
Abstract
Reactive compatibilization of immiscible polymer blends by in situ copolymer formation is reviewed using approximately 1,100 examples taken from both journal articles and patents. Selected references in English through approximately 2013 to early 2014 are included. Important chemical reactions are illustrated which are useful for copolymer formation across a melt-phase boundary during melt processing of the immiscible blends. Focus is on irreversible chemical reactions taking place within typical extrusion residence times for polymer processing. Examples of block, graft, cross-linked, and degradative copolymer formation are shown. The illustrated chemical reactions and processes are also generally useful for compatibilization of immiscible polymer blends either not illustrated or not yet conceived.
Formerly with Polymer Materials Laboratory, General Electric Co. Global Research Center, Schenectady, New York 12309
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Copolymer Formation
- Immiscible Polymer
- Blends Containing
- Cyclic Ortho Ester
- Selective Solvent Extraction
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Reactive compatibilization of immiscible polymer blends by in situ copolymer formation is reviewed using approximately 1,100 examples taken from both journal articles and patents. Selected references in English through approximately 2013 to early 2014 are included. Important chemical reactions are illustrated which are useful for copolymer formation across a melt-phase boundary during melt processing of the immiscible blends. Focus is on irreversible chemical reactions taking place within typical extrusion residence times for polymer processing. Examples of block, graft, cross-linked, and degradative copolymer formation are shown. The illustrated chemical reactions and processes are also generally useful for compatibilization of immiscible polymer blends either not illustrated or not yet conceived.
Commercial polymer products are frequently derived from blending two or more polymers to achieve a favorable balance of physical properties. As described in Chap. 2, “Thermodynamics of Polymer Blends” in this handbook, from the thermodynamic point of view, there are two basic types of polymer blends: miscible and immiscible. The vast majority of polymer pairs are immiscible. There are only a few commercially important polymer blends based on miscible or partially miscible (i.e., miscible within a low range of concentration) polymer pairs. It is seldom possible to mix two or more polymers and create a blend with useful properties. Instead, when preparing a new polymer blend from immiscible resins, it is necessary to devise a specific strategy for compatibilizing the mixture to provide for optimum physical performance and long-term stability. Although there do exist a very small number of commercial blends of immiscible polymers that are not compatibilized, most commercially available blends of immiscible polymers have been compatibilized by some specific mechanism.
The majority of polymer blends containing elastomeric, thermoplastic, and/or liquid crystalline polymers are processed by melt extrusion at some point in their history. After melt extrusion with intensive mixing, the morphology of an immiscible polymer blend on a microscopic scale will often consist of a dispersed phase of the more viscous polymer in a continuous matrix of the less viscous polymer (depending upon the relative amounts and viscosities of the two polymers in the blend). A good analogy from everyday experience is a dispersed mixture of viscous oil droplets in an immiscible water matrix.
The formation of optimum dispersed phase particle size and the long-term stabilization of the resulting blend morphology are critical if the blend is to have optimum properties and in particular good mechanical properties. If this morphology is not stabilized, then the dispersed phase may coalesce during any subsequent heat and/or high stress treatment, such as injection molding. Coalescence may result in gross-phase segregation of the two polymers and delamination on a macroscopic scale and/or brittleness or poor surface appearance in the final molded part. Good analogies from everyday experience would be the separation on standing of a not stabilized oil-in-water dispersion into two separate liquid phases. Therefore, an important aspect of all compatibilization strategies is the promotion of morphology stabilization. Morphology stabilization may be provided by sufficient interfacial adhesion and/or lowered interfacial tension between the two polymer phases.
Of the various compatibilization strategies that have been devised, an increasingly common method is either to add a block, graft, or cross-linked copolymer of the two (or more) separate polymers in the blend or to form such copolymers through covalent or ionic bond formation in situ during a reactive compatibilization step. The first of these methods was described in Chap. 4, “Interphase and Compatibilization by Addition of a Compatibilizer,” in this handbook, while the second method is the topic of this chapter.
The said copolymer can reside at the interface between the dispersed and matrix phases, acting as an emulsifying agent that effectively stabilizes the dispersed phase particles against coalescence and providing interfacial adhesion between dispersed and continuous phases in the solid state. In this manner such a copolymer can both promote optimum dispersed phase particle size formation during compounding and prevent phase coalescence of the dispersed phase during any subsequent heat treatment and/or high stress processes. Again, the analogy from everyday experience is the addition of a soap or other emulsifying agent to stabilize an oil-in-water emulsion. Often, as little as 0.5–2.0 wt% copolymer is sufficient to achieve morphology stabilization of an immiscible polymer blend. However, frequently higher amounts, for example, as much as 10–20 wt% copolymer, may be necessary to obtain optimum physical properties of the blend, e.g., impact strength.
The majority of commercially important, immiscible polymer blends rely for compatibilization on the presence of a copolymer of the blended polymers. However, such a copolymer is almost never synthesized in a separate step and then added as a distinct entity to the blend of immiscible polymers. Instead, a compatibilizing copolymer is most economically formed simultaneously with generation of morphology during extrusion processing, a process referred to as reactive compatibilization. The reactive compatibilization process is logically a subcategory of the broader class of interchain copolymer formation reactions performed by reactive extrusion (Brown 1992a), because there are other commercial reasons for preparing copolymers of immiscible polymers aside from using them as in situ generated compatibilizing agents for immiscible blends.
Copolymer formation by reactive compatibilization is a heterogeneous reaction taking place across a melt-phase boundary. Often this process occurs by direct reaction between chemical functionalities on some fraction of each of the two polymers. In some cases a third reactive species may be added to the blend to promote copolymer formation by one of several mechanisms.
Reactive compatibilization has at least two advantages:
-
1.
First, the compatibilizing copolymer is automatically formed at the interface between the two immiscible polymers where it is needed to stabilize morphology. In contrast, when a compatibilizing copolymer is added as a separate entity to a polymer blend, it must diffuse to the polymer-polymer interface to be effective for promoting morphology stabilization and interfacial adhesion between dispersed and continuous phases. However, that added copolymer may prefer to self-associate in micelles and form a separate phase that is useless for compatibilization.
-
2.
A second advantage of in situ copolymer formation is that the molecular weight of each of the two distinct polymeric segments in the copolymer is usually the same as that of the individual bulk polymer phase in which the segment must dissolve. Even approximate molecular weight matching between copolymer segment and bulk phase can result in optimum copolymer/bulk phase interaction for maximum interfacial adhesion. See, for example, Jiao et al. (1999).
2 Purpose
Only a relatively small number of chemical reactions have been devised to form a compatibilizing copolymer during extrusion processing. Therefore, a purpose of this chapter is to identify these different chemical reactions and give selected examples illustrating their scope to form block, graft, or cross-linked copolymers as compatibilizers for immiscible polymer blends. The emphasis is on copolymer formation during melt reaction occurring during development of morphology. With few exceptions, the examples are limited to processes that require mixing in the molten state. This includes processes run in single-screw or twin-screw extruders, or similar continuous or semicontinuous processing equipment, as well as in batch mixers.
The references in this review include both journal articles and selected published or issued patents. A large number of reactive compatibilization examples are found in industrial research and are documented mostly in patents. Patent references are included in this chapter if they reveal a novel compatibilization strategy apparently not otherwise documented until later in the journal literature. Numerous examples of industrial compatibilization methods have also been provided in a book based on the patent literature (Utracki 1998).
It is not the purpose of this chapter to describe “compatibilization” of layers of immiscible polymers in laminates. Strategies similar to those used to compatibilize intimately mixed polymer blends have also been used to prepare stable laminates, and in those cases, where a chemical reaction takes place between laminate layers, similar types of chemical reaction have been used. Nevertheless, laminate macroscopic morphology is essentially fixed, and formation of stable laminates is better treated as an adhesion problem. Similarly, composite compositions which may comprise a blend of one or more functionalized polymers with a second, less tractable component such as starch, lignin, clay, silica, POSS, carbon nanotubes, etc. are outside the scope of this chapter.
It is also not the purpose of this chapter to summarize examples of “compatible” polymer blends formed in a solution step involving dissolution of the polymer components, whether or not a chemical reaction takes place between them. In some cases, particularly when no reaction takes place, such blends are only “pseudo-stable,” since they may not have been processed above the Tg of one or both of the polymer components. Also, mixing in solution followed by devolatilization is rarely economical for practice in industry, particularly since many commercially important compatibilized polymer blends comprise at least one semicrystalline component (e.g., PA) which is poorly soluble in common solvents. There are included in the Tables a small number of examples of solution blended polymer blends when these complement similar examples prepared by melt processing.
It is also not the purpose of this chapter to describe examples of compatibilized polymer blends formed by polymerization of a monomer in the presence of a second polymer. In these cases, the growing polymer chain may react with functionality on the second polymer to form a certain fraction of compatibilizing copolymer.
The coverage of this chapter is arranged by binary polymer Blend Type, in alphabetical order of the first polymeric component. Thus, “polyamide blend” is the first category discussed herein. Subcategories within each Blend Type category are arranged by the specific chemical reactions that have been described in the literature for reactive compatibilization processes.
The emphasis is on illustrating the scope of these particular reactions and not on presenting every known example of a particular compatibilization strategy. For example, polyamide-polyolefin (PA-PO) blends compatibilized by reaction of PA amine end-groups with anhydride-functionalized PO have been studied in hundreds of different published examples, as have immiscible pairs of polyesters (or polycarbonate) compatibilized by transesterification reactions and polyester-polyolefin blends compatibilized using anhydride-grafted PO. Although these studies contribute to understanding the physics and property optimization of such blends, the underlying chemistry is basically the same in each case, and limitations of space preclude comprehensively listing all such examples. It should also be noted that many published studies listed herein actually represent a series of papers or patents. In some cases, only one paper or patent in the series is referenced. Also included in this chapter are some examples of blends which illustrate simple copolymer formation between two functionalized polymers although a corresponding blend with either of the unfunctionalized polymers is not exemplified, because it is perfectly possible that the said copolymer could be a compatibilizer in corresponding blends with one or both unfunctionalized polymers.
3 Definitions of Compatibilization and Polymer Alloys
As defined in the appendix “Dictionary of Terms Used in Polymer Science and Technology” in this handbook, compatibilization means “A process of modification of interfacial properties of an immiscible polymer blend, leading to creation of polymer alloy.” A polymer alloy in turn is defined as “An immiscible polymer blend having a modified interface and/or morphology,” whereas a polymer blend is simply “A mixture of at least two polymers or copolymers.” In other words, all polymer alloys are blends, but not all polymer blends are alloys. A somewhat more elaborate definition of a polymer alloy would describe a blend of at least two immiscible polymers stabilized either by covalent bond or ionic bond formation between phases or by attractive intermolecular interaction, e.g., dipole-dipole, ion-dipole, charge-transfer, hydrogen-bonding, van der Waals forces, etc. Only stabilization by covalent bond formation or ionic association (including acid-base and ion-neutral donor group interaction) is covered in this chapter.
Thermodynamic compatibility describes a miscible polymer blend that displays a single glass transition temperature, Tg, within the full range of composition of the two polymers. For the purposes of this chapter, we will be more concerned with Technological Compatibility. This term describes a polymer blend that does not separate into its individual components and does not lose useful technological properties over the expected lifetime of a molded part (which has been estimated by the Society of Plastics Engineers to be about 10 years) (Gaylord 1989; Rudin 1982). Taking a somewhat different view, Coran and Patel have defined compatibilization as a process for improving ultimate properties by making polymers in a blend less incompatible (Coran and Patel 1983b).
4 Types of Polymer Blends
The market for commercial polymer blends has grown steadily over the past four decades. A recent estimate of the polymer alloy/blend market by volume for 2012 was about 2.2 billion pounds. The market was projected to grow to about 2.6 billion pounds by 2018 (BCC Research 2013). The principal markets for all blends include the automotive industry; phone, computer, and other business machine housings; electrical components such as connectors; appliances; consumer products; recreational equipment; and construction and industrial applications.
Commercial activity is mirrored by technological activity. It was estimated that roughly 87,000 patents appeared worldwide on all aspects of polymer blends between 1970 and 1987 averaging almost 5,000 patents per year (Juliano 1988). The pace appears to have slowed little since then although the emphasis has changed from simple blends (e.g., binary blends with additives) to more complex compositions for specialty applications.
Common polymer blend building blocks arranged in a hierarchy of price and performance are shown in Fig. 19.2 of Chap. 19, “Commercial Polymer Blends” in this handbook. As the price gets higher, one is typically paying for higher heat stability and higher modulus. High performance thermoplastics such as PPS, PEI, and LCP and engineering thermoplastics such as PPE, PBT, and PC have high heat stability and are often designed to take the place of metals in typical applications. Lower modulus, commodity plastics such as PE, EPDM, and modified styrenics have lower heat stability and are often used in applications requiring high flexibility.
The goal of combining two or more polymers such as polymer pairs from those categories described above (e.g., an engineering thermoplastic plus a commodity polyolefin) is to achieve in the blend a combination of favorable properties from each polymer. Figure 5.1 shows idealized expected properties from blending two polymers that are either miscible (straight center line), immiscible and uncompatibilized (curved bottom line), or immiscible and compatibilized (curved top line). In the case of polymers that are miscible in all proportions, one can only hope to obtain in their blend an average of their physical properties depending upon the proportion of each polymer present. In a common example, the Tg of a miscible blend will vary linearly from that of the lower Tg polymer to that of the higher Tg polymer as the higher Tg polymer increases in proportion in the blend.

Potential effect on polymer blend properties as component concentration changes
When two immiscible polymers are blended without compatibilization, one generally obtains a mixture with physical properties worse than those of either individual polymer. Usually such a blend has poor structural integrity and poor heat stability since there is no mechanism for stabilizing a dispersion of one polymer in a matrix of the other. On a macroscopic scale, the blend may appear heterogeneous and in the extreme case grossly delaminated, e.g., in a molded part.
When two immiscible polymers are blended with compatibilization, one may expect a synergistic combination of properties derived from each polymer. A common example is a blend of a thermoplastic (to provide high heat stability) with an immiscible, rubbery impact modifier (to provide impact resistance), e.g., a rubber-toughened PA. A second common example is a blend of a semicrystalline thermoplastic with an amorphous thermoplastic. Because of their semicrystalline nature, polymers such as PA, PBT, PPS, or PP often have high chemical solvent resistance but low ductility, low dimensional stability, and low Tg. In contrast, amorphous polymers such as PPE, PEI, PC, and PE typically have poor solvent resistance and stress crack sensitivity but higher ductility, dimensional stability, and Tg. Often a binary blend includes a third, lower modulus polymer to provide optimum impact strength. A good example is a rubber-toughened blend of PPE with PA. In commercial examples, PPE and PA are combined in amounts such that PPE is the dispersed phase and PA is the continuous, matrix phase so that the blend possesses adequate solvent resistance (e.g., to common solvents used in automotive applications) but also higher heat stability compared to unmodified PA. Again, because of its inherent solvent resistance, this type of blend is prepared by melt processing and cannot economically be prepared by combining the components in solution.
Quite generally, the goal in preparing any polymer blend is to obtain one or all of the following benefits: higher heat distortion temperature (HDT), improved variable temperature impact resistance, solvent resistance, dimensional tolerance, higher flow, utilization of recycle/regrind, and lower cost.
5 Characteristics of Immiscible Polymer Blends
The general characteristics of immiscible polymer blends have been described in a large number of references (e.g., many of those listed in Sect. 5.6 on “General Stractegies for Compatibilization of Immiscible Polymer Blends”). Commercial polymer blends are most often prepared by some form of processing in a molten state, usually extrusion. Among the factors that determine which polymer will be the dispersed phase and the continuous matrix are the relative volume proportions and relative viscosities of the two polymers. During intensive mixing in a twin-screw or single-screw extruder, the less viscous molten polymer in a simple 1:1 mixture of two polymers will form an easily deformable matrix, while the more viscous polymer will form a difficultly deformable dispersed phase. Generally, the more viscous polymer will form the dispersed phase even in some cases when it represents more than 50 vol% of the blend.
Blend properties depend strongly on which polymer is the continuous phase. The majority of commercially important compatibilized blends of semicrystalline polymers with amorphous polymers are prepared in compositions such that the semicrystalline component is the matrix and the amorphous component is the dispersed phase. Such blends show adequate solvent resistance since in this morphology the surface consists largely of the dominant, matrix phase of semicrystalline polymer.
The formation of optimum dispersed phase particle size and the stabilization of the resulting blend morphology are critical if the blend is to have optimum properties and in particular good mechanical properties. Figure 5.2 shows a morphology generated by processing an uncompatibilized blend of PPE dispersed phase in a PBT matrix (Brown, unpublished results, 1988). Figure 5.2a shows that a reasonably uniform dispersion of PPE may be formed simply by suitable degree of mixing during extrusion. Figure 5.2b, however, demonstrates phase coalescence of PPE particles to form large, irregularly shaped islands when the extrudate of the uncompatibilized blend is molded at normal processing temperature. In this blend there is no interfacial adhesion between the two phases and, hence, no mechanism for morphology stabilization. Even in the presence of an impact modifier, the resulting molded parts are quite brittle since there are no uniform dispersed phase particles of proper size to dissipate impact energy.

Morphology of PPE dispersed phase in a PBT matrix (a) as extruded and (b) after molding
Table 5.1 shows further examples of dispersed phase coalescence in blends of PA as the dispersed phase in a less viscous PE or PS matrix. The data show that the mean PA particle size increases dramatically with simple heating under static conditions in the absence of any mechanism for morphology stabilization. The same coalescence can occur in molded parts of uncompatibilized polymer blends subjected to further thermal treatment after molding (e.g., in a paint drying oven). The mechanical properties of these blends are quite poor.
In summary, a frequent goal in making a technologically compatible blend of immiscible polymers is to stabilize an appropriate morphology of the dispersed phase polymer in the matrix polymer by promotion of interfacial adhesion and/or by lowering the interfacial tension. These conditions are critical for providing good mechanical properties, toughness, molded part dimensional integrity, and maximum solvent resistance in the blend.
6 General Strategies for Compatibilization of Immiscible Polymer Blends
Numerous reviews have been published which discuss general (or specific) aspects of strategies for compatibilization of immiscible polymer blends, including but not limited to those by Fink (2013), Imre and Pukanszky (2013) (bio-based and biodegradable polymer blends including reactive compatibilization), Karaağaç and Deniz (2013) (rubber based blends), Covas et al. (2011), Jiang et al. (2010) (reactive compatibilization), Nwabunma and Kyu (2008) (polyolefin blends), Robeson (2007), Yu et al. (2006) (polymer blends from renewable resources), Feldman (2005), Macosko et al. (2005) (reactive compatibilization), Mangaraj (2005) (recycling ground rubber waste), Harrats and Groeninckx (2004) (reactive compatibilization) Paul (2004) (reactive compatibilization), Platé et al. (2004) (theoretical considerations), Horák et al. (2002), Litmanovich et al. (2002) (theoretical considerations), Baker et al. (2001), Prut and Zelenetskii (2001), Bussink and van de Grampel (2000), Paul and Bucknall (2000), Fakirov (1999) (transreactions in condensation polymers), Shonaike and Simon (1999), Xanthos (1999) (polypropylene), Koning et al. (1998), Robeson et al. (1998) (PVAl-PO blends), Tran-Cong (1998), Al-Malaika (1997), Baranov (1997), Gao et al. (1997) (ionomer blends), Lohse et al. (1997), Datta and Lohse (1996), Utracki and Dumoulin (1995) (polypropylene), Folkes and Hope (1993), Brown (1992a) (reactive compatibilization). Liu and Baker (1992a) (reactive compatibilization), Elmendorf and Van der Vegt (1991), Xanthos and Dagli (1991) (reactive compatibilization), Menges (1989), Utracki (1989, 1998), Brown and Orlando (1988) (reactive compatibilization), Paul et al. (1988), Teyssie et al. (1988), Sperling (1987), Fox and Allen (1985), Solc (1981), Rudin (1980), Paul and Newman (1978), and Bucknall (1977). The basic strategies for compatibilization of two-phase polymer blends can be divided into at least four major categories.
6.1 Co-crystallization of Two Phases
This particular strategy is limited to those cases in which an immiscible polymer blend contains two semicrystalline polymers that can co-crystallize. Nadkarni and Jog (1989, 1991) have reviewed examples of this type of compatibilized blend. Co-crystallization may also occur as a secondary process in an intimately mixed blend containing a copolymer resulting in concomitant effects on blend properties as shown in a few of the examples of this review.
6.2 In Situ Immobilization of One Phase: Dynamic Vulcanization
In these examples a dispersed phase of a cross-linkable rubber is vulcanized in the presence of a matrix of a second, immiscible, non-vulcanizable polymer during the residence time of melt processing. Examples have also been reported in which a mixture of two vulcanizable polymers has been employed. Coran (1995) has summarized five key requirements for preparing optimum compositions by dynamic vulcanization:
-
1.
Good match between surface energies of the dispersed phase and the matrix
-
2.
Low entanglement molecular length (high entanglement density) of the rubber
-
3.
Crystalline plastic matrix
-
4.
Stable rubber and plastic at blend processing temperatures
-
5.
Availability of appropriate curing system for rubber under desired processing conditions
Coran and others have reviewed work in this area (Coran and Patel 1995, 1996, 2004; Karger-Kocsis 1999; Abdou-Sabet et al. 1996; Coran 1987, 1990, 1995).
Most examples of dynamically vulcanized blends do not involve covalent bond formation between the immiscible phases. However, other work has shown that covalent bond formation between phases in conjunction with dynamic vulcanization of one phase can lead to blends with improved properties in certain cases. This is particularly true in blends where certain of Coran’s five key requirements above are not met. Some examples of dynamically vulcanized blends that also feature copolymer formation between the two immiscible phases are summarized in this review under the appropriate categories.
6.3 Inclusion of a Third Material as a Compatibilizing Agent
6.3.1 Addition of a Separate Compatibilizing Agent
A separate compatibilizing agent included in a blend may be a third material not derived from either of the two immiscible polymers. Representative examples include certain plasticizers, random copolymers, and block copolymers, which may lower the interfacial tension between the two immiscible polymer components. Examples also exist where the separate compatibilizing agent is a chemically unreactive analog of one (or both) of the two immiscible polymers that has an attractive interaction with each polymer. In any case this is often a semiempirical compatibilization strategy since precedent may be the only basis for choosing an effective compatibilizer. Reviews on addition of this type of compatibilizing agent as a separate component to an immiscible polymer blend have appeared in addition to Chap. 4, “Interphase and Compatibilization by Addition of a Compatibilizer,” in this handbook. They include but are not limited to those listed above in Sect. 5.6 and those by Gaylord (1989), Xanthos (1988), and Paul (1978).
6.3.2 Inclusion of a Copolymer of the Two Immiscible Polymers
As stated earlier, a copolymer of the two immiscible polymers themselves would seem to be ideally suited to act as a compatibilizing agent for an immiscible blend. If the copolymer is at the interface of the two phases, then the segments of the copolymer dissolve in the respective bulk phases of the same identity. The copolymer acts as emulsifying agent for the blend resulting in reduced interfacial energy and improved interfacial adhesion.
Table 5.2 shows dramatic examples of the stabilization of dispersed phase morphology in the presence of a compatibilizing copolymer, in these cases formed through reaction of PA amine end-groups with anhydride-functionalized matrix polymer. In all examples, essentially no change in dispersed phase particle size occurs after annealing under static conditions for up to 90 min. The data shown in Table 5.2 should be compared with those presented in Table 5.1, where the dispersed phase mean dimensions were presented for similar, uncompatibilized blends.
There are two basic options for inclusion of a copolymer compatibilizer in a blend of immiscible polymers. First, the copolymer can be synthesized in a separate step followed by addition to the blend. One disadvantage is that this requires a new product synthesis with expensive and time-consuming process development, and, hence, a significant number of years before profitability, since scale of copolymer manufacture will be initially low.
A second and more important disadvantage is that adding the copolymer as a separate species to the blend requires that the copolymer diffuse to the phase interface of the immiscible polymers to be effective as a compatibilizer. Diffusion to the interface may not be efficient within the residence time of a typical extrusion blending process (usually 2–5 min). In addition, high concentrations of added copolymer may form micelles as a third, distinct phase that does not contribute to compatibilization (see, e.g., Jeon et al. 2005).
A third, potential disadvantage is that for optimum interfacial interaction, a copolymer synthesized in a separate reaction step must have carefully controlled segment lengths to best match the molecular weight of the bulk phase in which the segment must dissolve (see, e.g., Cercle and Favis 2012; see also Gani et al. 2010, for an approach to solving this dilemma). It is often desired to offer for different applications a series of commercial blends containing the same two polymers but with different molecular weights for the polymers in each blend. Any copolymer synthesis process would then have to be capable of producing a series of copolymers with a variety of controlled segment molecular weights for optimum compatibilization efficiency, an economically difficult task.
Commonly, the most economical and efficient process for including a copolymer in a blend of immiscible polymers is to form the copolymer in situ by a chemical reaction during the extrusion process during establishment of the immiscible phase morphology – the process known as reactive compatibilization. In summary the advantages of such a process include:
-
The copolymer is made only as needed and a separate copolymer commercialization process need not be developed.
-
The copolymer is formed directly at the phase interface where it can serve as a compatibilizer, and no diffusion process dependent on extruder residence time is involved.
-
The copolymer, except when formed in a degradative process, typically has segment molecular weights similar to the molecular weights of the bulk phases in which the segments must dissolve, which should promote optimum interaction between copolymer and bulk phases. This also facilitates commercialization of a series of blends containing polymers with quite different molecular weight since the copolymer formed in situ will usually have ideal segment molecular weights.
-
A disadvantage of forming copolymer in situ is that such a process often requires that each of the immiscible polymers bear an appropriate chemical functionality for reaction across a melt-phase boundary.
As practiced commercially, reactive compatibilization is a continuous extruder process with material residence time usually 1–5 min. Such a process permits large-scale preparation of a polymer blend as needed (“Just-In-Time” inventory control). Because reactive compatibilization involves a heterogeneous reaction across a phase boundary, the reaction is limited by the interfacial volume available at this phase boundary. Most often, twin-screw extruders (having screw diameter from about 20 to >120 mm) are employed. The screws are designed using an appropriate sequence of screw elements and auxiliary conditions (e.g., subsequent vacuum venting of volatiles) to promote generation of a large interfacial area for the desired chemical reaction to form copolymer.
7 Generic Processes and Specific Types of Reactions to Form Copolymer in a Reactive Compatibilization Process
When it is desired to form in situ a compatibilizing graft, block, or cross-linked copolymer, there are at least two distinct generic processes available for copolymer formation:
-
1.
Direct Reaction, wherein reactive functionalities on each of the two immiscible polymers react with each other across the melt-phase boundary.
-
2.
Addition of a Third, Reactive Species to effect or promote copolymer formation. This situation is a typical “three-body” reactive extrusion problem requiring that three chemical species (at least two of which are immiscible) react within the short residence time of extrusion processing. In some cases, the third, reactive species is simply a catalyst that activates functionality on one polymer for reaction with functionality on the second polymer. Such cases produce results similar to the direct reaction process. Frequently, however, the added reactive species is a coupling agent capable of reacting with each of the polymers individually (as opposed to reacting with and activating only one of the two polymers). If the coupling agent is preferentially soluble in one of the two phases (e.g., a polar coupling agent in the more polar polymer phase), it may give predominantly homogeneous reaction instead of promoting heterogeneous reaction (copolymer formation) across the melt-phase boundary. Hu et al. (1997) have studied one type of polymer melt reaction (carboxylic acid + epoxide) in which the kinetic efficiency depends upon the partition coefficient of reactant between two immiscible polymer phases. Such considerations must be applied to all reactive compatibilization processes involving three or more reactive species. Coupling agents are further discussed herein below.
As subcategories of the two generic processes, there are at least five specific processes for achieving interchain copolymer formation between two polymers during reactive compatibilization in an extruder. The following sections and their accompanying tables show these five processes starting with two idealized homopolymers, one derived from monomer “A” with structure AAAAAAAA and the other derived from monomer “B” with structure BBBBBBBB. Each process produces a specific type of copolymer compatibilizing agent by particular types of chemical reactions.
7.1 Compatibilization by a Redistribution Reaction to Produce Block and Random Copolymer: Reaction Type #1
As summarized in Table 5.3, redistribution reactions (often referred to as “transreactions”) occur by chemical interchange of block segments of one polymer chain for corresponding segments of a second polymer chain. Such reactions may be homogeneous (self-reaction) or heterogeneous. In the homogeneous case, the molecular weight distribution of a polymer may reach equilibrium. In the heterogeneous case, redistribution reactions can form a copolymer between two different polymers. This type of reaction is typically dependent upon reaction time and temperature and is not often used to form polymer alloys in a reactive compatibilization process because the time for forming a stable compatibilizing copolymer may be longer than a typical extrusion process time.
Redistribution reactions can occur by several different mechanisms. In one common example, nucleophilic end-groups of one polymer react with electrophilic linkages in the main chain of a second polymer resulting in chain cleavage (e.g., acidolysis or alcoholysis). The initial product is a block copolymer of the two polymers along with a lower molecular weight fragment of the second polymer. Since the initial block copolymer can participate in further redistribution reactions, the net product after sufficient time may be a random copolymer. The propensity to form random copolymer is further increased if both polymers have nucleophilic end-groups and also electrophilic linkages in the main chain that can participate in redistribution. Redistribution during thermal processing is also a common self-reaction in condensation polymers such as PA, PEST, and PC that often contain nucleophilic amine, hydroxy, or phenolic end-groups, along with electrophilic groups such as amide, ester, or carbonate linking the individual monomer units.
In common examples, essentially all of the polymer chains in each of the immiscible polymers are capable of participating in the copolymer-forming reaction by redistribution. This is in contrast to many other processes for in situ copolymer formation where only those few chains bearing reactive functionality participate. Unless the redistribution process is carefully controlled, it is difficult to stop the process to make stable, compatibilized polymer blends. If the reaction is thermally initiated, the blend processing temperatures and residence times must be strictly and reproducibly controlled within narrow limits to achieve reproducible properties. For prolonged reaction times at a temperature above that necessary to initiate the reaction, one may obtain a broad distribution of block lengths and eventually random copolymer. The random copolymer may not be as efficient a compatibilizer for the immiscible polymer blend as the block copolymer initially formed. More importantly, a high degree of random copolymer formation may destroy desirable properties in the polymer blend such as crystallinity in one of the polymers and, hence, solvent resistance in the final blend. In the extreme case, phase separation is lost and the mixture may become homogeneous and transparent. The problem of controlling the redistribution process does not necessarily stop at the manufacturing stage. After a compatibilized polymer blend leaves the manufacturer, it typically undergoes further thermal histories such as molding or paint-oven drying at the processing facilities of the final user. Continued redistribution reaction in the hands of a final user may cause deterioration and non-reproducibility in blend properties.
When applicable, a common method for controlling a redistribution process is to initiate the reaction with a catalyst. Control may then be achieved by quenching the catalyst at the desired extent of reaction. Certain types of redistribution catalyst may thermally decompose under controlled processing conditions that make quenching unnecessary. In these cases, a predominance of block copolymer may be formed that serves as an effective compatibilizer for an immiscible polymer blend. Just as importantly, only a relatively small fraction of the polymer chains may actually participate in the redistribution process so that phase separation and the properties attributable to the original sequence distribution may be maintained.
The redistribution reaction is a degradative process for making a compatibilizing copolymer. A common feature of all redistribution reactions to form copolymer between two different polymers is that the molecular weight of at least one segment of the initially formed block copolymer is less than that of the bulk polymer phase from which it is derived. Therefore, even when the redistribution process is carefully controlled to give predominantly block copolymer, the copolymer may not be as efficient a compatibilizer as a similar type of block copolymer formed by an end-group/end-group reaction (see herein below). With a low molecular weight block segment, one may have poor penetration into the corresponding bulk polymer phase and less than optimum interfacial adhesion between the immiscible phases with copolymer at the interface. As discussed before, an optimum interfacial adhesion is usually obtained when the segmental molecular weights of the block copolymer are similar to the molecular weights of the individual bulk polymer phases. A general review of “Interchange Reactions Involving Condensation Polymers” describes early work on redistribution reactions in the melt blends of polyesters, polyamides, and polyester + polyamide (Kotliar 1981).
7.2 Compatibilization by Graft Copolymer Formation: Reaction Type #2
Graft copolymer formation has been the most common method of forming a compatibilizing copolymer between two immiscible polymers during reactive compatibilization. As shown in Table 5.4, there are at least four processes for forming graft copolymer in a melt reaction. In the direct reaction process, the reaction occurs between one polymer containing reactive sites along its main chain and a second polymer with reactive sites only at end-groups. Depending upon stoichiometry and concentration of functional groups, copolymer structures of the general type 2a and 2b are obtained. In this particular type of graft copolymer formation, the average molecular weight of the copolymer is the simple sum of the average molecular weights of the two reacting species.
Graft copolymers may also be formed through reaction of a bi- or multifunctional coupling agent with one polymer containing reactive sites along its main chain and a second polymer with reactive sites only at end-groups (Type 2c). Typical coupling agents include multifunctional epoxy resins, oxazolines, carbodiimides, and isocyanates that react with nucleophilic end-groups of condensation polymers. The coupling agent is incorporated into the copolymer. When the coupling agent is an epoxide, a new secondary alcohol is formed when the epoxide ring is opened by a nucleophile. This alcohol may also be reactive to one or more polymeric components (particularly polyesters) similar to the reactivity of alcohol groups on phenoxy resin (see, e.g., Su et al. 1997).
A third, less common process for forming graft copolymer as shown in Table 5.4 is Type 2d. In this process, multiple reactive sites pendent on one polymer chain can bite into the linkages of a second type of polymer chain. The reaction creates a copolymer having segments with average molecular weight less than the sum of the two initial reacting species. This is a degradative method for forming a compatibilizing copolymer. A common example is the transesterification reaction between the poly(hydroxy ether) of bisphenol A (a phenoxy resin) and a polyester. Pendent hydroxy groups on phenoxy resin can undergo transesterification with ester linkages in the polyester chains resulting in graft copolymer formation accompanied by lower molecular weight polyester fragments. Because the molecular weight of the grafted polyester species is less than that of the homopolymer from which it was derived, the grafted chain segments may be below optimum molecular weight necessary for most efficient chain entanglement with the remaining homopolymer phase. This may result in less than optimum physical properties. Furthermore, if there is a large number of pendent reactive sites, then degradation of the second polymer may reach the point where it has lost the physical properties that made it useful for blending. Consequently, formation of a graft copolymer compatibilizing agent by a degradative process is not a common method for immiscible blend compatibilization.
Many commercial thermoplastics for high-impact strength applications are two-phase blends in which a higher modulus thermoplastic matrix is toughened by the presence of a lower modulus, dispersed phase polyolefin. In the majority of cases, such blends are compatibilized by graft copolymer formation between at least some fraction of the chains of the two immiscible polymers. Rubber-toughened PAs are the most common examples. In most of these cases, PA amine end-groups react with pendent anhydride or epoxy groups along the main chain of an immiscible rubbery polyolefin to form sufficient copolymer to compatibilize a dispersed polyolefin phase in a matrix phase of PA. Rubber-toughened PEST are also often compatibilized through graft copolymer formation formed through reaction between polyester acid end-groups and epoxy-functionalized polyolefins.
In rubber-toughened thermoplastic blends, the efficiency of compatibilization depends among other things upon sufficient concentrations of both thermoplastic reactive end-groups and polyolefin reactive pendent groups to give adequate levels of copolymer under the mixing, temperature, and residence time protocol of the extrusion process. Concentrations of thermoplastic reactive end-groups are usually controlled during the manufacturing process, e.g., through control of stoichiometry in condensation polymerization and/or through addition of reactive or unfunctionalized chain-capping agents either during synthesis or in a subsequent processing step.
Functionality in rubbery polyolefins is usually introduced in one of two ways:
-
1.
Copolymerization of olefin monomer(s) with another functionalized monomer (e.g., poly(ethylene-co-acrylic acid), poly(ethylene-co-glycidyl methacrylate), etc.)
-
2.
Graft functionalization of polyolefin in a separate processing step (e.g., polyethylene-g-maleic anhydride, polypropylene-g-maleic anhydride, etc.)
Graft functionalization may be performed either by reactive extrusion in the molten state (see, e.g., Brown 1992a), in solution, or by solid-state processes. In these cases, concentration of functionality is controlled by temperature, physical phase of the polymer substrate, stoichiometry of functionalization agent, and (optional) catalyst among other factors.
When graft functionalization is performed by extrusion, the removal of unbound functionalization agent from functionalized PO is critical for success of subsequent copolymer formation with reactive thermoplastic end-groups. Unbound functionality (e.g., free maleic anhydride) in the PO phase may tie up reactive end-groups of the thermoplastic resin during subsequent reactive compatibilization processing, making these end-groups unavailable for copolymer formation. For functionalization during a separate extruder grafting reaction, unbound functionalization agent is removed by efficient devolatilization, and for solution functionalization, by solubilization of excess functionalization agent during isolation of solid, functionalized polymer.
7.3 Compatibilization by Block Copolymer Formation: Reaction Type #3
Compatibilized polymer blends have been prepared through block copolymer formation between immiscible polymers. In the direct reaction process, during melt processing, the functionalized end-groups on some fraction of chains in each of the polymers react across a melt-phase boundary to form block copolymers. Depending upon stoichiometry, either A-B or A-B-A or both copolymer structures may be obtained as shown in Table 5.5 (Type 3a or 3b). The average molecular weight of the copolymer corresponds to the sum of the average molecular weights of the reacting polymers.
Block copolymers may also be formed through reaction of the end-group on one polymer with a condensing agent which activates that end-group for reaction with a nucleophilic end-group on a second immiscible polymer. Typical condensing agents include phosphite esters that react with acid and hydroxy end-groups on condensation polymers. A by-product from the condensing agent is always formed in the copolymer reaction and is often removed by devolatilization of the blend melt. Since the condensing agent is not incorporated into the copolymer, the process is similar to that shown in Table 5.5, Type 3a.
Block copolymers may also be formed through reaction of end-groups on each of the immiscible polymers with a coupling agent. Typical coupling agents are the same as for graft copolymer formation and include multifunctional epoxy resins, oxazolines, carbodiimides, and isocyanates that react with nucleophilic end-groups of condensation polymers. The coupling agent is incorporated into the copolymer. The process is shown in Table 5.5 (Type 3c). When the coupling agent is an epoxide, a new secondary alcohol is formed when the epoxide ring is opened by a nucleophile. This alcohol may also be reactive to one or more polymeric components (particularly polyesters) similar to the reactivity of alcohol groups on phenoxy resin.
Block copolymers may also be formed by a degradative process in which end-groups on one polymer undergo transreaction with linkages in the main chain of a second, immiscible polymer. A low molecular weight fragment of the second polymer is formed as by-product. The block copolymer has lower average molecular weight than the sum of the average molecular weights of the reactants. The process shown in Table 5.5 (Type 3d) is essentially the same as Reaction Type 1a in Table 5.3 terminating at the block copolymer. A typical copolymer architecture in this process is an A-B block. An A-B-A block can form if the degradable segment is further degraded through transreaction with another end-group-functionalized polymer.
7.4 Compatibilization by Covalently Cross-Linked Copolymer Formation: Reaction Type #4
As shown in Table 5.6, compatibilizing copolymers may be formed in situ by a covalent cross-linking process.
The cross-linking reactions have been performed by at least five processes. In Reaction Type 4a, direct cross-linking occurs by covalent bond formation between functionalities on each of the two immiscible polymers without degradation of either polymer. The cross-linking is most often performed by reaction of pendent, nucleophilic sites of one multifunctional polymer with pendent, electrophilic sites of the second multifunctional polymer. Common examples include reactions of pendent acid or amine nucleophiles on one functionalized polymer with pendent electrophilic groups such as epoxide, oxazoline, or ortho ester on a second functionalized polymer.
Covalent cross-linking reactions mediated by a third, added reagent may give the same type of copolymer structure as that which results from direct cross-linking reactions (Table 5.6, Type 4b and 4c). In this case, the added reagent may be a radical initiator or other type of activating agent such as a condensing agent. Such activating agents are not incorporated into the final copolymer. Radical initiators may promote radical formation on each of two immiscible polymers. A cross-linked copolymer results through radical-radical coupling between the two polymers at a melt-phase interface. Self-coupling of each polymer may compete with cross-coupling as is the case with most three-body reactions in which an added reagent is capable of reacting with each of the two immiscible polymers.
Covalent cross-linking by covalent bond formation arising from mechanochemical radical generation and recombination in the absence of an added radical initiator may also be performed (Table 5.6, Type 4d). The method is less frequently used than other cross-linking reaction types. When two immiscible polymers can both form radicals on their main chains in the absence of added radical initiator, then a copolymer results when radical sites on the two different polymers recombine at the phase interface. If radical formation occurs without chain degradation, then the copolymer becomes cross-linked as multiple sites on each chain participate in the reaction. Alternatively, one radical-forming polymer can form a cross-linked copolymer with a second polymer containing a radical trap such as an unsaturated site (e.g., EPDM). This cross-linking process may be difficult to control since it only stops when thermal and/or shear conditions are below some threshold level. Casale and Porter (1975, 1978) and La Mantia and Valenza (1994) have briefly reviewed mechanochemical radical generation and its use to form copolymers in immiscible blends. Ahn et al. (1995) have described radical generation and copolymer formation in immiscible polymer blends subjected to elastic strain pulverization in specially modified extruders building on earlier Russian work. Pulverization occurs in an extruder section kept below the polymer melting points. The compatibilizing copolymer formed in these cases may be a block, graft, or cross-linked copolymer depending upon the polymers involved. See also the more recent review by Beyer and Clausen-Schaumann (2005) concerning mechanochemical radical generation in polymers.
