
Abstract
Mitochondria are specialised organelles that are structurally and functionally integrated into cells in the vast majority of eukaryotes. They are the site of numerous enzymatic reactions, some of which are essential for life. The double lipid membrane of the mitochondrion, that spatially defines the organelle and is necessary for some functions, also creates a physical but semi-permeable barrier to the rest of the cell. Thus to ensure the biogenesis, regulation and maintenance of a functional population of proteins, an autonomous protein handling network within mitochondria is required. This includes resident mitochondrial protein translocation machinery, processing peptidases, molecular chaperones and proteases. This review highlights the contribution of proteases of the AAA+ superfamily to protein quality and activity control within the mitochondrion. Here they are responsible for the degradation of unfolded, unassembled and oxidatively damaged proteins as well as the activity control of some enzymes. Since most knowledge about these proteases has been gained from studies in the eukaryotic microorganism Saccharomyces cerevisiae, much of the discussion here centres on their role in this organism. However, reference is made to mitochondrial AAA+ proteases in other organisms, particularly in cases where they play a unique role such as the mitochondrial unfolded protein response. As these proteases influence mitochondrial function in both health and disease in humans, an understanding of their regulation and diverse activities is necessary.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mitochondrial Protein
- Protein Quality Control
- Yeast Mitochondrion
- Inter Membrane Space
- Matrix Compartment
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The highly compartmentalised nature of a eukaryotic cell brings about increased complexity relative to a prokaryotic cell, with respect to the biogenesis and regulation of its proteome. With the vast majority of eukaryotic cellular proteins encoded by the nuclear genome and synthesised on cytosolic ribosomes, many steps are required for their successful trafficking from the cytosol to the correct subcellular location and their subsequent maturation and folding. Furthermore, proteins regardless of their subcellular location, need to be removed in a regulated manner to allow their replenishment or to alter their concentration in the cell. The balance between protein biogenesis and degradation determines the concentration of a particular type of protein in the cell at any particular time. This is often referred to as the steady state level of a protein. An ability to regulate protein concentration in a post-translational manner permits a rapid and/or graded response to physiological and environmental stimuli. It can also ensure the correct subunit stoichiometry of protein complexes and the maintenance of functional populations of proteins through the removal of unfolded, misfolded or otherwise damaged proteins. But how can the concentration and integrity of intracellular proteins, located in various parts of the cell, be adequately regulated in a post-translational manner? In cells, protein degradation is achieved by the enzyme-mediated hydrolysis of peptide bonds between amino acid residues. This end-point in the life of a protein is attained by two main systems in eukaryotic cells; the lysosome/vacuole-mediated “acid cocktail” mechanism and the AAA+ protease-mediated “chamber of doom” mechanism. In the former case, portions of cellular content, organelles and individual proteins (in mammals) are delivered to the lysosome (vacuole in plants and fungi) for degradation; a process known as autophagy. In the lumen of this organelle, internalised cellular proteins are hydrolysed to small peptides and amino acids via a cocktail of exo- and endo-proteases optimally active in the acid environment of this compartment. The major autophagic pathways are macro-, micro- and chaperone-mediated autophagy and are reviewed elsewhere [1–3]. The second major mechanism of protein degradation is via the action of ATP-dependent proteases. In this case individual proteins are selectively delivered to large oligomeric proteases, unfolded (if required) and translocated into an internal catalytic chamber where hydrolysis of the proteins into short peptides takes place. The 26S proteasome is an example of an ATP-dependent protease in which substrates are tagged with ubiquitin for delivery and degradation [4]. Many cellular proteins located in the cytoplasm, nucleus, endoplasmic reticulum and the mitochondrial outer membrane, are degraded by the ubiquitin-proteasome pathway. However, this is not the only ATP-dependent mechanism of protein degradation operational in eukaryotic cells. Unique systems are also present in chloroplast, peroxisomes and mitochondria [5, 6]. The ATP-dependent proteases of mitochondria are the topic of this chapter.
Early research on mitochondrial protein degradation examined if mitochondrial proteins were turned over with the same or different half-lives. A fixed half-life of degradation for all proteins would be suggestive of a lysosomal mode of mitochondrial protein degradation. On the other hand, vastly different degradation half-lives would indicate a compartment based intrinsic capacity to degrade proteins selectively. Studies on protein turnover in rat liver mitochondria revealed that there was considerable protein-dependent variation in half-lives from hours to days [7, 8]. Also, it was observed that the products of mitochondrial translation in isolated rat liver mitochondria were rapidly degraded in the absence of cytoplasmic synthesised partner proteins [9]. Such findings gave support for an intrinsic protease activity in mitochondria, providing the impetus to search for the factor or factors responsible. Classical biochemical approaches using a range of additives (e.g. ATP) and protease inhibitors (e.g. phenylmethylsulfonyl fluoride, o-phenanthroline) were used to dissect the protease activity of mitochondria. In these studies, both endogenous (e.g. incompletely synthesised or uncomplexed mitochondrial DNA (mtDNA)-encoded protein subunits of the respiratory chain) and exogenous (e.g. casein) proteins were examined as substrates. Collectively, the research identified ATP-dependent metallo- and serine protease activities in both animal and fungal mitochondria [10–13]. Further biochemical and genetic analysis identified the distinct proteases responsible for these different activities within mitochondria [14–22].
This chapter highlights current knowledge regarding the role of ATP-dependent proteases in the biogenesis, quality control and regulation of the mitochondrial proteome largely but not exclusively in the single-cell eukaryotic organism Saccharomyces cerevisiae. The intrinsic ATP-dependent proteases of yeast mitochondria are known as i-AAA protease (intermembrane space active-ATPase associated with a variety of activities), m-AAA protease (matrix active-AAA) and Pim1p (proteolysis in mitochondria) also known as Lon. In higher eukaryotes the casein lytic protease XP (ClpXP) is also present in mitochondria [15, 23, 24]. Although not covered in this chapter, it should be noted that the cytoplasmic ubiquitin proteasome system contributes to the regulation of the mitochondrial outer membrane proteome [25, 26]. Also described in this chapter are instances where mitochondrial ATP-dependent proteases perform unique roles in animals (i.e. not present in yeast), for example Caenorhabditis elegans ClpXP, in the mitochondrial unfolded protein response (section “The Mitochondrial Unfolded Protein Response (UPRmt)”). We begin with an overview of mitochondrial function and protein biogenesis including translocation of nuclear-encoded and mtDNA-encoded proteins, their maturation by processing peptidases and their folding and assembly into active units.
Structure and Function of Mitochondria
Structurally, mitochondria are distinct double membrane bound organelles. They are dynamic however, undergoing constant fusion and division [27]. Functionally, many enzymes reside in mitochondria that are important for processes such as ATP production, fatty acid, amino acid and carbohydrate metabolism, and the synthesis of Fe-S clusters and heme. Depending on the organism, mitochondria also play key roles in cell signalling pathways such as programmed cell death, calcium homeostasis and innate immunity [28–30]. While mitochondria contain their own DNA, they are not self-sufficient because the organellar genome is generally very small. The vast majority of mitochondrial proteins are nuclear-encoded. Thus, full mitochondrial function requires the co-ordinated synthesis of its proteome expressed from two genomes. The retention of a small mitochondrial genome, a remnant of the ancient α-proteobacterium endosymbiont from which mitochondria evolved [31], means that the organelle must retain an ability (and all the necessary components) to carry out tasks such as DNA replication and repair, transcription, processing of RNA and protein synthesis. Many other proteins and complexes are required for the integrity of this organelle and its cooperation and communication with other cellular entities [32–38]. Around 1,000 different proteins are expected to perform all the necessary functions in S. cerevisiae mitochondria. Using a mass spectrometry approach, 750 different yeast mitochondrial proteins were identified following growth in YPG medium with an estimated 90% coverage of the proteome [39]. Further yeast mitochondrial proteins may exist under different growth or environmental conditions.
The Biogenesis of Mitochondrial Proteins
Synthesis and Transport of Mitochondrial Proteins
Nuclear-encoded mitochondrial proteins are synthesised on ribosomes in the cytosol and transported to the organelle; directed by targeting information contained within the protein. The nascent polypeptide may be assisted on its journey by molecular chaperones that act to maintain the protein in an import competent state, protecting against aggregation and premature degradation and in some cases delivering the protein directly to the import machinery [40]. The types of targeting signals that direct mitochondrial proteins to the correct location within the organelle can take a number of forms. However, the most common mechanism involves that of an N-terminal targeting signal, also known as a presequence. Around 70% of nuclear-encoded mitochondrial proteins in yeast possess an N-terminal targeting signal [41]. These generally range in size from 15 to 65 amino acids, carry a net positive charge and have a propensity to form amphipathic α-helices [41, 42]. Such signals direct the proteins to the mitochondrial matrix or, when combined with sorting signals, assist in directing proteins to the mitochondrial inner membrane or inter membrane space (IMS). The remaining ∼30% of mitochondrial proteins contain internal targeting signals. In these cases the targeting information may take the form of a signal motif or be spread throughout the protein [43]. For many proteins containing internal mitochondrial localisation signals, the precise nature of the targeting elements is not known.
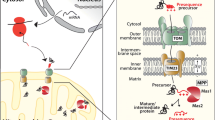
There are several different pathways of protein import into mitochondria involving specialised protein translocation machineries. However, most nuclear-encoded preproteins engage the central translocase of the outer membrane (TOM) complex to cross the outer lipid bilayer of mitochondria. Following import across the outer membrane, the preprotein interacts with pathway specific translocases and cofactors (for a brief overview see Fig. 9.1). For example, preproteins containing N-terminal targeting sequences transition to the translocase of the inner membrane 23 (TIM23) complex for import into the matrix, inner membrane or IMS. For a detailed description of the mechanisms and models of protein import see recent reviews by [43, 44] and references therein. Mitochondrial DNA of S. cerevisiae codes for seven inner membrane embedded proteins of the respiratory chain; cytochrome b (Cytb or Cob), cytochrome c oxidase subunits 1, 2 and 3 (Cox1, Cox2 and Cox3), and ATP synthase subunits 6, 8 and 9 (Atp6, Atp8 and Atp9). An eighth mitochondrial-encoded protein, Var1, is a ribosomal protein. The insertion of the mitochondria-encoded respiratory subunits is co-translational and mediated by the OXA translocase (Fig. 9.1). This consists of the membrane-embedded protein insertion complex formed by the Oxa1 protein, which directly contacts the emerging nascent polypeptide, and a number of other proteins that collectively contribute to ribosome positioning, translation regulation and insertion of protein transmembrane domains [45].

Major import and export pathways for the biogenesis of mitochondrial proteins. Preproteins synthesised in the cytosol are recognised by receptors exposed on the surface of mitochondria directing them to the outer membrane translocation channel formed by Tom40. As the import channel is narrow, the preproteins are imported in an unfolded state as linear polypeptides or loop structures. Once the lipid bilayer has been traversed the preprotein import pathways diverge. Presequence-containing preproteins interact sequentially with the IMS exposed receptors of the TOM and TIM23 complexes. They are then translocated across the inner membrane in a membrane potential-dependent manner via the voltage-activated cation-selective channel formed by Tim23. In the absence of a sorting signal, the emerging presequence interacts with the presequence associated motor (PAM) complex that mediates import via an ATP-driven binding and release mechanism. Some preproteins are translocated into the inner membrane (IM) and IMS via the TIM23 complex directed by a hydrophobic sorting signal situated directly behind the presequence. The presequence directs the preprotein to the TIM23 complex via the standard presequence pathway however a stop transfer signal leads to the arrest of the preprotein in the TIM23 complex. It then laterally transfers into the IM. A variation on this pathway is when laterally sorted presequence-containing proteins contain a protease cleavage site following the stop transfer signal. In such cases, endoprotease(s) cleave these preproteins near to their membrane anchor releasing the mature protein into the IMS. With respect to other pathways, both outer membrane β-barrel proteins and polytopic inner membrane proteins engage holder chaperones (small TIM complexes) in the IMS to assist their transition through this aqueous environment. While the β-barrel preproteins insert into the outer membrane (OM) aided by the sorting and assembly machinery (SAM) complex, polytopic inner membrane proteins are imported via the TIM22 complex in a membrane potential-dependent manner. For non-presequence containing preproteins destined for the IMS, import into this compartment occurs directly via the TOM complex. It seems however that a requisite factor for successful or efficient import, in these cases, is the presence of a binding partner, either a folding catalyst (e.g. Mia40) or a functional partner protein. Finally, mitochondrial inner membrane proteins that are encoded by mtDNA are imported in a co-translational manner mediated by the OXA complex
Maturation of Mitochondrial Proteins
An important aspect of mitochondrial protein biogenesis is the removal of N-terminal targeting and sorting signals by compartment specific peptidases [46]. The vast majority of presequence-containing mitochondrial matrix proteins are cleaved by the mitochondrial processing peptidase (MPP) [41]. This enzyme is a dimeric metalloendopeptidase consisting of α and β subunits [47]. The recent determination of the N-terminal sequences of yeast mitochondrial proteins (N-proteome) has clarified the substrate specificity of MPP [41]. The substrate specificity of MPP is characterised by a very high frequency (>75%) of Arg in the P2 position [48], as deduced from the N-proteome data (Fig. 9.2). This MPP substrate specificity is often referred to as the R-2 motif [42]. Residues most commonly found in the P1' position of MPP substrates are Tyr, Leu, Phe and Ala while Ser and Ala are most commonly found in the P2' position. For many mitochondrial proteins with a presequence, cleavage by MPP is the only matrix proteolytic maturation step (Fig. 9.2, Path I). However, for a subset of proteins, further proteolytic processing is required (Fig. 9.2, Path II and III). For example, in yeast mitochondria, 14 proteins have been identified as substrates of the peptidase octapeptidyl aminopeptidase 1 (Oct1) [41, 42, 49]. Following removal of the presequence by MPP, Oct1 cleaves eight amino acid residues from the N-terminus of some matrix proteins. The substrate specificity of Oct1 extends to P8 with Phe found in this position in the vast majority of cases (Fig. 9.2, Path II). The P1' residue in the MPP cleavage pattern becomes the P8 site for Oct1 recognition. The N-proteome study lead to the identification of a new player in mitochondrial matrix protein maturation, the metalloaminopeptidase termed intermediate cleaving peptidase of 55 kDa (Icp55). For a subset of mitochondrial matrix proteins, Icp55 cleaves a single amino acid residue from the N-terminus of proteins following cleavage of the mitochondrial targeting sequence by MPP (Fig. 9.2, Path III). Specifically, the P1' position in the MPP substrate recognition pattern becomes the P1 site for Icp55 recognition. With respect to substrate specificity, Icp55 appears to favour bulky hydrophobic amino acids Tyr, Phe and Leu in the P1 position with Tyr occurring most frequently. Ser is most frequently found in the P1' position, and to a lesser extent Ala and Thr. A quite different specificity for Icp55 has been reported [50] where the scissile bond sits three amino acid residues from the MPP cleavage site. Further analysis is required to fully understand the precise substrate specificity of Icp55. In the case where processing by either Icp55 or Oct1 is prevented (i.e. in gene deletion strains), the resulting intermediate proteins are moderately unstable relative to the mature protein [41, 49]. The intermediates resemble bacterial N-degrons; degradation signals which mediate the turnover of proteins by the N-end rule pathway [51–53]. This is discussed further in section “Activity Control of Mitochondrial Proteins by AAA+ Proteases”. Finally, the ATP-dependent m-AAA protease also plays a role in mitochondrial protein maturation [54–56]. Normally responsible for the complete degradation of proteins into peptides (see later), this maturation activity is unique and dependent on specific features of the substrate protein.

