
Abstract
Cells traverse the cell cycle through G1 → S → G2 → M phases, and then divide into two daughter cells, which then enter the next cycle or exit to a quiescent G0 phase. This process is tightly controlled by serine–threonine kinases named cyclin-dependent kinases (CDKs). CDKs, as catalytic subunits, become active only in association with their regulatory partner cyclins (e.g., cyclin D–CDK4/CDK6, cyclin E–CDK2, cyclin A–CDK2, cyclin B–CDK1, cyclin C–CDK3). Full activation of the cyclin–CDK holoenzymes requires phosphorylation at particular sites in CDKs. CDK activity is also negatively regulated by direct interaction with CDK inhibitors, which consist of two families, the inhibitor of CDK4 (INK4) family, which specifically inhibit cyclin D-associated kinases, and the kinase inhibitor protein (Cip/Kip) family, which inhibit most CDKs. Dysregulation of these genes (e.g., CDK inhibitors, cyclins, and CDKs themselves) is a common mechanism responsible for out-of-control cell growth, the main characteristic in cancer. Beyond cell cycle regulation, CDKs also play critical roles in gene transcription and neuronal function. In the former case, cyclin T–CDK9 and cyclin C–CDK8 are only involved in transcriptional regulation, whereas cyclin H–CDK7 is involved in regulation of both the cell cycle and transcription. In the latter case, so far CDK5 is the only characterized neuron-specific CDK that appears to function as a double-edged sword dependent on its binding partners (i.e., physiological p35/p39 vs pathological p25). Thus, CDKs are attractive targets for both cancer therapy and neuroprotection, and numerous pharmacological CDK inhibitors have been reported. One major challenge remains whether and how CDK(s) should be inhibited in either of the circumstances. This review summarizes current understanding and recent advances in this field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Cyclin-dependent kinases (CDKs) are a family of serine/threonine kinases that have been implicated in the regulation of cell cycle progression, transcription and neuronal function (Malumbres and Barbacid 2009). So far, about 20 mammalian CDKs have been identified, including CDK1–CDK19 (Malumbres and Barbacid 2005). The activity of most, if not all, CDKs requires the formation of holoenzymes consisting of both CDKs (catalytic subunits) and their partners, in most cases, cyclins (regulatory subunits) (Lapenna and Giordano 2009; Echalier et al. 2010). In words, binding of cyclins (or other partner proteins) is necessary for kinase activity of CDKs. Cyclin binding controls the substrate specificity of CDKs by providing targeting domains, which in turn determine their biological activity. To this end, at least 13 classes (A to L and T) of cyclins have been described so far (Malumbres and Barbacid 2005). Moreover, full activation of CDKs also requires phosphorylation of a threonine residue located between positions 159 and 174 within the T-loop of the kinase domain of CDKs, which is catalyzed by CDK-activating kinase (CAK; composed of CDK7 and cyclin H) (Larochelle et al. 2007). This crucial threonine residue is highly conserved from yeast to mammals. However, cyclin binding is most likely essential for the CAK-mediated phosphorylation of CDKs (Larochelle et al. 2007). Furthermore, the activity of CDKs can also be negatively regulated by phosphorylation of several residues in their active sites. As CDKs play different roles in the cell cycle, transcription, and neuronal function, they can be functionally divided into three classes, i.e., cell-cycle-regulatory, transcriptional, and neuron-specific CDKs.
2 Cell-Cycle-Regulatory CDKs
Cell cycle progression provides a mechanism which allows both normal and neoplastic cells to proliferate and grow. The cell cycle is divided into four distinct but tightly related phases, i.e., DNA synthesis (S phase) and mitosis (M phase), which are separated by two gaps (G1 and G2 phases) (Malumbres and Barbacid 2007). Following growth stimuli, cells traverse the cell cycle through G1 → S → G2 → M phases, and then divide to produce two daughter cells, which then enter G1 phase once again to initiate the next cycle or exit from the cell cycle into a quiescent G0 phase (Wesierska-Gadek et al. 2011). The G1 phase contains a transition point referred to as the restriction point which determines whether the cell cycle progression occurs independently of exogenous stimuli (Cicenas and Valius 2011).
Cell cycle progression is tightly controlled by the cyclin–CDK complex composed of cyclin and CDK in a 1:1 ratio. In a number of CDK complexes identified, CDK1, CDK2, CDK4, CDK6, cyclin A (A1 and A2), cyclin B (B1, B2, and B3), cyclin D (D1, D2, and D3), and cyclin E (E1 and E2) are directly involved in the cell cycle machinery (Canavese et al. 2012). CDKs are the catalytic subunits of the cyclin–CDK complexes, and their activity is regulated by several mechanisms, including their binding to the appropriate cyclin, their folding, and the phosphorylation of a threonine residue in a loop within their structure known as the T-loop (Merrick and Fisher 2012). CDKs are activated by their regulatory partners, members of the cyclin family. Binding of cyclins to this complex induces a conformational change in CDK structure producing a basal, active state (Knockaert et al. 2002). Cyclin–CDK complexes are activated by phosphorylation of CDKs at specific conserved threonine residues (e.g., Thr161 in CDK1, Thr160 in CDK2, Thr172 in CDK4, Thr177 in CDK6) within the T-loop of their kinase domains, a reaction catalyzed by CAK (cyclin H–CDK7 complex) (Larochelle et al. 2007). The activity of CDKs is also regulated by the (de)phosphorylation at conserved tyrosine and threonine residues (Thr14 and Tyr15 in CDK1 and CDK2). These critical residues are phosphorylated by the mixed-lineage kinases Wee1 and Myt1, rendering them inactive (Lapenna and Giordano 2009). The final activation of the cyclin–CDK holoenzymes occurs only after dephosphorylation of these residues, catalyzed by the dual-specificity phosphatases Cdc25s (Cdc25A, Cdc25B, and Cdc25C) (Lents et al. 2002). The activity of these phosphatases is regulated through their protein levels (e.g., Cdc25A) and/or intracellular location (e.g., Cdc25C), which is tightly regulated by proteins of the 14-3-3 family. The 14-3-3 bindings are triggered by phosphorylation of Cdc25s at multiple sites (e.g., Ser178, Thr507, Ser76, Ser123, Ser278, and Ser292 in Cdc25A; Ser309 and Ser361 in Cdc25B; Ser216 in Cdc25C), which in turn lead to βTrCP-dependent degradation via the ubiquitin–proteasome system (e.g., Cdc25A) or sequestration in the cytoplasm (e.g., Cdc25C) (Lapenna and Giordano 2009).
Cyclins are the regulatory components of the cyclin–CDK complexes. Their cellular levels fluctuate through the cell cycle, controlled by a finely tuned balance between de novo synthesis and degradation (Coudreuse and Nurse 2010). Cyclin expression determines a transition specifically from one phase to the next, as well as progression during a particular phase. Cyclins such as cyclins B, A, and E are predominantly regulated by an ubiquitin–proteasome-dependent degradation pathway (Coudreuse and Nurse 2010). These cyclins share a nine-residue sequence in the N-terminal region called the “destruction box,” which can be recognized by the enzyme ubiquitin ligase and resulting proteasomal degradation of the cyclins (Lapenna and Giordano 2009). In contrast, D-type cyclins are primarily regulated by transcriptional and translational mechanisms. The perfect timing of individual cyclin expression is controlled by the regulatory elements located in the gene promoters. Cyclins that are no longer needed undergo phosphorylation of specific residues, promoting their recruitment to the Skp1–Cullin–F-box protein or anaphase-promoting complex/cyclosome (APC/C) multiprotein complexes and subsequent degradation through the ubiquitin–proteasome system (Lapenna and Giordano 2009).
CDK activity is also negatively controlled by interactions with endogenous CDK inhibitors, which counterbalance cell cycle progression mediated by cyclin–CDK complexes. The CDK inhibitors are divided two families, the inhibitor of CDK4 (INK4) family including p16INK4a, p15INK4b, p18INK4c, and p19INK4d, which inhibit D-type cyclin-associated kinases (CDK4, CDK6), and the kinase inhibitor protein (Cip/Kip) family containing p21Cip1/Waf1, p27Kip1, and p57Kip2, which efficiently inhibit most CDKs, such as CDK2, CDK4, and CDK6 (Sandal 2002). The regulation of CDK activity by CDK inhibitors is an important mechanism in cell cycle progression after stimulation by mitogenic signals and particularly in tumorigenesis as one or multiple CDK inhibitors are often defected in human cancers. Protein levels of many CDK inhibitors are also regulated through transcriptional (e.g., promoter methylation) and posttranslational (e.g., phosphorylation, ubiquitin–proteasomal degradation) processes.
In mammalian cells, cell cycle progression is regulated by activation of CDKs in sequential order (Ortega et al. 2002) (Fig. 23.1): cyclin D–CDK4/CDK6 holoenzymes promote G1 progression, particularly passing of the restriction point, a point in the G1 phase at which the cell becomes “committed” to the cell cycle, after which extracellular mitogenic stimuli are no longer required. Then, cyclin E–CDK2 complexes act on the G1–S transition, followed by cyclin A–CDK2 on S phase progression. Last, cyclin B–CDK1 complexes control the G2–M transition, mitosis, and M phase exit. Thus, cyclins E, A, and B are expressed during the late G1, S, and G2 phases of the cell cycle, respectively.

Regulation of the cell cycle by cyclin-dependent kinases (CDK). pRb retinoblastoma protein
2.1 CDK4/CDK6
Cyclin D–CDK4/CDK6 complexes phosphorylate the retinoblastoma tumor suppressor protein (pRb; a primary member of the “pocket protein” family, which also contains p107 and p130), a key negative regulator of cell proliferation (Malumbres and Barbacid 2006). In quiescent cells and early G1 phase, pRb is dephosphorylated or hypophosphorylated, which halts cell cycle progression through interactions with the E2F family of transcription factors and thereby inhibition of their transactivation (Li et al. 2012). After phosphorylation by cyclin D–CDK4/CDK6, pRb is inactivated, releasing E2Fs from an inactive pRb–E2F complex (Yu et al. 2006). E2Fs are thus activated and bind to their heterodimeric partner DP-1, resulting in expression of genes responsible for S phase entry and progression, including cyclin E, which is required for CDK2 activation. Cyclin D–CDK4 can also phosphorylate the other “pocket protein” family members p130 and p107, which may then interact with certain E2Fs (e.g., E2F1 and E2F4) and mimic the function of pRb in RB1-null cells (Ciemerych et al. 2008).
Therefore, cyclin D–CDK4/CDK6 complexes are long been believed to be important, perhaps essential components of the core cell cycle apparatus for G1 progress. However, gene knockout of either CDK4 or CDK6, or both, as well as their partner cyclin D is not lethal in mice (Malumbres et al. 2004). Although CDK4 knockout results in insulin-deficient diabetes and partial sterility, mice lacking CDK4 are viable and CDK4−/− mouse embryonic fibroblasts (MEFs) proliferate normally. Moreover, CDK4 and CDK6 double-knockout mouse embryos display normal organogenesis at early stages and most cell types proliferate normally, although they die at late stages because of severe anemia. After serum stimulation, quiescent CDK4−⁄−/CDK6−⁄− cells are capable of entering S phase. Furthermore, cyclin D1/D2/D3 triple-knockout mouse embryos develop until mid/late gestation and die of heart abnormalities combined with a severe anemia (Kozar et al. 2004). Cyclin D1−/−/D2−/−/D3−/− MEFs proliferate almost normally but show increased requirement for mitogenic stimulation in cell cycle reentry (Kozar et al. 2004). As a conclusion, cyclin D–CDK4/CDK6 complexes are not as critical for cell cycle progression as previously thought, and these proteins are critically required for proliferation only in selected cell types, such as hematopoietic stem cells.
2.2 CDK2
Another important protein kinase involved in interphase progression is CDK2, which targets numerous substrates that are important in DNA replication and gene transcription (Horiuchi et al. 2012). CyclinE–CDK2 and cyclin A–CDK2 regulate the G1–S transition and S phase progression, respectively (Yu and Sicinski 2004). After being phosphorylated by cyclin D–CDK4/CDK6 in late G1 phase, pRb is further phosphorylated by cyclin E–CDK2, leading to complete inactivation of pRb (Ezhevsky et al. 2001), which is then able to drive the G1–S transition (Merrick et al. 2011). During S phase, cyclin A–CDK2 predominates and phosphorylates various protein substrates involved in DNA synthesis and replication (Wohlbold et al. 2012). Cyclin A–CDK2 also deactivates the E2F proteins, thereby shutting down E2F-dependent transcription (Morris et al. 2002). The active CDK2 complex persists in the nucleus throughout G2 phase. Activation of CDK2 complexes also requires dephosphorylation of Thr14 and Tyr15 by Cdc25s and phosphorylation of Thr160 by CAK (Sandal 2002). CDK2 is phosphorylated at Thr14 and Tyr15 by the dual-specificity kinases Wee1 and Myt1, resulting in inhibition of CDK2 kinase activity. Thr14 and Tyr15 are dephosphorylated by Cdc25s (particularly Cdc25A), leading to activation of both cyclin E–CDK2 and cyclin A–CDK2 (Lapenna and Giordano 2009).
The discovery that mouse embryos can develop normally after knockout of either CDK2 or cyclin E challenges the previous thought that cyclin E–CDK2 activity is strictly required for the cell cycle (Geng et al. 2003). Similarly to the case of cyclin D–CDK4/CDK6, CDK2 knockout mice are viable and develop normally (Ortega et al. 2003). CDK2−/− MEFs proliferate but delay entry into S phase. Quiescent CDK2−/− cells reenter the cell cycle without significant delay in response to stimulation with serum. Therefore, Cdk2 is not an essential gene in the mouse, although it is required for germ cell development and meiosis (Ortega et al. 2003). On the other hand, cyclin E1−/− and E2−/− mice develop normally, with the exception of deficient spermatogenesis in cyclin E−/− male mice (Yu and Sicinski 2004). Cyclin E1 and cyclin E2 double-knockout embryos die during mid-gestation, caused by placental abnormality (Yu and Sicinski 2004). MEFs from cyclin E-deficient embryos proliferate relatively normally during conditions of continuous cell cycling, but fail to reenter the cell cycle from the quiescent G0 state.
2.3 CDK1
Cyclin A–CDK2 and cyclin B–CDK1 govern the G2–M transition (Merrick and Fisher 2010b). The cyclin B–CDK1 complex also regulates the transition of cells into anaphase and through mitosis (Merrick and Fisher 2010a). CDK1 (previously referred to as cdc2) interacts primarily with cyclin B to regulate the G2–M transition (Nurse 2012). Expression of cyclin B is periodic. During interphase, protein levels of cyclin B gradually increase following G1, S and G2 phases, and reach a critical threshold at the end of G2 phase, which activates CDK1, thereby triggering onset of mitosis. During mitosis, cyclin B–CDK1 plays an essential role in control of cell division. The heterodimeric cyclin B–CDK1 complex is also known as maturation-promoting factor, mitosis-promoting factor, or M-phase-promoting factor (MPF) because of its functions in stimulation of the mitotic and meiotic cell cycle. MPF has been shown to execute all the events required to drive cell division (Merrick et al. 2008): (1) after being activated at the end of G2 phase via Thr14 and Tyr15 dephosphorylation by Cdc25, MPF drives the entry into mitosis from G2 phase by phosphorylating multiple proteins required for mitosis; (2) activated MPF also phosphorylates numerous proteins, such as nuclear lamins (A, B, and C), APC/C, nucleolin, condensins, histones (e.g., H1 and H3), and survivin (Lapenna and Giordano 2009), events critical for cell division. For example, MPF plays an important role in (1) depolymerization of nuclear lamina and breakdown of the nuclear envelope into small vesicles, through phosphorylation-dependent disassembly of the lamins that form an intermediate filament-type network (i.e., nuclear lamina) underlying the inner nuclear membrane; (2) spindle assembly through microtubule instability by targeting various microtubule-associated proteins; (3) chromosome condensation via phosphorylation of condensins; (4) Golgi apparatus and endoplasmic reticulum fragmentation by targeting Golgi matrix proteins such as GM130; and (5) prevention of apoptosis through survivin phosphorylation (Lapenna and Giordano 2009).
Among the substrates of MPF, APC/C drives progression into metaphase by ubiquitinating different regulatory proteins and the resulting proteasomal degradation (Lapenna and Giordano 2009). As the concentration of cyclin B–CDK1 increases, MPF promotes APC/C to polyubiquitinate cyclin B, leading to its degradation and thus disassembling MPF as a negative-feedback loop. Cyclin B degradation by APC/C begins shortly after the onset of anaphase and continues during the period of mitosis, when sister chromatids are separated and pulled toward opposite spindle poles (Lapenna and Giordano 2009).
MPF must be activated and inactivated for the cell to transition from G2 to M phase, and progression and accomplishment of mitosis. Whereas binding of cyclin B is essential for CDK1 activation, activity of MPF is also regulated by phosphorylation and dephosphorylation, as well as subcellular localization (Lapenna and Giordano 2009). Three residues on CDK1 are responsible for the G2–M transition. First, Thr161 must be phosphorylated by CAK (cyclin H–CDK7 complex), which occurs only when cyclin B binds to CDK1. Second, inhibitory phosphorylation of Thr14 and Tyr15 must be removed by Cdc25. During G1 and S phase, MPF is held in the inactive state by Myt1- and Wee1-mediated Thr14 and Tyr15 phosphorylation of CDK1. As Myt1 is a cell-membrane-associated protein kinase, it binds and phosphorylates CDK1 at both Thr14 and Tyr15, thereby sequestering CDK1 in the cytoplasm. Wee1 only phosphorylates Tyr15 and negatively regulates CDK1 activity in the nucleus. At the end of G2 phase, both Myt1 and Wee1 are inactivated (e.g., by MPF, as a positive-feedback loop), whereas a specific dual phosphatase, Cdc25, is activated (e.g., by MPF, representing another positive-feedback loop). Activated Cdc25 then dephosphorylates both Thr14 and Tyr15, activating CDK1 (Lapenna and Giordano 2009). Cdc25C was thought to be responsible for Thr14 and Tyr15 dephosphorylation of CDK1 for the G2–M transition. However, later reports revealed that overexpression of Cdc25A and Cdc25B, but not Cdc25C, may promote activation of CDK1. Furthermore, both Wee1 and Cdc25 are regulated by checkpoint kinase 1 (Chk1) and 14-3-3 through phosphorylation during interphase.
Some CDK complexes, e.g., cyclin A–CDK2 in S phase and cyclin B1–CDK1 in G2/M phase, are associated with the DNA replication competent complex, which may be directly involved in regulation of DNA replication (Hu and Moscinski 2011). Lastly, cyclin H–CDK7 (known as CAK) activates CDK1, CDK2, CDK4, and CDK6 via phosphorylation at specific threonine residues, events required for full activation of these CDKs.
In the traditional model, cyclin D–CDK4/CDK6 and cyclin E–CDK2 drive cells through interphase via the stepwise phosphorylation of pRb, whereas cyclin B–CDK1 acts primarily only in the G2–M transition. However, later studies revealed that CDK1 is also able to drive the G1–S transition (Santamaria et al. 2007), which challenges the traditional model and suggests that CDK1 may be a pluripotent kinase acting globally throughout the cell cycle (Hu and Moscinski 2011).
As described above, gene knockout of interphase CDKs, including CDK4/CDK6 and CDK2 and their partners cyclin D and cyclin E, is not lethal to mice and their MEFs can still proliferate in a relatively “normal” way (Barriere et al. 2007). These findings raise the possibility that other molecules may compensate for CDK2, CDK4, and CDK6. In CDK2−/− cells, cyclin E binds to CDK1 and forms an active complex, whereas knockdown of CDK1 by short hairpin RNA slows down S phase progression and significantly reduces cell proliferation (Santamaria et al. 2007). CDK2 knockout markedly increases the capability of CDK1 to mediate the G1–S transition (Ortega et al. 2003). Similar phenomena are also found in the case of cyclin D–CDK4/CDK6. In CDK4−/− cells, CDK1 interacts with D-type cyclins (Malumbres et al. 2004). Unlike CDK4/CDK6 and CDK2, CDK1 is essential for early stages of embryonic development. Although knockdown of CDK1 had no effect on interphase progression induced by CDK4 and CDK2 in primary MEFs, CDK1 deletion completely abrogated S phase entry in embryonic cells lacking CDK4/CDK6 and CDK2 (Santamaria et al. 2007). These findings suggesting that CDK1 is a pluripotent CDK that alone is sufficient to drive mammalian cell cycle progression, e.g., promoting entry into S phase as well as the G2–M transition (Hu and Moscinski 2011).
It was thought that only G1 phase CDKs (CDK4/CDK6 and CDK2) phosphorylate pRb. However, both cyclin D–CDK1 and cyclin E–CDK1 are able to phosphorylate pRb proteins in vitro. Actually, whereas inactivation or overexpression of cyclin D–CDK4/CDK6 does not affect pRb activities, increased CDK1 activity might be responsible for pRb phosphorylation (Santamaria et al. 2007). Moreover, CDK1 physically binds to and thus phosphorylates pRb. CDK1 is a target of E2F. In quiescent cells, p130–E2F4 complexes bind to the CDK1 promoter and negatively regulate transcription of CDK1, whereas E2F1, E2F2, and E2F3 bind to positive-regulatory site in the CDK1 promoter and thus induces CDK1 expression. In this context, CDK1–pRb–E2F represents a positive-feedback loop that may amplify CDK1-mediated cell proliferation, whereas inhibition of CDK1 expression may contribute to replication inhibition by pocket protein–E2F complexes.
The CDK inhibitor p21Cip was thought to bind and inhibit cyclin E–CDK2 and/or cyclin D–CDK4, thereby causing G1 arrest. However, p21Cip can directly bind to CDK1. After serum stimulation, both p21Cip and CDK1 locate predominantly to the nucleolus and the levels of the p21Cip–CDK1 complexes increase only in CDK2−/− MEFs, but not in wild-type cells (Martin et al. 2005). The p21Cip–CDK1 complex is likely responsible for cell cycle arrest at the G1–S transition in CDK2−/− cells. Moreover, p21Cip is also required for p53-mediated inhibition of CDK1 activity.
Another endogenous CDK inhibitor, p27Kip1, was identified as an inhibitor of cyclin E–CDK2 and cyclin D–CDK4. However, p27Kip1 can also directly bind to and inhibit CDK1 activity in CDK2−/− MEFs (Martin et al. 2005). Deletion of p27Kip1 significantly increases CDK1 activity, which promotes the G1–S transition (Martin et al. 2005). Therefore, p21Cip- or p27Kip1-induced growth inhibition is, at least in part, due to negative regulation of CDK1 activity.
2.4 CDK7
In addition to binding of cyclins, activation of cell-cycle-regulatory CDKs (e.g., CDK1, CDK2, CDK4/CDK6) also requires T-loop phosphorylation. The latter event is catalyzed by CAK (Larochelle et al. 2007). CAK is composed of the catalytic subunit CDK7 and two regulatory subunits, cyclin H and the RING finger protein ménage à trois 1 (Mat1) (Schneider et al. 2002). So far, the trimeric kinase cyclin H–CDK7–Mat1 is the only CAK identified in mammalian cells. CDK7 is activated via binding of cyclin H, whereas its substrate specificity is governed by Mat1 (Schneider et al. 2002).
The phosphorylation by cyclin H–CDK7 is required for activation of cell-cycle-regulatory CDKs in the timing of the transition from one phase to the next as well as progression during individual phases. For example, CDK7 appears to be required for both S phase entry and mitosis in human cancer cells (Wallenfang and Seydoux 2002). Cyclin H–CDK7 phosphorylates cell-cycle-regulatory CDKs at a conserved threonine (Thr161 in CDK1, Thr160 in CDK2, Thr172 in CDK4, and Thr177 in CDK6) located within their T-loop (Larochelle et al. 2007). The activating phosphorylation within the T-loop of CDKs results in a correct structural orientation of amino acids near the active site. This phosphorylation can be reversed by the CDK-associated protein phosphatase KAP, leading to deactivation of CDKs (Larochelle et al. 2007). Unlike in other cell-cycle-regulatory cyclin–CDK complexes, the protein levels of cyclin H and kinase activity of CDK7 do not fluctuate during the cell cycle, suggesting they have other functions beyond cell cycle regulation, such as in gene transcription (see Sect. 23.3).
Later evidence indicated that the absence of CAK activity is completely dispensable for global transcription mediated by RNA polymerase II (RNA pol II) (Ganuza et al. 2012), a well-established target of cyclin H–CDK7–Mat1. However, CDK7 deficiency results in severe mitotic defects in Caenorhabditis elegans and Drosophila melanogaster without concomitant loss of C-terminal domain (CTD) phosphorylation or transcriptional integrity, respectively (Ganuza et al. 2012). Loss of CDK7 impairs T-loop phosphorylation of cell-cycle-regulatory CDKs (e.g., CDK1 and CDK2), leading to cessation of cell division in vitro and early embryonic lethality in vivo (Ganuza et al. 2012). But it does not affect transcription mediated by RNA pol II, with the exception of E2F-controlled genes, indicating an indirect consequence of deficient CDK function (Ganuza et al. 2012). As a result, loss of CDK7 expression in adult mice has little effect on nonproliferating tissues, but leads to the premature onset of age-related phenotypes in proliferating tissues, most likely due to depletion of progenitor cells and exhaustion of their renewal capacity. In this context, deficiency of either Mat1 or cyclin H also results in early embryonic lethality in mice (Ganuza et al. 2012).
