
Abstract
Obesity is an emerging public health concern that has numerous secondary health consequences, including heart disease, high blood pressure, diabetes mellitus, osteoarthritis, and overall reduced quality of life. Historically, obesity has been viewed as increased body fat caused by overconsumption of food, combined with the sedentary lifestyle of modern society. Simply put, energy input exceeds energy output, creating an excess in fat mass. This viewpoint largely focuses on environmental and social factors in the obesity epidemic. However, it fails to take into account a growing body of evidence from several monogenetic human obesity disorders and mutant mouse and rat obesity models that indicate a profound role for genetic factors. Although most of these monogenetic human conditions are rare, it is clear that the study of their molecular and cellular etiology will offer insights into the mechanisms that regulate appetite and satiety. The objectives of this review are to discuss how mutations in genes required for the formation or function of the cilium result in obesity in human and mouse models and how the cilium may function to regulate appetite and satiation responses.
Authors Nicolas F. Berbari and Raymond C. Pasek contributed equally to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Obesity
- Ciliopathy
- Bardet-Biedl syndrome
- Alström syndrome
- Neuronal cilia
- Leptin
- Intraflagellar transport
- Mouse models
- Hedgehog
- Melanin concentrating hormone
- Hyperphagia
- Food anticipatory behavior
6.1 Ciliopathies Associated with Obesity
The cilium is a small microtubule based cellular appendage found on most mammalian cells where it plays a crucial role as a complex sensory and signaling center (for a review on cilia signaling see Berbari et al. 2009). There is now an emerging class of genetic diseases coined ciliopathies that is associated with dysfunction of the cilium. Interestingly, the clinical features of ciliopathies range from renal cysts in humans with polycystic kidney disease (PKD1, OMIM #601313) to the cystic kidneys, skeletal anomalies, neurodevelopmental defects, heart malformations and perinatal lethality associated with Meckel-Gruber Syndrome (MKS1, OMIM # 249000) (for an in-depth review on ciliopathies see Sharma et al. 2008). This broad scope of clinical features has been attributed to both the ubiquitous nature of the cilium and the types of mutations affecting its function. PKD, for example, is associated with mutations in signaling proteins that appear to have crucial roles in renal homeostasis, while the mutations in MKS are often associated with proteins found at the base of the cilium, where they are thought to play more general roles in cilia structural integrity and the regulation of cilia protein composition. Until relatively recently much of our knowledge of cilia has come from studies in model systems focusing on the process of building and maintaining the cilium, known as Intraflagellar Transport (IFT) (for a review on IFT machinery see Pedersen and Rosenbaum 2008). While the list of ciliopathies continues to grow at an incredible pace, our current understanding of how dysfunction of the cilium leads to some of the clinically observed phenotypes remains elusive. One of the clinical features where this is most evident is the hyperphagia-associated obesity that occurs in two ciliopathies, Alström Syndrome (ALMS, OMIM #203800) and Bardet-Biedl Syndrome (BBS, OMIM #209900). The proteins affected in both of these disorders are associated with the cilium and are required for normal cilia function or formation.
Bardet-Biedl Syndrome (BBS) is a group of rare genetically heterogeneous disorders resulting in an array of seemingly unrelated symptoms and progressive degenerative defects. A triad of symptoms including obesity, retinitis pigmentosa/retinal degeneration, and polydactyly remains the hallmark for diagnosing the disease, as was the case when it was independently classified by Georges Bardet and Arthur Biedl in the early twentieth century (Bardet 1920; Biedl 1922). Subsequent analysis has revealed that hypogonadism, renal dysfunction, and mental retardation are also highly prevalent among BBS patients. Nearly half of BBS patients are completely or partially anosmic and have deficits in thermosensation (Kulaga et al. 2004; Tan et al. 2007). Also of consequence are the secondary features of BBS, including diabetes mellitus, hypertension, and heart disease, which develop likely as a result of the obesity. Less commonly, BBS patients can have situs inversus, a defect often caused by dysfunctional cilia on the embryonic gastrulation stage structure called the node (Lorda-Sanchez et al. 2000; Deffert et al. 2007). Among North American and European populations, BBS is relatively uncommon with an estimated occurrence of 1 in 120,000 live births, while in Middle Eastern populations it has been reported to be as high as 1 in 13,500 (Klein and Ammann 1969; Croft et al. 1995; Beales et al. 1997). Although it is possible to detect the condition during gestation, patients are typically diagnosed when both obesity and retinal degeneration are apparent and polydactyly or mental deficits have been observed.
Confusion in diagnosing BBS exists, as a similar condition, Laurence-Moon Syndrome, is also a genetically-inherited disease in which patients present with retinitis pigmentosa and mental disability. This observation subsequently caused Solis-Cohens and Weiss to conclude that both diseases are synonymous and thus both disorders have been referred to as Laurence-Moon-Bardet-Biedl Syndrome. However, there is debate about the synonymous classification as the patients reported by Laurence and Moon displayed paraplegia of the lower extremities that is not typically associated with BBS. Furthermore, polydactyly and obesity are not generally traits of Laurence-Moon syndrome, while they are clinical hallmarks of BBS. Given that polydactyly is not fully penetrant among BBS patients, and that the obesity experienced by most BBS patients can be attenuated or eliminated with diet and exercise, the lack of these symptoms does not necessarily mean the two disorders are not allelic variations (Beales et al. 1999; Ghadami et al. 2000).
BBS has extensive genetic heterogeneity with 16 known loci (BBS1-BBS16, Table 6.1). Mutations in BBS1-10 are responsible for approximately 70% of known BBS cases, and even the identification of BBS11-16 only added fractional amounts to that number; thus it remains almost certain that other BBS genes have yet to be found (Chen et al. 2011; Stoetzel et al. 2006; Leitch et al. 2008; Chiang et al. 2006). Conversely, the genetic basis of Laurence-Moon is currently unknown, so it remains to be seen if the symptoms of Laurence-Moon are caused by mutations in BBS genes.
The mode of BBS inheritance is also complicated. Although originally believed to be a typical recessive Mendelian disorder, analysis by Katsanis et al. demonstrated this may not always be the case (Katsanis et al. 2001). In their study, the authors reported families where both affected and unaffected individuals could carry homozygous mutant alleles of BBS2. However, the affected individuals were also heterozygous for a mutation in a second BBS gene, BBS6. This phenomenon, which the authors named ‘triallelic inheritance’, demonstrates that manifestations of BBS phenotypes sometimes rely on mutations at a second locus and thus BBS may result from the overall genetic mutational load in a patient. However, analysis of the triallelic inheritance hypothesis in several other BBS cohorts did not reveal evidence of triallelism suggesting this may be an exception (Mykytyn et al. 2003; Abu-Safieh et al. 2012; Smaoui et al. 2006; Laurier et al. 2006). Information from these cohorts suggests that BBS is an autosomal recessive inherited disease. But these data cannot rule out possible contributions from unidentified BBS alleles and it remains possible that BBS can manifest through both an autosomal recessive fashion and triallelic inheritance.
The great degree of variability in both inheritance and symptoms presented by BBS patients leads to the question of whether there exists any correlation between the genes and mutations involved and the way the disease manifests itself. For example, while obesity and renal abnormalities are frequent, the degree of mental retardation or learning disabilities varies greatly, with patients from some families showing little or no mental deficits (Riise et al. 1997). Thus, the relationship between BBS mutations and the expressivity of traits has been of great interest, but largely remains inconclusive with limited genotype-phenotype correlation.
Birth weight in BBS patients is usually normal, with obesity developing during childhood continuing into adulthood (Beales et al. 1999). This observation indicates that the obese phenotype may not be a direct consequence of defects in development, but rather due to errors in energy metabolism or appetite regulation. Evidence for this hypothesis is found in the fact that when compared to BMI matched controls, BBS patients did not possess significant differences in body fat or resting metabolic rate (RMR) (Grace et al. 2003). Although this information indicates that body fat and RMR is the same, subsequent work demonstrated that circulating leptin and triglyceride levels were significantly higher in BBS patients compared to other BMI matched obese individuals, despite the fact that glucose tolerance and insulin resistance was comparable between the two groups (Feuillan et al. 2011). Leptin has a known role in suppressing appetite, and this finding supports the possibility that BBS patients have a higher degree of leptin resistance. Recent work in animal models have strongly implicated primary cilia as being necessary for regulating appetite, and that cilia regulated signaling can be disrupted in mouse models of BBS (Davenport et al. 2007; Weatherbee et al. 2009). However, the molecular mechanism causing this disease in BBS patients remains uncertain.