MPP-dependent pathways of mitochondrial protein maturation. Schematic indicating primary sequences of Pam16, Sdh1 and Mge1 precursors around the cleavage site (scissile bond) for the peptidase MPP and where relevant Oct1 and Icp55. Amino acid residues N-terminal of the scissile bond on the substrate are nominated P1, P2, P3 etc. and those residues C-terminal are nominated P1', P2', P3' etc. In Path I, cleavage of the presequence by MPP directly generates the mature protein without any further proteolytic processing as exemplified by the substrate Pam16. In Path II, MPP cleavage generates an immature intermediate species. This first cleavage event is necessary for subsequent recognition of the substrate by the peptidase Oct1. Cleavage of the substrate by Oct1 removes eight residues from the N-terminus of the protein to generate the mature form. The primary sequence of Sdh1 around the respective cleavage sites is shown in this example. Finally in Path III, MPP cleavage again generates an immature intermediate species however in this case it only differs from the mature protein by one residue. The final maturation step is mediated by the aminopeptidase Icp55. The sequence of Mge1 is shown in this example. Note the presequences and mature domains are not to scale. The length of presequences and mature protein are protein dependent and vary considerably
Some proteins that are sorted to the IMS via the presequence pathway are processed by the IMS peptidase (IMP) complex [43, 46]. Following lateral transfer into the inner membrane such preproteins are processed by MPP in the matrix to remove the presequence, then by the IMP complex on the IMS side of the membrane, releasing the protein from its transmembrane anchor and hence into the aqueous compartment. The signal sequence of the mtDNA-encoded protein Cox2 is also cleaved by the IMP complex [43]. Yeast Atp6 is another mtDNA-encoded protein synthesised with an N-terminal signal sequence, however, in this case, it is cleaved by a different protein, the metallopeptidase ATP synthase 23 (Atp23) [57, 58]. Yeast mitochondria also possess an inner membrane integrated serine endopeptidase of the rhomboid family known as processing of cytochrome c peroxidase (Pcp1). Two substrates have been described; cytochrome c peroxidase (Ccp1), and mitochondrial genome maintenance protein 1 (Mgm1), a mitochondrial outer membrane dynamin-like GTPase [59–63].
Formation of Functional Mitochondrial Proteins
Once proteins are imported into the mitochondrial matrix and processed they must fold into their unique three-dimensional structure to be functional. Such processes can occur spontaneously but in the crowded environment of cells or organelles, protein folding may be assisted by molecular chaperones. Chaperones capture folding intermediates preventing non-productive aggregation or rescuing kinetically trapped species [64]. The major chaperones of yeast mitochondria belong to the Hsp70 and Hsp60 protein families. Both types of chaperones are intrinsically tied to the import and folding reaction of mitochondrial precursor proteins [65, 66]. Mitochondrial Hsp70 (mtHsp70), encoded by the yeast gene SSC1 is a component of the mitochondrial import machinery for proteins of the matrix compartment, driving the full membrane translocation of these precursors and assisting their folding reaction. These folding reactions are performed in a close collaboration with the Hsp60 chaperone complex in the matrix. In addition, yeast mitochondria contain a member of the Hsp100/ClpB family, called Hsp78, which is missing in metazoan cells. Like its relatives in bacterial cells, Hsp78 is able to recover polypeptides from the aggregated state, promoting the recovery of mitochondrial functions after temperature stress [67]. Another special feature of yeast mitochondria is the presence of the chaperone component of the ClpXP protease (see below), called Mcx1 (for mitochondrial ClpX), although the corresponding protease component is missing [68, 69]. The function of this chaperone in yeast mitochondria is unknown. Finally, once the imported protein subunits are folded, the final steps in the biogenesis of proteins may be their oligomerisation, binding to prosthetic groups such as flavin, Fe-S clusters or heme, or assembly into multi-subunit structures such as the respiratory chain complexes. It should be noted that in this context, yeast mitochondria contain a specialised Hsp70 system consisting of the chaperone Ssq1 and its cofactors for the transfer of Fe-S cluster cofactors from their assembly scaffold protein to the respective apoenzymes [70]. Further post-translation modifications such as acetylation and phosphorylation may contribute to the activity of mitochondrial proteins [71, 72].
Mitochondrial Protein Quality Control (PQC) by Compartment Specific AAA+ Proteases
While many sophisticated proteinaceous molecular components and pathways mediate the synthesis, transport, folding and assembly of mitochondrial proteins, these are not fail-safe processes. ATP-dependent proteases therefore serve as ever-present organelle protein quality control (PQC) systems that constantly survey protein integrity. Collectively, they have the capacity to degrade unfolded/oxidatively damaged, immature and unassembled proteins. Together with molecular chaperones, these proteases ensure that a functional population of proteins is maintained in mitochondria. Mitochondria, as endosymbiotic organelles, contain many chaperones and proteases homologous to those found in bacteria. For example, major ATP-dependent proteases are represented with homologs of Lon, ClpXP and FtsH families found in mitochondria, while bacterial HslUV and ClpAP families are absent. There are however significant differences in the composition of mitochondrial proteases between eukaryotic organisms. While metazoan mitochondria contain a Lon protease, a ClpXP protease and at least two distinct AAA proteases of the FtsH family, the situation in yeast mitochondria [67] is somewhat different. Yeast mitochondria lack a Clp protease homolog, leaving Lon/Pim1 as the only ATP-dependent protease in the matrix. The main components of the yeast mitochondrial PQC system and their location are depicted in Fig. 9.3. The main functional distinction between these types of proteases is their substrate specificity – the membrane-integrated proteases preferentially degrade membrane proteins while the matrix proteases are responsible for the degradation of water soluble proteins.

Schematic overview of the PQC system in the matrix of yeast mitochondria. Misfolded proteins are recognized and bound primarily by the mitochondrial Hsp70 (mtHsp70) system that works together with its partner proteins Mge1 (nucleotide exchange factor) and Mdj1 (co-chaperone of the J-protein family). If unfolding efforts fail, the misfolded proteins are transferred to the matrix protease Pim1 that degrades the substrates to small oligopeptides in an ATP-dependent reaction. Pim1-dependent degradation is assisted by the Hsp100 chaperone Hsp78. If the maximum capacity of the PQC system is exceeded, misfolded polypeptides accumulate and eventually aggregate. Aggregated polypeptides may be resolubilised in a concerted process by Hsp78 and mtHsp70. Disaggregated polypeptides are either refolded or directly degraded by the Pim1/Lon protease. Membrane proteins of the IM are degraded by either of the two AAA proteases. The m-AAA protease, composed of the subunits Yta10 and Yta12 faces the matrix compartment while the i-AAA protease (a Yme1 homo-oligomer) faces the IMS
Like their bacterial counterparts, mitochondrial proteases are ATP-dependent self-compartmentalising complexes. That is, the catalytic residues responsible for the hydrolysis of peptide bonds line the internal walls of an enclosed aqueous degradation channel or chamber formed by a multi-subunit protease. Recognition of the protein targets is mediated by the ATPase component of the proteases and the degradation signals (degrons) displayed by the target protein. Also, the ATPase domain is responsible for the unfolding and translocation of the substrates into the internal proteolytic cavity. This chambered mechanism of protein degradation prevents the inadvertent unregulated hydrolysis of native proteins. The ATPase domains of these processive proteases are very similar to each other and to many other ATPases with diverse functions in cells including cellular regulation and PQC. These related proteins are all members of the ATPases associated with various cellular activities (AAA+) superfamily of proteins [73, 74]. As the mitochondrial ATP-dependent proteases Pim1, i-AAA, m-AAA and ClpXP are all members of the AAA+ superfamily of proteins, this group of proteases will herein be referred to as AAA+ proteases.
Pim1, the Soluble AAA+ Protease of the Yeast Mitochondrial Matrix
The most prominent member of the AAA+ proteases is the Lon protease from Escherichia coli, which gives its name to the protein family [75]. Pim1 in yeast mitochondria and the related Lon proteases from other eukaryotic organisms such as LONM in human mitochondria (also known as mitochondrial Lon peptidase 1 (LonP1)) and Lon1 and Lon4 from plant mitochondria [5] are highly conserved and, as far as it has been studied, also exhibit similar functions. Hence, Pim1 from the model organism S. cerevisiae commonly serves as the typical representative for the Lon-A type ATP-dependent proteases found in mitochondria.
Identification of Pim1 and Phenotype of pim1 Deletion Strain
Due to their endosymbiotic origin it had been assumed that mitochondria, similar to bacteria, contain proteases for the specific degradation of their endogenous proteins. Indeed, the first identification of an ATP-dependent proteolytic activity in mitochondria with properties similar to the bacterial protease Lon reach back to the early 1990s [16, 76]. The yeast gene responsible for this activity was cloned independently by two groups and the encoded protease was named Pim1 or Lon based on its high amino acid homology to the E. coli Lon protease [77, 78].
A deletion of the PIM1 gene in yeast resulted in severe consequences for the function of mitochondria. The main phenotype is a growth defect on non-fermentable carbon sources like glycerol or lactate [77]. The inability of ∆pim1 mitochondria to respire correlates with a non-functional mitochondrial genome (see section “Association with Mitochondrial DNA (mtDNA)”). Further examination demonstrated the lack of any ATP-dependent proteolysis activity in soluble mitochondrial extracts [78]. These experiments indicated that Pim1 represents the main proteolytic activity in the mitochondrial matrix. First direct demonstrations of its protease function were based on its ability to degrade radioactively labelled reporter proteins that were imported into isolated mitochondria [79]. Pim1-dependent degradation was shown to be restricted to soluble protein substrates located in the matrix compartment. In contrast, membrane associated substrates, even those facing the matrix compartment were preferentially degraded by the membrane-integrated m-AAA protease [80].
Structure-Function Relationships
The PIM1 gene of S. cerevisiae encodes a 1133-amino acid long protein with an overall sequence identity of 30–35% to bacterial family members. Pim1 is encoded in the nuclear genome and has to be imported into the mitochondria after it is synthesised on cytosolic ribosomes. Hence, it is characterised by a long N-terminal amino acid extension that serves as a mitochondrial targeting signal that is missing in its bacterial relatives. Similar to the other ATP-dependent proteases, Pim1 belongs to the AAA+ protein superfamily [81]. AAA+ protease complexes generally consist of a proteolytic subunit and a regulatory subunit that is responsible for substrate recognition and unfolding [82, 83]. Due to the high sequence conservation, most information on the catalytic mechanism of Pim1 has so far been deduced from the properties of its bacterial relative Lon. Similar to other AAA+ proteases, Pim1 consists of two major domains and also activities: a protease with broad specificity and an ATPase responsible for the recognition and/or unfolding of polypeptide substrates. A unique property of Lon-type proteases is that the ATPase domain and the proteolytic domain are located on a single polypeptide chain. An additional third N-terminal domain (N domain) essentially interacts with the potential substrate proteins in concert with the ATPase domain. The C-terminal peptidase contains the catalytic dyad consisting of a conserved serine and lysine residue [84]. The main difference to the bacterial proteases, apart from the targeting sequence, is the presence of three rather large hydrophilic inserts, one in the N-terminal part after the targeting sequence, one between amino acids 300 and 380 and the last between amino acids 840 and 900. Pim1 shares the presence of these inserts with its other eukaryotic homologs, although the sequence conservation in these regions is rather low. So far no data on the significance of these inserts is available but it could be speculated that they are connected with some eukaryotic-specific function.
