
Abstract
Hypoxia and ischemia resulting in reduced oxygen delivery to brain tissues is reported to cause neurodegeneration in both in vitro and in vivo models. Similar decrease in partial pressure of oxygen occurs on ascent to high altitude, a situation referred to as hypobaric hypoxia that limits oxygen availability to the brain. Our studies on human subjects reveal decrease in vigilance and response time along with decreased cerebral oxygenation which is both altitude and duration dependent. Alterations in evoked potentials and change in hedonic matrix were also observed following exposure to high altitude environment. Investigations in animal models exposed to simulated altitude showed occurrence of oxidative stress, neurodegeneration and memory impairment. This hypoxic response of neurons is multi-factorial and involves complex signaling pathways thereby limiting the therapeutic efficacy of several antioxidants in ameliorating hypobaric hypoxia–induced memory impairment. Animals exposed to hypobaric hypoxia show depletion in the antioxidant status along with increased free radical generation. Neuromorphological studies revealed neurodegeneration and dendritic atrophy in the hippocampus. Altered neurotransmitter synthesis, release and metabolism have also been observed along with occurrence of calcium overload in hypoxic neuronal cells. These changes in the hypoxic brain find an analogy with the aging related changes that include decreased conduction rate, generation of free radicals, protein oxidation and cellular apoptosis. Administration of N-acetyl cysteine to animals exposed to hypobaric hypoxia showed considerable improvement in memory functions along with decrease in free radical generation. Acetyl-L-Carnitine administration during hypobaric hypoxia also improved the cognitive capabilities in animal models. Our investigations revealed a multifactorial action of Acetyl-L-Carnitine that included improved mitochondrial bioenergetics, neurotrophin mediated signaling and antioxidant status. The implications of these compounds as anti aging and anti-senescence interventions, however, need to be investigated.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
12.1 Introduction
The brain is not only situated at the anterior most region of the body, but also regulates an array of physiological functions through its complex wired neuronal networks. It directly or indirectly influences the central, peripheral and autonomic responses which govern an individual’s psychological, physical and physiological responses to internal and external stimuli. The brain comprises of billions of neurons and glial cells that work in unison to form complex circuits for storing and processing of information. These closely coordinated miniature electrical circuits operate through tightly regulated opening and closing of ion channels which could be ligand-gated or voltage-gated. Maintenance of a potential gradient across the neuronal membrane which is imperative for transmission of impulse through these neuronal circuits involves utilization of large amount of ATP. This high energy requirement makes brain metabolically the most active organ in human body. This probably also explains the reason why neurodegenerative disorders like Alzheimer’s, Parkinson’s, Amyotrophic Lateral Sclerosis are all associated with aging brain . Derailments in higher order cognitive functions and early dementia are considered to be preliminary symptoms of an aging brain. Interestingly, aging is often associated with vascular dementia and reduced oxygen supply to the brain. Several psychological, physiological and biochemical manifestations during aging appear to be similar to hypoxia and ischemia. However, aging related neurodegeneration is a slow and progressive phenomenon spanning over years, while hypoxia is a more severe insult on the neurons resulting in their immediate death. Hence, understanding the mechanisms of hypoxic neurodegeneration which is a much severe stress in comparison to aging could surely provide some valuable insights into strategies for preventing neurodegeneration during aging. Besides that, it could also help in understanding the effect of episodes of hypoxic stress during young and adult stages of an individual’s life on neuronal aging at old age .