The other human ciliopathy associated with obesity is Alström syndrome (ALMS), which was first classified in 1959 (for a review of ALMS see Girard and Petrovsky 2011). Human ALMS patients manifest with several symptoms including obesity, retinitis pigmentosa, and hearing loss with a tendency towards shorter stature, and a disruption in the growth hormone/Insulin-like growth factor 1 signaling axis. They also exhibit phenotypes likely related to their obesity that include diabetes mellitus and elevated leptin levels when compared to unaffected individuals (Maffei et al. 2007). ALMS is a rare autosomal recessive disorder with an occurrence at less than 1 in 100,000 and is caused by mutations in the gene ALMS1 (Collin et al. 2002; Hearn et al. 2002). To date, 81 different disease causing mutations in ALMS1 have been reported (Joy et al. 2007; Marshall et al. 2007). The exact function of ALMS1 remains unknown, but clues to its possible cellular function were uncovered when it was found that ALMS1 is widely expressed and localizes to the centrosome and the basal body of the primary cilium in cultured human function cells (Hearn et al. 2005; Knorz et al. 2010). Interestingly, dermal fibroblasts derived from an ALMS patient had normal basal body localization and primary cilia assembly suggesting that ALMS1 might be involved in ciliary related signaling pathways, but not in establishing cilia architecture itself (Hearn et al. 2005). In contrast, knockdown of Alms1 by siRNA in mouse inner medullary collecting duct (mIMCD3) cells caused a stunted cilia phenotype, and also impaired their mechanical stimuli sensing abilities. This discrepancy in phenotype could be due to the nature of the mutation, which may not have caused a complete lack of protein function.
Although BBS and ALMS are relatively rare diseases, understanding how these proteins normally regulate satiation responses will provide important insights into molecular pathways that could be manipulated to control satiation and obesity.
6.2 Ciliopathy Mouse Models of Obesity
To better understand the causes of human obesity, genetically manipulated mouse models are continuously being developed with the ultimate goal of elucidating the molecular mechanisms driving the phenotype. Surprisingly, over the past decade the primary cilium has emerged as a key factor regulating satiation responses. The Bbs and Alms1 mouse models along with mutations affecting the Intraflagellar transport 88 (Ift88) gene have established a strong link between defects in cilia mediated sensory or signaling activity and obesity (Table 6.2). In this section, we will review data derived from several of the obesity mouse models that have been associated with ciliary dysfunction and highlight the proposed function of the affected proteins.
6.2.1 Bbs Mutant Mouse Models
Seminal work in the BBS field by Nachury et al. has shown that BBS proteins 1, 2, 4, 5, 7, 8, and 9 form a ∼450 kDa complex called the BBSome (Nachury et al. 2007). The BBSome is thought to function in transport of membrane along with specific transmembrane proteins to the cilium (Jin et al. 2010). Of these BBSome genes, mouse models of Bbs1, 2, 4, and 8 have been generated and characterized. The validity of utilizing mice to model human BBS was demonstrated when a knock-in allele of the Bbs1 M390R mutation, one of the most common single human BBS disease alleles, was created and replicated many of the human symptoms of BBS, including retinal degeneration, male infertility, and obesity (Davis et al. 2007). Strikingly, these hallmark features of BBS are shared phenotypes among other mouse models of BBS such as Bbs2 and Bbs4 mutants (Mykytyn et al. 2004; Nishimura et al. 2004) (Fig. 6.1a). Neurological defects were also observed. For example, disruption of Bbs1 or Bbs4 caused cilia loss on the olfactory epithelium, and the same report demonstrated partial or total anosmia in a cohort of human BBS patients. These same studies found a common social dominance defect among Bbs2 and Bbs4 mutant mice, in which the mutants were more submissive to control mice. Although the olfactory and behavior phenotypes may not directly be related to the obesity seen in these mice, it does reflect the importance of the BBSome genes in the regulation of behavior.
As in BBS patients, obesity is not present in young Bbs mutant mice. In fact, most Bbs mutant mice are initially runted, and it has been proposed that this is possibly due to olfactory defects that make it difficult for pups to accomplish nipple searching and suckling (Eichers et al. 2006). However, mutants eventually developed hyperphagia and became obese. The obesity phenotype also correlated with hyperleptinemia in Bbs1 M390R knock-in mice. More recently, Bbs8-null mice have been reported that also have defects in olfactory function, as has been shown in Bbs1 and 4 mutant mice. When these Bbs8 mutant mice were crossed to an olfactory receptor reporter line (M72TL), severe defects in the targeting of olfactory sensory neurons became apparent, and individual axonal fibers seemed to wander, instead of terminating at a single glomerulus as in the control mice (Tadenev et al. 2011). The axonal targeting defects reported in the Bbs8 mutant mice further confirm the importance of the BBSome in proper neuronal development and signaling.
Other genes involved in human BBS that do not encode direct BBSome components have been identified. These proteins share homology to chaperones, and there are indications that these too are necessary for normal activity of satiation pathways. These genes include BBS6, 10, and 12, and they encode proteins that may be necessary to stabilize the BBSome (Seo et al. 2010). Of these three, only a mouse mutant of Bbs6 (previously referred to as Mkks for its involvement in McKusick-Kaufman syndrome) has been reported. As seen in the other BBSome mutant mice, Bbs6 mutants display age dependent retinal degeneration and exhibit hyperphagic behavior leading to obesity with elevated leptin levels (Fath et al. 2005). Furthermore, male infertility was reported due to a failure in the formation of spermatozoa flagella, similar to the findings of Bbs1, 2, and 4 mutant mice.
Not all known BBS genes fall into the category of being a BBSome complex member or having chaperone-like properties. This includes the BBS3 gene that encodes the small GTPase ARL6. Mouse Bbs3 mutants exhibit both retinal degeneration and male infertility due to loss of sperm flagella (Zhang et al. 2011). In addition, severe hydrocephalus accompanied by altered beating of ependymal cilia was found, but no loss or obvious defects in primary cilia morphology were evident. Most strikingly however, was the apparent lack of an overt obesity phenotype in the Bbs3 mutants. Likewise, leptin levels in the mutants were not statistically different than controls (Zhang et al. 2011). Although the Bbs3 mutants do not have a significant increase in body weight, they do have an increase in the amount of gonadal and retroperitoneal fat. The reason that Bbs3 mutant mice do not display an obesity phenotype is unknown. It was proposed this may be due to the early onset hydrocephalus; however, it should be noted that obesity along with hydrocephalus is seen in some of the other Bbs models. The lack of an obesity phenotype could also be related to different functions of the Bbs proteins and differential effects they have on protein trafficking. Melanin concentrating hormone receptor1 (Mchr1) is a ciliary localized G protein coupled receptor (GPCR) known to have orexigenic effects. Intriguingly, in obese models such as Bbs2 and Bbs4 mutants, Mchr1 is not present in the cilium while it does localize to in the cilia of cultured neurons from Bbs3 mutants (Zhang et al. 2011). It is also important to note that Bbs3 is neither a BBSome complex member nor a BBS chaperone protein, raising the possibility that Bbs3 has functions independent of the other BBS proteins.
In some cases, previously identified genes are now being recognized as belonging to the BBS family. For example, mutations in the E3 ubiquitin ligase TRIM32 was identified in BBS patient (hence called BBS11) through the use of homozygosity mapping with SNP arrays (Chiang et al. 2006). Trim32/Bbs11 mutant mice display muscular dystrophy and a decreased concentration of neurofilaments, as well as a reduction in myelinated motoraxon diameters (Kudryashova et al. 2009, 2011). A small increase in body weight was found in the Bbs11 mice when compared to controls, but this was only a 10% difference at 8 weeks of age (Kudryashova et al. 2009). Much like Bbs3, Bbs11 is neither a BBSome complex member nor a BBS chaperon protein, and thus may also have independent or unique functions from the rest of the BBSome. However, it remains possible that the muscular dystrophy and reduction in motor axon myelination are precluding the emergence of an obesity phenotype. In addition, mutations in BBS11 can cause two distinct clinical disorders; BBS and limb-girdle muscular dystrophy type 2H (LGMD2H). BBS phenotypes were associated with N-terminal mutations (P130S) while LGMD2H appears to be caused by mutations in the C-terminal region (R394H, D487N, D588del, or T520TfsX13) that do not disrupt its ability to function in ubiquitination.
Studies of more recently identified BBS genes include MKS1/BBS13 and CEP290/BBS14. However, current reports utilize Bbs13 and Bbs14 mutant mice that are either embryonically lethal and/or not true genetic nulls, thus making the potential role of Bbs13 and 14 in obesity and appetite regulation ambiguous (Tadenev et al. 2011; Weatherbee et al. 2009; Lancaster et al. 2011). Regardless, the fact that not all Bbs mutant mice have the same phenotypes indicates complexity and diversity in the functions of the different BBS genes along with differential effects of the specific mutations on gene function.
