
Abstract
The potential energy surfaces (PES) and the corresponding spectroscopic constants describing the interaction between the Li +2 (X2Σ +g ) alkali dimer in its ground state and the xenon atom are evaluated very accurately including the three-body interactions. We have used an accurate ab initio approach based on nonempirical pseudopotential, parameterized l-dependent polarization potential, and an analytic potential form for the Li+Xe interaction. The potential energy surfaces of the interaction Li +2 (X2Σ +g )-Xe have been computed for a fixed distance of the Li +2 (X2Σ +g ) and for an extensive range of the remaining two Jacobi coordinates, R and γ. The use of the pseudopotential technique has reduced the number of active electrons of Li +2 (X2Σ +g )Xe complex to only one electron. The core-core interaction for Li+Xe is included using the (CCSD(T)) accurate potential of Lozeille et al. (Phys Chem Chem Phys 4:3601, 2002). This numerical potential is adjusted using the analytical form of Tang and Toennies. Moreover, the interaction forces and the potential anisotropy are analyzed in terms of Legendre polynomials analytical representation of the potential energy surface (PES). To our best knowledge, there are no experimental nor theoretical study on the collision between the Li +2 (X2Σ +g ) ionic alkali dimer and the xenon atom. These results are presented for the first time.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
During the past decade, the characterization of the structures and the stability of atomic and molecular clusters has been developed rapidly and become a current challenge of both experimental and theoretical research directed to attaining accurate descriptions of their nanoscopic properties. Special intense interest is focused on the study of the clusters involving helium and other noble gas atoms as components. These clusters constitute an important environment as a non homogenous quantum system that is fairly different from similar examples provided by a film on a solid surface or a macroscopic liquid with a free surface. In addition, they represent an ideal testing ground for many computational approaches [1–5], because the accurate knowledge of the relevant intermolecular forces between the solvent atoms and the dopants present in the cluster is an important prerequisite for the structural calculations, and, therefore, fairly simple components provide ideal model systems for the analysis of the influence of intermolecular interactions on the clusters properties [6–8]. In recent times, the structure and stability of the small clusters is the subject of few experimental and theoretical works because doped noble gas clusters present some additional interesting features, like the rapid heat transport generated inside the complex to the surface. Experimentally, Fuchs et al. [9] have examined the influence of the vibrational energy content of the Li2 molecules in collision with the helium atoms when producing initial-state-selected integral cross section. Various theoretical studies employed either ab initio or semiempirical potentials have been realized in the field of the interaction between neutral or ionic alkali molecules and a single noble gas atom, rare gas matrix, or droplets. In this context, Douady et al. [10] have performed a theoretical study of the Na +2 solvation in an argon matrix Arn. They have showed that the relatively strong interaction between the charged molecule and the Ar atoms favors trapping of the molecule inside the cluster rather than at the surface. Recently, an ab initio computed interaction forces are employed by Marinetti et al. [11] to describe the microsolvation of the Li +2 , Na +2 , and K +2 molecular ion in the helium clusters of small variable size. Bodo et al. [12–16] have investigated by Hartree-Fock calculations the potential energy surfaces for the ground electronic states of the alkali dimer Li2, Li +2 , Na +2 , and K +2 interacting with neutral helium. In all these systems, they found that the He atoms occupy the external sites along the molecular axis.
In this chapter, we present an ab initio study of the potential energy surface and stability of the Li +2 (X2Σ +g ) alkali dimer interacting with the xenon atom in different radial geometries and for six angles from 0° to 90°. In Sect. 16.2, the ab initio calculation method is presented. Section 16.3 reports the results of calculation and analysis of the interesting and unusual feature of the strong interaction and anisotropy of the potential. Finally, we present our conclusions in Sect. 16.4.
2 Method of Calculation
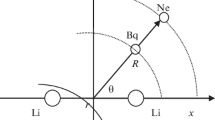
The potential energy surface is computed in Jacobi coordinates by fixing the internuclear distance of Li +2 (X2Σ +g ) ionic molecule at its equilibrium distance of 5.84 a.u. determined previously in our group by Bouzouita et al. [17]. We report in Fig. 16.1 a descriptive model of the coordinates of the Li +2 (X2Σ +g )Xe system. The distance R represents the separation between the xenon atom and the center of mass of the Li +2 (X2Σ +g ) ionic molecule, R a and R b are the separations between the Xe and the two Li+ cores, and γ is the angle between R and the Li +2 (X2Σ +g ) internuclear axis.