12.2 The Brain Function in Low Oxygen
The brain has the highest oxygen and glucose dependency and consumes 20 % of the total oxygen for generation of ATP through the aerobic mechanisms (Halliwell 1992). Decreased supply of oxygen to the brain during conditions of hypoxia and ischemia therefore results in neurodegeneration. A unique situation of global decrease in oxygen supply to the brain is encountered during ascent to high altitude. The reduction in partial pressure of oxygen on ascent to high altitude leads to decreased oxygen saturation of arterial blood and compromised oxygen delivery to tissues (Peacock and Jones 1997). This condition, referred to as hypobaric hypoxia , not only limits human performance (Pugh 1964; West 1988, 2002) but also triggers several physiological, sensory and neurobehavioral alterations (Houston et al. 1987; Hornbein 1992). The effects of hypoxia are greatly influenced by rate of ascent and duration of stay at high altitude. The range of altitude has been distinguished into: (1) Intermediate altitude (1,500–2,500 m), where physiological changes are detectable due to hypobaric hypoxia, but arterial oxygenation remains above 90 %. However, altitude illness is possible, (2) High altitude (2,500–3,500 m), where altitude illness is commonly observed due to rapid ascent above 2,500 m, (3) Very high altitude (3,500–5,800 m), where arterial oxygenation falls below 90 %. Altitude illness is common and marked hypoxemia can occur due to exercise, (4) Extreme altitude ( > 5,800 m), where successful acclimatization cannot be achieved, progressive deterioration follows and hypoxemia occurs at rest. It is assumed that long term stay for humans is not possible above 5,500 m, although moderate altitudes can sometimes be tolerated without supplementary oxygen (Hackett and Roach 2001). Beginning with the balloon flights in the latter half of the nineteenth century, extensive literature has described the subtle effects of hypoxia on the brain and CNS (West 2004). Much early work by McFarland (1937) documented the effect of hypoxia on mental performance at high altitude. On the basis of observations and tests of sensory, motor and cognitive function, McFarland observed that individuals taken rapidly (hours) to 4,000–4,500 m exhibited impairment in both simple and complex psychological performance. Motor functions, such as handwriting, were also impaired but to a lesser extent, and sensory modalities were affected little if at all. Investigators have documented decrements in performance on a variety of neuropsychometric tests for cognitive and motor functions after sudden exposure to even relatively moderate hypoxia (2,000–4,500 m) (Stickney and Van Liere 1953; Ernsting 1978).
Changes in a visual-positioning test performed during light work have also been reported at an altitude as low as 1,500 m (Denison et al. 1966). A study in humans has shown that 15 adults (29–37 years old), tested under high altitude conditions (4,500 and 5,050 m), displayed difficulties in recalling word lists, specifically those words that came early in the list (primacy effect). In this study, memory recall remained impaired 45 days after descent from high altitude (Pelamatti et al. 2003). There is further evidence that memory impairment may last several months after returning to the lowlands. A number of other studies have shown that verbal and visual short-term memory capacity and recall is impaired at altitudes starting at 2,500 m (Cavaletti et al. 1987; Regard et al. 1989; Hopkins et al. 1995). Acute exposure to hypoxia (few minutes) at altitudes above 6,500 m is known to cause severe neurobehavioral dysfunctions and loss of consciousness in non-acclimatized individuals. These studies suggest that a critical impairment in higher cognitive functions is the earliest and most insidious consequence of exposure to high altitude.
12.3 Neuronal Response to Hypoxia
Various cell types in the brain and CNS show differential susceptibility to hypoxic insult, with the neurons dying of hypoxia long before glial cells and among glia, oligodendrocytes before astrocytes (Wang et al. 2002). Rats exposed to hypobaric hypoxia reveal occurrence of oxidative stress, neuronal degeneration and dendritic atrophy (Titus et al. 2007; Maiti et al. 2006). The mechanisms pertaining to hypobaric hypoxia induced neurodegeneration appear to be multi-factorial and may involve oxidative stress, neurotransmitter alterations, altered bioenergetics, altered neuromorphology and disturbed ionic homeostasis. Several studies have indicated towards the occurrence of glutamate excitotoxicity in hypoxic and ischemic stress (Won et al. 2002; Hemi et al. 2003). Hypoxia has been reported to cause robust calcium influx into neuronal cells through the NMDA receptors, thus mediating excitotoxic cell death (Khodorov et al. 1996; Hota et al. 2008). Calcium mediated free radical generation through activation of PhospholipaseA2 (PLA2), Xanthine Oxidase and Monoamine Oxidase by Calcium Calmodulin complex has also been reported in the hippocampus following hypoxic insult (Barhwal et al. 2009a). Exposure to hypobaric hypoxia also results in alterations in cholinergic transmission and altered corticosterone that could contribute to the memory impairment (Hota et al. 2009; Muthuraju et al. 2009).