6.2.2 Mouse Model of Alström Syndrome
The other ciliopathy with obesity as a symptom is Alström syndrome. In contrast to BBS, Alström syndrome appears to be caused by mutations in a single gene ALMS1. The ALMS1 protein localizes to the basal body of ciliated cells, but the function of the protein is not certain (Hearn et al. 2005; Collin et al. 2005). Mouse models corresponding to Alström syndrome have also been reported and include a gene-trapped allele (Alms1 −/−) and a spontaneous mutant (fat aussie, foz) (Arsov et al. 2006; Collin et al. 2005). Mice lacking functional Alms1 are born at a normal weight much like their human counterparts. However, hyperphagic behavior and obesity ensue that is accompanied by hyperinsulinemia and type II diabetes. The Alms1 −/− mutant mice also have enlarged livers with the accumulation of lipid deposits, and the pancreas is hyperplastic. In addition to obesity, mice lacking Alms1 display male infertility, as well as retinal and cochlear defects, all of which are reminiscent of cilia associated defects in human patients.
6.2.3 Obesity in the Intraflagellar Transport Mutants
Cilia formation and maintenance, and possibly its signaling activity, is dependent on the intraflagellar transport (IFT) system to mediate bidirectional transport of proteins between the base and tip of the cilium. Null alleles of the Ift88 gene (originally referred to as Tg737 in mouse) caused early embryonically lethality and even hypomorphic alleles caused death prior to adulthood with systemic effects (Moyer et al. 1994; Murcia et al. 2000; Lehman et al. 2008). The necessity of cilia for normal mammalian development has made analyzing possible roles of the cilium in satiation and obesity difficult. This problem was circumvented with the creation of conditional alleles of Ift88 and the IFT motor Kif3a (Marszalek et al. 1999; Haycraft et al. 2007). Using a tamoxifen-inducible cre recombinase expressed from the actin promoter (CAGG-creERTM) (Davenport et al. 2007), cilia loss could be induced systemically in adult mice. This was found to cause hyperphagia within 3 weeks of inducing cilia loss and subsequently caused obesity. Furthermore, the obesity phenotype was prevented by maintaining adult conditional cilia mutant mice on a restricted diet, wherein they were provided the same daily amount of food as normal controls consumed. This observation indicates that the obesity phenotype is caused by the lack of a satiation response that leads to the overconsumption and not a general alteration in metabolic or locomotor activity.
The change in feeding behavior observed in the Ift88 and Kif3a conditional mutant mice led to the possibility that cilia on neurons may be responsible for the obesity phenotype. To test this hypothesis, conditional Ift88 and Kif3a mutant mice were crossed to synapsin1-cre mice to cause loss of cilia exclusively in neurons (Zhu et al. 2001). As with the systemically induced cilia mutants, neuronal specific cilia mutant mice became morbidly obese and strongly implicated a previously unappreciated role for neuronal cilia in regulating appetite (Fig. 6.1b).

Obese cilia mutant mouse models. (a) An obese Bbs4 −/− mutant (right) next to his wild-type littermate (left). (b) An obese conditional Ift88 mutant that has lost cilia throughout the central nervous system as the result of synapsin1-cre expression (right) next to a wild-type littermate (left)
The hypothalamus is a critically important signaling center of the brain known to regulate appetite. This action is done in large part by neurons that express either pro-opiomelanocortin (POMC) or agouti-related protein (AgRP) that release signaling factors ultimately suppressing or enhancing appetite, respectively (for a review see Mountjoy 2010). Importantly, hypothalamic neurons each possess a single primary cilium, although the function of the cilium on these neurons is largely unexplored. To address the role of neuronal cilia and appetite, Ift88 and Kif3a conditional mutants were crossed to POMC-cre or AgRP-cre expressing mice, to conditionally ablate cilia on POMC or AgRP expressing neurons, respectively (Xu et al. 2005b). By 6 weeks of age, both male and female POMC cilia mutant mice weighed significantly more than control mice, and continued to become morbidly obese into adulthood. This was not evident in the mice lacking cilia on AgRP neurons (Berbari and Yoder, unpublished data). Another observation that was reported in the POMC cilia mutant mice was an increase in the levels of leptin, fasting serum glucose, and insulin (Davenport et al. 2007). This was observed only in the obese state and not in mice kept lean by pair-feeding, indicating that these elevated levels were a secondary consequence of the obesity. This report was significant for providing some of the initial evidence indicating the importance of neuronal cilia in regulating obesity.
6.2.4 Other Obesity Mouse Models Associated with Ciliary Proteins
In addition to the Bbs, Alms1, and Ift88 mouse models there are several other mutant mouse strains supporting a connection between cilia and obesity. One prime example is a mutation in the type III adenylyl cyclase (ACIII). ACIII localizes to the primary cilia throughout the adult rodent brain (Bishop et al. 2007) and loss of ACIII causes anosmia and obesity by 3 months of age. Interestingly, even when ACIII mutants do not weigh significantly more than wildtype siblings, they have an increased level of serum leptin (Wang et al. 2009). It is interesting to note that a recent Genome Wide Association Study revealed there is a SNP near ACIII that is associated with obesity in humans (Hebebrand et al. 2010; Nordman et al. 2008).
Another example may be the tubby mouse that was first identified at the Jackson Laboratory as a spontaneous mutant causing a maturity-onset obesity phenotype (Coleman and Eicher 1990) and subsequently regenerated by gene targeting (Ashrafi et al. 2003). The tubby mutants have progressive loss of hearing and vision (Ohlemiller et al. 1995), similarities that are also shared with the Bbs and Alms1 mutant mice. The spontaneous tubby mouse possesses a single base pair mutation within a splice site of the gene Tub (named after the mutant mouse) resulting in the expression of an aberrant transcript. The functions of the Tub protein remain ambiguous, but it is found to be highly expressed in portions in the brain, including the arcuate nucleus of the hypothalamus (Kleyn et al. 1996). The Tub protein is dispensable in assembly of the cilia, and no defects in the cilia assembly process of intraflagellar transport (IFT) have been reported. Intriguingly, despite the fact that Tub has yet to be reported in mammalian primary cilia, a physical association between Tub and the IFT complex has been reported in an immortalized human cell line (Mukhopadhyay et al. 2010). Further evidence for a ciliary role of Tub can be found with the C. elegans homolog tub-1 that undergoes transport along the ciliary axoneme (Mukhopadhyay et al. 2005). Like the tubby mouse, C. elegans with a deletion of tub-1 show an increase in fat content, suggesting an evolutionarily conserved role of the gene in regulating fat storage. Other proteins in the tubby family of proteins have also been implicated as having ciliary roles. For example, tubby-like protein 3 (Tulp3) localizes to the primary cilia during mouse development, and is necessary for proper Shh signaling but its association with obesity has not yet been determined (Norman et al. 2009).
6.3 Potential Molecular Mechanisms of Ciliopathy Associated Obesity
The hyperphagia induced obesity is one of the more intriguing phenotypes of ciliopathies that remains to be fully explained. There are indeed several possibilities described in the literature as to how loss of the cilia could alter states of satiety and appetite. Here we review a few of the candidate molecular pathways that may play roles in obesity associated with cilia dysfunction. These possibilities include primary deficits in leptin signaling, altered G-protein coupled receptor (GPCR) signaling, and abnormal regulation of mTor and hedgehog signaling pathways (Table 6.3).
6.3.1 Cilia and the Leptin Signaling Axis
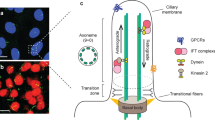
While the conditional allele of Ift88 implicated a role for neuronal cilia in satiety and more specifically a role for POMC neuronal cilia, this study did not specify a molecular framework for the underlying hyperphagia phenotype (Davenport et al. 2007). More recent data has aimed to accomplish this by showing that Bbs1, a component of the BBSome, directly binds to the leptin receptor and that BBS proteins may have a role in leptin receptor trafficking (Fig. 6.2a) (Seo et al. 2009). The initial identification of the leptin gene encoding a small protein hormone in the spontaneous obese mouse mutant ob/ob was the source of much excitement (Zhang et al. 1994). Importantly, leptin suppresses feeding activity and it is secreted into serum at levels proportionate to the amount of adipose tissue, the hormone’s primary source (Considine et al. 1996). Interestingly, these recent studies in Bbs2, Bbs4, and Bbs6 mutant mice also revealed that they are hyper-leptinemic and importantly, they fail to reduce food intake in response to IP or ICV injection of leptin (Rahmouni et al. 2008). Thus, defects in leptin signaling were thought to contribute directly to the obesity phenotype in BBS.