The assembly of the Pim1 oligomeric protease complex exhibits a special feature. After translocation of the polypeptide chain across the mitochondrial membranes, the presequence is first cleaved in a standard process after position 37 by MPP. However, functional activity of the protease requires the autocatalytic removal of an additional 61 amino acid long pro-region. This second endoproteolytic cleavage is performed by an intermolecular (and likely also intramolecular) reaction by the protease itself and closely coupled to the ATP-dependent assembly of the protease subunits [85].
Lon-type proteases are organized into large homo-oligomeric, ring-shaped protein complexes consisting of several subunits of ∼100 kDa each. While bacterial family members are usually composed of six subunits, the data available for Pim1 indicate a heptameric structure [86]. Up to now, no direct ultrastructural studies have been performed on Pim1 but it can be assumed that it is structurally organized in a similar way to its bacterial homologs. As described earlier, a general feature of ATP-dependent proteases is that they form a cylindrical complex with a rather large inner cavity. The active sites are oriented to the interior of this cavity thus forming a proteolytic chamber that is shielded from the environment. Substrates can only enter the proteolytic chamber by pore structures situated at the ends of the cylindrical structure. Substrate entry is usually tightly regulated. Recent structural studies of bacterial proteases related to Lon confirmed this overall structural arrangement [87].
Protease Classification and Cleavage Specificity
The presence of a proteolytic active site characterized by the amino acid Ser at position 1015 confirms that Pim1 as well as Lon are serine proteases with chymotrypsin-like cleavage specificity (proteolysis at regions rich in hydrophobic amino acids) [88]. Similar to other PQC proteases, the regulatory ATPase domain of Lon also seems to exhibit chaperone activity since overexpression of a proteolytically inactive mutant of Pim1 supported the assembly of mitochondrial respiratory complexes [89]. The structurally separated enzymatic domains of Pim1 permitted the functional relationship of the two different domains to be addressed by the recombinant generation of individual mutant constructs [90]. Single point mutations abolishing either ATPase or protease activity both destroyed proteolytic activity. Neither of these mutants were able to support respiration-dependent growth of the cells. When the ATPase or protease domains were expressed separately as individual proteins, neither allowed growth on non-fermentable carbon sources. Interestingly, co-expression of both domains as separate proteins restored full proteolytic and respiratory activities, indicating that both domains are able to functionally interact even as physically separate polypeptides, in order to fulfil the wild-type functions.
General Function of Pim1 in PQC
It was noted relatively early that proteins imported into the mitochondrial matrix compartment under in vitro conditions were susceptible to degradation by Pim1 [79, 80]. However, the preproteins used in these experiments represented artificial reporter constructs, which had only a minor structural or functional relationship to the native proteins present in mitochondria. Although ATP-dependent proteases like Pim1 are supposed to have a broad substrate specificity, the number of identified endogenous mitochondrial substrate proteins remained very small for a long time. Due to the restricted availability of antisera against mitochondrial proteins, the initial publications identified a subunit of the matrix processing peptidase (MPP-β) and the β-subunit of the mitochondrial ATP-synthase (F1β) as potential substrates [77]. Further progress was achieved by the first proteomic characterisation of mitochondrial protein turnover. Although the mitochondrial proteome is remarkably stable at least under normal conditions, these experiments revealed a set of novel protease substrates [91]. Depending on their behaviour these proteins could be sorted into three different groups: (i) proteins that were protease substrates under all conditions tested, (ii) proteins that remained stable at normal temperatures but became degraded at elevated temperatures, and (iii) proteins that were degraded only at normal temperatures. Proteins in the second group probably represent classical Pim1 substrates, conformationally labile polypeptides that become (partially) unfolded at elevated temperatures and need to be removed. However, the behaviour of group (iii) was counterintuitive at first glance. Later, experiments revealed that proteins of this group had a high tendency to aggregate at elevated temperatures. Typically, under stress conditions a competition between degradation and aggregation takes place (see below). Since aggregated polypeptides are essentially resistant to Pim1-dependent proteolysis, certain aggregation-prone polypeptides are apparently stabilized at higher temperatures although they have of course lost their activity. Interestingly, this hypothesis correlates well with the observation that the accumulation of electron-dense inclusions in the matrix compartment of ∆pim1 mitochondria [77] most likely represent aggregated damaged polypeptides that could not be removed due to the absence of the protease.
Role of Pim1 Under Oxidative Stress
Apart from elevated temperatures, mitochondria are also vunerable to another stress condition: oxidative stress. Mitochondria themselves are major generators of reactive oxygen species (ROS). In particular, ROS are produced by non-specific electron transfer reactions from components of the mitochondrial respiratory chain to molecular oxygen. Typical examples are superoxide radicals or hydrogen peroxide and its related reaction products [92, 93]. Mitochondrial ROS production is a normal side-reaction of ATP-production by oxidative phosphorylation but can be drastically elevated in certain pathological situations. Due to their high non-specific reactivity, ROS molecules tend to modify and thereby inactivate any type of macromolecule; nucleic acids, lipids and polypeptides. Hence, a major question concerning the role of the mitochondrial PQC system with its core component Pim1 is the impact of its protective function under oxidative stress conditions. This is of particular importance with regard to the multitude of human pathologies that involve some form of mitochondrial oxidative stress, most prominent examples are neurodegenerative diseases [94].
The consequences of oxidative stress conditions on the mitochondrial proteolytic system were studied using a quantitative proteomic approach, identifying proteins that were degraded after treatment of isolated mitochondria with different forms of ROS [95]. Here, three conditions were distinguished: elevated superoxide concentrations, treatment with external H2O2 and the artificial inhibition of the respiratory chain. An important observation of this study, was that the sensitivity of the various proteins to the different types of ROS stress was relatively specific. This implies that each of the various conditions leading to oxidative stress can have very different consequences on cellular functions. One class of mitochondrial proteins that have been identified as prominent substrates of ROS-induced proteolysis are enzymes that contain an Fe-S cluster as a cofactor. The Fe-S cluster seems to be very sensitive to modifications by ROS molecules and is likely to be destroyed under these conditions. As a consequence of the loss of their cofactor, the affected polypeptides become conformationally destabilized and will be recognized by the quality control proteases like Pim1. For example, the soluble and Fe-S containing enzyme aconitase (Aco1 in yeast), a component of the citrate cycle, was found as one of the most ROS-sensitive and degradation-prone proteins in the mitochondrial matrix [95, 96]. However, one has to take into account that the presence of a Fe-S cluster does not automatically turn a protein into a protease substrate. This is exemplified by subunit 2 of the succinate dehydrogenase complex (Sdh2), which did not show any alterations in protein levels after ROS stress.
ROS modifications represent covalent, and in most cases irreversible alterations in the affected molecules. Since refolding of ROS-damaged polypeptides, even with the help of chaperones, would not be possible, ATP-dependent proteases like Pim1 have to play a decisive role in the removal of ROS-modified proteins. Their removal is a key response of the cellular PQC system to many different pathological conditions [97, 98]. Interestingly, Pim1 is the only ATP-dependent protease in yeast that is significantly up-regulated under oxidative stress conditions [95]. In contrast, the main chaperones like Hsp70 and Hsp60 show only slightly elevated levels under heat stress but not after ROS treatment. The important protective function of Pim1 under oxidative stress is corroborated by an enhanced sensitivity of ∆pim1 cells to elevated levels of H2O2 in the growth medium.
Pim1 Cooperates with Molecular Chaperones
Substrate Capture
In general, protein degradation in the cellular context is governed by a fine-tuned interplay or cooperation of protease enzymes and molecular chaperones. Instead of focusing on the functions of the individual protein components, it is rather more appropriate to view this system as a functional network for PQC [67, 99]. Chaperones play important roles in recognising and binding to damaged (at least partially unfolded) polypeptide chains. The interaction with a chaperone component is therefore often the first step in preparation of the substrate for recognition by the protease. Cooperation of proteases with chaperones can be either very direct, i.e. the ClpP protease complexes with chaperones (e.g. ClpA or ClpX) to generate the ATP-dependent proteolytic machine or more indirect, when chaperones and proteases act in an independent but coordinated fashion. The latter case applies to the Pim1 protease. It has to be noted that the intrinsic chaperone-like activity of Pim1 is disregarded in this context (see also below). It has been demonstrated that Pim1 closely cooperates with the mitochondrial Hsp70 system in the matrix compartment [79, 80]. Efficient Pim1-dependent degradation of imported reporter proteins that tend to misfold is assisted by mtHsp70. The Hsp70-cochaperone Mdj1, a member of the DnaJ-like protein family, was shown to be closely involved in this function. The primary function of chaperones in this context seems to be the maintenance of the misfolded non-aggregated state of the substrate increasing the time available for safe removal.
Role in Aggregation Prevention
The matrix compartment of fungal mitochondria, like yeast, contains a chaperone that is typical for bacteria but not found in higher eukaryotic species [100]. This chaperone, named Hsp78, is a homolog of the bacterial ClpB protein that has been implicated in the cellular repair of protein aggregates [101]. Enzymes of the ClpB family are able to resolubilise aggregated polypeptides in a reaction that requires ATP and a close cooperation with the respective Hsp70 chaperone system [102]. Hsp78 has been shown to perform a similar role in the mitochondrial matrix [103, 104]. However, when analysing the degradation rates of imported reporter proteins it was observed that ∆hsp78 deletion mutants exhibited a significant defect in Pim1-dependent protein degradation [105]. This unprecedented cooperation between Hsp78 and Pim1 during degradation takes place independent of the aggregation state of the substrates and was also observed with destabilised mutants of an abundant endogenous substrate protein named acetohydroxyacid reductoisomerase (Ilv5) [106]. In support of this conclusion it was observed that the yield of disaggregated polypeptides was significantly increased in ∆pim1 mutant mitochondria [103], again indicating a close functional relationship between Hsp78 and Pim1. This relationship between a ClpB-type chaperone and a Lon-type protease seems to be a special feature of fungal mitochondria. It could be speculated that single cell eukaryotes are subjected to more extreme environmental temperature changes than multicellular eukaryotes and hence require a more efficient removal system for heat-denatured polypeptides.
All the intricate activities of the chaperone-protease network forming cellular PQC systems have two ultimate goals. One is the refolding of damaged or misfolded polypeptide chains in order to regain their enzymatic activities. A second equally important goal is the prevention of the accumulation of misfolded protein species that would otherwise form insoluble toxic aggregates [107]. In a recent study, the aggregation propensity of mitochondrial proteins under heat stress conditions was tested on a proteome scale [108]. In this context it is of particular interest that Pim1 was identified as a major mediator of mitochondrial aggregation reactions, exhibiting an even more significant protective impact than the chaperone proteins from the Hsp70 family. It was shown that the amount of aggregated polypeptides under heat stress was significantly increased in ∆pim1 mitochondria. Overexpression of Pim1, on the other hand, exhibited a pronounced protective role under these conditions. In the absence of a ClpP protease in yeast mitochondria, Pim1 is the only ATP-dependent protease that is able to degrade soluble substrate proteins in the mitochondrial matrix. Taken together, Pim1 fulfils a key role in the mitochondrial PQC system by removing misfolded proteins in the matrix compartment.
Recognition Motifs and Mechanism of Targeted Degradation
A major general question concerning the degradation of substrate polypeptides by ATP-dependent proteases is the mechanism of substrate selectivity. In principle, the substrate specificities of these proteases are very broad, enabling the proteolysis of any kind of protein substrate. However, in this case it is required that the proteolysis is restricted to short-lived or damaged proteins. As already described, substrate degradation is mainly regulated by the selective entry of polypeptide chains into the proteolytic chamber of the protease complex. The main proteolytic enzyme of the cell, the proteasome recognises a molecular tag on the substrate, a polyubiquitin chain, which mediates engagement of the target substrate with the protease and hence its subsequent degradation [109]. A related tagging system exists in Mycobacteria, where proteins can be tagged for degradation by an ubiquitin-like molecule known as prokaryotic ubiquitin like protein (pup) (for a recent review see [110]). Also, a different type of tagging system exists in bacteria whereby stalled translation products are tagged with an 11 amino acid sequence known as the SsrA tag that targets the abnormal protein for degradation by the ClpXP protease system [53, 111, 112]. Interestingly, a molecular tag system, as a means of substrate recognition, has not been identified for proteolytic reactions in mitochondria. In contrast, current evidence points to a mechanism where the conformational state of a polypeptide chain serves as the main criterion for its selection as a protease substrate. Since it was well established that newly imported reporter proteins containing a dihydrofolate reductase (DHFR) domain are “standard” substrates of the protease Pim1 in the matrix compartment, it was surprising that imported DHFR fusions with only a small N-terminal extension failed to be degraded [113]. A detailed analysis revealed that Pim1 requires an unstructured N-terminal segment of 50–60 amino acid residues in front of the DHFR domain to commence degradation. Pim1 is also able to recognize internal segments as long as they are exposed on the surface of the substrate protein [88]. In addition, it was observed that Pim1 was unable to degrade a folded DHFR domain despite its relatively low thermodynamic stability. Full degradation of DHFR fusion proteins was only possible if the folding state of DHFR was compromised by higher ambient temperatures or destabilizing mutations [113]. Hence, the substrate selectivity of Pim1 (Fig. 9.4) is based on the combination of two properties, (i) the requirement for an unstructured segment in the substrate for initiation of proteolysis and (ii) the low or even absent intrinsic unfolding capacity of Pim1. Taken together, these properties restrict proteolysis to unfolded or damaged polypeptide chains while folded and active enzymes remain largely resistant. Similar properties have been observed in the case of the bacterial protease FtsH [114] and also for the proteasome [115], suggesting that this mechanism represents a basic and probably evolutionary ancient process for substrate selectivity.