2.5 CDK3
In the concert of the cell cycle, the final missing piece of the puzzle is the regulatory mechanism for the G0–G1 transition, that is, reentry of quiescent G0 cells into the cell cycle. The importance of the G0–G1 transition is underscored by the fact that reentry of commonly quiescent cancer stem cells into the cell cycle is a major reason for recurrence or relapse of cancer after chemotherapy (Sage 2004).
Recent evidence revealing that cyclin C–CDK3 is responsible for the G0–G1 transition (Ren and Rollins 2004) is summarized as follows. In mammalian cells, (1) intracellular levels of cyclin C are high in G0 phase; (2) CDK3, rather than CDK8 (another CDK that is known to bind cyclin C) (Perez et al. 2009; Hoeppner et al. 2005) associates with cyclin C to drive cells from G0 phase to G1 phase by phosphorylation of certain targets, especially pRb (Ren and Rollins 2004); (3) cyclin C–CDK3 phosphorylates pRb at Ser807/811 in G0 phase (Ren and Rollins 2004; Hofmann and Livingston 1996); (4) phosphorylation of pRb by cyclin C–CDK3 is required for cells to exit G0 phase efficiently (Ren and Rollins 2004). Therefore, the G0–G1 transition is regulated through a process similar to the G1–S transition, which is controlled by cyclin E–CDK2, but involves an entirely different cyclin–CDK complex (Sage 2004).
2.6 CDK10
CDK10 (previously referred to as PISSLRE) is CDK1-related kinase and has been implicated in the regulation of the G2/M phase of the cell cycle (Kasten and Giordano 2001). CDK10 contains residues that are important regulatory sites in CDK1 and other CDKs, including tyrosine and threonine sites in the ATP-binding domain as well as a threonine residue corresponding to Thr161 of CDK1. As discussed already, the phosphorylation of these sites is critical for activation of CDKs, suggesting that CDK10 may be regulated in a similar fashion. One of CDK10’s partners is the transcription factor Ets2. CDK10 binds to the N-terminus of Ets2, thereby inhibiting Ets2 transactivation in mammalian cells (Kasten and Giordano 2001).
2.7 CDK14 and CDK15
CDK14 (PFTK1) binds to cyclin Y (Shu et al. 2007). CDK14 is expressed predominantly in mitosis, concurrent with the peak in cyclin Y levels, indicating its role in regulation of mitosis, probably via phosphorylating the substrates in the Wnt pathway.
By analogy, CDK15 (PFTK2) and cyclin Y-like 1 form a similar complex that may share substrate specificity with cyclin Y–CDK14 (Shu et al. 2007).
2.8 CDK16
CDK16–CDK18 are very similar and only differ within their N-terminals domains and CTDs. CDK16 can be detected in many tissues (Mikolcevic et al. 2012), particularly in testis and brain. In human cells, CDK16 is phosphorylated at several residues (e.g., Ser119 and Ser153) by protein kinase A (PKA), as well as other residues in the N- and C-terminal extensions (Mikolcevic et al. 2012). Phosphorylation of Ser119 and Ser153 promotes binding of 14-3-3, but the function of these phosphorylations and 14-3-3 binding remains to be defined. CDK16 is activated by membrane-associated cyclin Y, an event inhibited by Ser153 phosphorylation. CDK16 isolated from tissues (e.g., murine testis) is unphosphorylated, interacts with cyclin Y, and exhibits kinase activity (Drexler 1998). Thus, in contrast to other CDKs, the cyclin binding capacity of CDK16 is negatively regulated by phosphorylation. Although CDK16 activity is cell-cycle-related, it is uncertain whether CDK16 itself is involved in regulation of cell cycle progression (Drexler 1998). Interestingly, CDK16 knockout mice develop normally, but male mice are infertile, indicating the essential role of CDK16 in spermatogenesis (Drexler 1998).
3 Transcriptional CDKs
Transcription starts with the binding of specific transcription factors to their DNA binding sites in the promoter region of target genes, followed by recruitment of the Mediator complex and multiple general transcription factors (e.g., TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH), and RNA pol II, which together form the preinitiation or pre-elongation complexes (Wesierska-Gadek and Krystof 2009). After the recruitment of RNA pol II to the promoter regions by the general transcription factors, DNA surrounding the transcription start site is melted and allows the transcription initiation and elongation to occur (Wesierska-Gadek and Krystof 2009). After completion of one transcription cycle, RNA pol II is released from DNA. However, several general transcription factors (e.g., TFIIA, TFIID, TFIIE, TFIIH) and the Mediator remain on DNA, forming the scaffold complex, facilitating transcription reinitiation for subsequent cycles of transcription.
The second group of CDKs, functionally different from cell-cycle-regulatory CDKs, consists of kinases involved in the regulation of gene transcription. The well-known transcriptional CDKs are CDK7, CDK8, and CDK9 (Fig. 23.2). These transcriptional CDKs share several features. First, they are subunits of multiprotein transcription-regulatory complexes. CDK7 is a subunit of TFIIH, a general transcription factor component of the preinitiation complex (Glover-Cutter et al. 2009). CDK8 is a part of the CDK module of Mediator (Akoulitchev et al. 2000). CDK9 is the catalytic subunit of positive transcription elongation factor b (P-TEFb), a critical regulator of RNA pol II elongation (Price 2000). Second, they can phosphorylate specific serine residues in the CTD of RNA pol II (Larochelle et al. 2012). CDK7 phosphorylates Ser5 and Ser7, but its major contribution is Ser7 phosphorylation (Glover-Cutter et al. 2009). CDK8 phosphorylates Ser2 and Ser5 within the CTD repeats in vitro (Akoulitchev et al. 2000), but its in vivo contributions remain uncertain. CDK9 is the major Ser2 kinase, but it can also contribute to Ser5 phosphorylation (Larochelle et al. 2012). Third, unlike the cyclin partners of the cell-cycle-regulatory CDKs, the cyclin subunits of transcriptional CDKs—cyclin H for CDK7 (Glover-Cutter et al. (2009), cyclins T1 and T2 for CDK9 (Price 2000), and cyclin C for CDK8 (Barette et al. 2001)—do not exhibit significant oscillations in protein levels during the cell cycle. Last, in addition to cyclin binding, they are also regulated via other interactors, such as repression of CDK9 activity by hexamethylene bisacetamide inducible 1 (HEXIM1), or activation of CDK8 by association with MED12.

Regulation of gene transcription by CDKs. DSIF 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole-sensitivity-inducing factor HEXIM hexamethylene bisacetamide inducible, Mat1 ménage à trois 1, mRNA messenger RNA, NELF negative elongation factor, P-TEFb positive transcription elongation factor b, RNAP II RNA polymerase II, snRNA small nuclear RNA, TFIIH transcription factor IIH
3.1 CDK7
CDK7 is, so far, the only one atypical CDK that acts at the crossroad between the cell cycle and transcription (Wallenfang and Seydoux 2002; Ganuza et al. 2012). As discussed already, CDK7 plays a key role in cell cycle progression by phosphorylating multiple cell-cycle-regulatory CDKs; therefore, it is known as CAK. On the other hand, the cyclin H–CDK7–Mat1 complex is also a component of the general transcription factor IIH (TFIIH) (Glover-Cutter et al. 2009). The TFIIH holoenzyme consists of the cyclin H–CDK7–Mat1 complex (thus, it is also named TFIIK in this context) and at least six other proteins (XPB, XPD, p62, p55, p44, p34). TFIIK (or CAK) activation is controlled by phosphorylation-dependent binding of the regulatory partners (e.g., cyclin H), and is likely also regulated by other posttranslational modifications. To form a stable complex with its activating partner cyclin H, CDK7 must be phosphorylated at either Ser164 or Thr170 in its own T-loop, which cooperates with binding of Mat1 to stabilize the TFIIK complex. Moreover, binding of Mat1 and Thr170 phosphorylation markedly promotes the activity of CDK7 as a CTD kinase, whereas the substrate specificity of TFIIK for the CTD of RNA pol II requires association with TFIIH.
CTD phosphorylation by cyclin H–CDK7 is responsible for initiation of transcription (Glover-Cutter et al. 2009). The TFIIH complex contains catalytic activities of ATPase, helicase, and kinase, which are required for regulation of different events that control transcription. The helicase activity of TFIIH is involved in ATP-dependent promoter DNA opening, an event required for initiation of transcription. The kinase activity of CDK7 within TFIIH phosphorylates the CTD of RNA pol II, leading to promoter clearance, a step essential for the switch from initiation to elongation during transcription (Larochelle et al. 2012). In this context, the TFIIK complex associates with TFIIH and phosphorylates Ser5 and Ser7 in the CTD of the large subunit of RNA pol II. Ser5 phosphorylation is important for recruitment of chromatin-modifying factors and messenger RNA (mRNA) capping enzymes to the nascent transcript, and Ser7 phosphorylation is specifically involved in regulating small nuclear RNA (snRNA) expression. Moreover, the ability of CDK7 to phosphorylate the CTD is also regulated by phosphorylation of cyclin H. For example, cyclin C–CDK8 phosphorylates cyclin H, resulting in inhibition of the TFIIK activity, and CK2 phosphorylates cyclin H, leading to full activation of TFIIK.
Genetic inactivation of the CDK7 locus revealed that CDK7 is completely unnecessary for global transcription (Ganuza et al. 2012), but is essential for cell cycle regulation (see earlier). Instead, it only affects transcription of a specific cell-division gene cluster, which is less than 5 % of all transcripts (Patel and Simon 2010). Similarly, mouse cells with defective Mat1 also exhibit functional de novo transcription. Thus, CDK7 seems to be only required for the expression of a subset of genes. In addition, the CDK7 complex may also regulate gene expression by directly phosphorylating transcription factors, including retinoic acid receptor peroxisome-proliferator-activated receptor γ, to either enhance or repress their activity. Therefore, the repertoire of CDK7-responsive genes and the functional requirements for the cyclin H–CDK7–Mat1 complex likely vary in a cell-type-dependent manner.
3.2 CDK9
In the elongation phase of transcription, interplay between negative and positive elongation factors regulates the elongation potential of RNA pol II (Canduri et al. 2008). P-TEFb is the first and only known positive elongation factor, and is a complex composed of CDK9 and its partner cyclin T (Price 2000). P-TEFb preferentially phosphorylates Ser2, and most likely Ser5 as well, of the CTD, thereby activating RNA pol II, leading to promotion of transcriptional elongation (Marshall and Price 1995). This event is sensitive to 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), a well-known inhibitor of transcriptional elongation (Garriga and Grana 2004). Moreover, P-TEFb also phosphorylates multiple inhibitory proteins and releases their inhibition on transcription.
Immediately after initiation, transcription is paused via cooperative repression of RNA pol II by two negative elongation factors, DRB-sensitivity-inducing factor (DSIF) and negative elongation factor (NELF) (Peterlin and Price 2006). These factors only bind to the hypophosphorylated form of RNA pol II (IIa), and not to its hyperphosphorylated form (IIo). First, P-TEFb phosphorylates the negative elongation factors DSIF at its SPT5 subunit and NELF at its RD subunit. These phosphorylations release the transcription block of both DSIF and NELF on RNA pol II, permitting the transition into productive elongation (Garriga and Grana 2004). Second, P-TEFb hyperphosphorylates Ser2 of the CTD of RNA pol II that had been previously phosphorylated at Ser5 by CDK7 during transcription initiation. However, although it releases the CTD from the DNA and make it available for CDK9, CDK7-catalyzed Ser5 phosphorylation is most unlikely to be a prerequisite for efficient recognition of Ser2 by CDK9 (Cheng and Price 2007). Ser2 phosphorylation is essential not only for productive transcription elongation, but also for coupling pre-mRNA synthesis with splicing and polyadenylation (Garriga et al. 2010). Last, P-TEFb is recruited to certain promoters by particular transcription factors (e.g., NF-κB and Myc), which may determine the transcription selectivity of target genes.
Like most CDKs, activity of CDK9 relies on binding of its partner cyclin T. Three T-type cyclins have been identified in human cells, named cyclins T1, T2a, and T2b (Peng et al. 1998). The predominant form of human P-TEFb is composed of CDK9 and cyclin T1 (Cheng and Price 2007; Peng et al. 1998). The three T-type cyclins share a highly conserved N-terminus containing an 81 % identical cyclin box. Cyclins T2a and T2b are splice variants of a primary transcript. They share the first 642 amino acids, but cyclin T2b contains a larger CTD that is much less conserved (46 % identity) (Peng et al. 1998). A more distantly related cyclin, cyclin K, may also bind to CDK9 and form an active complex (Fu et al. 1999). Interestingly, in contrast to T-type cyclin—CDK9 complexes, cyclin K–CDK9 only activates transcription when tethered to RNA rather than DNA. Cyclin K lacks an essential region in its C-terminus, through which cyclin T1 recognizes the CTD of RNA pol II (Fu et al. 1999).
In addition to the known 42-kDa form of CDK9, another isoform (55 kDa) has been identified, and is transcribed from an alternative upstream promoter (Garriga and Grana 2004). The 55-kDa isoform also interacts with cyclin T1, and shares the ability to phosphorylate the CTD of RNA pol II in vitro as well as to stimulate transcription (Garriga and Grana 2004).
Like CDK2, full activation of CDK9 depends not only on cyclin binding but also on phosphorylation of Thr186 within the activation loop (Garriga and Grana 2004). However, unlike CDK2, the Thr186 phosphorylation of CDK9 seems not to be dependent on interaction with cyclin T1. Nevertheless, this phosphorylation causes conformational change in the activation loop, allowing CDK9 to recognize the substrate.
The ratio between the active and inactive forms of P-TEFb is tightly regulated by the actual transcriptional demand in the cell (Van Herreweghe et al. 2007). Cyclin T–CDK9 complexes are inactive when bound to 7SK snRNA and the hexamethylene bisacetamide inducible proteins HEXIM1 (or MAQ1) and/or HEXIM2 (Barboric et al. 2005; Li et al. 2005). The large amount of P-TEFb (e.g., about 50 % in HeLa cells) exists as the kinase-inactive 7SK–HEXIM–P-TEFb complex (termed small nuclear ribonucleoprotein, snRNP), which contains HEXIM1 (and/or HEXIM2) and 7SK snRNA (Barboric et al. 2005; Li et al. 2005). In humans, 7SK RNA is an abundant, RNA polymerase III synthesized snRNA (Yang et al. 2001b). The 7SK snRNA acts as a key regulator of mRNA production by controlling the activity of P-TEFb (Yang et al. 2001b; Nguyen et al. 2001). HEXIM1 and HEXIM2, in homodimeric or heterodimeric form, bind to a distal region of the 5′ hairpin of 7SK snRNA (Li et al. 2005; Michels et al. 2004; Egloff et al. 2006), resulting in a conformational change in HEXIM proteins that enables their C-terminal region to interact with cyclin T1 (Michels et al. 2004). The 3′ hairpin of 7SK snRNA then interacts with cyclin T1, leading to inactivation of P-TEFb (Van Herreweghe et al. 2007; Egloff et al. 2006). In doing so, the 7SK snRNA, in cooperation with the HEXIM proteins, sequesters P-TEFb into the kinase-inactive 7SK–HEXIM1–P-TEFb snRNP, and thus controls the nuclear level of active P-TEFb.
This inhibitory mechanism is abrogated as a result of activation of upstream signaling pathways. For example, inhibition of global transcription by actinomycin D or DRB, or irradiation with ultraviolet (UV) light, triggers rapid disruption of the 7SK–HEXIM–P-TEFb snRNP complex, thereby releasing P-TEFb and increasing the nuclear level of its active form (Krueger et al. 2010). In contrast, inhibition of cell growth can shift the P-TEFb equilibrium toward the 7SK–HEXIM–P-TEFb snRNP complex (Van Herreweghe et al. 2007). Therefore, the nuclear level of active P-TEFb is controlled by dynamic and reversible remodeling of 7SK snRNPs containing RNA helicase A and the heterogeneous nuclear ribonucleoproteins A1, A2/B1, R, and Q (Krueger et al. 2008).
Therefore, activity of the cyclin T–CDK9 complex (P-TEFb) is regulated by at least four mechanisms: (1) the protein levels of its CDK9 and T-type cyclin subunits (Marshall et al. 2005); (2) assembly of cyclin T and CDK9 (Garriga and Grana 2004); (3) inhibition of P-TEFb by 7SK snRNA and HEXIM proteins; and (4) posttranslational modification of P-TEFb components (e.g., Thr186 phosphorylation of CDK9) as well as other regulatory proteins (Fujinaga et al. 2012).
3.3 CDK8
CDK8 is a nuclear serine–threonine kinase that functions as a transcriptional regulator. CDK8 is a key component of the Mediator complex (Tsutsui et al. 2011). There are two distinct Mediator complexes for transcriptional regulation, a small complex that activates transcription via RNA pol II, and a large complex that generally represses transcription (Xu and Ji 2011). CDK8 is a subunit of the large Mediator complex (about 1.2 MDa) composed of 25–30 proteins and acts as a molecular bridge between DNA-binding transcription factors and RNA pol II (Fukasawa et al. 2012). CDK8 associates in a dynamic fashion with the Mediator complex as a four-subunit module containing CDK8, cyclin C, MED12, and MED13 (in a 1:1:1:1 stoichiometry, designed the cyclin C–CDK8 module) (Hoeppner et al. 2005). This module is conserved among eukaryotes, and phosphorylates the RNA pol II CTD (Gallorini et al. 2012). MED12 and cyclin C are required for the kinase activity of CDK8 toward the CTD of RNA pol II (Gallorini et al. 2012), whereas MED13 is necessary to recruit the cyclin C–CDK8 module to the small Mediator complex (Fukasawa et al. 2012). In turn, MED13 itself can be phosphorylated by CDK8 (Gallorini et al. 2012). Association of the cyclin C–CDK8 module with the core Mediator enables CDK8 to phosphorylate its substrates such as histone H3 on chromatin. Thus, kinase activity of CDK8 is regulated by association with other subunits of the cyclin C–CDK8 module, whereas its substrate accessibility is regulated via association with the core Mediator.
Cyclin C binds CDK8 and CDK3, which regulate mRNA transcription and the cell cycle, respectively. It is clear that CDK8 acts as a corepressor to negatively affect transcription (Akoulitchev et al. 2000). The inhibitory action of cyclin C–CDK8 is due to disruption of Mediator–RNA pol II interactions likely by MED12 and MED13 (Galbraith et al. 2010), but is independent of the kinase activity of CDK8. The cyclin C–CDK8 module induces conformational change of the core Mediator complex, which physically disrupts the interaction between RNA pol II and the small Mediator complex, thereby blocking the subsequent cycles of transcription (i.e., transcription reinitiation). However, since only a fraction of CDK8 is associated with Mediator, CDK8 may have roles outside this complex. Unlike CDK7 and CDK9, CDK8 prematurely phosphorylates the CTD, thereby preventing, rather than promoting, formation of a transcription initiation complex. In addition, cyclin C–CDK8 phosphorylates cyclin H, repressing CDK7 and thus transcription. Furthermore, CDK8 also phosphorylates certain gene-specific transcription factors and decreases their stability (Xu and Ji 2011). In summary, CDK8 negatively regulates transcription through at least two distinct mechanisms, i.e., via inhibition of the small Mediator complex, independent of its kinase activity (Galbraith et al. 2010), and as a kinase, by phosphorylating multiple transcription-regulatory proteins. As a consequence, these mechanisms cooperate to negatively control the rate of transcription reinitiation, thereby limiting the quantity of mRNA produced.
However, CDK8 may also play positive roles in transcription in certain circumstances at multiple stages of the transcription cycle (Galbraith et al. 2010). For example, CDK8 has been described as a coactivator in several molecular pathways, including β-catenin-, p53-, and SMAD-mediated signaling pathways, as well as thyroid-hormone-dependent transcription (Galbraith et al. 2010). Therefore, the net influence of CDK8 is determined by both substrate accessibility and gene-specific/stimulus-specific susceptibility. In this context, CDK8 can promote transcription by (1) promoting recruitment of coactivator and transactivation-coupled turnover of transcription factors, (2) cooperating with CDK7 to govern the transition from the preinitiation complex to the scaffold and back to the preinitiation complex during the transcription cycles (Galbraith et al. 2010), (3) facilitating the recruitment of P-TEFb in the elongation phase of transcription, and (4) contributing to chromatin modifications that correlate with transcriptional activation. Several phosphorylation targets of CDK8 have been identified, including the RNA pol II CTD, histone H3, subunits of general transcription factors, and certain transactivators (Galbraith et al. 2010). Thus, the role of CDK8 in transcription is most likely context-specific, dependent on the specific biological contexts and the identity of the transcription factors with which it interacts.
CDK8 knockout in mice is lethal prior to compaction and implantation at embryonic days (Xu and Ji 2011), suggesting a critical role of CDK8 for cell-fate determination in early embryos. Whereas the function and regulation (e.g., their upstream signals) of CDK8 and cyclin C in vivo are still poorly understood, the universal requirement of the CDK8 kinase function in various cellular and developmental contexts and the specific requirements for other conserved module members are unknown.
3.4 CDK11
CDK11 is a p110 and p58 PITSLRE protein kinase. Expression of the CDK11p110 isoform is ubiquitous and constant throughout the cell cycle (Trembley et al. 2004). In contrast, CDK11p58 is expressed and functions specifically in G2/M phase (Hu et al. 2003). During apoptosis, a third isoform, CDK11p46, is generated by caspase-dependent cleavage of CDK11p110 and CDK11p58, leaving the catalytic domain intact (Hu et al. 2003).
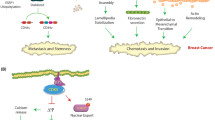
The CDK11p110-containing complexes influence both transcription and pre-mRNA splicing, suggesting that this CDK may help to link the two processes (Trembley et al. 2004; Loyer et al. 2005). There are approximately 30,000 genes in the human genome. However, the heterogeneity generated by this number of genes is still not enough to explain the complexity of humans. About 38–74 % of human genes are subject to alternative splicing (Hu et al. 2003), creating more proteomic variations. Moreover, mutations that affect splicing patterns are the underlying causes of some cancers and neurodegenerative diseases. It is estimated that about 15 % of disease-causing mutations in human genes involve misregulation of alternative splicing.
CDK11p110, complexed with cyclins L1 and L2 (Loyer et al. 2008, 2011), belongs to large protein complexes (1-2 MDa and 800 kDa) that contain transcription-related proteins such as RNA pol II, FACT (facilitates chromatin transcription), CK2, and TFIIF (Trembley et al. 2003). The kinase activity of CDK11p110 is functionally required for the regulation of the pre-mRNA splicing process. Several splicing-related factors (e.g., RNPS1 and 9G8) interact with CDK11p110 to regulate transcription and splicing (Loyer et al. 2008). RNPS1 is an SR protein that as a general activator of splicing and a component of the exon–exon junction complex promotes alternative splicing in a substrate-specific manner; 9G8 is another general splicing factor that promotes the nucleocytoplasmic export of mRNA.
3.5 CDK12 and CDK13
The functions of CDK12 (CrkRS) and CDK13 (CDC2L5), complexed with cyclins L1 and L2 (Chen et al. 2006, 2007a), were originally related to pre-mRNA splicing (Chen et al. 2006). However, later studies indicated that the endogenous human CDK12 and CDK13 associate with cyclin K, rather than cyclin L (Kohoutek and Blazek 2012). In humans, cyclin K binds CDK12 and CDK13 in two separate complexes (Kohoutek and Blazek 2012). CDK12 and CDK13 are 1,490 and 1,512 amino acid proteins, respectively, both of which contain a conserved central CTD kinase domain (Bartkowiak et al. 2010). Human CDK12 phosphorylate Ser2 in the CTD of RNA pol II in vitro and in vivo (Bartkowiak et al. 2010), whereas CDK13 also phosphorylates the CTD of RNA pol II at least in vitro (Kohoutek and Blazek 2012).
Depletion of cyclin K–CDK12, but not cyclin K–CDK13, results in decreased expression of predominantly long genes with high numbers of exons (Blazek et al. 2011). The most prominent group of downregulated genes are the DNA damage response genes, including BRCA1 (breast–ovarian cancer type susceptibility protein 1), ATR (ataxia telangiectasia and Rad3 related), FANCI, and FANCD2. Consistent with this, cells that lack cyclin K–CDK12 exhibit spontaneous DNA damage and are sensitive to a variety of DNA damaging agents (Blazek et al. 2011). The essential role of cyclin K–CDK12 is further supported by the fact that genetic inactivation of cyclin K in mice causes early embryonic lethality (Kohoutek and Blazek 2012). In conclusion, cyclin K–CDK12 maintains genomic stability via regulation of expression of DNA damage response genes. The function of cyclin K–CDK13 is still unknown.
3.6 CDK19
CDK19 (previously known as CDK8-like, CDK8L or CDC2L6) (Tsutsui et al. 2011) is also identified in the Mediator complex, and is very similar to CDK8, but is conserved only in vertebrates (Fukasawa et al. 2012). Although CDK19 was sporadically referred to as CDK11 (Tsutsui et al. 2011), it should not be confused with the “splicing kinase” CDK11.
Although CDK8 and CDK19 associate with seemingly identical Mediator complexes (Fukasawa et al. 2012), they are likely not functionally redundant. CDK19 forms the Mediator complexes independent of CDK8. In viral activator VP16-dependent transcriptional regulation, CDK8 supports transcriptional activation, whereas CDK19 represses it (Fukasawa et al. 2012). Both CDK8 and CDK9 bind to same genes regardless of whether they are CDK8’s or CDK19’s targets, suggesting that Mediator functions as a context-specific transcriptional regulator. CDK8 and CDK19 share the highly conserved kinase and cyclin binding domains, whereas they differ in the C-terminal regions (Tsutsui et al. 2011), which might alter access to substrates or incorporation into complexes. This may provide an explanation for their distinct (likely opposite) functions in regulation of transcription.
