
Abstract
Carbon allotropes, built up as hyper-structures of the classical diamond and having a high percentage of sp3 carbon atoms and pentagons, are generically called diamond D5. Four allotropes are discussed in this chapter: a spongy net; a dense hyper-diamond D5, with an “anti”-diamantane structure; the corresponding hyper-lonsdaleite; and a quasi-diamond which is a fivefold symmetry quasicrystal with “sin”-diamantane structure. Substructures of these allotropes are presented as possible intermediates in a lab synthesis, and their energetics evaluated at Hartree-Fock, DFT, and DFTB levels of theory. A topological description of these networks is also given.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
Nano-era, a period starting since 1985 with the discovery of C60, is dominated by the carbon allotropes, studied for applications in nanotechnology. Among the carbon structures, fullerenes (zero dimensional), nanotubes (one dimensional), graphenes (two dimensional), diamonds, and spongy nanostructures (three dimensional) were the most studied (Diudea and Nagy 2007). Inorganic compounds also attracted the attention of scientists. Recent articles in crystallography promoted the idea of topological description and classification of crystal structures (Blatov et al. 2004, 2007, 2009; Delgado-Friedrichs and O’Keeffe 2005).
Dendrimers are hyper-branched nanostructures, made by a large number of (one or more types) substructures called monomers, synthetically joined within a rigorously tailored architecture (Diudea and Katona 1999; Newkome et al. 1985; Tomalia 1993). They can be functionalized at terminal branches, thus finding a broad pallet of applications in chemistry, medicine, etc. (Tang et al. 1996; Pan et al. 2007).
Multi-tori are structures of high genera (Diudea 2005b, 2010b; Diudea and Nagy 2007), consisting of more than one tubular ring. Such structures can appear in spongy carbon or in zeolites (DeCarli and Jamieson 1961; Aleksenski et al. 1997; Krüger et al. 2005). Spongy carbon has recently been synthesized (Benedek et al. 2003; Barborini et al. 2002).
There are rigid monomers that can self-assemble in dendrimers, but the growing process stops rather at the first generation. At a second generation, yet the endings of repeat units are not free, they fit to each other, thus forming either an infinite lattice, if the monomer symmetry is octahedral, or a spherical multi-torus, if the symmetry is tetrahedral. The last one is the case of structures previously discussed by Diudea and Ilic (2011).
A detailed study on a multi-torus (Diudea 2010a; Diudea and Ilic 2011), built up by a tetrapodal monomer designed by Trs(P 4(T)) sequence of map operations (Diudea 2005a, b; Diudea et al. 2006) and consisting of all pentagonal faces, revealed its dendrimer-like structure (given as the number of monomer units added at each generation, in a dendrimer divergent synthesis, up to the 5th one): 1; 4; 12, 24, 12, 4. Starting with the second generation (i.e., the stage when first 12 monomers were added), pentagonal super-rings appear, leading finally to the multi-torus. The above sequence will be used to suggest a synthetic way to the multi-cage C57, which is the reduced graph of the above multi-torus and one of the main substructures of the~diamond D5.
This chapter is organized as follows: after a short introduction, the main substructures of the D5 diamonds are presented in Sect. 5.2, while the networks structure is detailed in Sect. 5.3. The next section provides a topological description of the nets, and computational details are given in Sect. 5.5. The chapter ends with conclusions and references.
5.2 Main Substructures of D5
Carbon allotropes, built up as hyper-structures of the classical diamond and having a high percentage of sp3 carbon atoms and pentagons, are generically called diamond D 5 (Diudea 2010a, b; Diudea and Nagy 2012; Diudea et al. 2012). The most important substructures, possible intermediates in the synthesis of D5, are detailed in the following.