Model for the mechanism of Pim1 substrate selectivity. Tightly folded substrates with no or short unstructured extensions are not cleaved by Pim1 (I). The substrate requires a 56-60 amino acid (aa) long unstructured segment for Pim1 to commence its processive degradation reaction. Due to its weak intrinsic unfolding capacity the tightly folded domains of the substrate cannot enter the proteolytic chamber, resulting in clipping of the protein to generate a few specific degradation fragments (II). Only damaged or unfolded polypeptide chains are completely degraded by Pim1 (III). The Pim1 complex and a DHFR fusion protein as substrate are roughly drawn to scale (based on EM pictures published in [86]) to visualize the dimensions of the protease system. ds, destabilised
This principle of substrate selectivity has been confirmed by the identification of endogenous mitochondrial substrate proteins [91]. A proteomic screen identified proteins containing cofactors or prosthetic groups as main degradation-prone targets of Pim1. In particular, enzymes with Fe-S cluster cofactors, like aconitase and its relatives, were susceptible to degradation. It is conceivable that an absence or the loss of a cofactor would lead to a conformational destabilization of either a part of, or the whole apoenzyme, which could then be recognized by the protease as a potential substrate. Another group of proteins found as potential degradation substrates were single subunits of larger oligomeric complexes. Best examples were the homo-oligomeric chaperone Hsp60 or the dimeric citrate cycle enzyme succinate thiolase. In this case the argument that applies is, that subunits without the respective partners most likely expose unstructured segments that become recognition sites for Pim1.
Additional Cellular Processes Involving Pim1 Function
Association with Mitochondrial DNA (mtDNA)
One of the main effects of a Pim1 mutation in yeast is the loss of mtDNA integrity resulting in a rho–, respiratory deficient phenotype. Elucidation of the biochemical mechanism underlying the lack of respiratory activity in ∆pim1 cells remains one of the more enigmatic phenomena concerning the cellular function of the protease Pim1. Interestingly, the general effect could be separated into two independent aspects by the identification of a suppressor mutation that retained the integrity of the mtDNA but still rendered the mitochondria respiratory deficient [116]. Using this tool, it could be shown that the proteolytic activity of Pim1 is required for the function of mRNA maturases that are required for the splicing of mitochondrially encoded transcripts, in particular for the expression of the respiratory chain components CoxI and Cob [116]. The precise involvement of Pim1 in this process has not been defined so far but it is conceivable that Pim1 might be required in a processing step during biogenesis of the respective enzymes. Another aspect of Pim1 function, contributing to the respiratory deficient phenotype, was the observation that Pim1 also assists the assembly of respiratory chain components through its chaperone activity, independent of its role in proteolysis [89].
Although these functions would readily explain the respiratory deficient phenotypes of pim1 mutants, Pim1 has also been shown to play a direct role in mtDNA maintenance. Several lines of evidence show a direct physical interaction of human LONM with the mitochondrial genome [117]. The accumulation of mtDNA lesions was also correlated with a role of LONM under oxidative stress conditions [118], although the details remain unclear. In this context it should be noted that many identified substrate proteins of Pim1 were also shown to be components of the mitochondrial nucleoid, Aco1 being one of the most prominent examples [119]. Integrity of the mitochondrial genome is also maintained if E. coli Lon is expressed in mitochondria instead of Pim1 at normal temperature, but not under mild heat stress conditions [120], suggesting that the role in mtDNA maintenance is connected to its proteolytic activity. Although many open questions remain, it can be stated that Pim1/Lon is a multifunctional protein contributing more to mitochondrial maintenance than just PQC.
Lifespan Regulation in Fungi
An important consequence of a successive accumulation of mitochondrial defects seems to be cellular ageing. There is an ongoing discussion on the molecular processes underlying the mitochondrial contribution to ageing processes, summarized under the key terms “mitochondrial free radical theory of aging” [121, 122]. This hypothesis states that during the lifespan of an organism, mitochondria-generated ROS leads to accumulated mitochondrial and cellular damage, resulting in a gradual decline in important functions like ATP synthesis, which usually contribute to cellular survival. Elevated ROS levels lead to various covalent modifications of polypeptide chains, negatively influencing enzyme activities or conformational states. Hence, it can be postulated that the effectiveness of the mitochondrial PQC system may have a significant influence on the lifespan determination of a cell. A direct connection between PQC and aging could be demonstrated in the fungal organism Podospora anserina [123], which is used as a model system for studying cellular aging processes. Here, overexpression of Lon resulted in a decreased amount of ROS-modified proteins, consequently a higher resistance against oxidative stress, essentially correlating with a longer lifespan of the cells. Also in P. anserina mitochondria, one of the major mitochondrial proteins affected by ROS-related proteolysis was the citrate cycle enzyme aconitase. Although there are many supporting observations for an involvement of mitochondrial PQC in aging processes, the cause-and-effect relationships in particular at a molecular level remain to be clarified.
AAA+ Proteases of the Yeast Mitochondrial Inner Membrane
Whereas the quality of yeast mitochondrial matrix proteins is controlled by Pim1, a different type of proteolytic machine is required for the quality control of inner membrane proteins. In fact, this function is performed by two structurally related inner membrane anchored ATP-dependent proteases, with catalytic domains on opposite sides of the membrane (Fig. 9.3) [14, 18–21, 124, 125]. These are known as i-AAA and m-AAA proteases, and belong to the M41 (FtsH family) of ATP-dependent metalloendopeptidases found in a wide range of bacteria, and chloroplast and mitochondria of eukaryotes [126]. E. coli FtsH and yeast mitochondrial i-AAA and m-AAA proteases serve as the typical representatives of the bacterial and eukaryotic members of this family. There are however unique oligomeric combinations of m-AAA protease in mammalian mitochondria and multiple FtsH proteases in plants [5, 127].
Structure-Function Relationships
All subunits of FtsH family proteins have the same core modular structure. They contain at least one transmembrane anchor in their N-terminal region, a conserved AAA domain followed by a C-terminal protease domain. The AAA domain, conserved in the AAA+ superfamily of proteins, contains the Walker A and Walker B motifs for ATP binding and hydrolysis respectively [14, 19, 21, 74, 125, 128–131]. They are also characterised by a consensus zinc-binding motif HEXXH in the protease domain where Glu is the predicted catalytic residue of the peptidase and the two His residues are expected to coordinate zinc. Indeed, mutation of the conserved Glu in subunits of i-AAA and m-AAA proteases abolished peptidase activity [14, 125, 128]. The active proteases are ring-shaped oligomeric assemblies of six subunits. The oligomeric arrangement creates a central translocation pore in the ATPase domain, which provides an aqueous path to the internal proteolytic chamber. Like all ATP-dependent proteases the ATPase activity of the protein is not required for peptidase activity per se, but rather to present the substrate to the peptidase domain via energy-dependent translocation into the internal proteolytic cavity where hydrolysis of peptide bonds occurs.
Despite a number of common features, the i-AAA and m-AAA proteases have unique quaternary and topological structures with respect to each other. The i-AAA protease is a homo-oligomeric ring-shaped protein anchored to the mitochondrial inner membrane by a single span transmembrane domain situated near its N-terminus with its catalytic domains (ATPase and peptidase) projecting into the mitochondrial IMS (Fig. 9.3) [18, 19, 125, 130]. The protein subunit that makes up the oligomer is known as yeast mitochondrial escape 1 (Yme1) [130, 132]. The m-AAA protease on the other hand is a hexameric hetero-oligomer of polypeptides, composed of yeast tat-binding analogs 10 and 12 (Yta10 and Yta12) [14, 129, 133] also known as ATPase family gene 3 (AFG3) and respiratory chain assembly 1 (Rca1) respectively [14, 128, 131]. It is anchored to the inner membrane by two transmembrane domains near the N-terminus of each subunit with the catalytic domains protruding into the matrix space [14, 20]. The topology and modular structure of i-AAA and m-AAA proteases are well adapted for a role in membrane PQC (see below). Due to the lipid-phase anchor, the ATPase domain of these proteins sit close to the membrane, followed by the peptidase domain including a C-terminal coiled-coil region [133]. Thus, the molecular motor (ATPase) of these proteases is in close proximity to target proteins in the inner membrane where they can extract them for delivery to the respective peptidase chambers [134]. Recent low resolution structural studies revealed a space between the ATPase domain and the transmembrane domain of the m-AAA protease, which is proposed to provide sufficient room for an unfolded substrate to dock onto a surface recognition site within the ATPase domain, allowing engagement with the protease for subsequent degradation [133]. A detailed description of the known structure-function relationships of these ATP-dependent membrane proteases can be found in [134].
General Functions of i-AAA and m-AAA Proteases in Inner Membrane PQC
The discovery of energy-dependent degradation of mtDNA-encoded inner membrane proteins [9, 12, 13] was suggestive of a membrane associated ATP-dependent protease [19]. This indeed turned out to be the case with both i-AAA and m-AAA proteases mediating the degradation of incompletely synthesised and unassembled mtDNA-encoded subunits of respiratory complexes [14, 19–21, 128]. For example, the first substrate of i-AAA protease to be identified was Cox2. When this integral inner membrane protein fails to assemble with nuclear-encoded co-subunits it is rapidly degraded [19, 135]. Likewise, m-AAA protease can degrade unassembled mtDNA encoded subunits Cox1, Cox3, Cob, Atp6, Atp8 and Atp9 [14, 124]. This activity of the FtsH family of mitochondrial inner membrane proteases means they help regulate the required stoichiometry of inner membrane protein complex subunits (the respiratory complexes). Thus, post-translation regulation of protein concentration ensures the inner membrane remains free of superfluous proteins thereby contributing an important PQC function. The i-AAA proteases Yme1 and PalAP of S. cerevisiae and P. anserina respectively, both play temperature-related roles in mitochondria, which may be linked to their PQC functions. Mutations in yeast yme1 cause a heat-sensitive respiratory growth defect [130] while a palAP deletion strain of P. anserina displays heat-sensitive developmental defects and a reduced lifespan at elevated growth temperatures [136]. A detailed description of the role of these proteases in protein activity control is provided in the following section.
Activity Control of Mitochondrial Proteins by AAA+ Proteases
Another important aspect of AAA+ protease function is their contribution to protein activity control. This can be the direct activation of proteins or the conditional degradation of non-damaged native proteins in order to positively or negatively modulate cellular or organellar pathways. This section describes interconnected processes and regulatory pathways in mitochondria in which AAA+ proteases play a role.
A Unique and Critical Role of m-AAA Protease in Mitochondrial Ribosome Biogenesis
As discussed earlier (section “Maturation of Mitochondrial Proteins”), most matrix-destined preproteins that are directed to this compartment are processed by MPP to remove the N-terminal presequence. However, a sub-population of yeast mitochondrial matrix proteins are processed at the N-terminus but do not appear to possess an MPP recognition motif [41, 54]. One such protein is mitochondrial ribosomal protein L32 (MrpL32). As the name indicates, MrpL32 is a component of the mitochondrial ribosome, specifically the large subunit. Its proteolytic maturation, which is necessary for its function, is highly specialised and mediated by the m-AAA protease [54, 56]. Thus, m-AAA protease can mediate the complete degradation of proteins into peptides and the selective maturation of a preprotein to generate the functionally active mature form. But how the m-AAA protease performs multiple and seemingly conflicting functions was a conundrum for a while. The answer however lies with the biogenesis pathway and structural properties of MrpL32. Yeast MrpL32 is synthesised as a 183 amino acid preprotein and directed to the matrix compartment by an N-terminal presequence. Once it is fully imported, it folds into its unique three-dimensional structure in a mechanism that requires the presence of the N-terminal region of the protein [54]. Based on comparison to the bacterial homolog, yeast MrpL32 is expected to form a globular domain at its C-terminus mediated by the metal binding twin Cys motif CxxC-x9-CxxC [54]. Like the bacterial counterpart, the N-terminal region is expected to be in an extended conformation. It is the folded state of the mitochondrial protein that is processed to its mature form by the m-AAA protease, which results in the removal of 71 amino acids from the N-terminus [54, 137, 138]. It is anticipated that the extended N-terminal region of MrpL32 is recognised by the m-AAA protease and feeds into the inner proteolytic chamber where the catalytic residues act on the polypeptide chain, hydrolysing it into peptides. The folded domain of MrpL32 blocks any further processing of the protein. Thus, the folding of MrpL32 permits maturation of the protein while averting complete degradation (Fig. 9.5). In such a mechanism, the middle region of MrpL32 is believed to act as a linker bridging the gap between the folded domain butting the external surface of the protease and the proteolytic residues of the inner chamber. Such a scenario is supported by experiments in which the N-terminal region of MrpL32 was extended in length (20 and 40 amino acids respectively) between the presequence and the middle region. Still these longer variants were cleaved to produce proteins of similar length to wild type MrpL32 [54].