12.4 The Aging Neuron
Aging has been classically considered as a process of slow deterioration of neuronal functions associated with degradation and altered recycling of long-lived proteins, macromolecular aggregates, and damaged intracellular organelles. Conversely, it is now evident that neuronal aging is a biological process tightly controlled by evolutionary highly conserved signaling pathways. Importantly, genetic mutations that enhance longevity significantly delay the loss of synaptic connectivity and, therefore, the onset of age-related brain disorders (Bano et al. 2011). The molecular mechanisms pertaining to neuronal degeneration are similar to hypoxic cell death in several aspects and involve dysregulation in calcium homeostasis , altered mitochondrial activity and cellular bioenergetics (Wang and Michaelis 2010) and alterations in cellular redox status such as increased generation of mitochondrial oxidants, altered GSH status, and increased protein oxidation . Cholinergic neurons of the basal forebrain complex have been described to undergo moderate degenerative changes during aging, resulting in cholinergic hypofunction that has been related to the progressing memory deficits with aging (Schliebs and Arendt 2006). Morphologically, both hypoxic insult and aging result in reduction of spine numbers and synaptic dysfunction (Hota et al. 2009; Bano et al. 2011). Aging however differs from hypoxic neurodegeneration on the basis of genetic determinants that serve as an internal trigger for cell death. Age -dependent accumulation of partially deleted mitochondrial DNA and altered transcriptome activity and synthesis of mitochondrial proteins have been suggested to contribute to aging and the development of age-associated diseases (Fukui and Moraes 2009).
12.5 Oxidative Stress in Hypoxia and Aging
The biological process of aging is associated with impairment of cellular bioenergetic function and increased oxidative stress (Escames et al. 2010; Floyd et al. 2002; Calabrese et al. 2001). The mitochondrion is considered to be the most important cellular organelle to contribute to the aging process, mainly through respiratory chain dysfunction and formation of reactive oxygen species , leading to damage to mitochondrial proteins, lipids and mitochondrial DNA (Paradies et al. 2011; Calabrese et al. 2001). In addition to the changes in electron transport chain , ions like calcium and iron also play a key role in mediating free radical generation and oxidative stress. Alteration of calcium homeostasis in the aging brain results in calcium ion sequestration into the mitochondria. This calcium overload perturbs the redox state of mitochondria and causes oxidative stress (Foster 2007; Toescu and Verkhratsky 2007; Biessels and Gispen 1996). Several studies indicate that the sensitivity of mitochondria to Ca2 +-induced PTP opening is greater in the aged compared to the young mature brain (Toman and Fiskum 2011). Post-translational modifications of proteins due to oxidative and nitrative stress have also been associated to the aging brain (Grimm et al. 2011) .
Oxidative stress and related biochemical factors that play a major role in aging and related neurodegenerative disorders also appear to influence the neuronal survival in hypoxia . Free radical generation and oxidative damage to bio-molecules have been invariably associated to hypoxic exposure. Studies conducted in both in vitro and in vivo models of hypoxia and ischemia show increase in lipid peroxidation and DNA damage (Hota et al. 2007, Barhwal et al. 2008). This is also associated with decrease in antioxidant enzyme activities and depletion of cellular antioxidants (Blum and Fridovich 1985). The oxidative stress in hypoxic brain is primarily attributed to glutamate excitotoxicity and deregulation of calcium ion homeostasis. Increased release of glutamate in the excitatory synapses along with upregulation of N-methyl-D-aspartate (NMDA) receptors results in robust influx of calcium into the cells (Hota et al. 2008). Increased expression of L type calcium channels during hypoxic exposure has also been reported to exert an additive calcium overload in neuronal cells (Barhwal et al. 2009a). The calcium in turn activates several pro-oxidant enzymes viz., xanthine oxidases, monoamine oxidases, cytosolic phospholipase A2 and cyclooxygenase (COX-2) leading to generation of free radicals. Calcium also mediates leakage of electrons from the mitochondria and opening of the permeability transition pore (PTP) that triggers apoptosis .
Though the mechanisms pertaining to free radical generation and oxidative stress appear to be similar in both aging and hypoxia, a presumed difference between the two is that oxidative stress is considered to be a cause of aging but a consequence of hypoxia. There has been a growing consensus regarding acceleration of aging due to lipid peroxidation and post translational modification of proteins. On the other hand, hypoxia has been portrayed as a trigger for oxidative stress that in turn causes neurodegeneration . However, with the recent reports on role of free radicals as signaling molecules and regulation of the expression of transcription factors and protein activity through oxidation and carboxylation, the notion on oxidative stress being a consequence needs to be redefined. This is evident from the fact that subunits of NMDA receptor that contribute to the calcium overload in neuronal cells during hypoxia are themselves regulated by a free radical mediated mechanism (Hota et al. 2010). Besides that, the fact that there is an increase in lipid peroxidation , protein oxidation , DNA damage and accumulation of oxidized biomolecules in neurons exposed to hypoxia also raises a concern that hypoxic exposure could accelerate aging.