5.2.1 Structure C57
Structure C57, above mentioned, can be “composed” by condensing four C20 cages so that they share a common vertex. Starting from a tetrahedral configuration, geometry optimization of C57, without symmetry constraints, leads to a structure with D 2d symmetry. The deformation occurs because of the degeneracy of the frontier orbitals. Maximal symmetry can be achieved by an octa-anionic form. This can be explained if we consider C57 consisting of two fragments: the core (in blue, Fig. 5.1), i.e., the centrohexaquinane C17 (Fig. 5.4 – Paquette and Vazeux 1981; Kuck 2006) which is capped by four acepentalene (Haag et al. 1996) fragments (consisting of only three-valence carbon atoms – marked in red, Fig. 5.1). A theoretical study (Zywietz et al. 1998) has shown the ground state of acepentalene C10H6 with 10π electrons has C s symmetry. Since in C57 the four acepentalene fragments are isolated from each other, their local geometry is close to the isolated acepentalene molecule. The dianion of acepentalene, with 12π electrons, is a stable and aromatic structure (C10H6 2−-C 3v) and has been isolated as salts (Haag et al. 1998). If two electrons are added for each acepentalene fragment, the geometry optimization resulted in a structure with tetrahedral symmetry C57 8−-T d.

C57 multi-cage in different views
Figure 5.2 summarizes the geometry and local ring aromaticity in the acepentalene fragments of C57 and its anion (in italics) compared to that of acepentalene (underlined values) and its dianion (in italics). It can be seen that in C57 the bonds are in general slightly longer than in its octa-anionic form, where the bond lengths are nearly uniform, ranging from 1.43 to 1.48 Ǻ. Notice the central bonds (AB = AC = 1.48) are much longer in C57 than in C10H6 2−, with implications in the pyramidalization of the central atom “A” (see below).

Bond lengths and NICS values (boldface) in the acepentalene fragment for C57-D 2d and C57 8−-T d (italics) and in C10H6-C s (underlined) and C10H6 2−-C 3v (italics) obtained at the B3LYP/6-311G(2d,p) level of theory
The NICS study revealed that, in C57 8−, the fragment is aromatic and nearly the same as the acepentalene dianion. However these rings are more antiaromatic in C57 than in C10H6-C s.
The local strain energy of the three-coordinated atoms, induced by deviation from planarity, was evaluated by the POAV theory (Haddon 1987, 1990) and is presented in Table 5.1. Notice that both in C57 and its anionic form there is a big strain on each atom compared to the isolated acepentalene. The central atom “A” has the largest strain and becomes a reactive site, particularly in case of C57 8−; this polar atom is then pushed away from the molecule, and therefore the C20 moieties have an elongated shape.
Strain relief could be achieved by partial or total hydrogenation (in general, exohedral derivatization). There are known examples of non-IPR fullerenes that are stabilized by hydrogenation/halogenation of their pentagon double/triple substructures (Wahl et al. 2006; Prinzbach et al. 2006; Chen et al. 2004; Fowler and Heine 2001; Han et al. 2008). Patterns appearing in the partially hydrogenated C57 structure are illustrated in Fig. 5.3.

Patterns of the partially hydrogenated C57 structure. The black dots correspond to the linking position of the hydrogen atoms in the acepentalene fragment
All possible isomers in the addition of hydrogen to C57 were checked: an even number of hydrogen atoms (with one exception) were added to each acepentalene fragment, from four up to ten (i.e., complete reduced species), only the lowest energy isomers being illustrated in Fig. 5.3. Exception was the case when added five hydrogen atoms and one electron, thus resulting an isomer with one aromatic pentagon in each acepentalene fragment. Both C57H24 and C57H32 have two isomers, with symmetries D 2d and S 4, respectively, and very close stability (the difference in their total energy is only 0.01 kcal/mol, while in the HOMO-LUMO gap is 0.05 eV). In the totally reduced species C57H40, the bond lengths are in the range of 1.52 (core)–1.56 Ǻ (periphery) compared to 1.55 Ǻ in the dodecahedrane, so that the C20 fragments regain a quasi-spherical shape. Single-point calculations for hydrogenated C57H n derivatives are listed in Table 5.2.
The stabilization by hydrogenation is more pregnant in case of C20; while dodecahedrane C20H20 was synthesized in amounts of grams, the efforts of scientists to prove the existence of the smallest fullerene C20 are well-known (Paquette and Balogh 1982; Prinzbach et al. 2006).