Covalent cross-linking reactions to form a compatibilizing copolymer may also be performed by addition of a coupling agent. Coupling agents react with the same type of functionality in each of the immiscible polymers and remain bound in the cross-linked product as linking agents. Examples include diepoxide reaction with pendent carboxylic acid groups on each of two immiscible polymers. In this case, the structure of the cross-linked copolymer is shown in Table 5.6 (Type 4e). Most commonly, coupling agents are multifunctional reagents with molecular weights less than about 1,000. When immiscible polymer pairs are employed, each bearing multiple pendent nucleophilic groups (such as carboxylic acids), then low molecular weight coupling agents such as bis-, tris, tetra-, and higher epoxides; bis-oxazolines; and other multifunctional electrophilic species are used. A low molecular weight coupling agent bearing multiple olefinic sites may be used to promote cross-linking in blends containing POs. Common examples include commercially available tris-acrylates and triallyl isocyanurate. Often these cross-linking reactions are performed in the presence of radical initiator. Self-coupling of each polymer may compete with cross-coupling as is the case with most three-body reactions in which an added reagent is capable of reacting with each of the two immiscible polymers.
7.5 Compatibilization by Ionic Interaction to Form Copolymer: Reaction Type #5
Immiscible polymer blends have been compatibilized through formation of a compatibilizing copolymer linked by ionic association instead of by covalent bonding. Although many examples have been published, most of these involve solution mixing of the two immiscible polymers (see Natansohn et al. 1990). Most examples given in this chapter describe only such polymer blends prepared by melt mixing.
In theory, the possible architectures of a compatibilizing copolymer arising from ionic association may be the same as all those architectures arising from covalent bond formation that were previously discussed. However, in practice only a small number of copolymer architectures have been reported for compatibilizing agents arising from ionic association. In the most common examples (Table 5.7; Type #5a), ionizable groups such as carboxylic, sulfonic, or phosphonic acid are present in low concentrations (e.g., about 5 % or less) on both polymers. The ionizable groups may be at least partially neutralized by a mono-, di-, or trivalent metal cation, such as Na+1, Zn+2, or Al+3. Multivalent cations may form a bridging linkage between the ionizable groups of the two immiscible polymers resulting in interchain copolymer formation by ion-ion association. Monovalent cations such as Na+1 or K+1 may also be used to promote association through ion-dipole association. With either type of cation, a morphology is formed in which there are concentrated domains of associated ionic species (ion clusters) in a matrix of the immiscible homopolymers.
In the first type of ionic association (Type #5a), the ionizable functionalities of the two polymers are located in the pendent side groups. These polymers are prepared either through copolymerization with ion-containing monomers (or latent ion-containing monomers) or through subsequent grafting with such monomers. Consequently, the resulting compatibilizing agents are most often cross-linked copolymers with the structure shown in Table 5.7 as Type 5a.
In a second type of ionic association (Type #5b) metal cations may mediate association between ionic groups on one polymer and neutral donor groups on a second immiscible polymer. Typical ionic groups are again carboxylic, sulfonic, and phosphonic acids. Neutral donor groups contain atoms, usually nitrogen or phosphorus, having unshared pairs of electrons capable of coordinating to metal cations. Such groups include pyridine, quinoline, and phenanthroline. These are usually introduced into a polymer via copolymerization with the vinyl analog, e.g., vinylpyridine. Again, the structure of the compatibilizing copolymer is usually cross-linked.
In a third type of ionic association (Type #5c), acidic groups such as carboxylic acids bound to one polymer may mediate interchain copolymer formation by protonation of basic groups on a second, immiscible polymer. In this case, the compatibilizing agent is again most often a cross-linked copolymer.
General discussions of the properties of polymers containing ionizable groups (ionomers) have been published (see, e.g., Kim et al. 2002; Hara and Sauer 1994; Lundberg 1987; Rees 1986). Ionic cross-links are usually thermally reversible, which may limit the usefulness of blends containing them in certain commercial applications. Since ionomers are initially self-associated through ionic bonds, thermal reversibility of ionic cross-links in the melt is necessary to overcome homogeneous, self-cross-linking within each homopolymer before heterogeneous, interchain cross-linking can occur. Often a high degree of plasticization of the ion-containing polymer melt is required so that high processing temperatures that might lead to polymer decomposition need not be used. In some cases, polymers containing masked ionomeric functionality, i.e., chemical groups that form ionic species during extrusion, have been used to form copolymers during reactive processing. Use of masked ionomers may require lower energy during extrusion since ionic self-association does not have to be overcome before interchain copolymer formation can occur.
8 Polyamide Blends
Examples of polyamide blends are listed in alphabetical order of the second polymer in the blend unless otherwise noted. When copolymer characterization was not performed, the structure of the compatibilizing copolymer is inferred from the functionality location on each of the two polymers. In some cases, more than one type of compatibilizing copolymer may have formed.
Many of the copolymer-forming reactions employed to compatibilize PA blends with a second immiscible polymer have been studied by Orr et al. (2001) who determined that the order of increasing reactivity in functionalized polymer pairs is acid/amine, hydroxyl/(anhydride or acid), aromatic amine/epoxy, aliphatic amine/epoxy, acid/oxazoline, acid/epoxy, aromatic amine/anhydride, and aliphatic amine/anhydride (most reactive).
8.1 Polyamide + Polyamide Blends
8.1.1 Copolymer Formation by Redistribution Reaction
Examples of copolymer formation by redistribution reactions (sometimes referred to as transreactions) in PA/PA blends are given in Table 5.8. In related work, Liu and Donovan (1995) failed to find evidence for transamidation in PA-6 blends with an aromatic polyamide during molding and annealing. Aspects of transreactions in PA/PA blends have been described in Eersels et al. (1999) and in portions of other chapters included in Fakirov (1999) (transreactions in condensation polymers).
8.1.2 Copolymer Formation by Amine + Carboxylic Acid Reaction: Blends Containing a Condensing Agent
Aharoni et al. (1984) and Aharoni (1983) have shown that blends of immiscible polyamides may be compatibilized through copolymer formation mediated by addition of a phosphite condensing agent. A block copolymer results when the phosphite-activated end-group of one PA reacts at the phase interface with a nucleophilic end-group on the second PA. The reaction also produces a secondary phosphite by-product. The relative proportions of copolymer vs. simple chain-extended PA may depend upon the relative solubility of the condensing agent in each of the immiscible polymer phases. For example, blends of 95-50 parts PA-6 were extruded using an SSE at 265–315 °C with 5-50 parts PA-11 (or PA-12 or PA-66 or PA-6T) in the presence of 0-1 part triphenyl phosphite or other trialkyl phosphite. Copolymer-containing blends were characterized by selective solvent extraction, FTIR,13C NMR, and31P NMR. Model compound studies were done to understand the mechanism of copolymer formation.
8.1.3 Copolymer Formation by Amine + Anhydride Reaction: Blends Containing a Coupling Agent
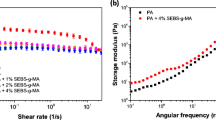
Xie and Yang (2004) have prepared blends of PA-6 (70 parts) and PA-12,12 (30 parts) through addition of SEBS-g-MA (15 wt%) which may serve as a coupling agent between the two PA. Blend characterization included SEM.
8.2 Polyamide + Polyester (or Polycarbonate) Blends
8.2.1 Copolymer Formation by Amine + Anhydride Reaction
Qu et al. (2008) reported blends of PA-6 with PET compatibilized through addition of ethylene-acrylate-maleic anhydride terpolymer.
John and Bhattacharya (2000) reported that PBT may be modified by extrusion with MA. The modified PBT forms compatibilized blends with PA-66 characterized by FTIR,13C NMR, SEM, and mechanical properties.
PA/PC ternary blends have also been compatibilized through copolymer formation between PA amine end-groups and anhydride-functionalized styrene copolymer. Kim et al. (1999a) employed SAN-co-MA in PA-6,12/PC blends. Lee et al. (1999b) and Horiuchi et al. (1996, 1997a, b, c) employed SEBS-g-MA with PA-6/PC blends.
PA/PC blends have also been compatibilized by block copolymer formation through reaction of PA amine end-groups with PC anhydride end-groups (Hathaway and Pyles 1988, 1989). PC phenolic end-groups were anhydride-functionalized by reaction with trimellitic anhydride acid chloride. Extruded blends of PA-6 and PC were characterized by selective solvent extraction and mechanical properties of test parts. An amorphous polyamide could also be compatibilized with PC using this strategy.
8.2.2 Copolymer Formation by Amine + Carboxylic Acid Reaction: Blends Containing a Condensing Agent
PA/PEST blends have been compatibilized through block copolymer formation mediated by addition of a phosphite condensing agent (Aharoni et al. 1984; Aharoni 1983; Aharoni and Largman 1983). Block copolymer results when the phosphite-activated end-group of a PEST (or PA) reacts with a nucleophilic end-group on a PA (or PEST) at the phase interface with generation of secondary phosphite by-product. The relative proportions of copolymer vs. simple chain-extended PA or PEST may depend upon the relative solubility of condensing agent in each of the immiscible polymer phases. For example, blends of 95-5 parts PA-6 (or PA-66 or PA-12) were extruded using an SSE at 265–315 °C with 5-95 parts PET (or PBT or PCT) in the presence of 0-1 part triphenyl phosphite or trialkyl phosphite. Copolymer-containing blends were characterized by morphology, viscosity, selective solvent extraction, FTIR,13C NMR, and31P NMR. Model compound studies were done to understand the mechanism of copolymer formation. Various other PEST and PA resins were also used.
8.2.3 Copolymer Formation by Carboxylic Acid + Epoxide Reaction: Blends Containing a Coupling Agent
As shown in Table 5.9, PA/PEST blends can be compatibilized through block copolymer formation mediated by addition of a multifunctional epoxide coupling agent. The coupling agent may react with nucleophilic end-groups on each of the two immiscible polymers at the phase interface to give a block copolymer containing the coupling agent as linking group. The relative proportions of copolymer vs. simple chain-extended PA or PEST may depend upon the relative solubility of coupling agent in each of the immiscible polymer phases. See, e.g., Jeziórska (2005) wherein a bis-oxazoline coupling agent was used, but the predominant reaction was PEST chain extension.
8.2.4 Copolymer Formation by Degradative Process
Examples of compatibilizing copolymer formation in PA/PEST blends by reaction of PA end-groups with PEST main-chain units by a degradative process (including PA/PC blends) are given in Table 5.10. The degradative process in this instance may also be considered a transreaction. Reviews of transreactions in PA/PEST blends include those described in chapters of Fakirov (1999) (transreactions in condensation polymers).
8.3 Polyamide + Polyesteramide LCP + Polyolefin Blends
8.3.1 Copolymer Formation by Amine + Anhydride Reaction
Seo (1997) prepared compatibilized PA blends with LCP polyesteramide (Hoechst Vectra® B950) in the presence of anhydride-functionalized polyolefin. Specifically, 60 parts PA-6 was mixed with 25 parts LCP and 15 parts EPDM-g-MA in a TSE at 290 °C. The blend was characterized by SEM, optical microscopy, Raman spectroscopy, mechanical properties, selective solvent extraction, and FTIR.
8.4 Polyamide + Polyester LCP + Polypropylene Blends
8.4.1 Copolymer Formation by Amine + Anhydride Reaction
Tjong and Meng (1997) have described PA/LCP polyester blends with improved properties through addition of anhydride-terminated PP-MA. A block copolymer may form between PA amine end-groups and anhydride-terminated PP. For example, 86 parts PA-6 was mixed in an internal mixer with 14 parts PP-MA at 220 °C followed by injection molding with 5-40 parts LCP (Hoechst Vectra® A950). The blends were characterized by torque rheometry, mechanical properties, DMA, and SEM.
8.5 Polyamide + Polyethersulfone Blends
Kanomata et al. (2011) prepared compatibilized blends of PA-6 and polyethersulfone using PES having hydroxyphenyl end-groups. Blends prepared in a Brabender mixer were characterized using TEM and torque rheometry in comparison with control blends.
Weber and Güntherberg (1999) have prepared compatibilized blends of PA and polyethersulfone in the presence of S-MA-(N-phenylmaleimide) terpolymer. In the examples, the PA was derived either from hexamethylenediamine-isophthalic acid or from hexamethylenediamine-caprolactam-terephthalic acid. Blends comprising PA, an amine-terminated polyethersulfone, and S-MA-(N-phenylmaleimide) terpolymer prepared in a Haake mixer were characterized using mechanical properties testing, selective solvent extraction, DSC, and Vicat B test. Blends optionally contained phenoxy resin.
8.6 Polyamide + Polyolefin Blends (Excepting Polypropylene)
Blends in these sections include those with either a single PA or a mixture of semicrystalline PAs or a mixture of amorphous and semicrystalline PAs.
8.6.1 Copolymer Formation by Amide-Ester Exchange
Graft copolymer compatibilizing agents have been prepared by direct reaction (Table 5.11) through amide-ester exchange reaction between polyamide amine end-groups and pendent ester groups on polyolefin copolymers such as EEA. In these cases, a low molecular weight alcohol is generated as a by-product.
8.6.2 Copolymer Formation by Amine + Anhydride Reaction
The most common method for compatibilizing PA/amorphous polyolefin blends involves graft copolymer formation by reaction of polyamide amine end-groups with pendent anhydride groups on an appropriate polyolefin to form a compatibilizing copolymer linked through an imide bond. Anhydride groups may be incorporated into suitable POs through grafting or through copolymerization with maleic anhydride, citraconic anhydride, itaconic anhydride, and congeners or through grafting or copolymerization with potentially latent anhydrides such as vicinal diacids or acid-esters including fumaric acid, maleic acid monoalkyl ester, and congeners. Selected examples are listed in Table 5.12.
8.6.3 Copolymer Formation by Amine + Carboxylic Acid Reaction
As shown by examples listed in Table 5.13, PA/PO blends have been compatibilized through block copolymer formation between PA amine end-groups and terminal carboxylic acid groups of polyolefins. PA blends with oxidized PE should fall into this category since carboxylic acid groups are believed to be located at PE chain ends (see El’darov et al. 1996).
Compatibilized PA/PO blends have also been prepared by graft copolymer formation between the amine end-groups of PA and PO pendent carboxylic acid groups to give a new amide linkage (Table 5.14). Water is the by-product of this reaction. The work by Aharoni is noteworthy in that it employs a condensing agent to effect copolymer formation between polyamide end-groups and pendent carboxylic acid groups on EAA.
Mascia and Hashim (1997, 1998) have prepared compatibilized blends of PA with PVDF by using carboxylic acid-functionalized PVDF. In an example, 20 parts PA-6 was combined with 80 parts PVDF-g-methacrylic acid (10 % MAA) in an internal mixer at 240 °C. The graft copolymer-containing blend was characterized by SEM, FTIR, mechanical properties, selective solvent extraction, and rheology. The effects of adding zinc acetate were studied.
8.6.4 Copolymer Formation by Amine or Carboxylic Acid + Epoxide Reaction
Table 5.15 shows examples of PA/PO blends compatibilized through graft copolymer formation between PA amine or carboxylic acid end-groups and pendent epoxy groups on polyolefins.
8.6.5 Copolymer Formation by Miscellaneous Reactions
Miscellaneous compatibilization methods belonging to this category are listed in Table 5.16. Chen and Wang (2001) reported that pan-milling a blend of PA-6 and PP resulted in chain scission and subsequent copolymer formation resulting in a blend with improved properties compared to the same blend prepared by extrusion. Li et al. (1993) have shown that PA-PO copolymer may be formed through displacement reaction between PA amine end-groups and benzylic bromide groups pendent on brominated poly(isobutylene-co-p-methylstyrene) (see also Bhadane et al. 2008, 2011; and Tsou et al. 2009, 2011). Coran and Patel (1983a) suggest that PA-PO copolymer may be formed by a displacement reaction between PA amine end-groups and PO chloride groups, simultaneous with dynamic vulcanization of the PO phase. Moncur (1982) reported that PA-PO copolymer may form through PA amine end-group displacement of chloride from a PO copolymer bearing reactive chloride.
8.7 Polyamide + Polyolefin + Polypropylene Blends
8.7.1 Copolymer Formation by Amine + Carboxylic Acid Reaction
Favis (1994) and Willis and Favis (1988) prepared compatibilized PA blends with PP and carboxylic acid-functionalized EMAA ionomer. Blends containing 90-10 parts PA-6, 0-30 parts EMAA ionomer, and 10-90 parts PP were combined in an internal mixer at 250 °C and characterized by torque rheometry and SEM. Dispersed phase particle size vs. interfacial modifier concentration was determined. Emulsification curves were constructed. Effects of mixing protocol on blend properties were studied. Blends were also prepared containing HDPE in place of PP.
8.7.2 Copolymer Formation by Amine + Anhydride Reaction
PA/PP blends have been compatibilized through graft copolymer formation between polyamide amine end-groups and pendent anhydride groups on a functionalized polyolefin as exemplified in Table 5.17.
Chen and White (1993) and Chen et al. (1988) have reported properties for blends containing 75-25 parts PA-6 (or PA-11), 25-75 parts LDPE (or HDPE), and 0-5 parts PP-MA. The blends were prepared in an SSE at 200–230 °C and characterized by mechanical properties and DSC. Morphology and capillary rheometry were done before and after annealing. Interfacial tension measurements for the blends were also reported.
8.7.3 Copolymer Formation by Amine or Carboxylic Acid + Epoxide Reaction
As shown in Table 5.18, PA/PP blends can be compatibilized through graft copolymer formation between polyamide amine end-groups and pendent epoxide groups on a functionalized PO. Since there are multiple epoxide sites on the polyolefin, some cross-linked copolymer may result as well if the PA is functionalized at both ends. The proportion of cross-linked copolymer formed also depends upon blend composition and processing conditions.
8.8 Polyamide + Polyolefin + Styrene Copolymer Blends
8.8.1 Copolymer Formation by Amide-Ester Exchange
Horak et al. (1997) prepared compatibilized PA blends with ABS in the presence of polyacrylate copolymer bearing pendent ester groups. A graft copolymer is obtained when PA amine end-groups undergo transreaction with the pendent ester groups. In an example, 50-45 parts PA-6 was mixed with 45-50 parts ABS and 0-10 parts MMA-co-AN (80 % MMA) in an internal mixer at 240 °C. The blend was characterized by SEM, WAXS, and mechanical properties. The effects of premixing PA + MMA-co-AN or of adding dibutyltin dilaurate or Ti(OBu)4 catalyst were examined.
8.8.2 Copolymer Formation by Amine + Anhydride Reaction
As shown in Table 5.19, PA/PO blends have been compatibilized through graft copolymer formation between polyamide amine end-groups and pendent anhydride groups on a functionalized styrene copolymer or alternatively through copolymer formation between PA and anhydride-functionalized PO in the presence of PS or a styrene copolymer.
8.8.3 Copolymer Formation by Amine or Carboxylic Acid + Epoxide Reaction
Huang et al. (2011) prepared blends of PA-6 with ABS in the presence of ethylene-acrylate-GMA copolymer. Characterization techniques included SEM, DSC, HDT, and mechanical properties.
Filippi et al. (2004) prepared blends of PA-6 with LDPE in the presence of SEBS-g-GMA. Inefficient compatibilization of PA/PE was observed in comparison to the use of SEBS-g-MA apparently due to cross-linking reactions involving both amine and carboxylic acid end-groups on PA.
An interesting study by Kudva et al. (1998) showed that PA-6/ABS blends were poorly compatibilized through graft copolymer formation between PA amine or carboxylic acid end-groups and MMA-co-GMA. Extensive characterization indicated that, although the epoxy-functionalized MMA is miscible with the SAN domain, the difunctionality of PA end-groups led to PA cross-linking rather than formation of a compatibilizing copolymer.
8.9 Polyamide + Polyphenylene Ether Blends
PA/PPE blends in these sections include those containing PPE-miscible PS or a functionalized PS. These blends often contain a rubbery impact modifier, such as SEBS.
8.9.1 Blends Containing PPE Melt Functionalized with Anhydride Groups: PA-PPE Copolymer Formation by Amine + Anhydride Reaction
Anhydride groups are readily introduced into polyphenylene ethers such as poly(2,6-dimethylphenylene ether) (PPE) by extrusion with maleic anhydride (MA) or congeners such as fumaric acid (FA), itaconic acid, citraconic acid, and related compounds. Evidence has been presented that such anhydride groups are predominantly located along the PPE main chain and with a fraction also at chain ends (Glans and Akkapeddi 1991b). This functionality distribution depends on the type and level of functionalization agent and on the mixing protocol during extrusion processing. PPE that has been melt functionalized, for example, with FA or MA reacts with PA to give predominantly graft copolymer by reaction of anhydride pendent groups on PPE with amine end-groups of PA. There is certainly a proportion of block copolymer formation as well since some anhydride functionality may be present at the chain ends on PPE. In the examples given in Table 5.20, PPE was functionalized in either a separate extrusion step before mixing with PA or in the front section of an extruder with downstream feeding of PA.
Son et al. (2000a, b) have studied the effect of processing conditions on the morphology of PA blends with PPE compatibilized through addition of MA.
PPE has also been melt functionalized with various esters of trimellitic anhydride substituted in the 4-position (Sivavec and M?cCormick 1991). A phosphite catalyst was used to promote transesterification and functionalization at PPE phenolic chain ends. As an example, 412 g PPE was extruded with 8 % 4-(o-carbophenoxyphenyl)-trimellitic anhydride and 0.5 % triphenyl phosphite on a TSE at 170–300 °C. Analysis of the extrudate showed 40 % carboxylation and 55 % capping of PPE phenolic end-groups. Extrusion of 49 parts capped PPE with 41 parts PA-66 and 10 parts SEBS provided molded test parts with Izod impact strength of 247 J/m. The PPE-PA copolymer in this case is primary block type. For melt functionalization of PPE with various trimellitamides and subsequent compatibilized blends with PA, see also Sivavec and Fukuyama (1992).
8.9.2 Blends Containing PPE Solution Functionalized with Anhydride Groups: PA-PPE Block Copolymer Formation by Amine + Anhydride Reaction
Campbell et al. (1990) have reported properties for compatibilized PA-PPE blends made using anhydride-terminated PPE. Anhydride-terminated PPE was made by capping PPE phenolic end-groups with trimellitic anhydride acid chloride in solution. A block copolymer may form between PA amine end-groups and PPE-anhydride during subsequent melt mixing. For example, a blend containing 49 parts PPE-anhydride, 41 parts PA-6, and 10 parts SEBS impact modifier was extruded using a TSE at 280 °C. The copolymer-containing blend was characterized by selective solvent extraction, mechanical properties, SEM, and TEM. Ductile-brittle transition temperatures were determined. The same anhydride-terminated PPE was also used to prepare compatibilized blends with PA-66 (Aycock and Ting 1986, 1987).
8.9.3 Blends Containing PPE Solution or Melt Functionalized with Aryloxy Triazine Groups: PA-PPE Copolymer Formation by Amine + Ester Exchange Reaction
PPE has been functionalized in solution with a variety of chloro aryloxy triazine derivatives in the presence of a base to provide a reactive diaryloxy triazine-capped PPE (Brown 1991b, 1993). Diaryloxy triazine-capped PPE can also be prepared by melt functionalization through transesterification of the cyanuric acid phenyl ester (i.e., aryloxy triazine) with PPE phenolic end-groups (Brown 1992b). Extrusion of the functionalized PPE with an amine-functionalized polymer such as amine-terminated PA results in formation of a compatibilizing block copolymer through displacement of at least one aryloxy group from the diaryloxy triazine end-cap. Typically, a 2,6-unfunctionalized aryloxy group is displaced from the triazine end-cap during extrusion in preference to displacement of more hindered 2,6-dimethylphenoxy PPE terminal unit. The reaction may also be viewed as an esteramide exchange reaction between a cyanuric acid ester (i.e., the diaryloxy triazine terminal group on PPE) and PA-amine end-group. In one example, 41 % PA-66 extruded with 49 % diaryloxy triazine-capped PPE, and 10 % impact modifier showed Izod impact strength of 753 J/m and tensile elongation of 122 % compared to 37 J/m and 11 % for the same blend containing unfunctionalized PPE. The effect of PA amine end-group concentration on blend properties was examined.
8.9.4 Blends Containing PPE Functionalized with Carboxylic Acid: Copolymer Formation by Amine + Carboxylic Acid Reaction
Yates and White (1989) introduced carboxylic acid functionality into PPE by metalation in solution with alkyl lithium and treatment with carbon dioxide. Extrusion of this functionalized PPE (45 parts) with PA-66 (45 parts) and SEBS (10 parts) provided a composition which showed eightfold improvement in impact strength compared to a similar blend made with unfunctionalized PPE.
8.9.5 Blends Containing Functionalized PPE: PA-PPE Copolymer Formation by Amine or Carboxylic Acid + Electrophile Reaction
PA/PPE blends have been compatibilized through copolymer formation between PA amine or carboxylic acid end-groups and an electrophile-functionalized PPE (Table 5.21). Typically, the PPE is functionalized in a separate reaction either in the melt or in solution to introduce an electrophilic moiety (such as epoxide, carbodiimide, cyclic ortho ester, imide, or the like) at a phenolic end-group or along the PPE main chain or both.
8.9.6 Blends Containing Unfunctionalized PPE: Copolymer Formation by Coupling Agent Addition
Unfunctionalized PPE has been compatibilized with immiscible PA by addition of a coupling agent capable of reacting with both PPE and PA end-groups. In one example, 4,4′-methylenediphenyl diisocyanate (1 part) was used to compatibilize PPE (49 parts) with PA-6 (41 parts) in the presence of an impact modifier. Properties of compatibilized blends were compared to those without coupling agent and without impact modifier (Pernice et al. 1993). Similar work was performed by Chiang et al. (1998). Chiang and Chang (1998) employed a multifunctional epoxy resin for the same purpose.
8.9.7 Blends Containing Unfunctionalized PPE + Functionalized PS: PA-PS Copolymer Formation by Miscellaneous Reactions
Unfunctionalized PPE may be compatibilized with immiscible PA by addition of functionalized polystyrene capable of forming copolymer with PA (Table 5.22). This is a common compatibilization strategy for PPE blends since both PS itself and functionalized polystyrenes with a relatively low level of functionality are miscible with PPE. The examples in the table include use of anhydride-, acid-, and epoxide-functionalized polystyrenes, all of which are capable of reacting with nucleophilic end-groups on PA to form a graft copolymer.
8.10 Polyamide + Polyphenylene Sulfide Blends
Gui et al. (2013) prepared blends of an elastomeric PA with PPS in the presence of an epoxy resin. Blend characterization techniques included rheology, FTIR, and mechanical properties.
Yamao and Kosaka (2000) have prepared compatibilized blends of PA-66 or PA-46 with an amine-functionalized PPS in the presence of either pyromellitic acid anhydride or a multifunctional epoxy resin as coupling agent. See also Ishio et al. (2011).
Blends containing PA and an anhydride-modified polyphenylene sulfide have been prepared by Kadoi et al. (1996). For example, PPS was extruded with either maleic anhydride, itaconic anhydride, or succinic anhydride to form a PPS shown to have carbonyl incorporation by FTIR after selective solvent extraction to remove unreacted anhydride. Blends of modified PPS and PA-66 were extruded at 290–310 °C and molded to provide test parts with improved properties compared to blends with unmodified PPS.
8.11 Polyamide + Polypropylene Blends
8.11.1 Copolymer Formation by Amine + Anhydride Reaction
Immiscible blends of PA and PP have been compatibilized through copolymer formation between PA amine end-groups and maleic anhydride-functionalized PP to form a new imide linkage (Table 5.23). The structure of maleic anhydride-functionalized PP has been discussed, e.g., by De Roover et al. (1995) and Sclavons et al. (1996). The authors demonstrated that the free radical initiated maleation of PP in the molten state leads to anhydride groups locating mainly at PP chain ends. Consequently, unless PP has been functionalized by a process that suppresses PP chain degradation, the reaction product of amine-terminated PA with MA-functionalized PP may be predominantly a block copolymer.
8.11.2 Copolymer Formation by Amine + Carboxylic Acid Reaction
As shown in Table 5.24, PA/PP blends can be compatibilized through graft copolymer formation between PA amine end-groups and pendent acid groups grafted at more than one site along the PP chain or with acid groups along a poly(acrylic acid) segment grafted at a single PP site.
8.11.3 Copolymer Formation by Amine or Carboxylic Acid + Epoxide or Oxazoline Reaction
Deng et al. (2007) compatibilized PA-66 blends with PP through addition of alpha-methylstyrene-GMA copolymer. Evidence was presented that the copolymer acts to functionalize PP in situ with GMA groups. PA-6 blends with PP or PE were also prepared using this method.
Pompe et al. (2002) have prepared compatibilized PA/PP blends using oxazoline-modified PP.
Zhang and Yin (1998), Xiaomin et al. (1997, 1998), and Zhang et al. (1996, 1997) have prepared compatibilized PA/PP blends by adding epoxide-grafted PP. Graft copolymers result from reaction between PA amine end-groups and pendent epoxide groups on PP (or with epoxide groups along a poly(GMA) segment grafted at a single PP site). For example, 100-15 parts PA-1010 was mixed with 0-85 parts PP and 0-25 parts PP-g-GMA in either an SSE or TSE at 200–210 °C. The resulting blends were characterized by selective solvent extraction, SEM, rheology, DSC, ESCA nitrogen analysis, FTIR, mechanical properties, and peel test. Since there may be multiple epoxide sites on the polypropylene, some cross-linked copolymer may result if the polyamide is functionalized at both ends. The proportion of cross-linked copolymer formed also depends upon blend composition and processing conditions.
8.11.4 Copolymer Formation by Miscellaneous Reactions
Lin and Isayev (2006) prepared blends of PA-6 and PP by treatment with high-intensity ultrasound during extrusion. Mechanical properties, crystallinity, and morphology were investigated. The competition between polymer degradation and partial in situ compatibilization was assessed.
8.12 Polyamide + Polypropylene + Styrene Copolymer Blends
8.12.1 Copolymer Formation by Amine + Anhydride Reaction
As shown by examples listed in Table 5.25, PA/PP blends have been compatibilized through graft copolymer formation between polyamide amine end-groups and pendent anhydride groups on a functionalized styrene copolymer.
8.13 Polyamide + Polysiloxane + Styrene Copolymer Blends
Maric et al. (2001) have studied PA blends with poly(dimethylsiloxane) (PDMS) either in binary blends of the functionalized polymers or in ternary blends with a functionalized styrene copolymer. The efficiency of copolymer formation concurrent with morphology development and stabilization was studied for reactions between PA-amine and PDMS-anhydride, between PA-amine and PDMS-epoxy, and between PA-carboxylic acid and PDMS-epoxy. The effects of relative melt viscosities on interfacial reactivity and resulting morphology were noted.
8.14 Polyamide + Polysulfone Blends
8.14.1 Copolymer Formation by Amine + Carboxylic Acid Reaction
PA blends with polysulfone have been compatibilized through graft copolymer formation between polyamide amine end-groups and pendent carboxylic acid groups on a polysulfone functionalized using 4,4′-bis(4-hydroxyphenyl)pentanoic acid (Marechal et al. 1998). Characterization was by mechanical properties and morphology. A similar strategy was used by Ibuki et al. (1999) who in addition studied anhydride-terminated or anhydride-grafted polysulfone. See also Charoensirisomboon et al. (1999a, b, 2000) and Koriyama et al. (1999) and further papers by these authors.
8.15 Polyamide + Polystyrene or Styrene Copolymer Blends
8.15.1 Copolymer Formation by Acid-Base Interaction
Villarreal et al. (2004) have prepared blends of PA-6 with PS compatibilized by addition of poly(styrene-co-sodium acrylate). Characterization techniques included mechanical properties and SEM. See also Rodríguez-Ríos et al. (2004) for related work.
Compatibilized blends of PA-6 with syndiotactic PS (sPS) were prepared by melt blending in the presence of sulfonated sPS (Li et al. 2002). Blends were characterized by morphology, mechanical properties, and DSC. Sulfonated sPS at a level of 20 wt% or less was reported to be miscible with sPS. For related work involving PA-6 and sulfonated PS, see Molnar and Eisenberg (1991, 1992).
8.15.2 Copolymer Formation by Amine + Anhydride Reaction
Immiscible blends of PA and PS or styrene copolymer have been compatibilized through graft copolymer formation between PA amine end-groups and anhydride-functionalized styrene copolymer to form a new imide linkage (Table 5.26).
PA/PS blends have also been compatibilized through block copolymer formation between amine-terminated PA and anhydride-terminated PS. Anhydride end-groups were introduced into PS through reaction of either anion-terminated PS or hydroxy-terminated PS with trimellitic anhydride acid chloride. For example, Park et al. (1992) blended 80 parts PA-6 with 10-16 parts PS and 4-10 parts anhydride-terminated PS in an internal mixer at 240 °C. The blends were characterized by torque rheometry, SEM, selective solvent extraction, DSC, morphological stability to annealing, and lap shear adhesion. The effect of mixing protocol on properties was studied. Properties were also compared to those for blends compatibilized by added PA-PS graft copolymer that had been synthesized in a separate step.
8.15.3 Copolymer Formation by Amine + Carboxylic Acid Reaction
Melt reactions between amine-terminated PA and carboxylic acid groups on styrene-acrylic acid copolymer have been demonstrated (Table 5.27). The initial reaction product is a graft copolymer, but longer reaction times may result in cross-linking, since the polyamides (PA-66 and PA-69) can have two amine end-groups per chain (Kuphal et al. 1991). Monoamine-terminated polyamides were reported in the same study to exhibit miscibility with SAA through hydrogen-bonding depending upon AA content. For PA-1010 blends with carboxylated PS, see Li and Li (1999).
Kausar et al. (2013) prepared blends of PS and amine-f-PS with an aramid prepared from 1,5-diaminonaphthalene and 1,4-phenylenediamine with isophthaloyl chloride. Morphological and thermophysical properties were investigated. Formation of an aramid-g-PS copolymer was proposed, perhaps through an amine-carboxylic acid reaction. For related blends of amine-f-PS and aramid, see Shabbir et al. (2008, 2010).
8.15.4 Copolymer Formation by Amine or Carboxylic Acid + Epoxide Reaction
Sun et al. (2005) prepared compatibilized blends of PA-6 with ABS-co-GMA. See also Singh and Gupta (2011).
Chen et al. (2003) have studied PA-6 blends with sPS compatibilized using S-co-GMA in a torque rheometer. Blends were characterized using SEM, mechanical properties, and DSC.
Chang and Hwu (1991) prepared compatibilized PA/PS blends through addition of epoxide-functionalized S-GMA copolymer. A graft copolymer results from reaction of pendent epoxide groups with either amine or acid end-groups on PA. A blend of 50 parts PA-66, 50 parts PS, and 0-0.5 parts S-GMA (3 % GMA) was prepared on a TSE and characterized by rheology, SEM, and mechanical properties. Since there are multiple epoxide sites on S-GMA, some cross-linked copolymer may result if the polyamide is functionalized at both ends. The proportion of cross-linked copolymer formed also depends upon blend composition and processing conditions. See also Martens et al. (2004).
8.15.5 Copolymer Formation by Amine or Carboxylic Acid + Isocyanate Reaction
Zhang et al. (2013a) prepared compatibilized PA/PS blends through addition of an isocyanate-functionalized PS. For example, blends comprising PA-6 and PS-co-(3-isopropenyl-α,α-dimethylbenzene isocyanate) were characterized using FTIR, DSC, and morphology determination. The effects of different functionalized PS loading and the isocyanate level in the PS copolymer were investigated.
Yin et al. (2009b) prepared blends of PA-6 with SEBS which had been functionalized with ε-caprolactam-blocked allyl (3-isocyanate-4-tolyl) carbamate.
8.15.6 Copolymer Formation by Amine or Carboxylic Acid + Oxazoline Reaction
As shown in Table 5.28, blends of PA and PS have been compatibilized through graft copolymer formation between PA amine or acid end-groups and oxazoline-functionalized styrene copolymer.
8.16 Polyamide + Polyurethane Blends
8.16.1 Copolymer Formation by Carboxylic Acid or Amine + Isocyanate Reaction: Blends Containing a Coupling Agent
PA/TPU blends have been compatibilized by addition of a bis-isocyanate coupling agent that is capable of reacting with nucleophilic end-groups on both polymers to form a block copolymer. Franke et al. (1993) have extruded PA-6 (20-0 parts) with polyester-urethane (78-100 parts) using a TSE at 230 °C in the presence of 0.5-2 parts diphenylmethane diisocyanate. The coupling agent was added downstream of the extruder feed throat. The blends were characterized by TEM, SEC, DSC, DMA, and FTIR.
9 Polyester Blends
Examples of polyester blends not shown in earlier sections are listed in alphabetical order of the second polymer in the blend unless otherwise noted. Polycarbonate blends are also included as polyesters. When copolymer characterization was not performed, the structure of the compatibilizing copolymer is inferred from the functionality location on each of the two polymers. In some cases, more than one type of compatibilizing copolymer may have formed.
Copolymer-forming reactions for compatibilizing immiscible blends, some of which are applicable to PEST blends, have been studied by Orr et al. (2001) who determined that the order of increasing reactivity in functionalized polymer pairs is acid/amine, hydroxyl/(anhydride or acid), aromatic amine/epoxy, aliphatic amine/epoxy, acid/oxazoline, acid/epoxy, aromatic amine/anhydride, and aliphatic amine/anhydride (most reactive).
For reviews of reactive compatibilization of poly(lactic acid) (sometimes referred to as polylactide) with other immiscible polymers, see Imre and Pukanszky (2013) and Liu and Zhang (2011).
9.1 Polyester + Polyester (or Polycarbonate) Blends
9.1.1 Copolymer Formation by Alcohol + Carboxylic Acid Reaction: Blends Containing a Condensing Agent
As shown in Table 5.29, blends of immiscible polyesters may be compatibilized through copolymer formation mediated by addition of a phosphite condensing agent. Block copolymer results when the phosphite-activated end-group of one PEST reacts with a nucleophilic end-group of another PEST. The reaction takes place at the phase interface. A secondary phosphite is a by-product. The relative proportions of copolymer vs. simple chain-extended PEST may depend upon the relative solubility of condensing agent in each of the immiscible polymer phases.
9.1.2 Copolymer Formation by Carboxylic Acid + Epoxide or Oxazoline or Isocyanate Reaction: Blends Containing a Coupling Agent
Blends of poly(lactic acid) and poly((butylene-adipate)-co-terephthalate) have been prepared with the addition of either 2,2′-(1,3-phenylene)-bis(2-oxazoline) or phthalic anhydride (Dong et al. 2013). Blends were characterized using DSC, SEM, and mechanical properties.
Polycarbonate blends with poly(lactic acid) have been compatibilized through addition of bis(isocyanate), bis(carbodiimide), oxazoline-f-PS, or epoxy resin (Wang et al. 2012e; Mukawa et al. 2011).
Blends of aliphatic-aromatic polyester (75-25 parts) and poly(lactic acid) (25-75 parts) have been compatibilized through extrusion with a copolymer of styrene, GMA, and isomethacrylates (0-5 parts) (Hale 2008). Polyesters included those derived from adipic acid-terephthalic acid-butanediol. Mechanical properties were greatly improved compared to those for blends with no compatibilizer.
Poly(ε-caprolactone) blends with poly(lactic acid) have been compatibilized in the presence of polyepoxide or either di- or trisisocyanate (Harada et al. 2008).
Harada et al. (2007) have prepared compatibilized blends of PLA with poly(butylene succinate) through addition of lysine trisisocyanate. Characterization techniques included MFR, mechanical properties, SEC, and laser scanning confocal microscopy.
The crystallization behavior of PTT-PC blends in the presence of either epoxy resin or EPDM-g-GMA has been studied by Xue et al. (2005).
PET-PEN blends have been compatibilized in the presence of a bis-oxazoline coupling agent (Yang et al. 2002b).
Ju et al. (2000) prepared compatibilized blends of polyarylate with an LCP in the presence of tetraglycidyl-4,4′-diaminodiphenyl methane coupling agent.
Chin and Chang (1997) and Chin et al. (1996) have compatibilized blends of immiscible PEST through addition of a multifunctional epoxide coupling agent capable of reacting with nucleophilic end-groups on each of the two immiscible PEST at the phase interface to give a block copolymer containing the coupling agent as linking group. In one example 100-85 parts PET was extruded using a TSE at 270–285 °C with 0-15 parts LCP (Hoechst Vectra® A900) and 0-2 parts tetrafunctional epoxy resin. The blends were characterized by torque rheometry, capillary rheometry, DSC, SEM, and FTIR. Mechanical properties were determined vs. composition and morphology. Ethyltriphenylphosphonium bromide was added as a catalyst to promote the reaction of polyester acid or alcohol end-groups with epoxy resin. The relative proportions of copolymer vs. simple chain-extended PEST depend at least partly upon the relative solubilities of coupling agent and catalyst in each of the immiscible polymer phases. See also Tjong and Meng (1999) and Dekkers et al. (1992).
9.1.3 Copolymer Formation by Radical Coupling
Krishnaswamy et al. (2013) prepared blends of poly(lactic acid) with poly(3-hydroxybutyrate-co-4-hydroxybutyrate) containing 17–40 wt% 4-hydroxybutyrate in the presence of radical initiator. Blends were characterized by melt strength, viscosity, and mechanical properties. Blend properties were compared to control blends without RI.
Wang et al. (2009a) have compatibilized the biodegradable polymers poly(l-lactic acid) and poly(butylene succinate) in the presence of DCP radical initiator (0.05–0.2 phr). For related work, see Lan et al. (2013).
For blends of poly(butylene succinate) with poly(hydroxybutyrate-co-hydroxyvalerate or poly(hydroxybutyrate) compatibilized in the presence of radical initiator, see Ma et al. (2012a).
Blends of poly(lactic acid) and poly(butylene-adipate-co-terephthalate) were prepared in the presence of varying concentrations of L101 RI by Coltelli et al. (2010). The blends were characterized by morphology, rheology, and mechanical properties.
Other radical-radical coupling reactions used to compatibilize blends of immiscible polyesters include those by Avella et al. (1996); Immirzi et al. (1994); and Cavallaro et al. (1993). Specifically, 70-30 parts poly(hydroxybutyrate-co-hydroxyvalerate) (4 mol% valerate) or poly(hydroxybutyrate) was mixed with 30-70 parts PCL in an internal mixer at 100 °C or 160 °C in the presence of 0-0.5 parts DCP or DBP radical initiator. Blends were characterized by SEM, mechanical properties, selective solvent extraction, and FTIR.