Coordinates of the Li +2 (X2Σ +g )Xe system used in the calculation
As in our previous work [17–21], Li +2 (X2Σ +g )Xe is treated as a one-electron system using the nonempirical pseudopotential proposed by Barthelat et al. [22] in its semilocal form. The Gaussian-type orbital basis set values on lithium and xenon are, respectively, (9s8p5d/8s6p3d) and (4s3p). The cutoff radii (in bohr) used for s, p, and d orbitals are, respectively, 1.434, 0.982, and 0.6 for Li [23] and 3.500148, 4.0, and 1.401128 for Xe [24]. The core dipole polarizability of Li+ and Xe atom are, respectively, 0.1915 [23] and 27.29 a.u. [25].
Using the nonempirical pseudopotential proposed by Barthelat et al. [22], the two Li+ ions and the Xe atom are treated as a three cores interacting with the alkali valence electron. Based on this approach, the total potential of the Li +2 (X2Σ +g ) system is a sum of three contributions: the valence electron-core interaction, core-core interactions, and the three-body interactions. In this context, the total potential is given by:
The three terms represent, respectively, the interaction between the valence electron and the ionic system Li 2+2 Xe, the core-core interactions, and finally the three-body interactions. The three terms are developed in the next subsections.
The \( {V_{{\text{L}}{{\text{i}}^{+} }{\text{Xe}}}} \) contribution is taken from the accurate and recent coupled cluster simple and double excitation (CCSD) calculations of Lozeille et al. [26]. For a better representation of the Li+Xe interaction in the region of interest for the Li +2 (X2Σ +g ) system, the numerical potential is fitted using the analytical form of Tang and Toennies [27]. Such potential contains the well-known long-range terms of van der Waals interactions and the usual exponential repulsive term. It is written as follows:
The parameters A eff, b, D 4, D 6, D 8, and D 10 are obtained by a square fitting using the numerical potential of Lozeille et al. [26]. These parameters are presented in Table 16.1. Figure 16.2 presents the Lozeille et al. [26] numerical potential (circles) of Li+Xe compared to the analytical one (solid line). The difference between our analytical potential and the numerical one does not exceed 1.9∙10−6 a.u., which corresponds to less than 1 cm−1.

Lozeille et al. [26] numerical potential of Li+Xe interaction compared to the analytical one
For the \( {V_{e - {\text{L}}{{\text{i}}_2}^{2 + }{\text{Xe}}}} \) contribution, we have performed a one-electron ab initio calculation where the two Li+ cores and the electron-xenon effects have been replaced by semilocal pseudopotentials [22].
The analytical formula for the three-body interactions is given by
where the angles \( {\gamma_a} \) and \( {\gamma_b} \) are formed between each dipole distance from the point-like charge (R a , R b ) and the line joining them (R ab ). Each dipole moment \( {\mu_a} \) can then be evaluated via the well-known formula: \( {\mu_a} = {{\alpha } \left/ {{R_a^2}} \right.} \) where α being the Xe polarizability.
3 Results and Discussions
3.1 Potential Energy Surfaces and Spectroscopic Constants
The potential energy surfaces for the Li +2 (X2Σ +g )Xe system have been computed as a function of the Jacobi coordinate V(R e, R, γ) and for six different angles \( \gamma \) and a fixed distance for the Li +2 (X2Σ +g ) ionic molecule corresponding to the equilibrium distance. The distance R is the separation between the xenon atom and the center of mass of the Li +2 (X2Σ +g ) ionic molecule, and γ is the angle between R and the Li +2 internuclear axis. These potential energy surfaces have been determined including the three-body interactions. The potential energy surfaces of the Li +2 (X2Σ +g )-Xe interactions corresponding to the six different angles are displayed on Figs. 16.3 and 16.4. Figure 16.3 presents the potential energy surfaces of the Li +2 (X2Σ +g )-Xe interactions corresponding to the six different angles without the three-body effects. First, we notice that all these curves tend to the same asymptotic limit. This limit, which equals −0.24597 a.u., is the energy of the Li +2 (X2Σ +g ) at its equilibrium distance (R e = 5.84 a.u.). Second, we remark that all the potential energy surfaces are attractive since they present minimums of lower energy relative to the asymptotic limit. In addition, these potential energy surfaces show that the Li +2 (X2Σ +g )-Xe interactions present an interesting and unusual feature of the strong interaction and anisotropy. The comparison between the potential energy surfaces shows that the attractive effects decrease their importance with respect to the attractive long-range interaction forces as one goes from γ = 0° to 90°. In addition, the geometry exhibiting the deepest well is obtained for a collinear orientation around γ = 0°. So, it is clear that the Xe atom would be linked at the extremity of the Li +2 (X2Σ +g ) alkali dimer. In fact, the Li +2 (X2Σ +g ) alkali dimer in its ground state can be roughly considered as an electron cloud located in the middle of the two Li+ cores. The short-range repulsion between the electron and Xe atom combined with the attraction between the cationic cores and Xe atom thus favors the positioning of the rare gas atom at one extremity of the Li +2 (X2Σ +g ) alkali dimer molecule. The potential energy surfaces including the three-body effects of the Li +2 (X2Σ +g )-Xe interactions are presented with the black dashed line in Fig. 16.4. We note that the three-body interactions decrease the interaction energy. This decrease is significant at distances close to the equilibrium distances.