Possible intermediates in the pathway to C57 molecule, starting from C17 considered the “seed” of D5, are presented in Fig. 5.4, while the single-point calculation data are shown in Table 5.3. These species could be used as derivatives (e.g., halogenated and hydrogenated ones) in the building of further structures. Their stability was evaluated as partially hydrogenated species, the red bonds in Fig. 5.4 being kept as double bonds. The vibrational spectra of these molecules evidenced a very rigid carbon skeleton, only the hydrogen atoms presenting intense signals. Of particular interest are the outer (red) bonds in C17, the length of which varying by the structure complexity. As the structure grows, an increase in their strain appears provoking an elongation of the mentioned bond. This can be observed in the increase of the total energy per carbon atom (and decrease of the gap energy) in the order C17 < C41 < C53. However, with further addition of C1/C2 fragments, finally leading to a periodic network (see below), the considered bonds are shortened progressively.

Structures in the pathway to C57 (1; 4; 12, 24, 12, 4); the number of atoms added shell by shell is given in brackets
A way from C57 to D5 could include C65 and C81 intermediates (see Fig. 5.5). The stability of these structures was evaluated as hydrogenated species (Table 5.3, the last two rows). The structure C81 (with a C57 core and additional 12 flaps) is the monomer of spongy D5 network (see below). Its stability is comparable to that of the reduced C17 seed (Table 5.3, first row) and also to that of the fully reduced C57 (Table 5.2, last row), thus supporting the viability of the spongy lattice.

Intermediate structures to D5 network
5.2.2 Hyper-Adamantane
Other substructures/intermediates, related to D5, could appear starting from C17. The seed C17 can dimerize (probably by a cycloaddition reaction) to C34H12 (Fig. 5.6), a C20 derivative bearing 2 × 3 pentagonal wings in opposite polar disposition. The dimer can further form an angular structure C51 (Fig. 5.6, right).

Steps to ada_20: C17 (left) dimerizes to C34H12 (middle) and trimerizes to C51H14 (right)
A linear analogue is energetically also possible. The angular tetramer C51 will compose the six edges of a tetrahedron in forming an adamantane-like ada_20_170, with six pentagonal wings (in red – Fig. 5.7, left) or without wings, as in ada_20_158 (Fig. 5.7, central). Energetic data for these intermediates are given in Table 5.4. The unit ada_20_158 consist of 12 × C20 cages, the central hollow of which exactly fitting the structure of fullerene C28. A complete tetrahedral ada_20_196 consist of 16 × C20 or 4 × C57 units. The hyper-adamantane is the repeating unit of the dense diamond D5 (see below). A corresponding ada_28_213 can be conceived starting from C28 (Fig. 5.7, right).

Adamantane-like structures: ada_20_170 (left), ada_20_158 (central), and ada_28_213 (right)
In the above symbols, “20” refers to C20, as the basic cage in the frame of dense diamond D5 (see below), while the last number counts the carbon atoms in the structures.
5.3 Diamond D5 Allotropes
Four different allotropes can be designed, as will be presented in the following.
5.3.1 Spongy Diamond D5
In spongy diamond D5 (Fig. 5.8), the nodes of the network consist of alternating oriented (colored in red/blue) C57 units; the junction between two nodes recalls a C20 cage. The translational cell is a cube of eight C57 entities. This network is a decoration of the P-type surface; it is a new 7-nodal 3,3,4,4,4,4,4-c net, group Fm-3m; point symbol for net: (53)16(55.8)36(56)17; stoichiometry (3-c)4(3-c)12(4-c)24(4-c)12 (4-c)12(4-c)4(4-c).

Spongy D5 (C57) triple periodic network
The density of the net varies around an average of d = 1.6 g/cm3, in agreement with the “spongy” structure illustrated in Fig. 5.8.
5.3.2 Diamond D5
The ada_20 units can self-arrange in the net of dense diamond D5 (Fig. 5.9, left). As any net has its co-net, the diamond D5_20 net has the co-net D5_28 (Fig. 5.9, right), with its corresponding ada_28_213 unit (Fig. 5.7, right). In fact it is one and the same triple periodic D5 network, built up basically from C20 and having as hollows the fullerene C28.

