
Abstract
Calcium and phosphorus are essential for a myriad of intracellular functions and also form the basis for the structural integrity of bone. As such, mechanisms have evolved to ensure exquisite control over their circulating concentrations. These concentrations are largely maintained by fluxes of these mineral ions across the intestine, kidney, and bone and are regulated by three major hormones, parathyroid hormone (PTH), the active form of vitamin D, 1,25-dihydroxyvitamin D, and fibroblast growth factor-23 (FGF23). Each hormone acts to directly influence mineral ion transport across intestine or kidney and may also regulate mineral ion entry into and out of bone. The production and secretion of each hormone may in turn be modulated by circulating concentrations of these mineral ions and by the action of the other hormones, producing a complex network of negative and positive feedback systems. Disruption of these homeostatic systems can produce dramatic disease profiles but improved understanding of the underlying molecular mechanisms may lead to more salutary approaches to therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Parathyroid hormone
- Vitamin D
- Fibroblast growth factor-23
- Calcium
- Phosphorus
- Kidney
- Intestine
- Bone
- Ion transport
- Feedback loops
1 Calcium and Phosphorus Balance
The skeleton, the gut, and the kidney each play a major role in assuring calcium (Ca++) homeostasis. Overall, in a typical individual if 1,000 mg of Ca++ are ingested in the diet per day, approximately 200 mg will be absorbed across the intestinal epithelium and about 200 mg excreted.
The traditional model of transcellular Ca++ transport consists of influx through an apical calcium channel (TRPV6), which provides the rate-limiting step, diffusion through the cytosol, and active extrusion at the basolateral membrane by a plasma membrane ATPase (PMCA1b) [1]. Although entry of Ca++ has been reported to involve TRPV6, other Ca++ channels may also be involved. Ca++ binding proteins including calmodulin and calbindin-D9k (CaBP9k) may be important for fine-tuning Ca++ channel activity, and in the cytosol, calbindin 9 k may “buffer” and/or mediate the transit of intracellular absorbed Ca++ to the basolateral membrane. An additional modulator of transcellular Ca++ transport is a Ca ATPase, PMCA1b, encoded by ATP2B1, which is important for the extrusion of Ca++ at the basolateral membrane to complete the transcellular transport of this ion. Increasing evidence also suggests the importance of paracellular Ca++ transport in Ca++ absorption via tight junctions.
The skeleton, where approximately 1,000 mg of calcium is stored, is the major Ca++ reservoir in the body. Skeletal Ca++ is stored mainly in the form of hydroxyapatite crystals, the major inorganic component of the mineralized bone matrix. Ordinarily as a result of normal bone turnover, approximately 500 mg of Ca++ is resorbed from the bone per day and the equivalent amount is accreted. Approximately 10 g of Ca++ will be filtered daily through the kidney and most will be reabsorbed, with about 200 mg being excreted in the urine. The normal 24-h urine excretion of Ca++ may however vary between 100 and 300 mg per day (2.5–7.5 mmoles per day).
The average consumption of phosphorus (Pi) (i.e., about 1,000–1,500 mg) in a Western diet is similar to that of Ca++ (about 1,000 mg); however, about 70 % of phosphorus is absorbed daily compared with only about 20–30 % of Ca++. Pi ingested through the diet is absorbed by the small intestine through sodium–phosphate cotransporters, as well as by sodium-independent diffusional absorption across intercellular spaces in the lumen. The skeleton also represents the largest reservoir in the body of Pi which is stored in an exact stoichiometry with Ca++ as hydroxyapatite crystals. The major control site for Pi homeostasis is the kidney where enhanced reabsorption can occur from the tubular lumen via sodium–phosphate cotransporters, NaPi2s (also known as SLC34As) that are expressed in the renal proximal convoluted tubule [2] (see also Chap. 6).