A specialised function of m -AAA protease in protein maturation required for mitochondrial ribosome biogenesis. Under normal conditions (Path I) the m-AAA protease cleaves an N-terminal presequence from folded ribosomal subunit MrpL32 via a partial degradation mechanism. The folded cysteine knot blocks complete processing of this protein and upon release from the protease it assembles into the large subunit of the ribosome. To date the route of entry of the substrate presequence into the proteolytic chamber has not been determined. In situations where the activity of m-AAA protease is compromised (e.g. gene deletion in yeast) or genetic mutation in humans, MrpL32 processing is inhibited and thus ribosome assembly and function is impaired (Path II). In the case that the folding of MrpL32 is disrupted (Path III) due to oxidative stress, the unfolded substrate is fully degraded and thus ribosome assembly and function is also impaired
Once folded and processed, MrpL32 assembles with the ribosome. Unprocessed MrpL32 on the other hand is unable to assemble into the mitochondrial ribosome and therefore cannot perform its function – resulting in a protein translation incompetent ribosomal complex (Fig. 9.5) [56]. Thus, yeast m-AAA protease plays a critical role in activity control, not only of MrpL32, but also of the ribosome. The crystal structure of a bacterial large ribosomal subunit provides an explanation for the inability of unprocessed yeast MrpL32 to assemble into the mitochondrial ribosome large complex. The N-terminal region (∼27 residues) of bacterial ribosome subunit L32 is buried, extending into the interior of the ribosome with the Cys metal binding motif residing on the ribosome surface [139]. The 71 amino acid presequence of MrpL32 cannot be accommodated in such a structural arrangement and thus it fails to assemble in the mitochondrial large ribosomal subunit. An incompletely assembled mitochondrial ribosome missing the MrpL32 subunit is inactive and yeast cannot synthesise mitochondrial-encoded polypeptides. Not surprisingly, mrpl32 null mutants are respiratory deficient. As m-AAA protease is responsible for the generation of assembly competent MrpL32, yeast lacking active m-AAA protease are also respiratory deficient, unable to synthesise subunits of the respiratory complexes encoded by mitochondrial DNA [56, 140].
Another potential post-translation regulatory mechanism involving MrpL32 and the m-AAA protease is oxidative stress sensing [54]. It appears that the cysteine fold of MrpL32 is sensitive to oxidative stress whereby folding is prevented. Unfolded MrpL32 is still recognised by the m-AAA protease but instead of undergoing productive maturation controlled by the folded domain, it is completely degraded into peptides. Whether this serves only as a PQC activity for the removal of a non-functional unfolded protein from mitochondria or additionally as a mechanism to regulate mitochondrial protein translation in response to the oxidative state, remains to be determined. The critical role of the m-AAA protease in mitochondria is exemplified by human diseases such as spinocerebellar ataxia (SCA28) and a form of hereditary spastic paraplegia. These diseases are caused by loss of function mutations in AFG3L2 (homolog of Yta10/Afg3) and paraplegin (homolog of yeast Yta12/Rca1) respectively, subunits of the human m-AAA protease [141, 142].
The Role of i-AAA Protease in Lipid Homeostasis
Recently, the role of i-AAA protease in quality control of lipid homeostasis proteins in the mitochondrial IMS was revealed [143]. Three membrane-associated IMS proteins Ups1, Ups2, and Ups3 collectively play a role in the metabolism of phosphatidylethanolamine and cardiolipin although they seem to contribute unique functions [144–146]. A loss of these proteins alters mitochondrial lipid composition and effects the stability of inner membrane protein complexes such as the TIM23 complex, as well as mitochondrial morphology [144–146]. Each of these three proteins forms a stable complex with a small IMS protein known as Mdm35 [143, 147]. This protein also acts to drive import of Ups proteins into the IMS upon translocation through the TOM complex. In the absence of Mdm35 (i.e. in mdm35 deletion strain), Ups1 and Ups2 are unstable and degraded more rapidly than in its presence [143]. This change in stability is attributed to the action of i-AAA protease and Atp23 [143]. In the case of uncomplexed Ups2, its degradation is mediated by i-AAA protease. Degradation of free Ups1 on the other hand is mediated by i-AAA protease and Atp23. It seems that the degradation of Ups proteins at least serves a PQC function to avoid the accumulation of non-functional protein. However, it is possible that the i-AAA protease plays a regulatory role in the activity control of Ups proteins and hence mitochondrial lipid homeostasis, for example by competing with Mdm35 for binding of Ups proteins. In such a situation adaptor proteins could play a leading role in the targeted delivery of the substrate, however it seems that putative i-AAA protease adaptor proteins Mgr1 and Mgr3 [148, 149] are not involved in i-AAA protease-mediated degradation of Ups1 and Ups2 [143]. A potential role of i-AAA protease and Atp23 in regulatory control of phosphatidylethanolamine and cardiolipin metabolism awaits further analysis. Regulation of lipid biogenesis is also required for normal function in E. coli and the i-AAA protease homolog FtsH plays a critical role. In this case, FtsH-mediated degradation modulates the levels of two enzymes, KdtA and LpxC in the lipopolysaccharide biosynthetic pathway (for a recent review see [126]).
Regulation of Metazoan Biosynthetic Pathways by Lon
Until very recently it seemed that the mitochondrial matrix protease Lon only contributed to PQC and not protein activity control. However, recent studies in human cells (and other animals) have revealed important contributions of LONM to the regulation of mitochondrial biogenesis and metabolic pathways. One of these pathways is the heme biosynthetic pathway. Mitochondria are the site of several steps of the cellular heme biosynthetic pathway. Heme is generated from substrates glycine and succinyl-CoA. The enzyme 5-aminolevulinic acid synthase (known as ALAS-1 in humans), catalyses the first of eight enzymatic steps. ALAS-1 activity, the rate-limiting step of the pathway, is regulated at many levels through expression and import control by a heme-mediated negative feedback mechanism. Heme inhibits transcription and import of the ALAS-1 precursor into mitochondria while enhancing mRNA degradation and turnover of the endogenous protein in mitochondria [150–153]. All of these effects lead to a decrease in the steady state levels of ALAS-1, allowing the levels of heme to be adjusted according to need. The ability to sense and regulate heme levels is important as both an excess or a deficiency is toxic. The proteolytic element of regulation allows a rapid post-translational adjustment of protein levels. Recently, it was revealed that LONM is responsible for the turnover of ALAS-1 in hepatic cells however the mechanism by which heme targets ALAS-1 for degradation is currently unknown. At this point, partial functional redundancy with other proteases, i.e. the mitochondrial ClpXP protease, have not been ruled out [152]. A regulatory role of Lon has also been identified in Drosophila Schneider cells where it degrades mitochondrial transcription factor A (TFAM) [154]. A major component of mitochondrial nucleoids, TFAM contributes to mtDNA maintenance and transcription [155]. Thus, proteolysis may also contribute to the transcriptional expression of the mitochondrial genome. Steroidogenic acute regulatory protein (StAR) is a protein found in mitochondria of mammalian cells in the adrenal cortex, gonads and placenta. It is a key enzyme in steroid hormone biogenesis from cholesterol. Due to its high turnover rates after import into the matrix compartment of mitochondria it has been suspected to be a substrate of LONM, a hypothesis which has been supported by experiments in vitro and in vivo examining murine StAR with mammalian LONM [156]. However, so far it is unclear if its degradation represents a PQC process to prevent the accumulation of large quantities of unwanted protein in mitochondria or has a more important role in regulation of steroid biogenesis. In mammals, LONM has also been implicated in the degradation of cytochrome oxidase subunit 4-1 (Cox4-1) under conditions of hypoxia permitting exchange with the low oxygen efficient isoform Cox4-2 [157]. However, direct degradation of Cox4-1 by LONM was not demonstrated in these studies and the addition of MG132, which is known to inhibit LONM as well as the proteasome [156], didn’t change the steady state levels of the protein [157]. Thus, further studies are required to understand the full contribution of mitochondrial matrix proteases to the elimination of hypoxia sensitive Cox4-1 and thus regulation of the cellular response to changes in oxygen concentrations.
Mitochondrial Protein Degradation via an N-End Rule Pathway?
Some mitochondrial precursor proteins are cleaved by Icp55 or Oct1, following MPP processing to generate the mature protein (see section “Maturation of Mitochondrial Proteins”). In the absence of Icp55 or Oct1, intermediate forms of the preprotein are unstable relative to mature counterparts and possess either Tyr, Phe or Leu at their N-terminus [41]. This is suggestive of an N-end rule pathway of protein degradation being operational in mitochondria. In bacteria, amino acid residues Phe, Leu, Tyr and Trp can act as destabilising signals when exposed at the N-terminus of a protein [158]. They mediate recognition of the protein substrate directly by the pathways degradation machinery and as such are referred to as primary destabilising residues [51, 52, 158]. Basic residues Arg and Lys and in a specific case Met are classified as secondary destabilising residues as they act as acceptors for the non-ribosomal attachment of primary destabilising residues Leu and Phe by the leucyl/phenylalanyl-tRNA protein transferase thereby tagging the protein for degradation [51, 52, 158, 159]. In E. coli, primary destabilising residues bind the adaptor protein ClpS and are directly delivered to the AAA+ protease ClpAP for degradation [159–162]. In the absence of mitochondrial peptidases Icp55 or Oct1, substrates retain N-terminal residues Tyr, Leu and Phe and are unstable. It is suspected that these proteins are removed by a proteolytic pathway but this has not been determined experimentally. Also, it is yet to be determined if this is a PQC mechanism to remove immature proteins from mitochondria or it serves as a regulatory mechanism whereby the activity of Icp55 and Oct1 or molecular components of the hypothetical degradation pathway are regulated to enable post-translational expression control of a subpopulation of mitochondrial proteins. In general the N-end rule pathway of degradation contributes to regulatory networks rather than PQC tasks [52, 163, 164]. Thus, it will be interesting to determine if such a pathway is present in mitochondria and what role it plays.
The Mitochondrial Unfolded Protein Response (UPRmt)
In addition to the contribution that AAA+ proteases make directly to protein quality and activity control, some members participate in stress response pathways. In particular, in metazoan mitochondria it has been established that the matrix peptidase ClpP is a core component of the mitochondrial unfolded protein response (see later). This signal transduction pathway allows mitochondria-to-nuclear communication ensuring that the capacity of the mitochondrial protein quality control machinery matches demand.
The constitutive expression of numerous members of the PQC network ensures that cells are under constant molecular surveillance for unfolded, damaged and misassembled proteins. Whilst this may contribute to protein homeostasis under normal cellular conditions, imbalances in the protein-folding environment can place additional demands on the chaperone and protease machinery. For example, when protein damage is amplified through stress or other physiological insults, the resulting substrate load may exceed the capacity of these systems. This leads to the accumulation of unfolded and aggregated proteins. If the unfolded protein stress is excessive or prolonged, cellular function may be compromised [107, 165, 166]. Thus, to alleviate the additional substrate burden, cells have evolved signalling mechanisms to upregulate members of their PQC networks [107, 166–172]. By enabling the cell to manage unfolded or damaged proteins more effectively, protein homeostasis is expected to be restored.
Elevated levels of misfolded proteins can arise through a variety of cellular stresses including errors in protein biogenesis, exposure to reactive oxygen species, metabolic deficiencies and thermal stress. Although perturbations in protein folding can occur broadly throughout the cell, in some situations, protein damage may be restricted to specific organelles. Importantly, cells have dedicated pathways for managing unfolded protein stress within their different subcellular compartments. While the mechanisms that the cell employs for sensing and responding to unfolded protein stress within the cytosol and endoplasmic reticulum are well characterised, the discovery of an equivalent regulatory pathway in metazoan mitochondria has been more recent [168, 169, 173].
Discovery of the Mitochondrial Unfolded Protein Response
Early indications of a mitochondrial specific stress response came from studies examining the regulation of chaperones in cells depleted of mtDNA. Using cultured mammalian cells, Hoogenraad and co-workers demonstrated that the loss of mtDNA resulted in the transcriptional activation and increased expression of mitochondrial chaperonins HSP60 and HSP10 [174]. Interestingly, as there were no detectable changes in the expression of selected cytosolic chaperones, this response was suggested to be compartment-specific. While the primary stress signal was not identified in this study, the authors proposed that the stress response could be linked to the accumulation of unfolded protein [174].
To establish whether the specific upregulation of mitochondrial chaperones was connected to protein misfolding within mitochondria, a mammalian cell-based model was developed for the overexpression of a folding deficient mutant of the mitochondrial protein, ornithine transcarbamylase (OTC) [169]. In comparison to cells expressing wild type OTC, the expression of the OTC mutant resulted in the selective induction of a number of mitochondrial stress proteins including HSP60, HSP10, mtDnaJ/Tid-1 (HSP40), and the CLPP peptidase. Consistent with a role in PQC, both HSP60 and CLPP co-immunoprecipitated with the OTC mutant, implicating them in the clearance of the substrate. Importantly, like other stress response pathways, the removal of non-native protein corresponded with the transcriptional attenuation of chaperone genes [169]. However, the protease responsible for degradation of mutant OTC is yet to be established.
Since the initial discovery of this unfolded protein response in mammals, an equivalent pathway has been identified in C. elegans. Like the observations made in mammalian cells, the loss of mtDNA resulted in the upregulation of genes encoding HSP60 and mtHSP70, but did change the expression profile of chaperones in other subcellular compartments [168]. A similar response was also observed upon inactivation of genes suggested to function in the biogenesis or turnover of mitochondrial proteins [168]. Taken together, these findings demonstrate that the selective upregulation of mitochondrial chaperones in both C. elegans and mammalian cells is triggered by disturbances in the mitochondrial protein folding environment. This pathway has been coined, the mitochondrial unfolded protein response (UPRmt). As yeast appear to lack a mitochondrial specific unfolded protein response, this section describes the pathway in metazoa. A key component of this signal transduction pathway in the nematode worm is the mitochondrial matrix protease ClpXP.