9.1.4 Copolymer Formation by Redistribution Reaction
Brief reviews covering redistribution reactions (often referred to as transesterification reactions or simply transreactions) in polyester and in polycarbonate binary blends have been prepared by Porter et al. (1989) and Porter and Wang (1992). Carrot et al. (2007) have surveyed more recent knowledge relating to PET/PC blends. Pesneau et al. (2001) have studied the relative effectiveness of different transesterification catalysts, finding that dibutyltin oxide had the highest activity of those studied. Other reviews of transreactions in polyester blends include Montaudo et al. (1999), Pilati et al. (1999), Economy et al. (1999), and also portions of other chapters in Fakirov (1999) (transreactions in condensation polymers).
Selected references for redistribution processes in PEST/PEST blends are listed in Table 5.30. Early studies of these processes focused on measuring the extent of redistribution under specific processing conditions rather than on producing compatibilized polymer blends with an attractive balance of properties. A number of other studies have reported the limits of miscibility for certain melt-mixed polyester pairs in the absence of transesterification – see, for example, the NMR study of PC/PET blends (Abis et al. 1994). Table 5.30 omits references in which transesterification in PEST/PEST blends is brought about under static conditions either by annealing or heating in a DSC chamber.
9.2 Polyester + Polyether Blends (Including Polycarbonate)
9.2.1 Copolymer Formation by Transesterification
A graft copolymer may be formed through transesterification between pendent hydroxy groups on phenoxy polyether resin and ester linkages in the chains of an immiscible polyester or polycarbonate phase (Table 5.31). Since the product is a graft copolymer accompanied by a low molecular weight fragment from the polyester, this is a degradative copolymer-forming process. The initial product of the transreaction is a graft copolymer as the alcohol reacts into the polyester chain. Longer reaction time may result in a cross-linked copolymer since the pendent polyester segment is capable of further reaction with OH on a different phenoxy chain. These types of blends have also been prepared by solution casting followed by annealing. Blends of LCP with phenoxy resin provide an example (Kodama 1992).
9.3 Polyester + Polyetherimide Blends
9.3.1 Copolymer Formation through Coupling Agent Addition
PEST blends with PEI (derived from 2,2-bis[4-(3,4-dicarboxyphenoxy)phenyl] propane dianhydride and meta-phenylene diamine) have been compatibilized in the presence of various multifunctional epoxy resins and a catalyst (Brown et al. 2000b). PEST types included mixtures of PCT and PETG; catalysts included sodium stearate. Mechanical properties and HDT were compared to properties for blends without either epoxy resin or catalyst.
Silvi et al. (1997) have compatibilized an immiscible blend of LCP and similar PEI through copolymer formation in the presence of a coupling agent. Representative coupling agents included PE-co-GMA and the o-cresol novolak reaction product with epichlorohydrin. Mechanical properties and HDT were compared to properties for blends without polyepoxide. PEI could be diluted with polyarylate, polyestercarbonate, PET, or PEN.
9.3.2 Copolymer Formation by Transreaction
A copolymer may be formed through transreaction between a bisphenol A polyestercarbonate resin and imide linkages in the chains of an immiscible PEI phase in the presence of a catalyst (Brown et al. 1998b). This is a degradative process initially forming block copolymer and eventually random copolymer as chains continue to react. The final product may be transparent. Various phosphite catalysts were used. PC and polyarylate could be used in place of polyestercarbonate. Bookbinder and Sybert (1992) prepared compatibilized blends of Hoechst Vectra® A950 with amine-terminated PEI. A copolymer may form through reaction of PEI amine groups with main-chain ester units of LCP in a degradative process.
9.4 Polyester + Polyethersulfone Blends
Kanomata et al. (2011) prepared compatibilized blends of either PBT or PET and polyethersulfone using PES having hydroxyphenyl end-groups. Blends prepared in a Brabender mixer were characterized using TEM and torque rheometry in comparison with control blends.
9.5 Polyester + Polyolefin Blends (Excepting Polypropylene)
9.5.1 Copolymer Formation by Alcohol + Anhydride Reaction
As shown in Table 5.32, PEST/PO blends have been compatibilized through copolymer formation between polyester alcohol end-groups and pendent anhydride functionality on a polyolefin such as maleic anhydride-grafted polyolefin. Because this alcohol-anhydride reaction is reversible with the equilibrium lying on the side of unreacted anhydride, only a relatively small amount of copolymer may be formed. Consequently, the dispersed polymer phase may not be well stabilized against coalescence upon further thermal treatment (for a discussion, see, e.g., Sun et al. (1996) and Boyer et al. (2005)). Alternatively, at least some copolymer may be formed by a degradative mechanism through transesterification between polyester main-chain linkages and a low concentration of pendent acid groups in anhydride functionalized polyolefin. In addition copolymer may possibly form through anhydride exchange between PEST-CO2H end-groups and PO-anhydride. Alternatively, it may also happen that compatibilization results from hydrogen-bonding interaction.
9.5.2 Copolymer Formation by Carboxylic Acid + Cyclic Ortho Ester or Epoxide Reaction
Table 5.33 shows examples of PEST/PO blends that have been compatibilized through graft copolymer formation either by reaction of polyester carboxylic acid end-groups with either pendent cyclic ortho ester or pendent epoxide groups on a polyolefin or by reaction of polycarbonate epoxide end-groups with carboxylic acid groups on a polyolefin. In any case, the copolymer is joined through a new ester linkage. Since there may be multiple electrophilic sites or multiple carboxylic acid sites on the polyolefin, some cross-linked copolymer may result if the polyester is functionalized at both ends. The proportion of cross-linked copolymer formed also depends upon blend composition and processing conditions.
When the new ester linkage is formed, a secondary alcohol group forms as well through ring opening of the epoxide group. The new secondary alcohol may also react with polyester main-chain linkages to form copolymer through a degradative transesterification process. Consequently, two very different types of copolymer may be formed in certain blends of this section.
Perret et al. (1996) have encapsulated a third polymer within a PO phase dispersed in a PEST matrix; the third polymer was PA-66, having higher Tm than the matrix. In this ternary blend, the epoxy-functionalized PO was capable of reacting with terminal functional groups on both of the other two polymers. The blend was formed either by preextrusion of PO with PA, followed by extrusion with PEST, or by feeding PO and PA to the feed throat of an extruder, then adding PEST downstream. The morphology showed a PEST matrix in which shells of PO surround cores of PA. The blend was an impact-modified PEST with higher impact strength than the corresponding blend containing epoxy-functionalized PO alone. One reason for the improvement may be that the volume fraction of PO impact modifier was effectively increased by inclusion of PA core. Copolymer formation between PEST and PO may also be combined with subsequent dynamic vulcanization of the PO phase in either the same or a separate processing step (Okamoto et al. 1994; Moffett and Dekkers 1992).
9.5.3 Copolymer Formation by Carboxylic Acid + Oxazoline Reaction
PEST/PO blends have been compatibilized through graft copolymer formation by reaction of polyester carboxylic acid end-groups with pendent oxazoline groups on an appropriate PO. The copolymer contains a new esteramide linkage. Wörner et al. (1997) have blended 0-20 parts oxazoline-functionalized rubbers containing more than one oxazoline group per chain with 100-80 parts acid-terminated PBT in an internal mixer at 240 °C. Oxazoline-functionalized B-AN or E-B-AN was used. The copolymer structure was initially a graft copolymer. However, if additional oxazoline groups on the rubber react with additional PBT acid groups, then a cross-linked copolymer structure can arise. The blends of Wörner et al. were characterized by torque rheometry, mechanical properties, DMA, SEM, DSC, and level of oxazoline groups on the rubber (fraction of nitrile groups on AN-containing rubber that had been converted to oxazoline groups). Improved blend properties were obtained through addition of a bis-oxazoline chain extender for PBT.
9.5.4 Copolymer Formation through Coupling Agent Addition
Zhang et al. (2013c) prepared compatibilized blends of poly(lactic acid) and EVAl in the presence of a multifunctional epoxy resin and zinc stearate. Characterization techniques included morphology, dynamic light scattering, and barrier properties.
Compatibilized blends of PBT with EVAc have been prepared through addition of 2,2′-(1,3-phenylene)-bis(2-oxazoline) coupling agent (Scaffaro and La Mantia 2006; Scaffaro et al. 2004). The effect of EAA addition was studied. Evidence was presented that chain extension of the reactive polymers by the coupling agent was negligible. Blends characterization included mechanical properties and morphology in comparison to uncompatibilized blends.
Zhang and Hourston (1999) prepared blends of PBT and either LDPE or EPDM in the presence of a bismaleimide in a Haake mixer. A copolymer formation mechanism involving radical coupling was proposed.
Okamoto and Inoue (1993) have compatibilized an immiscible blend of terminally functionalized PEST and PO through block copolymer formation in the presence of a coupling agent. For example, 80 parts hydroxy-terminated PCL was mixed in a custom melt reactor at 120 °C with 20 parts carboxyl-terminated butadiene oligomer (or carboxyl-terminated NBR) in the presence of 0-1 part aminopropyltriethoxysilane coupling agent. Morphology development could be followed through analysis of successive samples taken periodically from the reactor. Samples were characterized by light-scattering photometry, ellipsometric analysis, and GPC.
9.5.5 Copolymer Formation by Diels-Alder Reaction
The benzocyclobutene functionality can thermally form an intermediate that can function as the diene partner in a classical Diels-Alder ring-forming reaction with an olefin. When benzocyclobutene and olefin are located on two immiscible polymers, the reaction can in theory lead to copolymer formation. Dean (1993) has postulated that copolymer is formed when 50-15 parts benzocyclobutene-terminated polyarylate is mixed with olefin-containing EPDM in an internal mixer at 265 °C. Blends were characterized by mechanical properties and DMTA.
9.5.6 Copolymer Formation by Transesterification
Polyester-polyolefin copolymer compatibilizers have been made through catalyzed or thermal transesterification of polyester or polycarbonate or polyestercarbonate main-chain ester linkages with pendent ester or acid groups (acidolysis) in a polyolefin copolymer such as EVAc or EMAA (Table 5.34). In a separate example, polyester-polyolefin copolymer has been formed through transesterification between a carbonate ester linkage and an anhydride in a second polymer chain. All these examples represent degradative copolymer formation since the PEST chains are cleaved and the average molecular weight of the new copolymer is less than the sum of the average molecular weights of the two immiscible polymers.
In Table 5.34, Debier et al. (1995, 1997) present evidence that copolymer formation occurs in PC + PMMA blends through transesterification between PC carbonate ester linkages and acid groups produced by hydrolysis on PMMA. The table omits references in which transesterification in appropriate polyester-polyolefin blends is brought about under static conditions either by annealing or heating in a DSC chamber.
A block copolymer may be formed through transesterification between nucleophilic end-groups of one polymer and ester linkages in the chains of an immiscible polyester phase. Sek and Kaczmarczyk (1997) and Minkova et al. (1996) used oxidized PE as the acid-terminated polymer and an LCP with ester linkages susceptible to transesterification with PE acid end-groups. Since the product is a block copolymer accompanied by low molecular weight fragments from the polyester, this type of copolymer-forming reaction is degradative. The process is not redistributive since the PE chain cannot participate in redistribution. In an example, 50 parts LCP polyester (a copolymer of sebacic acid, dihydroxybiphenyl, and p-hydroxybenzoic acid) and 50 parts oxidized PE were mixed in an internal mixer at 240 °C. The blend was characterized by selective solvent extraction, FTIR, DSC, and SEM. Optionally, Ti(OBu)4 catalyst was added to promote reaction.
9.5.7 Copolymer Formation by Miscellaneous Reactions
Chen et al. (2013a) prepared blends of PCL with NR in the presence of peroxide which, in addition to cross-linking NR, served to enhance the compatibility of PCL and NR as shown by FTIR and SEM. The blend was studied as a shape-memory composition.
Hachemi et al. (2013) prepared blends of poly(lactic acid) with PVC in the presence of DCP RI and MA. Characterization techniques included DSC, TGA, SEM, and mechanical properties.
Gramlich et al. (2010) prepared blends of maleimide-terminated poly(lactic acid) with conjugated soybean oil. A compatibilizing copolymer was formed through Diels-Alder reaction between maleimide double bond and diene units in conjugated soybean chains. Blends were characterized by morphology and mechanical properties.
Pan et al. (2007) prepared compatibilized blends of PC with EMAc-g-GMA. Characterization concluded that copolymer was formed through reaction of PC phenolic end-groups and epoxy groups.
Jung et al. (2003) have prepared compatibilized blends of PET with LDPE comparing a variety of functionalized LDPEs as compatibilizing agents, including LDPE-co-AA, LDPE-f-MA, LDPE-f-GMA, and LDPE functionalized with either masked or naked NCO monomers.
Park et al. (1998b) have prepared compatibilized blends of PET with PE using PE grafted with 2-hydroxyethyl methacrylate-isophorone diisocyanate. See also Park et al. (2002), Bae et al. (2001), and Kim et al. (2000a, b). For PBT/ethylene-octene copolymer blends compatibilized using masked isocyanate, see Yin et al. (2009a).
Hourston et al. (1991) have prepared compositions of 60-0 parts PBT and 16-40 parts EPDM in the presence of 0-60 parts copolymer of PBT with maleate ester (3.5 % maleate) using a TSE at 255 °C. A compatibilizing copolymer was postulated from the cross-linking reaction between maleate olefinic groups and EPDM olefinic groups. Blends were characterized by mechanical properties and TEM. Model studies were performed to understand the cross-linking process. Blends were also prepared using an internal mixer at 250 °C.
9.6 Polyester + Polyolefin + Polypropylene Blends
9.6.1 Copolymer Formation by Carboxylic Acid + Epoxide Reaction
PEST/PP blends have been compatibilized through graft copolymer formation by reaction of polyester carboxylic acid end-groups with pendent epoxide groups on an appropriate PO with potentially some miscibility with PP (Table 5.35). The copolymer is joined through a new ester linkage. Some cross-linked copolymer may also form. When the new ester linkage is formed, a secondary alcohol group forms as well. The new secondary alcohol may also react with polyester main-chain linkages to form copolymer through a degradative transesterification process.
9.7 Polyester + Polyolefin + Styrene Copolymer Blends
9.7.1 Copolymer Formation by Alcohol + Anhydride Reaction
Examples of PEST/PO/styrene copolymer blend compatibilization in which a copolymer may be formed between polyester alcohol end-groups and pendent anhydride functionality on a styrene copolymer are shown in Table 5.36. Because the alcohol-anhydride reaction is reversible (with the equilibrium lying on the side of unreacted anhydride), only a relatively small amount of copolymer may be formed. In consequence, the dispersed phase polymer may not be well stabilized against coalescence upon further thermal treatment (for a discussion, see, e.g., Sun et al. (1996) and Boyer et al. (2005)). Alternatively, at least some copolymer may be formed by a degradative mechanism through transesterification between PEST main-chain linkages and a low concentration of pendent acid groups in anhydride-functionalized styrene copolymer or in anhydride-functionalized PO. In addition copolymer may possibly form through anhydride exchange between PEST-CO2H end-groups and either anhydride-functionalized styrene copolymer or anhydride-functionalized PO. Alternatively, it may also happen that compatibilization results from H-bonding interaction. For related work in PET/PP blends compatibilized using SEBS-g-MA (or PP-g-MA), see Papadopoulou and Kalfoglou (2000).
9.7.2 Copolymer Formation by Carboxylic Acid + Epoxide Reaction
Larocca et al. (2010) prepared blends of PBT and SAN in the presence of MMA-co-EA-co-GMA. Blend characterization techniques included morphology. PBT samples with different molecular weight were used to change the PBT/SAN viscosity ratio. In related work, AES was used in place of SAN (Larocca et al. 2005).
9.8 Polyester + Polyphenylene Ether Blends
PEST/PPE blends in these sections include those containing PPE-miscible PS or a functionalized PS. These blends often contain a rubbery impact modifier, such as SEBS, as well.
9.8.1 Blends Containing Functionalized PPE: PEST-PPE Copolymer Formation by Degradative Process
Furuta et al. (1994) have prepared compatibilized blends of LCP with an amine-functionalized PPE. For example, an LCP derived from p-acetoxybenzoic acid, terephthalic acid, isophthalic acid, and 4,4′-diacetoxydiphenyl was blended with an amine-functionalized PPE to provide a blend with improved properties compared to those with unfunctionalized PPE. PPE was melt functionalized with either p-aminostyrene, diallylamine, allylamine, or vinylimidazole. Other LCP were also used.
9.8.2 Blends Containing Functionalized PPE: PEST-PPE Copolymer Formation by Carboxylic Acid + Electrophile Reaction
PEST/PPE blends have been compatibilized through copolymer formation between polyester and a functionalized PPE (Table 5.37). Typically, the PPE is functionalized in a separate reaction either in the melt or in solution to introduce an electrophilic moiety (such as epoxide, carbodiimide, cyclic ortho ester, or the like) at a phenolic end-group or along the PPE main chain or both. The electrophilic moiety can react with PEST carboxylic acid end-groups in the melt resulting in a compatibilizing copolymer.
PEST-PPE blends have been compatibilized through block copolymer formation between carboxylic acid end-groups on polyesters, such as PBT, and epoxy-terminated PPE (Brown and Lowry 1992a). PPE was functionalized in solution with a variety of chloro-epoxy triazine derivatives in the presence of a base to provide a reactive epoxy triazine-capped PPE (Brown and Lowry 1992b; Yates et al. 1992). Melt functionalization of PPE was also possible (Brown et al. 2009). Representative chloro-epoxy triazine capping agents included 2-chloro-4-(2,4,6-trimethylphenoxy)-6-glycidoxy-1,3,5-triazine. For example, blends containing PBT, PPE-epoxide, and SEBS impact modifier were prepared using a TSE and characterized by selective solvent extraction, morphology, and mechanical properties. Numerous other polyesters, polyestercarbonates, and polyesteramides were used in these blends (see, e.g., Brown and Lowry 1992b).
PEST-PPE blends have also been compatibilized through block copolymer formation between carboxylic acid end-groups on polyesters, such as PBT, and PPE bearing reactive dialkylphosphatoethoxy triazine terminal groups (Phanstiel and Brown 1991, 1992). In this case PPE was functionalized in solution with a variety of chloro dialkylphosphatoethoxy triazine derivatives in the presence of a base to provide a reactive triazine-capped PPE. Representative chloro dialkylphosphatoethoxy triazine capping agents included 2-chloro-4-(2-di-n-butylphosphatoethoxy)-6-(2,6-xylenoxy)-1,3,5-triazine. For example, a blend containing 60 parts PBT, 30 parts functionalized PPE, and 10 parts impact modifier was prepared using a TSE. Molded test parts showed Izod impact strength of 860 J/m compared to 48 J/m for a similar blend containing unfunctionalized PPE. 2-Chloro-4-(2-chloroethoxy)-6-(2,4,6-trimethylphenoxy)-1,3,5-triazine or 2-chloro-4-(2-bromoethoxy)-6-(2,4,6-trimethylphenoxy)-1,3,5-triazine could be used in place of phosphatoethoxy triazine (Phanstiel and Brown 1993). See also Schmidhauser and Longley (1991).
PEST-PPE blends have also been compatibilized through block or graft copolymer formation between carboxylic acid end-groups on polyesters, such as PBT, and PPE bearing cyclic ortho ester groups. PPE could be either end-capped in solution using a chloro ortho ester triazine derivative such as 2-chloro-4-(2-methoxy-2-methyl-1,3-dioxolanyl)methoxy-6-phenoxy-1,3,5-triazine) (Khouri et al. 1992; Khouri and Phanstiel 1992) or functionalized in the melt using a graftable cyclic ortho ester such as 4-methacryloyloxymethyl-2-methoxy-2-methyl-1,3-dioxolane (Khouri et al. 1993). In either case, blends of various polyesters with cyclic ortho ester-functionalized PPE showed markedly better mechanical properties compared with control blends containing unfunctionalized PPE. Polyolefin grafted with cyclic ortho ester could also be used in PEST blends comprising PPE grafted with cyclic ortho ester (Khouri 1996).
9.8.3 Blends Containing Functionalized or Unfunctionalized PPE: PEST-PPE Copolymer Formation through Coupling Agent or Condensing Agent Addition
As illustrated in Table 5.38, coupling agents have been employed to compatibilize PEST-PPE blends through copolymer formation between PEST carboxylic acid end-groups and either PPE phenolic end-groups (unfunctionalized PPE) or PPE-g-carboxylic acid or PPE-g-anhydride (functionalized PPE). Numerous similar examples using epoxide coupling agents in PEST-PPE blends have been patented. Tang et al. (1992) have employed a phosphite condensing agent in similar manner to form compatibilizing copolymer. See also van Aert et al. (2001). Chen et al. (1993) employed a catalyst of boric acid with either polyphosphoric acid or sulfuric acid as a condensing agent to link PPE phenolic end-groups with LCP carboxylic acid end-groups.
9.8.4 Blends Containing Unfunctionalized PPE + Functionalized PS: PEST-PS Copolymer Formation by Carboxylic Acid + Epoxide Reaction
PEST/PPE blends have been compatibilized through graft copolymer formation between polyester and a PPE-miscible functionalized styrene copolymer (Table 5.39). In this instance, the reaction is between polyester carboxylic acid end-groups and pendent electrophilic groups on the styrene copolymer such as epoxide, oxazoline, cyclic ortho ester, and the like typically leading to a new ester linkage in the resulting copolymer. In the examples where the new ester linkage is formed from an epoxide group, a secondary alcohol group forms as well. The new secondary alcohol may also react with polyester main-chain linkages to form copolymer through a degradative transesterification process. Since there are typically multiple epoxide sites on the styrene copolymer, some cross-linked copolymer may result as well if the polyester is functionalized at both ends. The proportion of cross-linked copolymer formed also depends upon functionality concentration on both PEST and PS, blend composition, and processing conditions.
9.9 Polyester + Polyphenylene Sulfide Blends
Luo et al. (2013) employed a disulfide-modified PPS in compatibilized blends with LCP blend characterization techniques including rheological, mechanical, and thermal properties. Gopakumar et al. (1999) reported transesterification of thermotropic LCP with carboxylic acid-terminated PPS. Blends were characterized by morphology, mechanical properties, and DSC. Hanley et al. (1999) used a similar compatibilization strategy for blends of PET and carboxylic acid-terminated PPS. Chen et al. (1993) employed a catalyst of boric acid with either polyphosphoric acid or sulfuric acid as a condensing agent to link PPS thiol end-groups with LCP carboxylic acid end-groups.
9.10 Polyester + Polyphosphonate Blends
Kauth et al. (1988) have disclosed transesterification of aromatic polyesters and a polyphosphonate in a devolatilizing extruder. For example, a polyester derived from bisphenol A and 1:1 iso/terephthalic acid dissolved in dichloromethane/chlorobenzene was devolatilized along with a similar solution of a polyphosphonate derived from methanephosphonic acid and 4,4′-dihydroxybiphenyl in a TSE at 340 °C. The isolated transparent product had a single Tg by DSC and improved impact strength compared to test parts of the individual homopolymers. Polycarbonate and polyestercarbonates were also successfully transesterified with polyphosphonate using this procedure.
9.11 Polyester + Polypropylene Blends
9.11.1 Copolymer Formation by Alcohol + Anhydride Reaction
Bettini et al. (2013) prepared PET/PP blends in a TSE with addition of PP-g-MA, the latter grafted polymer prepared using MA alone or MA in the presence of styrene monomer. The extent of PP degradation and MA incorporation was measured as a function of styrene monomer level. The effect of styrene level in PP-g-(S)MA on subsequent blend properties was assessed using rheology, mechanical properties, and SEM. A similar study was performed by Khonakdar et al. (2013). See also Zhidan et al. (2011) for PET/PP/PP-g-MA blends incorporating recycle PET.
Akbari et al. (2007) prepared PET/PP blends using 5, 10, or 15 wt% PP-g-MA, the latter grafted polymer being prepared by solid-state grafting. Characterization methods included DSC, optical microscopy, SEM, and EDXA.
Xue et al. (2007a, b) investigated the effects of PP-g-MA addition for compatibilizing blends of PTT and PP. Rheological, morphological, thermal and mechanical properties were measured. For blends of poly(esteramide) LCP with PP-g-MA, see Seo et al. (2006).
9.11.2 Copolymer Formation by Carboxylic Acid + Epoxide Reaction
PEST/PP blends have been compatibilized through graft copolymer formation by reaction of polyester carboxylic acid end-groups with pendent epoxide groups on an appropriate PP or on a PO or styrene-polyolefin copolymer showing some miscibility with PP (Table 5.40). The copolymer is joined through a new ester linkage. When the new ester linkage is formed, a secondary alcohol group forms as well through ring opening of the epoxide. It is theoretically possible that the new secondary alcohol may also react with polyester main-chain linkages to form copolymer through a degradative transesterification process. Since there are multiple reactive sites on the epoxide-containing polymers, some cross-linked copolymer may result if the acid-containing polymer is functionalized at both ends. The proportion of cross-linked copolymer formed also depends upon blend composition and processing conditions. An example is also included in this section where a compatibilizing copolymer is postulated to form by reaction between acidic phenolic end-groups on polycarbonate and epoxide groups grafted to PP (Zhihui et al. 1998, 1997).
Using a TSE at 180 °C, Vainio et al. (1996b) have prepared blends of 42 parts poly(butyl acrylate-co-GMA) (2–5 mol% GMA) with 53-58 parts PP and 0-5 parts PP-g-AA (6 % AA) as a compatibilizer. A cross-linked copolymer may result through reaction of epoxide groups along the copolyester main chain with acid groups grafted at more than one site along the PP chain (or with acid groups along a poly(acrylic acid) segment grafted at a single PP site). The blends were characterized by DMTA, gel content, FTIR, and TEM. Other functionalized poly(butylacrylate) copolymers for use in these blends were also prepared by copolymerization with olefinic oxazoline-, amine-, carboxylic acid-, and hydroxyl-containing monomers.
9.11.3 Copolymer Formation by Carboxylic Acid + Oxazoline Reaction
Vainio et al. (1997, 1996a) have compatibilized PEST/PP blends by graft copolymer formation between acid-terminated polyester and oxazoline-grafted PP. Specifically, 30 parts PBT was mixed with 0-70 parts PP and 0-70 parts PP-g-oxazoline in an internal mixer at 250 °C or TSE at 240 °C. Blends were characterized by SEM, torque rheometry, DMA, and DSC. Oxazoline-functionalized PP was prepared by grafting PP with ricinol oxazoline maleinate in the presence of styrene monomer + RI. The inclusion of styrene monomer suppresses radical-induced decomposition of PP. Some cross-linked copolymer may also form in this blend if the polyester is acid-functionalized at both chain ends. See also Vocke et al. (1998) and Jeziórska (2001).
9.11.4 Copolymer Formation through Ionomeric Cross-Linking
Compatibilized blends of 90 parts PBT and 10 parts oxidized PP or its corresponding Na ionomer were prepared in TSE at 240 °C (Dang et al. 2005). Blends were characterized by mechanical properties, morphology, and MFR compared to blends with unfunctionalized PP.
9.11.5 Copolymer Formation by Radical Coupling
Li et al. (2012c) prepared blends of poly(lactic acid) with PP in the presence of radical initiator. Blend characterization techniques included DSC. The effects of different radical initiators and different concentrations of RI were studied.
9.12 Polyester + Polysulfone Blends
Zhang and He (2002) have compatibilized LCP polyester (Hoechst Vectra® B950) with polysulfone-g-MA, the functionalized polysulfone having been prepared in solution. Blend characterization techniques included XPS, DMA, morphology and melt viscosity.
9.13 Polyester + Styrene Copolymer Blends
9.13.1 Copolymer Formation by Alcohol + Anhydride or Carboxylic Acid Reaction
Studies have been made of PEST/styrene copolymer compatibilization in which a copolymer is formed between polyester alcohol end-groups and pendent anhydride functionality on a styrene copolymer (Table 5.41). Because the alcohol-anhydride reaction is reversible with the equilibrium lying on the side of unreacted anhydride, only a relatively small amount of copolymer may be formed. Thus, the dispersed phase polymer may not be well stabilized against coalescence upon further thermal treatment (for a discussion, see, e.g., Sun et al. 1996 and Boyer et al. 2005). Alternatively, at least some copolymer may be formed by a degradative mechanism through transesterification between polyester main-chain linkages and a low concentration of pendent acid groups in anhydride-functionalized styrene copolymer. See also the work of Wu et al. (2010) for blends comprising PBT and ultrafine, vulcanized ABS wherein a copolymer was postulated to form between PBT-OH groups and surface carboxyl groups on ABS.
9.13.2 Copolymer Formation by Carboxylic Acid + Epoxide Reaction
As the examples of Table 5.42 demonstrate, PEST/styrene copolymer blends may be compatibilized through graft copolymer formation by reaction of polyester carboxylic acid end-groups with pendent epoxide groups on an appropriate styrene copolymer. Also included in the table is an example of graft copolymer formation in PC-styrene copolymer blends through reaction of styrene copolymer carboxylic acid pendent groups with epoxide end-groups on an appropriately functionalized PC. In either case, the copolymer formed is joined through a new ester linkage. When the new ester linkage is formed, a secondary alcohol group is also formed through ring opening of the epoxide. The new secondary alcohol may also react with polyester main-chain linkages to generate copolymer through a degradative transesterification process. When there are multiple reactive sites on at least one of the two polymers, some cross-linked copolymer may result depending upon functionalized polymer type, blend composition, and processing conditions.
9.13.3 Copolymer Formation by Transreaction
Bolton et al. (2005) prepared a copolymer of S-AN and alpha-methyl-p-hydroxystyrene. The copolymer was shown to be an effective compatibilizer for PC/SAN blends extruded in the presence of tetraphenylphosphonium benzoate catalyst.
Su et al. (2001a, b) have prepared blends of PBT with hydroxy-functionalized polystyrenes which differed in the number of hydroxy groups per chain. The effects of triphenyl phosphite and titanium butoxide on PBT chain degradation and copolymer formation were studied. Blends were characterized by FTIR, DSC, GPC, morphology, rheology, and mechanical properties.
Wildes et al. (1999) have reported compatibilized blends of PC with amine-functionalized SAN.
In a study by Landry et al. (1994), 50 parts PBT (or PET) was extruded with 50 parts poly(vinylphenol) using a mini-SSE at 254 °C or 293 °C, followed by annealing at 265 °C or 290 °C. The blends were characterized by DSC. Copolymer formation evidently occurs through a degradative reaction between pendent phenolic groups on poly(vinylphenol) and ester linkages in the polyester main chain. A low molecular weight polyester fragment results from this process.
9.13.4 Copolymer Formation by Miscellaneous Reactions
Liu et al. (2011a) prepared compatibilized PBT-ASA blends by addition of epoxy resin coupling agent. Blends were characterized by mechanical properties and morphology (TEM and FESEM).
Lee and Park (2001) prepared compatibilized PC-PBT-PS blends through reacting PBT with oxazoline-functionalized PS. See Becker and Schmidt-Naake (2003) for compatibilized blends of PC with oxazoline-functionalized SAN and Becker and Schmidt-Naake (2004) for compatibilized blends of PC with oxazoline- or benzoxazole-functionalized ABS.
Lee and Park (2000) found evidence for copolymer formation in blends of anhydride-terminated PC and oxazoline-functionalized PS prepared in a Haake mixer. Blends were characterized by torque rheometry, SEM, FTIR, NMR, and mechanical properties. Anhydride-terminated PC was prepared by reaction of PC phenolic end-groups with trimellitic anhydride acid chloride (cf. Hathaway and Pyles 1988, 1989).
Lee et al. (2000) prepared compatibilized PET-PS blends through reacting PET with carbamate-functionalized PS, the carbamate serving as a masked isocyanate.
Ju and Chang (1999) prepared compatibilized PET-PS blends through blending PET and PS with SMA in the presence of a tetra-epoxide coupling agent.
Krabbenhoft (1986) prepared copolymers of PC with SEBS by extrusion with a disulfonyl azide. In one example, a blend of PC (1 part), SEBS (2 parts), and 4,4′-biphenyl disulfonyl azide was extruded on an SSE at 182 °C. This extrudate was melt blended with EEA and additional PC to give a blend comprising 82.5 % PC, 7.5 % SEBS, and 10 % EEA. Copolymer formation was shown by selective solvent extraction. Improved impact strength was seen in comparison to blends prepared without disulfonyl azide.
9.14 Polyester + Polyurethane Blends
Dogan et al. (2013) prepared blends of poly(lactic acid) with TPU through addition of 1,4-phenylene diisocyanate coupling agent. Characterization techniques included mechanical, thermal, morphological, and rheological properties.
Imre et al. (2013) also prepared compatibilized blends of poly(lactic acid) with polyurethane elastomer by coupling reaction under extrusion conditions. Copolymer formation was shown by SEM, AFM, DMA, and mechanical property measurement.
Archondouli et al. (2003) studied blends of PC with polyester-type polyurethane using SEM, mechanical properties, DMA, DSC, TGA, FTIR, and NMR. Formation of copolymer was shown by selective solvent extraction and spectroscopic analysis.
Samios et al. (2000) prepared PET blends with polyester polyurethane. Blends were characterized by mechanical properties, DMA, NMR, and morphology. An esteramide interchange reaction was proposed as the mechanism for compatibilizing copolymer formation.
10 Polyether or Polyphenylene Ether Blends
Examples of polyether blends not shown in earlier sections are listed in alphabetical order of the second polymer in the blend unless otherwise noted. Included in this section are polyphenylene ether blends not described in sections on PA, PEST, or PO. When copolymer characterization was not performed, the structure of the compatibilizing copolymer is inferred from the functionality location on each of the two polymers. In some cases, more than one type of compatibilizing copolymer may have formed.
10.1 Polyether + Polyolefin Blends
10.1.1 Copolymer Formation by Alcohol + Anhydride Reaction
Compatibilizing copolymers have been formed by direct reaction between pendent alcohol groups of phenoxy resin and pendent anhydride groups on MA-grafted polyolefins. Again, the initial product of the reaction was a graft copolymer, but longer reaction time resulted in a cross-linked copolymer since the pendent phenoxy chain is capable of further reaction with MA on a different polyolefin chain. Mascia and Bellahdeb (1994a, b) have blended 75-25 parts phenoxy resin with 25-75 parts EP-g-MA (0.7 % MA) in either an internal mixer or a TSE at 180–200 °C. The blends were characterized by selective solvent extraction, SEM, DSC, and rheology. Increased cross-linking was observed with increasing MA-to-phenoxy ratio and with the addition of either NaOEt or sodium benzoate (2 %). The resulting copolymers were used to compatibilize PET + HDPE blends.
10.1.2 Copolymer Formation by Alcohol + Ester Transreaction
Kim and Choi (1996b) have compatibilized blends of phenoxy resin with PMMA. Compatibilizing cross-linked copolymer resulted from transreaction between phenoxy pendent alcohol groups and pendent PMMA ester groups. Compositions of 100-0 parts phenoxy resin and 0-100 parts PMMA were prepared in an internal mixer at 240 °C. The blends were characterized by DSC, FTIR, mechanical properties, rheology, and DMA. Mascia and Bellahdeb (1994a, b) have compatibilized phenoxy polyether resin with an acid-functionalized polyacrylate resin through cross-linking. A base capable of forming alkoxide from the polyether hydroxy groups was used. Thus, 75-25 parts phenoxy resin and 25-75 parts E-tBA-AA terpolymer (or EMAA Na ionomer) were blended in an internal mixer or TSE at 180–200 °C in the presence of 2 parts NaOEt. Blends were characterized by selective solvent extraction, SEM, DSC, and rheology. The resulting cross-linked copolymers were used to compatibilize PET + HDPE.
10.1.3 Copolymer Formation by Miscellaneous Reactions
Frick et al. (2013) prepared compatibilized blends of PEEK and PTFE (component ratios 0-100 to 100-0) using melt-processable PTFE treated by electron beam radiation to introduce –COF and –COOH functional groups by chain scission. Blend characterization techniques included mechanical properties and morphology.
10.2 Polyether + Styrene Copolymer Blends
10.2.1 Copolymer Formation by Alcohol + Anhydride Reaction
Bayam and Yilmazer (2002) have reported compatibilization of poly(tetramethylene ether glycol) with SMA prepared in a batch mixer or in a TSE in the presence of zinc acetate hydrate. Blends were characterized using thermal, mechanical, morphological, and spectroscopic techniques. FTIR analysis indicated the presence of copolymer from ester formation.
10.2.2 Copolymer Formation by Alcohol + Epoxide Reaction
Compatibilizing copolymers have been formed by direct reaction between pendent alcohol groups of phenoxy resin and pendent epoxide groups on GMA-grafted styrene copolymer. Again, the initial product of the reaction was a graft copolymer, but longer reaction time resulted in a cross-linked copolymer since the pendent phenoxy chain was capable of further reaction with GMA. Furthermore, the new hydroxy group arising from ring-opened epoxy groups may also participate in cross-linking reactions. It is also possible that some cross-linked copolymer resulted from transesterification between pendent hydroxy groups on phenoxy resin and the GMA ester groups. Chen and Chang (1994) have prepared blends of 75-45 parts phenoxy resin and 25-50 parts ABS with addition of 0-10 parts SAN-GMA (5 % GMA) copolymer in an internal mixer or a TSE at 230 °C. The blends were characterized by torque rheometry, SEM, viscosity behavior, and mechanical properties. Sodium lauryl sulfonate (0.02-0.10 parts) was added as a catalyst to promote the copolymer-forming reaction.
10.2.3 Copolymer Formation by Amine + Anhydride Reaction
Schaefer et al. (1995) prepared blends of poly(tetrahydrofuran) with SMA in the presence of bis(amine)-terminated poly(tetrahydrofuran). Characterization techniques included FTIR and microscopy. Reaction rate constants were determined. The effects of temperature and poly(tetrahydrofuran) MW were studied.
10.3 Polyetherimide + Polyphenylene Ether Blends
PEI-PPE blends have also been compatibilized through copolymer formation between functionalized PPE and PEI in the presence of a multifunctional epoxy resin such as PE-co-GMA (Brown et al. 2000a). Functionalized PPE included PPE-g-FA. Catalysts such as sodium stearate were optionally added.
PEI-PPE blends have also been compatibilized through copolymer formation between anhydride-functionalized PPE such as PPE-g-FA and amine-terminated PEI (White et al. 1989). Amine-terminated PEI was prepared from bisphenol A-diphthalic acid anhydride and m-phenylene diamine, the latter used in excess. Copolymer analysis was by selective solvent extraction. Mechanical properties and morphologies were compared to blends with unfunctionalized PPE and PEI.
10.4 Polyetherimide + Polyphenylene Sulfide Blends
PEI-PPS blends have also been compatibilized through copolymer formation between amine-containing PEI and PPS in the presence of a multifunctional epoxy resin such as epoxy cresol novolak (Nazareth 1996).
10.5 Polyphenylene Ether + Polyphenylene Sulfide Blends
Some early work on copolymer-compatibilized PPE-PPS blends is summarized in Arashiro et al. (1992). PPE-PPS blends have also been compatibilized through copolymer formation between citric acid-functionalized PPE (e.g., 162 parts) and PPS (e.g., 225-275 parts) in the presence of a multifunctional epoxy resin (0-17.5 parts) and a minor amount of PBT (25-50 parts) (Brown et al. 1997a) or in the presence of a epoxy-functionalized PO and a catalyst (Brown et al. 2001) or in the presence of a bifunctional cyclic ortho ester compound (Brown et al. 1998a). See also Dekkers (1989) and Inoue et al. (1990). Okabe et al. (1989) have prepared compatibilized blends comprising PPE-g-MA and amine-functionalized PPS in the presence of 4,4′-diphenylmethane diisocyanate coupling agent.
PPE-PPS blends have also been compatibilized through copolymer formation between a PPS bearing nucleophilic functional groups and either epoxy-functionalized PPE (Han et al. 1992) or cyclic ortho ester-functionalized PPE (Brown et al. 1996). In the former case, PPE was functionalized by reaction with a chloro-epoxy triazine and, in the latter, by reaction with either a chloro (cyclic ortho ester) triazine or a graftable cyclic ortho ester.
Kubo and Masamoto (2002) have prepared compatibilized blends of PPE and PPS through addition of styrene-co-GMA.
10.6 Polyphenylene Ether + Polysiloxane Blends
PPE-polysiloxane blends have also been compatibilized through copolymer formation between amine-terminated polydimethylsiloxane and either epoxy-functionalized PPE (Blohm et al. 1995) or anhydride-functionalized PPE (Shea et al. 1989). Compatibilized blends have also been formed by extrusion of PPE with epoxy-functionalized polydimethylsiloxane (Cella et al. 2002) and by kneading of anhydride-functionalized PPE with carboxylic acid-functionalized polydimethylsiloxane in the presence of a diamine coupling agent (Moritomi and Iji 2002).
10.7 Polyphenylene Ether + Styrene Copolymer Blends
PPE is not miscible with SMA containing as much as 28 % MA (Witteler et al. 1993). To compatibilize these two resins, Koning et al. (1993b, 1996) have added a monoamine-terminated PS that can form a graft copolymer with SMA. Since the amine-terminated PS is miscible with PPE, compatibilized PPE-SMA blends are obtained. Specifically, 30 parts of unfunctionalized PPE was blended (internal mixer at 220 °C, or mini-SSE at 280 °C, or TSE at 326 °C) with 56 parts SMA (28 % MA) and 14 parts amine-functionalized PS. The blend was characterized by TEM, SEM, mechanical and thermal properties, DMA, and GPC copolymer detection. The effect of pre-reacting amine-terminated PS with SMA was studied. The blend properties were compared to those for uncompatibilized blends. Blends were also made containing ABS + SEBS. Further examples of compatibilizing copolymer formation in PPE-styrene copolymer blends are shown in Table 5.43.
11 Polyolefin Blends
Examples of polyolefin blends not shown in earlier sections are listed in alphabetical order of the second polymer in the blend unless otherwise noted. PVC is included as a polyolefin. When copolymer characterization was not performed, the structure of the compatibilizing copolymer is inferred from the functionality location on each of the two polymers. In some cases, more than one type of compatibilizing copolymer may have formed.