12.6 Nrf-2 Regulated Antioxidant Systems in Hypoxia and Aging
The fate of the neurons is dictated by an intricate balance between the oxidative stress and intracellular antioxidant systems in both hypoxia and aging . Overwhelming of the antioxidant defense systems by excess generation of free radicals acts as a trigger for the onset of aging and hypoxia mediated neurodegeneration. In recent years, it has been realized that peroxiredoxins may be the most important peroxide free radical removal systems (Rhee et al. 2005). They are a family of peroxidases that reduce H2O2 and organic peroxides. They are homodimers and contain no prosthetic group: the redox reactions are dependent on cysteine at the active sites. Thioredoxin-1 (Trx-1) is one such peroxiredoxin which is a small 12 kDa multifunctional protein having a redox-active disulfide/dithiol within its active site sequence, -Cys-Gly-Pro-Cys- and operates together with NADPH and thioredoxin reductase as a protein disulfide-reducing system (Holmgren 1985). In addition to peroxiredoxins, various phase II detoxification enzymes and antioxidants work together to reduce damage caused by oxidative stress . Many reports indicate that phase II detoxification enzymes and antioxidant genes are regulated by an antioxidant responsive element (ARE) , which is located within the promoter regions of these genes (Huang et al. 2000; Lee et al. 2003). The ARE activity is regulated by an array of transcription factors including NF-E2-related factor2 (Nrf2). Nrf2, belonging to the basic leucine zipper family of proteins, is an important candidate involved in the transcriptional regulation of ARE motifs (Itoh et al. 1997). The genes regulated by ARE include glutathione-S-transferase (GST), NAD(P)H quinone oxidoreductase-1 (NQO1), Heme oxygenase-1 (HO-1), Glutamate-cysteine ligase (GCL), ferritin-L, metallothionin-1 and UDP-glucuronyl transferase (UGT) (Favreau and Pickett 1995).
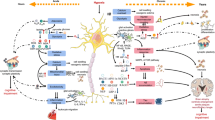
In unstressed cells, Nrf2 is sequestered in the cytosol by Keap1 which is crucial for targeting Nrf2 for ubiquitination and degradation. However, Nrf2 may also exist in the nucleus under homeostatic resting conditions for basal transcription of Nrf2 mediated genes. When cells are exposed to oxidative or electrophilic stress, Nrf2 appears to be liberated from the Keap1-Nrf2 complex and translocates into the nucleus (Nakaso et al. 2003; Kobayashi et al. 2006), thereby activating Nrf2 dependent gene transcription . Thus, Keap1 negatively regulates Nrf2 stability by targeting Nrf2 for ubiquitination by Cul3 and subsequent degradation by the proteasome pathway (Fig. 12.1).

Schematic diagram depicting regulatory role of Nrf2 during normoxic and oxidative stress. Supplmentation of ALCAR during hypoxia, results in degradation of Cul3 through pERK mediated mechanisms resulting in stabilization of Nrf2. (Barhwal et al. 2009)
Several antioxidant compounds like Acetyl-L-Carnitine provide neuroprotection in hypoxia by eliciting Nrf2 mediated transcription of ARE regulated antioxidant genes through a pERK mediated mechanism (Barhwal et al. 2009b). Acetyl-L-Carnitine has also been found to be effective in ameliorating aging related neuronal death. Nrf2 mediated improvement in D1 receptor function of renal neurons has also been reported in old rats subjected to exercise (Asghar et al. 2007). Similarly, phytochemicals like plumbagin significantly reduce the amount of brain damage and ameliorate associated neurological deficits in focal ischemic stroke by activating Nrf2/ARE pathway. In addition to augmenting the antioxidant status in neuronal cells, Nrf2 also regulates expression of xenobiotic detoxifying (phase II) enzymes (Zhang et al. 2012). Removal of the xenobiotic compounds, on the other hand, slows the aging of neuronal cells. Hence drugs mediating Nrf-2 upregulation could be beneficial in delaying aging and ameliorating hypoxia related cognitive impairments .
12.7 Mitochondrial Mechanisms of Neurodegeneration in Hypoxia and Aging
Both hypoxia and aging have been associated with cell death in several susceptible regions of the brain. Two distinct mechanisms of cell death have been characterized, i.e. apoptosis and necrosis (Kerr et al. 1972). Necrosis and apoptosis may occur either distinctly or simultaneously within a damaged region of the brain, and are related to the magnitude of the toxic stimuli. Acute insults such as hypoxia, stroke , trauma, and infection cause harsh, usually focal injuries to the central nervous system. In general, such severe injuries to the brain result in rapid necrosis in the core regions, although in most cases apoptosis is also observed (Honig and Rosenberg 2000).