Diamond D5_20 net and its co-net D5_28 represented as (k,k,k)-cubic domains: D5_20_3,3,3_860 (left) and D5_28_3,3,3_1022 (right); “k” is the number of repeating units on each edge of the domain
This dominant pentagon-ring diamond (Fig. 5.8) is the mtn triple periodic, three-nodal net, namely, ZSM-39, or clathrate II, of point symbol net: {5^5.6}12{5^6}5 and 2[512]; [512.64] tiling, and it belongs to the space group: Fd-3m. For all the crystallographic data, the authors acknowledge Professor Davide Proserpio, University of Milan, Italy.
Domains of this diamond network, namely, D5_20_3,3,3_860 and D5_28_3,3, 3_1022 co-net, were optimized at the DFTB level of theory (Elstner et al. 1998). Hydrogen atoms were added to the external carbon atoms of the network structures, in order to keep the charge neutrality and the sp3 character of the C–C bonds at the network surface. Energetically stable geometry structures were obtained in both cases, provided the same repeating unit was considered.
Identification of the equivalent carbon atoms in the neighboring units of the 3 × 3 × 3 super-cell along the main symmetry axes, envisaged a well-defined triclinic lattice, with the following parameters: a = b = c = 6.79 Å and α = 60°, β = 120°, γ = 120°, even the most symmetrical structure is fcc one. Density of the D5 network was calculated to be around 2.8 g/cm3.
Analyzing the C–C bond distances in these carbon networks, the values vary in a very narrow distance domain of 1.50–1.58 Å, suggesting all carbon atoms are sp3 hybridized. Considering the one-electron energy levels of the HOMO and LUMO, a large energy gap could be observed for both D5_20_860 net (E HOMO = −5.96 eV, E LUMO = +2.10 eV, ΔE HOMO−LUMO = 8.06 eV) and D5_28_1022 co-net (E HOMO = −6.06 eV, E LUMO = +2.45 eV, ΔE HOMO−LUMO = 8.51 eV) structures, which indicates an insulating behavior for this carbon network.
Structural stability of substructures related to the D5 diamond was evaluated both in static and dynamic temperature conditions by molecular dynamics MD (Kyani and Diudea 2012; Szefler and Diudea 2012). Results show that C17 is the most temperature resistant fragment. For a detailed discussion, see Chap. 7.
Note that the hypothetical diamond D5 is also known as fcc-C34 because of its face-centered cubic lattice (Benedek and Colombo 1996). Also note that the corresponding clathrate structures are known in silica synthetic zeolite ZSM-39 (Adams et al. 1994; Meier and Olson 1992; Böhme et al. 2007) and in germanium allotrope Ge(cF136) (Guloy et al. 2006; Schwarz et al. 2008) as real substances.
5.3.3 Lonsdaleite L5
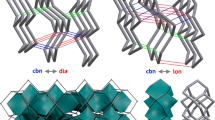
Alternatively, a hyper-lonsdaleite L5_28 network (Fig. 5.10, left) can be built (Diudea et al. 2011, 2012) from hyper-hexagons L5_28_134 (Fig. 5.10, right), of which nodes represent C28 fullerenes, joined by identifying the four tetrahedrally oriented hexagons of neighboring cages. The lonsdaleite L5_28/20 is a triple periodic network, partially superimposed to the D5_20/28 net. Energetic data for the structures in Fig. 5.10 are given in Table 5.4.

Lonsdaleite L5_28 net represented as L5_28_250 (side view – left); the substructure L5_28_134 is a hyper-hexagon of which nodes are C28 with additional two C atoms, thus forming a C20 core (top view – central); the L5_20 co-net (in red) superimposes partially over the net of D5_20 (side view – right) in the domain (k,k,2)
5.3.4 Quasi-Diamond D5
A fourth allotrope of D5 was revealed by Diudea (Chap. 19) as D5_sin quasicrystal diamond (Fig. 5.11), clearly different from the “classical” D5, named here D5_anti. The quasi-diamond D5_sin is a quasicrystal 27 nodal 3,4-c net, of the Pm group, with the point symbol: {53}18{55.6}18 {55.8}16{56}13. Substructures of this new allotrope are shown in the top of Fig. 5.11.