4 CDKs in Neuron Protection
It is generally believed that neurons are terminally differentiated cells. All CDKs, except CDK5—a neuron-specific CDK, are silenced in postmitotic neurons. However, CDKs appear to be deregulated in several neurodegenerative diseases (Fig. 23.3). Multiple cell-cycle-regulatory CDKs are also related to various pathways required for neuronal death after ischemia/hypoxic injury, particularly stroke. In general, “inappropriate” activation of cell-cycle-regulatory CDKs leads to neuronal death, rather than proliferation, in terminally differentiated (or postmitotic) neurons. Therefore, inhibition of CDKs (e.g., by the pan-CDK inhibitor flavopiridol) is generally neuron-protective. On the other hand, CDK5, complexed with its non-cyclin partner p35 or p39, is the only one postmitotic CDK that functions exclusively in the brain, and plays important functional roles in various aspects of nervous system development and functions, including neuronal migration, neuronal survival, dendritic spine formation, synaptogenesis, adult neurogenesis, neurotransmission, homeostatic plasticity, and learning and memory. In this case, pan-CDK inhibitors such as flavopiridol also inhibit CDK5, which may be harmful to the normal functions of neurons. In contrast, once bound to a smaller but stabler and mislocalized p25 form in some neurodegenerative diseases, CDK5 becomes a neuron death signal. Thus, it remains uncertain how and which CDKs should be used for the therapeutic purpose of neuron protection in ischemia/hypoxic injury versus neurodegeneration.

Dysregulation of CDKs in neurodegenerative diseases
4.1 CDK5: A Neuron-Specific CDK
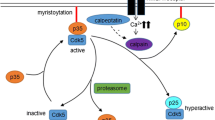
CDK5 is a peculiar proline-directed serine–threonine kinase. Unlike other CDKs, CDK5 is not directly involved in regulation of the cell cycle or transcription (Nikolic and Tsai 2000). This kinase is present mainly in postmitotic neurons and its activity is tightly regulated by the interaction with two non-cyclin regulatory components, p35 and p39 (Yamada et al. 2007; Hisanaga and Saito 2003). Kinase activity of CDK5 is mainly determined by the amount of p35 available, which is controlled by a balance between synthesis and degradation (Cruz and Tsai 2004a). CDK5 function is also regulated by phosphorylation, the effect of which effect CDK5 kinase activity is opposite to that of cell-cycle-regulatory CDKs.
CDK5 is a versatile protein kinase that normally regulates multiple neuronal processes such as migration, cortical layering, and synaptic plasticity (Cruz and Tsai 2004a; Hindley and Philpott 2012; Odajima et al. 2011; Cheung and Ip 2007; Cheung et al. 2007). CDK5 also plays an important role in both survival and death of neurons (Hisanaga and Asada 2012; Honma et al. 2003). The prosurvival activity of CDK5 is apparent in neurons when they are exposed to stress (Cheung and Ip 2004; Cheung et al. 2008), whereas long-term inactivation and/or hyperactivation of CDK5 triggers cell death as seen in neurodegenerative disorders (Hisanaga and Endo 2010). The prodeath activity of CDK5 is suppressed by its membrane association via myristoylation of p35 (Sato et al. 2007; Zhu et al. 2005). Thus, appropriate activity, localization, and regulation of CDK5 is critical for long-term survival of neurons, which is more than 80 years in humans (Asada et al. 2012).
Despite the pivotal role of CDK5 in CNS development, CDK5 dysregulation is significantly implicated in different neuronal diseases, such as Alzheimer’s disease, amyotrophic lateral sclerosis, Parkinson’s disease, and prion-related encephalopathies (Lopes and Agostinho 2011; Cheung et al. 2006). In these neurodegenerative conditions, CDK5 is overactivated and relocalized owing to association with p25, a truncated form of p35 (Sato et al. 2011; Kamei et al. 2007). The activator switching leads to a shift in the phosphorylation pattern of CDK5 (Cruz et al. 2003, 2006), with an alteration both in target specificity and activity, causing neuronal disorders (Lopes et al. 2007, 2010). For example, in Alzheimer’s disease and prion-related encephalopathies, two disorders that share clinical and neuropathological features, CDK5 dysregulation is a linking mechanism between the major neuropathological markers (i.e., amyloid plaques, tau hyperphosphorylation, and synaptic and neuronal loss) (Lopes and Agostinho 2011; Cruz and Tsai 2004b). Moreover, this kinase was shown to be involved in abortive cell cycle reentry (Lopes et al. 2009), a feature recently proposed as a possible step in the neuronal apoptosis mechanism of several neurological diseases (Cruz and Tsai 2004a).
4.2 Other CDKs
After death or injury, a neuron, as a terminally differentiated cell, generally cannot be repaired or replaced by neonatal neurons, owing to lack of the capability to grow. In neuronal development and disease, multiple pathways required for neuron death involve various cell cycle molecules (Greene et al. 2007). For example, activation of FOXO1 by CDK1 in cycling cells and postmitotic neurons causes neuronal degeneration (Yuan et al. 2008). On the other hand, microglia become active after injury of the CNS, releasing many types of inflammatory factors to promote neuron apoptosis and aggravate inflammatory injury of tissue; astrocytes proliferate to form a compact glial scar and secret axon regeneration inhibitors, which restrict regeneration of damaged axons and block repair of the structure and recovery of the function of neurons. These pathological processes are closely related to cell cycle regulation. Therefore, cell cycle regulation is related to neuroprotection in two aspects: boosting neuron regeneration and inhibiting activation of microglia and proliferation of astrocytes (Di Giovanni et al. 2005). Appropriate regulation of the cell cycle is an important strategy to protect neurons (Di Giovanni et al. 2005).
Mature neurons mostly rest in G0 phase (Lopes et al. 2009). However, they can reenter the cell cycle in certain pathological conditions such as neurodegeneration and cerebral ischemia (Meijer et al. 1997). The fact that the level of cyclin E is elevated in neurons of patients with Alzhemier’s disease suggests that cells progress through S phase (Odajima et al. 2011; Yang et al. 2001a). Likewise, the level of cyclin B1 is increased in hippocampus neurons of patients with vascular dementia, indicating that cells enter G2 phase (Husseman et al. 2000). Further, a modest increase in protein levels of cyclin D1 and CDK4 is observed in brain tissue of cerebral infarcted patients (Love 2003). Commonly, this cell cycle reentry leads to apoptosis, rather than proliferation, of neurons (Liu and Greene 2001b; Wartiovaara et al. 2002). In this context, expression of cyclin D–CDK4, a complex primarily responsible for the G1–S transition, is increased in ischemic cerebral tissue, accompanied by increased apoptosis of neurons (Liu and Greene 2001a; Becker and Bonni 2005). Activation of CDK1, complexed with cyclin B1, is not an indicator of mitosis in neurons. Instead, it prompts neuronal death by direct activation of the proapoptotic protein Bad, before entry to M phase (Konishi et al. 2002). The findings that CDKs are activated in neurons exposed to various apoptotic stimuli further support a notion that activation and expression of CDKs and their partner cyclins may be an important mechanism for neuron apoptosis. In addition to direct involvement in activation of the apoptotic signaling pathways in neurons, neurons may also undergo apoptosis because of abnormally entry to the cell cycle.
In response to injuries, nervous tissue undergoes a series of pathological processes, including ischemia, edema, toxicity of excitatory amino acids, and oxidative stress. However, activation of microglia and proliferation of astrocytes has received attention. Microglia release a number of inflammatory factors, such as TNFα, IL-1β, and IL-6, which play a significant role in initiating secondary injury. On the other hand, astrocytes exhibit marked morphological changes, including thicker neurites and cell hypertrophy, and form glial scar with enhanced expression of intermediate fiber (e.g., glial fibrillary acidic protein) and proliferation of microglia. In the early stage of injury, glial scar is composed of astrocytes, microglia, oligodendrocytes, and infiltrating macrophages; in the later stage, astrocytes substitute other components and become predominant. By quickly replacing damaged tissue, glial scar blocks axonal budding growth and hinders the formation of neural circuits, thus impeding reconstruction and recovery of the nervous tissue. The increase of the levels of cyclins and proliferating cell nuclear antigen (PCNA) in microglia and astrocytes after CNS injury indicates that gliocytes enter the cell cycle and thus undergo division and grow. A strikingly increased number of cyclin D1-, CDK4-, and PCNA-positive cells has been observed in the hippocampal CA1 area after transient total cerebral ischemia, and most of them are microglia and astrocytes (Kato et al. 2003). Granulocyte–macrophage colony-stimulating factor induces activation and proliferation of GMI-M6-3 microglia, in association with elevated levels of cyclins A, D1, and E, as well as a decreased level of p27Kip1 (Koguchi et al. 2003). After stimulation with serum, the number of astrocytes isolated from mouse cortex in S phase is increased by 224 % (Di Giovanni et al. 2005). High levels of cyclin D1 and PCNA are also noted in posttraumatic gliocytes. In vitro, astrocytes are activated and proliferate in response to injury, with a significant increase in 5-bromo-2′-deoxyuridine incorporation (Koguchi et al. 2003). After spinal injury, cyclin A, cyclin B, cyclin E, and PCNA are upregulated, accompanied by increased numbers of OX42/Ki67-positive and glial fibrillary acidic protein/PCNA-positive cells, indicating that gliocytes and astrocytes are activated and proliferate.
In a cerebral ischemia model, after the CDK inhibitor flavopiridol has been administered for 7–9 days after 4 h of global cerebral ischemia/reperfusion, the number of surviving neurons is increased in the hippocampal CA1 area and animal behavior is improved (Koguchi et al. 2003). In a craniocerebral trauma model, the CDK inhibitor roscovitine significantly reduces the number of PCNA-positive cells and the number of cells entering S phase, inhibits cell proliferation, reduces injury, and boosts recovery (Di Giovanni et al. 2005; Meijer et al. 1997). In a spinal injury model, the CDK inhibitor olomoucine prevents expression of cell-cycle-related protein, proliferation of microglia, and release of inflammatory factors, accompanied by reduced glial scarring. It also promotes production of chondroitin sulfate proteoglycan, a secreted factor that regulates cell division (Wartiovaara et al. 2002; Becker and Bonni 2005), neural migration (Konishi et al. 2002; Kato et al. 2003), and axon path finding, as well as stimulating neural stem cell survival. In doing so, olomoucine facilitates reconstruction and recovery (Tian et al. 2006; Tian et al. 2007). Moreover, in an in vitro cutting injury model of astrocytes, olomoucine suppresses cell proliferation and arrest cells in the G1/S and G2/M phases. In a model of photochemically induced ischemia, knockout of cyclin D1 inhibits proliferation of gliocytes, indicating that expression of cyclin D1 is required for division and proliferation of gliocytes (Zhu et al. 2007). In summary, CDK inhibition plays an important role in neuroprotection by at least two distinct but cooperative mechanisms (Di Giovanni et al. 2005): (1) reduction of neural injury and stimulation of neuron replacement and recovery and (2) prevention of inflammatory injury and glial scarring by inhibition of microglia and astrocyte proliferation and inflammatory factor production.
5 CDKs in Cancer Treatment
5.1 Cell-Cycle-Related Abnormalities in Cancer
Loss of cell cycle control provides a growth advantage to neoplastic cells, thus representing a classic feature of human cancer. Therefore, abnormalities in expression and/or activity of a variety of proteins that directly or indirectly involve the cell-cycle-regulatory machinery play essential roles in the pathogenesis of tumors. These abnormalities usually include loss/inactivation of endogenous CDK inhibitors, overexpression of CDK partner cyclins, amplification/active mutations of CDK genes, or their combination (Malumbres and Barbacid 2009). Such considerations provide a rationale for employing inhibitors of cell cycle progression as anticancer agents.
Aberrations in cell-cycle-regulatory molecules in human cancers occur most frequently in molecules associated with control of the G1 → S transition, a key step which determines initiation of the cell cycle. This signaling pathway is universally disrupted in human cancer even though most human malignancies retain wild-type RB1 (Li et al. 2012). In fact, dysregulation of the cyclin D–CDK4/CDK6/INK4/pRb/E2F signaling pathway has been identified in more than 80 % of human cancers (Cheung and Ip 2004). In human cancers, the main genetic alterations are deletions (biallelic or monoallelic) or 5′ CpG island methylation of p16INK4a and p15INK4b, whereas very few cases or cell lines had p18INK4c or p19INK4d deletions or hypermethylation (Drexler 1998). Among those, the commonest alteration is p16INK4a (CDK inhibitor 2A, a potent tumor suppressor) downregulation because of deletions of gene loci, loss-of-function point mutation, or epigenetic silencing (e.g., by hypermethylation in the promoter region) (Kohno and Yokota 2006; Chakravarti et al. 2007; Auerkari 2006). As a consequence, cancer cells grow uncontrollably owing to hyperactivation of cyclin D–CDK4/CDK6 activity (Yu et al. 2006). For example, whereas deletion of p16INK4a has been found in human primary myeloma cells (Tasaka et al. 1997), a more frequent abnormality in primary myeloma cells is inactivation of p16INK4a and p15INK4b genes by methylation (Chen-Kiang 2003; Ng et al. 1997). 5-CpG island hypermethylation of the p16INK4a locus has been reported in over 50 % of patients with myeloma and related disorders. However, it is uncertain whether hypermethylation of p16INK4a is correlated with a worse prognosis (Lesage et al. 2005; Galm et al. 2004). Inactivation of p18INK4c, such as biallelic deletion of p18INK4c and expression of a mutated p18INK4c fragment, is frequently found in myeloma cell lines (Kulkarni et al. 2002). The prognostic significance of such mutations in patients with myeloma is not known. Biallelic deletion of p18INK4c appears to be a late event of myeloma progression (Dib et al. 2006). Hypermethylation or deletion of p15INK4b has been reported in patients with myeloma (Chen-Kiang 2003; Ng et al. 1997), whereas high expression of p15INK4b is associated with diminished proliferative rate and more favorable prognosis in patients with myeloma (Sarasquete et al. 2006). Moreover, concurrent hypermethylation of p15INK4b and p16INK4a has been noted in a significant number of myeloma patients (Chim et al. 2003). Abnormalities in expression of the endogenous CDK inhibitors p15INK4b, p16INK4a, and p21Cip1/Waf1 also often occur in T cell lymphomas (Evens and Gartenhaus 2003).
Overexpression of cyclin D (primarily cyclin D1) is also common in a variety of human cancers (e.g., breast cancer, mantle cell lymphoma, and multiple myeloma) (Li et al. 2012). Aberrant overexpression of cyclin D1 usually stems from gene rearrangement [chromosome inversion or translocations, e.g., t(11p15;q13) and t(11;14)(q13;q32)], gene amplification, or alternative splicing (which generates a cyclin D1b transcript with constitutive nuclear localization and enhanced transforming capacity) (Lu et al. 2003; Carrere et al. 2005; Burd et al. 2006; Krieger et al. 2006; Knudsen et al. 2006). Cyclin D1 is a critical mediator of breast cancer induction by the oncogenes RAS and ERBB2 (Arnold and Papanikolaou 2005). Overexpression of cyclin D (primarily cyclin D1) is also a hallmark of mantle cell lymphoma (Oka et al. 1996). Chromosomal abnormalities are frequently found in multiple myeloma [e.g., t(11;14)(q13;q32) and t(4:14)(p16;q32)], and often involve cyclin D1 (11q13) (Lesage et al. 2005). Cyclin D1 overexpression is often accompanied by loss of p16INK4a, suggesting their possible cooperation in oncogenesis (Shapiro 2006). Overexpression of cyclin D1 results in activation of CDK4/CDK6 owing to an inappropriate increase in the amount of cyclin D–CDK4/CDK6 holoenzyme, and also leads to activation of cyclin E–CDK2 by sequestering Cip/Kip family CDK inhibitors (e.g., p21Cip1 and p27Kip1) in the cyclin D-dependent kinase complex (Shapiro 2006). Gene amplification and overexpression of cyclins D2 and D3 are also found in some cancers, including B cell malignancies (Delmer et al. 1995; Sonoki et al. 2001). For example, myeloma cells exhibit dysregulation of at least one of the three cyclins [cyclin D1 (11q13), cyclin D2 (MAF/16q23 or MAFB/20q11), or cyclin D3 (6p21)], whereas normal bone marrow plasma cells express low levels of cyclins D2 and D3, and little or no cyclin D1 (Bergsagel et al. 2005). Dysregulation of cyclin D is an early and unifying pathogenic event in myeloma. Patients whose cells exhibit dysregulation of cyclin D1 may have a particularly poor prognosis (Perez-Simon et al. 1998). Cyclin D1–CDK4 and cyclin D2–CDK6 pairing may be a critical determinant for cell cycle reentry and progression in expansion of self-renewing myeloma cells (Ely et al. 2005). Moreover, accumulating evidence also indicates that cyclin D may exert CDK-independent functions (e.g., by acting as a modulator of various transcription factors) in control of cell growth (Tashiro et al. 2007; Fu et al. 2004).
Amplification and point mutations (e.g., CDK4R24C with loss of INK4-binding ability) of the CDK4 gene have also been observed in human cancers (Wolfel et al. 1995). The central mechanism by which dysregulation of the cyclin D–CDK4/CDK6/INK4 pathway contributes to growth advantage of tumor cells involves “unscheduled” inactivation or inhibition of pocket proteins (e.g., pRb, and most likely p107 and p130 as well), resulting in the loss of their function as tumor suppressors. Loss of pRb or hyperactivation of CDK4/CDK6 is found in most human tumor cells (Shapiro 2006; van Deursen 2007). For example, partial or complete deletions in chromosome 13 (e.g., 13q14), which harbors the RB1 locus, have been reported in up to 30 % of myeloma patients and in up to 70 % of myeloma cell lines. In the remaining cases, pRb is predominantly phosphorylated (Urashima et al. 1996). Abnormalities in chromosome 13 have been associated with a particularly poor prognosis in patients with myeloma (Tricot et al. 1995). Co-expression of CDK4 with oncogenic Ras in normal human epidermal cells induces invasive neoplasia resembling human squamous cell carcinoma (Lazarov et al. 2002). Moreover, it has been found recently that kinase activity of cyclin D1–CDK4 is largely dispensable for normal development, whereas it is critically required for the initiation and maintenance of mammary carcinoma (Landis et al. 2006; Price 2000). Together, cyclin D–CDK4 and cyclin D–CDK6 are a very attractive therapeutic target (Lee and Sicinski 2006; Deshpande et al. 2005).
However, it is noteworthy that the linear model (cyclin D↑ and/or p16INK4a↓ → CDK4/CDK6↑ → pRb↓) has been challenged. For example, it has been found that the functions of cyclin D–CDK4/CDK6 can be recapitulated by either cyclin D–CDK2 or cyclin E–CDK2 (Horiuchi et al. 2012), both of which are able to phosphorylate pRb and induce cell proliferation (Barriere et al. 2007). In contrast, loss of CDK2 can also be recapitulated by CDK4, which can phosphorylate pRb even at CDK2-preferred sites, and cyclin E–CDK1 as well (Ortega et al. 2003). This model is further challenged by the recent findings from gene knockout mice (see earlier). In this context, CDK1 is able to replace functions of these interphase CDKs (CDK4/CDK6 and CDK2) by binding to their regulatory partners cyclin D and cyclin E in CDK4/CDK6 or CDK2 knockout mice. Nevertheless, overexpression or constitutive activation of CDK2 has been observed in some types of human cancers (Kohzato et al. 2001; Li et al. 2002; Dong et al. 2001). In addition, overexpression of cyclin A or cyclin E, overexpression/activated mutation of CDK1 or CDK7, and loss of Cip1/Kip family CDK inhibitors (e.g., p27Kip1, and most likely p21Cip1 as well) have also been reported in many types of human malignancies (Shapiro 2006). Furthermore, loss of endogenous CDK inhibitors (e.g., p27Kip1, p16INK4a, and possibly p21Cip1) is associated with poor outcome in patients with various cancers (Senderowicz and Sausville 2000).
CDK7 and CDK9 phosphorylate the CTD of RNA pol II, facilitating transcription and elongation. The inhibition of this phosphorylation blocks TP53-dependent and independent expression of CDKN1A (a gene encoding p21Cip1). CDK inhibition (e.g., by flavopiridol) stabilizes TP53, reducing expression of MTBP (mouse TP53 binding protein, transformed 3T3 cell double minute), an important negative regulator of the TP53 tumor suppressor. Overexpression of MTBP is related to several types of human cancer, such as breast cancer, tissue sarcomas, and osteosarcomas (Gallorini et al. 2012).
CDK8 and its regulatory partner cyclin C, two subunits of the Mediator complex, are frequently either mutated or amplified in a variety of human cancers (Xu and Ji 2011). CDK8 functions as an oncoprotein in melanoma and colorectal cancers. CDK8, as one of the most significant colorectal-cancer-associated genes, is the only one that exhibits frequent copy number gain in human colorectal cancers: the CDK8 gene is amplified in 47–76 % of colorectal adenocarcinoma patient samples, and the chromosomal region (13q12.13) that harbors CDK8 is gained in 60 % of colorectal cancers (Gallorini et al. 2012). Thus, CDK8 is a valuable molecular biomarker particularly for the prognosis of a subset of colon cancer patients. The dysregulation of CDK8 is significantly correlated with increased colon-cancer-specific mortality. CDK8 expression is also significantly associated with β-catenin activation in gastric adenocarcinoma (Xu and Ji 2011). Elevated expression of CDK8 predicts poor prognosis in gastric cancers (Firestein et al. 2008).
The gain of CDK8 activity is sufficient to transform 3T3 cells, whereas CDK8 activity is necessary for β-catenin-driven transformation of 3T3 cells. Conversely, knockdown of CDK8 significantly reduces tumor cell growth. CDK8 expression is correlated with high β-catenin activation, expression of tumor suppressor p53, and overexpression of fatty acid synthase (FASN), suggesting multiple functions of CDK8 in colorectal tumorigenesis. Indeed, CDK8 is identified as an oncoprotein that promotes the proliferation of colorectal cancer cells (Xu and Ji 2011).
Elevated expression of the CDK8 gene is reported to play a major role in promoting the proliferation of melanoma cells (Xu and Ji 2011), particularly in the subtype of vertical growth phase and metastatic melanomas that display loss of the histone variant macroH2A (Kapoor et al. 2010). Knockdown of either CDK8 or MED12 suppresses the proliferative advantage induced by macroH2A loss in melanoma cells, suggesting that the effect of CDK8 is dependent on the Mediator complex.
However, loss or reduction of expression of CDK8 is also found in a few types of cancers. For example, the CDK8 gene is deleted in esophageal squamous cell carcinoma (Xu and Ji 2011). Likewise, the expression of CDK8 is significantly reduced in bladder cancers. A CDK8 point mutation (D189N) is found in diverse tumor samples, and is likely to cause a loss of CDK8 kinase activity. This implies that CDK8 may not always behave as an oncoprotein in all human cancers, and its activity needs to be tightly regulated.
The CCNC gene (encoding cyclin C) is significantly upregulated in patients with gastric cancer, colorectal cancers, adenocarcinoma, leukemia, and lymphoma, as well as a few hepatoma cell lines (Xu and Ji 2011). However, the CCNC gene is also frequently deleted in a subset of acute lymphoblastic leukemia, osteosarcoma, and gastric cancers.
CDK10 is an important determinant of resistance to endocrine therapies for breast cancer, whereas CDK10 silencing increases Ets2-driven transcription of c-RAF, resulting in mitogen-activated protein kinase pathway activation and loss of tumor cell reliance on estrogen signaling (Iorns et al. 2008). Patients with estrogen-receptor-α-positive tumors that express low levels of CDK10 relapse early when receiving tamoxifen. Low levels of CDK10 are likely associated with methylation of the CDK10 promoter.
Collectively, these findings strongly support the notion that both cell-cycle-regulatory and transcriptional CDKs are attractive therapeutic targets in human cancer (Firestein et al. 2008).
5.2 Pharmacological CDK Inhibitors
CDKs and related molecules are very promising targets in the development of cancer therapeutics (Cicenas and Valius 2011; Canavese et al. 2012; Gallorini et al. 2012; Wesierska-Gadek and Maurer 2011). Among a variety of CDK inhibitors under development and evaluation, several (e.g., flavopiridol, CYC202, UCN-01, and BMS-387032) are currently undergoing clinical evaluation based on preclinical evidence of antitumor activity (Shapiro 2006; Dai and Grant 2004; Schwartz and Shah 2005; Benson et al. 2005). Flavopiridol, as a pan-CDK inhibitor, exerts multiple actions in tumor cells, including inhibition of both cell cycle and transcriptional CDKs (both CDK9 and CDK7), induction of apoptosis, and antiangiogenic activity. UCN-01 was initially developed as a protein kinase C (PKC) inhibitor, and was later found to act as a CDK inhibitor. However, its antitumor effects appear to be more closely related to inhibition of Chk1, leading to “unscheduled” activation of CDK1 and abrogation of G2/M and S checkpoints, as well as inhibition of the prosurvival 3-phosphoinositide-dependent protein kinase (PDK) 1/Akt pathway. CYC-202 and BMS-387032 have been developed as CDK2 inhibitors, but like most relatively specific inhibitors of CDK2, also inhibit CDK1. In addition, CYC-202 has been also found to inhibit cyclin T–CDK9 and cyclin H–CDK7, thereby blocking phosphorylation of the RNA pol II CTD, which is associated with transcriptional repression of proteins with short half-lives. It is noteworthy that genetic evidence suggests that inhibition of a single CDK (e.g., CDK2) may be insufficient to induce cell death or even prevent cell growth (Tetsu and McCormick 2003), and that inhibition of transcriptional and cell-cycle-regulatory CDKs may cooperate to induce lethality in tumor cells (Cai et al. 2006). Such findings suggest that highly specific CDK inhibitors may be suboptimal as anticancer agents (Merrick and Fisher 2012), and that factors other than or in addition to CDK inhibition very likely contribute to the lethal actions of these compounds (Sausville 2002).