Orientational features of the rigid rotor potential energy surfaces for six different angles (γ = 0°, 11°, 25.3°, 39.7°, 68.5°, and 90.0°) without the three-body effects

Effect of the three-body interaction on the potential energy surfaces (black dotted lines)
The spectroscopic constants corresponding to the equilibrium distance (R e), the well depth (D e), and the harmonicity frequency (ω e) of all potential energy surfaces without and with the three-body interactions are collected in Table 16.2. The analysis of these data shows that the equilibrium distance, the depth of the well, and the harmonicity frequency depend on the angle γ showing the strong anisotropy of the Li +2 (X2Σ +g )-Xe system. In fact, we remark that the well depth and the harmonicity frequency decrease when γ increases. The same remark is observed for the equilibrium distance for the lowest four angles γ, then it increases for γ = 68.5° and γ = 90.0°. As it seems from Table 16.2, the three-body interactions lead to a significant decrease in energy and to a small increase in equilibrium position. For example, the potential energy surface, for γ = 0°, exhibit the deepest well depth. This curve presents a well depth of 1,625 cm−1 located at the equilibrium distance of 8.54 a.u. without the three-body effects and a well depth of 1,240 cm−1 located at the equilibrium distance of 8.66 a.u. when the three-body effect is included.
3.2 Analysis of the Surface Anisotropy
To assess the main features for the orientational anisotropy in the RR (rigid rotor) interactions, we used the familiar multipolar expansion:
where R is the separation between the xenon atom and the center of mass of the Li +2 (X2Σ +g )Xe ionic molecule and γ is the angle between R and the Li +2 (X2Σ +g ) internuclear axis.
Figure 16.5 reports \( {V_\lambda }(R) \) the multipolar functions from \( \lambda = 0 \) to 5. These multipolar functions show different orientational anisotropy in the repulsive region and also differences in the long-range strength of the interaction. Only the multipolar function for \( \lambda = 1 \) exhibits a clear attractive well located at 8.97 a.u. The curves of the multipolar functions for \( \lambda = 3 \) and \( \lambda = 4 \) are repulsive, while those of \( \lambda = 0 \), \( \lambda = 2 \) , and \( \lambda = 5 \) are similar in shape and exhibit a small barrier. Furthermore, these \( {V_\lambda }(R) \) can be used in the standard close-coupling formulation of atom-rigid rotor collisions, as they facilitate the determination of the required matrix elements of the potential. Moreover, they will be used for exploring the structure of Li+Xen clusters.

Multipolar functions from \( \lambda = 0 \) to \( \lambda = 5 \)
4 Conclusion
In this work, we have evaluated the potential energy surfaces, including the three-body interactions, describing the interaction between the Li +2 (X2Σ +g ) alkali ionic dimer in its ground state and the xenon atom. We have used a standard quantum chemistry approach based on nonempirical pseudopotential, parameterized l-dependent polarization potential, and an analytic potential form for the Li+Xe interaction. The potential energy surfaces for the interaction Li +2 (X2Σ +g )-Xe have been computed for a fixed distance of the Li +2 (X2Σ +g ) and for an extensive range of the remaining two Jacobi coordinates, R and γ. In this context, the Li +2 (X2Σ +g )Xe is reduced to only one-electron system. The spectroscopic constants of these potential energy curves for fixed angles and varying R have been extracted. As it is expected, the potential energy surface of the Li +2 (X2Σ +g )-Xe system presents an interesting and unusual feature associated to the strong interaction and anisotropy. This anisotropy is demonstrated by writing the potential energy surface in terms of the Legendre polynomial multipolar expansion. It seems that the deepest well is associated with γ = 0°. We assume that the Xe atom would be attached at the extremity of the Li +2 (X2Σ +g ) alkali dimer. Moreover, the three-body interactions lead to a small decrease in energy. This decrease is remarkable close to the equilibrium distances.