Apoptosis is a tightly controlled cell death process involving definite enzyme cascades, which keeps the content of the dying cell intracellular. It is initiated by both physiological and pathological stimuli. On the contrary, necrosis refers to relatively uncontrolled cell death and is generally correlated with injury (Sastry and Rao 2005). The cell’s decision to die from necrosis or apoptosis is dictated at least in part by the abundance of intracellular energy stores. Whereas, apoptosis requires a minimal amount of intracellular ATP, necrosis is generally accompanied by its total depletion (Nicotera et al. 1998). Thus necrosis may be viewed as an accidental type of cell death. Necrosis is not genetically predetermined and normally occurs within a short period following a triggering insult (2–4 h). Necrosis has been invariably associated with immediate cell death following hypoxic or ischemic insult. Though it is not directly correlated to aging, necrotic cells can accelerate the aging process of neurons in their vicinity by causing neuroinflammation and oxidative stress .
Apoptosis , depending upon the origin of the activator molecule, is categorized into extrinsic and intrinsic pathways. In the extrinsic pathway (also known as “death receptor pathway”), apoptosis is triggered by an extracellular ligand-induced activation of death receptors at the cell surface. Such death receptors include the tumor necrosis factor (TNF) receptor-1, CD95/Fas (the receptor of CD95 L/FasL), as well as the TNF-related apoptosis inducing ligand (TRAIL) receptors-1 and −2. Several inflammatory cytokines belonging to the interlukin family also play a key role in mediating death receptor mediated apoptosis . Aging processes stimulate secretion of proinflammatory cytokines IL-1β and IL-18 that may contribute to age-related cognitive decline in the growing elderly population. Besides that, the beneficial or detrimental transcriptional response of the inflammatory mediator TNFα, that activates a signaling cascade involving NFκB translocation to the nucleus, is also governed by the age of the neurons (Patel and Brewer 2008).
In the intrinsic pathway (also called “mitochondrial pathway”), apoptosis results from an intracellular cascade of events in which mitochondrial permeabilization plays a crucial role (Scaffidi et al. 1998). Activation of a specific class of proteases, the caspases (“cysteine protease cleaving after Asp”), is required for the rapid and complete manifestation of apoptotic features. However, not all caspases are required for apoptosis and the process generally results from the activation of a limited subset of caspases, in particular, caspases-3, -6, and −7 (Fuentes-Prior and Salvesen 2004). These are the “executioner” caspases that mediate their effects by cleavage of specific substrates in the cell.
The release of pro-apoptotic factors occurs through the permeabilization of the mitochondrial membrane. The opening of the permeability transition pore causes swelling of the mitochondrial matrix, which results in mitochondrial uncoupling, rupturing of the mitochondrial outer membrane, and release of pro-apoptotic proteins into the cytosol leading to apoptosis (Yang and Cortopassi 1998). Altered calcium ion homeostasis resulting due to robust influx of extracellular calcium during hypoxic insult is known to play a key role in opening of the permeability transition pore. Aging and aging related neurodegenerative disorders have also been associated with calcium mediated toxicity and cellular apoptosis. The entry of calcium ion into the mitochondria occurs through an electrogenic uniporter, now known to be a channel, and is pumped out again by a Na +/Ca2 +antiporter (Gunter et al. 2000). The activity of the Na +/Ca2 +antiporter saturates as mitochondrial matrix calcium increases, whereas the uniporter acts as a channel and is thus not saturated with increasing extramitochondrial calcium concentration. Consequently, as the extramitochondrial calcium concentration increases beyond a certain value, the mitochondria can no longer regulate their matrix calcium concentration, and mitochondrial calcium overload ensues (Gunter et al. 2000). When the overload is accompanied by a combination of other factors, most notably oxidative stress , high phosphate concentrations and low adenine nucleotide concentrations, the mitochondria undergo a permeability transition i.e. a pore opens in their inner membrane, known as the ‘mitochondrial permeability transition pore’ (MPTP), causing the membrane to become nonspecifically permeable to any molecule less than 1.5 kDa in size. This results in proton leak, and thus the mitochondria become uncoupled and are no longer able to maintain a pH gradient or membrane potential. As a result, mitochondria not only become incapable of ATP synthesis, but also now actively degrade ATP, as the proton-translocating ATPase reverses. Left unchecked, this inevitably leads to a loss of metabolic and ionic integrity of the cells, and ultimately to cell death (Halestrap 2004). Altered cellular bioenergetics in aging related neurodegeneration is attributed to the decreased activity of Complexes I, II and IV leading to chronic inflammation and triggering of apoptotic cell death pathways (Menardo et al. 2012). Hypoxic stress has also been associated with reduced Complex I and Complex IV activity. Interestingly, supplementation of Acetyl-L-Carnitine that is known to improve mitochondrial biogenesis and ATP generation has been found to be beneficial in both hypoxia and aging.