5.2.1 Flavopiridol (Alvocidib)
Flavopiridol is a semisynthetic small-molecule derivative of rohitukine, an alkaloid isolated from Dysoxylum binectariferum (a plant indigenous to India). In preclinical studies, flavopiridol potently inhibited cell proliferation (IC50 = 66 nM) in all 60 NCI human tumor cell lines, with no obvious tumor-type selectivity (Senderowicz 2002). As the first clinically relevant CDK inhibitor, initial trials used a schedule of 24 or 72 h continuous infusion every 2 weeks. These schedules achieved concentrations capable of producing preclinical effects. For example, a 72-h infusion regimen produced a 271–415 nM steady-state plasma concentration (Colevas et al. 2002). However, prolonged infusion of flavopiridol proved largely inactive in trials involving several hematopoietic malignancies. Consequently, a bolus administration (1-h infusions for 1–5 days every 21 days) was designed to achieve higher plasma concentrations. Indeed, the 1-h infusion regimen resulted in 1.7–3.8 μM median maximum concentrations, reflecting postinfusion peak concentrations (Colevas et al. 2002), and a limited number of response in certain settings. Notably, clinically achievable concentrations by either continuous or bolus infusion exceeded the threshold for inhibition of CDKs and cell growth, and induction of apoptosis in preclinical studies. However, in striking contrast to its impressive activity in vitro and in various xenograft models (Zhai et al. 2002), the outcomes of most clinical trials were disappointing (Shapiro et al. 2001; Schwartz et al. 2001; Lin et al. 2002; Tan et al. 2002). The failure of flavopiridol to recapitulate its in vitro activity may stem from more than 90 % plasma protein binding and inadequate plasma concentrations of the free drug. In contrast, a variety of clinical trials have demonstrated that combinations of flavopiridol and either conventional chemotherapeutic agents (e.g., paclitaxel, fludarabine, cytosine arabinoside, and irinotecan/CPT-11) or novel signal transduction modulators may be more promising (Karp et al. 2005).
Very recently, a pharmacologically directed infusion schedule has been developed in which half of the flavopiridol dose is administered in 30 min and the other half in 4 h. This schedule was associated with very promising activity in patients with refractory chronic lymphocytic leukemia(Byrd et al. 2007). In fact, the major dose-limiting toxicity was tumor lysis syndrome. Trials are currently under way to evaluate this schedule in patients with other hematologic malignancies.
5.2.1.1 Cell Cycle Inhibition
Flavopiridol induces cell cycle arrest by targeting cell-cycle-regulatory CDKs. In in vitro studies using purified CDKs, flavopiridol has been shown to inhibit cyclin B-CDK1 (IC50 = 30–40 nM), cyclin A–CDK2 and cyclin E–CDK2 (IC50 = 100 nM), cyclin D–CDK4 (IC50 = 20–40 nM), cyclin D–CDK6 (IC50 = 60 nM), and cyclin H–CDK7 (IC50 = 110–300 nM) (Sedlacek 2001; Hardcastle et al. 2002). X-ray crystallographic analysis revealed that L868276 (a deschlorophenyl derivative of flavopiridol with approximately tenfold reduction in inhibitory activity toward CDKs) binds to the ATP-binding pocket of CDK2 (Senderowicz 2002; Hardcastle et al. 2002). In the structure of flavopiridol, the chloro group on the phenyl ring is able to make additional contacts with CDK2, which may explain the tenfold greater potency of flavopiridol compared with L868276. The overall molecular structure of CDKs is quite similar, and they share 40 % sequence homology, including the highly conserved catalytic core region of 300 residues. Flavopiridol directly inhibits the activity of most CDKs by occupying the ATP-binding site of these kinases, an effect that can be competitively blocked by excess ATP. Indeed, CDK1, CDK2, CDK4, and CDK7 in the soluble extracts from non-small-cell lung carcinoma have been shown to bind to immobilized flavopiridol in the absence of ATP but not in its presence. Furthermore, by inhibiting CAK (i.e., cyclin H–CDK7), flavopiridol also prevents phosphorylation at Thr160 and Thr161 of most CDKs (e.g., CDK1, CDK2, CDK4, and CDK6) (Senderowicz 2002), whereas these phosphorylations are necessary for full activation of the CDKs.
Inhibition of CDKs by flavopiridol leads to cell cycle arrest at the G1–S phase transition and the G2–M phase transition, as well as delay in S phase progression (Sedlacek 2001; Shapiro 2004). For example, flavopiridol can block G1 progression by inhibiting cyclin D–CDK4/CDK6, retard S phase progression, or arrest cells in G1 phase by inhibiting cyclin E–CDK2 and cyclin A–CDK2, and arrest cells in G2 phase by inhibiting cyclin A–CDK1 and cyclin B–CDK1. Moreover, flavopiridol also induces cell cycle arrest through transcriptional inhibition and downregulation of cyclin D1, although this action requires slightly higher drug concentrations (100–300 nM) than those necessary for inhibition of cell-cycle-regulatory CDKs (Dai and Grant 2004). It is noteworthy that CDK6 inhibition by flavopiridol seems to play a functional role in cell cycle arrest (e.g., G1 arrest) only in tumor cells lacking CDK4 (Sedlacek 2001). Nevertheless, the patterns of cell cycle arrest (e.g., G1/S arrest, G2/M arrest, or both) induced by flavopiridol (and other pan-CDK inhibitors) appears largely cell-type dependent.
As a first-generation CDK inhibitor, flavopiridol acts as a pan-CDK inhibitor. However, its inhibitory capacity is relatively selective for most CDKs but not for a specific CDK. Subsequently, efforts have been directed at identifying either structure-based synthetic/semisynthetic compounds or natural products that act on specific CDKs, such as CDK4, CDK1, or CDK2. As a consequence, more specifically selective CDK inhibitors have been developed, for example, inhibitors of CDK4/CDK6 (e.g., PD-0332991 and CINK4) (Hardcastle et al. 2002; Fry et al. 2001) and inhibitors of CDK2 and CDK1 which are significantly less potent against CDK4/CDK6, e.g., CYC202/(R)-roscovitine, BMS-387032, PNU-252808, AZ703, NU6102, and NU6140 (Dai and Grant 2003, 2003). A number of these compounds are currently under evaluation as antitumor agents in preclinical models and some are in early stage clinical trials. However, it is difficult to design and develop inhibitors specifically targeting only a single CDK, most likely owing to conservation of amino acids lining the ATP-binding pocket and the high structural homology shared by CDKs. Three-dimensional structural analysis of CDKs, particularly the CDK–inhibitor complex, has provided useful information for the development of novel CDK inhibitors. In particular, the crystal structures of CDK2 have been well established and used extensively for the synthesis of CDK2-specific inhibitors, as well as for evaluating CDK inhibitor potency and selectivity. For example, a new purine-based inhibitor has been described which is 1,000-fold more potent than the parent compound (K i of 6 nM for CDK2 and 9 nM for CDK1) (Davies et al. 2002). Similar strategies have been used to develop CDK4-specific inhibitors, by using structure-based information related to a CDK4–mimic CDK2 protein (Ikuta et al. 2001; Honma et al. 2001). Moreover, new approaches (e.g., affinity chromatography of immobilized inhibitors) have been established to identify the intracellular targets (selectivity) of individual CDK inhibitors. Clearly, selectivity is a key issue for the use of CDK inhibitors as pharmacological tools to demonstrate the function of CDKs. However, a key question remaining to be answered is whether inhibition of a specific CDK by a highly selective inhibitor, rather than inhibition of broad CDKs by a pan-CDK inhibitor such as flavopiridol, will be efficient in killing tumor cells in view of evidence that (1) tumor cells usually exhibit multiple genetic alterations and/or dysregulation of multiple signaling pathways related to cell cycle regulation and (2) there exists functional overlap and/or cross talk between different CDKs as well as CDKs and other proteins (e.g., their partner cyclins). Thus, it remains possible that the broad actions of a compound such as flavopiridol are beneficial for its antitumor activity.
5.2.1.2 Transcription Inhibition
Flavopiridol very potently represses transcription (IC50 < 10 nM) in vitro by blocking the transition into productive elongation mediated by RNA pol II, which is controlled by P-TEFb (cyclin T–CDK9) (Chao et al. 2000). Flavopiridol inhibits CTD kinase activity of RNA pol II with K i of 3 nM, a concentration significantly lower than that required for inhibition of most other CDKs (e.g., CDK1, CDK2, and CDK4 with K i values between 40 and 70 nM). Furthermore, unlike inhibition of other CDKs, inhibition of CDK9 by flavopiridol is noncompetitive with respect to ATP (Chao and Price 2001). A P-TEFb-immobilized assay demonstrates that flavopiridol (1:1 stoichiometry) remains bound even in the presence of high salt concentrations, suggesting that the apparent lack of competition with ATP could result from very tight binding between the drug and the enzyme. Indeed, recent structural information related to the binary CDK9–flavopiridol complex indicates that flavopiridol binds very tightly to the ATP-binding pocket of CDK9 with higher affinity than CDK2, even though, in contrast to the case of CDK2, no additional binding site has been identified for CDK9 (de Azevedo et al. 2002). In cells, flavopiridol inhibits transcription at concentrations far lower than those required to inhibit CDK1 and CDK2, even in the presence of physiological concentration of ATP (Dai and Grant 2004). Another potential target for transcriptional repression by flavopiridol is CDK7 (catalytic subunit of TFIIH). However, CDK7 inhibition requires higher concentrations of flavopiridol than are necessary for inhibition of CDK9 (Dai and Grant 2003). Therefore, inhibition of transcription by flavopiridol primarily stems from direct inhibition of CDK9.
In mammalian cells, DNA microarrays have shown that flavopiridol inhibits gene expression broadly, similar to the actions of general transcription inhibitors (e.g., actinomycin D and DRB) (Lu et al. 2004). However, at the protein level, flavopiridol primarily downregulates the expression of short-lived proteins, such as cyclin D1 and Mcl-1.
5.2.1.3 Cyclin D1 Downregulation
Cyclin D1 is a multifunctional protein that not only plays a critical role in regulation of the cell cycle (e.g., the G1–S transition) as a partner of CDK4/CDK6 (see earlier), but also acts as a transcriptional regulator by modulating the activity of several transcription factors (e.g., STAT3) which are CDK-independent, and may explain why cyclin D1 is involved not only in cell cycle progression but also in cell growth and survival (Tashiro et al. 2007; Fu et al. 2004). Recently, it was shown that cyclin D1 binds to the transcription factors STAT3 and NeuroD and inhibits their transcriptional activity, which may be related to modulation of cell differentiation (Coqueret 2002). It has been reported that cyclin D1 also interacts with histone deacetylases, and in doing so blocks access of transcription factors to the promoter and inhibits loading of the initiation complex (Fu et al. 2005). Cyclin D1, as an oncogene, also plays an important role in carcinogenesis, probably by driving cells into S phase and cooperating with various oncogenes (such as Myc and Ras) in malignant transformation (Rodriguez-Bravo et al. 2006). Rearrangement of the cyclin D1 locus and/or overexpression of cyclin D1 have been reported in many human tumors (Hosokawa and Arnold 1998).
Expression of cyclin D1 is growth-factor-dependent, and is regulated at the levels of transcriptional activation, protein degradation, or nuclear export. Mitogen induction of cyclin D1 generally relies on activation of the Ras/Raf/MEK/ERK pathway. Ras signaling and ERK activation promotes transcription of the cyclin D1 gene, probably through transcription factors (Jirmanova et al. 2002). In addition, the Ras signaling pathway is also necessary for associations between cyclin D1 and CDK4. A variety of transcription factors such as AP-1, STATs (STAT3 and STAT5), NF-κB, Egr-1, Ets, CREB, β-catenin, and certain nuclear receptors activate the cyclin D promoter (Lavoie et al. 1996). On the other hand, expression of cyclin D1 is subject to transcriptional inhibition by other factors, such as E2F1, JunB, INI1/hSNF5, peroxisome-proliferator-activated receptor (nuclear receptor), and calveolin-1 (Hulit et al. 2000). In the control of cell growth rate, the target of rapamycin/eIF4E signaling pathway may act upstream of cyclin D (Shi et al. 2005). The levels of both free and CDK-bound cyclin D1 are also regulated by proteasome-dependent degradation, causing rapid turnover (an approximate half-life of 20 min) of this protein (Diehl et al. 1997). The Ras signaling cascade can prevent ubiquitin–proteasome-dependent degradation of cyclin D1 (Shao et al. 2000). Following its association with CDK4, cyclin D1 is phosphorylated at Thr286 by glycogen synthetase kinase 3β, a kinase controlled by Akt through inhibitory phosphorylation (Takahashi-Yanaga et al. 2006). This event may represent a mechanism by which cyclin D1 is exported from the nucleus to the cytoplasm, resulting in its proteasomal degradation, thereby shutting down this signaling cascade. The pharmacological inactivation of the phosphatidylinositide 3-kinase (PI3K)/Akt pathway and transfection with a constitutively active form of Akt extends the half-life of cyclin D1 twofold to threefold (Radu et al. 2003).
Flavopiridol transcriptionally downregulates expression of cyclin D1 in multiple types of cancer cells. For example, exposure of MCF-7 breast cancer cells to flavopiridol results in a decline in cyclin D1 promoter activity, leading to a decrease in the mRNA and protein levels of cyclin D1 (Carlson et al. 1999). This effect is followed by a decline in the levels of cyclin D3, but not those of cyclin D2 and cyclin E, as well as loss of CDK4/CDK6 activity. In vivo, flavopiridol resulted in depletion of cyclin D1 in the HN12 tumor xenograft, whereas the levels of cyclin D3 and cyclin E remained constant (Patel et al. 1998). Cyclin D1 transcriptional repression may stem from inhibition of P-TEFb by flavopiridol. However, this hypothesis is not supported by observations that inhibition of cyclin D1 expression requires much higher concentrations of flavopiridol (100–1,000 nM) than are required for inhibition of P-TEFb activity. Other mechanisms may involve interruption of transcriptional regulation of cyclin D1 by a number of transcription factors, including positive regulators (e.g., STAT3 and NF-κB) and negative regulators (e.g., E2F1). In addition, flavopiridol can directly bind to duplex DNA with a range of equilibrium dissociation constant values similar to that for the DNA intercalators doxorubicin and pyrazoloacridine (Bible et al. 2000), which may affect the function of DNA as a transcriptional template.
Thus, administration of flavopiridol leads to cell cycle arrest through mechanisms related to inhibition of CDK activities, that is, by direct binding to the ATP-binding sites, by preventing phosphorylation of CDKs through inhibition of CAK (cyclin H–CDK7), or by transcriptional downregulation of cyclin D1. Transcriptional repression of cyclin D1 by flavopiridol may be particularly relevant in mantle cell lymphoma, in which cyclin D1 is overexpressed in 95 % of patients. Notably, flavopiridol has been reported to delay disease progression in a substantial fraction of patients with mantle cell lymphoma (Kouroukis et al. 2003).
5.2.1.4 Mcl-1 Downregulation
Recent interest has focused on the antiapoptotic protein Mcl-1 as a transcriptional target of flavopiridol. For example, in vitro treatment with flavopiridol induces declines in expression of Mcl-1 mRNA and/or protein levels, which precedes apoptosis, in a variety of cancer cells, including non-small-cell lung cancer cells, multiple myeloma cells, and freshly isolated CD5+/CD19+ cells from patients with B cell chronic lymphocytic leukemia, and CD138+ cells from patients with multiple myeloma (Pepper et al. 2001; Semenov et al. 2002). Downregulation of Mcl-1 has also been confirmed in vivo in primary leukemic cells from flavopiridol-treated acute myelogenous leukemia (AML) patients (Karp et al. 2005). H1299 (non-small-cell lung cancer) and NIH3T3 (transformed fibroblasts) cells constitutively expressing Mcl-1 are resistant to apoptosis induced by flavopiridol (Ma et al. 2003).
Flavopiridol induces Mcl-1 downregulation most likely by inhibition of P-TEFb (Dai and Grant 2004; Blagosklonny 2004). However, expression of the Mcl-1 gene is controlled by multiple signaling pathways. For example, it is negatively regulated by E2F1 through direct binding to the Mcl-1 promoter, and is positively regulated by the PI3K/Akt pathway, by the mitogen-activated protein kinase pathway, and by transcription factors such as STAT3 and CREB (Wang et al. 1999). Consequently, flavopiridol-mediated downregulation of Mcl-1 may be also related to other mechanisms, including accumulation of E2F1 and disruption of STAT3/DNA binding (Croxton et al. 2002; Lee et al. 2006; Aggarwal et al. 2006).
5.2.1.5 Apoptosis Induction
In multicellular organisms, cells engage an intrinsic mechanism of self-destruction designated programmed cell death or apoptosis, which is essential for maintaining tissue homeostasis. Tumor cells often have defects in the apoptosis-inducing pathway, resulting in the dysregulated expansion of a population of neoplastic cells, escape of cancer cells from surveillance by the immune system, and resistance to apoptosis induced by chemotherapy and radiotherapy. Initiation of apoptosis involves at least two distinct pathways: the extrinsic pathway, which is mediated by death receptors, and the intrinsic pathway, which is dependent on mitochondria (Strasser et al. 2000). In the former, apoptotic signaling is initiated by binding of members of the TNF family to death receptors, such as CD95 and TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2. When the death receptors are activated by TNF family ligands, their death domains attract the intracellular adaptor protein Fas-associated death domain (FADD), which, in turn, recruits the inactive form of certain initiator caspases (e.g., caspases 8 and 10) to the death-inducing signaling complex (DISC). At the DISC, procaspases 8 and 10 are cleaved and converted into active forms (Fulda and Debatin 2006). In type I cells, the DISC-activated caspase 8 is sufficient to trigger apoptosis directly, but in type II cells, the mitochondria-dependent pathway is required for amplification of initial apoptotic signals, which is linked by truncation/activation of Bid (a proapoptotic member of the Bcl-2 family) by active caspase 8 (Scaffidi et al. 1998). In the intrinsic pathway, death signals (e.g., DNA damage) lead to mitochondrial damage, probably mediated by caspase 2 activation (Lassus et al. 2002). Mitochondria release cytochrome c and other proapoptotic factors, such as AIF and second mitochondria-derived activator of caspase (Smac)/direct inhibitor of apoptosis (IAP) binding protein with low pI (DIABLO), from the intermembrane space to the cytosol, where cytochrome c forms a complex (known as the apoptosome) with apoptotic protease-activating factor 1 (APAF1), an inactive form of the initiator caspase (e.g., caspase 9), and ATP (Zou et al. 1999). In the apoptosome, procaspase 9 is cleaved and activated. For both pathways, once the initiator caspases (caspases 8 and 10 in the extrinsic pathway, and caspase 9 in the intrinsic pathway) have been activated, they further cleave and activate executioner caspases (e.g., caspases 3, 6, and 7). Activation of executioner caspases can further result in cleavage/activation of other caspases (including ones lying upstream) to amplify the death-signal cascade (Green and Kroemer 2004). Eventually, the activated executioner caspases cleave a number of cellular “death substrates,” leading to biochemical and morphological changes of apoptosis. Apoptotic pathways are tightly controlled by various proteins. For example, processing/activation of caspase 8 is inhibited by a protein referred to as FLIP (FLICE/caspase 8 inhibitory protein) through binding to DISC (Micheau 2003). Importantly, Bcl-2 family members, including antiapoptotic proteins (e.g., Bcl-2, Bcl-xL, Bcl-w, A1, and Mcl-1) and proapoptotic proteins (e.g., multidomain family Bax, Bak, and Bok; BH3-only family Bid, Bim, Bik, Bad, Bmf, Hrk, Noxa, and Puma), play critical roles in regulation of both the intrinsic and extrinsic pathways, primarily at the mitochondrial level (Zamzami and Kroemer 2001). Moreover, IAP protein family members (e.g., XIAP, cIAP1, cIAP2, NAIP, MLAIP, ILP2, livin/KIAP, apollon, and survivin) are antiapoptotic proteins that regulate apoptotic signaling mostly downstream of mitochondria. Most members of the IAP family directly bind to and inhibit the active form of both initiator (e.g., caspase 9) and executioner (e.g., caspases 3, 6, and 7) caspases by promoting their degradation through the ubiquitin–proteasome pathway (Vaux and Silke 2005). In turn, IAPs are inhibited by mitochondria-releasing Smac/DIABLO (Verhagen and Vaux 2002).
It has been well documented that flavopiridol induces apoptosis in a broad spectrum of malignant cells. For example, in vitro, 6–48 h exposure to 100–400 nM flavopiridol induces apoptosis in a variety of tumor cells, including leukemia, lymphoma, head and neck squamous cell carcinoma (HNSCC), breast cancer, non-small-cell lung cancer, prostate carcinoma, gastric carcinoma, esophageal carcinoma, and bladder carcinoma (Shapiro 2004). Human leukemia cells, regardless of their origins (i.e., cultured cell lines or freshly isolated primary cells from patients) or subtype (myeloid, B cell, or T cell type), are the most sensitive to induction of apoptosis by flavopiridol (Kitada et al. 2000). Notably, flavopiridol can also induce apoptosis in tumor cells that are resistant to DNA-damaging agents and radiation (Sedlacek 2001; Shapiro 2004). In vivo, treatment with flavopiridol (5 mg/kg intraperitoneally daily for 5 days) induced apoptosis in the HNSCC xenograft HN12 as detected by terminal deoxynucleotidyl transferase mediated dUTP-biotin nick end labeling (TUNEL), with significant reduction (60–70 %) in tumor size (Patel et al. 1998).
The mechanisms by which flavopiridol induces apoptosis have been extensively studied. First, flavopiridol is able to induce apoptosis in tumor cells in which caspase 8 is absent (Achenbach et al. 2000). Moreover, neither the pharmacological caspase 8 inhibitor IETD-FMK nor transfection of the viral caspase 8 inhibitor CrmA is able to block flavopiridol-induced cytochrome c release and apoptosis (Decker et al. 2001). These findings suggest that the extrinsic pathway is not primarily involved in flavopiridol-induced apoptosis, despite the fact that cleavage of caspase 8 and Bid has been observed after exposure to flavopiridol (Achenbach et al. 2000).
Flavopiridol induces apoptosis in resting tumor cells which exhibit sensitivities similar to those of proliferating cells, even in the same cell lines (Dai and Grant 2004; Sedlacek 2001), arguing against the possibility that the cytotoxicity of flavopiridol stems from inhibition of CDKs involved in cell cycle regulation. However, direct binding of flavopiridol to duplex DNA may provide an explanation for the ability of flavopiridol to kill noncycling (resting) cancer cells (Bible et al. 2000). Moreover, no significant difference in the cytotoxic activity of flavopiridol has been found between cells expressing pRb versus those defective in pRb expression, even though flavopiridol treatment induces hypophosphorylation of pRb (Cartee et al. 2001). Moreover, certain cell lines that lack detectable pRb expression exhibit more pronounced apoptosis following flavopiridol treatment (Dai et al. 2006). Treatment of H1299 non-small-cell lung cancer cells with flavopiridol (200 nM) results in the rapid elevation of E2F1 levels followed by apoptosis, whereas H1299 cells with deletion of E2F1 through RNA interference or murine embryo fibroblasts deficient in E2F1 are less susceptible but not completely resistant to the cytotoxicity of flavopiridol (Ma et al. 2003). It is known that E2F1 mediates cell death through both p14ARF/MDM2/p53-dependent and p14ARF/MDM2/p53-independent pathways. In most cases, flavopiridol has little or no effect on p53 levels, and its cytotoxic activity appears to be independent of the genetic status of p53 (Reed 2003). There is no direct evidence for the notion that transcriptional downregulation of cyclin D1 contributes to the cytotoxicity of flavopiridol, although repression of cyclin D1 expression by an antisense oligonucleotide approach triggers apoptosis in carcinoma cells (Dai et al. 2006). In contrast, overexpression of cyclin D1 sensitizes human pRb-null myeloma cells (e.g., U266) to flavopiridol (Dai et al. 2006).