11.1 Polyolefin + Polyolefin Blends (Excepting Polypropylene)
11.1.1 Copolymer Formation by Alcohol + Anhydride Reaction
Yang et al. (2013) prepared compatibilized blends of EVAl with maleic anhydride-grafted ethylene-octene copolymer (0–25 wt%). Blends were characterized using mechanical, thermal, FTIR, and morphological techniques.
Wang et al. (2007c) prepared blends of HDPE and EVAl compatibilized through addition of HDPE-g-MA. Characterization methods included mechanical, thermal, and rheological properties.
Boyer et al. (2005) have reported that hydroxy-functionalized oligomers have little tendency to form a stable graft to PP-g-MA under the conditions studied.
Lee and Kim (1998) prepared compatibilized blends of EVAl with LDPE by addition of LDPE-g-MA (1–12 phr).
Schmukler et al. (1986a, b) prepared compatibilized blends of PVAl with an anhydride-functionalized polyethylene. For example, 50 wt% PVAl was reacted at 325 °C in a Brabender mixer with 50 wt% HDPE grafted with 1.5 wt% methylbicyclo(2.2.1)-hept-5-ene-2,3-dicarboxylic anhydride. Characterization showed evidence for copolymer formation and no gross phase separation.
11.1.2 Copolymer Formation by Carboxylic Acid + Epoxide Reaction
As shown in Table 5.44, immiscible blends containing at least two polyolefins have been compatibilized through cross-linked copolymer formation between carboxylic acid groups at multiple sites on one polyolefin and epoxide groups at multiple sites on another polyolefin.
11.1.3 Copolymer Formation through Coupling Agent Addition
Examples in Table 5.45 show use of coupling agents that can, in theory, react at sites along the main chain of each of the two immiscible polyolefins. The work of Inoue (1994a, b) and of Naskar et al. (1994) involve concomitant dynamic vulcanization of an elastomeric phase with an added cross-linking agent.
11.1.4 Copolymer Formation through Ionomeric Cross-Linking Mediated by Metal Cations
Examples of some immiscible polyolefin blends that have been compatibilized through ionomeric cross-linking between ionic groups on each polyolefin are listed in Table 5.46.
11.1.5 Copolymer Formation Involving Mechanochemical Radical Formation
Radicals may be formed on polyolefins under high-shear mixing conditions in a process termed mechanochemical radical generation. Two immiscible polyolefins may both be subject to mechanochemical radical generation during extrusion processing. In blends of PO that are not prone to degradation upon radical formation, a cross-linked copolymer may result from the recombination of radicals from each polymer. As a result, a compatibilized blend with improved physical properties may be obtained. In blends of PO subject to chain scission under high-shear mixing conditions, radical recombination reactions may lead to block and eventually random copolymer formation similar to other degradative copolymer formation processes. The examples listed in Table 5.47 show cross-linked copolymer formation in PVC blends with PO. For a general review of polymer reactions under the action of mechanical forces, see Prut (2009). Other aspects of mechanochemical processes in polymers may be found in Beyer and Clausen-Schaumann (2005). For a study of similarities between mechanochemical and high-energy radiation processes in polymer processing, see, e.g., Smith et al. (2001).
Casale and Porter (1975) reported that copolymer formation between NBR and PVC may occur via mechanochemical radical generation on each polymer followed by recombination. The proposed mechanism was based on earlier studies by Akutin (1968). Later, Manoj et al. (1993b) postulated another mechanism for the same system involving hydrolysis of nitrile groups to amide or carboxylic acid followed by displacement of allylic chloride on PVC by amide-NH2 or acid-OH.
11.1.6 Copolymer Formation by Miscellaneous Reactions
Song et al. (2012a) prepared blends of PMMA with various functionalized poly(propylene-ethylene) copolymers. Blends were characterized by mechanical, morphological, and adhesion tests. Compatibility was found to increase in the order of unfunctionalized poly(propylene-ethylene), MA-grafted poly(propylene-ethylene), hydroxy-grafted poly(propylene-ethylene), and secondary amine-grafted poly(propylene-ethylene) which latter species exhibited the best compatibilization efficiency.
Lopattananon et al. (2007) have prepared blends of maleated natural rubber and carboxylated nitrile rubber in the presence of zinc acetate to form a compatibilizing copolymer through ionic cross-links. The effect of different maleation levels was studied. Blend properties were compared to those for blends with unfunctionalized rubbers.
Oliveira et al. (2004) prepared compatibilized blends of EPDM and NBR using mercapto-modified EPDM with oxazoline-functionalized NBR. Mercapto-modified EVAc was also used. Blends were characterized by techniques including mechanical properties and morphology.
Soares et al. (2004) prepared blends of partially hydrolyzed EVAc and NBR using oxazoline-functionalized NBR. Blends were characterized by techniques including mechanical properties, morphology, selective solvent extraction, and FTIR.
Dalai and Wenxiu (2002b, c) have reported blends of either HDPE or LDPE with EVAc effected by radiation cross-linking. Blend characterization techniques included morphology and thermal properties.
Nugay and Nugay (2000) prepared blends of poly(methyl methacrylate)-b-poly(4-vinylpyridine) with poly(acrylic acid), possibly compatibilized through copolymer formation by ion-neutral donor group association.
Some unusual ester cross-linking reactions have been proposed as copolymer-forming processes for immiscible PO blends (Table 5.48). Alternative mechanisms include consideration of the possible contributions of simpler processes such as IPN formation, radical-radical coupling, or ionomeric cross-linking.
11.1.7 Copolymer Formation through Radical Initiator Addition
As shown in the examples listed in Table 5.49, immiscible blends of polyolefins have been compatibilized by copolymer formation brought about by radical coupling in the presence of a radical initiator. The work of Kim et al. (1997a) and Xu et al. (1997) involved copolymer formation between immiscible rubber and plastic phases simultaneous with the dynamic vulcanization of the elastomeric phase.
11.1.8 Copolymer Formation by Transesterification of Pendent Ester Groups
Immiscible polyacrylates have been compatibilized through transesterification between pendent ester groups on one polyacrylate with pendent ester groups on another polyacrylate. Selected examples are listed in Table 5.50.
11.2 Polyolefin + Polyoxymethylene Blends
Compatibilized blends of hydroxylated polyoxymethylene (polyacetal) with carboxylic acid-functionalized PP have been prepared (Chen et al. 1991). The formation of ester linkages between the polymers was proposed. For example, blends comprising hydroxylated polyoxymethylene and muconic acid-grafted PP were made into film by calendering at 200 °C to provide compositions with markedly improved mechanical properties compared to similar blends containing unfunctionalized PP or unfunctionalized polyoxymethylene.
11.3 Polyolefin + Polyphenylene Ether Blends
11.3.1 Blends Containing Functionalized PPE: PO-PPE Copolymer Formation by Carboxylic Acid + Cyclic Ortho Ester or Epoxide Reaction
As shown in Table 5.51, immiscible blends containing a functionalized polyolefin and a functionalized polyphenylene ether have been compatibilized through cross-linked or graft copolymer formation between carboxylic acid groups at multiple sites on one polymer and either cyclic ortho ester groups or epoxide groups at sites on another polymer.
11.3.2 Blends Containing Functionalized PPE: PO-PPE Copolymer Formation through Coupling Agent Addition
Togo et al. (1988) compatibilized PP with PPE by combining carboxylic acid-functionalized PP with carboxylic acid-functionalized PPE in the presence of a diamine. For example, a 1:1 mixture of PP-g-MA and PPE-g-MA was melt kneaded with 1 wt% p-phenylenediamine at 270 °C. The product was formed into sheet with high tensile strength. PP was grafted with MA in the presence of styrene. BPA or a polyepoxide resin was also used in place of diamine.
11.4 Polyolefin + Polyphenylene Ether + Styrene Copolymer Blends
11.4.1 Blends Containing Unfunctionalized PPE + Functionalized PS: PO-PS Copolymer Formation by Carboxylic Acid + Epoxide Reaction
Immiscible blends containing a polyolefin, an unfunctionalized polyphenylene ether and a functionalized styrene copolymer, have been compatibilized through cross-linked copolymer formation between carboxylic acid groups at multiple sites on polyolefin and epoxide groups at multiple sites on styrene copolymer. Fujii and Ting (1988) reported compatibilized blends of EMAA (15 parts), PPE (47 parts), HIPS (30 parts), SEBS (8 parts), and S-co-GMA (1 part) prepared on a TSE at 288 °C. Characterization included mechanical properties vs. blends without functionalized polymers. Abe et al. (1984) prepared blends of EVAc-co-GMA (1 part; 8 wt% VAc; 4 wt% GMA) with SMA (1 part; 8 wt% MA) in a kneader at 200 °C and characterized the copolymer by selective solvent extraction. Blends of the copolymer with PPE/PS had greatly improved mechanical properties compared to blends without copolymer.
11.4.2 Blends Containing Unfunctionalized PPE + Functionalized PS: PO-PS Copolymer Formation by Carboxylic Acid + Oxazoline Reaction
Hohlfeld (1986) prepared compatibilized blends of EAA and unfunctionalized PPE by extrusion with styrene-isopropenyl oxazoline copolymer (S-IPO). In one example, a blend of EAA (30 parts), PPE (35 parts), and S-IPO (35 parts) was mixed in a Brabender mixer at 280 °C. Torque level was higher and mechanical properties were significantly improved compared to blends with PS used in place of S-IPO. See also Xu et al. (1999b) for a study of similar blends.
11.4.3 Copolymer Formation through Ionomeric Cross-Linking Mediated by Metal Cations
Immiscible polyolefin impact modifiers have been compatibilized with PPE/PS blends by employing a polyolefin sulfonate ionomer and also including PS-sulfonate ionomer in the blend (Table 5.52). Alternatively, PPE-sulfonate ionomer could be used in place of PS-sulfonate ionomer. In these compositions, the polyolefin ionomer can cross-link with PS ionomer through metal cations. The PS-sulfonate ionomer is at least partially miscible with PS, which in turn is miscible with PPE. Simple ionomers have been used (Golba and Seeger 1987; Campbell et al. 1986, 1989). In similar blends, a masked ionomer, such as a polyolefin phosphonate ester, could also be used (Brown and McFay 1986, 1987). The masked ionomer is melt processable but generates a polyolefin phosphonate ionomer through ester hydrolysis in situ during extrusion in the presence of zinc stearate. The polyolefin phosphonate ionomer can cross-link with PS sulfonate ionomer that is miscible with PS which in turn is miscible with PPE.
11.5 Polyolefin + Polyphenylene Sulfide
11.5.1 Copolymer Formation by Nucleophile + Epoxide Reaction
Immiscible blends containing an epoxy-functionalized polyolefin and a polyphenylene sulfide have been compatibilized by Oyama et al. (2011) through graft copolymer formation between epoxide groups at multiple sites on either EMA-GMA or E-GMA-g-PMMA and nucleophilic end-groups such as amine, carboxylic acid, or thiophenol on PPS. Blend characterization techniques included morphology and mechanical properties.
Horiuchi and Ishii (2000) prepared PPS blends with LDPE-g-MA and PE-f-GMA in a TSE. Blend characterization techniques included morphology, TEM, EELS, DSC, tribological tests, and mechanical properties.
Hwang et al. (1998) effected graft copolymer formation between epoxide groups at multiple sites on PE-co-GMA and nucleophilic end-groups on PPS (see also Bailly et al. (1997) and Sugie et al. (1985)). SEBS impact modifier was also used in these blends. Han (1991a, b) employed amine- and carboxylic acid-terminated PPS in similar blends. Numerous similar examples of this compatibilization strategy have appeared, as documented, for example, in patents assigned to Toray Industries.
11.5.2 Copolymer Formation by Miscellaneous Reactions
Lehmann (2011) prepared compatibilized blends of PPS and PTFE (component ratios 0-100 to 100-0) using melt-processable PTFE treated by radiation to introduce –COF and –COOH functional groups by chain scission. It was proposed that functional groups on PTFE may react with thiol end-groups on PPS. Blend characterization techniques included mechanical and tribological properties.
11.6 Polyolefin + Polypropylene Blends
11.6.1 Copolymer Formation by Alcohol + Anhydride Reaction
Cascone et al. (2001) prepared compatibilized blends of PP-g-MA with poly(vinyl butyral) having different content of vinyl alcohol units. Demarquette and Kamal (1998) have prepared compatibilized blends of EVAl and PP through inclusion of PP-g-MA (0.1 mol% MA). Characterization methods included SEM, ESCA, and interfacial tension measurement. Tselios et al. (1998) have prepared compatibilized blends of LDPE and PP through inclusion of PP-g-MA (0.8 mol% MA) and EVAl (7.5 mol% vinyl alcohol). Blends were characterized using SEM, torque rheometry, mechanical properties, FTIR, and micro-Raman spectroscopy.
11.6.2 Copolymer Formation by Amine + Anhydride or Amine + Carboxylic Acid Reaction
Coran and Patel (1983b) have shown that the mechanical properties of dynamically vulcanized NBR-PP blends can be improved through copolymer formation between the two immiscible polymers concurrent with vulcanization. In the first example in Table 5.53, block copolymer resulted from reaction of amine-terminated NBR with anhydride-terminated PP. The latter was prepared through functionalization of PP with MA in the presence of radical initiator. In the second example, a block copolymer may have resulted from reaction of acid-terminated NBR with a primary amine-terminated PP. The latter was prepared in a prior reaction between maleic anhydride-terminated PP and triethylenetetramine. It is also possible that the block copolymer may be linked through ionomeric association resulting from protonation of PP-amine with NBR acid.
11.6.3 Copolymer Formation by Carboxylic Acid + Epoxide Reaction
A compatibilizing copolymer may be formed through reaction between carboxylic acid groups grafted onto a PO chain and acrylate epoxide groups grafted onto a PP chain. In an internal mixer, Liu et al. (1993) have prepared compositions comprising 20 parts NBR-g-AA, 0-75 parts PP, and 0-25 parts PP-g-GMA (0.8 % GMA). The blends were characterized by torque rheometry, SEM, FTIR, and mechanical properties. Control blends were made using either unfunctionalized NBR or PP with different GMA levels. Although the exact structure of the copolymer is not known, it is convenient to consider that the reaction leads to a cross-linked copolymer based on the assumption that the acrylate epoxide groups are truly grafted onto the PP chains and are not all at terminal sites. Ao et al. (2006) compatibilized EPDM/PP blends using epoxidized EPDM and PP-g-AA.
Interestingly, Wang et al. (2012b) have reported that solid-state chlorination of PP in the presence of GMA results in high levels of GMA grafting. The PP-g-GMA was shown to form compatibilized blends with hydroxy-terminated butadiene-acrylonitrile rubber.
11.6.4 Copolymer Formation by Carboxylic Acid + Oxazoline Reaction
A compatibilizing copolymer may be formed through reaction of carboxylic acid groups grafted onto a PO chain and olefinic oxazoline groups grafted onto a PP chain. Again, it is reasonable to consider that this copolymer may be cross-linked based on the assumption that the oxazoline groups are truly grafted along the PP chains and are not all at terminal sites. Liu and Baker (1994) and Liu et al. (1993) have prepared PO-PP blends containing 20 parts NBR-g-AA, 0-80 parts PP, and 80-0 parts PP-g-IPO (0.2 % grafted isopropenyl oxazoline) in an internal mixer. The blends were characterized by torque rheometry, SEM, FTIR, and mechanical properties. Control blends were made using either unfunctionalized NBR or different IPO levels on PP.
11.6.5 Copolymer Formation through Coupling Agent Addition
As shown in Table 5.54, PO/PP blends have been compatibilized by copolymer formation formed through addition of a coupling agent such as a tris-acrylate and a radical initiator that can react with both PO and PP in an immiscible blend. Although PP may have at most one unsaturated site per chain (see Coran and Patel 1983b), the immiscible rubber phase may have multiple reactive sites. Depending upon stoichiometry of coupling agent and reactant polymers, the reaction may lead to a cross-linked copolymer. In the examples from Coran and coworkers, an additional coupling agent could be used to dynamically vulcanize the elastomeric phase following copolymer formation with the PP matrix.
11.6.6 Copolymer Formation by Displacement Reaction
Solid-state chlorination of PP may lead to low levels of chlorine incorporation along the PP chain with minimal PP molecular weight degradation. Coran and Patel (1983b) have blended (internal mixer at 190 °C) 50 parts chlorinated PP with 45 parts NBR containing 5 parts amine-terminated NBR. The blend was characterized by mechanical properties and compared to those for blends using unfunctionalized PP. Compatibilization resulted from copolymer formation through displacement reaction of chloride by amine groups. The blend was prepared in the presence of a vulcanization agent (3.75 % of dimethylol phenol plus 0.5 % of SnCl2) to cause concomitant vulcanization of the rubber phase.
11.6.7 Copolymer Formation through Radical Coupling
PO/PP blends have been compatibilized by addition of a radical initiator. Early examples were summarized in a review by Teh et al. (1994); additional examples are listed in Table 5.55. PP can undergo chain scission in the presence of radical initiator to give a terminal radical site. Coupling of this PP radical with a PE radical located on the PE chain may initially yield a graft copolymer. Since the new copolymer may participate in further coupling reactions, a cross-linked copolymer may eventually be formed depending upon reaction time and radical concentration among other factors.
EPDM/PP blends have been compatibilized by exposure to high-intensity ultrasonic waves during extrusion (Feng and Isayev 2004; Chen and Li 2005), as have blends of PP with natural rubber (Oh et al. 2003). HDPE/PP (or HDPE/ground tire rubber) blends have been compatibilized through copolymer formation promoted by gamma irradiation (Sonnier et al. 2010). Dalai and Wenxiu (2002a) have reported blends of PP and EVAc with copolymer formation effected by radiation cross-linking. Blend characterization techniques included morphology and thermal properties.
11.7 Polyolefin + Polypropylene + Styrene Copolymer Blends
Dharmarajan et al. (1995) have prepared compatibilized blends of PP/styrene copolymer with or without functionalized PO. Blends of 100-0 parts PP, 0-100 parts SMA, 0-15 parts EP-g-(primary amine) (0.3 mol% amine), and 0-5 parts PP-(secondary amine) (0.4 wt% amine) were combined in an internal mixer at 220 °C. Blends were characterized by FTIR, DMTA, TEM, rheology, mechanical properties, lap shear adhesion, and paint adhesion. Properties were compared for blends containing either of the two amine-functionalized polymers alone. Reaction of EP-g-(primary amine) with SMA should result in a cross-linked copolymer because EP-g-(primary amine) contains randomly distributed amine functionality in the backbone. The secondary amine-terminated PP was prepared by first extruding PP with MA to form predominantly anhydride-terminated PP, followed by extrusion with N-methyl-1,3-propanediamine to give the secondary amine-terminated PP through reaction of the primary amine of the diamine with the PP-anhydride. Dharmarajan et al. suggested that PP-MA is mostly end-group functionalized with a smaller portion of main-chain-functionalized species as the result of trapping PP radicals before chain scission. Chains with combinations of end-group and main-chain functionalization should also be present since the authors report that their PP-MA contains a significant amount of highly functionalized PP-MA oligomer. Therefore, in these examples, the copolymers prepared from PP-MA and SMA may consist of a mixture of cross-linked and grafted species.
11.8 Polyolefin + Polysiloxane Blends
11.8.1 Copolymer Formation by Amine + Anhydride Reaction
DeLeo et al. (2011) and DeLeo and Velankar (2008) prepared compatibilized blends of polyisoprene-PDMS (70-30 and 30-70) with addition of 0.1–3.0 wt% copolymer of MA-f-polyisoprene and amine-f-PDMS. Characterization methods included optical microscopy and rheology.
Kole et al. (1995) postulated the formation of a cross-linked copolymer through reaction between amine groups distributed along the main chain of a polysiloxane and anhydride groups distributed along the main chain of a PO. A unique feature of this example is the use of acrylamide-grafted siloxane rubber as the source of amine groups. Amine groups alpha to carbonyl groups (as in acrylamide) are much less nucleophilic than typical amine functionality. In an example, 50/50 blends of EPDM-g-MA (1 % MA) with acrylamide-grafted silicone rubber were mixed in an internal mixer at 35 °C, 70 °C, or 150 °C. The blends were characterized by DMA, FTIR, mechanical properties, solvent swelling, and TGA.
11.8.2 Copolymer Formation Involving Mechanochemical Radical Formation
As shown in Table 5.56, copolymer formation was postulated between mechanochemically generated radicals at sites on EMAc and vinyl-functionalized PDMS during melt processing (Santra et al. 1993a, b). The substitution pattern of vinyl groups on PDMS was not reported. Assuming that the vinyl groups are distributed along the PDMS chains (and not present only as end-groups), then the compatibilizing copolymer formed is a cross-linked copolymer. EMAc-PDMS copolymer formed in situ has also been used to compatibilize PDMS with thermoplastic polyurethane (Santra et al. 1995).
11.8.3 Copolymer Formation by Acid-Base Interaction
Blends of carboxylic acid-terminated polybutadiene and amine-terminated PDMS have been prepared by Fleischer et al. (1994). A copolymer with block-like structure was postulated to form. Blends were characterized using pendant drop tensiometry and FTIR.
11.8.4 Copolymer Formation by Miscellaneous Reactions
PMMA/polydimethylsiloxane blends have been compatibilized in the presence of methacryloxypropyl trimethylsiloxane (dos Anjos et al. 2010). Formation of covalent bonds between PMMA and silane coupling agent was examined using FTIR. Modulus and tensile strength were improved in the compatibilized blends.
11.9 Polyolefin + Polystyrene or Styrene Copolymer Blends (Including Polypropylene)
An interesting publication by Martini et al. (2006) describes a method for separating a reactively compatibilized PP/PS blend into its individual polymeric components for analysis. The method involved use of high-temperature, high-pressure, near-critical solvent extraction with n-alkane. The effects of different n-alkanes and different temperatures and the influence of blend morphology and composition on separation efficiency were studied. The compatibilizing copolymer could be isolated and quantified using this procedure.
11.9.1 Copolymer Formation by Acid-Base Interaction
Compatibilized blends of ethylene-methacrylic acid copolymer and PS were prepared by Kim et al. (1998) through addition of S-co-4-vinylpyridine. Similarly, blends of poly(isobutyl methacrylate) were compatibilized with poly(styrene-co-methacrylic acid) using poly(isobutyl methacrylate-co-2-(N,N-dimethylamino)ethyl methacrylate) or poly(isobutyl methacrylate-co-4-vinylpyridine) (Habi and Djadoun 1999). Turcsáyii (1995) has reported compatibilized blends of PE-g-(N-vinylimidazole) with acrylic acid-modified PP.
11.9.2 Copolymer Formation by Alcohol + Anhydride Reaction
Tang et al. (2002) prepared blends of PS and ethylene-vinyl acetate-vinyl alcohol in the presence of SMA using a TSE. Characterization techniques included morphology, mechanical properties, and FTIR.
Tselios et al. (1997) have compatibilized PO/styrene copolymer blends through cross-linked copolymer formation between PO alcohol groups and anhydride groups on styrene copolymer. Specifically, 50 parts EVAl (1.6–7.5 % VAl) was mixed with 50 parts SMA (8.4–14.7 mol% MA) in an internal mixer at 200 °C. The blends were characterized by torque rheometry, FTIR, DSC, TGA, selective solvent extraction, and mechanical properties as a function of mole ratio alcohol to anhydride. Blend properties were compared to those with EVAc in place of EVAl.
11.9.3 Copolymer Formation by Amine + Anhydride Reaction
As shown in Table 5.57, PO/styrene copolymer blends have been compatibilized through cross-linked copolymer formation between amine-functionalized PO and anhydride-functionalized styrene copolymer.
Dharmarajan et al. (1995) have compatibilized PP/styrene copolymer blends by formation of a graft copolymer through reaction of secondary amine-terminated PP (0.4 wt% amine) with SMA. The secondary amine-terminated PP was prepared by first extruding PP with MA to form anhydride-terminated PP followed by extrusion with N-methyl-1,3-propanediamine to give the secondary amine-terminated PP through reaction of the primary amine of the diamine with the PP-anhydride. Blends within the range PP to SMA from 0-100 to 100-0, containing 0-5 parts amine-terminated PP, were prepared in an internal mixer at less than 220 °C. They were characterized by FTIR, DMTA, TEM, mechanical properties, rheology, lap shear adhesion, and paint adhesion. Some cross-linked copolymer may be present if the PP-MA contains more than one anhydride group. A similar compatibilization strategy was used (Datta et al. 1993a) wherein EP-g-MA or PP-g-MA was extruded with an excess of diaminopropane to yield an amine-functionalized PO which was used in blends with SMA and also in combination with an engineering thermoplastic such as PPE, PBT, and SAN (Dekoninck 1993).
11.9.4 Copolymer Formation by Carboxylic Acid + Cyclic Ortho Ester or Epoxide Reaction
PO/styrene copolymer blends have been compatibilized through cross-linked or graft copolymer formation between acid-functionalized PO and epoxide-functionalized PS or vice versa (Table 5.58). Also, acid-functionalized styrene copolymer has been compatibilized with PO grafted with cyclic ortho ester. Anhydride-functionalized polymer is also effective in these blends since some acid groups are present from ring-opened anhydride. Epoxide groups are most frequently introduced into PO or PS by copolymerization with GMA, while cyclic ortho ester groups are introduced by grafting or copolymerization with olefinic cyclic ortho ester.
11.9.5 Copolymer Formation by Carboxylic Acid + Oxazoline Reaction
PO/styrene copolymer blends have been compatibilized through cross-linked copolymer formation between acid-functionalized PO and oxazoline-functionalized PS (Table 5.59). Anhydride-functionalized PO is also effective in these blends since some acid groups may be present from ring-opened anhydride. Oxazoline groups are most frequently introduced into PS by copolymerization of styrene with isopropenyl oxazoline (IPO).
Sundararaj et al. (1995) have prepared blends containing PP-MA and oxazoline-functionalized PS. A graft copolymer may form through reaction between pendent oxazoline groups on PS and terminal acid groups (from some hydrolysis of anhydride groups) on PP. Specifically, 80 parts S-IPO (1 % IPO) was blended with 20 parts PP-MA (0.1 % MA) in either an internal mixer at 200 °C or in a TSE. The blends were characterized by selective solvent extraction and SEM. Morphology development in the different mixing equipment was studied in both reactive and nonreactive blends (i.e., unfunctionalized PS and PP).
11.9.6 Copolymer Formation through Coupling Agent Addition
PO/styrene copolymer blends have been compatibilized by cross-linked copolymer formation in the presence of coupling agent as shown in Table 5.60.
In addition, PP/styrene copolymer blends have also been compatibilized by addition of a bismaleimide coupling agent capable of reacting with both polymers. For example, Inoue (1994a, b) has prepared blends containing 80 parts PP and 20 parts SIS (or SBS) in the presence of 0-0.3 parts m-phenylene bismaleimide coupling agent using a TSE at 210 °C. The blends were characterized by mechanical properties, melt flow rate, DSC, morphology, and selective solvent extraction. Evidence was presented for PP-SIS copolymer formation.
Bromobutyl rubber and SMA blends have been compatibilized through addition of a bifunctional coupling agent (Willis et al. 1990). The authors assumed that a low molecular weight amino alcohol reacted with rubber bromo groups to form a quaternary ammonium salt. The resulting alcohol-functionalized rubber could form cross-linked copolymers through reaction of hydroxy groups with anhydride of SMA. Blends of 30-5 parts bromobutyl rubber with 70-95 parts SMA and 0-8 parts 2-dimethylaminoethanol coupling agent were prepared in an internal mixer at 150–200 °C, followed by TSE processing at 200 °C. The blends were characterized by FTIR, SEM, and mechanical properties. The effects of premixing rubber with amine and the effects of processing conditions were studied. Model reactions for the proposed copolymer-forming reaction were carried out in solution.
11.9.7 Copolymer Formation by Friedel-Crafts Coupling
The Friedel-Crafts alkylation reaction is one of the oldest known methods for attaching an alkyl group to an aromatic ring. The process typically proceeds by reaction between an alkyl cation precursor and an aromatic compound in the presence of a Lewis acid catalyst. In one example, Sun and Baker (1997) obtained copolymer in blends of 80-20 parts LLDPE with 20-80 parts PS and 0.3 parts aluminum chloride. The proposed mechanism involved cation formation on PE through Lewis acid-catalyzed degradation, followed by attachment of PE cation to aromatic rings of PS. Blends were prepared in an internal mixer at 180 °C and characterized by selective solvent extraction, GPC, SEM, FTIR, and mechanical properties. The effects of added styrene monomer were also studied. This process is an example of degradative copolymer formation since the PE graft segment attached to the PS chain has lower molecular weight than the PE phase from which it was derived. Although the initial copolymer formed in this process is a graft copolymer, the grafted PE segments may still be capable of reacting with catalyst with subsequent cation formation and attachment to a different PS chain leading potentially to a cross-linked copolymer. For related work, see also Sun et al. (1998), Diaz et al. (2002, 2005, 2007), Guo et al. (2007), Liu et al. (2009b), Jian-Ping et al. (2011), and Shahbazi et al. (2012). Li et al. (2009a, b, 2011b) employed aluminum chloride in compatibilized blends of PP and PS. A potential issue with any Friedel-Crafts process employed to form a compatibilizing copolymer is removal of residual catalyst from the blend to prevent adverse effects on blend properties during the lifetime of any formed plastic part.
11.9.8 Copolymer Formation by Ion-Neutral Donor Group Association
Compatibilized blends of 77 parts EPDM-SO3Zn salt and 9 parts S-co-4-vinylpyridine with 4 parts zinc stearate (ZnSt) plasticizer were prepared in an internal mixer at 200 °C (Lundberg et al. 1988; Agarwal et al. 1987; Peiffer et al. 1986). The blends were characterized by FTIR, DMA, melt viscosity, DSC, and SEM. Mechanical properties were compared to blends containing unfunctionalized PS or containing EPDM-SO3Na or Mg salts. A copolymer linked by ion-neutral donor group cross-links may form between sulfonate anion and pyridine nitrogen mediated by Zn cation. Related blends comprising sulfonated EPDM and styrene/maleimide/2-vinyl pyridine copolymer have been described by Dean (1986).
11.9.9 Copolymer Formation by Radical Coupling
As shown in Table 5.61, PO blends with either PS or styrene copolymer have been compatibilized through radical coupling reaction. In these examples, radicals were generated either through addition of radical initiators, through addition of a peroxide-containing polymer, through addition of an azide species, through use of ultrasonic oscillation during extrusion, through mechanochemical processing, or through preirradiation.
11.9.10 Copolymer Formation by Thiol-Alkene Coupling
In a series of papers by Soares and coworkers (see, e.g., Soares et al. 2001), compatibilized blends have been prepared through copolymer formation between a thiol-functionalized polymer (also termed a mercapto-functionalized polymer) and a second polymer comprising a double bond (alkene). In a specific example, styrene-butadiene copolymer/EVAc blends with different ratios of components were compatibilized through addition of mercapto-modified EVAc. Evidence for copolymer formation came from FTIR and DMA. Morphology, mechanical properties, and DSC results were also reported.
11.9.11 Copolymer Formation by Transesterification
Hu and Lambla (1995) have blended EMAc (90-65 parts) with monohydroxy-terminated PS (10-35 parts) in an internal mixer at 180–220 °C in the presence of dibutyltin dilaurate or dibutyltin oxide catalyst. A compatibilizing copolymer arises from transesterification between pendent ester groups of EMAc and terminal hydroxy groups of PS. The effects on blend properties of PS molecular weight were reported. The effects of processing conditions and addition of solvent on conversion kinetics were studied.
11.10 Polyolefin + Polyurethane Blends
Ma et al. (2012b) reported compatibilized PVDF-TPU blends comprising PVDF-g-acrylic acid. Farah and Lerma (2010) prepared hydroxy-functionalized PP by reaction of PP-g-MA with 2-aminoethanol and used this functionalized PP in blends with TPU and unfunctionalized PP. Ethylene-octene copolymer was also used in place of PP.
Wang et al. (2007a) prepared compatibilized ethylene-octene copolymer blends with polyurethane using maleated ethylene-octene copolymer and amine-functionalized polyurethane. However, a study by Stutz et al. (1996) found no evidence for copolymer formation between thermoplastic polyurethane and either EAA or SMA under the specific conditions studied.
Qureshi (2009) prepared PP/TPU blends compatibilized using an amine-functionalized PP, which functionalized PP had been prepared in a separate step by extrusion of PP-g-MA with either hexamethylenediamine or dodecamethylenediamine. Blends characterization techniques included SEM, rheology, and mechanical properties.
Lu and Macosko (2004) and Lu et al. (2003) have prepared compatibilized blends of polyurethane with functionalized PP characterizing the blends by rheology, DMA, tensile properties, and morphology. Primary and secondary amine-functionalized PP were more efficient compatibilizers than was PP-g-MA. A degradative mechanism for copolymer formation involving polyurethane chain cleavage was postulated. See also Kobayashi et al. (2011) for related PE/TPU blends.
With regard to reactively compatibilized TPU blends, Lu et al. (2002) determined the relative reactivity of various functionalities toward TPU using model compounds. The ranking of relative reactivity was found to be primary amine (most reactive) > secondary amine >> hydroxyl ∼ carboxylic acid ∼ anhydride >> epoxide (least reactive).
12 Polyphenylene Sulfide Blends
Examples of polyphenylene sulfide blends not shown in other sections are listed in alphabetical order of the second polymer in the blend unless otherwise noted. Included in this section are polyphenylene sulfide blends not containing PA, PEST, or PO. When copolymer characterization was not performed, the structure of the compatibilizing copolymer is inferred from the functionality location on each of the two polymers. In some cases, more than one type of compatibilizing copolymer may have formed.
12.1 Polyphenylene Sulfide + Polysiloxane Blends
12.1.1 Copolymer Formation by Amine + Epoxide Reaction
Compatibilized blends of polyarylene sulfide with epoxy-functionalized polydimethylsiloxane have been prepared by Han (1994). An amine-terminated polysiloxane was functioned in solution with a chloro-epoxy triazine. Blends of 95:5 PPS:polysiloxane were extruded at 130–290 °C to provide compositions with markedly improved mechanical properties compared to a similar blend containing unfunctionalized polysiloxane.
12.2 Polyphenylene Sulfide + Styrene Copolymer Blends
Compatibilized blends of polyarylene sulfide with SEBS-g-MA have been prepared by Hisamatsu et al. (2000). Possibly an amine-terminated PPS reacts with anhydride to form a compatibilizing copolymer. Blend properties were measured as a function of MA content on SEBS. Nam et al. (2003) prepared compatibilized blends of PPS with ABS-g-MA in a TSE. Blends were characterized using optical microscopy, SEM, FTIR, DMA, and heat distortion temperature.
13 Polystyrene or Styrene Copolymer Blends
Examples of polystyrene blends not shown in earlier sections are listed in alphabetical order of the second polymer in the blend unless otherwise noted.
13.1 Polystyrene + Styrene Copolymer Blends
Xu et al. (1999a) prepared compatibilized blends of PS and the Zn salt of sulfonated PS by addition of poly(styrene-b-4-vinylpyridine) diblock copolymer. Characterization methods included SEM, DSC, SAXS, and FTIR. The effect of block copolymer level was studied. Evidence was found for Zn-mediated cross-linking between sulfonate groups and pyridine nitrogen.
Taubitz et al. (1988e) prepared ABS-g-GMA by reactive extrusion and used the product to make compatibilized blends with carboxylic acid-terminated PS. In one example, ABS-g-GMA (40 parts; 1.5 % GMA) was extruded with carboxylic acid-terminated PS (40 parts) on a TSE at 210 °C. The product showed only 15 % unbound PS by selective solvent extraction and GPC. For comparison blends prepared with unfunctionalized ABS showed 96–100 % unbound PS.
13.2 Polystyrene + Polyurethane Blends
Cassu and Felisberti (2001) prepared compatibilized PS/polyurethane blends by reactive extrusion in the presence of SMA. Blends were characterized by rheology, solubility tests, GPC, and SEM to confirm the presence of copolymer.
13.3 Styrene Copolymer + Polysiloxane Blends
Livengood et al. (2002) prepared compatibilized blends of amine-terminated polydimethylsiloxane and SMA by extrusion at 135–210 °C. The products were formulated into toner compositions with improved properties.
14 Summary
This chapter has not presented every known example of a reactive compatibilization strategy, nor has it included every known polymer that has been compatibilized in an immiscible blend with one or more other polymers. However, the compatibilization strategies presented herein illustrate broadly general methods which may be applied to new polymer blends or applied to known polymer blends for a higher return on cost vs. performance ratio.
The papers cited in this chapter reach similar conclusions concerning the effects on immiscible blend properties of a copolymer formed by reactive compatibilization. In virtually all cases, the generated blend morphology shows a smaller dispersed phase particle size than that observed in the absence of copolymer formation. In the majority of cases, this morphology is stabilized against agglomeration and coalescence of the dispersed phase during subsequent thermal processing. The specific morphology size distribution and its stability result in improved mechanical properties, not observed in uncompatibilized blends. A few efforts have been made to characterize the phase interface and to correlate the interfacial thickness with the level of copolymer formed. However, in most older papers, the existence of copolymer is simply inferred from secondary evidence and its level is not quantified.
Except for a few notable cases, there was seldom consideration in these papers about the architectures of copolymers generated and their possible effects on compatibilization efficiency and morphological stability. Furthermore, since morphological stability alone may not result in optimum physical properties, it is sometimes not possible to know if optimum physical properties have been obtained. In numerous papers, clearly more than one type of copolymer architecture can form. For example, in acid-epoxide reactions, a new secondary alcohol may form that may possibly equilibrate into polyester constituting one blend component (see, e.g., Su et al. 1997). Only a few papers discuss the consequences of using di- vs. mono-end-group-functionalized polymers and their possible effects on generating graft vs. cross-linked copolymers. In many papers, the level and type of functionality on the reacting polymers are not specified.
There are numerous cited examples of blends where at least one polymeric component is vulcanizable. Such systems may be quite difficult to analyze after processing. In some cases, formation of an interpenetrating network (IPN) may be mistaken for covalently bonded copolymer. This is particularly so when no deliberate effort has been made to promote vulcanization and its occurrence was adventitious. Selective solvent extraction experiments to detect copolymer can be misinterpreted if some polymer is physically occluded in a matrix of the second polymer.
Some polymers contain reactive functionality but are also themselves subject to mechanochemical radical generation. When such polymers are blended under high-shear mixing with a second functionalized polymer, the architectures of formed copolymer may derive both from the primary, expected reaction and also from an unexpected, radical-radical coupling process.
In some cited examples, a third, multifunctional reagent is added that reacts with both polymers, for example, as a coupling agent. The relative solubility of many such reagents in particular polymers is often unknown. In immiscible blends, the reagent may segregate into one phase and selectively couple or cross-link that phase, in competition with the desired interpolymer reaction at the phase interface. Again, IPN formation may result, leading to confusion when interpreting selective solvent extraction data. For some types of reagents the problem of phase segregation can be solved by preparing a masterbatch in which a slight stoichiometric excess of the reagent is added to one of the polymers. The reagent caps the reactive functionality, but it does not couple or chain extend the polymer in which it is selectively soluble. Thus, reaction of the newly functionalized polymer at a phase interface with a second polymer is facilitated when the two polymers are blended. This strategy often works when using a coupling agent and in certain cases when using a condensing agent but not always when using a nonselective activating agent. However, relative stoichiometries of reacting species are often not considered in older cited examples.
There have been only a few studies of kinetics of copolymer-forming reactions under melt processing conditions because it is quite difficult to generate these data. In the papers cited in this chapter, there are many examples where not only the level of copolymer formed but also the architecture of the copolymer strongly depends on the melt processing time. Many of the blends are “living” in the sense that more copolymer would be generated with additional time. In contrast, some types of copolymer linkage (e.g., the reaction product of an alcohol and a cyclic anhydride) may degrade, and copolymer may be lost with additional processing time or at higher processing temperature. Different copolymer architecture may form with additional processing time, particularly when a redistribution reaction is possible as either the primary or as the secondary reaction (e.g., when a reactive alcohol is generated from epoxide ring opening with acid). It is usually difficult to isolate and characterize a copolymer from a melt-processed polymer blend. Model studies of copolymer formation between immiscible polymers have been performed either in solution (where there is unlimited interfacial volume for reaction) or using hot-pressed films of the polymers (where the interfacial volume for reaction is strictly controlled at a fixed phase interface). Model studies using low molecular weight analogs of the reactive polymers are useful, but their applicability to high molecular weight reacting systems may be limited. Nevertheless, excellent insight into reactive compatibilization processes has been obtained in recent years (such as through bilayer film studies) in publications including, but limited to, those by Scott and Macosko (1994b) (model experiments for interfacial reaction between SMA and either PA-11 or amine-terminated butadiene-acrylonitrile copolymer during reactive polymer blending); Merfeld et al. (1998) (interfacial thickness in bilayers of PPE and styrenics copolymers), Karim et al. (1999) (transesterification at a polymer blend layer examined by multiple physical techniques), Hayashi et al. (2000) (study of interfacial reaction between PA and anhydride-terminated polysulfone by neutron reflectivity and small-angle neutron scattering), Schulze et al. (2000) (reaction kinetics of end-functionalized chains at an amino-terminated PS/anhydride-terminated PMMA interface), Koulic et al. (2001) (premade vs. in situ formed compatibilizer at the PS/PMMA interface), Schulze et al. (2001) (measuring copolymer formation from end-functionalized chains at anhydride-terminated PMMA and amino-terminated PS interface using forward recoil spectrometry and SEC/fluorescence detection), Cheng et al. (2003) (computational modeling of reactive extrusion process), Yeung and Herrmann (2003) (computational modeling of reactive extrusion process), Coote et al. (2003) (neutron reflectometry investigation of polymer-polymer reactions at the interface between immiscible polymers), Jones et al. (2003) (effect of thermodynamic interactions on reactions of anhydride-terminated PMMA and amino-terminated PS in bilayer film), Harton et al. (2005) (diffusion-controlled reactive coupling at polymer-polymer interfaces), Kho et al. (2005) (morphological development at a bilayer film interface of PMMA-GMA/PS-COOH under electric field), Yu et al. (2005) (interfacial reaction kinetics at a PA-6/SMA reactive interface), Zhang et al. (2005) (interfacial morphology development during reactive coupling of anhydride-terminated PMMA and amino-terminated PS in bilayer film), Kim et al. (2006) (bilayer film study of COOH-terminated PS and PMMA-GMA reaction), Chi et al. (2007) (kinetics of interfacial reaction between carboxylic acid-terminated PB and amino-terminated PDMS studied by interfacial tension measurements), Wang et al. (2010a) (investigation of reactive polymer-polymer interface using nanomechanical mapping), and Wang et al. (2013b) (interfacial interchange reaction between PC and amorphous PA).