12.8 Role of Antioxidant Supplementation in Preventing Hypoxic Neurodegeneration
Antioxidant supplementation has been a widely accepted prophylactic strategy for both hypoxia and aging. Antioxidants like carnosine, melatonin and herbal extracts of gingko have been reported to reduce aging related changes in the brain. Antioxidants like N-Acetyl Cysteine and Acetyl-L-Carnitine , on the other hand, have been reported to ameliorate hypoxia induced neurodegeneration . Studies carried out by Barhwal et al. (2007) have shown improved working and reference memory of hypoxic rats supplemented with Acetyl-L-Carnitine. The nootrophic effect of Acetyl-L-Carnitine is due to its ability to augment NGF-TrkA mediated neurotrophin signaling mechanisms and stabilization of Nrf-2 through ERK mediated mechanisms . This probably explains the role of Acetyl-L-Carnitine as an antioxidant despite its inability to directly quench the free radicals. Besides that, Acetyl-L-Carnitine also mediates mitochondrial biogenesis resulting in buffering of calcium ions into nonfunctional mitochondria during hypoxic stress . Alpha lipoic acid and ascorbic acid supplementation also protect neurons from oxidative stress mediated cell death by directly quenching the free radicals. However, the efficacy of these antioxidants in delaying aging related cognitive impairment and prevention of age related neurodegenerative disorders remains to be conclusively proved for human population .
12.9 Implications of Hypoxic Research for Aging Related Cognitive Impairment
Though the causative factors for hypoxia and aging related cognitive impairment appear to differ distinctly, the down stream events resulting in compromised neuronal activity during both these conditions are similar to a great extent. The pathophysiology in both these conditions is invariably associated with oxidative stress , altered calcium ion homeostasis and mitochondrial dysfunction . Inflammatory responses and activation of microglia play a key role in both aging and hypoxic neuronal damage. At the molecular level, the hypoxia inducible factor (HIF) is the master regulator for hypoxia-induced gene expression. Recent studies displayed age-related changes in the HIF system that might explain reduced ability to cope with hypoxia in elderly. Conversely, oxidative damage to sub cellular components following hypoxic insult could accelerate aging and age associated neurodegenerative disorders . Future research on the effect of hypoxia on genomic and proteomic changes in the neuronal cells and the effect of these changes on aging could surely help in identifying prophylactic and therapeutic targets for delaying aging and age related cognitive impairment.
References
Asghar M, George L, Lokhandwala MF (2007) Exercise decreases oxidative stress and inflammation and restores renal dopamine D1 receptor function in old rats. Am J of Physiol Renal Physiol, 293:F914–9
Bano D, Agostini M, Melino G, Nicotera P (2011) Aging, neuronal connectivity and brain disorders: an unsolved ripple effect. Mol Neurobiol 43:124–130
Barhwal K, Hota SK, Baitharu I, Prasad D, Singh SB, Ilavazhagan G (2009a) Isradipine antagonizes hypobaric hypoxia induced CA1 damage and memory impairment: complementary roles of L-type calcium channel and NMDA receptors. Neurobiol Dis 34:230–244
Barhwal K, Hota SK, Jain V, Prasad D, Singh SB, Ilavazhagan G (2009b) Acetyl-l-carnitine prevents hypobaric hypoxia--induced spatial memory impairment through extracellular related kinase--mediated nuclear factor erythroid 2-related factor 2 phosphorylation. Neuroscience 161:501–514
Barhwal K, Hota SK, Prasad D, Singh SB, Ilavazhagan G (2008) Hypoxia induced deactivation of NGF mediated ERK ½ signaling in hippocampal cell culture: neuroprotection with Acetyl-L-Carnitine. J Neurosci Res 86:2705–2721
Barhwal K, Singh SB, Hota SK, Jayalakshmi K, Ilavazhagan G (2007) Acetyl-L-Carnitine ameliorates hypobaric hypoxic impairment and spatial memory deficits in Rats. Eur J Pharmacol 570:97–107
Biessels G, Gispen WH (1996) The calcium hypothesis of brain aging and neurodegenerative disorders: significance in diabetic neuropathy. Life Sci 59:379–387
Blum J, Fridovich I (1985) Inactivation of glutathione peroxidase by superoxide radical. Arch Biochem Biophys 240:500–508