Attention has recently focused on transcriptional downregulation of proteins involved in the regulation of apoptosis, which most is likely a central theme underlying the induction of apoptosis by flavopiridol. In this context, Mcl-1 is an important target (see earlier). In addition, flavopiridol also downregulates many other antiapoptotic proteins. For example, administration of flavopiridol results in decreased expression of Bcl-2 in several cell lines, such as B cell leukemia, ovarian carcinoma, prostate carcinoma, and multiple myeloma cells (Semenov et al. 2002). However, flavopiridol-induced apoptosis appears largely independent of Bcl-2 inasmuch as flavopiridol kills tumor cells displaying Bcl-2 overexpression, an event that confers resistance to conventional chemotherapeutic agents (Lavoie et al. 1996). Moreover, neither ectopic overexpression nor antisense-oligonucleotide-mediated downregulation of Bcl-2 affects flavopiridol-induced cell killing (Lavoie et al. 1996). However, human leukemia cells displaying ectopic expression of N-terminal phosphorylation loop-deleted Bcl-2 (amino acids 32–80, a region known to negatively regulate its function) are highly resistant to flavopiridol-mediated cleavage of Bid, cytochrome c release, activation of caspases, degradation of poly(ADP-ribose) polymerase, and apoptosis (Decker et al. 2002), indicating that posttranslational modification(s) (e.g., phosphorylation) of Bcl-2 rather than transcriptional regulation may be involved in flavopiridol-induced apoptosis. Exposure to flavopiridol also results in downregulation of Bcl-xL and XIAP in various types of cancer cells, events likely associated with inhibition of NF-κB (Kim et al. 2003; Takada and Aggarwal 2004). In addition, downregulation of other antiapoptotic molecules (e.g., BAG-1, a regulator of the Hsp70 family that confers resistance to apoptosis induced by a variety of stimuli) has also been reported in B cell chronic lymphocytic leukemia cells exposed to flavopiridol (Kitada et al. 2000).
5.2.1.6 Other Mechanisms
In several systems, it has been reported that flavopiridol has a significant antiangiogenic activity, which indicates that inhibition of tumor angiogenesis could play a considerable role in the antitumor effects of flavopiridol (Newcomb 2004). The antiangiogenic activity of flavopiridol may be related to the ability of flavopiridol to induce apoptosis in both resting and proliferating endothelial cells through an unknown mechanism which is independent of expression ofCDKs (e.g., CDK1 and CDK2) (Brüsselbach et al. 1998). Indeed, endothelial cells are more sensitive to flavopiridol than other normal cells, such as fibroblasts, bone marrow cells, and peripheral lymphocytes, but are less sensitive than most tumor cells. However, inhibition of vascular endothelial growth factor (VEGF) expression could play an important role in the antiangiogenic effects of flavopiridol. VEGF is an angiogenic factor which is critical for cancer progression and metastasis. In human peripheral blood mononuclear cells and human neuroblastoma cells, it has been shown that flavopiridol completely blocks hypoxia-induced VEGF mRNA transcription and downregulates VEGF protein levels by dramatically decreasing VEGF mRNA stability (Newcomb et al. 2005).
It is also been reported that flavopiridol significantly inhibits rabbit muscle glycogen phosphorylases (a and b) (Oikonomakos et al. 2000). With use of immobilized flavopiridol, glycogen phosphorylases have been identified as flavopiridol-binding proteins (Kaiser et al. 2001). Treatment of A549 non-small-cell lung cancer cells with flavopiridol results in an increase in glycogen accumulation (Kaiser et al. 2001). Further studies showed that flavopiridol inhibits glycogen phosphorylase by directly binding to the inhibitor site in these proteins (Oikonomakos et al. 2000). These findings raise the possibility that interference with glucose homeostasis may also contribute to the antitumor effects of flavopiridol.
5.2.2 UCN-01
UCN-01 (7-hydroxystaurosporine, NSC638850, or KW-2401; Kyowa Hakka Kogyo), a derivative of the nonspecific PKC inhibitor staurosporine (a natural product isolated from Streptomyces staurosporeus), was originally developed as a selective PKC inhibitor. It has also been reported to inhibit several CDKs. However, recent studies have shown that UCN-01 exerts other antitumor effects, including inhibition of Chk1, which results in “inappropriate” activation of CDKs and abrogation of DNA-damage-induced cell cycle checkpoints, as well as interference with the PDK1/Akt survival pathway, thus promoting induction of apoptosis. These effects are largely independent of PKC inhibition. UCN-01 displays antitumor activity in in vitro systems and in in vivo xenograft models involving multiple human tumor types, with greater antitumor effects observed with longer administration intervals (e.g., 72 h in in vitro systems). Initial clinical trials of UCN-01 involved a 72-h continuous infusion schedule every 2 weeks (Fuse et al. 2005). Unexpectedly, the plasma half-life (approximately 30 days) of UCN-01 in patients was observed to be 100-fold longer than that observed in preclinical models. It was subsequently shown that UCN-01 extensively binds to plasma α1-acidic glycoprotein in humans, which accounts for the unique clinical pharmacological behavior of UCN-01 (Dai and Grant 2004; Fuse et al. 2005). On the basis of these findings, further clinical trials are being conducted using modified UCN-01 schedules (e.g., a 36-h continuous infusion every 4 weeks). Such schedules result in a mean UCN-01 half-life of approximately 588 h with peak plasma concentrations of total drug ranging from 30 to 40 μM, with approximately 100 nM concentrations of free UCN-01 detected in saliva (Fuse et al. 2005). Significantly, such concentrations are in excess of those necessary to inhibit Chk1. Several responses have been observed in patients with melanoma and refractory anaplastic large-cell lymphoma. In addition, several phase I trials with shorter schedules (e.g., 3-h infusion) are currently ongoing as combination regimens involving DNA-damaging agents (Dees et al. 2005; Vogel et al. 2007; Sampath et al. 2006).
5.2.2.1 PKC Inhibition
UCN-01 selectively inhibits Ca2+-dependent PKC isozymes (e.g., PKCα, PKCβ, and PKCγ; IC50 = 4–30 nM), and less potently inhibits Ca2+-independent PKC isozymes (IC50 ~ 500 nM) (Hofmann 2004). However, it exerts no effect on the atypical PKCs (e.g., PKCδ). In clinical trials, a clear decrease in the level of the phosphorylated cytoskeletal membrane protein adducin, a specific substrate phosphorylated by PKC, was observed in tumor and bone marrow samples following UCN-01 administration (Dai and Grant 2004). However, PKC inhibition appears to be unrelated to various actions of UCN-01, including antiproliferative activity, interference with cell cycle progression, and induction of apoptosis.
5.2.2.2 CDK Inhibition
UCN-01 can either inhibit or activate CDKs. Crystal structure analysis has shown that UCN-01 binds to active cyclin A–phospho-CDK2 (Johnson et al. 2002). It has been noted that UCN-01 induces G1 cell cycle arrest at low concentrations (IC50 = 100–300 nM) (Akiyama et al. 1997). However, this effect seems unrelated to direct inhibition of CDKs, as UCN-01 inhibits CDK1 and CDK2 in vitro only at higher concentrations (IC50 = 300–600 nM). In HNSCC cells, UCN-01 treatment results in G1 block, a phenomenon most likely secondary to depletion of cyclin D3 and induction of the endogenous CDK inhibitors p21Waf1 and p27Kip1 (Patel et al. 2002). Similar alterations have been observed in HNSCC xenograft (Patel et al. 2002).
5.2.2.3 Chk1 Inhibition
In normal cells, DNA damage generally induces G1 arrest mediated by accumulation/activation of p53, a major component of the G1 checkpoint machinery (Zhou and Bartek 2004; Bartek and Lukas 2003). In contrast, p53-defective tumor cells primarily arrest in S or G2 phase in the checkpoint response to DNA damage. As most (e.g., more than 50%) human tumors lack p53 function, G2 and S checkpoints play key roles in tumor cell responses to DNA damage. UCN-01 has been found to abrogate the G2 checkpoint selectively in p53-defective cells with 100,000-fold greater (IC50 ~ 50 nM) potency compared with caffeine. Chk1 has been defined as a major target in UCN-01-mediated G2 abrogation (Tse et al. 2007; Reinhardt et al. 2007; Vogel et al. 2007). Crystal structure analysis demonstrated that UCN-01 binds the ATP-binding pocket in the Chk1 kinase domain, and the hydroxy group in the lactam moiety of UCN-01 interacts with the ATP-binding pocket, providing a basis for the greater selectivity of UCN-01 toward Chk1 compared with staurosporine and its analogue SB218078 (Zhao et al. 2002).
Pharmacological concentrations of UCN-01 inhibit the activity of both Chk1 and checkpoint kinase 2 (Chk2) immunoprecipitated from human tumor cells, which may account for the observation that UCN-01 abrogates IR-induced p53-independent G2 arrest, whereas Chk1 activity remains unchanged (Yu et al. 2002b). UCN-01 was also shown to block Cdc25C phosphorylation mediated by another kinase, C-TAK1, which inhibits Cdc25C constitutively in the absence of DNA damage (Karlsson-Rosenthal and Millar 2006; Kohn et al. 2002). Therefore, regardless of which kinase is responsible for the phosphorylation/inactivation of Cdc25C, inhibition of this event by UCN-01 results in “inappropriate activation” of CDK1 that drives tumor cells through mitosis prior to repair of DNA damage, resulting in apoptosis (Callegari and Kelly 2007; Harrison and Haber 2006). Plasma samples isolated from patients who received UCN-01 were found to induce 40–70 % abrogation in an ex vivo G2 checkpoint assay (Kawabe 2004).
UCN-01 has also been reported to abrogate the S phase checkpoint (Gottifredi and Prives 2005), but the mechanisms responsible for this event appear to be complex. In p53 mutant tumor cells, low concentrations of UCN-01 causes S phase cells to progress to G2 phase before undergoing mitosis and cell death, whereas high concentrations (approximately 500 nM) lead to rapid premature mitosis and death of S phase cells. The latter event may stem from rapid Cdc25C activation by C-TAK1 inhibition (Kohn et al. 2002). IR-induced S checkpoint response can be divided into fast (less than 2 h) and slow (more than 1–6 h) processes. The ataxia telangiectasia mutated (ATM)-dependent pathway controls only the fast response, whereas the slow response is controlled by an ATM-independent pathway involving Chk1 (Dai and Grant 2004). These results are consistent with observations that UCN-01 abolishes the UV-induced S checkpoint response through inhibition of ATR-dependent Chk1 activation (Heffernan et al. 2002).
These findings have created a theoretical basis for developing a therapeutic strategy in which UCN-01 may sensitize tumor cells (particularly p53-defective cells) to DNA-damaging agents and radiation by abrogating the G2 and/or S checkpoints. It is noteworthy that the checkpoint abrogation effects of UCN-01 are manifested at lower drug concentrations (e.g., IC50 ~ 50 nM for G2 checkpoint abrogation) than those responsible for cytotoxicity or inhibition of cell proliferation.
5.2.2.4 PDK1/Akt Inhibition
The PI3K/Akt cascade is a critical signaling pathway in cell survival mediated by many growth factors and cytokines. Phosphorylation of Akt at Thr308 is catalyzed by PDK1 and phosphorylation at Ser473 is catalyzed by PDK2. UCN-01 directly inhibits upstream Akt kinase PDK1 with IC50 < 33 nM in in vitro and in vivo assays, whereas enforced expression of PDK1 restores Akt kinase activity (Sato et al. 2002). Crystal structure analysis demonstrated that UCN-01 binds to the kinase domain of PDK1 more specifically than staurosporine (Komander et al. 2003). Overexpression of active Akt diminishes the cytotoxic effects of UCN-01, indicating that inhibition of the PDK1/Akt pathway is attributed to the antitumor activity of this agent (Sato et al. 2002).
5.2.2.5 Apoptosis Induction
UCN-01 induces apoptosis with IC50 values of 100–1,000 nM in a panel of HNSCC cell lines in vitro and in HN12 xenograft in vivo, and exhibits enhanced cytotoxicity in cells displaying mutant p53 (Patel et al. 2002). Although the mechanism underlying UCN-01-induced apoptosis is still unknown, several potential targets have been postulated. First, inhibition of PDK1/Akt has been directly related to the cytotoxicity of UCN-01 (Sato et al. 2002). Second, as CDK1 is identified as a proapoptotic mediator (Castedo et al. 2002), Chk1/Chk2–Cdc25C-mediated CDK1 activation, particularly under “inappropriate” circumstances, may contribute to induction of apoptosis by UCN-01 (see earlier). Finally, UCN-01-induced apoptosis has been associated with downregulation of antiapoptotic proteins, such as Mcl-1, XIAP, BAG-1, and Bcl-2 (Zhao et al. 2002).
5.2.3 CYC202
CYC202 [(R)-roscovitine, seliciclib; Cyclacel] is a substituted purine analogue derived from 6-dimethylaminopurine and isopentenyladenine. In vitro kinase assays using purified recombinant kinases have revealed that CYC202 inhibits CDK2 (cyclin E–CDK2, IC50 = 100 nM; cyclin H–CDK7, IC50 = 490 nM; cyclin A–CDK2, IC50 = 710 nM), and less potently CDK1 (cyclin B–CDK1, IC50 = 2.69 μM), but neither CDK4 (cyclin D1–CDK4, IC50 = 14.21 μM) nor other kinases (e.g., PKA and PKC) (Benson et al. 2005; McClue et al. 2002). Like most CDK inhibitors, CYC202 inhibits CDKs by competing with ATP for its CDK binding site (Tang et al. 2005; Bach et al. 2005). In vitro evaluation of antitumor activity demonstrated the cytotoxicity of CYC202 (average IC50 = 15.2 μM) against a panel of 19 human tumor cell lines, including those with cisplatin- and doxorubincin-resistant phenotypes, independent of p53 status and cell cycle alterations (McClue et al. 2002). In vivo administration of CYC202 resulted in a significant antitumor effect and reduction in tumor growth rate in mouse xenograft bearing human colorectal carcinoma and uterine cancer (McClue et al. 2002; Raynaud et al. 2005). On the basis of these findings, CYC202, the first oral bioavailable CDK inhibitor, has entered phase I clinical trials in patients with advanced solid tumors (Benson et al. 2007). These studies revealed that maximum plasma concentrations of more than 2,000 ng/ml at day 1 and day 7 were achievable at 800 mg/kg twice a day for 7 days, a dose in the range of the IC50 values reported for seliciclib in vitro activity and without dose-limiting toxicity (Benson et al. 2007).
The anticancer activity of CYC202 has been related to (1) inhibition of cell-cycle-regulatory CDKs (e.g., CDK1, CDK2, and CDK7) and downregulation of cyclin D1, leading to a reduction in pRb phosphorylation at multiple sites and cell cycle arrest in G1, S, and G2–M phases (Whittaker et al. 2004; Lacrima et al. 2005, 2007); (2) inhibition of transcriptional CDKs (e.g., CDK9 in particular, and CDK7), resulting in decrease/inactivation of RNA pol II and transcriptional repression of short-lifetime proteins such as cyclin D1, cyclin A, cyclin B1, as well as Mcl-1 and XIAP (Whittaker et al. 2004; Lacrima et al. 2005; Hahntow et al. 2004); (3) more importantly, induction of apoptosis in tumor cells while largely sparing normal cells (Alvi et al. 2005), which is most likely related to downregulation of antiapoptotic proteins, particularly Mcl-1 (Zhang et al. 2002; Maccallum et al. 2005; Rossi et al. 2006); and (4) lowering the threshold of cancer cells to cytotoxic agents or other novel agents (Maccallum et al. 2005; Coley et al. 2007a, b; Ribas et al. 2006).
5.2.4 BMS-387032 (SNS-032)
A series of compounds derived from 2-acetamidothiazolythioacetic ester have been discovered and optimized as small-molecule inhibitors of cyclin E–CDK2. Among these, BMS-387032 has been identified as an oral bioavailable CDK2 inhibitor. This compound selectively inhibits CDK2 (IC50 = 48 nM, 10-fold and 20-fold selective over cyclin B–CDK1 and cyclin D–CDK4, respectively) (Misra et al. 2004). X-ray crystallographic analysis demonstrated that these compounds bind to the active ATP-binding site of the CDK2 protein. BMS-387032 displays marked antiproliferative activity, with IC50 = 95 nM in A2780 ovarian carcinoma cells (Misra et al. 2004). Similar effects were observed in a panel of tumor cell lines in vitro. BMS-387032 has demonstrated significant antitumor activity in vivo in both a murine tumor model and human tumor xenograft models (Misra et al. 2004). This systematic investigation led to a phase I clinical trial of BMS-387032 (Senderowicz 2003). Initial results from the clinical trial showed some objective tumor responses and good tolerability.
These effects in all likelihood stem from rapid induction of apoptosis and cell cycle arrest (Dai and Grant 2004; Senderowicz 2003). BMS-387032 induces E2F1 but diminishes E2F4 levels, whereas E2F1-deficient fibroblasts are less sensitive to this agent (Ma et al. 2004). A similar phenomenon has been observed in human breast cancer cells, in which treatment with BMS-387032 leads to stabilization of E2F1 (Ma and Cress 2007). Moreover, this event is accompanied by a significant increase in the p57 mRNA and protein levels, whereas p57-deficient cells are more sensitive to BMS-387032-induced apoptosis, indicating that this event may serve to limit E2F1-mediated cell death. In human lung carcinoma cells, BMS-387032 has been found to block IL-1β-induced expression as well as steady-state mRNA levels of cyclooxygenase 2, a protein providing a survival advantage to transformed cells through the inhibition of apoptosis, increased attachment to the extracellular matrix, increased invasiveness, and stimulation of angiogenesis, indicating a novel target for BMS-387032 (Mukhopadhyay et al. 2006).
5.2.5 SCH 727965 (Dinaciclib)
SCH 727965 is an extremely potent and selective CDK inhibitor, which in vitro inhibits the activity of multiple CDKs at low nanomolar levels (cyclin A–CDK2, IC50 = 1 nM; p35–CDK5, IC50 = 1 nM; cyclin T–CDK9, IC50 = 1 nM; cyclin B–CDK1, IC50 = 4 nM) (Parry et al. 2010). SCH 727965 exhibits superior activity with an improved therapeutic index. In cell-based assays, SCH 727965 completely suppressed pRb phosphorylation, which correlated with apoptosis onset and total inhibition of 5-bromo-2′-deoxyuridine incorporation in 106 tumor cell lines (including 60 lines from NCI) with diverse origin and genetic background. SCH 727965 induces regression of established solid tumors in a range of mouse models, associated with modulation of pharmacodynamic biomarkers (e.g., S807/S811 phosphorylated pRb) in skin punch biopsies and rapidly reversible, mechanism-based effects on hematologic parameters. Apoptosis occurs prior to cell cycle arrest after exposure to SCH 727965, suggesting that inhibition of transcriptional CDK9 plays an important role in killing tumor cells. SCH 727965 is in multiple phase 1 and 2 trials in patients with advanced cancers, including solid tumors (e.g., breast and non-small-cell lung cancers) and hematopoietic malignancies (e.g., non Hodgkin’s lymphoma, multiple myeloma, chronic lymphocytic leukemia), administered as an intravenous infusion on days 1, 8, and 15 of each 28-day cycle (Johnson et al. 2012; Feldmann et al. 2011; Bates et al. 2011; Fu et al. 2011).
5.3 Molecular Mechanism of Action Driven Combinational Targeted Therapy
In view of the molecular mechanism of action (MMOA) of agents that target CDK-regulating events such as the cell cycle and transcription, efforts to combine CDK inhibitors with other targeted agents in various types of human cancer have been the focus of attention. Some representative cases are summarized as in the following sections.
5.3.1 Flavopiridol
Synergistic interactions between flavopiridol and TRAIL have been described by several groups. For example, it is reported that synergism between flavopiridol and TRAIL in human leukemia cells stems from downregulation of XIAP (Rosato et al. 2004). Such studies provide a rationale for future attempts to combine CDK inhibitors with TRAIL in leukemia and lymphoma therapy.
The preclinical studies demonstrate that flavopiridol synergistically enhances anticancer activity of pan-Bcl-2 antagonists (or BH3 mimetics) such as HA14-1 (Pei et al. 2004) and obatoclax (Chen et al. 2012). The MMOA includes reactive oxygen species generation dependent activation of the stress-related JNK pathway (Pei et al. 2004), and imbalance between antiapoptotic (e.g., downregulation of Mcl-1 and Bcl-xL) and proapoptotic (e.g., upregulation of the BH3-only protein Bim, NBK/Bik, Noxa) machinery (Chen et al. 2012).
There is also evidence that flavopiridol interacts synergistically with histone deacetylase inhibitors (HDACIs) to induce apoptosis in human leukemia cells. This concept was driven by the findings that interference with p21Cip expression and resulting cell differentiation (e.g., induced by phorbol 12-myristate 13-acetate) by flavopiridol results in a marked increase in apoptosis (Cartee et al. 2002). For example, a highly synergistic interaction between flavopiridol and the HDACI vorinostat (suberoylanilide hydroxamic acid) was observed in human leukemia cell lines as well as primary AML blasts (Almenara et al. 2002). This interaction stemmed in part from flavopiridol-mediated inhibition of p21CIP1 induction, an event known to promote the lethality of HDACIs. On the basis of these findings, a phase I trial of vorinostat, administered at 200 mg per os three times daily for 14 days in conjunction with flavopiridol administered as a 1-h infusion daily (days 1–5) in patients with refractory AML/high-risk myelodysplastic syndrome has been initiated and is ongoing, along with plans to incorporate the new infusional flavopiridol schedule. Moreover, mechanistic studies on interactions between flavopiridol and HDACIs have also led to new insight into a critical role of the NF-κB signaling pathway in tumor cell response to HDACIs (Dai et al. 2003b; Gao et al. 2004). Further, it has been found that disruption of HDACI-induced NF-κB activation strikingly increases the lethality of HDACIs (Dai et al. 2005a, 2011b). Based on these findings, multiple clinical trials have been initiated to test this novel therapeutic strategy (Dai et al. 2008a, 2011c).
On the basis of evidence of synergism between flavopiridol and the Bcr/Abl kinase inhibitor imatinib mesylate in chronic myeloid leukemia cells, including some resistant to imatinib (Kasten and Giordano 2001), a phase I trial of imatinib mesylate and flavopiridol has been initiated. Although the maximum tolerated dose for this combination was not identified, further efforts in this direction have been deferred in light of the introduction of second-generation Bcr/Abl kinase inhibitors (e.g., dasatinib, nilotinib).
Finally, on the basis of evidence of synergistic interactions between flavopiridol and the proteasome inhibitor bortezomib in malignant hematopoietic cells (Dai et al. 2003c, 2004b), a phase I trial has been initiated in patients with refractory multiple myeloma and indolent no-Hodgkin’s lymphoma in which escalating doses of flavopiridol and bortezomib are given as an intravenous infusion on days 1, 4, 8, and 11 of a 21-day cycle. The regimen has proven to be well tolerated, and the maximum tolerated dose has not yet been reached. Notably, several patients who either longer responded to bortezomib have responded to the combination of bortezomib and flavopiridol (Holkova et al. 2011). Two complete responses (12 %) and five partial responses (31 %) were observed at the maximum tolerated dose (overall response rate of 44 %) (Holkova et al. 2011).
5.3.2 CYC202
CYC202 interacted synergistically with bortezomib in human multiple myeloma cells (Raje et al. 2005), but whether the mechanism underlying this interaction is the same as that responsible for flavopiridol/bortezomib synergism in human leukemia cells, and whether such findings can be extended to leukemia and lymphoma, remains to be determined.
Synergy between roscovitine and the pan-HDACI LAQ824 has been described in human leukemia cells, and this interaction was related to downregulation of Mcl-1, p21CIP1, and XIAP, as well as induction of oxidative injury (Rosato et al. 2005).
Roscovitine displays highly synergistic interactions with the Bcl-2 antagonist ABT-737 in human leukemia cells (Chen et al. 2007b). The mechanism responsible for this interaction was determined to be roscovitine-mediated Mcl-1 downregulation, which cooperated with disruption of the function of Bcl-xL by ABT-737 to unleash Bak and activate Bax. In view of evidence of in vivo activity of agents such as ABT-737 in murine models of lymphoma (Oltersdorf et al. 2005), the concept of combining Bcl-2 antagonists with CDK inhibitors warrants further attention.
It is also noted that the PI3K/Akt pathway plays a functional role in regulating the apoptotic response of human leukemia cells to pharmacological CDK inhibitors (including roscovitine and flavopiridol), whereas combined interruption of CDK- and PI3K-related pathways significantly increases therapeutic activity in hematological malignancies (Yu et al. 2003).
5.3.3 UCN-01
In addition to combination strategies involving UCN-01 and conventional chemotherapy (Harvey et al. 2001), investigations of UCN-01 have focused on interactions with inhibitors of the Ras/Raf/MEK1/2/ERK1/2 pathway. Exposure of human leukemia or multiple myeloma cells to UCN-01 results in activation of MEK1/2/ERK1/2, and interference with the latter process, i.e., by MEK1/2 inhibitors such as PD184352, results in a dramatic increase in apoptosis (Dai et al. 2001, 2002b; Yu et al. 2002a). Similar synergistic interactions between MEK1/2 inhibitors and UCN-01 were observed in human solid tumors, including breast cancer, prostate carcinoma (McKinstry et al. 2002; Hawkins et al. 2005; Hamed et al. 2008), and glioblastoma (Tang et al. 2012a). These events were associated with enhanced activation of CDK1 (Pei et al. 2006), consistent with the ability of UCN-01 to inhibit Chk1. However, this regimen is also able to kill cytokinetically quiescent (G0/G1) human malignant cells (Pei et al. 2011). Such a finding argues against a notion that the activity of this combinational therapy is only restricted to cycling cells. It also raises the possibility that this regimen may be active against cytokinetically quiescent cancer stem cells.