Diamond D5-related structures
5.4 Topological Description
Topology of diamond D5, namely, spongy D5 (Fig. 5.8) and D5_anti (Fig. 5.9), is presented in Tables 5.5 and 5.6, respectively: formulas to calculate the number of atoms, number of rings R, and the limits (at infinity) for the ratio of sp3 C atoms over the total number of atoms and also the ratio R[5] over the total number of rings are given function of k that is the number of repeating units in a cuboid (k,k,k). One can see that, in an infinitely large net, the content of sp3 carbon approaches 0.77 in case of spongy net while it is unity in case of dense diamond D5.
5.5 Computational Methods
Geometry optimizations were performed at the Hartree-Fock (HF) and density functional (DFT) levels of theory using the standard polarized double-zeta 6-31G(d,p) basis set. For DFT calculations, the hybrid B3LYP functional was used. Harmonic vibrational frequencies were calculated for all optimized structures at the same level of theory to ensure that true stationary points have been reached. Symmetry was used to simplify calculations after checking the optimizations without symmetry constraints resulted in identical structures. The following discussion only considers the singlet states.
To investigate the local aromaticity, NICS (nucleus-independent chemical shift) was calculated on the DFT optimized geometries. NICS was measured in points of interest using the GIAO (Gauge-Independent Atomic Orbital) method at GIAO-B3LYP/6-311G(2d,p)//B3LYP/6-311G(2d,p). Calculations were performed using the Gaussian 09 package (Gaussian 09 2009).
For larger structures, geometry optimization was performed at SCC-DFTB level of theory (Elstner et al. 1998) by using the DFTB+ program (Aradi et al. 2007) with the numerical conjugated gradient method.
Strain energy, induced by deviation from planarity, appearing in such nanostructures, was evaluated by the POAV theory (Haddon 1987, 1990), implemented in our JSChem software (Nagy and Diudea 2005).
Topological data were calculated by our NanoStudio software (Nagy and Diudea 2009).
5.6 Conclusions
Four allotropes of the diamond D5 were discussed in this chapter: a spongy net; a dense hyper-diamond D5, with an “anti”-diamantane structure; the corresponding hyper-lonsdaleite; and a quasi-diamond which is a fivefold symmetry quasicrystal with “sin”-diamantane structure. The main substructures of these allotropes were presented as possible intermediates in a lab synthesis on the ground of their energetics, evaluated at Hartree-Fock, DFT, and DFTB levels of theory. A topological description of these networks, made in terms of the net parameter k, supports the generic name diamond D5 given to these carbon allotropes; among these, the spongy and quasi-diamond represent novel networks of D5.
References
Adams GB, O’Keeffe M, Demkov AA, Sankey OF, Huang Y-M (1994) Wide-band-gap Si in open fourfold-coordinated clathrate structures. Phys Rev B 49(12):8048–8053
Aleksenski AE, Baidakova MV, Vul’ AY, Davydov VY, Pevtsova YA (1997) Diamond-graphite phase transition in ultradisperse-diamond clusters. Phys Solid State 39:1007–1015
Aradi B, Hourahine B, Frauenheim T (2007) DFTB+, a sparse matrix-based implementation of the DFTB method. J Phys Chem A 111:5678–5684
Barborini E, Piseri P, Milani P, Benedek G, Ducati C, Robertson J (2002) Negatively curved spongy carbon. Appl Phys Lett 81:3359–3361
Benedek G, Colombo L (1996) Hollow diamonds from fullerenes. Mater Sci Forum 232:247–274
Benedek G, Vahedi-Tafreshi H, Barborini E, Piseri P, Milani P, Ducati C, Robertson J (2003) The structure of negatively curved spongy carbon. Diam Relat Mater 12:768–773