Calabrese V, Scapagnini G, Giuffrida Stella AM, Bates TE, Clark JB (2001) Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res 26:739–64
Cavaletti G, Moroni R, Garavaglia P, Tredici G (1987) Brain damage after high-altitude climbs without oxygen. Lancet 10:101
Denison DM, Ledwith F, Poulton EC (1966) Complex reaction times at simulated cabin altitudes of 5,000 feet and 8,000 feet. AeMed 37:1010–1013
Ernsting J (1978) Prevention of hypoxia--acceptable compromises. Aviat Space Environ Med, 49:495–502
Escames G, López A, García JA, García L, Acuña-Castroviejo D, García JJ, López LC (2010) The role of mitochondria in brain aging and the effects of melatonin. Curr Neuropharmacol 8:182–93
Favreau LV, Pickett CB (1995) The rat quinone reductase antioxidant response element. Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines. J Biol Chem 270:24468–24474
Floyd RA, Hensley K (2002) Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23:795–807
Foster TC (2007) Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 6:319–325. (Review)
Fuentes-Prior P, Salvesen GS (2004) The protein structures that shape caspase activity, specificity, activation and inhibition. J Biochem (Tokyo) 384:201–232
Fukui H, Moraes CT (2009) Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet 18:1028–1036
Grimm S, Hoehn A, Davies KJ, Grune T (2011) Protein oxidative modifications in the aging brain: consequence for the onset of neurodegenerative disease. Free Radic Res 45:73–88
Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K (2000) Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28:285–296
Hackett PH, Roach RC (2001) High-altitude illness. N Engl J Med 345:107–114
Halestrap AP (2004) Mitochondrial permeability: dual role for the ADP/ATP translocator? Nature 430:1
Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59:1609–1623
Hemi T, Ikeda M, Yasuhara T, Murota S (2003) Oxidative neuronal death caused by glutamate uptake inhibition in cultured hippocampal neurons. J Neurosci Res 71: 679–688
Holmgren A (1985) Thioredoxin. Annu Rev Biochem 54:237–271
Honig LS, Rosenberg RN (2000) Apoptosis and neurologic disease. Am J Med 108:317–330
Hopkins RO, Kesner RP, Goldstein M (1995) Item and order recognition memory in subjects with hypoxic brain injury. Brain Cogn 27:180–201
Hornbein TF (1992) Long term effects of high altitude on brain function. Int J Sports Med 1:S43-S45
Sunil Kumar Hota, Kalpana Barhwal Hota, Dipti Prasad, Govindasamy Ilavazhagan, Shashi Bala Singh (2010) Oxidative-stress-induced alterations in Sp factors mediate transcriptional regulation of the NR1 subunit in hippocampus during hypoxia. Free Radic Biol Med 49:178–191
Hota SK, Barhwal K, Baitharu I, Prasad D, Singh SB, Ilavazhagan G (2009) Bacopa monniera leaf extract ameliorates hypobaric hypoxia induced spatial memory impairment. Neurobiol Dis 34:23–39
Hota SK, Barhwal K, Singh SB, Sairam M, Ilavazhagan G (2008) NR1 and GluR2 expression mediates excitotoxicity in chronic hypobaric hypoxia. J Neurosci Res 86:1142–1152
Hota SK, Barhwal K, Singh SB, Ilavazhagan G (2007) Differential temporal response of hippocampus, cortex and cerebellum to hypobaric hypoxia: a biochemical approach. Neurochem Int 51:384–390
Houston CS, Sutton JR, Cymerman A, Reeves JT (1987) Operation Everest II: man at extreme altitude. J Appl Physiol 63:877–882
Huang HC, Nguyen T, Pickett CB (2000) Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Nat Acad Sci U S A 97:12475–12480
Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–22
Kerr J F, Wyllie A H Currie A R (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257
Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N (1996) Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Letters 397:230–234
Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M (2006) Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol Cell Biol 26:221–229
Lee JM, Shih AY, Murphy TH, Johnson JA (2003) NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem 278:37948–37956
Menardo J, Tang Y, Ladrech S, Lenoir M, Casas F, Michel C, Bourien J, Ruel J, Rebillard G, Maurice T, Puel JL, Wang J (2012) Oxidative stress, inflammation, and autophagic stress as the key mechanisms of premature age-related hearing loss in SAMP8 mouse cochlea. Antioxid Redox Signal 16:263–74