Lethality of the UCN-01/MEK1/2 inhibitor regimen primarily involved activation of the intrinsic, mitochondrial pathway likely via upregulation of the BH3-only protein Bim (Pei et al. 2007), and was substantially blocked in cells overexpressing Bcl-2 or Bcl-xL. However, lethality of the regimen was restored in leukemia cells by agents capable of activating the extrinsic, apoptotic pathway, e.g., TRAIL (Dai et al. 2003a). Similar events were observed in human hematopoietic malignant cells exposed to UCN-01 in conjunction with agents targeting Ras, such as farnesyltransferase inhibitor (Dai et al. 2005b, 2008b; Pei et al. 2005), statins (Dai et al. 2007), or Src (Dai et al. 2011a; Mitchell et al. 2011). UCN-01 activity was also shown to be dramatically enhanced by the mammalian target of rapamycin inhibitor rapamycin (Hahn et al. 2005), NF-κB inhibitors (Dai et al. 2004a), or poly(ADP-ribose) polymerase inhibitors (Tang et al. 2012b). Combining UCN-01 with multiple agents (particularly those targeting the Src/Ras/Raf/MEK/ERK pathway) is a good example for development of an MMOA-driven rationale, that is, to combine the cell cycle modulators with other targeted agents that block the activation of the compensatory survival pathway in tumor cells exposed to the former, an important mechanism responsible for acquired drug resistance toward most anticancer agents, including the novel small-molecule inhibitors (Dai and Grant 2010a, b; Dent et al. 2011). This concept is further supported by later observations that combining inhibitors of Aurora kinases that regulate cell cycle transit from G2 phase through cytokinesis with other targeted agents (e.g., HDACIs) markedly increases the anticancer activity (Dai et al. 2008c; Nguyen et al. 2011).
6 Future Perspectives
Targeting cell-cycle-regulatory, transcriptional, neural CDKs is a highly attractive approach in cancer treatment as well as neuron protection, in which dysregulation of CDKs or their regulatory partners (e.g., cyclins, cyclin-like proteins, and endogenous CDK inhibitors) appears to play an important role in disease pathogenesis and prognosis (Dai and Grant 2004). The recent advances in methods allows us to gain deeper insight into the MMOA of pharmacological CDK inhibitors as well as other cell cycle modulators (Dai and Grant 2011). These MMOA are way beyond the original consideration of CDK inhibition (Dai et al. 2002a), but are related to multiple novel mechanistic aspects, including apoptosis induction, transcription inhibition, downregulation of various short-lived proteins (e.g., cyclin D1, Mcl-1, and VEGF), and antiangiogenesis. (Dai and Grant 2006). Better understanding of MMOA has been providing fundamental power to drive recent rapid development of new-generation, more potent and more selective small-molecule CDK inhibitors. Several of them, such as CYC202 (seliciclib), BMS-387032 (SNS-032), SCH 727965 (dinaciclib), PD-0332991, P-276-00, R-547, AZD5438, ZK 204709, PHA-848125, and AT7519 have entered clinical trials, and many others are in preclinical development with considerable interest.
Single-agent activity of CDK inhibitors in cancer has been limited to date, although many of them appear to have significant activity in preclinical settings. Significantly, the MMOA-driven rationale of combining CDK inhibitors with conventional chemotherapeutic drugs and, importantly, other novel targeted agents offers the potential for enhanced tumor-selective cytotoxicity and circumvention of drug resistance. In fact, the ultimate role of CDK inhibitors as anticancer therapeutics may be as modulators or sensitizers for conventional chemotherapy and/or novel agents. In a context related to this, one overarching question is whether to target single or multiple CDKs for cancer therapy. Despite great efforts, the development of monospecific CDK inhibitors has not succeed so far in cancer treatment. Neoplastic cells are characterized by uncontrolled proliferation due to constitutive activation of cell-cycle-regulatory CDKs (e.g., CDK4/CDK6, CDK2, and probably CDK1 as well) owing to abnormalities of their moderators (e.g., cyclins and endogenous CDK inhibitors). Moreover, rapid growth of transformed cells requires continuous transcriptional activity of RNA pol II to ensure de novo protein synthesis via gene expression. Therefore, therapeutic intervention that interrupts cell cycle progression and global transcription by inhibition of both cell-cycle-regulatory and transcriptional CDKs is a very sound rationale in the treatment of cancer (Wesierska-Gadek and Kramer 2012). Therefore, the highly heterogeneous characteristics of human cancer may not only necessitate the pleiotropic effects of pan-CDK inhibitors, but may also warrant the simultaneous interference with multiple pathways by combining CDK inhibitors with conventional chemotherapy as well as other targeted agents.
Most, if not all, currently available CDK inhibitors hit at least CDKs. Theoretically, targeting a single CDK may improve the selectivity of therapy, thus minimizing toxicity by preventing healthy cells from experiencing undesired side effects. In the case of neuron protection, although “inappropriate” activation of cell-cycle-regulatory CDKs generally leads to neuronal death (Rashidian et al. 2005), global inhibition of multiple CDKs by pan-CDK inhibitors may be not beneficial, and is probably harmful, because they also inhibit CDK5, the only postmitotic neural CDK, which is critical for various aspects of nervous system development and functions (e.g., neuronal migration, neuronal survival, dendritic spine formation, synaptogenesis, adult neurogenesis, neurotransmission, homeostatic plasticity, and learning and memory) (Ou et al. 2010; Park et al. 2011). Thus, it is plausible that the monospecific CDK inhibitors might be more beneficial in the treatment of disorders such as cardiac hypertrophy and neurodegenerative diseases, in which a single CDK is dysregulated. In this context, therapeutic inhibition of the hyperactivated CDK9 or CDK5 may be sufficient to reduce the enhanced transcription required for hypertrophic cardiomyocytes or to prevent extensive neuron apoptosis in neurodegenerative diseases, respectively.
In summary, pan-CDK inhibitors might be more effective in the treatment of cancer, as multiple, rather than single, pathways are deregulated in malignancies. In contrast, inhibition of a single CDK (e.g., CDK4 or CDK5) may provide more benefit for neuron protection in stroke or neurodegeneration, respectively. However, since the normal and abnormal roles of CDK5 are dependent on its non-cyclin partners, p35/p39 versus p25, it will be ideal to develop small-molecule inhibitors that specifically target p25–CDK5, rather than p35/p39–CDK5, for treatment of neurodegenerative diseases. Therefore, therapeutic development and application of CDK inhibitors, like that of most targeted agents, should be tailored to the individual patient by use of genetic and other information, a model termed “personalized medicine” (Roychowdhury et al. 2011).
References
Achenbach TV, Muller R, Slater EP (2000) Bcl-2 independence of flavopiridol-induced apoptosis. Mitochondrial depolarization in the absence of cytochrome c release. J Biol Chem 275:32089–32097
Aggarwal BB, Sethi G, Ahn KS, Sandur SK, Pandey MK, Kunnumakkara AB, Sung B, Ichikawa H (2006) Targeting signal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: modern target but ancient solution. Ann N Y Acad Sci 1091:151–169
Akiyama T, Yoshida T, Tsujita T, Shimizu M, Mizukami T, Okabe M, Akinaga S (1997) G1 phase accumulation induced by UCN-01 is associated with dephosphorylation of Rb and CDK2 proteins as well as induction of CDK inhibitor p21/Cip1/WAF1/Sdi1 in p53-mutated human epidermoid carcinoma A431 cells. Cancer Res 57:1495–1501
Akoulitchev S, Chuikov S, Reinberg D (2000) TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 407(6800):102–106
Almenara J, Rosato R, Grant S (2002) Synergistic induction of mitochondrial damage and apoptosis in human leukemia cells by flavopiridol and the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA). Leukemia 16:1331–1343
Alvi AJ, Austen B, Weston VJ, Fegan C, MacCallum D, Gianella-Borradori A, Lane DP, Hubank M, Powell JE, Wei W, Taylor AM, Moss PA, Stankovic T (2005) A novel CDK inhibitor, CYC202 (R-roscovitine), overcomes the defect in p53-dependent apoptosis in B-CLL by down-regulation of genes involved in transcription regulation and survival. Blood 105:4484–4491
Arnold A, Papanikolaou A (2005) Cyclin D1 in breast cancer pathogenesis. J Clin Oncol 23:4215–4224
Asada A, Saito T, Hisanaga S (2012) Phosphorylation of p35 and p39 by Cdk5 determines the subcellular location of the holokinase in a phosphorylation-site-specific manner. J Cell Sci 125:3421–3429
Auerkari EI (2006) Methylation of tumor suppressor genes p16(INK4a), p27(Kip1) and E-cadherin in carcinogenesis. Oral Oncol 42:5–13
Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, Baratte B, Koken M, Coburn SP, Tang L, Jiang T, Liang DC, Galons H, Dierick JF, Pinna LA, Meggio F, Totzke F, Schachtele C, Lerman AS, Carnero A, Wan Y, Gray N, Meijer L (2005) Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem 280:31208–31219
Barboric M, Kohoutek J, Price JP, Blazek D, Price DH, Peterlin BM (2005) Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J 24:4291–4303
Barette C, Jariel-Encontre I, Piechaczyk M, Piette J (2001) Human cyclin C protein is stabilized by its associated kinase cdk8, independently of its catalytic activity. Oncogene 20:551–562
Barriere C, Santamaria D, Cerqueira A, Galan J, Martin A, Ortega S, Malumbres M, Dubus P, Barbacid M (2007) Mice thrive without Cdk4 and Cdk2. Mol Oncol 1:72–83
Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3(5):421–429
Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL (2010) CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev 24:2303–2316
Bates DJ, Salerni BL, Lowrey CH, Eastman A (2011) Vinblastine sensitizes leukemia cells to cyclin-dependent kinase inhibitors, inducing acute cell cycle phase-independent apoptosis. Cancer Biol Ther 12:314–325
Becker EB, Bonni A (2005) Beyond proliferation–cell cycle control of neuronal survival and differentiation in the developing mammalian brain. Semin Cell Dev Biol 16:439–448
Benson C, Kaye S, Workman P, Garrett M, Walton M, De BJ (2005) Clinical anticancer drug development: targeting the cyclin-dependent kinases. Br J Cancer 92:7–12
Benson C, White J, De BJ, O’Donnell A, Raynaud F, Cruickshank C, McGrath H, Walton M, Workman P, Kaye S, Cassidy J, Gianella-Borradori A, Judson I, Twelves C (2007) A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-roscovitine), administered twice daily for 7 days every 21 days. Br J Cancer 96:29–37
Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr (2005) Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 106:296–303
Bible KC, Bible RH Jr, Kottke TJ, Svingen PA, Xu K, Pang YP, Hajdu E, Kaufmann SH (2000) Flavopiridol binds to duplex DNA. Cancer Res 60:2419–2428
Blagosklonny MV (2004) Flavopiridol, an inhibitor of transcription: implications, problems and solutions. Cell Cycle 3:1537–1542
Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, Peterlin BM (2011) The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 25:2158–2172
Brüsselbach S, Nettelbeck DM, Sedlacek HH, Muller R (1998) Cell cycle-independent induction of apoptosis by the anti-tumor drug flavopiridol in endothelial cells. Int J Cancer 77:146–152
Burd CJ, Petre CE, Morey LM, Wang Y, Revelo MP, Haiman CA, Lu S, Fenoglio-Preiser CM, Li J, Knudsen ES, Wong J, Knudsen KE (2006) Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci USA 103:2190–2195
Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, Fischer B, Moran M, Blum KA, Rovin B, Brooker-McEldowney M, Broering S, Schaaf LJ, Johnson AJ, Lucas DM, Heerema NA, Lozanski G, Young DC, Suarez JR, Colevas AD, Grever MR (2007) Flavopiridol administered using a pharmacologically derived schedule is associated with marked clinical efficacy in refractory, genetically high-risk chronic lymphocytic leukemia. Blood 109:399–404
Cai D, Latham VM Jr, Zhang X, Shapiro GI (2006) Combined depletion of cell cycle and transcriptional cyclin-dependent kinase activities induces apoptosis in cancer cells. Cancer Res 66:9270–9280
Callegari AJ, Kelly TJ (2007) Shedding light on the DNA damage checkpoint. Cell Cycle 6(6):660–666
Canavese M, Santo L, Raje N (2012) Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther 13:451–457
Canduri F, Perez PC, Caceres RA, de Azevedo WFJ (2008) CDK9 a potential target for drug development. Med Chem 4:210–218
Carlson B, Lahusen T, Singh S, Loaiza-Perez A, Worland PJ, Pestell R, Albanese C, Sausville EA, Senderowicz AM (1999) Down-regulation of cyclin D1 by transcriptional repression in MCF-7 human breast carcinoma cells induced by flavopiridol. Cancer Res 59:4634–4641
Carrere N, Belaud-Rotureau MA, Dubus P, Parrens M, de MA, Merlio JP (2005) The relative levels of cyclin D1a and D1b alternative transcripts in mantle cell lymphoma may depend more on sample origin than on CCND1 polymorphism. Haematologica 90:854–885
Cartee L, Wang Z, Decker RH, Chellappan SP, Fusaro G, Hirsch KG, Sankala HM, Dent P, Grant S (2001) The cyclin-dependent kinase inhibitor (CDKI) flavopiridol disrupts phorbol 12-myristate 13-acetate-induced differentiation and CDKI expression while enhancing apoptosis in human myeloid leukemia cells. Cancer Res 61:2583–2591
Cartee L, Smith R, Dai Y, Rahmani M, Rosato R, Almenara J, Dent P, Grant S (2002) Synergistic induction of apoptosis in human myeloid leukemia cells by phorbol 12-myristate 13-acetate and flavopiridol proceeds via activation of both the intrinsic and tumor necrosis factor-mediated extrinsic cell death pathways. Mol Pharmacol 61:1313–1321
Castedo M, Perfettini JL, Roumier T, Kroemer G (2002) Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ 9:1287–1293
Chakravarti A, DeSilvio M, Zhang M, Grignon D, Rosenthal S, Asbell SO, Hanks G, Sandler HM, Khor LY, Pollack A, Shipley W (2007) Prognostic value of p16 in locally advanced prostate cancer: a study based on Radiation Therapy Oncology Group Protocol 9202. J Clin Oncol 25:3082–3089
Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH (2000) Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem 275:28345–28348
Chao SH, Price DH (2001) Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem 276:31793–31799
Chen HH, Wang YC, Fann MJ (2006) Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol Cell Biol 26:2736–2745
Chen HH, Wong YH, Geneviere AM, Fann MJ (2007a) CDK13/CDC2L5 interacts with L-type cyclins and regulates alternative splicing. Biochem Biophys Res Commun 354:735–740
Chen S, Dai Y, Harada H, Dent P, Grant S (2007b) Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 67:782–791
Chen S, Dai Y, Pei XY, Myers J, Wang L, Kramer LB, Garnett M, Schwartz DM, Su F, Simmons GL, Richey JD, Larsen DG, Dent P, Orlowski RZ, Grant S (2012) CDK inhibitors upregulate BH3-only proteins to sensitize human myeloma cells to BH3 mimetic therapies. Cancer Res 72:4225–4237
Cheng B, Price DH (2007) Properties of RNA polymerase II elongation complexes before and after the P-TEFb-mediated transition into productive elongation. J Biol Chem 282:21901–21912
Chen-Kiang S (2003) Cell-cycle control of plasma cell differentiation and tumorigenesis. Immunol Rev 194:39–47
Cheung ZH, Ip NY (2004) Cdk5: mediator of neuronal death and survival. Neurosci Lett 361:47–51
Cheung ZH, Ip NY (2007) The roles of cyclin-dependent kinase 5 in dendrite and synapse development. Biotechnol J 2:949–957
Cheung ZH, Fu AK, Ip NY (2006) Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron 50:13–18
Cheung ZH, Chin WH, Chen Y, Ng YP, Ip NY (2007) Cdk5 is involved in BDNF-stimulated dendritic growth in hippocampal neurons. PLoS Biol 5:e63
Cheung ZH, Gong K, Ip NY (2008) Cyclin-dependent kinase 5 supports neuronal survival through phosphorylation of Bcl-2. J Neurosci 28:4872–4877
Chim CS, Fung TK, Liang R (2003) Disruption of INK4/CDK/Rb cell cycle pathway by gene hypermethylation in multiple myeloma and MGUS. Leukemia 17:2533–2535
Cicenas J, Valius M (2011) The CDK inhibitors in cancer research and therapy. J Cancer Res Clin Oncol 137:1409–1418
Ciemerych MA, Yu Q, Szczepanska K, Sicinski P (2008) CDK4 activity in mouse embryos expressing a single D-type cyclin. Int J Dev Biol 52:299–305
Colevas D, Blaylock B, Gravell A (2002) Clinical trials referral resource. Flavopiridol. Oncology (Williston Park) 16:1204–1212, 1214
Coley HM, Shotton CF, Kokkinos MI, Thomas H (2007a) The effects of the CDK inhibitor seliciclib alone or in combination with cisplatin in human uterine sarcoma cell lines. Gynecol Oncol 105:462–469
Coley HM, Shotton CF, Thomas H (2007b) Seliciclib (CYC202; r-roscovitine) in combination with cytotoxic agents in human uterine sarcoma cell lines. Anticancer Res 27:273–278
Coqueret O (2002) Linking cyclins to transcriptional control. Gene 299:35–55
Coudreuse D, Nurse P (2010) Driving the cell cycle with a minimal CDK control network. Nature 468:1074–1079
Croxton R, Ma Y, Song L, Haura EB, Cress WD (2002) Direct repression of the Mcl-1 promoter by E2F1. Oncogene 21:1359–1369
Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40(3):471–483
Cruz JC, Tsai LH (2004a) A Jekyll and Hyde kinase: roles for Cdk5 in brain development and disease. Curr Opin Neurobiol 14:390–394
Cruz JC, Tsai LH (2004b) Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol Med 10:452–458
Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH (2006) p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci 26:10536–10541
Dai Y, Grant S (2003) Cyclin-dependent kinase inhibitors. Curr Opin Pharmacol 3:362–370
Dai Y, Grant S (2004) Small molecule inhibitors targeting cyclin-dependent kinases as anticancer agents. Curr Oncol Rep 6:123–130
Dai Y, Grant S (2006) CDK inhibitor targets: a hit or miss proposition? Cyclin-dependent kinase inhibitors kill tumor cells by downregulation of anti-apoptotic proteins. Cancer Biol Ther 5:171–173
Dai Y, Grant S (2010a) New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res 16:376–383
Dai Y, Grant S (2010b) Targeting Chk1 in the replicative stress response. Cell Cycle 9:1025
Dai Y, Grant S (2011) Methods to study cancer therapeutic drugs that target cell cycle checkpoints. Methods Mol Biol 782:257–304
Dai Y, Yu C, Singh V, Tang L, Wang Z, McInistry R, Dent P, Grant S (2001) Pharmacological inhibitors of the mitogen-activated protein kinase (MAPK) kinase/MAPK cascade interact synergistically with UCN-01 to induce mitochondrial dysfunction and apoptosis in human leukemia cells. Cancer Res 61:5106–5115
Dai Y, Dent P, Grant S (2002a) Induction of apoptosis in human leukemia cells by the CDK1 inhibitor CGP74514A. Cell Cycle 1:143–152
Dai Y, Landowski TH, Rosen ST, Dent P, Grant S (2002b) Combined treatment with the checkpoint abrogator UCN-01 and MEK1/2 inhibitors potently induces apoptosis in drug-sensitive and -resistant myeloma cells through an IL-6-independent mechanism. Blood 100:3333–3343
Dai Y, Dent P, Grant S (2003a) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) promotes mitochondrial dysfunction and apoptosis induced by 7-hydroxystaurosporine and mitogen-activated protein kinase kinase inhibitors in human leukemia cells that ectopically express Bcl-2 and Bcl-xL. Mol Pharmacol 64:1402–1409
Dai Y, Rahmani M, Grant S (2003b) An intact NF-kappaB pathway is required for histone deacetylase inhibitor-induced G1 arrest and maturation in U937 human myeloid leukemia cells. Cell Cycle 2:467–472
Dai Y, Rahmani M, Grant S (2003c) Proteasome inhibitors potentiate leukemic cell apoptosis induced by the cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK- and NF-kappaB-dependent process. Oncogene 22:7108–7122
Dai Y, Pei XY, Rahmani M, Conrad DH, Dent P, Grant S (2004a) Interruption of the NF-kappaB pathway by Bay 11–7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood 103:2761–2770
Dai Y, Rahmani M, Pei XY, Dent P, Grant S (2004b) Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms. Blood 104:509–518
Dai Y, Rahmani M, Dent P, Grant S (2005a) Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol 25:5429–5444
Dai Y, Rahmani M, Pei XY, Khanna P, Han SI, Mitchell C, Dent P, Grant S (2005b) Farnesyltransferase inhibitors interact synergistically with the Chk1 inhibitor UCN-01 to induce apoptosis in human leukemia cells through interruption of both Akt and MEK/ERK pathways and activation of SEK1/JNK. Blood 105:1706–1716
Dai Y, Hamm TE, Dent P, Grant S (2006) Cyclin D1 overexpression increases the susceptibility of human U266 myeloma cells to CDK inhibitors through a process involving p130-, p107- and E2F-dependent S phase entry. Cell Cycle 5:437–446
Dai Y, Khanna P, Chen S, Pei XY, Dent P, Grant S (2007) Statins synergistically potentiate 7-hydroxystaurosporine (UCN-01) lethality in human leukemia and myeloma cells by disrupting Ras farnesylation and activation. Blood 109:4415–4423
Dai Y, Chen S, Kramer LB, Funk VL, Dent P, Grant S (2008a) Interactions between bortezomib and romidepsin and belinostat in chronic lymphocytic leukemia cells. Clin Cancer Res 14:549–558
Dai Y, Chen S, Pei XY, Almenara JA, Kramer LB, Venditti CA, Dent P, Grant S (2008b) Interruption of the Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA damage in vitro and in vivo in human multiple myeloma cells. Blood 112:2439–2449
Dai Y, Chen S, Venditti CA, Pei XY, Nguyen TK, Dent P, Grant S (2008c) Vorinostat synergistically potentiates MK-0457 lethality in chronic myelogenous leukemia cells sensitive and resistant to imatinib mesylate. Blood 112:793–804
Dai Y, Chen S, Shah R, Pei XY, Wang L, Almenara JA, Kramer LB, Dent P, Grant S (2011a) Disruption of Src function potentiates Chk1-inhibitor-induced apoptosis in human multiple myeloma cells in vitro and in vivo. Blood 117:1947–1957
Dai Y, Chen S, Wang L, Pei XY, Funk VL, Kramer LB, Dent P, Grant S (2011b) Disruption of IkappaB kinase (IKK)-mediated RelA serine 536 phosphorylation sensitizes human multiple myeloma cells to histone deacetylase (HDAC) inhibitors. J Biol Chem 286:34036–34050
Dai Y, Chen S, Wang L, Pei XY, Kramer LB, Dent P, Grant S (2011c) Bortezomib interacts synergistically with belinostat in human acute myeloid leukaemia and acute lymphoblastic leukaemia cells in association with perturbations in NF-kappaB and Bim. Br J Haematol 153(2):222–235
Davies TG, Bentley J, Arris CE, Boyle FT, Curtin NJ, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Hardcastle IR, Jewsbury P, Johnson LN, Mesguiche V, Newell DR, Noble ME, Tucker JA, Wang L, Whitfield HJ (2002) Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor. Nat Struct Biol 9:745–749
de Azevedo WFJ, Canduri F, da Silveira NJ (2002) Structural basis for inhibition of cyclin-dependent kinase 9 by flavopiridol. Biochem Biophys Res Commun 293:566–571
Decker RH, Dai Y, Grant S (2001) The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in human leukemia cells (U937) through the mitochondrial rather than the receptor-mediated pathway. Cell Death Differ 8:715–724
Decker RH, Wang S, Dai Y, Dent P, Grant S (2002) Loss of the Bcl-2 phosphorylation loop domain is required to protect human myeloid leukemia cells from flavopiridol-mediated mitochondrial damage and apoptosis. Cancer Biol Ther 1:136–144
Dees EC, Baker SD, O’Reilly S, Rudek MA, Davidson SB, Aylesworth C, Elza-Brown K, Carducci MA, Donehower RC (2005) A phase I and pharmacokinetic study of short infusions of UCN-01 in patients with refractory solid tumors. Clin Cancer Res 11:664–671
Delmer A, Ajchenbaum-Cymbalista F, Tang R, Ramond S, Faussat AM, Marie JP, Zittoun R (1995) Overexpression of cyclin D2 in chronic B-cell malignancies. Blood 85:2870–2876
Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S (2011) CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv 11:133–140
Deshpande A, Sicinski P, Hinds PW (2005) Cyclins and cdks in development and cancer: a perspective. Oncogene 24:2909–2915
Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI (2005) Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc Natl Acad Sci USA 102:8333–8338
Dib A, Peterson TR, Raducha-Grace L, Zingone A, Zhan F, Hanamura I, Barlogie B, Shaughnessy J Jr, Kuehl WM (2006) Paradoxical expression of INK4c in proliferative multiple myeloma tumors: bi-allelic deletion vs increased expression. Cell Div 1:23
Diehl JA, Zindy F, Sherr CJ (1997) Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev 11:957–972
Dong Y, Sui L, Tai Y, Sugimoto K, Tokuda M (2001) The overexpression of cyclin-dependent kinase (CDK) 2 in laryngeal squamous cell carcinomas. Anticancer Res 21:103–108
Drexler HG (1998) Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia 12:845–859
Echalier A, Endicott JA, Noble ME (2010) Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim Biophys Acta 1804:511–519
Egloff S, Van HE, Kiss T (2006) Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol Cell Biol 26:630–642
Ely S, Di LM, Niesvizky R, Baughn LB, Cho HJ, Hatada EN, Knowles DM, Lane J, Chen-Kiang S (2005) Mutually exclusive cyclin-dependent kinase 4/cyclin D1 and cyclin-dependent kinase 6/cyclin D2 pairing inactivates retinoblastoma protein and promotes cell cycle dysregulation in multiple myeloma. Cancer Res 65:11345–11353
Evens AM, Gartenhaus RB (2003) Molecular etiology of mature T-cell non-Hodgkin’s lymphomas. Front Biosci 8:d156–d175
Ezhevsky SA, Ho A, Becker-Hapak M, Davis PK, Dowdy SF (2001) Differential regulation of retinoblastoma tumor suppressor protein by G(1) cyclin-dependent kinase complexes in vivo. Mol Cell Biol 21:4773–4784
Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H, Fendrich V, Rajeshkumar NV, Matsui W, Brossart P, Hidalgo M, Bannerji R, Maitra A, Nelkin BD (2011) Cyclin-dependent kinase inhibitor dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther 12:598–609
Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, Chheda MG, Tamayo P, Finn S, Shrestha Y, Boehm JS, Jain S, Bojarski E, Mermel C, Barretina J, Chan JA, Baselga J, Tabernero J, Root DE, Fuchs CS, Loda M, Shivdasani RA, Meyerson M, Hahn WC (2008) CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455:547–551
Fry DW, Bedford DC, Harvey PH, Fritsch A, Keller PR, Wu Z, Dobrusin E, Leopold WR, Fattaey A, Garrett MD (2001) Cell cycle and biochemical effects of PD 0183812. A potent inhibitor of the cyclin D-dependent kinases CDK4 and CDK6. J Biol Chem 276:16617–16623
Fu TJ, Peng J, Lee G, Price DH, Flores O (1999) Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J Biol Chem 274:34527–34530
Fu M, Wang C, Li Z, Sakamaki T, Pestell RG (2004) Minireview: cyclin D1: normal and abnormal functions. Endocrinology 145:5439–5447
Fu M, Rao M, Bouras T, Wang C, Wu K, Zhang X, Li Z, Yao TP, Pestell RG (2005) Cyclin D1 inhibits peroxisome proliferator-activated receptor gamma-mediated adipogenesis through histone deacetylase recruitment. J Biol Chem 280:16934–16941
Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer J, Altiok S, Pledger WJ (2011) The cyclin-dependent kinase inhibitor SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma cells. Mol Cancer Ther 10:1018–1027
Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM (2012) PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res 40:9160–9170
Fukasawa R, Tsutsui T, Hirose Y, Tanaka A, Ohkuma Y (2012) Mediator CDK subunits are platforms for interactions with various chromatin regulatory complexes. J Biochem 152:241–249
Fulda S, Debatin KM (2006) Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 25:4798–4811
Fuse E, Kuwabara T, Sparreboom A, Sausville EA, Figg WD (2005) Review of UCN-01 development: a lesson in the importance of clinical pharmacology. J Clin Pharmacol 45:394–403
Galbraith MD, Donner AJ, Espinosa JM (2010) CDK8: a positive regulator of transcription. Transcription 1:4–12
Gallorini M, Cataldi A, di Giacomo V (2012) Cyclin-dependent kinase modulators and cancer therapy. Biodrugs 26:377–391
Galm O, Wilop S, Reichelt J, Jost E, Gehbauer G, Herman JG, Osieka R (2004) DNA methylation changes in multiple myeloma. Leukemia 18:1687–1692
Ganuza M, Saiz-Ladera C, Canamero M, Gomez G, Schneider R, Blasco MA, Pisano D, Paramio JM, Santamaria D, Barbacid M (2012) Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J 31:2498–2510
Gao N, Dai Y, Rahmani M, Dent P, Grant S (2004) Contribution of disruption of the nuclear factor-kappaB pathway to induction of apoptosis in human leukemia cells by histone deacetylase inhibitors and flavopiridol. Mol Pharmacol 66:956–963
Garriga J, Grana X (2004) Cellular control of gene expression by T-type cyclin/CDK9 complexes. Gene 337:15–23
Garriga J, Xie H, Obradovic Z, Grana X (2010) Selective control of gene expression by CDK9 in human cells. J Cell Physiol 222:200–208
Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P (2003) Cyclin E ablation in the mouse. Cell 114:431–443
Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL (2009) TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol 29:5455–5464
Gottifredi V, Prives C (2005) The S phase checkpoint: when the crowd meets at the fork. Semin Cell Dev Biol 16:355–368
Green DR, Kroemer G (2004) The pathophysiology of mitochondrial cell death. Science 305:626–629
Greene LA, Liu DX, Troy CM, Biswas SC (2007) Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim Biophys Acta 1772:392–401
Hahn M, Li W, Yu C, Rahmani M, Dent P, Grant S (2005) Rapamycin and UCN-01 synergistically induce apoptosis in human leukemia cells through a process that is regulated by the Raf-1/MEK/ERK, Akt, and JNK signal transduction pathways. Mol Cancer Ther 4:457–470
Hahntow IN, Schneller F, Oelsner M, Weick K, Ringshausen I, Fend F, Peschel C, Decker T (2004) Cyclin-dependent kinase inhibitor roscovitine induces apoptosis in chronic lymphocytic leukemia cells. Leukemia 18:747–755
Hamed H, Hawkins W, Mitchell C, Gilfor D, Zhang G, Pei XY, Dai Y, Hagan MP, Roberts JD, Yacoub A, Grant S, Dent P (2008) Transient exposure of carcinoma cells to RAS/MEK inhibitors and UCN-01 causes cell death in vitro and in vivo. Mol Cancer Ther 7:616–629
Hardcastle IR, Golding BT, Griffin RJ (2002) Designing inhibitors of cyclin-dependent kinases. Annu Rev Pharmacol Toxicol 42:325–348
Harrison JC, Haber JE (2006) Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 40:209–235
Harvey S, Decker R, Dai Y, Schaefer G, Tang L, Kramer L, Dent P, Grant S (2001) Interactions between 2-fluoroadenine 9-beta-D-arabinofuranoside and the kinase inhibitor UCN-01 in human leukemia and lymphoma cells. Clin Cancer Res 7:320–330
Hawkins W, Mitchell C, McKinstry R, Gilfor D, Starkey J, Dai Y, Dawson K, Ramakrishnan V, Roberts JD, Yacoub A, Grant S, Dent P (2005) Transient exposure of mammary tumors to PD184352 and UCN-01 causes tumor cell death in vivo and prolonged suppression of tumor regrowth. Cancer Biol Ther 4:1275–1284
Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK (2002) An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol 22:8552–8561
Hindley C, Philpott A (2012) Co-ordination of cell cycle and differentiation in the developing nervous system. Biochem J 444:375–382
Hisanaga S, Saito T (2003) The regulation of cyclin-dependent kinase 5 activity through the metabolism of p35 or p39 Cdk5 activator. Neurosignals 12:221–229
Hisanaga S, Endo R (2010) Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J Neurochem 115:1309–1321
Hisanaga S, Asada A (2012) Cdk5-induced neuronal cell death: the activation of the conventional Rb-E2F G 1 pathway in post-mitotic neurons. Cell Cycle 11:2049
Hoeppner S, Baumli S, Cramer P (2005) Structure of the mediator subunit cyclin C and its implications for CDK8 function. J Mol Biol 350:833–842
Hofmann J (2004) Protein kinase C isozymes as potential targets for anticancer therapy. Curr Cancer Drug Targets 4:125–146
Hofmann F, Livingston DM (1996) Differential effects of cdk2 and cdk3 on the control of pRb and E2F function during G1 exit. Genes Dev 10:851–861
Holkova B, Perkins EB, Ramakrishnan V, Tombes MB, Shrader E, Talreja N, Wellons MD, Hogan KT, Roodman GD, Coppola D, Kang L, Dawson J, Stuart RK, Peer C, Figg WD Sr, Kolla S, Doyle A, Wright J, Sullivan DM, Roberts JD, Grant S (2011) Phase I trial of bortezomib (PS-341; NSC 681239) and alvocidib (flavopiridol; NSC 649890) in patients with recurrent or refractory B-cell neoplasms. Clin Cancer Res 17:3388–3397
Honma T, Hayashi K, Aoyama T, Hashimoto N, Machida T, Fukasawa K, Iwama T, Ikeura C, Ikuta M, Suzuki-Takahashi I, Iwasawa Y, Hayama T, Nishimura S, Morishima H (2001) Structure-based generation of a new class of potent Cdk4 inhibitors: new de novo design strategy and library design. J Med Chem 44:4615–4627
Honma N, Asada A, Takeshita S, Enomoto M, Yamakawa E, Tsutsumi K, Saito T, Satoh T, Itoh H, Kaziro Y, Kishimoto T, Hisanaga S (2003) Apoptosis-associated tyrosine kinase is a Cdk5 activator p35 binding protein. Biochem Biophys Res Commun 310:398–404
Horiuchi D, Huskey NE, Kusdra L, Wohlbold L, Merrick KA, Zhang C, Creasman KJ, Shokat KM, Fisher RP, Goga A (2012) Chemical-genetic analysis of cyclin dependent kinase 2 function reveals an important role in cellular transformation by multiple oncogenic pathways. Proc Natl Acad Sci USA 109:E1019–E1027
Hosokawa Y, Arnold A (1998) Mechanism of cyclin D1 (CCND1, PRAD1) overexpression in human cancer cells: analysis of allele-specific expression. Genes Chromosomes Cancer 22:66–71
Hu X, Moscinski LC (2011) Cdc2: a monopotent or pluripotent CDK? Cell Prolif 44:205–211
Hu D, Mayeda A, Trembley JH, Lahti JM, Kidd VJ (2003) CDK11 complexes promote pre-mRNA splicing. J Biol Chem 278:8623–8629
Hulit J, Bash T, Fu M, Galbiati F, Albanese C, Sage DR, Schlegel A, Zhurinsky J, Shtutman M, Ben-Ze’ev A, Lisanti MP, Pestell RG (2000) The cyclin D1 gene is transcriptionally repressed by caveolin-1. J Biol Chem 275:21203–21209
Husseman JW, Nochlin D, Vincent I (2000) Mitotic activation: a convergent mechanism for a cohort of neurodegenerative diseases. Neurobiol Aging 21:815–828
Ikuta M, Kamata K, Fukasawa K, Honma T, Machida T, Hirai H, Suzuki-Takahashi I, Hayama T, Nishimura S (2001) Crystallographic approach to identification of cyclin-dependent kinase 4 (CDK4)-specific inhibitors by using CDK4 mimic CDK2 protein. J Biol Chem 276:27548–27554
Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt AN, Crook T, Lord CJ, Ashworth A (2008) Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell 13:91–104
Jirmanova L, Afanassieff M, Gobert-Gosse S, Markossian S, Savatier P (2002) Differential contributions of ERK and PI3-kinase to the regulation of cyclin D1 expression and to the control of the G1/S transition in mouse embryonic stem cells. Oncogene 21:5515–5528
Johnson LN, De ME, Brown NR, Song H, Barford D, Endicott JA, Noble ME (2002) Structural studies with inhibitors of the cell cycle regulatory kinase cyclin-dependent protein kinase 2. Pharmacol Ther 93:113–124
Johnson AJ, Yeh YY, Smith LL, Wagner AJ, Hessler J, Gupta S, Flynn J, Jones J, Zhang X, Bannerji R, Grever MR, Byrd JC (2012) The novel cyclin-dependent kinase inhibitor dinaciclib (SCH727965) promotes apoptosis and abrogates microenvironmental cytokine protection in chronic lymphocytic leukemia cells. Leukemia
Kaiser A, Nishi K, Gorin FA, Walsh DA, Bradbury EM, Schnier JB (2001) The cyclin-dependent kinase (CDK) inhibitor flavopiridol inhibits glycogen phosphorylase. Arch Biochem Biophys 386:179–187
Kamei H, Saito T, Ozawa M, Fujita Y, Asada A, Bibb JA, Saido TC, Sorimachi H, Hisanaga S (2007) Suppression of calpain-dependent cleavage of the CDK5 activator p35 to p25 by site-specific phosphorylation. J Biol Chem 282:1687–1694
Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, Polsky D, Osman I, Garcia BA, Hernando E, Bernstein E (2010) The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468:1105–1109
Karlsson-Rosenthal C, Millar JB (2006) Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol 16:285–292
Karp JE, Passaniti A, Gojo I, Kaufmann S, Bible K, Garimella TS, Greer J, Briel J, Smith BD, Gore SD, Tidwell ML, Ross DD, Wright JJ, Colevas AD, Bauer KS (2005) Phase I and pharmacokinetic study of flavopiridol followed by 1-beta-D-arabinofuranosylcytosine and mitoxantrone in relapsed and refractory adult acute leukemias. Clin Cancer Res 11:8403–8412
Kasten M, Giordano A (2001) Cdk10, a Cdc2-related kinase, associates with the Ets2 transcription factor and modulates its transactivation activity. Oncogene 20:1832–1838
Kato H, Takahashi A, Itoyama Y (2003) Cell cycle protein expression in proliferating microglia and astrocytes following transient global cerebral ischemia in the rat. Brain Res Bull 60:215–221
Kawabe T (2004) G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther 3:513–519
Kim DM, Koo SY, Jeon K, Kim MH, Lee J, Hong CY, Jeong S (2003) Rapid induction of apoptosis by combination of flavopiridol and tumor necrosis factor (TNF)-alpha or TNF-related apoptosis-inducing ligand in human cancer cell lines. Cancer Res 63:621–626
Kitada S, Zapata JM, Andreeff M, Reed JC (2000) Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood 96:393–397
Knockaert M, Greengard P, Meijer L (2002) Pharmacological inhibitors of cyclin-dependent kinases. Trends Pharmacol Sci 23:417–425
Knudsen KE, Diehl JA, Haiman CA, Knudsen ES (2006) Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 25:1620–1628
Koguchi K, Nakatsuji Y, Okuno T, Sawada M, Sakoda S (2003) Microglial cell cycle-associated proteins control microglial proliferation in vivo and in vitro and are regulated by GM-CSF and density-dependent inhibition. J Neurosci Res 74:898–905
Kohn EA, Ruth ND, Brown MK, Livingstone M, Eastman A (2002) Abrogation of the S phase DNA damage checkpoint results in S phase progression or premature mitosis depending on the concentration of 7-hydroxystaurosporine and the kinetics of Cdc25C activation. J Biol Chem 277:26553–26564
Kohno T, Yokota J (2006) Molecular processes of chromosome 9p21 deletions causing inactivation of the p16 tumor suppressor gene in human cancer: deduction from structural analysis of breakpoints for deletions. DNA Repair (Amst) 5:1273–1281
Kohoutek J, Blazek D (2012) Cyclin K goes with Cdk12 and Cdk13. Cell Div 7:12
Kohzato N, Dong Y, Sui L, Masaki T, Nagahata S, Nishioka M, Konishi R, Tokuda M (2001) Overexpression of cyclin E and cyclin-dependent kinase 2 is correlated with development of hepatocellular carcinomas. Hepatol Res 21:27–39
Komander D, Kular GS, Bain J, Elliott M, Alessi DR, van Aalten DM (2003) Structural basis for UCN-01 (7-hydroxystaurosporine) specificity and PDK1 (3-phosphoinositide-dependent protein kinase-1) inhibition. Biochem J 375:255–262
Konishi Y, Lehtinen M, Donovan N, Bonni A (2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9:1005–1016
Kouroukis CT, Belch A, Crump M, Eisenhauer E, Gascoyne RD, Meyer R, Lohmann R, Lopez P, Powers J, Turner R, Connors JM (2003) Flavopiridol in untreated or relapsed mantle-cell lymphoma: results of a phase II study of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 21:1740–1745
Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P (2004) Mouse development and cell proliferation in the absence of D-cyclins. Cell 118:477–491
Krieger S, Gauduchon J, Roussel M, Troussard X, Sola B (2006) Relevance of cyclin D1b expression and CCND1 polymorphism in the pathogenesis of multiple myeloma and mantle cell lymphoma. BMC Cancer 6:238
Krueger BJ, Jeronimo C, Roy BB, Bouchard A, Barrandon C, Byers SA, Searcey CE, Cooper JJ, Bensaude O, Cohen EA, Coulombe B, Price DH (2008) LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res 36:2219–2229
Krueger BJ, Varzavand K, Cooper JJ, Price DH (2010) The mechanism of release of P-TEFb and HEXIM1 from the 7SK snRNP by viral and cellular activators includes a conformational change in 7SK. PLoS One 5:e12335
Kulkarni MS, Daggett JL, Bender TP, Kuehl WM, Bergsagel PL, Williams ME (2002) Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: ectopic p18 expression inhibits growth and induces apoptosis. Leukemia 16:127–134
Lacrima K, Valentini A, Lambertini C, Taborelli M, Rinaldi A, Zucca E, Catapano C, Cavalli F, Gianella-Borradori A, Maccallum DE, Bertoni F (2005) In vitro activity of cyclin-dependent kinase inhibitor CYC202 (seliciclib, R-roscovitine) in mantle cell lymphomas. Ann Oncol 16:1169–1176
Lacrima K, Rinaldi A, Vignati S, Martin V, Tibiletti MG, Gaidano G, Catapano CV, Bertoni F (2007) Cyclin-dependent kinase inhibitor seliciclib shows in vitro activity in diffuse large B-cell lymphomas. Leuk Lymphoma 48:158–167
Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW (2006) Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 9:13–22
Lapenna S, Giordano A (2009) Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 8:547–566
Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP (2007) Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell 25:839–850
Larochelle S, Amat R, Glover-Cutter K, Sanso M, Zhang C, Allen JJ, Shokat KM, Bentley DL, Fisher RP (2012) Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol 19:1108–1115
Lassus P, Opitz-Araya X, Lazebnik Y (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297:1352–1354
Lavoie JN, Rivard N, L’Allemain G, Pouyssegur J (1996) A temporal and biochemical link between growth factor-activated MAP kinases, cyclin D1 induction and cell cycle entry. Prog Cell Cycle Res 2:49–58
Lazarov M, Kubo Y, Cai T, Dajee M, Tarutani M, Lin Q, Fang M, Tao S, Green CL, Khavari PA (2002) CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat Med 8:1105–1114
Lee YM, Sicinski P (2006) Targeting cyclins and cyclin-dependent kinases in cancer: lessons from mice, hopes for therapeutic applications in human. Cell Cycle 5:2110–2114
Lee YK, Isham CR, Kaufman SH, Bible KC (2006) Flavopiridol disrupts STAT3/DNA interactions, attenuates STAT3-directed transcription, and combines with the Jak kinase inhibitor AG490 to achieve cytotoxic synergy. Mol Cancer Ther 5:138–148
Lents NH, Keenan SM, Bellone C, Baldassare JJ (2002) Stimulation of the Raf/MEK/ERK cascade is necessary and sufficient for activation and Thr-160 phosphorylation of a nuclear-targeted CDK2. J Biol Chem 277:47469–47475
Lesage D, Troussard X, Sola B (2005) The enigmatic role of cyclin D1 in multiple myeloma. Int J Cancer 115:171–176
Li KK, Ng IO, Fan ST, Albrecht JH, Yamashita K, Poon RY (2002) Activation of cyclin-dependent kinases CDC2 and CDK2 in hepatocellular carcinoma. Liver 22:259–268
Li Q, Price JP, Byers SA, Cheng D, Peng J, Price DH (2005) Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem 280:28819–28826
Li M, Lockwood W, Zielenska M, Northcott P, Ra YS, Bouffet E, Yoshimoto M, Rutka JT, Yan H, Taylor MD, Eberhart C, Hawkins CE, Lam W, Squire JA, Huang A (2012) Multiple CDK/CYCLIND genes are amplified in medulloblastoma and supratentorial primitive neuroectodermal brain tumor. Cancer Genet 205:220–231
Lin TS, Howard OM, Neuberg DS, Kim HH, Shipp MA (2002) Seventy-two hour continuous infusion flavopiridol in relapsed and refractory mantle cell lymphoma. Leuk Lymphoma 43:793–797
Liu DX, Greene LA (2001a) Neuronal apoptosis at the G1/S cell cycle checkpoint. Cell Tissue Res 305:217–228
Liu DX, Greene LA (2001b) Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 32(3):425–438
Lopes JP, Oliveira CR, Agostinho P (2007) Role of cyclin-dependent kinase 5 in the neurodegenerative process triggered by amyloid-beta and prion peptides: implications for Alzheimer’s disease and prion-related encephalopathies. Cell Mol Neurobiol 27:943–957
Lopes JP, Oliveira CR, Agostinho P (2009) Cdk5 acts as a mediator of neuronal cell cycle re-entry triggered by amyloid-beta and prion peptides. Cell Cycle 8:97–104
Lopes JP, Oliveira CR, Agostinho P (2010) Neurodegeneration in an Abeta-induced model of Alzheimer’s disease: the role of Cdk5. Aging Cell 9(1):64–77
Lopes JP, Agostinho P (2011) Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol 94:49–63
Love S (2003) Neuronal expression of cell cycle-related proteins after brain ischaemia in man. Neurosci Lett 353:29–32
Loyer P, Trembley JH, Katona R, Kidd VJ, Lahti JM (2005) Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 17:1033–1051
Loyer P, Trembley JH, Grenet JA, Busson A, Corlu A, Zhao W, Kocak M, Kidd VJ, Lahti JM (2008) Characterization of cyclin L1 and L2 interactions with CDK11 and splicing factors: influence of cyclin L isoforms on splice site selection. J Biol Chem 283:7721–7732
Loyer P, Busson A, Trembley JH, Hyle J, Grenet J, Zhao W, Ribault C, Montier T, Kidd VJ, Lahti JM (2011) The RNA binding motif protein 15B (RBM15B/OTT3) is a functional competitor of serine-arginine (SR) proteins and antagonizes the positive effect of the CDK11p110-cyclin L2alpha complex on splicing. J Biol Chem 286:147–159
Lu F, Gladden AB, Diehl JA (2003) An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res 63:7056–7061
Lu X, Burgan WE, Cerra MA, Chuang EY, Tsai MH, Tofilon PJ, Camphausen K (2004) Transcriptional signature of flavopiridol-induced tumor cell death. Mol Cancer Ther 3:861–872
Ma Y, Cress WD (2007) Transcriptional upregulation of p57 (Kip2) by the cyclin-dependent kinase inhibitor BMS-387032 is E2F dependent and serves as a negative feedback loop limiting cytotoxicity. Oncogene 26:3532–3540
Ma Y, Cress WD, Haura EB (2003) Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol Cancer Ther 2:73–81
Ma Y, Freeman SN, Cress WD (2004) E2F4 deficiency promotes drug-induced apoptosis. Cancer Biol Ther 3:1262–1269
Maccallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR (2005) Seliciclib (CYC202, R-roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res 65:5399–5407
Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M (2004) Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118:493–504
Malumbres M, Barbacid M (2005) Mammalian cyclin-dependent kinases. Trends Biochem Sci 30:630–641
Malumbres M, Barbacid M (2006) Is Cyclin D1-CDK4 kinase a bona fide cancer target? Cancer Cell 9:2–4
Malumbres M, Barbacid M (2007) Cell cycle kinases in cancer. Curr Opin Genet Dev 17:60–65
Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9:153–166
Marshall NF, Price DH (1995) Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 270:12335–12338
Marshall RM, Salerno D, Garriga J, Grana X (2005) Cyclin T1 expression is regulated by multiple signaling pathways and mechanisms during activation of human peripheral blood lymphocytes. J Immunol 175:6402–6411
Martin A, Odajima J, Hunt SL, Dubus P, Ortega S, Malumbres M, Barbacid M (2005) Cdk2 is dispensable for cell cycle inhibition and tumor suppression mediated by p27(Kip1) and p21(Cip1). Cancer Cell 7:591–598
McClue SJ, Blake D, Clarke R, Cowan A, Cummings L, Fischer PM, MacKenzie M, Melville J, Stewart K, Wang S, Zhelev N, Zheleva D, Lane DP (2002) In vitro and in vivo antitumor properties of the cyclin dependent kinase inhibitor CYC202 (R-roscovitine). Int J Cancer 102:463–468
McKinstry R, Qiao L, Yacoub A, Dai Y, Decker R, Holt S, Hagan MP, Grant S, Dent P (2002) Inhibitors of MEK1/2 interact with UCN-01 to induce apoptosis and reduce colony formation in mammary and prostate carcinoma cells. Cancer Biol Ther 1:243–253
Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243:527–536
Merrick KA, Larochelle S, Zhang C, Allen JJ, Shokat KM, Fisher RP (2008) Distinct activation pathways confer cyclin-binding specificity on Cdk1 and Cdk2 in human cells. Mol Cell 32:662–672
Merrick KA, Fisher RP (2010a) A virtual cycle: theory and experiment converge on the exit from mitosis. F1000 Biol Rep 2:33