Blatov VA, Carlucci L, Ciani G, Proserpio DM (2004) Interpenetrating metal-organic and inorganic 3D networks: a computer-aided systematic investigation. Part I. Analysis of the Cambridge structural database. CrystEngComm 6:377–395
Blatov VA, Delgado-Friedrichs O, O’Keeffe M, Proserpio DM (2007) Three-periodic nets and tilings: natural tilings for nets. Acta Crystallogr Sect A Found Crystallogr 63(5):418–425
Blatov VA, O’Keeffe M, Proserpio DM (2009) Vertex-, face-, point-, Schläfli-, and Delaney-symbols in nets, polyhedra and tilings: recommended terminology. CrystEngComm 12(1):44–48
Böhme B, Guloy A, Tang Z, Schnelle W, Burkhardt U, Baitinger M, Grin Y (2007) Oxidation of M4Si4 (M = Na, K) to clathrates by HCl or H2O. J Am Chem Soc 129:5348–5349
Chen Z, Heine T, Jiao H, Hirsch A, Thiel W, Schleyer PVR (2004) Theoretical studies on the smallest fullerene: from monomer to oligomers and solid states. Chem Eur J 10(4):963–970
DeCarli PS, Jamieson JC (1961) Formation of diamond by explosive shock. Science 133:1821–1822
Delgado-Friedrichs O, O’Keeffe M (2005) Crystal nets as graphs: terminology and definitions. J Solid State Chem 178(8):2480–2485
Diudea MV (ed) (2005a) Nanostructures, novel architecture. NOVA Scientific Publishers, New York
Diudea MV (2005b) Nanoporous carbon allotropes by septupling map operations. J Chem Inf Model 45:1002–1009
Diudea MV (2010a) Diamond D5, a novel allotrope of carbon. Studia Univ Babes Bolyai Chemia 55(4):11–17
Diudea MV (2010b) Nanomolecules and nanostructures-polynomials and indices, MCM, No. 10. University of Kragujevac, Serbia
Diudea MV, Ilić A (2011) All-pentagonal face multi tori. J Comput Theor Nanosci 8:736–739
Diudea MV, Katona G (1999) Molecular topology of dendrimers. In: Newkome GA (ed) Adv Dendritic Macromol 4(1999):135–201
Diudea MV, Nagy CL (2007) Periodic nanostructures. Springer, Dordrecht
Diudea MV, Nagy CL (2012) C20-related structures: diamond D5. Diam Relat Mater 23:105–108
Diudea MV, Ştefu M, John PE, Graovac A (2006) Generalized operations on maps. Croat Chem Acta 79:355–362
Diudea MV, Nagy CL, Ilic A (2011) Diamond D5, a novel class of carbon allotropes. In: Putz MV (ed) Carbon bonding and structures. Carbon materials: chemistry and physics, vol 5. Springer, Dordrecht, pp 273–289
Diudea MV, Nagy CL, Bende A (2012) On diamond D5. Struct Chem 23:981–986
Elstner M, Porezag D, Jungnickel G, Elsner J, Haugk M, Frauenheim T, Suhai S, Seifert G (1998) Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys Rev B 58:7260–7268
Fowler PW, Heine T (2001) Stabilisation of pentagon adjacencies in the lower fullerenes by functionalization. J Chem Soc Perkin Trans 2:487–490
Gaussian 09 Rev. A.1, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian Inc, Wallingford
Guloy A, Ramlau R, Tang Z, Schnelle W, Baitinger M, Grin Y (2006) A quest-free germanium clathrate. Nature 443:320–323
Haag R, Schröder D, Zywietz T, Jiao H, Schwarz H, Von Schleyer PR, de Meijere AT (1996) The long elusive acepentalene – experimental and theoretical evidence for its existence. Angew Chem 35:1317–1319
Haag R, Schüngel F-M, Ohlhorst B, Lendvai T, Butenschön H, Clark T, Noltemeyer M, Haumann T, Boese R, de Meijere A (1998) Syntheses, structures, and reactions of highly strained dihydro- and tetrahydroacepentalene derivatives. Chem Eur J 4:1192–1200
Haddon RC (1987) Rehybridization and π-orbital overlap in nonplanar conjugated organic molecules: π-orbital axis vector (POAV) analysis and three-dimensional hückel molecular orbital (3D-HMO) theory. J Am Chem Soc 109:1676–1685