Components of the UPRmt Signalling Pathway in C. elegans
The mitochondrial unfolded protein response (UPRmt) is defined by the ability of cells to selectively alter the level of PQC machinery in response to protein misfolding within mitochondria [168, 169]. Importantly, for target genes in the nucleus to be up-regulated in response to an extrinsic mitochondrial stress, a mechanism for communication between the two organelles must exist. To dissect this pathway and identify factors involved in relaying the stress signal from the mitochondrion to the nucleus, Ron and colleagues performed a series of gene silencing and deletion studies in C. elegans [165]. For these experiments, transgenic C. elegans reporter strains harbouring a temperature sensitive mutation (zc32 II) were used. At non-permissive temperatures, these animals experience unfolded protein stress within the mitochondrion, which leads to the transcriptional upregulation of a chaperone-GFP reporter (consisting of a DNA fragment containing the promoter region and the coding sequence for the first few amino acid residues of a mitochondrial chaperone) [23, 165]. Genes whose inactivation caused a reduction in reporter activity were predicted to encode proteins that contribute to UPRmt signalling [165]. Using this model, a number of cellular components that are critical for UPRmt signalling were identified [23, 24, 165]. These proteins can be broadly classified into two groups: those that are involved in the sensing and transmission of stress signals from the mitochondria (ClpX1, ClpX2, ClpP, HAF-1), and those that adjust their nuclear distribution or expression profiles to promote chaperone gene activation (ZC376.7, DVE-1, UBL-5).
The mitochondrial matrix ClpP protease together with its anticipated cognate partners, ClpX1 (encoded by KO7A3.3) and ClpX2 (encoded by D2030.2), [175] are thought to be responsible for the initial activation of the UPRmt signalling pathway [23, 24]. The C. elegans ClpXP machinery has been implicated in the ATP-dependent proteolysis of unfolded substrates [24]. While evidence suggests that the ClpP homologue forms homo-oligomeric complexes composed of 14 subunits, little is known about the composition of the ClpX oligomer [23, 24]. In vivo, a hexameric form of ClpX is likely to be involved in both substrate recognition and docking to the ClpP protease. Thus, different compositions or oligomeric arrangements of ClpX1 and ClpX2 could potentially serve as a mechanism for regulating substrate detection and delivery. In this regard, it is interesting to note that the loss of either ClpX1 or ClpX2 alone, did not inhibit UPRmt signalling. This functional redundancy suggests that the substrate specificity of ClpX1 and ClpX2 is overlapping [24]. Although the substrates of ClpX are currently unknown, their delivery to the ClpP protease appears to be critical for transmission of the UPRmt stress signal.
The requirement for ClpP in an upstream regulatory role was established through a series of knock-down experiments that examined the position of ClpP relative to other components within the signalling network. Consistent with a position near the top of the signalling hierarchy, depletion of ClpP in stressed worms prevented the activation, recruitment and nuclear redistribution of all known UPRmt transcription factors [23, 24, 165]. To elucidate the mechanism by which ClpP controls the signalling pathway, a chemical inhibitor of ClpP was employed. These studies revealed that the intrinsic proteolytic activity of ClpP was an essential element in UPRmt signal progression [23]. Not surprisingly, this raised questions regarding the identity of the signalling molecules generated by ClpP degradation, and the mechanisms by which these molecules transduce the signal. Although a number of scenarios are possible, there is evidence to suggest that peptides generated from the proteolysis of matrix proteins may be involved in at least one arm of the stress response pathway [24].
The discovery of HAF-1 as a component of the UPRmt signalling pathway suggested that peptides generated from ClpXP proteolysis may be involved in signal transmission. HAF-1 is a member of the ABC transporter family, which appears to be located in the mitochondrial inner membrane [24]. In a process that is thought to be mechanistically equivalent to that reported for the yeast Mdl1p homologue, HAF-1 actively transports peptides from the matrix into the IMS. Peptides are then thought to diffuse across the outer mitochondrial membrane into the cytosol [176, 177]. Importantly, in contrast to wild type worms experiencing stress, deletion of haf-1 resulted in reduced mitochondrial peptide efflux and attenuated mitochondrial chaperone gene induction [24]. Based on these findings, a model that integrates ClpP-mediated proteolysis with transmission of the stress signal across the mitochondrial membranes has been proposed (Fig. 9.6). Although HAF-1 has been shown to transport a broad spectrum of peptides derived from a range of mitochondrial matrix proteins, the precise nature of the stress signal is currently unknown [24]. It is not known whether signal transmission is dependent upon a unique type of peptide, or whether the rate of peptide flux is a contributing factor. Further, a role for HAF-1 in the transport of free prosthetic groups cannot be excluded. Although additional work is required to elucidate the sensory mechanisms within the cytosol, it appears that the introduction of these peptides or prosthetic groups into the signalling pathway results in the activation of downstream transcription factors. Specifically, the ZC376.7 (bZIP) transcription factor is recruited into the UPRmt pathway in a process that is dependent upon both ClpP and HAF-1. Under normal cellular conditions, ZC376.7 is located in the cytosol. Upon induction of UPRmt however, the protein translocates to the nucleus, where it accumulates. Although the gene targets of ZC376.7 are currently unknown, its translocation to the nucleus is closely linked to the upregulation of mitochondrial chaperone genes [24].

The mitochondrial unfolded protein response in C. elegans. Depicted are the known components and model of mitochondria-to-nuclear signalling pathway of the unfolded protein response. A disruption to protein homeostasis within the mitochondrial matrix leads to the proteolytic processing of an unknown substrate(s) by ClpXP. This might be the promiscuous degradation of any unfolded protein or the regulated degradation of specific proteins that are conditional substrates under stress conditions. The products of ClpXP-mediated protein degradation are peptides (red and black lines) but for cofactor-bound substrates, prosthetic groups would also be released (orange square). These peptides and/or prosthetic groups could act as signals to initiate the pathway outside of mitochondria following transport through HAF-1 and pores in the outer membrane. Other mechanisms may exist for a signalling path independent of HAF-1. The redistribution of pathway transcription factors initiated by the released signalling molecule(s) leads to the transcriptional up-regulation of specific genes, such as those encoding mitochondrial chaperones Hsp60 and Hsp70, that act to restore mitochondrial protein homeostasis
Whilst the incorporation of HAF-1 into the UPRmt signalling pathway provides a feasible model for transmission of the stress signal across the mitochondrial membranes, current evidence suggests that an alternative mechanism for UPRmt activation may also be at play. In what appears to be a separate arm of the response, ClpP has been shown to transmit signals independently of HAF-1 (Fig. 9.6). While the loss of ClpP prevented the nuclear redistribution of the transcription factor DVE-1 under conditions of mitochondrial stress, the loss of HAF-1 had no effect [23, 24]. Although the signalling mechanisms that govern this response are currently unknown, the redistribution of DVE-1 within the nucleus correlates with its binding to chaperone gene promoters [23]. Importantly, to enhance the transcription of chaperone genes, DVE-1 is proposed to function with a small ubiquitin-like protein called UBL-5 [23]. Upon induction of UPRmt, UBL-5 is upregulated and accumulates within the nucleus [165]. As demonstrated through pull-down experiments, this enrichment enhances the formation of stable UBL-5/DVE-1 complexes [23]. Interestingly, as DVE-1 redistribution and promoter binding can occur independently of UBL-5 [23], it is tempting to speculate that UBL-5 may be recruited into transcription complexes as a downstream event.
The above findings support a model whereby the stress-induced nuclear reorganisation of DVE-1 and UBL-5 serves as a mechanism for upregulating mitochondrial chaperone genes. Importantly however, within this model, the transcriptional induction of UBL-5 also requires regulation. In what appears to be an amplification circuit, RNAi knock-down studies revealed that the transcriptional induction of UBL-5 is also dependent upon DVE-1. It is currently unclear whether DVE-1 is directly involved in ubl-5 gene activation, or whether it exerts this control via an indirect mechanism [23]. Moreover, in worms experiencing mitochondrial stress, the loss of HAF-1 also circumvented ubl-5 gene activation [24]. Thus, the upregulation of UBL-5 also requires a mediator that is transmitted via the HAF-1 signalling pathway. While the factors that contribute to this event are currently unknown, it is possible to speculate that ZC376.7 may fulfil this role. Further analysis of this transcription factor and the identification of additional UPRmt signalling components may assist in elucidating this mechanism.
What Is the UPRmt Signalling Factor Generated by ClpXP Action?
The ability of cells to selectively respond to protein conformational stress within specific subcellular compartments is a conserved paradigm that exists in both prokaryotic and eukaryotic organisms [166, 170, 175, 176, 178]. These response pathways are underpinned by signalling cascades that are tailored towards sensing unfolded protein stress and transmitting stress signals to designated transcription factors. Interestingly, a common theme shared by many of these response pathways is the participation of cellular proteases in regulating stress signal transmission [166, 178]. The discovery that ClpXP plays a pivotal role in controlling the mitochondrial unfolded protein response in C. elegans provides yet another example of how proteolytic processing contributes to signal transduction [23, 24]. Although significant progress has been made in identifying components of the C. elegans UPRmt signalling pathway, a number of key elements have yet to be defined. In particular, the substrates that undergo ClpXP-mediated proteolysis and the type of the signalling molecules that are generated through this process require elucidation. Importantly, given that HAF-1 homologues have been implicated in the transport of both peptides and prosthetic groups [176, 177, 179], a range of potential signalling mechanisms can be conceived. Based on current knowledge of the UPRmt signalling pathway (Fig. 9.6), a number of hypothetical models describing the contribution that ClpXP could make to stress signal transmission have been proposed [23, 175, 176, 178]. It has been suggested that ClpXP could function in the promiscuous degradation of unfolded and/or misfolded proteins that arise from perturbations in mitochondrial protein folding. Transduction of the stress signal could involve a peptide/cofactor receptor in the cytosol, or may rely on a sensor molecule that monitors the rate of peptide efflux [23, 175, 176]. It is also possible that some matrix proteins may be conditional substrates of ClpXP – displaying specific recognition motifs under conditions of stress. In this scenario, a specific peptide or prosthetic group could be liberated from the protein following its targeted degradation. Again, signal transduction may rely on a specific cytosolic receptor that recognises mitochondrially-derived ligands [23, 175, 176].
The above models not only provide insight into the mechanisms that ClpXP may employ, but also pave the way for future studies. The identification of ClpXP substrates and the molecules involved in downstream signalling may assist in further defining the molecular mechanisms that control the C. elegans UPRmt signalling pathway. In addition, understanding the sensory components within the cytosol may also shed light on the mechanism by which ClpP is able to transmit signals independently of HAF-1. Whilst it is possible that ABC transporters that are similar to HAF-1 may have escaped detection in original screens, the existence of an alternative signalling route that also negotiates the mitochondrial membrane barrier cannot be discounted.
Despite C. elegans and mammalian UPRmt pathways being functionally equivalent, the transcription factors that control these stress response pathways appear to be quite different [169, 180]. Whilst studies have addressed the mechanisms by which UPRmt gene targets are selectively upregulated in response to mitochondrial stress [169, 181], the identity of the stress sensor component has remained elusive in mammalian mitochondria.
Future Perspectives
Most of our understanding of mitochondrial AAA+ proteases has been achieved via genetic, biochemical and proteomic studies, particularly in relation to PQC functions. A detailed understanding of the recognition motifs in substrates and substrate docking sites on mitochondrial AAA+ proteases will help to refine their substrate repertoire. It is anticipated that much will be revealed about the substrates of these proteases in higher eukaryotes where cell type dependent processes take place. Research on the role of AAA+ proteases in the mitochondrial unfolded protein response, cell death and aging are just a few areas that are expected to expand in the near future. An understanding of the mitochondrial AAA+ proteases in the context of human mitochondrial diseases will be important in the development of successful therapies for disease intervention.