Potential ciliary signaling pathways necessary for appetite regulation. (a) Depicts the leptin receptor interacting with Bbs1 of the BBSome near the base of the cilia where it has been proposed to be available for leptin activation and subsequent phospho-Stat3 induction and translocation to the nucleus. (b) A depiction of cilia specific GPCRs such as Mchr1, Sstr3, Drd1, and 5HT6, and their potential effector ACIII and G proteins such as Gs/q/i,o. (c) The Hedgehog signaling pathway, with patched repressing smoothened translocation into the cilium until ligand stimulation, upon which Gli transcription factors are processed from the inhibitor to the activator forms followed by subsequent translocation to the nucleus
The excitement surrounding leptin’s initial discovery was attenuated when it was determined that nearly all obese mice and humans have markedly elevated levels of circulating leptin, yet do not have normal leptin-mediated repression of appetite (Considine et al. 1996; Maffei et al. 1995). This barrier phenomenon is known as leptin resistance, the mechanism of which remains an active area of research. Thus, in obesity research, one challenge is determining whether leptin resistance is a primary cause or a consequence of the obesity. One approach used to overcome this situation is to decrease the amount of adipose tissue, and consequently the levels of circulating leptin, through caloric restriction. Interestingly, when this was performed on the BBS mutant mouse models to maintain body weight and leptin levels as seen in controls, they were still resistant to leptin, suggesting leptin signaling defects are a primary cause of the phenotype (Seo et al. 2009). However, the study did not take into account a phenomenon called food anticipatory behavior wherein the mice alter their meal structure in response to the calorie restriction such that they consume nearly all of their calories within the first few hours of food access (for a review on anticipatory feeding behavior and methods see Mistlberger 2009). Interestingly, during this entrained period the mice consume nearly the same amount of food as they were given during calorie restriction, even when they are given ad libitum amounts of food. This entrained behavior persists for over a week and during this period the mice do not reduce food intake in response to leptin and thus appear leptin resistant (Berbari and Yoder unpublished). This feeding behavior resulting from the anticipation of food is in large part thought to be the result of a feeding clock, somewhat analogous to but distinct from the circadian clock (Mistlberger 2009). It remains to be seen whether either Bbs or Ift conditional mice would respond to leptin when both body weight and anticipatory feeding behavior are experimentally controlled. This would require testing animals for leptin sensitivity after deterioration of the food anticipatory behavior.
The BBS studies utilized congenital mutants and reported a loss of approximately 20% of the leptin responsive pro-opiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamus (Seo et al. 2009). Thus, the authors propose that improper leptin receptor trafficking in POMC neurons of the arcuate nucleus leads to their inability to sense leptin and thus mediate its anorectic effects. While several possibilities exist for the loss of 20% of POMC neurons in Bbs mice, it is of note that alteration in the Foxo1/Insulin signaling pathway have resulted in similar changes in adult POMC neuronal populations (Plum et al. 2012). Loss of POMC neurons in Bbs models could be the result of neurodevelopmental changes or possibly maintenance of POMC neuron population through altered adult neurogenesis that lead to the hyperphagic phenotype. Similarly, the obesity phenotype observed in the hyperphagic Ift88 conditional POMC-cre model could be a hypothalamic developmental phenotype. However, the rapid onset of obesity upon ubiquitous loss of cilia induced by the actin promoter driving cre line (CaGG-CreER) in adult mice suggests that cilia play a direct role in appetite and satiety. What is needed is an investigation using inducible cilia mutants in POMC neurons or other neuronal populations implicated in feeding behavior in order to elucidate the role of primary cilia signaling in appetite and satiety.
6.3.2 Cilia and Melanin-Concentrating Hormone Pathway
While both genetic models and biochemical approaches have informed our current understanding of BBS the precise mechanism behind BBS-associated obesity is unknown. Many of the BBS proteins form a protein complex (BBSome) that is involved in proper cilia membrane formation (Nachury et al. 2007). Indeed there is evidence that the BBSome may be a membrane vesicle coat complex that is critical for establishing and maintaining the cilia membrane’s signaling capabilities by directing specific receptors to this compartment (Jin et al. 2010). Furthermore, Bbs mutant mouse models appear to improperly localize several cilia-specific GPCRs, and most relevant with regard to obesity is the melanin-concentrating hormone receptor 1 (Mchr1) (Berbari et al. 2008). Mchr1 couples through Gαi to reduce cAMP and decreases the frequency of spontaneous action potentials in hypothalamus (Gao and van den Pol 2001, 2002). Mch injections induce feeding behavior while Mchr1 mutant mice are resistant to diet induced obesity (Gomori et al. 2003; Chen et al. 2002). Intriguingly, Mchr1 fails to localize normally in neuronal cilia of Bbs2, Bbs3, and Bbs4 mutant mice (Berbari et al. 2008; Zhang et al. 2011). Thus in both Ift88 and Bbs obese mutants, Mchr1 fails to be properly localized creating a defect in Mchr1 signaling, possibly leading to the hyperphagic behavior in these models. Both pharmacological and genetic agonism of the Mchr1 pathway are associated with hyperphagia while antagonism is associated with anorectic behavior, as such antagonism of this receptor has been of interest to the pharmaceutical industry (Qu et al. 1996; Borowsky et al. 2002; Ludwig et al. 2001; Shimada et al. 1998; Chen et al. 2002). However, assuming that the obese phenotype behind both Bbs and Ift models is driven by a similar molecular pathway, then one would have to propose that in the absence of the cilium or the ability to reach the cilium that the Mchr1 pathway is ectopically activated or not efficiently desensitized after activation.
While there is circumstantial evidence for Mchr1 cilia mis-localization driving hyperphagia in cilia mutant mouse models, it is interesting to note that there is an emerging list of GPCRs that preferentially localize to neuronal cilia in different regions of the brain (Fig. 6.2b). Some of these neuronal cilia specific GPCRs include somatostatin receptor 3 (Sstr3), serotonin receptor 6 (5HT6), and dopamine receptor 1(Drd1) (Handel et al. 1999; Schulz et al. 2000; Hamon et al. 1999; Brailov et al. 2000; Marley and von Zastrow 2010; Domire et al. 2011). While the significance of localizing these receptors within the ciliary compartment remain unknown, it is enticing to speculate that perhaps they may play a role in appetite and satiety, especially when one considers that the somatostatin, serotonin, and dopaminergic systems have all been implicated in either reward or feeding behaviors directly (Vijayan and McCann 1977; Aponte et al. 1984; Pollock and Rowland 1981; Salamone et al. 1990).
6.3.3 Cilia and the Mammalian Target of Rapamycin (mTOR) Pathway
Another pathway that may be involved in neuronal cilia regulation of satiation is the mammalian target of rapamycin (mTOR) pathway. mTOR signaling is complex and involves many factors (for an in depth review of mTOR and disease see Dazert and Hall 2011). mTOR is a serine/threonine protein kinase which as its name implies can be inhibited by the antifungal rapamycin. It has been established as a regulator/coordinator of cellular metabolic activity that responds to both the energy and stress levels experienced by the cell. It carries out these regulatory roles by participating in two protein complexes, the rapamycin-sensitive mTOR Complex 1 and the rapamycin-insensitive mTORC2. In general mTORC1 regulates translational control and mTORC2 is involved in cytoskeleton organization. While the functions of mTOR and its interactors have been determined in considerable detail at the genetic and cellular levels, the effects of mTOR signaling on the organismal level continue to emerge. Interestingly, there are several reports associating the cilium or its signaling proteins with overactivation of mTOR activity or in changes in the cytoskeleton and cell size (Sharma et al. 2011; Bell et al. 2011; Boehlke et al. 2010). In addition, rapamycin is able to partially rescue renal cystic disease in mouse models of PKD, further supporting a connection between cilia and mTOR (Shillingford et al. 2006, 2010). While the in vivo relevance of cilia and mTOR signaling with regards to the obesity phenotype in cilia mutants remains to be determined, it is of note that mTOR signaling within the hypothalamus has been associated with obesity in other animal models (Cota et al. 2006). It will be interesting to determine if mTOR signaling activity within adult neurons is regulated through neuronal cilia and influences feeding behavior.