It is to be hoped that future work on reactive compatibilization will continue to combine the excellent materials science that has been done to date with additional investigations of the more exact nature of chemical processes occurring and their quantitative effect on blend properties. Such knowledge of the chemistry, coupled to fluid mechanics and morphology development models, would provide powerful tools for optimization of known and invention of new reactive compatibilization processes to prepare commercially valuable polymer blends.
15 Cross-References
Crystallization, Micro- and Nano-structure, and Melting Behavior of Polymer Blends
High Performance Polymer Alloys and Blends for Special Applications
Interphase and Compatibilization by Addition of a Compatibilizer
Mechanical Properties of Polymer Blends
Polyethylenes and Their Blends
References
M. Abbate, E. Martuscelli, G. Ragosta, G. Scarinzi, J. Mater. Sci. 26, 1119 (1991)
M. Abbate, V. Di Liello, E. Martuscelli, P. Musto, G. Ragosta, Polymer 33, 2940 (1992)
S. Abdou-Sabet, R.C. Puydak, P. Rader, Rubber Chem. Technol. 69, 476 (1996)
K. Abe, S. Yamauchi, A. Ohkubo, US Patent 4,460,743, 17 July 1984 to Mitsubishi Petrochemical
H. Abe, T. Nishio, Y. Suzuki, T. Sanada, S. Hosoda, T. Okada, Eur. Pat. Appl., 295,103, 12 Dec 1988 to Sumitomo Chemical
L. Abis, R. Braglia, I. Camurati, E. Merlo, K.M. Natarajan, D. Elwood, S.G. Mylonakis, J. Appl. Polym. Sci. 52, 1431 (1994)
D. Abraham, K.E. George, D.J. Francis, Angew. Makromol. Chem. 200, 15 (1992)
D. Abraham, K.E. George, D.J. Francis, J. Appl. Polym. Sci. 67, 789 (1998)
I.A. Abu-Isa, C.B. Jaynes, J.F. O’Gara, J. Appl. Polym. Sci. 59, 1957 (1996)
A.M. Aerdts, G. Groeninckx, H.F. Zirkzee, H.A.M. van Aert, J.M. Geurts, Polymer 38, 4247 (1997)
M. Afshari, R. Kotek, M.H. Kish, H.N. Dast, B.S. Gupta, Polymer 43, 1331 (2002)
P.K. Agarwal, I. Duvdevani, D.G. Peiffer, R.D. Lundberg, J. Polym. Sci. B Polym. Phys. 25, 839 (1987)
P. Agrawal, S.I. Oliveira, E.M. Araujo, J.A. Tomas, J. Mater. Sci. 42, 5007 (2007)
S.M. Aharoni, Polym. Bull. 10, 210 (1983)
S.M. Aharoni, T. Largman, US Patent 4,417,031, 22 Nov 1983, to Allied
S.M. Aharoni, W.B. Hammond, J.S. Szobota, D. Masilamani, J. Polym. Sci. A Polym. Chem. 22, 2567 (1984)
D. Ahn, K. Khait, M.A. Petrich, J. Appl. Polym. Sci. 55, 1431 (1995)
A. Ajji, Polym. Eng. Sci. 35, 64 (1995)
M. Akbari, A. Zadhoush, M. Haghighat, J. Appl. Polym. Sci. 104, 3986 (2007)
M.K. Akkapeddi, J.C. Haylock, J.A. Gervasi, US Patent 4,847,322, 11 July 1989, to Allied-Signal
M.K. Akkapeddi, B. Van Buskirk, A.C. Brown, US Patent 5,162,440, 10 Nov 1992, to Allied-Signal
M.K. Akkapeddi, B. Van Buskirk, C.D. Mason, S.S. Chung, X. Swamikannu, Polym. Eng. Sci. 35, 72 (1995)
M.S. Akutin, A.V. Melik-Kasumov, R.V. Torneu, V.N. Kotrelev, Plast. Massy 12, 45 (1968)
A. Alexandrova, A. Cabrera, M.A. Hernandez, M.J. Cruz, M.J.M. Abadie, O. Manero, D. Likhatchev, Polymer 43, 5397 (2002)
S. Al-Malaika (ed.), Reactive Modifiers for Polymers (Springer, New York, 1997)
S. Al-Malaika, K. Artus, J. Appl. Polym. Sci. 69, 1933 (1998)
S. Al-Malaika, W. Kong, Polymer 46, 209 (2005)
E. Amendola, C. Carfagna, P. Netti, L. Nicolais, S. Saiello, J. Appl. Polym. Sci. 50, 83 (1993)
J. An, J. Ge, Y. Liu, J. Appl. Polym. Sci. 60, 1803 (1996)
J.C. Angola, Y. Fujita, T. Sakai, T. Inoue, J. Polym. Sci. B Polym. Phys. 26, 807 (1988)
Anonymous, GB Patent 998,439, 14 July 1965, to DuPont
P. Antony, S.K. De, Polymer 40, 1487 (1999)
P. Antony, S. Bandyopadhyay, S.K. De, Polymer 41, 787 (2000)
U. Anttila, K. Hakala, T. Helaja, B. Löfgren, J. Seppälä, J. Polym. Sci. A Polym. Chem. 37, 3099 (1999)
Y.H. Ao, S.L. Sun, Z.Y. Tan, C. Zhou, H.X. Zhang, J. Appl. Polym. Sci. 102, 3949 (2006)
Y.H. Ao, K. Tang, N. Xu, H.-D. Yang, H.-X. Zhang, Polym. Bull. 59, 279 (2007a)
Y.H. Ao, S.L. Sun, Z.Y. Tan, C. Zhou, N. Xu, K. Tang, H.D. Yang, H.X. Zhang, Polym. Bull. 58, 447 (2007b)
M. Aramaki, T. Saitou, T. Maekawa, US Patent 6,794,463, 21 Sept 2004, to Asahi
N. Aranburu, J.I. Eguiazábal, J. Appl. Polym. Sci. 127, 5007 (2013)
Y. Arashiro, S. Yamauchi, H. Ohmura, F. Yamada, M. Kihira, Eur. Pat. Appl. 472,960, 4 Mar 1992 to Mitsubishi Petrochemical
E.M. Araujo, E. Hage Jr., A.J.F. Carvalho, J. Appl. Polym. Sci. 87, 842 (2003a)
E.M. Araujo, E. Hage Jr., A.J.F. Carvalho, J. Mater. Sci. 38, 3515 (2003b)
I. Aravind, A. Boumod, Y. Grohens, S. Thomas, Ind. Eng. Chem. Res. 49, 3873 (2010a)
I. Aravind, S. Jose, K.H. Ahn, S. Thomas, Polym. Eng. Sci. 50, 1945 (2010b)
P.S. Archondouli, N.K. Kalfoglou, Polymer 42, 3489 (2001)
P.S. Archondouli, J.K. Kallitsis, N.K. Kalfoglou, J. Appl. Polym. Sci. 88, 612 (2003)
A. Argoud, L. Trouillet-Fonti, S. Ceccia, P. Sotta, Eur. Polym. J. 50, 177 (2014)
R. Armat, A. Moet, Polymer 34, 977 (1993)
R.G. Armstrong, US Patent 3,373,222, 12 Mar 1968, to Continental Can
A. Aróstegui, J. Nazábal, Polym. Adv. Technol. 14, 400 (2003a)
A. Aróstegui, J. Nazábal, Polym. J. 35, 56 (2003b)
L. Arruabarrena, M.E. Munoz, J.J. Pena, A. Santamaria, Polym. Commun. 27, 92 (1986)
E. Ashcraft, H. Ji, J. Mays, M. Dadmun, ACS Appl. Mater. Interfaces 1, 2163 (2009)
K.B. Atwood, M.J. Brown, D.C. Urian, US Patent Appl. 20070232170, 4 Oct 2007, to DuPont
M. Avella, B. Immirzi, M. Malinconico, E. Martuscelli, M.G. Volpe, Polym. Int. 39, 191 (1996)
D.F. Aycock, S.-P. Ting, US Patent 4,600,741, 15 July 1986, to General Electric
D.F. Aycock, S.-P. Ting, US Patent 4,642,358, 10 Feb 1987, to General Electric
S.C.E. Backson, R.W. Richards, S.M. King, Polymer 40, 4205 (1999)
T.-Y. Bae, K.-Y. Park, D.-H. Kim, K.-D. Suh, J. Appl. Polym. Sci. 81, 1056 (2001)
S.-L. Bai, G.-T. Wang, J.-M. Hiver, C. G’Sell, Polymer 45, 3063 (2004)
C.M.E. Bailly, J.P. Shaw, C.-F.R. Hwang, US Patent 5,681,893, 28 Oct 1997, to General Electric
W.E. Baker, M. Saleem, Polymer 28, 2057 (1987a)
W.E. Baker, M. Saleem, Polym. Eng. Sci. 27, 1634 (1987b)
W.E. Baker, C.E. Scott, G.-H. Hu (eds.), Reactive Polymer Blending (Hanser/Gardner Publications, Cincinnati, 2001)
G.P. Balamurugan, S.N. Maiti, Polym. Eng. Sci. 48, 2482 (2008a)
G.P. Balamurugan, S.N. Maiti, Polym. Test. 27, 752 (2008b)
L.L. Ban, M.J. Doyle, M.M. Disko, G.R. Smith, Polym. Commun. 29, 163 (1988)
L.L. Ban, M.J. Doyle, M.M. Disko, G. Braun, G.R. Smith, Subinclusion morphology in compatibilised polymer blends, in Integration of Fundamental Polymer Science and Technology, ed. by P.J. Lemstra, L.A. Kleintjens (Elsevier Applied Science, New York, 1989), p. 117
L. Barangi, F.A. Taromi, H. Nazockdast, Macromol. Symp. 274, 166 (2008)
A.O. Baranov, A.V. Kotova, A.N. Zelenetskii, E.V. Prut, Russ. Chem. Rev. 66, 877 (1997)
A. Bassani, L.A. Pessan, J. Appl. Polym. Sci. 86, 3466 (2002)
A. Bassani, L.A. Pessan, J. Appl. Polym. Sci. 88, 1081 (2003)
D. Basu, A. Banerjee, J. Appl. Polym. Sci. 64, 1485 (1997)
G. Bayam, U. Yilmazer, J. Appl. Polym. Sci. 83, 2148 (2002)
G. Bayam, U. Yilmazer, M. Xanthos, J. Appl. Polym. Sci. 80, 790 (2001)
BCC Research (2013), cited at http://www.bccresearch.com/market-research/plastics/engineering-resins-polymer-alloys-blends-pls020c.html
H.G. Becker, G. Schmidt-Naake, J. Appl. Polym. Sci. 90, 2322 (2003)
H.G. Becker, G. Schmidt-Naake, Chem. Eng. Technol. 27, 909 (2004)
O. Becker, G.P. Simon, T. Rieckmann, J.S. Forsythe, R.F. Rosu, S. Völker, J. Appl. Polym. Sci. 83, 1556 (2002)
J.J. Belles Jr., H.R. Bhattacharjee, Y.P. Khanna, R. Kumar, US Patent 5,055,509, 8 Oct 1991, to Allied-Signal
S.I. Belousov, Y.K. Godovskii, V.V. Matveev, I.V. Sapozhnikova, A.E. Chalykh, Polym. Sci. USSR 34, 254 (1992)
P.L. Beltrame, A. Castelli, M.D. Pasquantonio, M. Canetti, A. Seves, J. Appl. Polym. Sci. 60(579) (1996)
G.A. Berkstresser IV, B.D. Mullen, P.D. Sybert, US Patent 7,323,535, 29 Jan 2008, to General Electric
C. Berti, V. Bonora, F. Pilati, Makromol. Chem. 193, 1679 (1992a)
C. Berti, V. Bonora, F. Pilati, Makromol. Chem. 193, 1665 (1992b)
S.H.P. Bettini, L.C. de Mello, P.A.R. Munoz, A. Ruvolo-Filho, J. Appl. Polym. Sci. 127, 1001 (2013)
M.K. Beyer, H. Clausen-Schaumann, Chem. Rev. 105, 2921 (2005)
P.A. Bhadane, A.H. Tsou, J. Cheng, B.D. Favis, Macromolecules 41, 7549 (2008)
P.A. Bhadane, A.H. Tsou, J. Cheng, M. Ellul, B.D. Favis, Polymer 52, 5107 (2011)
H.R. Bhattacharjee, Y.P. Khanna, US Patent 4,946,909, 7 Aug 1990, to Allied-Signal
A.K. Bhattacharya, R.N. Santra, V.K. Tikku, G.B. Nando, J. Appl. Polym. Sci. 55, 1747 (1995)
A.R. Bhattacharyya, A.K. Ghosh, A. Misra, Polymer 42, 9143 (2001)
A.R. Bhattacharyya, A.K. Ghosh, A. Misra, K.-J. Eichhorn, Polymer 46, 1661 (2005)
A.K. Bhowmick, T. Chiba, T. Inoue, J. Appl. Polym. Sci. 50, 2055 (1993)
J.-E. Bidaux, G.D. Smith, N. Bernet, J.-A.E. Manson, J. Hilborn, Polymer 37, 1129 (1996)
I. Blanco, G. Cicala, C.L. Restuccia, A. Latteri, S. Battiato, A. Scamporrino, F. Samperi, Polym. Eng. Sci. 52, 2498 (2012)
M.L. Blohm, S.B. Brown, G.T. Seeger, P.P. Anderson, US Patent 5,385,984, 31 Jan 1995, to General Electric
C.C. Bohn, S.C. Manning, R.B. Moore, J. Appl. Polym. Sci. 79, 2398 (2001)
D.H. Bolton, D.M. Derikart, N. Kohncke, P. Moulinie, Eur. Pat. Appl. 1,153,981, 15 June 2005, to Bayer
D.C. Bookbinder, P.D. Sybert, US Patent 5,135,990, 4 Aug 1992, to General Electric
R.J.M. Borggreve, R.J. Gaymans, Polymer 30, 63 (1989)
R.J.M. Borggreve, R.J. Gaymans, A.R. Luttmer, Makromol. Chem. Makromol. Symp. 16, 195 (1988a)
R.J.M. Borggreve, R.J. Gaymans, J. Schuijer, J.F. Ingen Housz, Polymer 28, 1489 (1988b)
R.J.M. Borggreve, R.J. Gaymans, H.M. Eichenwald, Polymer 30, 78 (1989a)
R.J.M. Borggreve, R.J. Gaymans, J. Schuijer, Polymer 30, 71 (1989b)
C. Boyer, B. Boutevin, J.J. Robin, Polym. Degrad. Stab. 90, 326 (2005)
D. Braun, W. Illing, Angew. Makromol. Chem. 154, 179 (1987)
S.B. Brown, US Patent 4,994,525, 19 Feb 1991a, to General Electric
S.B. Brown, US Patent 4,997,885, 5 Mar 1991b, to General Electric
S.B. Brown, Reactive extrusion: a survey of chemical reactions of monomers and polymers during extrusion processing, chapter 4, in Reactive Extrusion Principles and Practice, ed. by M. Xanthos (Hanser Publishers, New York, 1992a), p. 75
S.B. Brown, in Symposium on Blends of Amorphous and Crystalline Polymers sponsored by the Division of Polymer Chemistry, 204th ACS Meeting, Washington, DC, 24 Aug 1992b
S.B. Brown, US Patent RE. 34,364, 31 Aug 1993, to General Electric
S.B. Brown, R.C. Lowry, US Patent 5,089,566, 18 Feb 1992a, to General Electric
S.B. Brown, R.C. Lowry, US Patent 5,096,979, 17 Mar 1992b, to General Electric
S.B. Brown, D.J. McFay, Polym. Prepr. Am. Chem. Soc. Div. Polym. Chem. 27(1), 333 (1986)
S.B. Brown, D.J. McFay, US Patent 4,680,329, 14 July 1987, to General Electric
S.B. Brown, C.M. Orlando, Reactive extrusion, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, vol. 14, 2nd edn. (Wiley, New York, 1988), pp. 169–189
S.B. Brown, R.J. Gambale, L.L. McCracken, US Patent 5,030,693, 9 July 1991a, to General Electric
S.B. Brown, J.S. Trent, J.C. Golba Jr., R.C. Lowry, US Patent 5,041,504, 20 Aug 1991b, to General Electric
S.B. Brown, C.-F.R. Hwang, F.F. Khouri, S.T. Rice, J.J. Scobbo Jr., J.B. Yates, US Patent 5,504,165, 2 Apr 1996, to General Electric
S.B. Brown, K.H. Dai, C.-F.R. Hwang, S.T. Rice, J.J. Scobbo Jr., J.B. Yates, US Patent 5,612,401, 18 Mar 1997a, to General Electric
S.B. Brown, C.-F.R. Hwang, F.F. Khouri, S.T. Rice, J.J. Scobbo Jr., J.B. Yates, US Patent 5,698,632, 16 Dec 1997b, to General Electric
S.B. Brown, C.-F.R. Hwang, S.T. Rice, J.J. Scobbo Jr., J.B. Yates, F.F. Khouri, US Patent 5,837,758, 17 Nov 1998a, to General Electric
S.B. Brown, S.M. Cooper, B.A. Giles, US Patent 5,852,085, 22 Dec 1998b, to General Electric
S.B. Brown, J.R. Campbell, C.-F.R. Hwang, S.T. Rice, P.A. Rodgers, J.J. Scobbo Jr., J.B. Yates, US Patent 5,939,490, 17 Aug 1999, to General Electric
S.B. Brown, B.A. Giles, Y. Jin, D. Nazareth, S.T. Rice, R. Puyenbroek, B.J. McKinley, US 6,166,137, 26 Dec 2000a, to General Electric
S.B. Brown, Y. Jin, J.F. Kelley, J. Liao, M.T. Takemori, R.L. Utley, US Patent 6,150,473, 21 Nov 2000b, to General Electric
S.B. Brown, C.-F.R. Hwang, H. Ishida, J.J. Scobbo Jr., J.B. Yates III, US Patent 6,303,708, 16 Oct 2001, to General Electric
S.B. Brown, H.-P. Brack, J.A. Cella, D. Karlik, US Patent 7,547,749, 16 June 2009, to SABIC Innovative Plastics
C.B. Bucknall, Toughened Plastics (Applied Science, London, 1977)
S. Bum, Kil, O. Ok, Park, K.H. Yoon, J. Appl. Polym. Sci. 82, 2799 (2001)
G. Burgisi, M. Paternoster, N. Peduto, A. Saraceno, J. Appl. Polym. Sci. 66, 777 (1997)
J. Bussink, H.T. van de Grampel, Polymer blends, in Ullman’s Encyclopedia of Industrial Chemistry. In: B. Elvers (ed.) (Wiley-VCH Verlag, Weinheim, 2000) doi:10.1002/14356007.a21_273; published on-line
X. Cai, G. Wu, J. Macromol. Sci. B Phys. 53, 347 (2014)
L. Caligiuri, P. Stagnaro, B. Valenti, G. Canalini, Eur. Polym. J. 45, 217 (2009)
J.R. Campbell, US Patent 4,980,415, 25 Dec 1990, to General Electric
J.R. Campbell, P.M. Conroy, R.A. Florence, Polym. Prepr. Am. Chem. Soc. Div. Polym. Chem. 27(1), 331 (1986)
J.R. Campbell, R.E. Williams, S.B. Brown, P.M. Conroy, R.A. Florence, US Patent 4,840,982, 20 June 1989, to General Electric
J.R. Campbell, S.Y. Hobbs, T.J. Shea, V.H. Watkins, Polym. Eng. Sci. 30, 1056 (1990)
J.R. Campbell, T.J. Shea, S.Y. Hobbs, S.B. Brown, US Patent 5,068,286, 26 Nov 1991, to General Electric
L. Canfora, S. Filippi, F.P. La Mantia, Polym. Eng. Sci. 44, 1732 (2004)
L.B. Canto, E. Hage Jr., L.A. Pessan, J. Appl. Polym. Sci. 102, 5795 (2006)
E. Carone Jr., U. Kopcak, M.C. Goncalves, S.P. Nunes, Polymer 41, 5929 (2000)
C. Carrot, S. Mbarek, M. Jaziri, Y. Chalamet, C. Raveyre, F. Prochazka, Macromol. Mat. Eng. 292, 693 (2007)
T.L. Carte, A. Moet, J. Appl. Polym. Sci. 48, 611 (1993)
H. Cartier, G.-H. Hu, Polym. Eng. Sci. 39, 996 (1999)
A. Casale, R.S. Porter, Adv. Polym. Sci. 17, 1 (1975)
A. Casale, R.S. Porter, Polymer Stress Reactions, vol. 1 (Academic, New York, 1978), p. 226 ff
E. Cascone, D.J. David, M.L. DiLorenzo, F.E. Karasz, W.J. MacKnight, E. Martuscelli, M. Raimo, J. Appl. Polym. Sci. 82, 2934 (2001)
P. Cassagnau, A. Michel, Polym. Eng. Sci. 34, 1011 (1994)
P. Cassagnau, M. Bert, V. Verney, A. Michel, Polymer 34, 124 (1993)
S.N. Cassu, M.I. Felisberti, J. Appl. Polym. Sci. 82, 2514 (2001)
M. Castellano, E. Marsano, A. Turturro, M. Canetti, J. Appl. Polym. Sci. 122, 698 (2011)
P. Cavallaro, B. Immirzi, M. Malinconico, E. Martuscelli, M.G. Volpe, Angew. Makromol. Chem. 210, 129 (1993)
A. Cecere, R. Greco, G. Ragosta, G. Scarinzi, A. Taglialatela, Polymer 31, 1239 (1990)
J.A. Cella, J. Liska, V.L. Ulery, G.W. Yeager, S.A. Nye, H. Guo, N. Sing, US Patent 6,339,131, 15 Jan 2002, to General Electric
C. Cercle, B.D. Favis, Polymer 53, 4338 (2012)
M.F. Champagne, M.A. Huneault, C. Roux, W. Peyrel, Polym. Eng. Sci. 39, 976 (1999)
M.F. Champagne, A. Helmert, M.M. Dumoulin, Polym. Mater. Sci. Eng. 83, 465 (2000)
K. Chandramouli, S.A. Jabarin, Adv. Polym. Technol. 14, 35 (1995)
D.-Y. Chang, F.-C. Chang, J. Appl. Polym. Sci. 56, 1015 (1995)
F.-C. Chang, Y.-C. Hwu, Polym. Eng. Sci. 31, 1509 (1991)
P. Charoensirisomboon, T. Chiba, S.I. Solomko, T. Inoue, M. Weber, Polymer 40, 6803 (1999a)
P. Charoensirisomboon, T. Chiba, K. Torikai, H. Saito, T. Ougizawa, T. Inoue, M. Weber, Polymer 40, 6965 (1999b)
P. Charoensirisomboon, T. Chiba, T. Inoue, M. Weber, Polymer 41, 5977 (2000)
S.-H. Chen, F.-C. Chang, J. Appl. Polym. Sci. 51, 955 (1994)
Y. Chen, H. Li, Polymer 46, 7707 (2005)
G. Chen, J. Liu, J. Appl. Polym. Sci. 74, 857 (1999)
Y. Chen, Q. Wang, Polym. Int. 50, 966 (2001)
C.C. Chen, J.L. White, Polym. Eng. Sci. 33, 923 (1993)
C.C. Chen, E. Fontan, K. Min, J.L. White, Polym. Eng. Sci. 28, 69 (1988)
P.N. Chen Sr., A. Forschrim, M. Glick, M. Jaffe, US Patent 5,070,144, 3 Dec 1991, to Hoechst Celanese
P. Chen Sr., V. Sullivan, T. Dolce, M. Jaffe, US Patent 5,182,334, 26 Jan 1993, to Hoechst Celanese
G. Chen, J. Yang, J.J. Liu, J. Appl. Polym. Sci. 71, 2017 (1999)
G. Chen, S. Guo, H. Li, J. Appl. Polym. Sci. 86, 23 (2002)
B. Chen, T. Tang, S. Xu, X. Zhang, B. Huang, Polymer Journal 35, 141 (2003)
L. Chen, H.-Z. Huang, Y.-Z. Wang, J. Jow, K. Su, Polymer 50, 3037 (2009)
W.-C. Chen, S.-M. Lai, M.Y. Chang, Z.-C. Liao, J. Macromol. Sci. B Phys. (2013a). doi:10.1080/00222348.2013.860304; published online
Y. Chen, C. Xu, X. Liang, L. Cao, J. Phys. Chem. B 117, 10619 (2013b)
M.H. Cheng, A.C. Balazs, C. Yeung, V.V. Ginzburg, J. Chem. Phys. 118, 9044 (2003)
P.C. Cheung, S.T. Balke, Ind. Eng. Chem. Res. 36, 1191 (1997)
M.-F. Cheung, A. Golovoy, R.O. Carter, H. van Oene, Ind. Eng. Chem. Res. 28, 476 (1989)
P. Cheung, D. Suwanda, S.T. Balke, Polym. Eng. Sci. 30, 1063 (1990)
C. Chevallier, F. Becquart, C. Benoit, J.-C. Majeste, M. Taha, Polym. Eng. Sci. (2013). doi:10.1002/pen.23816; published online
C. Chi, Y.T. Hu, A. Lips, Macromolecules 40, 6665 (2007)
C.-R. Chiang, F.-C. Chang, J. Appl. Polym. Sci. 61, 2411 (1996)
C.-R. Chiang, F.-C. Chang, Polymer 38, 4807 (1997)
C.-R. Chiang, F.C. Chang, J. Polym. Sci. B Polym. Physics 36, 1805 (1998)
C.-R. Chiang, M.-Y. Ju, F.-C. Chang, Polym. Eng. Sci. 38, 622 (1998)
H.-C. Chin, F.-C. Chang, Polymer 38, 2947 (1997)
H.-C. Chin, K.-C. Chiou, F.-C. Chang, J. Appl. Polym. Sci. 60, 2503 (1996)
V. Chiono, S. Filippi, H. Yordanov, L. Minkova, P. Magagnini, Polymer 44, 2423 (2003)
K.-C. Chiou, F.-C. Chang, J. Polym. Sci. B Polym. Phys. 38, 23 (2000)
Y.-P. Chiou, D.-Y. Chang, F.-C. Chang, Polymer 37, 5653 (1996a)
Y.-P. Chiou, K.-C. Chiou, F.-C. Chang, Polymer 37, 4099 (1996b)
K.-C. Chiou, S.-C. Wu, H.-D. Wu, F.-C. Chang, J. Appl. Polym. Sci. 74, 23 (1999)
K. Cho, F. Li, Macromolecules 31, 7495 (1998)
K. Cho, K.H. Seo, T.O. Ahn, Polymer Journal 29, 987 (1997)
K. Cho, K.H. Seo, T.O. Ahn, J. Appl. Polym. Sci. 68, 1925 (1998)
I. Chodak, I. Chorvath, Makromol. Chem. Macromol. Symp. 75, 167 (1993)
I. Chodak, I. Janigova, A. Romanov, Makromol. Chem. 192, 2791 (1991)
I. Chodak, H. Repin, W. Bruis, I. Janigova, Macromol. Symp. 112, 159 (1996)
G.D. Choi, S.H. Kim, W.H. Jo, M.S. Rhim, J. Appl. Polym. Sci. 55, 561 (1995)
S.H. Choi, I. Cho, K.U. Kim, Polymer Journal 31, 828 (1999)
W.-M. Choi, C.-I. Park, O.O. Park, J.-G. Lim, J. Appl. Polym. Sci. 85, 2084 (2002)
J.-H. Choi, H.-G. Kim, D.-H. Han, J.-C. Lim, D.-H. Oh, K.-E. Min, J. Appl. Polym. Sci. 101, 1 (2006)
N.R. Choudhury, A.K. Bhowmick, J. Appl. Polym. Sci. 38, 1091 (1989)
R. Chowdhury, M.S. Banerji, K. Shivakumar, J. Appl. Polym. Sci. 104, 372 (2007)
S. Cimmino, L. D’Orazio, R. Greco, G. Maglio, M. Malinconico, C. Mancarella, E. Matuscelli, R. Palumbo, G. Ragosta, Polym. Eng. Sci. 24, 48 (1984)
S. Cimmino, F. Coppola, L. D’Orazio, R. Greco, G. Maglio, M. Malinconico, C. Mancarella, E. Matuscelli, G. Ragosta, Polymer 27, 1874 (1986)
A. Colbeaux, F. Fenouillot, J.-F. Gérard, M. Taha, H. Wautier, J. Appl. Polym. Sci. 93, 2237 (2004)
A. Colbeaux, F. Fenouillot, J.-F. Gérard, M. Taha, H. Wautier, J. Appl. Polym. Sci. 95, 312 (2005)
F.L. Colhoun, M.E. Stewart, S. Weinhold, R.D. Peters, R.L. Martin, US Patent Appl. 20110318519, 29 Dec 2011, to Grupo Petrotemex
S. Collins, S.K. Peace, R.W. Richards, W.A. MacDonald, P. Mills, S.M. King, Polymer 42, 7695 (2001)
M.-B. Coltelli, E. Passaglia, F. Ciardelli, Polymer 47, 85 (2006)
M.-B. Coltelli, S. Bianchi, M. Aglietto, Polymer 48, 1276 (2007)
M.-B. Coltelli, M. Aglietto, F. Ciardelli, Eur. Polym. J. 44, 1512 (2008)
M.-B. Coltelli, S. Bronco, C. Chinea, Polym. Degrad. Stab. 95, 332 (2010)
M.L. Coote, D.H. Gordon, L.R. Hutchings, R.W. Richards, R.M. Dalgliesh, Polymer 44, 7689 (2003)
A.Y. Coran, in Thermoplastic Elastomers, ed. by N.R. Legge, G. Holden, H.E. Schroeder (Hanser Publishers, New York, 1987), pp. 133–161, chapter 7
A.Y. Coran, Rubber Chem. Technol. 63, 599 (1990)
A.Y. Coran, Rubber Chem. Technol. 68, 351 (1995)
A.Y. Coran, R. Patel, Rubber Chem. Technol. 56, 210 (1983a)
A.Y. Coran, R. Patel, Rubber Chem. Technol. 56, 1045 (1983b)
A.Y. Coran, R. Patel, in Polypropylene: Structure, Blends, and Composites, ed. by J. Karger-Kocsis (Chapman and Hall Publishers, London, 1995), pp. 162–201, chapter 6
A.Y. Coran, R. Patel, in Thermoplastic Elastomers, ed. by G. Holden, N.R. Legge, R.P. Quirk, H.E. Schroeder, 2nd edn. (Hanser/Gardner Publishers, Cincinnati, 1996), pp. 153–190, chapter 7
A.Y. Coran, R. Patel, in Thermoplastic Elastomers, ed. by G. Holden, H.R. Kricheldorf, R.P. Quirk, 3rd edn. (Carl Hanser Verlag, Munich, 2004), pp. 143–182, chapter 7
A.Y. Coran, R. Patel, D. Williams-Headd, Rubber Chem. Technol. 58, 1014 (1985)
D.A. Costa, C.M.F. Oliveira, J. Appl. Polym. Sci. 69, 857 (1998)
D.A. Costa, C.M.F. Oliveira, J. International, Polym. Mater. 51, 393 (2002)
G. Costa, D. Meli, Y. Song, A. Turturro, B. Valenti, M. Castellano, L. Falqui, Polymer 42, 8035 (2001)
J.A. Covas, A.V. Machado, Int. Polym. Process. 20, 121 (2005)
J.A. Covas, L.A. Pessan, A.V. Machado, N.M. Larocca, Polymer blend compatibilization by copolymers and functional polymers, chapter 7, in Encyclopedia of Polymer Blends, ed. by A.I. Isayev, S. Palsule, vol. 2 (Wiley, New York, 2011), p. 315
A. Crespy, B. Joncour, J.P. Prevost, J.P. Cavrot, C. Caze, Eur. Polym. J. 22, 505 (1986)
A. Crespy, C. Caze, D. Coupe, P. Dupont, J.P. Cavrot, Polym. Eng. Sci. 32, 273 (1992)
J.J. Crevecoeur, L. Nelissen, M.C.M. van der Sanden, P.J. Lemstra, H.J. Mencer, A.H. Hogt, Polymer 36, 753 (1995)
J.-F. Croteau, G.V. Laivins, J. Appl. Polym. Sci. 39, 2377 (1990)
J. Curry, P. Andersen, Adv. Polym. Technol. 11, 3 (1991/1992)
D. Curto, A. Valenza, F.P. La Mantia, J. Appl. Polym. Sci. 39, 865 (1990)
L. D’Orazio, C. Mancarella, E. Martuscelli, J. Mater. Sci. 23, 161 (1988)
S.C.G. Da Costa, M.I. Felisberti, J. Appl. Polym. Sci. 72, 1835 (1999)
S.C.G. Da Costa, M.D.C. Goncalves, M.I. Felisberti, J. Appl. Polym. Sci. 72, 1827 (1999)
S.S. Dagli, K.M. Kamdar, Polym. Eng. Sci. 34, 1709 (1994)
S.S. Dagli, M. Xanthos, J.A. Biesenberger, Polym. Eng. Sci. 34, 1720 (1994)
S. Dalai, C. Wenxiu, J. Appl. Polym. Sci. 86, 3420 (2002a)
S. Dalai, C. Wenxiu, J. Appl. Polym. Sci. 86, 1296 (2002b)
S. Dalai, C. Wenxiu, J. Appl. Polym. Sci. 86, 553 (2002c)
M. Daneshvar, M. Masoomi, J. Appl. Polym. Sci. 124, 2048 (2012)
V.A. Dang, D. Dong, T.T.M. Phan, C.Q. Song, US Patent 6,887,940, 3 May 2005, to Basell Poliolefine Italia
S. Datta, D.J. Lohse, Polymeric Compatibilizers (Hanser/Gardner Publications, Cincinnati, 1996)
S. Datta, N. Dharmarajan, J.-M. Dekoninck, D.A. White, US Patent 5,225,483, 6 July 1993a, to Exxon
S. Datta, N. Dharmarajan, G. Ver Strate, L. Ban, Polym. Eng. Sci. 33, 721 (1993b)
S. Datta, P.P. De, S.K. De, J. Appl. Polym. Sci. 61, 1839 (1996)
A. De Loor, P. Cassagnau, A. Michel, B. Vergnes, J. Appl. Polym. Sci. 53, 1675 (1994)
A. De Loor, P. Cassagnau, A. Michel, B. Vergnes, J. Appl. Polym. Sci. 63, 1385 (1997)
S. De Petris, P. Laurienzo, M. Malinconico, M. Pracella, M. Zendron, J. Appl. Polym. Sci. 68, 637 (1998)
B. De Roover, M. Sclavons, V. Carlier, J. Devaux, R. Legras, A. Momtaz, J. Polym. Sci. A Polym. Chem. 33, 829 (1995)
B. De Roover, J. Devaux, R. Legras, J. Polym. Sci. A Polym. Chem. 35, 901 (2000)
B.D. Dean, US Patent 4,568,724, 4 Feb 1986, to Atlantic Richfield
B.D. Dean, J. Appl. Polym. Sci. 47, 2013 (1993)
D. Debier, J. Devaux, R. Legras, J. Polym. Sci. A Polym. Chem. 33, 407 (1995)
D. Debier, J. Devaux, R. Legras, J. Polym. Sci. B Polym. Phys. 35, 749 (1997a)
D. Debier, S. Vanclooster, J. Devaux, R. Legras, J. Polym. Sci. B Polym. Phys. 35, 735 (1997b)
K. Dedecker, G. Groeninckx, Polymer 39, 4985 (1998)
K. Dedecker, G. Groeninckx, J. Appl. Polym. Sci. 73, 889 (1999)
M.E.J. Dekkers, Eur. Pat. Appl. 341,421, 15 Nov 1989, to General Electric
M.E.J. Dekkers, D.K. Yoshimura, M.P. Laughner, US Patent 5,171,778, 15 Dec 1992, to General Electric
J.-M. Dekoninck, US Patent 5,244,971, 14 Sept 1993, to Exxon
C. DeLeo, S. Velankar, J. Rheol. 52, 1385 (2008)
C. DeLeo, K. Walsh, S. Velankar, J. Rheol. 55, 713 (2011)
N.R. Demarquette, M.R. Kamal, J. Appl. Polym. Sci. 70, 75 (1998)
J. Deng, S. Liang, C. Zhang, W. Yang, Macromol. Rapid Commun. 28, 2163 (2007)
N. Dharmarajan, S. Datta, Polymer 33, 3848 (1992)
N. Dharmarajan, S. Datta, G. Ver Strate, L. Ban, Polymer 36, 3849 (1995)
M.F. Diaz, S.E. Barbosa, N.J. Capiati, Polymer 43, 4851 (2002)
M.F. Diaz, S.E. Barbosa, N.J. Capiati, Polymer 46, 6096 (2005)
M.F. Diaz, S.E. Barbosa, N.J. Capiati, Polymer 48, 1058 (2007)
K. Dijkstra, J. ter Laak, R.J. Gaymans, Polymer 35, 315 (1994)
Q. Ding, W. Dai, J. Appl. Polym. Sci. 107, 3804 (2008)
Y. Ding, Z. Xin, Y. Gao, X. Xu, J. Yin, G. Costa, L. Falqui, B. Valenti, Macromol. Mat. Eng. 288, 446 (2003)
I.H. Do, L.K. Yoon, B.K. Kim, H.M. Jeong, Eur. Polym. J. 32, 1387 (1996)
S.K. Dogan, E.A. Reyes, S. Rastogi, G. Ozkoc, J. Appl. Polym. Sci. (2013). doi:10.1002/app.40251; published online