References
Todde V, Veenhuis M, van der Klei IJ (2009) Autophagy: principles and significance in health and disease. Biochim Biophys Acta 1792(1):3–13
Wong E, Cuervo AM (2010) Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol 2(12):a006734
Yang Z, Klionsky DJ (2009) An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 335:1–32
Buchberger A (2013) Roles of Cdc48 in regulated protein degradation in yeast. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Subcell Biochem 66:195–222
Kwasniak M, Pogorzelec L, Migdal I, Smakowska E et al (2012) Proteolytic system of plant mitochondria. Physiol Plant 145(1):187–195
Ezaki J, Kominami E, Ueno T (2011) Peroxisome degradation in mammals. IUBMB Life 63(11):1001–1008
Druyan R, DeBernard B, Rabinowitz M (1969) Turnover of cytochromes labeled with delta-aminolevulinic acid-3H in rat liver. J Biol Chem 244(21):5874–5878
Swick RW, Rexroth AK, Stange JL (1968) The metabolism of mitochondrial proteins. 3. The dynamic state of rat liver mitochondria. J Biol Chem 243(13):3581–3587
Wheeldon LW, Dianoux AC, Bof M, Vignais PV (1974) Stable and labile products of mitochondrial protein synthesis in vitro. Eur J Biochem 46(1):189–199
Desautels M, Goldberg AL (1982) Demonstration of an ATP-dependent, vanadate-sensitive endoprotease in the matrix of rat liver mitochondria. J Biol Chem 257(19):11673–11679
Desautels M, Goldberg AL (1982) Liver mitochondria contain an ATP-dependent, vanadate-sensitive pathway for the degradation of proteins. Proc Natl Acad Sci U S A 79(6):1869–1873
Kalnov SL, Novikova LA, Zubatov AS, Luzikov VN (1979) Participation of a mitochondrial proteinase in the breakdown of mitochondrial translation products in yeast. FEBS Lett 101(2):355–358
Yasuhara T, Mera Y, Nakai T, Ohashi A (1994) ATP-dependent proteolysis in yeast mitochondria. J Biochem 115(6):1166–1171
Arlt H, Tauer R, Feldmann H, Neupert W et al (1996) The YTA10-12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell 85(6):875–885
Kang SG, Ortega J, Singh SK, Wang N et al (2002) Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J Biol Chem 277(23):21095–21102
Kutejova E, Durcova G, Surovkova E, Kuzela S (1993) Yeast mitochondrial ATP-dependent protease: purification and comparison with the homologous rat enzyme and the bacterial ATP-dependent protease La. FEBS Lett 329(1–2):47–50
Kuzela S, Goldberg AL (1994) Mitochondrial ATP-dependent protease from rat liver and yeast. Methods Enzymol 244:376–383
Leonhard K, Herrmann JM, Stuart RA, Mannhaupt G et al (1996) AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J 15(16):4218–4229
Nakai T, Yasuhara T, Fujiki Y, Ohashi A (1995) Multiple genes, including a member of the AAA family, are essential for degradation of unassembled subunit 2 of cytochrome c oxidase in yeast mitochondria. Mol Cell Biol 15(8):4441–4452
Pajic A, Tauer R, Feldmann H, Neupert W et al (1994) Yta10p is required for the ATP-dependent degradation of polypeptides in the inner membrane of mitochondria. FEBS Lett 353(2):201–206
Pearce DA, Sherman F (1995) Degradation of cytochrome oxidase subunits in mutants of yeast lacking cytochrome c and suppression of the degradation by mutation of yme1. J Biol Chem 270(36):20879–20882
Watabe S, Kimura T (1985) ATP-dependent protease in bovine adrenal cortex. Tissue specificity, subcellular localization, and partial characterization. J Biol Chem 260(9):5511–5517
Haynes CM, Petrova K, Benedetti C, Yang Y et al (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13(4):467–480
Haynes CM, Yang Y, Blais SP, Neubert TA et al (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37(4):529–540
Heo JM, Rutter J (2011) Ubiquitin-dependent mitochondrial protein degradation. Int J Biochem Cell Biol 43(10):1422–1426
Karbowski M, Youle RJ (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23(4):476–482
Scott I, Youle RJ (2010) Mitochondrial fission and fusion. Essays Biochem 47:85–98
Drago I, Pizzo P, Pozzan T (2011) After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J 30(20):4119–4125
Smith DJ, Ng H, Kluck RM, Nagley P (2008) The mitochondrial gateway to cell death. IUBMB Life 60(6):383–389
West AP, Shadel GS, Ghosh S (2011) Mitochondria in innate immune responses. Nat Rev Immunol 11(6):389–402
Gray MW, Burger G, Lang BF (2001) The origin and early evolution of mitochondria. Genome Biol 2(6):reviews 1018.1–reviews 1018.5
Friedman JR, Lackner LL, West M, DiBenedetto JR et al (2011) ER tubules mark sites of mitochondrial division. Science 334(6054):358–362
Harner M, Korner C, Walther D, Mokranjac D et al (2011) The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J 30(21):4356–4370
Hoppins S, Collins SR, Cassidy-Stone A, Hummel E et al (2011) A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J Cell Biol 195(2):323–340
Kornmann B, Currie E, Collins SR, Schuldiner M et al (2009) An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325(5939):477–481
Kornmann B, Osman C, Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A 108(34):14151–14156
Stroud DA, Oeljeklaus S, Wiese S, Bohnert M et al (2011) Composition and topology of the endoplasmic reticulum-mitochondria encounter structure. J Mol Biol 413(4):743–750
von der Malsburg K, Muller JM, Bohnert M, Oeljeklaus S et al (2011) Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell 21(4):694–707
Sickmann A, Reinders J, Wagner Y, Joppich C et al (2003) The proteome of Saccharomyces cerevisiae mitochondria. Proc Natl Acad Sci U S A 100(23):13207–13212
Young JC, Hoogenraad NJ, Hartl FU (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112(1):41–50
Vogtle FN, Wortelkamp S, Zahedi RP, Becker D et al (2009) Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell 139(2):428–439
Gakh O, Cavadini P, Isaya G (2002) Mitochondrial processing peptidases. Biochim Biophys Acta 1592(1):63–77
Chacinska A, Koehler CM, Milenkovic D, Lithgow T et al (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138(4):628–644
Mokranjac D, Neupert W (2010) The many faces of the mitochondrial TIM23 complex. Biochim Biophys Acta 1797(6–7):1045–1054
Ott M, Herrmann JM (2010) Co-translational membrane insertion of mitochondrially encoded proteins. Biochim Biophys Acta 1803(6):767–775
Mossmann D, Meisinger C, Vogtle FN (2012) Processing of mitochondrial presequences. Biochim Biophys Acta 1819(9–10):1098–1106
Taylor AB, Smith BS, Kitada S, Kojima K et al (2001) Crystal structures of mitochondrial processing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9(7):615–625
Rawlings ND, Morton FR, Kok CY, Kong J et al (2008) MEROPS: the peptidase database. Nucleic Acids Res 36(Database issue):320–325
Vogtle FN, Prinz C, Kellermann J, Lottspeich F et al (2011) Mitochondrial protein turnover: role of the precursor intermediate peptidase Oct1 in protein stabilization. Mol Biol Cell 22(13):2135–2143
Naamati A, Regev-Rudzki N, Galperin S, Lill R et al (2009) Dual targeting of Nfs1 and discovery of its novel processing enzyme, Icp55. J Biol Chem 284(44):30200–30208
Dougan DA, Truscott KN, Zeth K (2010) The bacterial N-end rule pathway: expect the unexpected. Mol Microbiol 76(3):545–558
Varshavsky A (2011) The N-end rule pathway and regulation by proteolysis. Protein Sci 20(8):1298–1345
Gur E, Ottofuelling R, Dougan DA (2013) Machines of destruction – AAA+ proteases and the adaptors that control them. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Subcell Biochem 66:3–33
Bonn F, Tatsuta T, Petrungaro C, Riemer J et al (2011) Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J 30(13):2545–2556
Koppen M, Bonn F, Ehses S, Langer T (2009) Autocatalytic processing of m-AAA protease subunits in mitochondria. Mol Biol Cell 20(19):4216–4224
Nolden M, Ehses S, Koppen M, Bernacchia A et al (2005) The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell 123(2):277–289
Osman C, Wilmes C, Tatsuta T, Langer T (2007) Prohibitins interact genetically with Atp23, a novel processing peptidase and chaperone for the F1Fo-ATP synthase. Mol Biol Cell 18(2):627–635
Zeng X, Neupert W, Tzagoloff A (2007) The metalloprotease encoded by ATP23 has a dual function in processing and assembly of subunit 6 of mitochondrial ATPase. Mol Biol Cell 18(2):617–626
Esser K, Tursun B, Ingenhoven M, Michaelis G et al (2002) A novel two-step mechanism for removal of a mitochondrial signal sequence involves the mAAA complex and the putative rhomboid protease Pcp1. J Mol Biol 323(5):835–843
Herlan M, Vogel F, Bornhovd C, Neupert W et al (2003) Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and of mitochondrial DNA. J Biol Chem 278(30):27781–27788
McQuibban GA, Saurya S, Freeman M (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423(6939):537–541
Sesaki H, Southard SM, Yaffe MP, Jensen RE (2003) Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell 14(6):2342–2356
Tatsuta T, Augustin S, Nolden M, Friedrichs B et al (2007) m-AAA protease-driven membrane dislocation allows intramembrane cleavage by rhomboid in mitochondria. EMBO J 26(2):325–335
Hartl FU, Bracher A, Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475(7356):324–332
Voos W, Rottgers K (2002) Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim Biophys Acta 1592(1):51–62
Marom M, Azem A, Mokranjac D (2011) Understanding the molecular mechanism of protein translocation across the mitochondrial inner membrane: still a long way to go. Biochim Biophys Acta 1808(3):990–1001
Voos W (2009) Mitochondrial protein homeostasis: the cooperative roles of chaperones and proteases. Res Microbiol 160(9):718–725
Truscott KN, Lowth BR, Strack PR, Dougan DA (2010) Diverse functions of mitochondrial AAA+ proteins: protein activation, disaggregation, and degradation. Biochem Cell Biol 88(1):97–108
van Dyck L, Dembowski M, Neupert W, Langer T (1998) Mcx1p, a ClpX homologue in mitochondria of Saccharomyces cerevisiae. FEBS Lett 438(3):250–254
Craig EA, Marszalek J (2002) A specialized mitochondrial molecular chaperone system: a role in formation of Fe/S centers. Cell Mol Life Sci 59(10):1658–1665
Reinders J, Sickmann A (2007) Proteomics of yeast mitochondria. Methods Mol Biol 372:543–557
Sliwa D, Dairou J, Camadro JM, Santos R (2012) Inactivation of mitochondrial aspartate aminotransferase contributes to the respiratory deficit of yeast frataxin-deficient cells. Biochem J 441(3):945–953
Erzberger JP, Berger JM (2006) Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct 35:93–114
Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure–diverse function. Genes Cells 6(7):575–597
Van Melderen L, Aertsen A (2009) Regulation and quality control by Lon-dependent proteolysis. Res Microbiol 160(9):645–651
Goldberg AL (1992) The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur J Biochem 203(1–2):9–23
Suzuki CK, Suda K, Wang N, Schatz G (1994) Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science 264(5161):891
Van Dyck L, Pearce DA, Sherman F (1994) PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. J Biol Chem 269(1):238–242
Wagner I, Arlt H, van Dyck L, Langer T et al (1994) Molecular chaperones cooperate with PIM1 protease in the degradation of misfolded proteins in mitochondria. EMBO J 13(21):5135–5145
Savel’ev AS, Novikova LA, Kovaleva IE, Luzikov VN et al (1998) ATP-dependent proteolysis in mitochondria. m-AAA protease and PIM1 protease exert overlapping substrate specificities and cooperate with the mtHsp70 system. J Biol Chem 273(32):20596–20602
Hanson PI, Whiteheart SW (2005) AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6(7):519–529
Striebel F, Kress W, Weber-Ban E (2009) Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr Opin Struct Biol 19(2):209–217
Sauer RT, Baker TA (2011) AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612
Botos I, Melnikov EE, Cherry S, Tropea JE et al (2004) The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J Biol Chem 279(9):8140–8148
Wagner I, van Dyck L, Savel’ev AS, Neupert W et al (1997) Autocatalytic processing of the ATP-dependent PIM1 protease: crucial function of a pro-region for sorting to mitochondria. EMBO J 16(24):7317–7325
Stahlberg H, Kutejova E, Suda K, Wolpensinger B et al (1999) Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc Natl Acad Sci U S A 96(12):6787–6790
Cha SS, An YJ, Lee CR, Lee HS et al (2010) Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber. EMBO J 29(20):3520–3530
Ondrovicova G, Liu T, Singh K, Tian B et al (2005) Cleavage site selection within a folded substrate by the ATP-dependent lon protease. J Biol Chem 280(26):25103–25110
Rep M, van Dijl JM, Suda K, Schatz G et al (1996) Promotion of mitochondrial membrane complex assembly by a proteolytically inactive yeast Lon. Science 274(5284):103–106
van Dijl JM, Kutejova E, Suda K, Perecko D et al (1998) The ATPase and protease domains of yeast mitochondrial Lon: roles in proteolysis and respiration-dependent growth. Proc Natl Acad Sci U S A 95(18):10584–10589
Major T, von Janowsky B, Ruppert T, Mogk A et al (2006) Proteomic analysis of mitochondrial protein turnover: identification of novel substrate proteins of the matrix protease pim1. Mol Cell Biol 26(3):762–776
Moradas-Ferreira P, Costa V, Piper P, Mager W (1996) The molecular defences against reactive oxygen species in yeast. Mol Microbiol 19(4):651–658
Raha S, Robinson BH (2000) Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci 25(10):502–508
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97(6):1634–1658
Bender T, Leidhold C, Ruppert T, Franken S et al (2010) The role of protein quality control in mitochondrial protein homeostasis under oxidative stress. Proteomics 10(7):1426–1443
Bota DA, Davies KJ (2002) Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol 4(9):674–680