6.3.4 Hedgehog Signaling and the Cilium
The final pathway that we will discuss with regard to neuronal cilia and obesity is the hedgehog (Hh) pathway. Hh and its role in cilia and neuronal development is reviewed in Chap. 2 of this work by Mariani and Caspary. Several groups have demonstrated that canonical hedgehog signaling in mammalian cells utilizes the ciliary compartment. This is best demonstrated by the transient localization of several of the pathway components to the cilium and altered pathway activity when the cilium has been disrupted (reviewed in detail by Goetz and Anderson 2010). With regard to hedgehog signaling and neuronal cilia, there is a consensus emerging that primary cilia within the adult central nervous system sense hedgehog ligand and mediate the process of adult neurogenesis (Breunig et al. 2008; Han et al. 2008). Furthermore when neuronal cilia-mediated hedgehog signaling is altered in a gain of function fashion it can result in medulloblastoma and when it is disrupted in a loss of function fashion in the developing brain it is associated with a range of neurodevelopmental phenotypes (Chizhikov et al. 2007; Han et al. 2009).
If altered hedgehog signaling and cilia mutations have such profound effects on the adult and developing nervous system, how may they account for the hyperphagia associated obesity in adults? In both Ift conditional and Bbs models the possibility that altered hedgehog signaling in the developing hypothalamus can lead to obesity in adulthood has yet to be thoroughly investigated. For example, POMC-cre conditional Ift88 mutant models appear normal other than the onset of hyperphagia and obesity, but the potential for a developmental phenotype remains. This becomes important when the expression pattern of POMC is taken into account. POMC is known to be expressed in places outside of the arcuate nucleus, of the hypothalamus such as the nucleus tract solitarius of the hindbrain, and the anterior and intermediate lobe of the pituitary in neonatal animals (Xu et al. 2005a). This becomes particularly important when the crucial role of hedgehog not only in the developing neural tube but also in the developing hypothalamus is taken into account (Szabo et al. 2009; Alvarez-Bolado et al. 2012). To address these potentials both Ift and Bbs conditional models need to be tested with inducible POMC-cre deletion. These experiments would also be useful in assessing whether different molecular mechanisms may be involved in driving obesity in Bbs and Ift mouse models.
The possibility remains that hedgehog signaling, which is required for adult neurogenesis is disrupted, thus contributing to hyperphagia. There are also reports of adult neurogenesis within the hypothalamus (Kokoeva et al. 2005; Xu et al. 2005c; Pierce and Xu 2010; Lee et al. 2012). Perhaps this process is compromised in CAGG-creER;Ift88 conditional mutants. However, hyperphagic behavior is observed within 3 weeks of cilia loss in these mice and thus it may not have sufficient time to be a result of altered adult neurogenesis.
Finally a third potential for a non-canonical form of hedgehog signaling exists in the adult hypothalamus that requires neuronal cilia. The relevance of a non-canonical Hh pathway emerges when the expression pattern of certain pathway components in the adult brain is analyzed. For example, the Hh receptor, Patched, is expressed in regions of the brain that do not co-express the Hh effector Smoothened (Traiffort et al. 1998, 1999). This incongruence in pathway component expression pattern is especially true with regard to the hypothalamus (for a review of hedgehog in the adult brain see Traiffort et al. 2010). Furthermore it has been shown that hedgehog can directly alter neuronal activity (Bezard et al. 2003; Pascual et al. 2005). In developing spinal neurons, Hh stimulation causes a transient increase in Ca2+ activity that was dependent on Smo and Gαi (Belgacem and Borodinsky 2011). Since hedgehog pathway components such as Patched are expressed in the adult hypothalamus, it is feasible that cilia may alter satiation responses through regulation of neuronal firing activity (Traiffort et al. 1998, 1999). Exploring whether loss of cilia alters this increase in Ca2+ in response to Hh in POMC neurons could prove to be a very fruitful avenue of investigation to connect cilia dysfunction to abnormal satiation.
6.4 Non-Mammalian Ciliopathy Models of BBS
Although more distantly related to human beings than mice, non-mammalian models have proven to be invaluable to the study of the role of human cilia and their relation to disease. This is particularly evident in the study of the assembly and maintenance of the cilium through IFT, and how disruption of this event can lead to certain phenotypes. This process, referred to as intraflagellar transport (IFT), was largely characterized biochemically using the small green algae, Chlamydomonas, and genetically using C. elegans (for an in depth review see Pedersen and Rosenbaum 2008). In this section we focus solely on the genes and proteins known to be associated with the obesity phenotype observed in ciliopathies, as such it will largely focus on the functional roles of the BBS genes in both Chlamydomonas reinhardtii and Caenorhabditis elegans, two of the most well studied non-mammalian organisms in regards to cilia/flagella biology
While Chlamydomonas has served as good model for biochemical purification of flagellar and IFT components it has also proven useful for comparative genomics studies in discovering new ciliopathy genes, such as BBS5 (Kulaga et al. 2004). Through the use of this simple model, elegant studies have begun to shed new light on how the BBS proteins may function as modulators of ciliary signaling and even serve as structural components of the transition zone (Lechtreck et al. 2009; Craige et al. 2010).
Much of what we know about the molecular motors that mediate cilia formation and maintenance has come from studies visualizing IFT movement in the cilia of C. elegans. In C. elegans cilia of the sensory neurons it has been demonstrated that both BBS7 and BBS8 serve as adaptors to the IFT complexes and their cargoes (Blacque et al. 2004), however, whether they play similar roles in mammalian systems has not been determined. Interestingly, it has been shown that C. elegans ciliary morphology can change dependent on cilia-mediated signaling and that the phenotypes of bbs mutant worms can be ameliorated by altering the downstream second messengers (Tan et al. 2007; Mukhopadhyay et al. 2008; Mok et al. 2011). Recent work has also suggested that altered neuroendocrine signaling and exocytosis of factors such as insulin drives many of the phenotypes observed in bbs mutant worms (Lee et al. 2011). Interestingly, another study points to more general roles for bbs proteins in cilia membrane homeostasis (Kaplan et al. 2012). Although the invertebrates lack many of the organ systems present in mammals, it is clear that both C. elegans and Chlamydomonas models offer advantages in both cost, time and in some instances genetic tractability. These models will continue to provide insights into the fundamental processes that are mediated by the ciliopathy proteins and the cilium and thus further serve to inform our understanding of complex phenotypes such as feeding behavior and the regulation of appetite and satiety.
6.5 Conclusion
In summary, remarkable progress has been made in the past 20 years demonstrating the clinic importance of the cilium in may tissues and developmental processes. Despite this progress, there remain several key questions that must be addressed before we can understand the molecular and cellular mechanisms responsible. Hopefully, as research on the rare ciliopathies advances we will gain an understanding of fundamental processes such as satiety and appetite that we can then apply to direct therapeutic strategies for an exceedingly common clinical feature such as obesity.