2 Hormones Regulating Ca++ and Pi Homeostasis
2.1 Parathyroid Hormone (PTH)
PTH secretion from the parathyroid gland and parathyroid cell proliferation are inhibited by serum Ca++acting via a Ca++-sensing receptor (CaSR) [3], and PTH gene transcription [4] and parathyroid cell proliferation [5, 6] may be inhibited by the active form of vitamin D, 1,25-dihydroxyvitamin D [1,25(OH)2D], acting via the vitamin D receptor (VDR). Although the major glandular form of PTH is an 84 amino acid peptide, virtually all of the biological activity resides within its amino (NH2)-terminal domain [7, 8]. Intracellular degradation of PTH(1–84) within the parathyroid cell, which is enhanced by high ambient calcium concentrations, provides a means of modulating the fraction of secreted hormone that is PTH(1–84) (see Chap. 4 for details). The NH2-terminal domain interacts in target tissues with a classical G protein-coupled receptor (GPCR) termed the PTH/PTHrP receptor type 1, or PTHR1 [9, 10]. PTHR1 couples to several G protein subclasses, including Gs, Gq/11, and G12/13, resulting in the activation of many pathways, although the best studied are the adenylate cyclase (AC) and phospholipase C (PLC) pathways.
In the kidney, Ca++ reabsorption primarily occurs in the distal tubules and collecting ducts [11]. Ca++ ions cross the apical membrane from the tubular lumen via TRPV5, a highly selective Ca++channel, and are then transported across the basolateral membrane into the blood system by the sodium/calcium exchanger 1 (NCX1) and a plasma membrane ATPase. PTH regulates the expression of TRPV5 and NCX1 [12] as well as their activity. The PTH regulation of at least TRPV5 appears to occur through protein kinase C (PKC) [13]. TRPV5 is also regulated through the PKC-signaling pathway by the Ca++ sensing receptor (CaSR) [14] (see also Chap. 5).
Pi reabsorption across the apical membrane of renal proximal tubules mainly occurs through two sodium-dependent phosphate cotransporters, NaPi-2a and NaPi-2c, that are exclusively expressed in the brush border membrane of the proximal tubules [15] (see also Chap. 6). PTH increases Pi excretion in the proximal tubule mainly by reducing the levels of these transporters. Thus, PTH binding to PTHR1 at either apical or basolateral membrane results in removal of the transporters from the brush border membrane via clathrin-coated pits [16, 17]. PTH activation of apical PTHR1 leads to phospholipase C (PLC)–protein kinase C (PKC) stimulation mediated by sodium–hydrogen exchanger regulatory factor (NHERF), whereas activation of basolateral PTHR1 utilizes the AC/protein kinase A (PKA) pathway [18]. Following endocytosis NaPi-2 is eventually transported to the lysosomes for degradation.
PTH also stimulates the conversion of 25-hydroxyvitamin D [25(OH)D] to 1,25(OH)2D in the kidney by transcriptional activation of the gene encoding the 25-hydroxyvitamin D-1α hydroxylase [1(OH)ase] enzyme (CYP27B1) apparently by the AC/PKA pathway [19] (see also Chap. 11).
The bone undergoes constant remodeling in response to endocrine, autocrine/paracrine, and intracrine signals and bone mineral is maintained through a balance between bone formation and resorption. PTH binds to PTHR1 on cells of the osteoblastic lineage [20] including progenitor cells, osteoblasts, and osteocytes and can stimulate a variety of factors ultimately leading to increased proliferation of mesenchymal stem cells, such that these cells are committed into the osteoblast lineage, to enhance osteoblast differentiation and activity with new bone matrix production [21] and ultimately mineralization of bone tissue. However, osteoblastic cells also produce the TNF-related cytokine, receptor activator of nuclear factor κ-B (RANK) ligand (RANKL), a critical stimulator of osteoclast production and action, as well as the soluble RANKL decoy receptor, osteoprotegerin (OPG) [22]. PTH enhances production of RANKL and inhibits production of OPG leading to increased osteoclastogenesis and osteoclastic bone resorption [23]. Bone resorption leads to the release of Ca++ and Pi as a result of the degradation of hydroxyapatite, and it is the resorptive effect of PTH which plays a major role in mineral and particularly Ca++ homeostasis
2.2 Vitamin D
2.2.1 Metabolism of Vitamin D