Bota DA, Davies KJ (2001) Protein degradation in mitochondria: implications for oxidative stress, aging and disease: a novel etiological classification of mitochondrial proteolytic disorders. Mitochondrion 1(1):33–49
Nystrom T (2005) Role of oxidative carbonylation in protein quality control and senescence. EMBO J 24(7):1311–1317
Bukau B, Weissman J, Horwich A (2006) Molecular chaperones and protein quality control. Cell 125(3):443–451
Leonhardt SA, Fearson K, Danese PN, Mason TL (1993) HSP78 encodes a yeast mitochondrial heat shock protein in the Clp family of ATP-dependent proteases. Mol Cell Biol 13(10):6304–6313
Liberek K, Lewandowska A, Zietkiewicz S (2008) Chaperones in control of protein disaggregation. EMBO J 27(2):328–335
Weibezahn J, Tessarz P, Schlieker C, Zahn R et al (2004) Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119(5):653–665
von Janowsky B, Major T, Knapp K, Voos W (2006) The disaggregation activity of the mitochondrial ClpB homolog Hsp78 maintains Hsp70 function during heat stress. J Mol Biol 357(3):793–807
Germaniuk A, Liberek K, Marszalek J (2002) A bichaperone (Hsp70-Hsp78) system restores mitochondrial DNA synthesis following thermal inactivation of Mip1p polymerase. J Biol Chem 277(31):27801–27808
Rottgers K, Zufall N, Guiard B, Voos W (2002) The ClpB homolog Hsp78 is required for the efficient degradation of proteins in the mitochondrial matrix. J Biol Chem 277(48):45829–45837
Bateman JM, Iacovino M, Perlman PS, Butow RA (2002) Mitochondrial DNA instability mutants of the bifunctional protein Ilv5p have altered organization in mitochondria and are targeted for degradation by Hsp78 and the Pim1p protease. J Biol Chem 277(49):47946–47953
Morimoto RI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22(11):1427–1438
Bender T, Lewrenz I, Franken S, Baitzel C et al (2011) Mitochondrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol Biol Cell 22(5):541–554
Elsasser S, Finley D (2005) Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 7(8):742–749
Samanovic M, Li H, Darwin KH (2013) The Pup-Proteasome system of Mycobacterium tuberculosis. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Subcell Biochem 66:267–295
Gottesman S, Roche E, Zhou Y, Sauer RT (1998) The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev 12(9):1338–1347
Moore SD, Sauer RT (2007) The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem 76:101–124
von Janowsky B, Knapp K, Major T, Krayl M et al (2005) Structural properties of substrate proteins determine their proteolysis by the mitochondrial AAA+ protease Pim1. Biol Chem 386(12):1307–1317
Herman C, Prakash S, Lu CZ, Matouschek A et al (2003) Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol Cell 11(3):659–669
Prakash S, Matouschek A (2004) Protein unfolding in the cell. Trends Biochem Sci 29(11):593–600
van Dyck L, Neupert W, Langer T (1998) The ATP-dependent PIM1 protease is required for the expression of intron-containing genes in mitochondria. Genes Dev 12(10):1515–1524
Liu T, Lu B, Lee I, Ondrovicova G et al (2004) DNA and RNA binding by the mitochondrial lon protease is regulated by nucleotide and protein substrate. J Biol Chem 279(14):13902–13910
Lu B, Yadav S, Shah PG, Liu T et al (2007) Roles for the human ATP-dependent Lon protease in mitochondrial DNA maintenance. J Biol Chem 282(24):17363–17374
Chen XJ, Wang X, Butow RA (2007) Yeast aconitase binds and provides metabolically coupled protection to mitochondrial DNA. Proc Natl Acad Sci U S A 104(34):13738–13743
Teichmann U, van Dyck L, Guiard B, Fischer H et al (1996) Substitution of PIM1 protease in mitochondria by Escherichia coli Lon protease. J Biol Chem 271(17):10137–10142
Balaban RS, Nemoto S, Finkel T (2005) Mitochondria, oxidants, and aging. Cell 120(4):483–495
Luce K, Weil AC, Osiewacz HD (2010) Mitochondrial protein quality control systems in aging and disease. Adv Exp Med Biol 694:108–125
Luce K, Osiewacz HD (2009) Increasing organismal healthspan by enhancing mitochondrial protein quality control. Nat Cell Biol 11(7):852–858
Guelin E, Rep M, Grivell LA (1996) Afg3p, a mitochondrial ATP-dependent metalloprotease, is involved in degradation of mitochondrially-encoded Cox1, Cox3, Cob, Su6, Su8 and Su9 subunits of the inner membrane complexes III, IV and V. FEBS Lett 381(1–2):42–46
Weber ER, Hanekamp T, Thorsness PE (1996) Biochemical and functional analysis of the YME1 gene product, an ATP and zinc-dependent mitochondrial protease from S. cerevisiae. Mol Biol Cell 7(2):307–317
Okuno T, Ogura T (2013) FtsH protease-mediated regulation of various cellular functions. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Subcell Biochem 66:53–69
Koppen M, Metodiev MD, Casari G, Rugarli EI et al (2007) Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol 27(2):758–767
Guelin E, Rep M, Grivell LA (1994) Sequence of the AFG3 gene encoding a new member of the FtsH/Yme1/Tma subfamily of the AAA-protein family. Yeast 10(10):1389–1394
Schnall R, Mannhaupt G, Stucka R, Tauer R et al (1994) Identification of a set of yeast genes coding for a novel family of putative ATPases with high similarity to constituents of the 26S protease complex. Yeast 10(9):1141–1155
Thorsness PE, White KH, Fox TD (1993) Inactivation of YME1, a member of the ftsH-SEC18-PAS1-CDC48 family of putative ATPase-encoding genes, causes increased escape of DNA from mitochondria in Saccharomyces cerevisiae. Mol Cell Biol 13(9):5418–5426
Tzagoloff A, Yue J, Jang J, Paul MF (1994) A new member of a family of ATPases is essential for assembly of mitochondrial respiratory chain and ATP synthetase complexes in Saccharomyces cerevisiae. J Biol Chem 269(42):26144–26151
Thorsness PE, Fox TD (1993) Nuclear mutations in Saccharomyces cerevisiae that affect the escape of DNA from mitochondria to the nucleus. Genetics 134(1):21–28
Lee S, Augustin S, Tatsuta T, Gerdes F et al (2011) Electron cryomicroscopy structure of a membrane-anchored mitochondrial AAA protease. J Biol Chem 286(6):4404–4411
Gerdes F, Tatsuta T, Langer T (2012) Mitochondrial AAA proteases – towards a molecular understanding of membrane-bound proteolytic machines. Biochim Biophys Acta 1823(1):49–55
Pearce DA, Sherman F (1995) Diminished degradation of yeast cytochrome c by interactions with its physiological partners. Proc Natl Acad Sci U S A 92(9):3735–3739
Weil A, Luce K, Drose S, Wittig I et al (2011) Unmasking a temperature-dependent effect of the P. anserina i-AAA protease on aging and development. Cell Cycle 10(24):4280–4290
Graack HR, Bryant ML, O’Brien TW (1999) Identification of mammalian mitochondrial ribosomal proteins (MRPs) by N-terminal sequencing of purified bovine MRPs and comparison to data bank sequences: the large subribosomal particle. Biochemistry 38(50):16569–16577
Graack HR, Wittmann-Liebold B (1998) Mitochondrial ribosomal proteins (MRPs) of yeast. Biochem J 329(Pt 3):433–448
Harms J, Schluenzen F, Zarivach R, Bashan A et al (2001) High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell 107(5):679–688
Arlt H, Steglich G, Perryman R, Guiard B et al (1998) The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. EMBO J 17(16):4837–4847
Casari G, De Fusco M, Ciarmatori S, Zeviani M et al (1998) Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 93(6):973–983
Di Bella D, Lazzaro F, Brusco A, Plumari M et al (2010) Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 42(4):313–321
Potting C, Wilmes C, Engmann T, Osman C et al (2010) Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J 29(17):2888–2898
Osman C, Haag M, Potting C, Rodenfels J et al (2009) The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J Cell Biol 184(4):583–596
Sesaki H, Dunn CD, Iijima M, Shepard KA et al (2006) Ups1p, a conserved intermembrane space protein, regulates mitochondrial shape and alternative topogenesis of Mgm1p. J Cell Biol 173(5):651–658
Tamura Y, Endo T, Iijima M, Sesaki H (2009) Ups1p and Ups2p antagonistically regulate cardiolipin metabolism in mitochondria. J Cell Biol 185(6):1029–1045
Tamura Y, Iijima M, Sesaki H (2010) Mdm35p imports Ups proteins into the mitochondrial intermembrane space by functional complex formation. EMBO J 29(17):2875–2887
Dunn CD, Lee MS, Spencer FA, Jensen RE (2006) A genomewide screen for petite-negative yeast strains yields a new subunit of the i-AAA protease complex. Mol Biol Cell 17(1):213–226
Dunn CD, Tamura Y, Sesaki H, Jensen RE (2008) Mgr3p and Mgr1p are adaptors for the mitochondrial i-AAA protease complex. Mol Biol Cell 19(12):5387–5397
Dailey TA, Woodruff JH, Dailey HA (2005) Examination of mitochondrial protein targeting of haem synthetic enzymes: in vivo identification of three functional haem-responsive motifs in 5-aminolaevulinate synthase. Biochem J 386(Pt 2):381–386
Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, Meyer UA, Spiegelman BM (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 122(4):505–515
Tian Q, Li T, Hou W, Zheng J et al (2011) Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. J Biol Chem 286(30):26424–26430
Yoshino K, Munakata H, Kuge O, Ito A et al (2007) Haeme-regulated degradation of delta-aminolevulinate synthase 1 in rat liver mitochondria. J Biochem 142(4):453–458
Matsushima Y, Goto Y, Kaguni LS (2010) Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM). Proc Natl Acad Sci U S A 107(43):18410–18415
Hallberg BM, Larsson NG (2011) TFAM forces mtDNA to make a U-turn. Nat Struct Mol Biol 18(11):1179–1181
Granot Z, Kobiler O, Melamed-Book N, Eimerl S et al (2007) Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by Lon protease: the unexpected effect of proteasome inhibitors. Mol Endocrinol 21(9):2164–2177
Fukuda R, Zhang H, Kim JW, Shimoda L et al (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129(1):111–122
Tobias JW, Shrader TE, Rocap G, Varshavsky A (1991) The N-end rule in bacteria. Science 254(5036):1374–1377
Ninnis RL, Spall SK, Talbo GH, Truscott KN et al (2009) Modification of PATase by L/F-transferase generates a ClpS-dependent N-end rule substrate in Escherichia coli. EMBO J 28(12):1732–1744
Erbse A, Schmidt R, Bornemann T, Schneider-Mergener J et al (2006) ClpS is an essential component of the N-end rule pathway in Escherichia coli. Nature 439(7077):753–756
Schmidt R, Zahn R, Bukau B, Mogk A (2009) ClpS is the recognition component for Escherichia coli substrates of the N-end rule degradation pathway. Mol Microbiol 72(2):506–517
Schuenemann VJ, Kralik SM, Albrecht R, Spall SK et al (2009) Structural basis of N-end rule substrate recognition in Escherichia coli by the ClpAP adaptor protein ClpS. EMBO Rep 10(5):508–514
Dougan DA, Micevski D, Truscott KN (2012) The N-end rule pathway: From recognition by N-recognins, to destruction by AAA+ proteases. Biochim Biophys Acta 1823(1):83–91
Sriram SM, Kim BY, Kwon YT (2011) The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol 12(11):735–747
Benedetti C, Haynes CM, Yang Y, Harding HP et al (2006) Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174(1):229–239
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529
Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU (2010) Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2(12):a004390
Yoneda T, Benedetti C, Urano F, Clark SG et al (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 117(Pt 18):4055–4066
Zhao Q, Wang J, Levichkin IV, Stasinopoulos S et al (2002) A mitochondrial specific stress response in mammalian cells. EMBO J 21(17):4411–4419
Barchinger SE, Ades SE (2013) Regulated proteolysis: control of the Escherichia coli σE-dependent cell envelope stress response. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Dordrecht, pp xxx–xxx
Micevski D, Dougan DA (2013) Proteolytic regulation of stress response pathways in Escherichia coli. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Subcell Biochem 66:105–128
Molière N, Turgay K (2013) General and regulatory proteolysis in Bacillus subtilis. In: Dougan DA (ed) Regulated proteolysis in microorganisms. Springer, Subcell Biochem 66:73–103
Buchberger A, Bukau B, Sommer T (2010) Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell 40(2):238–252
Martinus RD, Garth GP, Webster TL, Cartwright P et al (1996) Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur J Biochem 240(1):98–103
Kirstein-Miles J, Morimoto RI (2010) Peptides signal mitochondrial stress. Cell Metab 11(3):177–178
Haynes CM, Ron D (2010) The mitochondrial UPR – protecting organelle protein homeostasis. J Cell Sci 123(Pt 22):3849–3855
Young L, Leonhard K, Tatsuta T, Trowsdale J et al (2001) Role of the ABC transporter Mdl1 in peptide export from mitochondria. Science 291(5511):2135–2138
Truscott KN, Bezawork-Geleta A, Dougan DA (2011) Unfolded protein responses in bacteria and mitochondria: a central role for the ClpXP machine. IUBMB Life 63(11):955–963
Shirihai OS, Gregory T, Yu C, Orkin SH et al (2000) ABC-me: a novel mitochondrial transporter induced by GATA-1 during erythroid differentiation. EMBO J 19(11):2492–2502
Horibe T, Hoogenraad NJ (2007) The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One 2(9):e835
Aldridge JE, Horibe T, Hoogenraad NJ (2007) Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2(9):e874
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Voos, W., Ward, L.A., Truscott, K.N. (2013). The Role of AAA+ Proteases in Mitochondrial Protein Biogenesis, Homeostasis and Activity Control. In: Dougan, D. (eds) Regulated Proteolysis in Microorganisms. Subcellular Biochemistry, vol 66. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5940-4_9
Download citation
DOI: https://doi.org/10.1007/978-94-007-5940-4_9
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5939-8
Online ISBN: 978-94-007-5940-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)