References
Abu-Safieh L, Al-Anazi S, Al-Abdi L, Hashem M, Alkuraya H, Alamr M, Sirelkhatim MO, Al-Hassnan Z, Alkuraya B, Mohamed JY, Al-Salem A, Alrashed M, Faqeih E, Softah A, Al-Hashem A, Wali S, Rahbeeni Z, Alsayed M, Khan AO, Al-Gazali L, Taschner PE, Al-Hazzaa S, Alkuraya FS (2012) In search of triallelism in Bardet-Biedl syndrome. Eur J Hum Genet 20:420–427
Alvarez-Bolado G, Paul FA, Blaess S (2012) Sonic hedgehog lineage in the mouse hypothalamus: from progenitor domains to hypothalamic regions. Neural Dev 7:4
Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N (2003) Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 425:628–633
Aponte G, Leung P, Gross D, Yamada T (1984) Effects of somatostatin on food intake in rats. Life Sci 35:741–746
Arsov T, Silva DG, O’Bryan MK, Sainsbury A, Lee NJ, Kennedy C, Manji SS, Nelms K, Liu C, Vinuesa CG, de Kretser DM, Goodnow CC, Petrovsky N (2006) Fat aussie – a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol 20:1610–1622
Ashrafi K, Chang FY, Watts JL, Fraser AG, Kamath RS, Ahringer J, Ruvkun G (2003) Genome-wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421:268–272
Badano JL, Ansley SJ, Leitch CC, Lewis RA, Lupski JR, Katsanis N (2003) Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet 72:650–658
Bardet G (1920) Sur un syndrome d’obesite’ congenitale avec polydactylie et retinite pigmentaire (contribution a l’etude des formes cliniques de l’obesite hypophysaire). University of Paris, Paris
Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA (1997) Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families. J Med Genet 34:92–98
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA (1999) New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 36:437–446
Belgacem YH, Borodinsky LN (2011) Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci USA 108:4482–4487
Bell PD, Fitzgibbon W, Sas K, Stenbit AE, Amria M, Houston A, Reichert R, Gilley S, Siegal GP, Bissler J, Bilgen M, Chou PC, Guay-Woodford L, Yoder B, Haycraft CJ, Siroky B (2011) Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J Am Soc Nephrol 22:839–848
Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K (2008) Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci USA 105:4242–4246
Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK (2009) The primary cilium as a complex signaling center. Curr Biol 19:R526–R535
Bezard E, Baufreton J, Owens G, Crossman AR, Dudek H, Taupignon A, Brotchie JM (2003) Sonic hedgehog is a neuromodulator in the adult subthalamic nucleus. FASEB J 17:2337–2338
Biedl A (1922) Geschwisterpaar mit adipose-genitaler Dystrophie. Dtsch Med Wochenschr 48:1630
Bishop GA, Berbari NF, Lewis J, Mykytyn K (2007) Type III adenylyl cyclase localizes to primary cilia throughout the adult mouse brain. J Comp Neurol 505:562–571
Blacque OE, Reardon MJ, Li C, McCarthy J, Mahjoub MR, Ansley SJ, Badano JL, Mah AK, Beales PL, Davidson WS, Johnsen RC, Audeh M, Plasterk RH, Baillie DL, Katsanis N, Quarmby LM, Wicks SR, Leroux MR (2004) Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev 18:1630–1642
Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Godel M, Muller K, Herbst M, Hornung M, Doerken M, Kottgen M, Nitschke R, Igarashi P, Walz G, Kuehn EW (2010) Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol 12:1115–1122
Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C (2002) Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med 8:825–830
Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Verge D (2000) Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res 872:271–275
Breunig JJ, Sarkisian MR, Arellano JI, Morozov YM, Ayoub AE, Sojitra S, Wang B, Flavell RA, Rakic P, Town T (2008) Primary cilia regulate hippocampal neurogenesis by mediating sonic hedgehog signaling. Proc Natl Acad Sci USA 105:13127–13132
Chen Y, Hu C, Hsu CK, Zhang Q, Bi C, Asnicar M, Hsiung HM, Fox N, Slieker LJ, Yang DD, Heiman ML, Shi Y (2002) Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology 143:2469–2477
Chen J, Smaoui N, Hammer MB, Jiao X, Riazuddin SA, Harper S, Katsanis N, Riazuddin S, Chaabouni H, Berson EL, Hejtmancik JF (2011) Molecular analysis of Bardet-Biedl syndrome families: report of 21 novel mutations in 10 genes. Invest Ophthalmol Vis Sci 52:5317–5324
Chiang AP, Nishimura D, Searby C, Elbedour K, Carmi R, Ferguson AL, Secrist J, Braun T, Casavant T, Stone EM, Sheffield VC (2004) Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am J Hum Genet 75:475–484
Chiang AP, Beck JS, Yen HJ, Tayeh MK, Scheetz TE, Swiderski RE, Nishimura DY, Braun TA, Kim KY, Huang J, Elbedour K, Carmi R, Slusarski DC, Casavant TL, Stone EM, Sheffield VC (2006) Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci USA 103:6287–6292
Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ (2007) Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci 27:9780–9789
Coleman DL, Eicher EM (1990) Fat (fat) and tubby (tub): two autosomal recessive mutations causing obesity syndromes in the mouse. J Hered 81:424–427
Collin GB, Marshall JD, Ikeda A, So WV, Russell-Eggitt I, Maffei P, Beck S, Boerkoel CF, Sicolo N, Martin M, Nishina PM, Naggert JK (2002) Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet 31:74–78
Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, Murray SA, Zheng QY, Smith RS, Nishina PM, Naggert JK (2005) Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet 14:2323–2333
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL et al (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334:292–295
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ (2006) Hypothalamic mTOR signaling regulates food intake. Science 312:927–930
Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB (2010) CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol 190:927–940
Croft JB, Morrell D, Chase CL, Swift M (1995) Obesity in heterozygous carriers of the gene for the Bardet-Biedl syndrome. Am J Med Genet 55:12–15
Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17:1586–1594
Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, Philp AR, Searby CC, Andrews MP, Thompson S, Berry CJ, Thedens DR, Yang B, Weiss RM, Cassell MD, Stone EM, Sheffield VC (2007) A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci USA 104:19422–19427
Dazert E, Hall MN (2011) mTOR signaling in disease. Curr Opin Cell Biol 23:744–755
Deffert C, Niel F, Mochel F, Barrey C, Romana C, Souied E, Stoetzel C, Goossens M, Dollfus H, Verloes A, Girodon E, Gerard-Blanluet M (2007) Recurrent insertional polydactyly and situs inversus in a Bardet-Biedl syndrome family. Am J Med Genet A 143:208–213
Domire JS, Green JA, Lee KG, Johnson AD, Askwith CC, Mykytyn K (2011) Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci 68:2951–2960
Eichers ER, Abd-El-Barr MM, Paylor R, Lewis RA, Bi W, Lin X, Meehan TP, Stockton DW, Wu SM, Lindsay E, Justice MJ, Beales PL, Katsanis N, Lupski JR (2006) Phenotypic characterization of Bbs4 null mice reveals age-dependent penetrance and variable expressivity. Hum Genet 120:211–226
Fath MA, Mullins RF, Searby C, Nishimura DY, Wei J, Rahmouni K, Davis RE, Tayeh MK, Andrews M, Yang B, Sigmund CD, Stone EM, Sheffield VC (2005) Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet 14:1109–1118
Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, Zheng YC, Caruso RC, Brooks BP, Johnston JJ, Yanovski JA, Biesecker LG (2011) Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance. J Clin Endocrinol Metab 96:E528–E535
Gao XB, van den Pol AN (2001) Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol 533:237–252
Gao XB, van den Pol AN (2002) Melanin-concentrating hormone depresses L-, N-, and P/Q-type voltage-dependent calcium channels in rat lateral hypothalamic neurons. J Physiol 542:273–286
Ghadami M, Tomita HA, Najafi MT, Damavandi E, Farahvash MS, Yamada K, Majidzadeh AK, Niikawa N (2000) Bardet-Biedl syndrome type 3 in an Iranian family: clinical study and confirmation of disease localization. Am J Med Genet 94:433–437
Girard D, Petrovsky N (2011) Alstrom syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol 7:77–88
Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11:331–344
Gomori A, Ishihara A, Ito M, Mashiko S, Matsushita H, Yumoto M, Ito M, Tanaka T, Tokita S, Moriya M, Iwaasa H, Kanatani A (2003) Chronic intracerebroventricular infusion of MCH causes obesity in mice. Melanin-concentrating hormone. Am j Physiol Endocrinol Metab 284:E583–E588
Grace C, Beales P, Summerbell C, Jebb SA, Wright A, Parker D, Kopelman P (2003) Energy metabolism in Bardet-Biedl syndrome. Int J Obes Relat Metab Disord 27:1319–1324
Hamon M, Doucet E, Lefevre K, Miquel MC, Lanfumey L, Insausti R, Frechilla D, Del Rio J, Verge D (1999) Antibodies and antisense oligonucleotide for probing the distribution and putative functions of central 5-HT6 receptors. Neuropsychopharmacology 21:68S–76S
Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A (2008) Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 11:277–284
Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15:1062–1065
Handel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, Hollt V (1999) Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 89:909–926
Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK (2007) Intraflagellar transport is essential for endochondral bone formation. Development 134:307–316
Hearn T, Renforth GL, Spalluto C, Hanley NA, Piper K, Brickwood S, White C, Connolly V, Taylor JF, Russell-Eggitt I, Bonneau D, Walker M, Wilson DI (2002) Mutation of ALMS1, a large gene with a tandem repeat encoding 47 amino acids, causes Alstrom syndrome. Nat Genet 31:79–83
Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, Wilson DI (2005) Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes 54:1581–1587
Hebebrand J, Volckmar AL, Knoll N, Hinney A (2010) Chipping away the ‘missing heritability’: GIANT steps forward in the molecular elucidation of obesity – but still lots to go. Obes Facts 3:294–303
Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV (2010) The conserved Bardet-Biedl syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141:1208–1219
Joy T, Cao H, Black G, Malik R, Charlton-Menys V, Hegele RA, Durrington PN (2007) Alstrom syndrome (OMIM 203800): a case report and literature review. Orphanet J Rare Dis 2:49
Kaplan OI, Doroquez DB, Cevik S, Bowie RV, Clarke L, Sanders AA, Kida K, Rappoport JZ, Sengupta P, Blacque OE (2012) Endocytosis genes facilitate protein and membrane transport in C. elegans sensory cilia. Curr Biol 22:451–460
Katsanis N, Ansley SJ, Badano JL, Eichers ER, Lewis RA, Hoskins BE, Scambler PJ, Davidson WS, Beales PL, Lupski JR (2001) Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 293:2256–2259
Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, Wallingford JB (2010) Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science 329:1337–1340
Klein D, Ammann F (1969) The syndrome of Laurence-Moon-Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci 9:479–513
Kleyn PW, Fan W, Kovats SG, Lee JJ, Pulido JC, Wu Y, Berkemeier LR, Misumi DJ, Holmgren L, Charlat O, Woolf EA, Tayber O, Brody T, Shu P, Hawkins F, Kennedy B, Baldini L, Ebeling C, Alperin GD, Deeds J, Lakey ND, Culpepper J, Chen H, Glucksmann-Kuis MA, Carlson GA, Duyk GM, Moore KJ (1996) Identification and characterization of the mouse obesity gene tubby: a member of a novel gene family. Cell 85:281–290
Knorz VJ, Spalluto C, Lessard M, Purvis TL, Adigun FF, Collin GB, Hanley NA, Wilson DI, Hearn T (2010) Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol Biol Cell 21:3617–3629
Kokoeva MV, Yin H, Flier JS (2005) Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science 310:679–683
Kudryashova E, Wu J, Havton LA, Spencer MJ (2009) Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum Mol Genet 18:1353–1367
Kudryashova E, Struyk A, Mokhonova E, Cannon SC, Spencer MJ (2011) The common missense mutation D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in knock-in mice resulting in a Trim32-null phenotype. Hum Mol Genet 20:3925–3932
Kulaga HM, Leitch CC, Eichers ER, Badano JL, Lesemann A, Hoskins BE, Lupski JR, Beales PL, Reed RR, Katsanis N (2004) Loss of BBS proteins causes anosmia in humans and defects in olfactory cilia structure and function in the mouse. Nat Genet 36:994–998
Lancaster MA, Gopal DJ, Kim J, Saleem SN, Silhavy JL, Louie CM, Thacker BE, Williams Y, Zaki MS, Gleeson JG (2011) Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat Med 17:726–731
Laurier V, Stoetzel C, Muller J, Thibault C, Corbani S, Jalkh N, Salem N, Chouery E, Poch O, Licaire S, Danse JM, Amati-Bonneau P, Bonneau D, Megarbane A, Mandel JL, Dollfus H (2006) Pitfalls of homozygosity mapping: an extended consanguineous Bardet-Biedl syndrome family with two mutant genes (BBS2, BBS10), three mutations, but no triallelism. Eur J Hum Genet 14:1195–1203
Lechtreck KF, Johnson EC, Sakai T, Cochran D, Ballif BA, Rush J, Pazour GJ, Ikebe M, Witman GB (2009) The Chlamydomonas reinhardtii BBSome is an IFT cargo required for export of specific signaling proteins from flagella. J Cell Biol 187:1117–1132
Lee BH, Liu J, Wong D, Srinivasan S, Ashrafi K (2011) Hyperactive neuroendocrine secretion causes size, feeding, and metabolic defects of C. elegans Bardet-Biedl syndrome mutants. PLoS Biol 9:e1001219
Lee DA, Bedont JL, Pak T, Wang H, Song J, Miranda-Angulo A, Takiar V, Charubhumi V, Balordi F, Takebayashi H, Aja S, Ford E, Fishell G, Blackshaw S (2012) Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nat Neurosci 15(5):700–702
Lehman JM, Michaud EJ, Schoeb TR, Aydin-Son Y, Miller M, Yoder BK (2008) The Oak Ridge Polycystic Kidney mouse: modeling ciliopathies of mice and men. Dev Dyn 237:1960–1971
Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S, Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL, Badano JL, Katsanis N (2008) Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet 40:443–448
Lorda-Sanchez I, Ayuso C, Ibanez A (2000) Situs inversus and hirschsprung disease: two uncommon manifestations in Bardet-Biedl syndrome. Am J Med Genet 90:80–81
Ludwig DS, Tritos NA, Mastaitis JW, Kulkarni R, Kokkotou E, Elmquist J, Lowell B, Flier JS, Maratos-Flier E (2001) Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J Clin Invest 107:379–386
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S et al (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1:1155–1161
Maffei P, Boschetti M, Marshall JD, Paisey RB, Beck S, Resmini E, Collin GB, Naggert JK, Milan G, Vettor R, Minuto F, Sicolo N, Barreca A (2007) Characterization of the IGF system in 15 patients with Alstrom syndrome. Clin Endocrinol (Oxf) 66:269–275
Marley A, von Zastrow M (2010) DISC1 regulates primary cilia that display specific dopamine receptors. PLoS One 5:e10902
Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, Tavares P, Vettor R, Veronese C, Martin M, So WV, Nishina PM, Naggert JK (2007) Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum Mutat 28:1114–1123
Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS (1999) Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA 96:5043–5048
Mistlberger RE (2009) Food-anticipatory circadian rhythms: concepts and methods. Eur J Neurosci 30:1718–1729
Mok CA, Healey MP, Shekhar T, Leroux MR, Heon E, Zhen M (2011) Mutations in a guanylate cyclase GCY-35/GCY-36 modify Bardet-Biedl syndrome-associated phenotypes in Caenorhabditis elegans. PLoS Genet 7:e1002335
Mountjoy KG (2010) Functions for pro-opiomelanocortin-derived peptides in obesity and diabetes. Biochem J 428:305–324
Moyer JH, Lee-Tischler MJ, Kwon HY, Schrick JJ, Avner ED, Sweeney WE, Godfrey VL, Cacheiro NL, Wilkinson JE, Woychik RP (1994) Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 264:1329–1333
Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA (2005) C. elegans tubby regulates life span and fat storage by two independent mechanisms. Cell Metab 2:35–42
Mukhopadhyay S, Lu Y, Shaham S, Sengupta P (2008) Sensory signaling-dependent remodeling of olfactory cilia architecture in C. elegans. Dev Cell 14:762–774
Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK (2010) TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev 24:2180–2193
Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP (2000) The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 127:2347–2355
Mykytyn K, Braun T, Carmi R, Haider NB, Searby CC, Shastri M, Beck G, Wright AF, Iannaccone A, Elbedour K, Riise R, Baldi A, Raas-Rothschild A, Gorman SW, Duhl DM, Jacobson SG, Casavant T, Stone EM, Sheffield VC (2001) Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet 28:188–191
Mykytyn K, Nishimura DY, Searby CC, Shastri M, Yen HJ, Beck JS, Braun T, Streb LM, Cornier AS, Cox GF, Fulton AB, Carmi R, Luleci G, Chandrasekharappa SC, Collins FS, Jacobson SG, Heckenlively JR, Weleber RG, Stone EM, Sheffield VC (2002) Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet 31:435–438
Mykytyn K, Nishimura DY, Searby CC, Beck G, Bugge K, Haines HL, Cornier AS, Cox GF, Fulton AB, Carmi R, Iannaccone A, Jacobson SG, Weleber RG, Wright AF, Riise R, Hennekam RC, Luleci G, Berker-Karauzum S, Biesecker LG, Stone EM, Sheffield VC (2003) Evaluation of complex inheritance involving the most common Bardet-Biedl syndrome locus (BBS1). Am J Hum Genet 72:429–437
Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, Braun T, Casavant T, Stone EM, Sheffield VC (2004) Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA 101:8664–8669
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129:1201–1213
Nishimura DY, Searby CC, Carmi R, Elbedour K, Van Maldergem L, Fulton AB, Lam BL, Powell BR, Swiderski RE, Bugge KE, Haider NB, Kwitek-Black AE, Ying L, Duhl DM, Gorman SW, Heon E, Iannaccone A, Bonneau D, Biesecker LG, Jacobson SG, Stone EM, Sheffield VC (2001) Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Hum Mol Genet 10:865–874
Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, Carmi R, Stone EM, Sheffield VC (2004) Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA 101:16588–16593
Nishimura DY, Swiderski RE, Searby CC, Berg EM, Ferguson AL, Hennekam R, Merin S, Weleber RG, Biesecker LG, Stone EM, Sheffield VC (2005) Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum Genet 77:1021–1033
Nordman S, Abulaiti A, Hilding A, Langberg EC, Humphreys K, Ostenson CG, Efendic S, Gu HF (2008) Genetic variation of the adenylyl cyclase 3 (AC3) locus and its influence on type 2 diabetes and obesity susceptibility in Swedish men. Int J Obes (Lond) 32:407–412
Norman RX, Ko HW, Huang V, Eun CM, Abler LL, Zhang Z, Sun X, Eggenschwiler JT (2009) Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum Mol Genet 18:1740–1754
Ohlemiller KK, Hughes RM, Mosinger-Ogilvie J, Speck JD, Grosof DH, Silverman MS (1995) Cochlear and retinal degeneration in the tubby mouse. Neuroreport 6:845–849
Pascual O, Traiffort E, Baker DP, Galdes A, Ruat M, Champagnat J (2005) Sonic hedgehog signalling in neurons of adult ventrolateral nucleus tractus solitarius. Eur J Neurosci 22:389–396
Pedersen LB, Rosenbaum JL (2008) Chapter two intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol 85:23–61
Pierce AA, Xu AW (2010) De novo neurogenesis in adult hypothalamus as a compensatory mechanism to regulate energy balance. J Neurosci 30:723–730
Plum L, Lin HV, Aizawa KS, Liu Y, Accili D (2012) InsR/FoxO1 signaling curtails hypothalamic POMC neuron number. PLoS One 7:e31487
Pollock JD, Rowland N (1981) Peripherally administered serotonin decreases food intake in rats. Pharmacol Biochem Behav 15:179–183
Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek R, Kanarek R, Maratos-Flier E (1996) A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 380:243–247
Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, Nishimura DY, Sheffield VC (2008) Leptin resistance contributes to obesity and hypertension in mouse models of Bardet-Biedl syndrome. J Clin Invest 118:1458–1467
Riise R, Andreasson S, Borgastrom MK, Wright AF, Tommerup N, Rosenberg T, Tornqvist K (1997) Intrafamilial variation of the phenotype in Bardet-Biedl syndrome. Br J Ophthalmol 81:378–385
Salamone JD, Zigmond MJ, Stricker EM (1990) Characterization of the impaired feeding behavior in rats given haloperidol or dopamine-depleting brain lesions. Neuroscience 39:17–24
Schaefer E, Zaloszyc A, Lauer J, Durand M, Stutzmann F, Perdomo-Trujillo Y, Redin C, Bennouna Greene V, Toutain A, Perrin L, Gerard M, Caillard S, Bei X, Lewis RA, Christmann D, Letsch J, Kribs M, Mutter C, Muller J, Stoetzel C, Fischbach M, Marion V, Katsanis N, Dollfus H (2011) Mutations in SDCCAG8/NPHP10 cause Bardet-Biedl syndrome and are associated with penetrant renal disease and absent polydactyly. Mol Syndromol 1:273–281
Schulz S, Handel M, Schreff M, Schmidt H, Hollt V (2000) Localization of five somatostatin receptors in the rat central nervous system using subtype-specific antibodies. J Physiol Paris 94:259–264
Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC (2009) Requirement of Bardet-Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet 18(7):1323–1331
Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, Sheffield VC (2010) BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci USA 107:1488–1493
Sharma N, Berbari NF, Yoder BK (2008) Ciliary dysfunction in developmental abnormalities and diseases. Curr Top Dev Biol 85:371–427
Sharma N, Kosan ZA, Stallworth JE, Berbari NF, Yoder BK (2011) Soluble levels of cytosolic tubulin regulate ciliary length control. Mol Biol Cell 22:806–816
Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T (2006) The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 103:5466–5471
Shillingford JM, Piontek KB, Germino GG, Weimbs T (2010) Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol 21:489–497
Shimada M, Tritos NA, Lowell BB, Flier JS, Maratos-Flier E (1998) Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature 396:670–674
Slavotinek AM, Stone EM, Mykytyn K, Heckenlively JR, Green JS, Heon E, Musarella MA, Parfrey PS, Sheffield VC, Biesecker LG (2000) Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet 26:15–16
Smaoui N, Chaabouni M, Sergeev YV, Kallel H, Li S, Mahfoudh N, Maazoul F, Kammoun H, Gandoura N, Bouaziz A, Nouiri E, M’Rad R, Chaabouni H, Hejtmancik JF (2006) Screening of the eight BBS genes in Tunisian families: no evidence of triallelism. Invest Ophthalmol Vis Sci 47:3487–3495
Stoetzel C, Laurier V, Davis EE, Muller J, Rix S, Badano JL, Leitch CC, Salem N, Chouery E, Corbani S, Jalk N, Vicaire S, Sarda P, Hamel C, Lacombe D, Holder M, Odent S, Holder S, Brooks AS, Elcioglu NH, Da Silva E, Rossillion B, Sigaudy S, de Ravel TJ, Alan Lewis R, Leheup B, Verloes A, Amati-Bonneau P, Megarbane A, Poch O, Bonneau D, Beales PL, Mandel JL, Katsanis N, Dollfus H (2006) BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 38(5):521–524
Stoetzel C, Muller J, Laurier V, Davis EE, Zaghloul NA, Vicaire S, Jacquelin C, Plewniak F, Leitch CC, Sarda P, Hamel C, de Ravel TJ, Lewis RA, Friederich E, Thibault C, Danse JM, Verloes A, Bonneau D, Katsanis N, Poch O, Mandel JL, Dollfus H (2007) Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 80:1–11
Stubdal H, Lynch CA, Moriarty A, Fang Q, Chickering T, Deeds JD, Fairchild-Huntress V, Charlat O, Dunmore JH, Kleyn P, Huszar D, Kapeller R (2000) Targeted deletion of the tub mouse obesity gene reveals that tubby is a loss-of-function mutation. Mol Cell Biol 20:878–882
Szabo NE, Zhao T, Cankaya M, Theil T, Zhou X, Alvarez-Bolado G (2009) Role of neuroepithelial Sonic hedgehog in hypothalamic patterning. J Neurosci 29:6989–7002
Tadenev AL, Kulaga HM, May-Simera HL, Kelley MW, Katsanis N, Reed RR (2011) Loss of Bardet-Biedl syndrome protein-8 (BBS8) perturbs olfactory function, protein localization, and axon targeting. Proc Natl Acad Sci USA 108:10320–10325
Tan PL, Barr T, Inglis PN, Mitsuma N, Huang SM, Garcia-Gonzalez MA, Bradley BA, Coforio S, Albrecht PJ, Watnick T, Germino GG, Beales PL, Caterina MJ, Leroux MR, Rice FL, Katsanis N (2007) From the cover: loss of Bardet-Biedl syndrome proteins causes defects in peripheral sensory innervation and function. Proc Natl Acad Sci USA 104:17524–17529
Traiffort E, Charytoniuk DA, Faure H, Ruat M (1998) Regional distribution of Sonic Hedgehog, patched, and smoothened mRNA in the adult rat brain. J Neurochem 70:1327–1330
Traiffort E, Charytoniuk D, Watroba L, Faure H, Sales N, Ruat M (1999) Discrete localizations of hedgehog signalling components in the developing and adult rat nervous system. Eur J Neurosci 11:3199–3214
Traiffort E, Angot E, Ruat M (2010) Sonic Hedgehog signaling in the mammalian brain. J Neurochem 113:576–590
Vijayan E, McCann SM (1977) Suppression of feeding and drinking activity in rats following intraventricular injection of thyrotropin releasing hormone (TRH). Endocrinology 100:1727–1730
Wang Z, Li V, Chan GC, Phan T, Nudelman AS, Xia Z, Storm DR (2009) Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS One 4:e6979
Weatherbee SD, Niswander LA, Anderson KV (2009) A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet 18:4565–4575
Wong ST, Trinh K, Hacker B, Chan GC, Lowe G, Gaggar A, Xia Z, Gold GH, Storm DR (2000) Disruption of the type III adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron 27:487–497
Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS (2005a) Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol 3:e415
Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS (2005b) PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest 115:951–958
Xu Y, Tamamaki N, Noda T, Kimura K, Itokazu Y, Matsumoto N, Dezawa M, Ide C (2005c) Neurogenesis in the ependymal layer of the adult rat 3rd ventricle. Exp Neurol 192:251–264
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Zhang Q, Nishimura D, Seo S, Vogel T, Morgan DA, Searby C, Bugge K, Stone EM, Rahmouni K, Sheffield VC (2011) Bardet-Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS-associated phenotypes and Bbs3 unique phenotypes. Proc Natl Acad Sci USA 108:20678-20683
Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF (2001) Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev 15:859–876
Acknowledgements
We thank Yoder lab members for suggestions and critical review of the chapter. This work was supported in part by NIH RO1 DK075996 to BKY. NFB was supported by NRSA F32 DK088404, RCP was supported by T32 GM008111 to (Dr. Yoder).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Berbari, N.F., Pasek, R.C., Yoder, B.K. (2013). Neuronal Cilia and Obesity. In: Tucker, K., Caspary, T. (eds) Cilia and Nervous System Development and Function. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5808-7_6
Download citation
DOI: https://doi.org/10.1007/978-94-007-5808-7_6
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5807-0
Online ISBN: 978-94-007-5808-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)