W. Dong, B. Zou, P. Ma, W. Liu, X. Zhou, D. Shi, Z. Ni, M. Chen, Polym. Int. 62, 1783 (2013)
D.S.C. Dos Anjos, E.C.V. Revoredo, A. Galembeck, Polym. Eng. Sci. 50, 606 (2010)
P. Doshev, D. Tomova, A. Wutzler, H.-J. Radusch, J. Polym. Eng. 25, 375 (2011)
R. Dou, W. Wang, Y. Zhou, L.-P. Li, L. Gong, B. Yin, M.-B. Yang, J. Appl. Polym. Sci. 129, 253 (2013)
M. Dröscher, H. Jadamus, US Patent 4,760,115, 26 July 1988, to Huls
J.F. Dunphy, US Patent 4,851,473, 25 July 1989, to DuPont
J. Duvall, C. Sellitti, C. Myers, A. Hiltner, E. Baer, J. Appl. Polym. Sci. 52, 207 (1994a)
J. Duvall, C. Sellitti, C. Myers, A. Hiltner, E. Baer, J. Appl. Polym. Sci. 52, 195 (1994b)
J. Duvall, C. Sellitti, V. Topolkaraev, A. Hiltner, E. Baer, C. Myers, Polymer 35, 3948 (1994c)
J. Economy, L.A. Schneggenburger, D. Frich, in Transreactions in Condensation Polymers, ed. by S. Fakirov (Wiley-VCH, Weinheim, 1999), p. 195
K.L.L. Eersels, G. Groeninckx, Polymer 37, 983 (1996)
K.L.L. Eersels, G. Groeninckx, J. Appl. Polym. Sci. 63, 573 (1997)
K.L.L. Eersels, G. Groeninckx, M.H.J. Koch, H. Reynaers, Polymer 39, 3893 (1998)
K.L.L. Eersels, A.M. Aerdts, G. Groeninckx, in Transreactions in Condensation Polymers, ed. by S. Fakirov (Wiley-VCH, Weinheim, 1999), p. 267
J.I. Eguiazábal, J. Nazábal, Makromol. Chem. Macromol. Symp. 20/21, 255 (1988)
J.I. Eguiazábal, J. Nazábal, J. Mater. Sci. 25, 1522 (1990)
J.I. Eguiazábal, M.J. Fernandez-Berridi, J.J. Iruin, I. Maiza, J. Appl. Polym. Sci. 59, 329 (1996)
E.G. El’darov, F.V. Mamedov, V.M. Gol’dberg, G.E. Zaikov, Polym. Degrad. Stab. 51, 271 (1996)
J.J. Elmendorf, A.K. Van der Vegt, Fundamentals of morphology formation in polymer blending, chapter 6, in Two-Phase Polymer Systems, ed. by L.A. Utracki (Hanser Publishers, New York, 1991), pp. 165–184
M. Enna, K. Fujihara, Y. Tsugane, Y. Aso, R. Seki, Eur. Pat. Appl. 2,631,270, 28 Aug 2013 to Mitsui Chemicals
B.N. Epstein, US Patent 4,174,358, 13 Nov 1979, to DuPont
I. Espinasse, P. Cassagnau, M. Bert, A. Michel, J. Appl. Polym. Sci. 54, 2083 (1994)
E. Espinosa, M.J. Fernandez-Berridi, I. Maiza, M. Valero, Polymer 34, 382 (1993)
M. Evstatiev, N. Nicolov, S. Fakirov, Polymer 37, 4455 (1996)
A. Fainleib, O. Grigoryeva, O. Starostenko, I. Danilenko, L. Bardash, Macromol. Symp. 202, 117 (2003)
S. Fakirov (ed.), Transreactions in Condensation Polymers (Wiley-VCH, Weinheim, 1999)
Z. Fang, C. Xu, S. Bao, Y. Zhao, Polymer 38, 131 (1997)
H. Farah, F. Lerma Jr., Eur. Pat. Appl. 1,235,879, 25 Aug 2010, to Dow Global Technologies
M. Farmahini-Farahani, S.H. Jafari, H.A. Khonakdar, F. Böhme, H. Komber, A. Yavari, M. Tarameshlou, Polym. Int. 57, 612 (2008)
B.D. Favis, Polymer 35, 1552 (1994)
R. Fayt, P. Teyssie, J. Polym. Sci. C Polym. Lett. 27, 481 (1989)
D. Feldman, J. Macromol. Sci. A Pure Appl. Chem. 42, 587 (2005)
W. Feng, A.I. Isayev, Polym. Eng. Sci. 44, 2019 (2004)
Y. Feng, Y. Hu, J. Yin, G. Zhao, W. Jiang, Polym. Eng. Sci. 53, 389 (2013a)
Y. Feng, G. Zhao, J. Yin, W. Jiang, Polym. Int. (2013b). doi:10.1002/pi.4632; published online
M.J. Fernandez-Berridi, J.J. Iruin, I. Maiza, Polymer 36, 1357 (1995)
S. Filippi, V. Chiono, G. Polacco, M. Paci, L.I. Minkova, P. Maganini, Macromol. Chem. Phys. 203, 1512 (2002)
S. Filippi, H. Yordanov, L. Minkova, G. Polacco, M. Talarico, Macromol. Mat. Eng. 289, 512 (2004)
S. Filippi, L. Minkova, N. Dintcheva, P. Narducci, P. Maganini, Polymer 46, 8054 (2005)
J.K. Fink, Reactive Polymers Fundamentals and Applications : A Concise Guide to Industrial Polymers, 2nd edn. (Elsevier, Waltham, 2013)
M. Fiorini, C. Berti, V. Ignatov, M. Toselli, F. Pilati, J. Appl. Polym. Sci. 55, 1157 (1995)
M. Fiorini, F. Pilati, C. Berti, M. Toselli, V. Ignatov, Polymer 38, 413 (1997)
C.A. Fleischer, A.R. Morales, J.T. Koberstein, Macromolecules 27, 379 (1994)
M.J. Folkes, P.S. Hope, Polymer Blends and Alloys (Blackie Academic, London, 1993)
M.W. Fowler, W.E. Baker, Polym. Eng. Sci. 28, 1427 (1988)
D.W. Fox, R.B. Allen, Compatibility, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, vol. 3, 2nd edn. (Wiley, New York, 1985), p. 772
C. Franke, P. Pötschke, M. Ratzsch, G. Pompe, K. Sahre, D. Voigt, A. Janke, Angew. Makromol. Chem. 206, 21 (1993)
O. Franzheim, T. Rische, M. Stephan, W.J. MacKnight, Polym. Eng. Sci. 40, 1143 (2000)
D.L. Freitag, L. Bottenbruch, M. Schmidt, US Patent 4,533,702, 6 Aug 1985, to Bayer
M. Freluche, I. Iliopoulos, J.J. Flat, A.V. Ruzette, L. Leibler, Polymer 46, 6554 (2005)
A. Frick, D. Sich, G. Heinrich, D. Lehmann, U. Gohs, C. Stern, J. Appl. Polym. Sci. 128, 1815 (2013)
H.G. Fritz, Q. Cai, U. Bölz, R. Anderlik, Macromol. Symp. 83, 93 (1994)
Y. Fu, H. Song, C. Zhou, H. Zhang, S. Sun, Polym. Bull. 70, 1853 (2013)
S. Fujii, S.-P. Ting, US Patent 4,728,461, 1 Mar 1988 to General Electric
S. Fujii, H. Ishida, M. Morioka, A. Saito, R. van der Meer, US Patent 4,873,276, 10 Oct 1989, to General Electric
Y. Fujita, M. Sakuma, K. Kitano, M. Sakaizawa, Y. Yagi, N. Yamamoto, T. Yokokura, Eur. Pat. Appl. 235,876, 9 Sept 1987, to Tonen Sekiyu
N. Furgiuele, A.H. Lebovitz, K. Khait, J.M. Torkelson, Macromolecules 33, 225 (2000)
M. Furuta, I. Murase, T. Yamaguchi, US Patent 5,278,254, 11 Jan 1994, to Sumitomo
R. Gadekar, A. Kulkarni, J.P. Jog, J. Appl. Polym. Sci. 69, 161 (1998)
R.R. Gallucci, R. van der Meer, R.W. Avakian, US Patent 4,873,286, 10 Oct 1989, to General Electric
L. Gani, S. Tence-Girault, M. Millequant, S. Bizet, L. Leibler, Macromol. Chem. Phys. 211, 736 (2010)
Z. Gao, A. Molnar, A. Eisenberg, Blend compatibilization, in Ionomers: Synthesis, Structure, Properties and Applications, ed. by M.R. Tant, K.A. Mauritz, G.L. Wilkes (Blackie Academic and Professional, London, 1997), p. 390
M. García, J.I. Eguiazábal, J. Nazábal, J. Appl. Polym. Sci. 81, 121 (2001)
E. Gattiglia, F.P. La Mantia, A. Turturro, A. Valenza, Polym. Bull. 21, 47 (1989a)
E. Gattiglia, A. Turturro, E. Pedemonte, J. Appl. Polym. Sci. 38, 1807 (1989b)
E. Gattiglia, A. Turturro, E. Pedemonte, G. Dondero, J. Appl. Polym. Sci. 41, 1411 (1990)
E. Gattiglia, A. Turturro, F.P. La Mantia, A. Valenza, J. Appl. Polym. Sci. 46, 1887 (1992)
N.G. Gaylord, J. Macromol. Sci. Chem. A26, 1211 (1989)
A. Ghaffar, C. Sadrmohaghegh, G. Scott, Polym. Degrad. Stab. 3, 341 (1980–1981)
D. Ghidoni, E. Bencini, R. Nocci, J. Macromol. Sci. 31, 95 (1996)
J.H. Glans, M.K. Akkapeddi, US Patent 5,037,897, 6 Aug 1991a, to Allied-Signal
J.H. Glans, M.K. Akkapeddi, Macromolecules, 24, 383 (1991b)
J.C. Golba Jr., G.T. Seeger, Plast. Eng. 43, 57 (1987)
A. Golovoy, M.F. Cheung, H. van Oene, Polym. Eng. Sci. 27, 1642 (1987)
A. Golovoy, M.-F. Cheung, K.R. Carduner, M.J. Rokosz, Polym. Eng. Sci. 29, 1226 (1989)
A.C.O. Gomes, B.G. Soares, M.G. Oliveira, L.A. Pessan, C.M. Paranhos, J. Appl. Polym. Sci. 127, 2192 (2013)
A. Gonzalez-Montiel, H. Keskkula, D.R. Paul, Polymer 36, 4621 (1995a)
A. Gonzalez-Montiel, H. Keskkula, D.R. Paul, Polymer 36, 4605 (1995b)
A. Gonzalez-Montiel, H. Keskkula, D.R. Paul, Polymer 36, 4587 (1995c)
A. Gonzalez-Montiel, H. Keskkula, D.R. Paul, J. Polym. Sci. B Polym. Phys. 33, 1751 (1995d)
A. Gonzalez-Montiel, L.F. Santos, E.S. Guerra, US Patent Appl. 20130144009, 6 June 2013, to CID Centro de Investigacion y Desarrollo Tecnologico SA de CV
R. Gonzalez-Nunez, B.D. Favis, P.J. Carreau, Polym. Eng. Sci. 33, 851 (1993)
T.G. Gopakumar, S. Ponrathnam, A. Lele, C.R. Rajan, A. Fradet, Polymer 40, 357 (1999)
D. Graebling, M. Lambla, H. Wautier, J. Appl. Polym. Sci. 66, 809 (1997)
W.M. Gramlich, M.L. Robertson, M.A. Hillmyer, Macromolecules 43, 2313 (2010)
T.S. Grant, D.V. Howe, US Patent 4,740,552, 26 Apr 1988, to Borg-Warner
T.S. Grant, R.L. Jalbert, US Patent 4,826,933, 2 May 1989, to Borg-Warner Chemicals
T.S. Grant, R.L. Jalbert, D. Whalen, US Patent 4,732,938, 22 Mar 1988, to Borg-Warner
K.G. Gravalos, J.K. Kallitsis, N.K. Kalfoglou, Polymer 36, 1393 (1995)
R. Greco, M. Malinconico, E. Matuscelli, G. Ragosta, G. Scarinzi, Polymer 28, 1185 (1987)
I. Grof, M.M. Sain, O. Durcova, J. Appl. Polym. Sci. 44, 1061 (1992)
G. Guerrica-Echevarría, J.I. Eguiazábal, J. Nazábal, Eur. Polym. J. 43, 1027 (2007)
H. Gui, T. Zhou, L. Li, T. Zhou, F. Liu, Y. Zhan, A. Zhang, J. Appl. Polym. Sci. 130, 3411 (2013)
Z. Guo, L. Tong, Z. Xu, Z. Fang, Polym. Eng. Sci. 47, 951 (2007)
A. Habi, S. Djadoun, Eur. Polym. J. 35, 483 (1999)
R. Hachemi, N. Belhaneche-Bensemra, V. Massardier, J. Appl. Polym. Sci. (2013). doi:10.1002/app.40045; published online
M. Hajian, C. Sadrmohaghegh, G. Scott, Eur. Polym. J. 20, 135 (1984)
W.R. Hale, US Patent 7,368,503, 6 May 2008, to Eastman Chemical
W. Hale, J.-H. Lee, H. Keskkula, D.R. Paul, Polymer 40, 3621 (1999a)
W. Hale, H. Keskkula, D.R. Paul, Polymer 40, 365 (1999b)
W.J.L.A. Hamersma, R.W. Avakian, C.M.E. Bailly, US Patent 5,011,889, 30 Apr 1991, to General Electric
C.Y. Han, US Patent 4,792,586, 20 Dec 1988, to General Electric
C.Y. Han, PCT Intl. Appl. WO 91/018054, 28 Nov 1991a, to General Electric
C.Y. Han, PCT Intl. Appl. WO 91/018055, 28 Nov 1991b, to General Electric
C.Y. Han, US Patent 5,324,796, 28 June 1994, to General Electric
C.Y. Han, W.L. Gately, US Patent 4,689,372, 25 Aug 1987, to General Electric
C.Y. Han, S.B. Brown, E.W. Walles, T. Takekoshi, A.J. Caruso, US Patent 5,122,578, 16 June 1992, to General Electric
S.Y. Han, T.O. Ahn, H.M. Jeong, J. Polym. Eng. 21, 421 (2011)
U.A. Handge, A. Galeski, S.C. Kim, D.J. Dijkstra, C. Gotz, F. Fischer, G.T. Lim, V. Alstadt, C. Gabriel, M. Weber, H. Steininger, J. Appl. Polym. Sci. 124, 740 (2012)
S.J. Hanley, J.J. Rafalko, K.A. Steele, H.C. Linstid, T.J. Dolce, L.H. Sperling, J. Polym. Sci. A Polym. Chem. 37, 3473 (1999)
M. Hara, J.A. Sauer, J. Macromol. Chem. Rev. Macromol. Chem. Phys. C34, 325 (1994)
M. Harada, T. Ohya, K. Iida, H. Hayashi, K. Hirano, H. Fukuda, J. Appl. Polym. Sci. 106, 1813 (2007)
M. Harada, K. Iida, K. Okamoto, H. Hayashi, K. Hirano, Polym. Eng. Sci. 48, 1359 (2008)
C. Harrats, G. Groeninckx, in Modification and Blending of Synthetic and Natural Macromolecules, NATO Science Series, ed. by F. Ciardelli, S. Penczek, vol. 175 (Kluwer, Dordrecht, 2004), p. 155
C. Harrats, T. Omonov, G. Groeninckx, P. Moldenaers, Polymer 45, 8115 (2004)
S.E. Harton, F.A. Stevie, H. Ade, Macromolecules 38, 3543 (2005)
N.G. Harvey, N.M. Bortnick, M.P. Hallden-Abberton, R.B. Queenan, T.D. Goldman, Eur. Pat. Appl. 0500361, 26 Aug 1992, to Rohm and Haas
F. Hassouna, J.-M. Raquez, F. Addiego, P. Dubois, V. Toniazzo, D. Ruch, Eur. Polym. J. 47, 2134 (2011)
S.J. Hathaway, R.A. Pyles, US Patent 4,732,934, 22 Mar 1988, to General Electric
S.J. Hathaway, R.A. Pyles, US Patent 4,800,218, 24 Jan 1989, to General Electric
M. Hayashi, T. Hashimoto, H. Hasegawa, M. Takenaka, H. Grüll, A.R. Esker, M. Weber, S.K. Satija, C.C. Han, M. Nagao, Macromolecules 33, 8375 (2000)
M. Heino, P. Hietaoja, J. Seppälä, T. Harmia, K. Friedrich, J. Appl. Polym. Sci. 66, 2209 (1997a)
M. Heino, J. Kirjava, P. Hietaoja, J. Seppälä, J. Appl. Polym. Sci. 65, 241 (1997b)
L.R. Hepp, Eur. Pat. Appl. 149,192, 24 July 1985, to General Electric
W.L. Hergenrother, M.G. Matlock, R.J. Ambrose, US Patent 4,427,828, 24 Jan 1984, to Firestone Tire & Rubber
W.L. Hergenrother, M.G. Matlock, R.J. Ambrose, US Patent 4,508,874, 2 Apr 1985, to Firestone Tire & Rubber
R. Hettema, J. Van Tol, L.P.B.M. Janssen, Polym. Eng. Sci. 39, 1628 (1999)
P.T. Hietaoja, R.M. Holsti-Miettinen, J.V. Seppälä, O.T. Ikkala, J. Appl. Polym. Sci. 54, 1613 (1994)
T. Hisamatsu, S. Nakano, T. Adachi, M. Ishikawa, K. Iwakura, Polymer 41, 4803 (2000)
D. Hlavata, Z. Krulis, Z. Horak, F. Lednicky, J. Hromadkova, Macromol. Symp. 176, 93 (2001)
J.-C. Ho, K.-H. Wei, Polymer 40, 717 (1999)
S.Y. Hobbs, R.C. Bopp, V.H. Watkins, Polym. Eng. Sci. 23, 380 (1983)
R.W. Hohlfeld, US Patent 4,590,241, 20 May 1986, to Dow
M. Hölderle, M. Bruch, H. Lüchow, W. Gronski, R. Mülhaupt, J. Polym. Sci. A Polym. Chem. 36, 1821 (1998)
R.M. Holsti-Miettinen, J.V. Seppälä, O.T. Ikkala, Polym Eng. Sci. 32, 868 (1992)
R.M. Holsti-Miettinen, J.V. Seppälä, O.T. Ikkala, I.T. Reima, Polym Eng. Sci. 34, 395 (1994)
R.M. Holsti-Miettinen, M. Heino, J.V. Seppälä, J. Appl. Polym. Sci. 57, 573 (1995)
S.M. Hong, S.S. Hwang, Y. Seo, I.J. Chung, K.U. Kim, Polym. Eng. Sci. 37, 646 (1997)
S.M. Hong, H.O. Yoo, S.S. Hwang, K.J. Ihn, C.H. Lee, Polym. J. 33, 457 (2001)
S.M. Hong, S.S. Hwang, J.S. Choi, H.J. Choi, J. Appl. Polym. Sci. 101, 1188 (2006)
I. Hopfe, G. Pompe, K.-J. Eichhorn, Polymer 38, 2321 (1997)
Z. Horak, Z. Krulis, J. Baldrian, I. Fortelný, D. Konecny, Polym. Netw. Blends 7, 43 (1997)
Z. Horák, I. Fortelný, J. Kolařík, D. Hlavatá, A. Sikora, Polymer blends, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, 4th edn. (Wiley, New York, 2002)
S. Horiuchi, Y. Ishii, Polym. J. 32, 339 (2000)
S. Horiuchi, N. Matchariyakul, K. Yase, T. Kitano, H.K. Choi, Y.M. Lee, Polymer 37, 3065 (1996)
S. Horiuchi, N. Matchariyakul, K. Yase, T. Kitano, Macromolecules 30, 3664 (1997a)
S. Horiuchi, N. Matchariyakul, K. Yase, T. Kitano, H.K. Choi, Y.M. Lee, Polymer 38, 6317 (1997b)
S. Horiuchi, N. Matchariyakul, K. Yase, T. Kitano, H.K. Choi, Y.M. Lee, Polymer 38, 59 (1997c)
D.J. Hourston, S. Lane, H.X. Zhang, J.P.C. Bootsma, D.W. Koetsier, Polymer 32, 1140 (1991)
Z. Hrnjak-Murgic, L. Kratofil, Z. Jelcic, J. Jelencic, Z. Janovic, Intl. Polym. Process. 2004/02, 139 (2004)
G.-H. Hu, M. Lambla, J. Polym. Sci. A Polym. Chem. 33, 97 (1995)
G.-H. Hu, Y.-J. Sun, M. Lambla, J. Appl. Polym. Sci. 61, 1039 (1996)
G.-H. Hu, S. Triouleyre, M. Lambla, Polymer 38, 545 (1997)
G. Hu, B. Wang, X. Zhou, Mater. Lett. 58, 3457 (2004)
J.-M. Huang, J. Appl. Polym. Sci. 88, 2247 (2003)
C.-C. Huang, F.-C. Chang, Polymer 38, 4287 (1997a)
C.-C. Huang, F.-C. Chang, Polymer 38, 2135 (1997b)
J.J. Huang, D.R. Paul, Polymer 47, 3505 (2006)
Z.H. Huang, L.H. Wang, Makromol. Chem. Rapid Commun. 7, 255 (1986)
Y. Huang, Y. Liu, C. Zhao, J. Appl. Polym. Sci. 69, 1505 (1998)
J.-M. Huang, M.-F. You, F.-C. Chang, J. Appl. Polym. Sci. 82, 3321 (2001)
J.-M. Huang, M.-Y. Ju, C.-J. Hung, W.-C. Luoh, F.-C. Chang, J. Appl. Polym. Sci. 87, 967 (2003)
J.J. Huang, H. Keskkula, D.R. Paul, Polymer 45, 4203 (2004)
J.J. Huang, H. Keskkula, D.R. Paul, Polymer 47, 639 (2006a)
J.J. Huang, H. Keskkula, D.R. Paul, Polymer 47, 624 (2006b)
J.-W. Huang, C.-C. Chang, C.-C. Kang, M.-Y. Yeh, Thermochim. Acta 468, 66 (2008)
B. Huang, D. Li, Z. Li, X. Li, W. Zhou, J. Appl. Polym. Sci. 122, 586 (2011)
C.-J. Hung, H.-J. Chuang, F.-C. Chang, J. Appl. Polym. Sci. 107, 831 (2008)
C.-F.R. Hwang, J.J. Scobbo Jr., J.B. Yates, US Patent 5,583,179, 10 Dec 1996, to General Electric
C.-F.R. Hwang, J.J. Scobbo Jr., S.B. Brown, US Patent 5,723,542, 3 Mar 1998, to General Electric
S.W. Hwang, H.C. Ryu, S.W. Kim, H.Y. Park, K.H. Seo, J. Appl. Polym. Sci. 125, 2732 (2012)
J. Ibuki, P. Charoensirisomboon, T. Chiba, T. Ougizawa, T. Inoue, M. Weber, E. Koch, Polymer 40, 647 (1999)
F. Ide, A. Hasegawa, J. Appl. Polym. Sci. 18, 963 (1974)
V. Ignatov, C. Carraro, V. Tartari, R. Pippa, F. Pilati, C. Berti, M. Toselli, M. Fiorini, Polymer 37, 5883 (1996)
V. Ignatov, C. Carraro, V. Tartari, R. Pippa, M. Scapin, F. Pilati, C. Berti, M. Toselli, M. Fiorini, Polymer 38, 201 (1997a)
V. Ignatov, C. Carraro, V. Tartari, R. Pippa, M. Scapin, F. Pilati, C. Berti, M. Toselli, M. Fiorini, Polymer 38, 195 (1997b)
D.J. Ihm, J.L. White, J. Appl. Polym. Sci. 60, 1 (1996)
O.T. Ikkala, R.M. Holsti-Miettinen, J. Seppälä, J. Appl. Polym. Sci. 49, 1165 (1993)
I. Iliopoulos, L. Leibler, M. Freluche, J.-J. Flat, P. Gerard, Macromol. Symp. 245–246, 371 (2006)
B. Immirzi, M. Malinconico, E. Martuscelli, M.G. Volpe, Macromol. Symp. 78, 243 (1994)
B. Imre, B. Pukanszky, Eur. Polym. J. 49, 1215 (2013)
B. Imre, D. Bedo, A. Domjan, P. Schon, J. Vancso, B. Pukanszky, Eur. Polym. J. 49, 3104 (2013)
T. Inoue, J. Appl. Polym. Sci. 54, 723 (1994a)
T. Inoue, J. Appl. Polym. Sci. 54, 709 (1994b)
T. Inoue, T. Suzuki, J. Appl. Polym. Sci. 56, 1113 (1995)
T. Inoue, T. Suzuki, J. Appl. Polym. Sci. 59, 1443 (1996)
K. Inoue, A. Saito, M. Morioka, Eur. Pat. Appl. 368,413, 16 May 1990, to GE Plastics Japan
M. Ishikawa, M. Sugimoto, T. Inoue, J. Appl. Polym. Sci. 62, 1495 (1996)
A. Ishio, K. Saitoh, S. Kobayashi, US Patent 8,076,423, 13 Dec 2011, to Toray Industries
B. Jacques, R. Legras, J. Devaux, E. Nield, Makromol. Chem. Macromol. Symp. 75, 231 (1993)
B. Jacques, J. Devaux, R. Legras, E. Nield, Polymer 37, 4085 (1996a)
B. Jacques, J. Devaux, R. Legras, E. Nield, Polymer 37, 1189 (1996b)
B. Jacques, J. Devaux, R. Legras, E. Nield, J. Polym. Sci. A Polym. Chem. 34, 1189 (1996c)
B. Jacques, J. Devaux, R. Legras, E. Nield, Polymer 38, 5367 (1997)
S.H. Jafari, P. Pötschke, M. Stephan, H. Warth, H. Alberts, Polymer 43, 6985 (2002a)
S.H. Jafari, P. Pötschke, M. Stephan, G. Pompe, H. Warth, H. Alberts, J. Appl. Polym. Sci. 84, 2753 (2002b)
S.-H. Jafari, H.A. Khonakdar, A. Asadinezhad, L. Haussler, F. Bohme, H. Komber, Macromol. Chem. Phys. 210, 1291 (2009)
R.L. Jalbert, T.S. Grant, US Patent 4,654,405, 31 Mar 1987, to Borg-Warner Chemicals
S.C. Jana, N. Patel, D. Dharaiya, Polymer 42, 8681 (2001)
M. Jaziri, N. Barhoumi, V. Massardier, F. Melis, J. Appl. Polym. Sci. 107, 3451 (2008)
H.K. Jeon, J.K. Kim, Macromolecules 31, 9273 (1998a)
H.K. Jeon, J.K. Kim, Polymer 39, 6227 (1998b)
H.K. Jeon, J.K. Kim, Macromolecules 33, 8200 (2000)
H.K. Jeon, O.H. Taek, J.K. Kim, Polymer 42, 3259 (2001)
H.K. Jeon, B.J. Feist, S.B. Koh, K. Chang, C.K. Macosko, R.P. Dion, Polymer 45, 197 (2004a)
H.K. Jeon, C.W. Macosko, B. Moon, T.R. Hoye, Z. Yin, Macromolecules 37, 2563 (2004b)
H.K. Jeon, J. Zhang, C.K. Macosko, Polymer 46, 12422 (2005)
R. Jeziórska, Macromol. Symp. 170, 21 (2001)
R. Jeziórska, Polym. Degrad. Stab. 90, 224 (2005)
C. Jiang, S. Filippi, P. Magagnini, Polymer 44, 2411 (2003)
G. Jiang, H. Wu, S. Guo, Polym. Eng. Sci. 50, 2273 (2010)
J. Jiang, L. Su, K. Zhang, G. Wu, J. Appl. Polym. Sci. 128, 3993 (2013)
X. Jian-Ping, C. Jian-Ding, C. Ming-Lian, J. Appl. Polym. Sci. 119, 2961 (2011)
J. Jiao, E.J. Kramer, S. de Vos, M. Moller, C. Koning, Polymer 40, 3585 (1999)
W.H. Jo, H.C. Kim, Polym. Bull. 27, 465 (1992)
W.H. Jo, C.D. Park, M.S. Lee, Polymer 37, 1709 (1996)
J. John, M. Bhattacharya, Polym. Int. 49, 860 (2000)
T.D. Jones, J.S. Schulze, C.W. Macosko, T.P. Lodge, Macromolecules 36, 7212 (2003)
S. Jose, P.S. Thomas, S. Thomas, J. Karger-Kocsis, Polymer 47, 6328 (2006a)
S. Jose, B. Francis, S. Thomas, J. Karger-Kocsis, Polymer 47, 3874 (2006b)
S. Jose, S.V. Nair, S. Thomas, J. Karger-Kocsis, J. Appl. Polym. Sci. 99, 2640 (2006c)
M.-Y. Ju, F.-C. Chang, J. Appl. Polym. Sci. 73, 2029 (1999)
M.-Y. Ju, M.-Y. Chen, F.-C. Chang, Macromol. Chem. Phys. 201, 2298 (2000)
P.C. Juliano, General Electric Co., personal communication based on literature survey, 1988
J.-B. Jun, J.-G. Park, K.-D. Suh, J. Appl. Polym. Sci. 74, 465 (1999)
W.-C. Jung, K.-Y. Park, J.-Y. Kim, K.-D. Suh, J. Appl. Polym. Sci. 88, 2622 (2003)
P. Juntuek, C. Ruksakulpiwat, P. Chumsamrong, Y. Ruksakulpiwat, J. Appl. Polym. Sci. 125, 745 (2012)
S. Kadoi, H. Yabe, K. Kobayashi, US Patent 5,488,084, 30 Jan 1996, to Toray Industries
N.K. Kalfoglou, D.S. Skafidas, D.D. Sotiropoulou, Polymer 35, 3624 (1994)
N.K. Kalfoglou, D.S. Skafidas, J.K. Kallitsis, J.-C. Lambert, L. Van der Stappen, Polymer 36, 4453 (1995)
N.K. Kalfoglou, D.S. Skafidas, J.K. Kallitsis, Polymer 37, 3387 (1996)
W. Kamoun, S. Salhi, B. Rousseau, R. El Gharbi, A. Fradet, Macromol. Chem. Phys. 207, 2042 (2006)
T.-K. Kang, Y. Kim, G. Kim, W.-J. Cho, C.-S. Ha, Polym Eng. Sci. 37, 603 (1997)
A. Kanomata, K. Yamauchi, S. Horiuchi, H. Sakata, S. Honda, I. Asano, US Patent Appl. 20110311816, 22 Dec 2011, to Toray Industries
B. Karaağaç, V. Deniz, in Advances in Elastomers I, ed. by P.M. Visakh, S. Thomas, A.K. Chandra, A.P. Mathew (Springer, Berlin/Heidelberg, 2013), p. 263
J. Karger-Kocsis, Thermoplastic rubbers via dynamic vulcanization, chapter 5, in Polymer Blends and Alloys, ed. by G.O. Shonaike, G.P. Simon (Marcel Dekker, New York, 1999), pp. 125–154
A. Karim, J.F. Douglas, S.K. Satja, C.C. Han, R.J. Goyette, Macromolecules 32, 1119 (1999)
H. Kasahara, K. Fukuda, H. Suzuki, US Patent 4,339,376, 13 July 1982, to Asahi-Dow
H. Kasahara, K. Tazaki, K. Fukuda, H. Suzuki, US Patent 4,421,892, 20 Dec 1983, to Asahi-Dow
A. Kausar, S. Zulfiqar, M.I. Sarwar, J. Appl. Polym Sci. (2013). doi:10.1002/app.39954; published online
H. Kauth, K. Reinking, D. Freitag, US Patent 4,782,123, 1 Nov 1988, to Bayer
J. Kawada, M. Mouri, O. Watanabe, A. Usuki, M. Kito, A. Amari, O. Kito, PCT Intl. Appl. WO 2013/191307, 27 Dec 2013, to Toyota
A. Kaya, G. Pompe, U. Schulze, B. Voit, J. Pionteck, J. Appl. Polym Sci. 86, 2174 (2002)
Y. Kayano, H. Keskkula, D.R. Paul, Polymer 38, 1885 (1997)
I. Kelnar, M. Stephan, L. Jakisch, I. Fortelný, J. Appl. Polym. Sci. 66, 555 (1997)
I. Kelnar, M. Stephan, L. Jakisch, I. Fortelný, J. Appl. Polym. Sci. 74, 1404 (1999)
Y.P. Khanna, US Patent 4,861,838, 29 Aug 1989, to Allied-Signal
Y.P. Khanna, E.A. Turi, S.M. Aharoni, T. Largman, US Patent 4,417,032, 22 Nov 1983, to Allied Corporation
D.H. Kho, S.H. Chae, U. Jeong, H.Y. Kim, J.K. Kim, Macromolecules 38, 3820 (2005)
H.A. Khonakdar, S.H. Jafari, S. Mirzadeh, M.R. Kalaee, D. Zare, M.R. Saehb, J. Vinyl & Additive Tech. 19, 25 (2013)
F.F. Khouri, US Patent 5,231,132, 27 July 1993, to General Electric
F.F. Khouri, US Patent 5,567,780, 22 Oct 1996, to General Electric
F.F. Khouri, J.R. Campbell, US Patent 5,691,411, 25 Nov 1997, to General Electric
F.F. Khouri, R.J. Halley, J.B. Yates III, US Patent 5,247,006, 21 Sept 1993, to General Electric
F.F. Khouri, O. Phanstiel, US Patent 5,162,448, 10 Nov 1992, to General Electric
F.F. Khouri, G.J. Stoddard, US Patent 5,391,616, 21 Feb 1995a, to General Electric
F.F. Khouri, G.J. Stoddard, US Patent 5,393,833, 28 Feb 1995b, to General Electric
F.F. Khouri, S.B. Brown, J.T. Jackman, US Patent 5,115,042, 19 May 1992, to General Electric
F.F. Khouri, R.J. Halley, J.B. Yates III, US Patent 5,281,667, 25 Jan 1994, to General Electric
B.K. Kim, C.H. Choi, J. Appl. Polym. Sci. 60, 2199 (1996a)
B.K. Kim, C.H. Choi, Polymer 37, 807 (1996b)
B.K. Kim, I.H. Do, J. Appl. Polym. Sci. 61, 439 (1996)
J.W. Kim, S.C. Kim, Polym. Adv. Technol. 2, 177 (1991)
J.K. Kim, H. Lee, Polymer 37, 305 (1996)
B.K. Kim, S.J. Park, J. Appl. Polym. Sci. 43, 357 (1991)
B.K. Kim, S.Y. Park, S.J. Park, Eur. Polym. J. 27, 349 (1991)
B.K. Kim, Y.M. Lee, H.M. Jeong, Polymer 34, 2075 (1993)
K.-H. Kim, W.-J. Cho, C.-S. Ha, J. Appl. Polym. Sci. 59, 407 (1996)
J.K. Kim, S. Kim, C.E. Park, Polymer 38, 2155 (1997a)
S. Kim, J.K. Kim, C.E. Park, Polymer 38, 2113 (1997b)
S. Kim, J.K. Kim, C.E. Park, Polymer 38, 1809 (1997c)
D.H. Kim, W.H. Jo, S.C. Lee, H.C. Kim, J. Appl. Polym Sci. 69, 807 (1998)
Y.H. Kim, M. Kikuchi, S. Akiyama, K. Sho, S. Izawa, Polymer 40, 5273 (1999a)
S.H. Kim, S.M. Hong, S.S. Hwang, H.O. Yoo, J. Appl. Polym Sci. 74, 2448 (1999b)
J.K. Kim, D.K. Yi, H.K. Jeon, C.E. Park, Polymer 40, 2737 (1999c)
D.H. Kim, K.Y. Park, K.-D. Suh, J.-Y. Kim, J. Macromol. Sci. A Pure Appl. Chem. 37, 1141 (2000a)
D.H. Kim, K.Y. Park, J.-Y. Kim, K.-D. Suh, J. Appl. Polym Sci. 78, 1017 (2000b)
S.J. Kim, B.-S. Shin, J.-L. Hong, W.-J. Cho, C.-S. Ha, Polymer 42, 4073 (2001a)
J.H. Kim, W.S. Lyoo, W.S. Ha, J. Appl. Polym. Sci. 82, 159 (2001b)
J.-S. Kim, M. Luqman, J.-M. Song, Ionomers, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, 4th edn. (Wiley, New York, 2002)
S.J. Kim, C.-J. Kang, S.R. Chowdhury, W.-J. Cho, C.-S. Ha, J. Appl. Polym. Sci. 89, 1305 (2003)
H.Y. Kim, H.J. Kim, J.K. Kim, Polymer Journal 38, 1165 (2006)
D.P.R. Kint, A.M. de Ilarduya, A. Sansalvado, J. Ferrer, S. Munoz-Guerra, J. Appl. Polym. Sci. 90, 3076 (2003)
J. Kirjava, T. Rundqvist, R. Holsti-Miettinen, M. Heino, T. Vainio, J. Appl. Polym. Sci. 55, 1069 (1995)
Y. Kobayashi, T. Itoh, T. Inoue, Eur. Pat. Appl. 282,052, 14 Sept 1988, to Mitsubishi Petrochemical
S. Kobayashi, J. Song, H.C. Silvis, C.W. Macosko, M.A. Hillmyer, Ind. Eng. Chem. Res. 50, 3274 (2011)
M. Kodama, Polym. Eng. Sci. 32, 267 (1992)
M.I. Kohan, W.H. Martin, C.K. Rosenbaum, US Patent 3,676,400, 11 July 1972, to DuPont
S. Kole, S. Roy, A.K. Bhowmick, Polymer 36, 3273 (1995)
C. Komalan, K.E. George, S. Jacob, S. Thomas, Polym. Adv. Technol. 19, 351 (2008)
Y. Kong, J.N. Hay, Polymer 43, 1805 (2002)
C. Koning, R. Fayt, W. Bruls, L. Vondervoort, T. Rauch, P. Teyssie, Makromol. Chem. Macromol. Symp. 75, 159 (1993a)
C. Koning, A. Ikker, R. Borggreve, L. Leemans, M. Möller, Polymer 34, 4410 (1993b)
C. Koning, W. Bruis, F. Op Den Buijsch, L. Vondervoort, Macromol. Symp. 112, 167 (1996)
C. Koning, M. van Duin, C. Pagnoulle, R. Jérôme, Prog. Polym. Sci. 23, 707 (1998)
E.V. Konyukhova, S.I. Belousov, Y.V. Kuz’mina, N.P. Bessonova, Y.K. Godovsky, Polym. Sci., Ser. A 36(9), 1209 (1994).
