterns of TPAV1/TiO2 catalysts are shown in Fig. 4.1. The XRD pattern of bulk TPA and TPAV1 is also shown for the sake of comparison. The characteristic Keggin peaks of pure TPA are obtained at a 2θ of 10.4°, 25.3° and 34.6° [24]. These characteristic lines are present even after incorporation of vanadium into the primary structure of TPA suggesting the presence of intact Keggin structure. The catalysts with low content of TPAV1 did not reveal any diffraction peaks of crystalline TPAV1. They display mainly the patterns of titania support. This might be due to the well dispersion of Keggin units on titania support. The XRD pattern related to Keggin ion is visible for the catalysts with high TPAV1 content (>20 wt%).

X-ray diffraction patterns of TPAV1/TiO2 catalysts
The FT-IR patterns of the catalysts are shown in Fig. 4.2. The IR pattern of TPA and TPAV1 is shown in the inset of Fig. 4.2. The TPA exhibited four bands in the region of 1,100–500 cm−1 with the main bands at 1,081, 986, 890 and 800 cm−1 which are assigned to the stretching vibrations of P–O, W=Ot, W–Oc–W and W–Oe–W, respectively, related to Keggin ion [25]. The IR spectra imply that the Keggin structure was intact after the incorporation of V in the tungsten matrix of 12-tungstophosphoric acid. With the substitution of one V atom for W in the primary structure of the oxoanion (TPAV1 catalyst), the P–O and W=O bands shifted towards lower wave number due to a reduced structural symmetry [23]. This splitting suggests the incorporation of vanadium into Keggin matrix. Therefore, XRD and FT-IR results are in agreement with each other.

FT-infrared patterns of TPAV1/TiO2 catalysts
Figure 4.3 shows the Raman spectra of the catalysts. The pure TPA exhibited characteristic Raman bands at 1,006, 991 and 905 cm−1 related to W=Ot symmetric and asymmetric vibrations of Keggin ion [26]. Similar bands were observed after substitution of vanadium in TPA. This shows vanadium incorporation into the Keggin matrix of TPA. The supported catalyst mainly exhibits Raman peaks at 394, 511 and 635 cm−1 related to the characteristic peaks of TiO2 [27]. The titania peaks are dominating in the supported catalyst and a small peak at 1,007 cm−1 corresponding to the Keggin ion of TPAV1 was observed. This indicates that TPAV1 is well dispersed with retention of Keggin structure on titania support.

Raman spectra of bulk (a) TPAV1 and(b) 20 %TPAV1/TiO2
The H2–TPR technique provides important information about the reducibility of individual species, making it a useful tool to characterise the heteropoly acid catalysts. The TPR pattern of 10–25 wt% of TPAV1/TiO2 catalysts is depicted in Fig. 4.4. Two types of reduction peaks were observed in all catalysts. Generally heteropoly acids start decomposition above 500 °C and the reduction peaks endorsed to the reduction of metal oxides generated during H2–TPR experiment. The first reduction peak observed at around 400–700 °C centred at 603 °C corresponds to reduction of segregated vanadium oxide species [21, 28]. L. Chen et al. observed that the TPR profile of V2O5–WO3 supported on titania the reduction peak at around 520 °C mentioned the reduction of V (V)–V (III) [2]. With the increase of loading from 10 to 25 wt%, the hydrogen consumption of vanadium increased and reduction peak slightly shifted to lower temperature. The high temperature reduction peak originated due to the reduction of product WO3 generated by the decomposition of TPAV1. As previously mentioned [29] it is speculated that the high temperature reduction peak corresponds to the reduction of W (VI) to W (0).

H2–TPR of (a) 10 wt% TPAV1/TiO2, (b) 15 wt% TPAV1/TiO2, (c) 20 wt% TPAV1/TiO2, (d) 25 wt% TPAV1/TiO2
XPS is used to investigate the binding energies of the surface elements provides the information about the chemical state of individual elements. Figure 4.5 shows the XPS measurement of the highly active 20 wt% TPAV1/TiO2 catalyst. The binding energy of P 2p in core level at 133.9 eV. The value of the position of P 2p photo peak confirms that phosphorous is phosphate [30]. The BEs for the Ti 2p core level are observed at 458.2 and 464 eV corresponding to Ti 2p3/2 and Ti 2p1/2, suggesting the oxidation state of titanium is Ti+4 [31]. Two well-resolved peaks of the O 1s core level perceived at 530.1 and 531.5 eV. The binding energy of the O 1s peak at 530.1 eV is assigned to TiO2. The second peak related to O 1s of W–O–W [32]. The measured binding energies of W 4f showed four photo electron peaks at 34.2, 35.5, 36.6 and 37.3 eV. The binding energy values at 35.5 and 36.6 eV assigned to W 4f7/2 and W 4f5/2, respectively. These are related to characteristic band of Keggin ion due to spin–orbit splitting. These values are in agreement with literature data [33]. It has been reported that binding energy values at 34.2 and 37.3 related to the presence of water on the surface [32]. The BE of the V 2p3/2 core level at 517.2 eV is characteristic of V+5 ions [34].