Merrick KA, Fisher RP (2010b) Putting one step before the other: distinct activation pathways for Cdk1 and Cdk2 bring order to the mammalian cell cycle. Cell Cycle 9:706–714
Merrick KA, Wohlbold L, Zhang C, Allen JJ, Horiuchi D, Huskey NE, Goga A, Shokat KM, Fisher RP (2011) Switching Cdk2 on or off with small molecules to reveal requirements in human cell proliferation. Mol Cell 42:624–636
Merrick KA, Fisher RP (2012) Why minimal is not optimal: driving the mammalian cell cycle–and drug discovery–with a physiologic CDK control network. Cell Cycle 11:2600–2605
Micheau O (2003) Cellular FLICE-inhibitory protein: an attractive therapeutic target? Expert Opin Ther Targets 7:559–573
Michels AA, Fraldi A, Li Q, Adamson TE, Bonnet F, Nguyen VT, Sedore SC, Price JP, Price DH, Lania L, Bensaude O (2004) Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J 23:2608–2619
Mikolcevic P, Sigl R, Rauch V, Hess MW, Pfaller K, Barisic M, Pelliniemi LJ, Boesl M, Geley S (2012) Cyclin-dependent kinase 16/PCTAIRE kinase 1 is activated by cyclin Y and is essential for spermatogenesis. Mol Cell Biol 32:868–879
Misra RN, Xiao HY, Kim KS, Lu S, Han WC, Barbosa SA, Hunt JT, Rawlins DB, Shan W, Ahmed SZ, Qian L, Chen BC, Zhao R, Bednarz MS, Kellar KA, Mulheron JG, Batorsky R, Roongta U, Kamath A, Marathe P, Ranadive SA, Sack JS, Tokarski JS, Pavletich NP, Lee FY, Webster KR, Kimball SD (2004) N-(Cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-Dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J Med Chem 47:1719–1728
Mitchell C, Hamed HA, Cruickshanks N, Tang Y, Bareford MD, Hubbard N, Tye G, Yacoub A, Dai Y, Grant S, Dent P (2011) Simultaneous exposure of transformed cells to SRC family inhibitors and CHK1 inhibitors causes cell death. Cancer Biol Ther 12:215–228
Morris MC, Gondeau C, Tainer JA, Divita G (2002) Kinetic mechanism of activation of the Cdk2/cyclin A complex. Key role of the C-lobe of the Cdk. J Biol Chem 277:23847–23853
Mukhopadhyay P, Ali MA, Nandi A, Carreon P, Choy H, Saha D (2006) The cyclin-dependent kinase 2 inhibitor down-regulates interleukin-1beta-mediated induction of cyclooxygenase-2 expression in human lung carcinoma cells. Cancer Res 66:1758–1766
Newcomb EW (2004) Flavopiridol: pleiotropic biological effects enhance its anti-cancer activity. Anticancer Drugs 15:411–419
Newcomb EW, Ali MA, Schnee T, Lan L, Lukyanov Y, Fowkes M, Miller DC, Zagzag D (2005) Flavopiridol downregulates hypoxia-mediated hypoxia-inducible factor-1alpha expression in human glioma cells by a proteasome-independent pathway: implications for in vivo therapy. Neuro Oncol 7:225–235
Ng MH, Chung YF, Lo KW, Wickham NW, Lee JC, Huang DP (1997) Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood 89:2500–2506
Nguyen VT, Kiss T, Michels AA, Bensaude O (2001) 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414:322–325
Nguyen T, Dai Y, Attkisson E, Kramer L, Jordan N, Nguyen N, Kolluri N, Muschen M, Grant S (2011) HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin Cancer Res 17:3219–3232
Nikolic M, Tsai LH (2000) Activity and regulation of p35/Cdk5 kinase complex. Methods Enzymol 325:200–213
Nurse P (2012) Finding CDK: linking yeast with humans. Nat Cell Biol 14:776
Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, Geng Y, Gawrzak S, Quadros IM, Newman J, Das M, Jecrois ME, Yu Q, Li N, Bienvenu F, Moss SJ, Greenberg ME, Marto JA, Sicinski P (2011) Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell 21:655–668
Oikonomakos NG, Schnier JB, Zographos SE, Skamnaki VT, Tsitsanou KE, Johnson LN (2000) Flavopiridol inhibits glycogen phosphorylase by binding at the inhibitor site. J Biol Chem 275:34566–34573
Oka K, Ohno T, Yamaguchi M, Mahmud N, Miwa H, Kita K, Shiku H, Shirakawa S (1996) PRAD1/cyclin D1 gene overexpression in mantle cell lymphoma. Leuk Lymphoma 21:37–42
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435:677–681
Ortega S, Malumbres M, Barbacid M (2002) Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta 1602:73–87
Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M (2003) Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35:25–31
Ou CY, Poon VY, Maeder CI, Watanabe S, Lehrman EK, Fu AK, Park M, Fu WY, Jorgensen EM, Ip NY, Shen K (2010) Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell 141:846–858
Park M, Watanabe S, Poon VY, Ou CY, Jorgensen EM, Shen K (2011) CYY-1/cyclin Y and CDK-5 differentially regulate synapse elimination and formation for rewiring neural circuits. Neuron 70:742–757
Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, Wang Y, Oft M, Chen T, Kirschmeier P, Lees EM (2010) Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 9:2344–2353
Patel SA, Simon MC (2010) Functional analysis of the Cdk7.cyclin H.Mat1 complex in mouse embryonic stem cells and embryos. J Biol Chem 285:15587–15598
Patel V, Senderowicz AM, Pinto D Jr, Igishi T, Raffeld M, Quintanilla-Martinez L, Ensley JF, Sausville EA, Gutkind JS (1998) Flavopiridol, a novel cyclin-dependent kinase inhibitor, suppresses the growth of head and neck squamous cell carcinomas by inducing apoptosis. J Clin Invest 102:1674–1681
Patel V, Lahusen T, Leethanakul C, Igishi T, Kremer M, Quintanilla-Martinez L, Ensley JF, Sausville EA, Gutkind JS, Senderowicz AM (2002) Antitumor activity of UCN-01 in carcinomas of the head and neck is associated with altered expression of cyclin D3 and p27(KIP1). Clin Cancer Res 8:3549–3560
Pei XY, Dai Y, Grant S (2004) The small-molecule Bcl-2 inhibitor HA14-1 interacts synergistically with flavopiridol to induce mitochondrial injury and apoptosis in human myeloma cells through a free radical-dependent and Jun NH2-terminal kinase-dependent mechanism. Mol Cancer Ther 3:1513–1524
Pei XY, Dai Y, Rahmani M, Li W, Dent P, Grant S (2005) The farnesyltransferase inhibitor L744832 potentiates UCN-01-induced apoptosis in human multiple myeloma cells. Clin Cancer Res 11:4589–4600
Pei XY, Li W, Dai Y, Dent P, Grant S (2006) Dissecting the roles of checkpoint kinase 1/CDC2 and mitogen-activated protein kinase kinase 1/2/extracellular signal-regulated kinase 1/2 in relation to 7-hydroxystaurosporine-induced apoptosis in human multiple myeloma cells. Mol Pharmacol 70:1965–1973
Pei XY, Dai Y, Tenorio S, Lu J, Harada H, Dent P, Grant S (2007) MEK1/2 inhibitors potentiate UCN-01 lethality in human multiple myeloma cells through a Bim-dependent mechanism. Blood 110:2092–2101
Pei XY, Dai Y, Youssefian LE, Chen S, Bodie WW, Takabatake Y, Felthousen J, Almenara JA, Kramer LB, Dent P, Grant S (2011) Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2. Blood 118:5189–5200
Peng J, Zhu Y, Milton JT, Price DH (1998) Identification of multiple cyclin subunits of human P-TEFb. Genes Dev 12:755–762
Pepper C, Thomas A, Hoy T, Fegan C, Bentley P (2001) Flavopiridol circumvents Bcl-2 family mediated inhibition of apoptosis and drug resistance in B-cell chronic lymphocytic leukaemia. Br J Haematol 114:70–77
Perez PC, Caceres RA, Canduri F, de Azevedo WFJ (2009) Molecular modeling and dynamics simulation of human cyclin-dependent kinase 3 complexed with inhibitors. Comput Biol Med 39:130–140
Perez-Simon JA, Garcia-Sanz R, Tabernero MD, Almeida J, Gonzalez M, Fernandez-Calvo J, Moro MJ, Hernandez JM, San Miguel JF, Orfao A (1998) Prognostic value of numerical chromosome aberrations in multiple myeloma: a FISH analysis of 15 different chromosomes. Blood 91:3366–3371
Peterlin BM, Price DH (2006) Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23:297–305
Price DH (2000) P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol 20:2629–2634
Radu A, Neubauer V, Akagi T, Hanafusa H, Georgescu MM (2003) PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol Cell Biol 23:6139–6149
Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, Shiraishi N, Chauhan D, Munshi NC, Green SR, Anderson KC (2005) Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood 106:1042–1047
Rashidian J, Iyirhiaro G, Aleyasin H, Rios M, Vincent I, Callaghan S, Bland RJ, Slack RS, During MJ, Park DS (2005) Multiple cyclin-dependent kinases signals are critical mediators of ischemia/hypoxic neuronal death in vitro and in vivo. Proc Natl Acad Sci USA 102:14080–14085
Raynaud FI, Whittaker SR, Fischer PM, McClue S, Walton MI, Barrie SE, Garrett MD, Rogers P, Clarke SJ, Kelland LR, Valenti M, Brunton L, Eccles S, Lane DP, Workman P (2005) In vitro and in vivo pharmacokinetic-pharmacodynamic relationships for the trisubstituted aminopurine cyclin-dependent kinase inhibitors olomoucine, bohemine and CYC202. Clin Cancer Res 11:4875–4887
Reed JC (2003) Apoptosis-targeted therapies for cancer. Cancer Cell 3(1):17–22
Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB (2007) p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11:175–189
Ren S, Rollins BJ (2004) Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 117:239–251
Ribas J, Boix J, Meijer L (2006) (R)-Roscovitine (CYC202, seliciclib) sensitizes SH-SY5Y neuroblastoma cells to nutlin-3-induced apoptosis. Exp Cell Res 312:2394–2400
Rodriguez-Bravo V, Guaita-Esteruelas S, Florensa R, Bachs O, Agell N (2006) Chk1- and claspin-dependent but ATR/ATM- and Rad17-independent DNA replication checkpoint response in HeLa cells. Cancer Res 66:8672–8679
Rosato RR, Dai Y, Almenara JA, Maggio SC, Grant S (2004) Potent antileukemic interactions between flavopiridol and TRAIL/Apo2L involve flavopiridol-mediated XIAP downregulation. Leukemia 18:1780–1788
Rosato RR, Almenara JA, Maggio SC, Atadja P, Craig R, Vrana J, Dent P, Grant S (2005) Potentiation of the lethality of the histone deacetylase inhibitor LAQ824 by the cyclin-dependent kinase inhibitor roscovitine in human leukemia cells. Mol Cancer Ther 4:1772–1785
Rossi AG, Sawatzky DA, Walker A, Ward C, Sheldrake TA, Riley NA, Caldicott A, Martinez-Losa M, Walker TR, Duffin R, Gray M, Crescenzi E, Martin MC, Brady HJ, Savill JS, Dransfield I, Haslett C (2006) Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat Med 12:1056–1064
Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, Barrette T, Everett J, Siddiqui J, Kunju LP, Navone N, Araujo JC, Troncoso P, Logothetis CJ, Innis JW, Smith DC, Lao CD, Kim SY, Roberts JS, Gruber SB, Pienta KJ, Talpaz M, Chinnaiyan AM (2011) Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med 3:111ra121
Sage J (2004) Cyclin C makes an entry into the cell cycle. Dev Cell 6:607–608
Sampath D, Cortes J, Estrov Z, Du M, Shi Z, Andreeff M, Gandhi V, Plunkett W (2006) Pharmacodynamics of cytarabine alone and in combination with 7-hydroxystaurosporine (UCN-01) in AML blasts in vitro and during a clinical trial. Blood 107:2517–2524
Sandal T (2002) Molecular aspects of the mammalian cell cycle and cancer. Oncologist 7:73–81
Santamaria D, Barriere C, Cerqueira A, Hunt S, Tardy C, Newton K, Caceres JF, Dubus P, Malumbres M, Barbacid M (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448:811–815
Sarasquete ME, Garcia-Sanz R, Armellini A, Fuertes M, Martin-Jimenez P, Sierra M, Del Carmen CM, Alcoceba M, Balanzategui A, Ortega F, Hernandez JM, Sureda A, Palomera L, Gonzalez M, San Miguel JF (2006) The association of increased p14ARF/p16INK4a and p15INK4a gene expression with proliferative activity and the clinical course of multiple myeloma. Haematologica 91:1551–1554
Sato S, Fujita N, Tsuruo T (2002) Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene 21:1727–1738
Sato K, Zhu YS, Saito T, Yotsumoto K, Asada A, Hasegawa M, Hisanaga S (2007) Regulation of membrane association and kinase activity of Cdk5-p35 by phosphorylation of p35. J Neurosci Res 85:3071–3078
Sato K, Minegishi S, Takano J, Plattner F, Saito T, Asada A, Kawahara H, Iwata N, Saido TC, Hisanaga S (2011) Calpastatin, an endogenous calpain-inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. J Neurochem 117:504–515
Sausville EA (2002) Complexities in the development of cyclin-dependent kinase inhibitor drugs. Trends Mol Med 8:S32–S37
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J 17:1675–1687
Schneider E, Kartarius S, Schuster N, Montenarh M (2002) The cyclin H/cdk7/Mat1 kinase activity is regulated by CK2 phosphorylation of cyclin H. Oncogene 21:5031–5037
Schwartz GK, Shah MA (2005) Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol 23:9408–9421
Schwartz GK, Ilson D, Saltz L, O’Reilly E, Tong W, Maslak P, Werner J, Perkins P, Stoltz M, Kelsen D (2001) Phase II study of the cyclin-dependent kinase inhibitor flavopiridol administered to patients with advanced gastric carcinoma. J Clin Oncol 19:1985–1992
Sedlacek HH (2001) Mechanisms of action of flavopiridol. Crit Rev Oncol Hematol 38:139–170
Semenov I, Akyuz C, Roginskaya V, Chauhan D, Corey SJ (2002) Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leuk Res 26:271–280
Senderowicz AM (2002) The cell cycle as a target for cancer therapy: basic and clinical findings with the small molecule inhibitors flavopiridol and UCN-01. Oncologist 7:12–19
Senderowicz AM (2003) Small-molecule cyclin-dependent kinase modulators. Oncogene 22:6609–6620
Senderowicz AM, Sausville EA (2000) Preclinical and clinical development of cyclin-dependent kinase modulators. J Natl Cancer Inst 92:376–387
Shao J, Sheng H, DuBois RN, Beauchamp RD (2000) Oncogenic Ras-mediated cell growth arrest and apoptosis are associated with increased ubiquitin-dependent cyclin D1 degradation. J Biol Chem 275:22916–22924
Shapiro GI (2004) Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res 10:4270s–4275s
Shapiro GI (2006) Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 24:1770–1783
Shapiro GI, Supko JG, Patterson A, Lynch C, Lucca J, Zacarola PF, Muzikansky A, Wright JJ, Lynch TJ Jr, Rollins BJ (2001) A phase II trial of the cyclin-dependent kinase inhibitor flavopiridol in patients with previously untreated stage IV non-small cell lung cancer. Clin Cancer Res 7:1590–1599
Shi Y, Sharma A, Wu H, Lichtenstein A, Gera J (2005) Cyclin D1 and c-myc internal ribosome entry site (IRES)-dependent translation is regulated by AKT activity and enhanced by rapamycin through a p38. J Biol Chem 280:10964–10973
Shu F, Lv S, Qin Y, Ma X, Wang X, Peng X, Luo Y, Xu BE, Sun X, Wu J (2007) Functional characterization of human PFTK1 as a cyclin-dependent kinase. Proc Natl Acad Sci USA 104:9248–9253
Sonoki T, Harder L, Horsman DE, Karran L, Taniguchi I, Willis TG, Gesk S, Steinemann D, Zucca E, Schlegelberger B, Sole F, Mungall AJ, Gascoyne RD, Siebert R, Dyer MJ (2001) Cyclin D3 is a target gene of t(6;14)(p21.1;q32.3) of mature B-cell malignancies. Blood 98:2837–2844
Strasser A, O’Connor L, Dixit VM (2000) Apoptosis signaling. Annu Rev Biochem 69:217–245
Takada Y, Aggarwal BB (2004) Flavopiridol inhibits NF-kappaB activation induced by various carcinogens and inflammatory agents through inhibition of IkappaBalpha kinase and p65 phosphorylation: abrogation of cyclin D1, cyclooxygenase-2, and matrix metalloprotease-9. J Biol Chem 279:4750–4759
Takahashi-Yanaga F, Mori J, Matsuzaki E, Watanabe Y, Hirata M, Miwa Y, Morimoto S, Sasaguri T (2006) Involvement of GSK-3beta and DYRK1B in differentiation-inducing factor-3-induced phosphorylation of cyclin D1 in HeLa cells. J Biol Chem 281:38489–38497
Tan AR, Headlee D, Messmann R, Sausville EA, Arbuck SG, Murgo AJ, Melillo G, Zhai S, Figg WD, Swain SM, Senderowicz AM (2002) Phase I clinical and pharmacokinetic study of flavopiridol administered as a daily 1-hour infusion in patients with advanced neoplasms. J Clin Oncol 20:4074–4082
Tang L, Li MH, Cao P, Wang F, Chang WR, Bach S, Reinhardt J, Ferandin Y, Galons H, Wan Y, Gray N, Meijer L, Jiang T, Liang DC (2005) Crystal structure of pyridoxal kinase in complex with roscovitine and derivatives. J Biol Chem 280:31220–31229
Tang Y, Dai Y, Grant S, Dent P (2012a) Enhancing CHK1 inhibitor lethality in glioblastoma. Cancer Biol Ther 13:379–388
Tang Y, Hamed HA, Poklepovic A, Dai Y, Grant S, Dent P (2012b) Poly(ADP-ribose) polymerase 1 modulates the lethality of CHK1 inhibitors in mammary tumors. Mol Pharmacol 82:322–332
Tasaka T, Berenson J, Vescio R, Hirama T, Miller CW, Nagai M, Takahara J, Koeffler HP (1997) Analysis of the p16INK4A, p15INK4B and p18INK4C genes in multiple myeloma. Br J Haematol 96:98–102
Tashiro E, Tsuchiya A, Imoto M (2007) Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci 98:629–635
Tetsu O, McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3:233–245
Tian DS, Yu ZY, Xie MJ, Bu BT, Witte OW, Wang W (2006) Suppression of astroglial scar formation and enhanced axonal regeneration associated with functional recovery in a spinal cord injury rat model by the cell cycle inhibitor olomoucine. J Neurosci Res 84:1053–1063
Tian DS, Xie MJ, Yu ZY, Zhang Q, Wang YH, Chen B, Chen C, Wang W (2007) Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res 1135:177–185
Trembley JH, Hu D, Slaughter CA, Lahti JM, Kidd VJ (2003) Casein kinase 2 interacts with cyclin-dependent kinase 11 (CDK11) in vivo and phosphorylates both the RNA polymerase II carboxyl-terminal domain and CDK11 in vitro. J Biol Chem 278:2265–2270
Trembley JH, Loyer P, Hu D, Li T, Grenet J, Lahti JM, Kidd VJ (2004) Cyclin dependent kinase 11 in RNA transcription and splicing. Prog Nucleic Acid Res Mol Biol 77:263–288
Tricot G, Barlogie B, Jagannath S, Bracy D, Mattox S, Vesole DH, Naucke S, Sawyer JR (1995) Poor prognosis in multiple myeloma is associated only with partial or complete deletions of chromosome 13 or abnormalities involving 11q and not with other karyotype abnormalities. Blood 86:4250–4256
Tse AN, Carvajal R, Schwartz GK (2007) Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res 13:1955–1960
Tsutsui T, Fukasawa R, Tanaka A, Hirose Y, Ohkuma Y (2011) Identification of target genes for the CDK subunits of the Mediator complex. Genes Cells
Urashima M, Ogata A, Chauhan D, Vidriales MB, Teoh G, Hoshi Y, Schlossman RL, DeCaprio JA, Anderson KC (1996) Interleukin-6 promotes multiple myeloma cell growth via phosphorylation of retinoblastoma protein. Blood 88:2219–2227
van Deursen JM (2007) Rb loss causes cancer by driving mitosis mad. Cancer Cell 11:1–3
Van Herreweghe E, Egloff S, Goiffon I, Jady BE, Froment C, Monsarrat B, Kiss T (2007) Dynamic remodelling of human 7SK snRNP controls the nuclear level of active P-TEFb. EMBO J 26:3570–3580
Vaux DL, Silke J (2005) IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol 6:287–297
Verhagen AM, Vaux DL (2002) Cell death regulation by the mammalian IAP antagonist Diablo/Smac. Apoptosis 7:163–166
Vogel C, Hager C, Bastians H (2007) Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res 67:339–345
Wallenfang MR, Seydoux G (2002) cdk-7 Is required for mRNA transcription and cell cycle progression in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99:5527–5532
Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF (1999) The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol 19:6195–6206
Wartiovaara K, Barnabe-Heider F, Miller FD, Kaplan DR (2002) N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J Neurosci 22:815–824
Wesierska-Gadek J, Kramer MP (2012) The impact of CDK inhibition in human malignancies associated with pronounced defects in apoptosis: advantages of multi-targeting small molecules. Future Med Chem 4:395–424
Wesierska-Gadek J, Krystof V (2009) Selective cyclin-dependent kinase inhibitors discriminating between cell cycle and transcriptional kinases: future reality or utopia? Ann N Y Acad Sci 1171:228–241]
Wesierska-Gadek J, Maurer M (2011) Promotion of apoptosis in cancer cells by selective purine-derived pharmacological CDK inhibitors: one outcome, many mechanisms. Curr Pharm Des 17:256–271
Wesierska-Gadek J, Maurer M, Zulehner N, Komina O (2011) Whether to target single or multiple CDKs for therapy? That is the question. J Cell Physiol 226:341–349
Whittaker SR, Walton MI, Garrett MD, Workman P (2004) The cyclin-dependent kinase inhibitor CYC202 (R-roscovitine) inhibits retinoblastoma protein phosphorylation, causes loss of cyclin D1, and activates the mitogen-activated protein kinase pathway. Cancer Res 64:262–272
Wohlbold L, Merrick KA, De S, Amat R, Kim JH, Larochelle S, Allen JJ, Zhang C, Shokat KM, Petrini JH, Fisher RP (2012) Chemical genetics reveals a specific requirement for cdk2 activity in the DNA damage response and identifies nbs1 as a cdk2 substrate in human cells. PLoS Genet 8:e1002935
Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De PE, Hankeln T, Meyer zum Buschenfelde KH, Beach D (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269:1281–1284
Xu W, Ji JY (2011) Dysregulation of CDK8 and Cyclin C in tumorigenesis. J Genet Genomics 38:439–452
Yamada M, Saito T, Sato Y, Kawai Y, Sekigawa A, Hamazumi Y, Asada A, Wada M, Doi H, Hisanaga S (2007) Cdk5–p39 is a labile complex with the similar substrate specificity to Cdk5–p35. J Neurochem 102:1477–1487
Yang Y, Geldmacher DS, Herrup K (2001a) DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci 21:2661–2668
Yang Z, Zhu Q, Luo K, Zhou Q (2001b) The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414:317–322
Yu Q, Sicinski P (2004) Mammalian cell cycles without cyclin E-CDK2. Cell Cycle 3:292–295
Yu C, Dai Y, Dent P, Grant S (2002a) Coadministration of UCN-01 with MEK1/2 inhibitors potently induces apoptosis in BCR/ABL+ leukemia cells sensitive and resistant to ST1571. Cancer Biol Ther 1:674–682
Yu Q, La RJ, Zhang H, Takemura H, Kohn KW, Pommier Y (2002b) UCN-01 inhibits p53 up-regulation and abrogates gamma-radiation-induced G(2)-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res 62:5743–5748
Yu C, Rahmani M, Dai Y, Conrad D, Krystal G, Dent P, Grant S (2003) The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res 63:1822–1833
Yu Q, Sicinska E, Geng Y, Ahnstrom M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stal O, Sicinski P (2006) Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9:23–32
Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, Schaefer EM, Bonni A (2008) Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science 319:1665–1668
Zamzami N, Kroemer G (2001) The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol 2:67–71
Zhai S, Senderowicz AM, Sausville EA, Figg WD (2002) Flavopiridol, a novel cyclin-dependent kinase inhibitor, in clinical development. Ann Pharmacother 36:905–911
Zhang B, Gojo I, Fenton RG (2002) Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 99:1885–1893
Zhao B, Bower MJ, McDevitt PJ, Zhao H, Davis ST, Johanson KO, Green SM, Concha NO, Zhou BB (2002) Structural basis for Chk1 inhibition by UCN-01. J Biol Chem 277:46609–46615
Zhou BB, Bartek J (2004) Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer 4:216–225
Zhu YS, Saito T, Asada A, Maekawa S, Hisanaga S (2005) Activation of latent cyclin-dependent kinase 5 (Cdk5)-p35 complexes by membrane dissociation. J Neurochem 94:1535–1545
Zhu Z, Zhang Q, Yu Z, Zhang L, Tian D, Zhu S, Bu B, Xie M, Wang W (2007) Inhibiting cell cycle progression reduces reactive astrogliosis initiated by scratch injury in vitro and by cerebral ischemia in vivo. Glia 55:546–558
Zou H, Li Y, Liu X, Wang X (1999) An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274:11549–11556
Acknowledgments
This work was supported by two RO1 grants (CA100866 and CA93738), a Multiple Myeloma SPORE award (1 P50 CA142509-01) and its Developmental Research Program subaward (29859/98018093) from the National Cancer Institute, and an award (6181-10) from the Leukemia and Lymphoma Society of America.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Dai, Y., Chen, S., Yi, L., Xu, M. (2013). Targeting the Cell Cycle for Cancer Treatment and Neuroprotection. In: Resende, R., Ulrich, H. (eds) Trends in Stem Cell Proliferation and Cancer Research. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6211-4_23
Download citation
DOI: https://doi.org/10.1007/978-94-007-6211-4_23
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6210-7
Online ISBN: 978-94-007-6211-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)