Maiti P, Singh SB, Sharma AK, Muthuraju S, Ilavazhagan G, Banerjee PK (2006) Hypobaric hypoxia induces oxidative stress in rat brain. Neurochem Int 49:709–716
McFarland RA (1937) Pscycho-physiological studies at high altitude in the Andes II: sensory and motor responses during acclimatization. Comp Psychol 23:227–258
Muthuraju S, Maiti P, Solanki P, Sharma AK, Amitabh, Singh SB, Prasad D, Ilavazhagan G (2009) Acetylcholinesterase inhibitors enhance cognitive functions in rats following hypobaric hypoxia. Behav Brain Res 203:1–14
Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K (2003) PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Lett 546:181–184
Nicotera P, Leist M, Ferrando-May E (1998) Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett 102:139–142
Paradies G, Petrosillo G, Paradies V, Ruggiero FM (2011) Mitochondrial dysfunction in brain aging: role of oxidative stress and cardiolipin. Neurochem Int 58:447–57
Patel JR, Brewer GJ (2008) Age-related differences in NFkappaB translocation and Bcl-2/Bax ratio caused by TNFalpha and Abeta42 promote survival in middle-age neurons and death in old neurons. Exp Neurol 213:93–100
Peacock AJ, Jones PL (1997) Gas exchange at extreme altitude: results from the British 40th Anniversary Everest Expedition. Eur Respir J 10:1439–1444
Pelamatti G, Pascotto M, Semenza C (2003) Verbal free recall in high altitude: proper names vs. common names. Cortex 39:97–103
Pugh LG (1964) Man at high altitude: studies carried out in the Himalaya. Sci Basis Med Annu Rev 32–54
Regard M, Oelz O, Brugger P, Landis T (1989) Persistent cognitive impairment in climbers after repeated exposure to extreme altitude. Neurology 39:210–213
Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS (2005) Controlled elimination of intracellular H2O2: regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal 7:619–626
Sastry PS, Rao KS (2000) Apoptosis and the nervous system. J Neurochem 74:1–20
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli KJ, Debatin KM, Krammer PH, Peter ME (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J 17:1675–1687
Schliebs R, Arendt T (2006) The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J Neural Transm 113:1625–1644
Stickney JC, Van Liere EJ (1953) Acclimatization to low oxygen tension. Physiol Rev 33:13–34
Titus AD, Shankaranarayana Rao BS, Harsha HN, Ramkumar K, Srikumar BN, Singh SB, Chattarji S, Raju TR (2007) Hypobaric hypoxia-induced dendritic atrophy of hippocampal neurons is associated with cognitive impairment in adult rats. Neuroscience 145:265–278
Toescu EC, Verkhratsky A (2007) The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 6:267–273 (Review)
Toman J, Fiskum G (2011) Influence of aging on membrane permeability transition in brain mitochondria. J Bioenerg Biomembr 43:3–10
Wang X, Michaelis EK (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 30:2–12
Wang JY, Shum AY, Wang JY (2002) Hypoxia/reoxygenation induces cell injury via different mechanisms in cultured rat cortical neurons and glial cells. Neurosci Lett 322:187–191
West JB (1988) High points in the physiology of extreme altitude. Adv Exp Med Biol 227:1–15
West JB (2002) Unexplained severe fatigue and lassitude at high altitude. High Alt Med Biol 3:237–241
West JB (2004) Paralysis and blindness during a balloon ascent to high altitude. High Alt Med Biol 5:453–456
Won SJ, Kim DY, Gwag BJ (2002) Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol 35:67–86
Yang JC, Cortopassi GA (1998) Induction of the mitochondrial permeability transition causes release of the apoptogenic factor cytochrome c. Free Radic Biol Med 24:624–631
Zhang H, Liu H, Davies KJ, Sioutas C, Finch CE, Morgan TE, Forman HJ (2012) Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic Biol Med [Epub ahead of print]
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Hota, K., Hota, S., Singh, S. (2012). Neurodegeneration in Hypoxia: Implications in Aging. In: Thakur, M., Rattan, S. (eds) Brain Aging and Therapeutic Interventions. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5237-5_12
Download citation
DOI: https://doi.org/10.1007/978-94-007-5237-5_12
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5236-8
Online ISBN: 978-94-007-5237-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)