XPS spectra of 20 % TPAV1/TiO2
The characterisation results support the intact Keggin ion of TPAV1/TiO2. TPAV1 is well dispersed on titania and the catalyst with 20 wt% showed optimum dispersion. The catalyst with 20 % TPAV1/TiO2 showed high activity mainly due to the presence of well dispersed TPAV1.
The most active 20 wt% TPAV1/TiO2 catalyst was further studied to evaluate the reaction parameters. The reaction was carried using different solvents and the results are shown in Table 4.3. The reaction was found to proceed more selectively in polar solvents and ineffective in nonpolar solvents. In the absence of solvent, the reaction did not proceed. Acetonitrile was found to be solvent of choice for this reaction.
The effect of styrene to H2O2 mol ratio on activity was studied and the results are shown in Table 4.4. The reaction was carried out by varying mole ratio of styrene to H2O2 from 1:1 to 1:3. The increase in concentration of H2O2 conversion of styrene and selectivity to benzaldehyde was increased. Almost quantitative yields are obtained with styrene to H2O2 mol ratio of 1:3. About 97 % conversion of styrene and 85 % selectivity to benzaldehyde were obtained at this molar ratio.
The effect of reaction temperature on oxidative cleavage of styrene was also studied. The best results were obtained when the reaction was carried at room temperature. With the increase in reaction temperature, the conversion of styrene did not vary considerably, and at the same time, the selectivity to benzaldehyde was decreased. This is due to the further oxidation of benzaldehyde to benzoic acid, a stable oxidation product.
The scope of the reaction for various styrene derivatives is explored with the optimised reaction conditions, and the results are presented in Table 4.5. The reaction proceeds smoothly with electron donating and withdrawing groups on aromatic ring (entries 1–9). With α-methyl styrene (entry 10), 98 % conversion and 97 % selectivity towards acetophenone were obtained. The reaction with cinnamaldehyde (entry 12) resulted about 50 % conversion with 96 % selectivity to benzaldehyde. When stilbene (entry 13) was used, the activity of the catalyst is low with only 30 % conversion and 96 % selectivity to benzaldehyde. The low activity for this olefin might be due to its steric hindrance. The present catalyst is also active for oxidative cleavage of aliphatic olefins. Aliphatic olefin 1-hexene gave 20 % conversion and 95 % selectivity to pentanal, whereas 3-methyl-1-pentene afforded 20 % conversion with 90 % selectivity to 2-methylbunal.
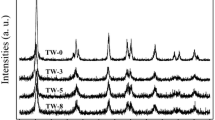
The catalyst was studied for its reusability and results obtained by recycling the catalyst are summarised in Table 4.6. Even after the third cycle, the catalyst showed almost consistent activity and selectivity. The FT-IR and Raman spectra of used catalyst are shown in Fig. 4.6. The characterisation results suggest the presence of intact Keggin ion structure of TPAV1 on titania for the used catalyst. The recycling results reiterate the heterogeneous nature of the catalysts for the oxidative cleavage of olefins. A separate experiment was carried to prove the heterogeneous nature of the catalyst. A styrene conversion of 50 % with about 87 % selectivity to benzaldehyde was obtained for a 4 h course of the reaction in the presence of 20 wt% TPAV1/TiO2. After quick removal of the catalyst by filtration, the reaction was further carried out for 4 h under the same conditions. There was no appreciable increase in conversion (50 %) and selectivity (86.5 %). This result reveals that the oxidation of styrene took place over the catalyst.