This simple model and also the produced analytical potential energy surface (PES) will be used to explore the structure, the geometry, and the stability of Li +2 -Xen clusters.
References
Toennies JP, Vilesov AF (1998) Ann Rev Phys Chem 49:1
Toennies JP, Vilesov AF, Whaley KB (2001) Phys Today 54:31
Stienkemeier F, Vilesov AF (2001) J Chem Phys 115:10119
Toennies JP, Vilesov AF (2004) Angew Chem Int Ed 43:2622
Stienkemeier F, Lehmann KK (2006) J Phys B: Atom Mol Opt Phys 39:R127
Buchachenko A, Halberstadt N, Lepetit B, Roncero O (2003) Int Rev Phys Chem 22:153
García-Vela A (1998) J Chem Phys 108:5755
Slavíček P, Roeselová M, Jungwirth P, Schmidt B (2001) J Chem Phys 114:1539
Fuchs M, Toennies JP (1986) J Chem Phys 85:7062
Douady J, Jacquet E, Giglio E, Zanuttini D, Gervais B (2008) J Chem Phys 129:184303
Marinetti F, Uranga-Piňa L, Coccia E, López-Durán D, Bodo E, Gianturco FA (2007) J Phys Chem A 111:12289
Bodo E, Yurtsever E, Yurtsever M, Gianturco FA (2006) J Chem Phys 124:074320
Bodo E, Gianturco FA, Yurtsever E, Yurtsever M (2005) Mol Phys 103:3223
Bodo E, Sebastianelli F, Gianturco FA, Yurtsever E, Yurtsever M (2003) J Chem Phys 120:9160
Bodo E, Gianturco FA, Sebastianelli F, Yurtsever E, Yurtsever M (2004) Theor Chem Acc 112:263
Bodo E, Gianturco FA, Yurtsever E (2005) J Low Temp Phys 138:259
Bouzouita H, Ghanmi C, Berriche H (2006) J Mol Struct (THEOCHEM) 777:75
Berriche H (2003) J Mol Struct (THEOCHEM) 663:101
Berriche H, Ghanmi C, Ben Ouada H (2005) J Mol Spectr 230:161
Ghanmi C, Berriche H, Ben Ouada H (2006) J Mol Spectr 235:158
Berriche H, Ghanmi C, Farjallah M, Bouzouita H (2008) J Comp Method Sci Eng 8:297
Barthelat JC, Durand P (1975) Theor Chim Acta 38:283: (1978) Gazz Chim Ital 108:225
Müller W, Flesh J, Meyer W (1984) J Chem Phys 80:3297
Foucrault M, Millié P, Daudey JP (1992) J Chem Phys 96:1257
Soldán P, Lee EPF, Wright TG (2001) Phys Chem Chem Phys 3:4661
Lozeille J, Winata E, Soldán P, Lee EPF, Viehland LA, Wright TG (2002) Phys Chem Chem Phys 4:3601
Tang KT, Toennies JP (1984) J Chem Phys 80:3726
Acknowledgment
This work has been supported by the Advanced Materials Center and KACST through the Long-Term Comprehensive National Plan for Science, Technology and Innovation Program (Project no. 10-ADV1164-07).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this paper
Cite this paper
Saidi, S., Ghanmi, C., Hassen, F., Berriche, H. (2012). Ab initio Study of the Potential Energy Surface and Stability of the Li2 +(X2Σg +) Alkali Dimer in Interaction with a Xenon Atom. In: Nishikawa, K., Maruani, J., Brändas, E., Delgado-Barrio, G., Piecuch, P. (eds) Quantum Systems in Chemistry and Physics. Progress in Theoretical Chemistry and Physics, vol 26. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5297-9_16
Download citation
DOI: https://doi.org/10.1007/978-94-007-5297-9_16
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5296-2
Online ISBN: 978-94-007-5297-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)