H. Koriyama, H.T. Oyama, T. Ougizawa, T. Inoue, M. Weber, E. Koch, Polymer 40, 6381 (1999)
A.M. Kotliar, J. Polym. Sci. Macromol. Rev. 16, 367 (1981)
C. Koulic, Z. Yin, C. Pagnoulle, B. Gilbert, R. Jérôme, Polymer 42, 2947 (2001)
C. Koulic, G. Francois, R. Jérôme, Macromolecules 37, 5317 (2004)
E.G. Koulouri, K.G. Gravalos, J.K. Kallitsis, Polymer 37, 2555 (1996)
E.G. Koulouri, K.G. Gravalos, J.K. Kallitsis, Polymer 38, 4185 (1997)
E.G. Koulouri, J.K. Kallitsis, G. Hadziioannou, Macromolecules 32, 6242 (1999a)
E.G. Koulouri, E.C. Scourlis, J.K. Kallitsis, Polymer 40, 4887 (1999b)
H.O. Krabbenhoft, US Patent 4,579,905, 1 Apr 1986, to General Electric
L.J. Kratofil, A. Pticek, Z. Hrnjak-Murgic, J. Jelencic, M. Mlinac-Misak, J. Elastomers & Plastics 39, 371 (2007)
R.K. Krishnaswamy, J. Van Walsem, O.P. Peoples, Y. Shabtai, A.R. Padwa, PCT Intl. Appl. WO 2013/184822, 12 Dec 2013, to Metabolix
K. Kubo, J. Masamoto, J. Appl. Polym Sci. 86, 3030 (2002)
R.A. Kudva, H. Keskkula, D.R. Paul, Polymer 39, 2447 (1998)
N. Kunimune, K. Yamada, Y.W. Leong, S. Thumsorn, H. Hamada, J. Appl. Polym. Sci. 120, 50 (2011)
J.A. Kuphal, L.H. Sperling, L.M. Robeson, J. Appl. Polym. Sci. 42, 1525 (1991)
P. Kurian, K.E. George, D.J. Francis, Eur. Polym. J. 28, 113 (1992)
T. Kurian, S. Datta, D. Khastgir, P.P. De, D.K. Tripathy, S.K. De, D.G. Peiffer, Polymer 37, 4787 (1996)
F.P. La Mantia, Adv. Polym. Technol. 12, 47 (1993)
F.P. La Mantia, A. Valenza, Angew. Makromol. Chem. 216, 45 (1994)
F.P. La Mantia, M. Vinci, F. Pilati, Polymer Recycling 1, 33 (1994)
F.P. La Mantia, L. Canfora, N.T. Dintcheva, Polym. Eng. Sci. 45, 1297 (2005)
C. Lacroix, M. Bousmina, P.J. Carreau, B.D. Favis, A. Michel, Polymer 37, 2939 (1996a)
C. Lacroix, M. Bousmina, P.J. Carreau, M.F. Llauro, R. Petiaud, A. Michel, Polymer 37, 2949 (1996b)
G.V. Laivins, Macromolecules 22, 3974 (1989)
M. Lambla, M. Seadan, Polym. Eng. Sci. 32, 1687 (1992)
M. Lambla, M. Seadan, Macromol. Symp. 69, 99 (1993)
X. Lan, X. Li, Z. Liu, Z. He, W. Yang, M. Yang, J. Macromol. Sci. A Pure Appl. Chem. 50, 2013 (2013)
M.R. Landry, D.J. Massa, C.J.T. Landry, D.M. Teegarden, R.H. Colby, T.E. Long, P.M. Henrichs, J. Appl. Polym. Sci. 54, 991 (1994)
N.M. Larocca, E. Hage Jr., L.A. Pessan, J. Polym. Sci. B Polym. Phys. 43, 1244 (2005)
N.M. Larocca, E.N. Ito, C.T. Rios, L.A. Pessan, R.E.S. Bretas, E. Hage Jr., J. Polym. Sci. B Polym. Phys. 48, 2274 (2010)
A. Lazzeri, M. Malanima, M. Pracella, J. Appl. Poly. Sci. 74, 3455 (1999)
A.H. Lebovitz, K. Khait, J.M. Torkelson, Macromolecules 35, 9716 (2002)
S.Y. Lee, C. Kim, J. Appl. Poly. Sci. 68, 1245 (1998)
S. Lee, O.O. Park, J. Appl. Poly. Sci. 77, 1338 (2000)
S. Lee, O.O. Park, Polymer 42, 6661 (2001)
P.-C. Lee, W.-F. Kuo, F.-C. Chang, Polymer 35, 5641 (1994)
C.W. Lee, S.H. Ryu, H.S. Kim, J. Appl. Polym. Sci. 64, 1595 (1997)
C.-H. Lee, S.K. Lee, S.-W. Kang, S.-H. Yun, J. Ho Kim, S. Choe, J. Appl. Polym. Sci. 73, 841 (1999a)
C.H. Lee, Y.M. Lee, H.K. Choi, S. Horiuchi, T. Kitano, Polymer 40, 6321 (1999b)
J.-S. Lee, K.-Y. Park, D.-J. Yoo, K.-D. Suh, J. Polym. Sci. B Polym. Phys. 38, 1396 (2000)
S. Lee, J.S. Park, H. Lee, J. Appl. Poly. Sci. 82, 1725 (2001)
A. Legros, P.J. Carreau, B.D. Favis, A. Michel, Polymer 35, 758 (1994)
A. Legros, P.J. Carreau, B.D. Favis, A. Michel, Polymer 38, 5085 (1997)
D. Lehmann, US Patent Appl. 20110040017, 17 Feb 2011, to Leibniz-Institut fuer Polymerforschung
D. Lehmann, B. Kluepfel, US Patent Appl. 20100249333, 30 Sept 2010, to Leibniz-Institut fuer Polymerforschung
Y. Lei, Q. Wu, C.M. Clemons, W. Guo, J. Appl. Polym. Sci. 113, 1710 (2009)
H. Li, Z. Li, Poly. Int. 48, 124 (1999)
S.-C. Li, L.-N. Lu, J. Appl. Polym. Sci. 108, 3559 (2008)
Y. Li, H. Shimizu, Eur. Polym. J. 45, 738 (2009)
D. Li, A.F. Yee, K.W. Powers, H.-C. Wang, T.C. Yu, Polymer 34, 4471 (1993)
H. Li, T. Chiba, N. Higashida, Y. Yang, T. Inoue, Polymer 38, 3921 (1997)
Q.F. Li, D.G. Kim, D. Wu, K. Lu, R.G. Jin, Polym. Eng. Sci. 41, 2155 (2001)
H.-M. Li, Z.-G. Shen, F.-M. Zhu, S.-A. Lin, Eur. Polym. J. 38, 1255 (2002)
J. Li, G. Ma, J. Sheng, J. Macromol. Sci. B Phys. 48, 979 (2009a)
J. Li, G. Ma, J. Sheng, J. Macromol. Sci. B Phys. 48, 471 (2009b)
R. Li, X. Zhang, L. Zhou, J. Dong, D. Wang, J. Appl. Polym. Sci. 111, 826 (2009c)
S.-C. Li, L.-N. Lu, W. Zeng, J. Appl. Polym. Sci. 112, 3341 (2009d)
D. Li, B. Shentu, Z. Weng, J. Macromol. Sci. B Phys. 50, 2050 (2011a)
J. Li, G. Ma, J. Sheng, J. Macromol. Sci. B Phys. 50, 741 (2011b)
Y. Li, D. Wang, J.-M. Zhang, X.-M. Xie, Polym. Bull. 66, 841 (2011c)
L.-P. Li, B. Yin, Y. Zhou, L. Gong, M.-B. Yang, B.-H. Xie, C. Chen, Polymer 53, 3043 (2012a)
F. Li, J. Ashbaugh, D. Rauscher, R. Dotter, PCT Intl. Appl. WO 2012112266, 23 Aug 2012b, to Fina Technology
F. Li, J. Ashbaugh, D. Rauscher, R. Dotter, PCT Intl. Appl. WO 2012112263, 23 Aug 2012c, to Fina Technology
F. Li, T.J. Coffy, M. Daumerie, US Patent Appl. 20120080822, 5 Apr 2012d, to Fina Technology
B. Liang, L. Pan, J. Appl. Polym. Sci. 54, 1945 (1994)
Z. Liang, H.L. Williams, J. Appl. Polym. Sci. 44, 699 (1992)
S. Lim, J.L. White, Polym Eng. Sci. 34, 221 (1994)
H. Lin, A.I. Isayev, J. Appl. Polym. Sci. 102, 2643 (2006)
O. Lin, A.F. Yee, J. Mater. Sci. 32, 3961 (1997)
J.S. Lin, E.Y. Sheu, Y.H.R. Jois, J. Appl. Polym. Sci. 55, 655 (1995)
A.D. Litmanovich, N.A. Platé, Y.V. Kudryavtsev, Prog. Polym. Sci. 27, 915 (2002)
N.C. Liu, W.E. Baker, Adv. Polym. Technol. 11, 249 (1992a)
N.C. Liu, W.E. Baker, Polym. Eng. Sci. 32, 1695 (1992b)
N.C. Liu, W.E. Baker, Polymer 35, 988 (1994)
Y. Liu, J.A. Donovan, Polymer 36, 4797 (1995)
H. Liu, J. Zhang, J. Polym. Sci. B Polym. Phys. 49, 1051 (2011)
N.C. Liu, W.E. Baker, K.E. Russell, J. Appl. Polym. Sci. 41, 2285 (1990)
N.C. Liu, H.Q. Xie, W.E. Baker, Polymer 34, 4680 (1993)
W.-B. Liu, W.-F. Kuo, C.-J. Chiang, F.-C. Chang, Eur. Polym. J. 32, 91 (1996)
H. Liu, T. Xie, Y. Zhang, Y. Ou, X. Fang, G. Yang, Polym. J. 36, 754 (2004)
H. Liu, T. Xie, Y. Zhang, Y. Ou, G. Yang, Polym. J. 38, 21 (2006)
Y. Liu, W. Guo, Z. Su, B. Li, C. Wu, J. Macromol. Sci. B Phys. 48, 414 (2009a)
Y. Liu, Q. Shi, Z. Ke, L. Yin, J. Yin, Polym. Bull. 63, 411 (2009b)
Y. Liu, Z. Su, W. Guo, B. Li, C. Wu, J. Macromol. Sci. B Phys. 49, 615 (2010a)
H. Liu, F. Chen, B. Liu, G. Estep, J. Zhang, Macromolecules 43, 6058 (2010b)
S. Liu, S. Qin, Z. Luo, J. Yu, J. Guo, M. He, J. Macromol. Sci. B Phys. 50, 1780 (2011a)
H. Liu, W. Song, F. Chen, L. Guo, J. Zhang, Macromolecules 44, 1513 (2011b)
H. Liu, L. Guo, X. Guo, J. Zhang, Polymer 53, 272 (2012a)
Z. Liu, Y. Deng, Y. Han, M. Chen, S. Sun, C. Cao, C. Zhou, H. Zhang, Ind. Eng. Chem. Res. 51, 9235 (2012b)
N.C. Liu, S. Lin, C. Zhou, W. Yu, Polymer 54, 310 (2013a)
H. Liu, X. Guo, W. Song, J. Zhang, Ind. Eng. Chem. Res. 52, 4787 (2013b)
B.P. Livengood, B.W. Baird, G.P. Marshall, US Patent 6,350,552, 26 Feb 2002, to Lexmark
D.-W. Lo, C.-R. Chiang, F.-C. Chang, J. Appl. Polym. Sci. 65, 739 (1997)
D.J. Lohse, T.P. Russell, L.H. Sperling (eds.), Interfacial Aspects of Multicomponent Polymer Materials (Plenum Press, New York, 1997)
N. Lopattananon, A. Kraibut, R. Sangjan, M. Seadan, J. Appl. Polym. Sci. 105, 1444 (2007)
S. Lopez-Quintana, C. Rosales, I. Gobernado-Mitre, J.C. Merino, J.M. Pastor, J. Appl. Polym. Sci. 107, 3099 (2008)
W. Loyens, G. Groeninckx, Polymer 43, 5679 (2002)
W. Loyens, G. Groeninckx, Polymer 44, 4929 (2003a)
W. Loyens, G. Groeninckx, Polymer 44, 123 (2003b)
Q.-W. Lu, C.W. Macosko, Polymer 45, 1981 (2004)
M. Lu, H. Keskkula, D.R. Paul, Polymer 34, 1874 (1993)
M. Lu, H. Keskkula, D.R. Paul, Polym. Eng. Sci. 34, 33 (1994)
M. Lu, H. Keskkula, D.R. Paul, J. Appl. Polym. Sci. 58, 1175 (1995)
M. Lu, H. Keskkula, D.R. Paul, J. Appl. Polym. Sci. 59, 1467 (1996)
Q.-W. Lu, T.R. Hoye, C.W. Macosko, J. Polym. Sci. A Polym. Chem. 40, 2310 (2002)
Q.-W. Lu, C.W. Macosko, J. Horrion, Macromol. Symp. 198, 221 (2003)
X. Lu, H. Zhang, Y. Zhang, J. Appl. Polym. Sci. (2013a). doi:10.1002/app.40064; published online
X. Lu, H. Zhang, Y. Zhang, J. Appl. Polym. Sci. 130, 4587 (2013b)
S. Lumlong, K. Kuboyama, T. Chiba, T. Ougizawa, Macromol. Symp. 233, 17 (2006)
R.D. Lundberg, Ionic polymers, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, vol. 8, 2nd edn. (Wiley, New York, 1987), p. 393
R.D. Lundberg, I. Duvdevani, D.G. Peiffer, P.K. Agarwal, in Advances in Polymer Blends and Alloys Technology, ed. by M.A. Kohudic, vol. 1 (Technomic Publishing, Lancaster, 1988), p. 131
R. Luo, X. Zhao, P.C. Yung, US Patent Appl. 20130069001, 21 Mar 2013, to Ticona
J.M. Lusinchi, B. Boutevin, N. Torres, J.J. Robin, J. Appl. Polym. Sci. 79, 874 (2000)
S.-P. Lyu, J.J. Cernohous, F.S. Bates, C.W. Macosko, Macromolecules 32, 106 (1999)
P. Ma, D.G. Hristova-Bogaerds, P.J. Lemstra, Y. Zhang, S. Wang, Macromol. Mat. and Eng. 297, 402 (2012a)
H. Ma, Z. Xiong, F. Lu, C. Li, Y. Yang, Macromol. Mat. and Eng. 297, 136 (2012b)
C.-T. Maa, F.-C. Chang, J. Appl. Polym. Sci. 49, 913 (1993)
A.V. Machado, J.A. Covas, M. van Duin, J. Polym. Sci. A Polym. Chem. 37, 1311 (1999)
A.V. Machado, J.A. Covas, M. van Duin, Polym. Eng. Sci. 42, 2032 (2002)
W.J. MacKnight, R.W. Lenz, P.V. Musto, R.J. Somani, Polym. Eng. Sci. 25, 1124 (1985)
C.W. Macosko, H.K. Jeon, T.R. Hoye, Progress in Polymer Science 30, 939 (2005)
D. Mader, J. Kressler, R. Mülhaupt, J. Appl. Polym. Sci. 73, 1685 (1999)
R. Magaraphan, R. Skularriya, S. Kohjiya, J. Appl. Polym. Sci. 105, 1914 (2007)
B. Majumdar, H. Keskkula, D.R. Paul, Polymer 35, 5468 (1994a)
B. Majumdar, H. Keskkula, D.R. Paul, Polymer 35, 5453 (1994b)
B. Majumdar, H. Keskkula, D.R. Paul, Polymer 35, 3164 (1994c)
B. Majumdar, H. Keskkula, D.R. Paul, Polymer 35, 1399 (1994d)
B. Majumdar, H. Keskkula, D.R. Paul, Polymer 35, 1386 (1994e)
B. Majumdar, H. Keskkula, D.R. Paul, J. Appl. Polym. Sci. 54, 339 (1994f)
B. Majumdar, H. Keskkula, D.R. Paul, N.G. Harvey, Polymer 35, 4263 (1994g)
B. Majumdar, D.R. Paul, A.J. Oshinski, Polymer 38, 1787 (1997)
A. Mallick, D.K. Tripathy, S.K. De, J. Appl. Polym. Sci. 50, 1627 (1993)
A. Mallick, A.K. Bhattacharya, B.R. Gupta, D.K. Tripathy, S.K. De, J. Appl. Polym. Sci. 65, 135 (1997)
D. Mangaraj, Rubber Chem. Technol. 78, 536 (2005)
N.R. Manoj, P.P. De, S.K. De, J. Appl. Polym. Sci. 49, 133 (1993a)
N.R. Manoj, P.P. De, S.K. De, Rubber Chem. Technol. 66, 550 (1993b)
N.R. Manoj, P.P. De, S.K. De, D.G. Peiffer, J. Appl. Polym. Sci. 53, 361 (1994)
C. Marco, G. Ellis, M.A. Gomez, J.G. Fatou, J.M. Arribas, I. Campoy, A. Fontecha, J. Appl. Polym. Sci. 65, 2665 (1997)
P. Marechal, G. Coppens, R. Legras, J.-M. Dekoninck, J. Polym. Sci. A Polym. Chem. 33, 757 (1995)
P. Marechal, T. Chiba, T. Inoue, M. Weber, E. Koch, Polymer 39, 5655 (1998)
M. Maric, N. Ashurov, C.W. Macosko, Polym. Eng. Sci. 41, 631 (2001)
M.M. Martens, R. Koshida, J.L. Bohan, W.R. Fielding, US Patent 6,774,174, 10 Aug 2004, to DuPont
P. Martin, J. Devaux, R. Legras, M. van Grup, M. van Duin, Polymer 42, 2463 (2001)
P. Martin, C. Gallez, J. Devaux, R. Legras, L. Leemans, M. van Grup, M. van Duin, Polymer 44, 5251 (2003)
P. Martin, C. Maquet, R. Legras, C. Bailly, L. Leemans, M. van Grup, M. van Duin, Polymer 45, 5111 (2004a)
P. Martin, C. Maquet, R. Legras, C. Bailly, L. Leemans, M. van Grup, M. van Duin, Polymer 45, 3277 (2004b)
P. Martin, J. Devaux, R. Legras, L. Leemans, M. van Grup, M. van Duin, J. Appl. Polym. Sci. 91, 703 (2004c)
J.M. Martinez, J.I. Eguiazábal, J. Nazábal, J. Appl. Polym. Sci. 45, 1135 (1992)
J.M. Martinez, J. Nazábal, J.I. Eguiazábal, J. Appl. Polym. Sci. 51, 223 (1994)
J.G. Martinez, R. Benavides, C. Guerrero, J. Appl. Polym. Sci. 104, 560 (2007)
R.E. Martini, S. Barbosa, E. Brignole, Ind. Eng. Chem. Res. 45, 3393 (2006)
E. Martuscelli, F. Riva, C. Sellitti, C. Silvestre, Polymer 26, 270 (1985)
L. Mascia, F. Bellahdeb, Adv. Polym. Technol. 13, 99 (1994a)
L. Mascia, F. Bellahdeb, Adv. Polym. Technol. 13, 37 (1994b)
L. Mascia, K. Hashim, J. Appl. Polym. Sci. 66, 1911 (1997)
L. Mascia, K. Hashim, Polymer 39, 369 (1998)
L. Mascia, A. Ophir, J. Appl. Polym. Sci. 81, 2972 (2001)
K. Mashita, T. Fujii, T. Omae, US Patent 4,780,505, 25 Oct 1988 to Sumitomo
K. Mashita, T. Fujii, T. Oomae, US Patent 5,004,782, 2 Apr 1991 to Sumitomo
M. Mawatari, T. Itoh, S. Tsuchikawa, S. Kimura, US Patent 4,839,425, 13 June 1989, to Japan Synthetic Rubber
J. Mayumi, H. Omori, Eur. Pat. Appl. 268,981, 1 June 1988 to Mitsubishi Petrochemical
R.M. Medina, D. Likhatchev, A. Alexandrova, A. Sanchez-Solis, O. Manero, Polymer 45, 8517 (2004)
N. Mekhilef, A.A. Kadi, A. Ajji, Polym. Eng. Sci. 32, 894 (1992)
T.J.A. Melo, S.V. Canevarolo, Polym. Eng. Sci. 45, 11 (2005)
L.C. Mendes, P.S.C. Pereira, V.D. Ramos, Macromol. Symp. 299–300, 183 (2011)
G. Menges, Macromol. Chem. Macromol. Symp. 23, 13 (1989)
G.D. Merfeld, A. Karim, B. Majumdar, S.K. Satija, D.R. Paul, J. Polym. Sci. B Polym. Phys. 36, 3115 (1998)
R.B. Mesrobian, P.E. Sellers, D. Adomaitis, US Patent 3,373,224, 12 Mar 1968, to Continental Can
D.M. Miley, J. Runt, Polymer 33, 4643 (1992)
L.I. Minkova, T. Miteva, D. Sek, B. Kaczmarczyk, P.L. Magagnini, M. Paci, F.P. La Mantia, R. Scaffaro, J. Appl. Polym. Sci. 62, 1613 (1996)
L. Minkova, H. Yordanov, S. Filippi, Polymer 43, 6195 (2002)
L. Minkova, H. Yordanov, S. Filippi, N. Grizzuti, Polymer 44, 7925 (2003)
A. Misra, G. Sawhney, R.A. Kumar, J. Appl. Polym. Sci. 50, 1179 (1993)
Y. Mizuno, T. Maruyama, S. Yachigo, US Patent 5,019,615, 28 May 1991, to Sumitomo
M.J. Modic, L.A. Pottick, Polym. Eng. Sci. 33, 819 (1993)
A.J. Moffett, M.E.J. Dekkers, Polym. Eng. Sci. 32, 1 (1992)
S. Mohanty, G.B. Nando, Polymer 38, 1395 (1997)
S. Mohanty, S. Roy, R.N. Santra, G.B. Nando, J. Appl. Polym. Sci. 58, 1947 (1995)
S. Mohanty, G.B. Nando, K. Vijayan, N.R. Neelakanthan, Polymer 37, 5387 (1996)
S. Mohanty, K. Vijayan, N.R. Neelkantan, G.B. Nando, Polym. Eng. Sci. 37, 1395 (1997)
A. Molnar, A. Eisenberg, Polym. Commun 32, 370 (1991)
A. Molnar, A. Eisenberg, Polym. Eng. Sci. 32, 1665 (1992)
M.V. Moncur, US Patent 4,350,794, 21 Sept 1982, to Monsanto
I. Mondragon, J. Nazábal, Polym. Eng. Sci. 25, 178 (1985)
I. Mondragon, J. Nazábal, J. Appl. Polym. Sci. 32, 6191 (1986)
I. Mondragon, J. Nazábal, J. Mater. Sci. Lett. 6, 698 (1987)
I. Mondragon, M. Gaztelumendi, J. Nazábal, Polym. Eng. Sci. 26, 1478 (1986)
I. Mondragon, P.M. Remiro, J. Nazabal, Eur. Polym. J. 23, 125 (1987)
I. Mondragon, M. Gaztelumendi, J. Nazábal, Polym. Eng. Sci. 28, 1126 (1988)
G. Montaudo, C. Puglisi, F. Samperi, J. Polym. Sci. A Polym. Chem. 31, 13 (1993)
G. Montaudo, C. Puglisi, F. Samperi, J. Polym. Sci. A Polym. Chem. 32, 15 (1994)
G. Montaudo, C. Puglisi, F. Samperi, Macromolecules 31, 650 (1998a)
G. Montaudo, C. Puglisi, F. Samperi, J. Polym. Sci. A Polym. Chem. 36, 1873 (1998b)
G. Montaudo, C. Puglisi, F. Samperi, in Transreactions in Condensation Polymers, ed. by S. Fakirov (Wiley-VCH, Weinheim, 1999), p. 159
B. Moon, T.R. Hoye, C.W. Macosko, Macromolecules 34, 7941 (2001)
S. Moritomi, M. Iji, US Patent 6,339,131, 2 July 2002, to Sumitomo
Y. Moriya, N. Suzuki, Y. Okada, US Patent 4,753,990, 28 June 1988 to Nippon Oil and Fats
Y. Moriya, N. Suzuki, H. Goto, US Patent 4,839,423, 13 June 1989 to Nippon Oil and Fats
N. Moussaif, C. Pagnoulle, R. Jérôme, Polymer 41, 5551 (2000)
M. Mukawa, Y. Hayata, A. Nodera, H. Yasuda, K. Watanabe, US Patent 7,994,239, 9 Aug 2011, to Idemitsu Kosan
O.K. Muratoglu, A.S. Argon, R.E. Cohen, M. Weinberg, Polymer 36, 4771 (1995a)
O.K. Muratoglu, A.S. Argon, R.E. Cohen, M. Weinberg, Polymer 36, 921 (1995b)
L.E. Murch, US Patent 3,845,163, 29 Oct 1974, to DuPont
N.S. Murthy, H. Minor, M.K. Akkapeddi, B. Van Buskirk, J. Appl. Polym. Sci. 41, 2265 (1990)
V.M. Nadkarni, J.P. Jog, Crystallization of polymers in thermoplastic blends and alloys, in Handbook of Polymer Science and Technology, ed. by N.P. Cheremisinoff, vol. 4 (Marcel Dekker, New York, 1989), pp. 81–120
V.M. Nadkarni, J.P. Jog, Crystallization behavior in polymer blends, chapter 8, in Two-Phase Polymer Systems, ed. by L.A. Utracki (Hanser Publishers, New York, 1991), pp. 213–239
K. Nakamura, K. Kometani, A. Koshino, K. Horiuchi, PCT Intl. Appl. WO 88/00953, 11 Feb 1988, to Toray Industries
C. Nakason, M. Jarnthong, A. Kaesaman, S. Kiatkamjornwong, J. Appl. Polym. Sci. 109, 2694 (2008)
O. Nakazima, S. Izawa, US Patent 4,863,996, 5 Sept 1989, to Asahi
J.-D. Nam, J. Kim, S. Lee, Y. Lee, C. Park, J. Appl. Polym. Sci. 87, 661 (2003)
M. Narathichat, C. Kummerlöwe, N. Vennemann, C. Nakason, J. Appl. Polym. Sci. 121, 805 (2011)
N. Naskar, T. Biswas, D.K. Basu, J. Appl. Polym. Sci. 52, 1007 (1994)
A. Natansohn, R. Murali, A. Eisenberg, CHEMTECH 20, 418 (1990)
D. Nazareth, Eur. Pat. Appl. 697,442, 21 Feb 1996, to General Electric
W. Neilinger, D. Wittman, U. Westeppe, L. Bottenbruch, J. Kirsch, H.-J. Fullmann, Eur. Pat. Appl. 291,796, 11 Nov 1988, to Bayer
W. Neugebauer, M. Bartmann, U. Kowalczik, J. Mugge, US Patent 5,039,746, 13 Aug 1991, to Huels
N. Nugay, T. Nugay, Eur. Polym. J. 36, 1027 (2000)
D. Nwabunma, T. Kyu (eds.), Polyolefin Blends (Wiley-Interscience, Hoboken, 2008)
J. Oderkerk, G. Groeninckx, Polymer 43, 2219 (2002)
T.S. Oh, J.H. Ryou, Y.S. Chun, W.N. Kim, Polym. Eng. Sci. 37, 838 (1997)
J.S. Oh, A.I. Isayev, M.A. Rogunova, Polymer 44, 2337 (2003)
B. Ohlsson, H. Hassander, B. Tornell, Polymer 39, 6705 (1998a)
B. Ohlsson, H. Hassander, B. Tornell, Polymer 39, 4715 (1998b)
Y. Ohmura, S. Maruyama, H. Kawasaki, US Patent 4,339,555, 13 July 1982, to Mitsubishi Chemical Industries
V. Ojijo, S.S. Ray, R. Sadiku, ACS Appl. Mater. Interfaces 5, 4266 (2013)
M. Okabe, A. Amagai, H. Eto, Y. Tanaka, UK Patent GB 2,218,996, 29 Nov 1989, to Mitsubishi Gas Chemical
O. Okada, H. Keskkula, D.R. Paul, Polymer 40, 2699 (1999)
M. Okamoto, T. Inoue, Polym. Eng. Sci. 33, 175 (1993)
M. Okamoto, T. Inoue, Polymer 35, 257 (1994)
M. Okamoto, T. Kotaka, Polymer 38, 1357 (1997)
T. Okamoto, K. Yasue, T. Marutani, Y. Fukushima, US Patent 4,804,707, 14 Feb 1989, to Unitika
M. Okamoto, K. Shiomi, T. Inoue, Polymer 35, 4618 (1994)
M.G. Oliveira, A.C. Gomes, C.O. Ana, M.S.M. Almeida, B.G. Soares, Macromol. Chem. Phys. 205, 465 (2004)
E.J. Olivier, US Patent 4,594,386, 10 June 1986a, to Copolymer Rubber & Chemical
E.J. Olivier, PCT Intl. Appl. WO 86/04076, 17 July 1986b, to Copolymer Rubber & Chemical
T.S. Omonov, C. Harrats, G. Groeninckx, Polymer 46, 12322 (2005)
T.S. Omonov, C. Harrats, G. Groeninckx, P. Moldenaers, Polymer 48, 5289 (2007)
Z. Oommen, S.R. Zachariah, S. Thomas, G. Groeninckx, P. Moldenaers, J. Mewis, J. Appl. Polym. Sci. 92, 252 (2004)
C.A. Orr, J.J. Cernohous, P. Guegan, A. Hirao, H.K. Jeon, C.W. Macosko, Polymer 42, 8171 (2001)
A. Oshima, H. Ishida, T. Shinohara, US Patent 5,780,548, 14 July 1998, to General Electric
A.J. Oshinski, H. Keskkula, D.R. Paul, Polymer 33, 284 (1992a)
A.J. Oshinski, H. Keskkula, D.R. Paul, Polymer 33, 268 (1992b)
A.J. Oshinski, H. Keskkula, D.R. Paul, Polymer 37, 4919 (1996a)
A.J. Oshinski, H. Keskkula, D.R. Paul, Polymer 37, 4909 (1996b)
A.J. Oshinski, H. Keskkula, D.R. Paul, Polymer 37, 4891 (1996c)
A.J. Oshinski, H. Keskkula, D.R. Paul, J. Appl. Polym. Sci. 61, 623 (1996d)
Y. Otawa, A. Uchiyama, K. Hiraoka, K. Okamoto, S. Shimizu, Eur. Pat. Appl. 269,274, 1 June 1988, to Mitsui Petrochemical Industries
C.-F. Ou, J. Appl. Polym. 68, 1591 (1998)
C.-F. Ou, C.-C. Lin, J. Appl. Polym. 61, 1455 (1996a)
C.-F. Ou, C.-C. Lin, J. Appl. Polym. 61, 1379 (1996b)
C.-F. Ou, M.S. Chao, S.L. Huang, J. Appl. Polym. 74, 2727 (1999)
K.J. Oxby, M. Maric, Macromol. Reaction Eng. (2013). doi: 10.1002/mren.201200081; published online
H.T. Oyama, Polymer 50, 747 (2009)
H.T. Oyama, M. Matsushita, M. Furuta, Polym. J. 43, 991 (2011)
A.R. Padwa, Polym. Eng. Sci. 32, 1703 (1992)
C. Pagnoulle, R. Jérôme, Macromolecules 34, 965 (2001a)
C. Pagnoulle, R. Jérôme, Polymer 42, 1893 (2001b)
C. Pagnoulle, C. Koning, L. Leemans, R. Jérôme, Macromolecules 33, 6275 (2000a)
C. Pagnoulle, P. Martin, R. Jérôme, Macromol. Chem. Phys. 201, 2181 (2000b)
F.-C. Pai, H.-H. Chu, S.-M. Lai, J. Polym. Eng. 31, 463 (2011)
L. Pan, T. Chiba, T. Inoue, Polymer 42, 8825 (2001)
L. Pan, T. Inoue, H. Hayami, S. Nishikawa, Polymer 43, 337 (2002)
M.-M. Pan, B. Yin, W. Yang, Y. Zhao, M.-B. Yang, J. Macromol. Sci. B Phys. 46, 1267 (2007)
C.P. Papadopoulou, N.K. Kalfoglou, Polymer 41, 2543 (2000)
D.S. Park, S.H. Kim, J. Appl. Polym. Sci. 87, 1842 (2003)
S.J. Park, B.K. Kim, H.M. Jeong, Eur. Polym. J. 26, 131 (1990)
I. Park, J.W. Barlow, D.R. Paul, J. Polym. Sci. B Polym. Phys. 30, 1021 (1992)
C.D. Park, W.H. Jo, M.S. Lee, Polymer 37, 3055 (1996)
K.Y. Park, S.H. Park, K.-D. Suh, J. Appl. Polym. Sci. 66, 2183 (1997)
S.H. Park, G.J. Lee, S.S. Im, K.D. Suh, Polym. Eng. Sci. 38, 1420 (1998)
K.Y. Park, S.-S. Lee, J.-Y. Kim, K.-D. Suh, J. Macromol. Sci. A Pure Appl. Chem. 39, 787 (2002)
R. Patel, D.A. Ruehle, J.R. Dorgan, P. Halley, D. Martin, Polym. Eng. Sci. (2013). doi:10.1002/pen.23692; published online
D.R. Paul, Interfacial agents for polymer blends, in Polymer Blends, ed. by D.R. Paul, S. Newman, vol. 2 (Academic, New York, 1978), pp. 35–62
D.R. Paul, in Modification and Blending of Synthetic and Natural Macromolecules, NATO Science Series, ed. by F. Ciardelli, S. Penczek, vol. 175 (Kluwer, Dordrecht, 2004), p. 293
D.R. Paul, C.B. Bucknall (eds.), Polymer Blends: Formulation and Performance, vols. 1 & 2 (Wiley, New York, 2000)
D.R. Paul, S. Newman (eds.), Polymer Blends, vols. 1 & 2 (Academic, New York, 1978)
D.R. Paul, J.W. Barlow, H. Keskkula, Polymer blends, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, vol. 12 (Wiley, New York, 1988), p. 417
F. Pazzagli, M. Pracella, Macromol. Symp. 149, 225 (2000)
D.G. Peiffer, I. Duvdevani, P.K. Agarwal, R.D. Lundberg, J. Polym. Sci. C Polym. Lett. 24, 581 (1986)
M. Penco, M.A. Pastorini, E. Occhiello, F. Garbassi, R. Braglia, G. Giannotta, J. Appl. Polym. Sci. 57, 329 (1995)
M. Penco, A. Lazzeri, T.V. Phuong, P. Cinelli, PCT Intl. Appl. WO 2012025907, 1 Mar 2012, to Universita’ di Pisa
Y. Peng, W. Guo, P. Zhu, C. Wu, J. Appl. Polym. Sci. 109, 483 (2008)
R. Pernice, C. Berto, A. Moro, R. Pippa, US Patent 5,210,125, 11 May 1993, to ECP Enichem Polimeri
H. Pernot, M. Baumert, F. Court, L. Leibler, Nat. Mater. 1, 54 (2002)
P. Perret, A. Bouilloux, M. Hert, I. Leclere, Macromol. Symp. 112, 183 (1996)
P.J. Perron, E.A. Bourbonais, PCT Intl. Appl. WO 88/06174, 14 Sept 1988 to Dexter
I. Pesneau, M.F. Llauro, M. Gregoire, A. Michel, J. Appl. Polym. Sci. 65, 2457 (1997)
I. Pesneau, M. Gregoire, A. Michel, J. Appl. Polym. Sci. 79, 1556 (2001)
S.V. Phadke, US Patent 4,757,112, 12 July 1988a, to Copolymer Rubber & Chemical
S.V. Phadke, Eur. Pat. Appl. 279,502, 24 Aug 1988b, to Copolymer Rubber & Chemical
T.T.M. Phan, A.J. DeNicola Jr., L.S. Schadler, J. Appl. Polym. Sci. 68, 1451 (1998)
O. Phanstiel, S.B. Brown, US Patent 5,010,144, 23 Apr 1991, to General Electric
O. Phanstiel, S.B. Brown, US Patent 5,089,567, 18 Feb 1992, to General Electric
O. Phanstiel, S.B. Brown, US Patent 5,210,191, 11 May 1993, to General Electric
Y. Pietrasanta, J.-J. Robin, N. Torres, B. Boutevin, Macromol. Chem. Phys. 200, 142 (1999)
J. Piglowski, M. Trelinska-Wlaźlak, B. Paszak, J. Macromol. Sci. B Phys. 38, 515 (1999)
J. Piglowski, I. Gancarz, M. Wlaźlak, Polymer 41, 3671 (2000)
F. Pilati, E. Marianucci, C. Berti, J. Appl. Polym. Sci. 30, 1267 (1985)
F. Pilati, M. Fiorini, C. Berti, in Transreactions in Condensation Polymers, ed. by S. Fakirov (Wiley-VCH, Weinheim, 1999), p. 79
L.Z. Pillon, L.A. Utracki, Polym. Eng. Sci. 24, 1300 (1984)
L.Z. Pillon, J. Lara, D.W. Pillon, Polym. Eng. Sci. 27, 984 (1987a)
L.Z. Pillon, L.A. Utracki, D.W. Pillon, Polym. Eng. Sci. 27, 562 (1987b)
L.A. Pinheiro, G.-H. Hu, L.A. Pessan, S.V. Canevarolo, Polym. Eng. Sci. 48, 806 (2008)
J. Pionteck, P. Poetschke, U. Schulze, N. Proske, A. Kaya, H. Zhao, H. Malz, Macromol. Symp. 214, 279 (2004)
N.A. Platé, A.D. Litmanovich, Y.V. Kudryavtsev, in Modification and Blending of Synthetic and Natural Macromolecules, NATO Science Series, ed. by F. Ciardelli, S. Penczek, vol. 175 (Kluwer, Dordrecht, 2004), p. 241
G. Pompe, L. Häubler, J. Polym. Sci. B Polym. Phys. 35, 2161 (1997)
G. Pompe, L. Häubler, W. Winter, J. Polym. Sci. B Polym. Phys. 34, 211 (1996)
G. Pompe, P. Pötschke, J. Pionteck, J. Appl. Polym. Sci. 86, 3445 (2002)
R.S. Porter, L.-H. Wang, Polymer 33, 2019 (1992)
R.S. Porter, J.M. Jonza, M. Kimura, C.R. Desper, E.R. George, Polym. Eng. Sci. 29, 55 (1989)
C.S. Powell, D.S. Kalika, Polymer 41, 4651 (2000)
M. Pracella, D. Chionna, Macromol. Symp. 198, 161 (2003)
M. Pracella, D. Chionna, Macromol. Symp. 218, 173 (2004)
M. Pracella, L. Rolla, D. Chionna, A. Galeski, Macromol. Chem. Phys. 203, 1473 (2002)
C.F. Pratt, PCT Intl. Appl. WO 88/07065, 22 Sept 1988, to General Electric
C.F. Pratt, S.V. Phadke, E.J. Olivier, PCT Intl. Appl. WO 88/05452, 28 July 1988, to General Electric
E.V. Prut, Polym. Sci. Ser. D 2, 1 (2009)
E.V. Prut, A.N. Zelenetskii, Russ. Chem. Rev. 70, 65 (2001)
M. Psarski, M. Pracella, A. Galeski, Polymer 41, 4923 (2000)
R. Qi, J. Nie, C. Zhou, D. Mao, B. Zhang, J. Appl. Polym. Sci. 102, 6081 (2006)
C. Qu, R. Su, Q. Zhang, R. Du, Q. Fu, Polym. Int. 57, 139 (2008)
W. Qui, K. Mai, K. Fang, Z. Li, H. Zeng, J. Appl. Polym. Sci. 71, 847 (1999)
R. Quintana, O. Persenaire, L. Bonnaud, P. Dubois, Y. Lemmouchi, PCT Intl. Appl. WO 2012131370, 4 Oct 2012, to British American Tobacco
N.M. Qureshi, US Patent Appl. 20090163663, 25 June 2009, to Escalator Handrail Co.