(a) Raman spectra and (b) FT-IR pattern of fresh and used catalysts
6 Conclusions
In conclusion a simple and highly efficient catalyst for the selective oxidative cleavage of olefins to carbonyl compounds at room temperature was demonstrated. The activity of the catalyst depends on vanadium present in the primary structure of Keggin ion rather than secondary structure. The activity also depends on the dispersed amount of TPAV1 on support. This catalyst is effective for oxidation of various styrene derivatives. The present catalyst is inexpensive, noncorrosive, environmentally friendly and easy to recover and recyclable without loss of activity and selectivity.
References
Kuhn FE, Fischer RW, Herrmann WA, Weskamp T (2004) In: Beller M, Bolm C (eds) Transition metals for organic synthesis. Wiley-VCH, Weinheim
Hudlicky M (1990) Oxidations in organic chemistry, American Chemical Society Monograph 186. American Chemical Society, Washington, DC
Larock RC (1999) Comprehensive organic transformations, 2nd edn. Wiley–VCH, New York
Bailey PS (1982) Ozonation in organic chemistry. Academic, New York
Criegee R (1975) Angew Chem Int Ed Engl 14:745–752
Koike K, Inoue G, Fukuda T (1999) J Chem Eng Jpn 32:295–299
Ogle RA, Schumacher JL (1998) Process Saf Prog 17:127–133
Wang A, Jiang H (2010) J Org Chem 75:2321–2326
Xing D, Guan B, Cai G, Fang Z, Yang L, Shi Z (2006) Org Lett 8:693–696
Yang D, Zang C (2001) J Org Chem 66:4814–4818
Sharma P, Patel A (2009) J Mol Catal A Chem 299:37–43
Pathan S, Patel A (2011) Dalton Trans 40:348–355
Hu J, Li K, Li W, Ma F, Guo Y (2009) Appl Catal A Gen 364:211–220
Maurya MR, Bisht M, Avecilla F (2011) J Mol Catal A Chem 344:18–27
Paradeshi SK, Pawar RY (2011) J Mol Catal A Chem 334:35–43
Kozhevnikov IV (1995) Catal Rev Sci Eng 37:311–352
Lim SS, Kim YH, Park GI, Lee WY, Song IK, Youn HK (1999) Catal Lett 60:199–204
Mori H, Mizuno N, Misono M (1990) J Catal 131:133–142
Nagaraju P, Pasha N, Sai Prasad PS, Lingaiah N (2007) Green Chem 9:1126–1129
Venkateswara Rao KT, Rao PSN, Nagaraju P, Sai Prasad PS, Lingaiah N (2009) J Mol Catal A Chem 303:84–89
Lingaiah N, Mohan Reddy K, Nagaraju P, Sai Prasad PS, Wachs IE (2008) J Phys Chem C 112:8294–8300
Tsigdinos GA, Hallada CJ (1968) Inorg Chem 7:437–441
Lingaiah N, Molinari JE, Wachs IE (2009) J Am Chem Soc 131:15544–15554
Dias JA, Osegovic JP, Drago RS (1998) J Catal 183:83–90
Thouvenot R, Fournier M, Franck R, Rocchiccioli - Deltcheff C (1984) Inorg Chem 23:598–605
Rocchiccioli-deltcheff C, Fournier M, Franck R, Thouvenot R (1983) Inorg Chem 22:207–216
Mallick K, Witcomb MJ, Scurrell MS (2004) Appl Catal A Gen 259:163–168
Kompio PGWA, Brückner A, Hipler F, Auer G, Löffler E, Grünert W (2012) J Catal 286:237–247. doi: 10.1016/j.jcat.2011.11.008
Chena L, Li J, Gea M (2011) Chem Eng J 170:531–537
Damyanova S, Fierro JLG, Sobrados I, Sanz J (1990) Langmuir 15:469–476
Damyanova S, Fierro J (1998) Chem Mater 10:871–879
Jalil PA, Faiz M, Tabet N, Hamdan NM, Hussain Z (2003) J Catal 217:292–297
Xu L, Li W, Hu J, Li K, Yang X, Ma F, Guo Y, Yu X, Guo Y (2009) J Mater Chem 19:8571–8579
Delichere P, Bere EK, Abon M (1998) Appl Catal A 172:295–309
Acknowledgement
One of the authors, KTVR, thanks the Council of Scientific and Industrial Research (CSIR), India, for the financial support in the form of Senior Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Panch Tattva Publishers, Pune, India
About this chapter
Cite this chapter
Lingaiah, N., Rao, K.T.V., Prasad, P.S.S. (2013). Vanadium-Substituted Tungstophosphoric Acid Supported on Titania: A Heterogeneous Catalyst for Selective Oxidative Cleavage of Olefins to Carbonyl Compounds at Room Temperature. In: Patel, A. (eds) Environmentally Benign Catalysts. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6710-2_4
Download citation
DOI: https://doi.org/10.1007/978-94-007-6710-2_4
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6709-6
Online ISBN: 978-94-007-6710-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)