Haddon RC (1990) Measure of nonplanarity in conjugated organic molecules: which structurally characterized molecule displays the highest degree of pyramidalization? J Am Chem Soc 112:3385–3389
Han X, Zhou S-J, Tan Y-Z, Wu X, Gao F, Liao Z-J, Huang R-B, Feng Y-Q, Lu X, Xie S-Y, Zheng L-S (2008) Crystal structures of saturn-like C50Cl10 and pineapple-shaped C64Cl4: geometric implications of double- and triple-pentagon-fused chlorofullerenes. Angew Chem Int Ed 47:5340–5343
Krüger A, Kataoka F, Ozawa M, Fujino T, Suzuki Y, Aleksenskii AE, Vul’ AYA, Ōsawa E (2005) Unusually tight aggregation in detonation nanodiamond: identification and disintegration. Carbon 43:1722–1730
Kuck D (2006) Functionalized aromatics aligned with the three Cartesian axes: extension of centropolyindane chemistry. Pure Appl Chem 78:749–775
Kyani A, Diudea MV (2012) Molecular dynamics simulation study on the diamond D5 substructures. Central Eur J Chem 10(4):1028–1033
Meier WM, Olson DH (1992) Atlas of zeolite structure types, 3rd edn. Butterworth-Heinemann, London
Nagy CL, Diudea MV (2005) JSChem. Babes–Bolyai University, Cluj
Nagy CL, Diudea MV (2009) NANO-Studio software. Babes-Bolyai University, Cluj
Newkome GR, Yao Z, Baker GR, Gupta VK (1985) Micelles. Part 1. Cascade molecules: a new approach to micelles. A [27]-arborol. J Org Chem 50:2003–2004
Pan BF, Cui DX, Xu P, Huang T, Li Q, He R, Gao F (2007) Cellular uptake enhancement of polyamidoamine dendrimer modified single walled carbon nanotubes. J Biomed Pharm Eng 1:13–16
Paquette LA, Balogh DW (1982) An expedient synthesis of 1,16-dimethyldodecahedrane. J Am Chem Soc 104:774–783
Paquette LA, Vazeux M (1981) Threefold transannular epoxide cyclization. Synthesis of a heterocyclic C17-hexaquinane. Tetrahedron Lett 22:291–294
Prinzbach H, Wahl F, Weiler A, Landenberger P, Wörth J, Scott LT, Gelmont M, Olevano D, Sommer F, Issendoef B (2006) C20 carbon clusters: fullerene-boat-sheet generation, mass selection, photoelectron characterization. Chem Eur J 12:6268–6280
Schwarz U, Wosylus A, Böhme B, Baitinger M, Hanfland M, Grin Y (2008) A 3D network of four-bonded germanium: a link between open and dense. Angew Chem Int Ed 47:6790–6793
Szefler B, Diudea MV (2012) On molecular dynamics of the diamond D5 seed. Struct Chem 23(3):717–722
Tang MX, Redemann CT, Szoka FC Jr (1996) In vitro gene delivery by degraded polyamidoamine dendrimers. Bioconjug Chem 7:703–714
Tomalia DA (1993) Starburst™/cascade dendrimers: fundamental building blocks for a new nanoscopic chemistry set. Aldrichim Acta 26:91–101
Wahl F, Weiler A, Landenberger P, Sackers E, Voss T, Haas A, Lieb M, Hunkler D, Worth J, Knothe L, Prinzbach H (2006) Towards perfunctionalized dodecahedranes – en route to C20 fullerene. Chem Eur J 12:6255–6267
Zywietz TK, Jiao H, Schleyer PR, de Meijere A (1998) Aromaticity and antiaromaticity in oligocyclic annelated five-membered ring systems. J Org Chem 63:3417–3422
Acknowledgments
CL Nagy acknowledges the financial support of the Sectorial Operational Programme for Human Resources Development 2007–2013, cofinanced by the European Social Fund, under the project number POSDRU 89/1.5/S/60189 with the title “Postdoctoral Programs for Sustainable Development in a Knowledge Based Society.”
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Nagy, C.L., Diudea, M.V. (2013). Diamond D5 . In: Diudea, M., Nagy, C. (eds) Diamond and Related Nanostructures. Carbon Materials: Chemistry and Physics, vol 6. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6371-5_5
Download citation
DOI: https://doi.org/10.1007/978-94-007-6371-5_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6370-8
Online ISBN: 978-94-007-6371-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)