H.-J. Radusch, J. Ding, M. Schulz, Angew. Makromol. Chem. 204, 177 (1993)
P. Ramesh, S.K. De, J. Appl. Polym. Sci. 50, 1369 (1993)
H. Raval, S. Devi, Y.P. Singh, M.H. Mehta, Polymer 32, 493 (1991)
H.B. Ravikumar, C. Ranganathaiah, G.N. Kumaraswamy, M.V. Deepa Urs, J.H. Jagannath, A.S. Bawa, S. Thomas, J. Appl. Polym. Sci. 100, 740 (2006)
R.W. Rees, Reversible crosslinking, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, vol. 4, 2nd edn. (Wiley, New York, 1986), p. 395
P.M. Remiro, J. Nazábal, J. Appl. Polym. Sci. 42, 1639 (1991a)
P.M. Remiro, J. Nazábal, J. Appl. Polym. Sci. 42, 1475 (1991b)
J. Ren, H. Wang, L. Jian, J. Zhang, S. Yang, J. Macromol. Sci. B Phys. 47, 712 (2008)
P.N. Richardson, US Patent 4,283,502, 11 Aug 1981, to DuPont
D.H. Roberts, R.C. Constable, S. Thiruvengada, Polym. Eng. Sci. 37, 1421 (1997)
L.M. Robeson, J. Appl. Polym. Sci. 30, 4081 (1985)
L.M. Robeson, Polymer Blends : A Comprehensive Review (Hanser/Gardner Publications, Cincinnati, 2007)
L.M. Robeson, A. Famili, J.F. Nangeroni, in Science and Technology of Polymers and Advanced Materials, ed. by P.N. Prasad, J.E. Mark, S.H. Kandil, Z.H. Kafafi (Springer, New York, 1998), p. 9
J.L. Rodriguez, J.I. Eguiazábal, J. Nazábal, Polym. J. 28, 501 (1996)
H. Rodríguez-Ríos, S.M. Nuño-Donlucas, J.E. Puig, R. González-Núñez, P.C. Schulz, J. Appl. Polym. Sci. 91, 1736 (2004)
S. Rooj, V. Thakur, U. Gohs, U. Wagenknecht, A.K. Bhowmick, G. Heinrich, Polym. Adv. Technol. 22, 2257 (2011)
J. Rösch, Polym. Eng. Sci. 35, 1917 (1995)
J. Rösch, R. Mülhaupt, Makromol. Chem. Rapid Commun. 14, 503 (1993)
J. Rösch, R. Mülhaupt, Polym. Bull. 32, 697 (1994)
J. Rösch, R. Mülhaupt, J. Appl. Polym. Sci. 56, 1599 (1995a)
J. Rösch, R. Mülhaupt, J. Appl. Polym. Sci. 56, 1607 (1995b)
J. Rösch, R. Mülhaupt, G.H. Michler, Macromol. Symp. 112, 141 (1996)
M.J. Roura, US Patent 4,346,194, 24 Aug 1982, to DuPont
M.J. Roura, US Patent 4,478,978, 23 Oct 1984, to DuPont
A. Roychoudhury, P.P. De, J. Appl. Polym. Sci. 63, 1761 (1997)
A. Rudin, J. Macromol. Chem. Rev. Macromol. Chem. C19, 267 (1980)
A. Rudin, The Elements of Polymer Science and Engineering (Academic, New York, 1982)
R.K. Sadi, R.S. Kurusu, G.J.M. Fechine, N.R. Demarquette, J. Appl. Polym. Sci. 123, 3511 (2012)
S. Safapour, M. Seyed-Esfahani, F. Auriemma, O.R. de Ballesteros, P. Vollaro, R. Di Girolamo, C. De Rosa, A. Khosroshahi, Polymer 51, 4340 (2010)
C. Sailer, U.A. Handge, Macromol. Symp. 254, 217 (2007a)
C. Sailer, U.A. Handge, Macromolecules 40, 2019 (2007b)
C. Sailer, U.A. Handge, Macromolecules 41, 4258 (2008)
M. Saleem, W.E. Baker, J. Appl. Polym. Sci. 39, 655 (1990)
P. Sambaru, S.A. Jabarin, Polym. Eng. Sci. 33, 827 (1993)
C.K. Samios, K.G. Gravalos, N.K. Kalfoglou, Eur. Polym. J. 36, 937 (2000)
F. Samperi, M. Montaudo, C. Puglisi, R. Alicata, G. Montaudo, Macromolecules 36, 7143 (2003a)
F. Samperi, C. Puglisi, R. Alicata, G. Montaudo, J. Polym. Sci. A Polym. Chem. 41, 2778 (2003b)
F. Samperi, M.S. Montaudo, C. Puglisi, S. Di Giorgi, G. Montaudo, Macromolecules 37, 6449 (2004)
F. Samperi, M.S. Montaudo, S. Battiato, D. Carbone, C. Puglisi, J. Polym. Sci. A Polym. Chem. 48, 5135 (2010)
A. Sanchez, C. Rosales, E. Laredo, A.J. Muller, M. Pracella, Macromol. Chem. Phys. 202, 2461 (2001)
M.S. Sanchez, V. Mathot, G. Groeninckx, W. Bruls, Polymer 47, 5314 (2006)
A. Sanchez-Solis, F. Calderas, O. Manero, Polymer 42, 7335 (2001)
H. Sano, H. Ohno, Eur. Pat. Appl. 268,280, 25 May 1988, to Mitsubishi Petrochemical
R.N. Santra, S. Roy, A.K. Bhowmick, G.B. Nando, Polym. Eng. Sci. 33, 1352 (1993a)
R.N. Santra, B.K. Samantaray, A.K. Bhowmick, G.B. Nando, J. Appl. Polym. Sci. 49, 1145 (1993b)
R.N. Santra, S. Roy, V.K. Tikku, G.B. Nando, Adv. Polym. Technol. 14, 59 (1995)
S.N. Sathe, S. Devi, G.S.S. Rao, K.V. Rao, J. Appl. Polym. Sci. 61, 97 (1996)
F.H. Sawden, Eur. Pat. Appl. 274,424, 13 July 1988, to DuPont Canada
R. Scaffaro, F.P. La Mantia, Macromol. Chem. Phys. 207, 265 (2006)
R. Scaffaro, F.P. La Mantia, L. Canfora, G. Polacco, S. Filippi, P. Maganini, Polymer 44, 6951 (2003)
R. Scaffaro, F.P. La Mantia, C. Castronovo, Macromol. Chem. Phys. 205, 1402 (2004)
R. Schaefer, J. Kressler, R. Neuber, R. Muelhaupt, Macromolecules 28, 5037 (1995)
S. Schlag, J. Rösch, Chr. Friedrich, Polym. Bull. 30, 603 (1993)
J.C. Schmidhauser, K.L. Longley, Tetrahedron Lett. 32, 7155 (1991)
S. Schmukler, M. Shida, J. Machonis Jr., US Patent 4,600,746, 15 July 1986a to Norchem
S. Schmukler, M. Shida, J. Machonis Jr., US Patent 4,575,532, 11 Mar 1986b to Norchem
N.R. Schott, B. Sanderford, Coat. Plast. Prepr. Am. Chem. Soc. Div. Org. Coat. Plast. Chem. 37(2), 73 (1977)
J.E. Schuetz, R.W. Hohlfeld, B.C. Meridith, US Patent 4,864,002, 5 Sept 1989, to Dow
J.S. Schulze, J.J. Cernohous, A. Hirao, T.P. Lodge, C.W. Macosko, Macromolecules 33, 1191 (2000)
J.S. Schulze, B. Moon, T.P. Lodge, C.W. Macosko, Macromolecules 34, 200 (2001)
M. Sclavons, V. Carlier, B. De Roover, P. Franquinet, J. Devaux, R. Legras, J. Appl. Polym. Sci. 62, 1205 (1996)
C.E. Scott, C.W. Macosko, Polymer 35, 5422 (1994a)
C.E. Scott, C.W. Macosko, J. Polym. Sci. B Polym. Phys. 32, 205 (1994b)
C.E. Scott, C.W. Macosko, Polymer 36, 461 (1995a)
C.E. Scott, C.W. Macosko, Int. Polym. Process. 10, 36 (1995b)
D. Sek, B. Kaczmarczyk, Polymer 38, 2925 (1997)
Y. Seo, J. Appl. Polym. Sci. 64, 359 (1997)
Y. Seo, T.H. Ninh, Polymer 45, 8573 (2004)
Y. Seo, S.S. Hwang, K.U. Kim, J. Lee, S.I. Hong, Polymer 34, 1667 (1993)
Y. Seo, T.H. Ninh, S.M. Hong, S. Kim, T.J. Kang, H. Kim, J. Kim, Langmuir 22, 3062 (2006)
T. Serhatkulu, B. Erman, I. Bahar, S. Fakirov, M. Evstatiev, D. Sapundjieva, Polymer 36, 2371 (1995)
G. Serpe, J. Jarrin, F. Dawans, Polym. Eng. Sci. 30, 553 (1990)
S. Shabbir, S. Zulfiqar, Z. Ahmad, M.I. Sarwar, Polym. Eng. Sci. 48, 1793 (2008)
S. Shabbir, S. Zulfiqar, S.I. Shah, Z. Ahmad, M.I. Sarwar, J. Phys. Chem. B 114, 13241 (2010)
K. Shahbazi, M.K.R. Aghjeh, F. Abbasi, M.P. Meran, M.M. Mazidi, Polym. Bull. 69, 241 (2012)
H. Shariatpanahi, H. Nazokdast, B. Dabir, K. Sadaghiani, M. Hemmati, J. Appl. Polym. Sci. 86, 3148 (2002)
T.J. Shea, J.R. Campbell, D.M. White, L.A. Socha, US Patent 4,814,392, 21 Mar 1989, to General Electric
T.J. Shea, A.J. Moffett, J.R. Campbell, M.E.J. Dekkers, F.F. Khouri, US Patent 5,132,361, 21 July 1992, to General Electric
Q. Shi, P. Stagnaro, C.-L. Cai, J.-H. Yin, G. Costa, A. Turturro, J. Appl. Polym. Sci. 110, 3963 (2008)
H. Shi, D. Shi, X. Wang, L. Yin, J. Yin, Y.-W. Mai, Polymer 51, 4958 (2010)
M. Shibayama, K. Uenoyama, J. Oura, S. Nomura, T. Iwamoto, Polymer 36, 4811 (1995)
N. Shibuya, K. Kosegaki, Eur. Pat. Appl. 248,526, 9 Dec 1987 to Mitsubishi Petrochemical
N. Shibuya, K. Takagi, S. Hattori, T. Kobayashi, H. Sano, Eur. Pat. Appl. 270,796, 15 June 1988a, to Mitsubishi Petrochemical
N. Shibuya, Y. Sobajima, H. Sano, US Patent 4,743,651, 10 May 1988b, to Mitsubishi Petrochemical
Y.-T. Shieh, T.-N. Liao, F.-C. Chang, J. Appl. Polym. Sci. 79, 2272 (2001)
M. Shimano, S. Moritomi, US Patent Appl. 20100261846, 14 Oct 2010, to Sumitomo Chemical
T. Shiraki, F. Hayano, H. Morita, US Patent 4,628,072, 9 Dec 1986, to Asahi
T. Shiraki, F. Hayano, H. Morita, US Patent 4,657,970, 14 Apr 1987a, to Asahi
T. Shiraki, F. Hayano, H. Morita, US Patent 4,657,971, 14 Apr 1987b, to Asahi
G.O. Shonaike, G.P. Simon (eds.), Polymer Blends and Alloys (Marcel Dekker, New York, 1999)
S. Shulin, C. Zhuo, S. Lili, Z. Huixuan, J. Appl. Polym. Sci. 121, 909 (2011)
M. Shuster, M. Narkis, A. Siegmann, J. Appl. Polym. Sci. 52, 1383 (1994)
N. Silvi, S.B. Brown, M.H. Giammattei, K.L. Howe, US Patent 5,939,490, 27 May 1997, to General Electric and DuPont
W.M. Sims, US Patent 3,966,839, 29 June 1976, to Foster Grant
H. Singh, N.K. Gupta, J. Polym. Res. 18, 1365 (2011)
A.K. Singh, R. Prakash, D. Pandey, J. Phys. Chem. B 115, 1601 (2011)
A.K. Singh, R. Prakash, D. Pandey, RSC Adv. 2, 10316 (2012)
T.M. Sivavec, S.J. McCormick, US Patent 5,066,719, 19 Nov 1991, to General Electric
T.M. Sivavec, S.M. Fukuyama, US Patent 5,140,077, 18 Aug 1992, to General Electric
W.A. Smith, J.W. Barlow, D.R. Paul, J. Appl. Polym. Sci. 26, 4233 (1981)
A.P. Smith, R.J. Spontak, H. Ade, Polym. Degrad. Stab. 72, 519 (2001)
B.G. Soares, F.F. Alves, M.G. Oliveira, A.C.F. Moreira, F.G. Garcia, M.F.S. Lopes, Eur. Polym. J. 37, 1577 (2001)
B.G. Soares, M.S.M. Almeida, P.I.C. Guimaraes, Eur. Polym. J. 40, 2185 (2004)
K. Solc (ed.), Polymer Compatibility and Incompatibility. Principles and Practice (Harwood Academic Publishers, New York, 1981)
Y. Son, R.A. Weiss, Polym. Eng. Sci. 41, 329 (2001)
Y. Son, R.A. Weiss, Polym. Eng. Sci. 42, 1322 (2002)
Y. Son, K.H. Ahn, K. Char, Polym. Eng. Sci. 40, 1376 (2000a)
Y. Son, K.H. Ahn, K. Char, Polym. Eng. Sci. 40, 1385 (2000b)
Z. Song, W.E. Baker, J. Appl. Polym. Sci. 44, 2167 (1992)
J. Song, C.M. Thurber, S. Kobayashi, A.M. Baker, C.W. Macosko, H.C. Silvis, Polymer 53, 3636 (2012a)
W. Song, H. Liu, F. Chen, J. Zhang, Polymer 53, 2476 (2012b)
R. Sonnier, V. Massardier, L. Clerc, J.M. Lopez-Cuesta, A. Bergeret, J. Appl. Polym. Sci. 115, 1710 (2010)
L.H. Sperling, Microphase structure, in Encyclopedia of Polymer Science and Engineering, ed. by J.I. Kroschwitz, vol. 9, 2nd edn. (Wiley, New York, 1987), p. 770
F. Speroni, Macromol. Symp. 78, 299 (1994)
M.J. Stachowski, A.T. DiBenedetto, Polym. Eng. Sci. 37, 252 (1997)
M.-D. Stelescu, Macromol. Symp. 263, 70 (2008)
M.E. Stewart, A.J. Cox, D.M. Naylor, Polymer 34, 4060 (1993a)
M.E. Stewart, S.E. George, R.L. Miller, D.R. Paul, Polym. Eng. Sci. 33, 675 (1993b)
H. Stutz, P. Pötschke, U. Mierau, Macromol. Symp. 112, 151 (1996)
K.-F. Su, K.-H. Wei, J. Appl. Polym. Sci. 56, 79 (1995)
C.C. Su, E.M. Woo, C.-Y. Chen, R.-R. Wu, Polymer 38, 2047 (1997)
W.-Y. Su, K. Min, R.P. Quirk, Polymer 42, 5121 (2001a)
W.-Y. Su, Y. Wang, K. Min, R.P. Quirk, Polymer 42, 5107 (2001b)
S. Su, Q. Li, Y. Liu, H. Xu, W. Guo, C. Wu, J. Macromol. Sci. B Phys. 48, 823 (2009)
T. Sugie, R. Ishikawa, F. Kobata, US Patent 4,528,346, 9 July 1985, to Dainippon Ink and Chemicals
Y.-J. Sun, W.E. Baker, J. Appl. Polym. Sci. 65, 1385 (1997)
Y.-J. Sun, G.-H. Hu, M. Lambla, H.K. Kotlar, Polymer 37, 4119 (1996)
Y.-J. Sun, R.J.G. Willemse, T.M. Liu, W.E. Baker, Polymer 39, 2201 (1998)
S.L. Sun, X.Y. Xu, H.D. Yang, H.X. Zhang, Polymer 46, 7632 (2005)
S.L. Sun, Z.Y. Tan, M.Y. Zhang, H.D. Yang, H.X. Zhang, Polymer International 55, 834 (2006)
S. Sun, Z. Chen, H. Zhang, Polym. Bull. 61, 443 (2008)
S. Sun, M. Zhang, H. Zhang, X. Zhang, J. Appl. Polym. Sci. 122, 2992 (2011)
S. Sun, F. Zhang, Y. Fu, C. Zhou, H. Zhang, J. Macromol. Sci. B Phys. 52, 861 (2013)
U. Sundararaj, C.W. Macosko, A. Nakayama, T. Inoue, Polym. Eng. Sci. 35, 100 (1995)
T. Suzuki, H. Tanaka, T. Nishi, Polymer 30, 1287 (1989)
R.T. Swiger, L.A. Mango, DE Patent 2,722,270, 1 Dec 1977, to General Electric
R.T. Swiger, P.C. Juliano, US Patent 4,147,740, 3 Apr 1979, to General Electric
P.D. Sybert, C.Y. Han, S.B. Brown, D.J. McFay, W.L. Gately, J.A. Tyrell, R.A. Florence, US Patent 5,015,698, 14 May 1991, to General Electric
A. Tabtiang, R.A. Venables, Polymer 43, 4791 (2002)
M. Taheri, J. Morshedian, H. Ali Khonakdar, J. Appl. Polym. Sci. 119, 1417 (2011)
Y. Takeda, D.R. Paul, Polymer 32, 2771 (1991)
Y. Takeda, H. Keskkula, D.R. Paul, Polymer 33, 3394 (1992a)
Y. Takeda, H. Keskkula, D.R. Paul, Polymer 33, 3173 (1992b)
N.C.B. Tan, S.-K. Tai, R.M. Briber, Polymer 37, 3509 (1996)
R.T.H. Tang, M.C. Bochnik, F. Mares, S. Arnold, US Patent 5,124,411, 23 June 1992, to Allied-Signal
T. Tang, Z. Lei, X. Zhang, H. Chen, B. Huang, Polymer 36, 5061 (1995)
T. Tang, Z. Lei, B. Huang, Polymer 37, 3219 (1996)
L.W. Tang, K.C. Tam, C.Y. Yue, X. Hu, Y.C. Lam, L. Li, J. Appl. Polym. Sci. 85, 209 (2002)
V. Tanrattanakul, A. Hiltner, E. Baer, W.G. Perkins, F.L. Massey, A. Moet, Polymer 38, 4117 (1997a)
V. Tanrattanakul, A. Hiltner, E. Baer, W.G. Perkins, F.L. Massey, A. Moet, Polymer 38, 2191 (1997b)
S.B. Tatum, D. Cole, A.N. Wilkinson, J. Macromol. Sci. B Phys. 39, 459 (2000)
C. Taubitz, E. Seiler, K. Boehlke, H. Gausepohl, B. Ostermayer, Eur. Pat. Appl. 285,966, 12 Oct 1988a, to BASF
C. Taubitz, E. Seiler, K. Boehlke, B. Ostermayer, H. Gausepohl, Eur. Pat. Appl. 285,969, 12 Oct 1988b, to BASF
C. Taubitz, E. Seiler, K. Boehlke, B. Ostermayer, H. Gausepohl, Eur. Pat. Appl. 285,975, 12 Oct 1988c, to BASF
C. Taubitz, E. Seiler, K. Boehlke, B. Ostermayer, H. Gausepohl, V. Muench, Eur. Pat. Appl. 285,976, 12 Oct 1988d, to BASF
C. Taubitz, E. Seiler, R. Bruessau, D. Wagner, Eur. Pat. Appl. 285,968, 12 Oct 1988e, to BASF
C. Taubitz, E. Seiler, H. Gausepohl, K. Boehlke, R. Bueschl, Eur. Pat. Appl. 289,780, 9 Nov 1988f, to BASF
C. Taubitz, E. Seiler, J. Hambrecht, K. Mitulla, K. Boehlke, US Patent 4,959,415, 25 Sept 1990, to BASF
C. Taubitz, E. Seiler, K. Boehlke, D. Wagner, US Patent 5,053,458, 1 Oct 1991, to BASF
J.W. Teh, A. Rudin, Polym. Eng. Sci. 31, 1033 (1991)
J.W. Teh, A. Rudin, Polym. Eng. Sci. 32, 1678 (1992)
J.W. Teh, A. Rudin, J.C. Keung, Adv. Polym. Technol. 13, 1 (1994)
P. Teyssie, R. Fayt, R. Jérôme, Macromol. Chem. Macromol. Symp. 16, 41 (1988)
S. Thomas, G. Groeninckx, Polymer 40, 5799 (1999)
J.-M. Thomassin, S. Lenoir, J. Riga, R. Jérôme, C. Detrembleur, Biomacromolecules 8, 1171 (2007)
E.J. Tijsma, L. van der Does, A. Bantjes, I. Vulic, G.H.W. Buning, Makromol. Chem. Macromol. Symp. 75, 193 (1993)
E.J. Tijsma, L. van der Does, A. Bantjes, I. Vulic, G.H.W. Buning, Macromol. Chem. Phys. 195, 1577 (1994)
S.C. Tjong, Y.Z. Meng, Polymer 38, 4609 (1997)
S.C. Tjong, Y.Z. Meng, J. Appl. Polym. Sci. 74, 1827 (1999)
S.C. Tjong, R.K.Y. Li, Y.Z. Meng, J. Appl. Polym. Sci. 67, 521 (1998)
S. Togo, A. Amagai, Y. Kondo, T. Yamada, Eur. Pat. Appl. 268,486, 25 May 1988, to Mitsubishi Gas Chemical
R.T. Tol, G. Groeninckx, I. Vinckier, P. Moldenaers, J. Mewis, Polymer 45, 2587 (2004)
R.T. Tol, V.B.F. Mathot, G. Groeninckx, Polymer 46, 383 (2005)
Q. Tran-Cong, Phase separation and morphology of chemically reacting polymer blends, in Structures and Properties of Multiphase Polymeric Materials, ed. by T. Araki, Q. Tran-Cong, M. Shibayama (Marcel Dekker, New York, 1998), p. 155
V.J. Triacca, S. Ziaee, J.W. Barlow, H. Keskkula, D.R. Paul, Polymer 32, 1401 (1991)
C.-H. Tsai, F.-C. Chang, J. Appl. Polym. Sci. 61, 321 (1996)
C. Tselios, D. Bikiaris, J. Prinos, C. Panayioutou, J. Appl. Polym. Sci. 64, 983 (1997)
C. Tselios, D. Bikiaris, V. Maslis, C. Panayioutou, Polymer 39, 6807 (1998)
A.H. Tsou, B.D. Favis, Y. Hara, P.A. Bhadane, Y. Kirino, Macromol. Chem. Phys. 210, 340 (2009)
A.H. Tsou, Y. Soeda, Y. Hara, M.B. Measmer, US Patent 8,021,730, 20 Sept 2011, to Exxonmobil and Yokohama Rubber
B. Turcsáyii, J. Macromol. Sci. A Pure Appl. Chem. 32, 255 (1995)
A. Tynys, U. Hippi, J. Seppälä, J. Appl. Polym. Sci. 86, 1886 (2002)
S. Ueda, H. Harada, K. Yoshida, K. Kasei, US Patent 4,772,664, 20 Sept 1988, to Asahi
K. Ueno, T. Maruyama, US Patent 4,315,086, 9 Feb 1982a, to Sumitomo Chemical
K. Ueno, T. Maruyama, US Patent 4,338,410, 6 July 1982b, to Sumitomo Chemical
L.A. Utracki, Polymer Blends and Alloys (Hanser Publishers, New York, 1989)
L.A. Utracki, Commercial Polymer Blends (Chapman and Hall, London, 1998)
L.A. Utracki, M.M. Dumoulin, Polypropylene alloys and blends with thermoplastics, in Polypropylene Structure, Blends and Composites, ed. by J. Karger-Kocsis (Chapman and Hall, London, 1995), pp. 50–94
T. Vainio, G.-H. Hu, M. Lambla, J.V. Seppälä, J. Appl. Polym. Sci. 61, 843 (1996a)
T. Vainio, H. Jukarainen, J.V. Seppälä, J. Appl. Polym. Sci. 59, 2095 (1996b)
T. Vainio, G.-H. Hu, M. Lambla, J.V. Seppälä, J. Appl. Polym. Sci. 63, 883 (1997)
A. Valenza, D. Acierno, Eur. Polym. J. 30, 1121 (1994)
A. Valenza, F.P. La Mantia, E. Gattiglia, A. Turturro, Int. Polym. Process. 9, 240 (1994)
A. Valenza, G. Carianni, L. Mascia, Polym. Eng. Sci. 38, 452 (1998)
M. Valero, J.J. Iruin, E. Espinosa, M.J. Fernandez-Berridi, Polym. Commun. 31, 127 (1990)
H.A.M. Van Aert, G.J.M. van Steenpaal, L. Nelissen, P.J. Lemstra, J. Liska, C. Bailly, Polymer 42, 2803 (2001)
P. Van Ballegooie, A. Rudin, Polym. Eng. Sci. 28, 1434 (1988)
A.C.M. Van Bennekom, D.T. Pluimers, J. Bussink, R.J. Gaymans, Polymer 38, 3017 (1997)
R. Van der Meer, J.B. Yates, PCT Intl. Appl. WO 87/00540, 29 Jan 1987, to General Electric
A. Van der Wal, J.J. Mulder, J. Oderkerk, R.J. Gaymans, Polymer 39, 6781 (1998)
M. Van Duin, M. Aussems, R.J.M. Borggreve, J. Polym. Sci. A Polym. Chem. 36, 179 (1998)
M.E. Villarreal, M. Tapia, S.M. Nuño-Donlucas, J.E. Puig, J. Gonzalez-Nunez, J. Appl. Polym. Sci. 92, 2545 (2004)
T. Vivier, M. Xanthos, J. Appl. Polym. Sci. 54, 569 (1994)
C. Vocke, U. Anttila, M. Heino, P. Hietaoja, J. Seppälä, J. Appl. Polym. Sci. 70, 1923 (1998)
C. Vocke, U. Anttila, J. Seppälä, J. Appl. Polym. Sci. 72, 1443 (1999)
J. Vogel, C. Heinze, Angew. Makromol. Chem. 207, 157 (1993)
H.J. Vroomans, Eur. Pat. Appl. 286,734, 19 Oct 1988, to Stamicarbon
D. Wang, X.-M. Xie, Polymer 47, 7859 (2006)
X.-H. Wang, H.-X. Zhang, Z.-G. Wang, B.-Z. Jiang, Polymer 38, 1569 (1997)
Z. Wang, C.-M. Chan, S.H. Zhu, J. Shen, Polymer 39, 6801 (1998a)
X.-H. Wang, H.-X. Zhang, W. Jiang, Z.-G. Wang, C.-H. Liu, H.-J. Liang, B.-Z. Jiang, Polymer 39, 2697 (1998b)
X. Wang, H. Li, E. Ruckenstein, Polymer 42, 9211 (2001)
C. Wang, J.X. Su, J. Li, H. Yang, Q. Zhang, R.N. Du, Q. Fu, Polymer 47, 3197 (2006)
J.S. Wang, X.D. Chen, B.Y. Mai, M.Q. Zhang, M.Z. Rong, J. Appl. Polym. Sci. 105, 1309 (2007a)
H.-G. Wang, L.-Q. Jian, B.-L. Pan, J.-Y. Zhang, S.-R. Yang, H.-G. Wang, Polym. Eng. Sci. 47, 738 (2007b)
Q. Wang, R. Qi, Y. Shen, Q. Liu, C. Zhou, J. Appl. Polym. Sci. 106, 3220 (2007c)
R. Wang, S. Wang, Y. Zhang, C. Wan, P. Ma, Polym. Eng. Sci. 49, 329 (2009a)
P. Wang, K. Meng, H. Cheng, S. Hong, J. Hao, C.C. Han, H. Haeger, Polymer 50, 2154 (2009b)
D. Wang, S. Fujinami, H. Liu, K. Nakajima, T. Nishi, Macromolecules 43, 5521 (2010a)
S. Wang, B. Li, Y. Zhang, J. Appl. Polym. Sci. 118, 3545 (2010b)
S. Wang, B. Li, Y. Zhang, Plast. Rubber Compos. 39, 379 (2010c)
S. Wang, B. Li, Y. Zhang, Polym. Polym. Compos. 18, 219 (2010d)
D. Wang, Y. Li, X.-M. Xie, B.-H. Guo, Polymer 52, 191 (2011)
S. Wang, C. Lin, H. Sun, F. Chen, J. Li, S. Guo, Polym. Eng. Sci. 52, 338 (2012a)
S. Wang, L. Shao, Z. Song, J. Zhao, Y. Feng, J. Appl. Polym. Sci. 124, 4827 (2012b)
Q. Wang, Y. Jiang, L. Li, P. Wang, Q. Yang, G. Li, J. Macromol. Sci. B Phys. 51, 96 (2012c)
H. Wang, Q. Qian, X. Jiang, X. Liu, L. Xiao, B. Huang, Q. Chen, J. Appl. Polym. Sci. (2012d). doi:10.1002/app.36984; published online
Y. Wang, S.M. Chiao, T.-F. Hung, S.-Y. Yang, J. Appl. Polym. Sci. (2012e). doi:10.1002/app.36920; published online
Q. Wang, J. Zhu, P. Wang, L. Li, Q. Yang, Y. Huang, J. Appl. Polym. Sci. 124, 5064 (2012f)
W. Wang, D.K. Knoeppel, F. Li, J.M. Sosa, US Patent Appl. 20130005852, 3 Jan 2013a, to Fina Technology
M. Wang, G. Yuan, C.C. Han, Polymer 54, 3612 (2013b)
M. Weber, N. Güntherberg, US Patent 5,907,101, 25 May 1999, to BASF
M. Weber, H. Honl, P. Ittemann, W. Haensel, US Patent Appl. 20100036043, 11 Feb 2010, to BASF
K.-W. Wei, J.-C. Ho, J. Appl. Polym. Sci. 63, 1527 (1997)
K.-W. Wei, K.-F. Su, J. Appl. Polym. Sci. 59, 787 (1996)
K.-W. Wei, W.-J. Hwang, H.-L. Tyan, Polymer 37, 2087 (1996)
K.-W. Wei, H.-C. Jang, J.-C. Ho, Polymer 38, 3521 (1997)
Q. Wei, D. Chionna, E. Galoppini, M. Pracella, Macromol. Chem. Phys. 204, 1123 (2003)
Q. Wei, D. Chionna, M. Pracella, Macromol. Chem. Phys. 206, 777 (2005)
K.A. Weiss, US Patent 4,816,515, 28 Mar 1989, to General Electric
J.L. White, K. Min, in Comprehensive Polymer Science, ed. by S.L. Aggarwal, vol. 7 (Pergamon Press, New York, 1989), p. 285
D.M. White, L.A. Socha, R. van der Meer, PCT Intl. Appl. WO 89/000179, 12 Jan 1989, to General Electric
G. Wildes, H. Keskkula, D.R. Paul, J. Polym. Sci. B Polym. Phys. 37, 71 (1999)
H.M. Wilhelm, M.I. Felisberti, J. Appl. Polym. Sci. 85, 847 (2002a)
H.M. Wilhelm, M.I. Felisberti, J. Appl. Polym. Sci. 86, 366 (2002b)
A.N. Wilkinson, S.B. Tattum, A.J. Ryan, Polymer 38, 1923 (1997)
A.N. Wilkinson, M.L. Clemens, V.M. Harding, Polymer 45, 5239 (2004)
A.N. Wilkinson, E.M.I. Nita, M.L. Clemens, E. Jobstl, J.P.A. Fairclough, J. Macromol. Sci. B Phys. 44, 1087 (2005)
J.M. Willis, B.D. Favis, Polym. Eng. Sci. 28, 1416 (1988)
J.M. Willis, B.D. Favis, J. Lunt, Polym. Eng. Sci. 30, 1073 (1990)
J.M. Willis, B.D. Favis, C. Lavallee, J. Mater. Sci. 28, 1749 (1993)
C. Wippler, Polym. Eng. Sci. 33, 347 (1993)
H. Witteler, G. Lieser, M. Dröscher, Makromol. Chem. Rapid Commun. 14, 401 (1993)
S.-C. Wong, Y.-W. Mai, Polymer 40, 1553 (1999)
S.-C. Wong, Y.-W. Mai, Polymer 41, 5471 (2000)
E.M. Woo, S.-S. Hou, D.-H. Huang, L.-T. Lee, Polymer 46, 7425 (2005)
C. Wörner, P. Müller, R. Mülhaupt, J. Appl. Polym. Sci. 66, 633 (1997)
S. Wu, Polym. Eng. Sci. 27, 335 (1987)
C.-J. Wu, J.-F. Kuo, C.-Y. Chen, Polym. Eng. Sci. 33, 1329 (1993)
C.-J. Wu, J.-F. Kuo, C.-Y. Chen, E. Woo, J. Appl. Polym. Sci. 52, 1695 (1994)
J.-Y. Wu, W.-C. Lee, W.-F. Kuo, H.-C. Kao, M.-S. Lee, J.-L. Lin, Adv. Polym. Technol. 14, 47 (1995)
D. Wu, X. Wang, R. Jin, Eur. Polym. J. 40, 1223 (2004)
D. Wu, X. Wang, R. Jin, J. Appl. Polym. Sci. 99, 3336 (2006a)
Y. Wu, Y. Yang, B. Li, Y. Han, J. Appl. Polym. Sci. 100, 3187 (2006b)
F. Wu, T. Xie, G. Yang, Polym. Bull. 65, 731 (2010)
W. Wu, C. Wan, H. Zhang, Y. Zhang, J. Appl. Polym Sci. (2013). doi:10.1002/pen.40272; published online
M. Xanthos, Polym. Eng. Sci. 28, 1392 (1988)
M. Xanthos, in Polypropylene, ed. by J. Karger-Kocsis (Kluwer, Dordrecht, 1999), p. 694
M. Xanthos, S.S. Dagli, Polym. Eng. Sci. 31, 929 (1991)
M. Xanthos, J.F. Parmer, M.L. LaForest, G.R. Smith, J. Appl. Polym. Sci. 62, 1167 (1996)
Z. Xiaomin, W. Dongmei, Y. Zhihui, Y. Jinghua, J. Appl. Polym. Sci. 62, 67 (1996)
Z. Xiaomin, Y. Zhihui, N. Tainhai, Y. Jinghua, Polymer 38, 5905 (1997)
Z. Xiaomin, L. Gang, W. Dongmei, Y. Zhihui, Y. Jinghua, L. Jingshu, Polymer 39, 15 (1998)
T. Xie, G. Yang, J. Appl. Polym. Sci. 93, 1446 (2004)
X.-M. Xie, X. Zheng, Mater. Design 22, 11 (2000)
X.-M. Xie, Y.-Y. Liu, B.-H. Guo, J. Feng, T. Ishikawa, T. Morinaga, J. Appl. Polym. Sci. 82, 1284 (2001a)
W.B. Xie, K.C. Tam, C.Y. Yue, X. Hu, Y.C. Lam, L. Li, J. Appl. Polym. Sci. 82, 477 (2001b)
B.-H. Xie, M.-B. Yang, S.-D. Li, Z.-M. Li, J.-M. Feng, J. Appl. Polym. Sci. 88, 398 (2003)
T. Xie, H. Wu, W. Bao, S. Guo, Y. Chen, H. Huang, H. Chen, S.-Y. Lai, J. Jow, J. Appl. Polym. Sci. 118, 1846 (2010)
C. Xu, Z. Fang, J. Zhong, Angew. Makromol. Chem. 212, 45 (1993)
C. Xu, Z. Fang, J. Zhong, Polymer 38, 155 (1997)
S. Xu, T. Tang, B. Chen, B. Huang, Polymer 40, 2239 (1999a)
S. Xu, H. Zhao, T. Tang, L. Dong, B. Huang, Polymer 40, 1537 (1999b)
X.Y. Xu, S.L. Sun, Z.C. Chen, H.X. Zhang, J. Appl. Polym. Sci. 109, 2482 (2008)
C. Xu, Z. Fang, Y. Chen, Polym. Eng. Sci. (2013a). doi:10.1002/pen.23789; published online
M. Xu, W. Qiu, G. Qiu, J. Macromol. Sci. B Phys. 52, 155 (2013b)
M.L. Xue, Y.-L. Yu, J. Sheng, H.H. Chuah, J. Macromol. Sci. B Phys. 44, 531 (2005)
M.L. Xue, Y.-L. Yu, H.H. Chuah, J. Macromol. Sci. B Phys. 46, 603 (2007a)
M.L. Xue, Y.-L. Yu, H.H. Chuah, G.X. Qiu, J. Macromol. Sci. B Phys. 46, 387 (2007b)
S. Yamao, W. Kosaka, US Patent 6,037,422, 14 Mar 2000, to Idemitsu Petrochemical
L.-T. Yan, J. Sheng, Polymer 47, 2894 (2006)
H. Yang, M. Lai, W. Liu, C. Sun, J. Liu, J. Appl. Polym. Sci. 85, 2600 (2002a)
H. Yang, J. Ma, W. Li, B. Liang, Y. Yu, Polym. Eng. Sci. 42, 1629 (2002b)
H. Yang, X. Cao, Y. Ma, J. An, Y. Ke, X. Liu, F. Wang, Polym. Eng. Sci. 52, 481 (2012)
L. Yang, A. Zhang, L. Wang, R. Chen, Y. Zeng, W. Wu, Polym. Eng. Sci. 53, 2093 (2013)
Z. Yao, J.-M. Sun, Q. Wang, K. Cao, Ind. Eng. Chem. Res. 51, 751 (2012)
J.B. Yates, US Patent 4,755,566, 5 July 1988, to General Electric
J.B. Yates, T.J. Ullman, US Patent 4,745,157, 17 May 1988, to General Electric
J.B. Yates, D.M. White, US Patent 4,859,739, 22 Aug 1989, to General Electric
J.B. Yates, S.B. Brown, R.C. Lowry, J.C. Blubaugh, D.F. Aycock, US Patent 5,115,043, 19 May 1992, to General Electric
J.T. Yeh, C.C. Fan-Chiang, J. Appl. Polym. Sci. 66, 2517 (1997)
J.T. Yeh, C.C. Fan-Chiang, S.-S. Yang, J. Appl. Polym. Sci. 64, 1531 (1997)
C. Yeung, K.A. Herrmann, Macromolecules 36, 229 (2003)
Z. Yin, C. Koulic, C. Pagnoulle, R. Jérôme, Macromolecules 34, 5132 (2001)
Z. Yin, C. Koulic, C. Pagnoulle, R. Jérôme, Langmuir 19, 453 (2003a)
Z. Yin, C. Koulic, C. Pagnoulle, R. Jérôme, Macromol. Symp. 198, 197 (2003b)
B. Yin, Y. Zhao, M.-M. Pan, M.-B. Yang, Polym. Adv. Technol. 18, 439 (2007)
L. Yin, D. Shi, Y. Liu, J. Yin, Polym. Int. 58, 919 (2009a)
L. Yin, J. Yin, D. Shi, S. Luan, Eur. Polym. J. 45, 1554 (2009b)
K. Yonekura, A. Uchiyama, A. Matsuda, US Patent 4,785,045, 15 Nov 1988, to Mitsui Petrochemical Industries
L.K. Yoon, C.H. Choi, B.K. Kim, J. Appl. Polym. Sci. 56, 239 (1995)
K.H. Yoon, S.C. Lee, I.H. Park, H.M. Lee, O.O. Park, T.W. Son, Polymer 38, 6079 (1997)
K.H. Yoon, H.W. Lee, O.O. Park, Polymer 41, 4445 (2000)
D.W. Yu, M. Xanthos, C.G. Gogos, Adv. Polym. Technol. 10, 163 (1990)
D.W. Yu, M. Xanthos, C.G. Gogos, Adv. Polym. Technol. 11, 295 (1992)
D.W. Yu, M. Xanthos, C.G. Gogos, J. Appl. Polym. Sci. 52, 99 (1994)
Z.-Z. Yu, Y.-C. Ou, G.-H. Hu, J. Appl. Polym. Sci. 69, 1711 (1998)
L. Yu, G. Simon, R.A. Shanks, M.R. Nobile, J. Appl. Polym. Sci. 77, 2229 (2000)
Z.-Z. Yu, M.-S. Yang, S.-C. Dai, Y.-W. Mai, J. Appl. Polym. Sci. 93, 1462 (2004)
X. Yu, Y. Wu, B. Li, Y. Han, Polymer 46, 3337 (2005)
L. Yu, K. Dean, L. Li, Prog. Polym. Sci. 31, 576 (2006)
S. Yukioka, T. Inoue, Polymer 35, 1182 (1994)
J. Zhang, J. He, Polymer 43, 1437 (2002)
H.X. Zhang, D.J. Hourston, J. Appl. Polym. Sci. 71, 2049 (1999)
X.Q. Zhang, Y. Son, J. Appl. Polym. Sci. 89, 2502 (2003)
X. Zhang, J. Yin, Polym. Eng. Sci. 37, 197 (1997)
X. Zhang, J. Yin, Macromol. Chem. Phys. 199, 2631 (1998)
X. Zhang, Z. Yin, J. Yin, J. Appl. Polym. Sci. 62, 893 (1996)
X. Zhang, X.L. Li, D. Wang, Z. Yin, J. Yin, J. Appl. Polym. Sci. 64, 1489 (1997)
H. Zhang, R.A. Weiss, J.E. Kuder, D. Cangiano, Polymer 41, 3069 (2000)
J. Zhang, T.P. Lodge, C.W. Macosko, Macromolecules 38, 6586 (2005)
H. Zhang, Y. Zhang, W. Guo, D. Xu, C. Wu, J. Appl. Polym. Sci. 109, 3546 (2008)
J. Zhang, L. Luo, S. Lyu, J. Schley, B. Pudil, M. Benz, A. Buckalew, K. Chaffin, C. Hobot, R. Sparer, J. Appl. Polym. Sci. 117, 2153 (2010)
J. Zhang, Y. Li, Y. Zhu, M. Cui, X. Jiang, J. Polym. Eng. 32, 487 (2012)
C.-L. Zhang, T. Zhang, L.-F. Feng, J. Appl. Polym. Sci. (2013a). doi:10.1002/app.39972; published online
X. Zhang, Y. Li, L. Han, C. Han, K. Xu, C. Zhou, M. Zhang, L. Dong, Polym. Eng. Sci. 53, 2498 (2013b)
W. Zhang, Z. Gui, C. Lu, S. Cheng, D. Cai, Y. Gao, Mater. Lett. 92, 68 (2013c)
L. Zhaohui, X. Zhang, S. Tasaka, N. Inagaki, Mater. Lett. 48, 81 (2001)
L. Zhidan, S. Juncai, C. Chao, Z. Xiuju, J. Appl. Polym. Sci. 121, 1972 (2011)
Y. Zhihui, Z. Xiaomin, Z. Yajie, Y. Jinghua, J. Appl. Polym. Sci. 63, 1857 (1997)
Y. Zhihui, Z. Yajie, Z. Xiaomin, Y. Jinghua, Polymer 39, 547 (1998)
Z.F. Zhou, N.C. Liu, H. Huang, Polymer 45, 7109 (2004)
W.-H. Zhou, S.-W. Ye, Y.-W. Chen, Y.-L. Huang, S.-S. Yuan, J. Macromol. Sci. B Phys. 51, 2361 (2012)
Y. Zhou, W. Wang, R. Dou, L.-P. Li, B. Yin, M.-B. Yang, Polym. Eng. Sci. 53, 1845 (2013)
X. Zhu, Z. Yang, S. Wu, J. Thermoplast. Compos. Mater. 23, 351 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Abbreviations
- AA
-
Acrylic acid
- ABS
-
Acrylonitrile-butadiene-styrene terpolymer
- AES
-
Terpolymer from acrylonitrile, ethylene-propylene elastomer and styrene
- AFM
-
Atomic force microscopy
- AN
-
Acrylonitrile
- ASA
-
Acrylonitrile-styrene-acrylate terpolymer
- ATRP
-
Atom transfer radical polymerization
- B
-
Butadiene
- BA
-
n-Butyl acrylate
- tBA
-
t-Butyl acrylate
- BPA
-
Bisphenol A
- CHP
-
Cumene hydroperoxide
- CL
-
Caprolactone
- DBP
-
Dibenzoyl peroxide
- DCP
-
Dicumyl peroxide
- DEM
-
Diethylmaleate
- DMA
-
Dynamic mechanical analysis
- DMTA
-
Dynamic mechanical thermal analysis
- DSC
-
Differential scanning calorimetry
- E
-
Ethylene
- EA
-
Ethylacrylate
- EAA
-
Ethylene acrylic acid copolymer
- EB
-
Ethylene-butene copolymer
- EBA
-
Ethylene butyl acrylate copolymer
- EDXA
-
Energy dispersive X-ray analysis
- EEA
-
Ethylene ethylacrylate copolymer
- EELS
-
Electron energy loss spectroscopy
- E-GMA
-
Ethylene glycidyl methacrylate copolymer
- EMA
-
Ethylene methyl acrylate copolymer
- EMAA
-
Ethylene-methacrylic acid copolymer
- EMAc
-
Ethylene methyl acrylate copolymer
- EMM
-
Ethylene methyl methacrylate copolymer
- ENR
-
Epoxidized natural rubber
- EP
-
Ethylene-propylene copolymer
- EPDM
-
Ethylene-propylene-diene modified rubber
- ESCA
-
Electron spectroscopy for chemical analysis
- ETFE
-
Ethylene-tetrafluoroethylene copolymer
- EVAc
-
Ethylene-vinyl acetate copolymer
- EVAl
-
Ethylene-vinyl alcohol copolymer
- -f-
-
Functionalized (or functionalized with)
- FA
-
Fumaric acid
- FESEM
-
Field emission scanning electron microscopy
- FTIR
-
Fourier transform infrared spectroscopy
- GMA
-
Glycidyl methacrylate
- GPC
-
Gel permeation chromatography
- HDPE
-
High density polyethylene
- HDT
-
Heat distortion temperature
- HIPS
-
High-impact polystyrene
- I
-
Isoprene
- IA
-
Isobutyl acrylate
- IM
-
Impact modifier
- IPO
-
Isopropenyl oxazoline
- IV
-
Intrinsic viscosity
- L101
-
Luperox® 101 (2,5-di-(t-butylperoxy)-2,5-dimethylhexane)
- L130
-
Luperox® 130 (2,5-dimethyl-2,5-di-(t-butylperoxy) hexyne-3)
- LCP
-
Liquid crystalline polymer(s) (polyester-type unless noted)
- LDPE
-
Low-density polyethylene
- LLDPE
-
Linear low-density polyethylene
- MA
-
Maleic anhydride
- MAA
-
Methacrylic acid
- MAc
-
Methyl acrylate
- MALDI
-
Matrix-assisted laser desorption/ionization mass spectrometry
- MDPE
-
Medium density polyethylene
- MFI
-
Melt flow index
- MFR
-
Melt flow rate
- MMA
-
Methyl methacrylate
- MS
-
Mass spectrometry
- MW
-
Molecular weight
- MWD
-
Molecular weight distribution
- NA
-
Nadic anhydride
- NBR
-
Nitrile-butadiene rubber
- NMR
-
Nuclear magnetic resonance spectroscopy
- NR
-
Natural rubber
- PA
-
Polyamide(s)
- PB
-
Polybutadiene
- PBN
-
Polybutylene naphthalate
- PBT
-
Polybutylene terephthalate(s)
- PC
-
Bisphenol A polycarbonate
- PCE
-
Polycarbonate ester copolymer
- PCL
-
Polycaprolactone
- PCT
-
Poly(cyclohexanedimethanol terephthalate)
- PDMS
-
Polydimethylsiloxane
- PE
-
Polyethylene
- PE-g-MA
-
Maleic anhydride-grafted polyethylene
- PEEK
-
Polyetheretherketone
- PEG
-
Polyethylene glycol
- PEI
-
Polyetherimide
- PEN
-
Polyethylene naphthalate
- P-E-P
-
Propylene-ethylene-propylene block copolymer
- PES
-
Polyethersulfone
- PEST
-
Polyester(s)
- PET
-
Polyethylene terephthalate(s)
- PETG
-
Polyethylene terephthalate glycol modified (glycol is typically cyclohexanedimethanol)
- Phenoxy
-
Copolymer of BPA and epichlorohydrin
- Phr
-
Parts per 100 parts resin
- PLA
-
Poly(lactic acid)
- PMMA
-
Poly(methyl methacrylate)
- PMP
-
Poly-4-methylpentene-1
- PO
-
Polyolefin(s)
- PP
-
Polypropylene
- PPE
-
Polyphenylene ether(s)
- PPS
-
Polyphenylene sulfide
- PPVL
-
Polypivalolactone
- PS
-
Polystyrene
- PTFE
-
Polytetrafluoroethylene
- PTT
-
Poly(trimethylene terephthalate)
- PVAc
-
Polyvinyl acetate
- PVAl
-
Polyvinyl alcohol
- PVC
-
Polyvinylchloride
- PVDF
-
Polyvinylidene difluoride
- RI
-
Radical initiator
- S
-
Styrene
- SAA
-
Styrene-acrylic acid copolymer
- SALS
-
Small-angle light scattering
- SAN
-
Styrene-acrylonitrile copolymer
- SAXS
-
Small-angle X-ray scattering
- SB
-
Styrene-butadiene copolymer
- SBS
-
Styrene-butadiene-styrene copolymer
- SEBS
-
Styrene-(ethylene/butylene)-styrene copolymer
- SEC
-
Size-exclusion chromatography
- SEM
-
Scanning electron microscopy
- SEP
-
Styrene (ethylene-propylene) block copolymer
- SI
-
Styrene-isoprene copolymer
- S-IPO
-
Styrene-isopropenyl oxazoline copolymer
- SIS
-
Styrene-isoprene-styrene copolymer
- SMA
-
Styrene-co-maleic anhydride copolymer
- sPS
-
Syndiotactic polystyrene
- SSE
-
Single-screw extruder
- Tg
-
Glass transition temperature
- Tm
-
Melting temperature
- TAIC
-
Triallyl isocyanurate
- TBAB
-
Tetrabutylammonium bromide
- TEM
-
Transmission electron microscopy
- TGA
-
Thermogravimetric analysis
- TPE
-
Thermoplastic elastomer
- TPP
-
Triphenyl phosphate
- TPPite
-
Triphenyl phosphite
- TPU
-
Thermoplastic polyurethane
- TSE
-
Twin-screw extruder
- UHMWPE
-
Ultra high molecular weight polyethylene
- ULDPE
-
Ultra low-density polyethylene
- UV
-
Ultraviolet
- VLDPE
-
Very-low-density polyethylene
- VTMS
-
Vinyl trimethoxysilane
- WAXD
-
Wide-angle X-ray diffraction
- WAXS
-
Wide-angle X-ray scattering
- XPS
-
X-ray photoelectron spectroscopy
- ZnSt
-
Zinc stearate
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this entry
Cite this entry
Brown, S.B. (2014). Reactive Compatibilization. In: Utracki, L., Wilkie, C. (eds) Polymer Blends Handbook. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6064-6_7
Download citation
DOI: https://doi.org/10.1007/978-94-007-6064-6_7
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6063-9
Online ISBN: 978-94-007-6064-6
eBook Packages: Chemistry and Materials ScienceReference Module Physical and Materials ScienceReference Module Chemistry, Materials and Physics