Vitamin D can be obtained as vitamin D3 (cholecalciferol), via UV-light irradiation of a skin precursor, 7-dehydrocholesterol [24], or can be ingested from the diet as vitamin D3 or as the plant-derived sterol vitamin D2 (ergocalciferol). Vitamin D is then transported to the liver, bound to a plasma vitamin D binding protein (DBP) [25], where it is hydroxylated at the C-25 position of the side chain to produce 25(OH)D, the most abundant circulating form of vitamin D [26]. The final step in the activation to the hormonal form, 1,25(OH)2D, occurs mainly, but not exclusively, in the kidney via a tightly regulated 1α-hydroxylation reaction catalyzed by a mitochondrial enzyme 1(OH)ase, or CYP27B1 [27]. The renal CYP27B1 gene is stimulated by PTH and hypocalcemia and inhibited by hyperphosphatemia thus sensing the need for mineral homeostasis. The renal CYP27B1 gene is also product-inhibited by 1,25(OH)2D, which acts via a short negative feedback loop to limit its own production [28]. 1,25(OH)2D circulates bound to vitamin D binding protein (DBP), to exert its endocrine actions in various target tissues. Extrarenal 1α-hydroxylation has also been described resulting in the production of 1,25(OH)2D, which can act locally in an intracrine mode [29]. The 1,25(OH)2D-mediated endocrine or intracrine signal may be terminated in all target cells via the catalytic action of CYP24A1, an enzyme that initiates the process of 1,25(OH)2D catabolism [30]. The CYP24A1 gene is transcriptionally activated by 1,25(OH)2D [31]. This feed forward induction of 1,25(OH)2D catabolism therefore prevents hypervitaminosis D.
2.2.2 Actions of Vitamin D
The active metabolite, 1,25(OH)2D, functions by initially binding to the VDR. The ligand-activated VDR interacts with the retinoid X receptor (RXR) to form a heterodimer that binds to vitamin D responsive elements in the region of genes directly regulated by 1,25(OH)2D [32]. By recruiting complexes of either coactivators or corepressors, ligand-activated VDR–RXR modulates the transcription of genes encoding proteins that carry out the functions of vitamin D.
Under conditions of low dietary Ca++, the 1,25(OH)2D/VDR system induces TRPV6 [33] and vitamin D-dependent calcium binding protein 9K (CaBPD9k) [34], to promote transcellular intestinal calcium absorption; the extrusion of calcium at the basolateral membrane is likely constitutive in part and is executed by the Ca ATPases PMCA1b/PMCA2c but could be amplified via induction of these molecules by 1,25(OH)2D. 1,25(OH)2D also increases expression of claudins 2 and 12 to possibly promote paracellular Ca++ entry [35]. Ca++ absorption under normal dietary conditions postnatally does not absolutely require TRPV6 and CaBPD9K.
Pi is predominantly absorbed in the intestine via paracellular mechanisms. Although 1,25(OH)2D may also increase intestinal Pi absorption by increasing expression of NaPi2b [36], because Pi is abundant in the diet, the Pi absorption effect of 1,25(OH)2D may not be as profound as the effect on Ca++ transport. The 1,25(OH)2D/VDR system can promote bone resorption [37–39], suggesting that at least part of its skeletal action is catabolic. In support of this, the 1,25(OH)2D/VDR system enhances the expression of RANKL in osteoblastic cells to stimulate bone resorption through osteoclastogenesis [40]. Furthermore, osteoprotegerin, the soluble decoy receptor for RANKL that tempers its activity, is repressed by the 1,25(OH)2D/VDR system, thus amplifying the bioeffect of RANKL.
The action of the 1,25(OH)2D/VDR system in the parathyroid gland to suppress PTH synthesis [4] effectively limits PTH-induced bone-resorbing activity and restricts PTH stimulation of renal CYP27B1 thereby preventing hypercalcemia.
2.3 Fibroblast Growth Factor 23 (FGF23)
Fibroblast growth factors (FGFs) are a large superfamily of peptides that act mainly as paracrine/autocrine substances to exert a broad range of biological functions in development and organogenesis. They act by binding and activation of FGF receptor (FGFR) tyrosine kinases [41, 42]. The unique FGF19 subfamily consists of FGF19, FGF21, and FGF23 [43] which act as hormones to regulate energy and mineral metabolism (see also Chap. 6).
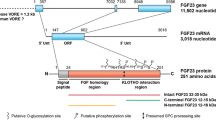
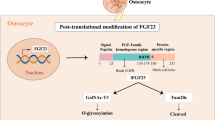
FGF23 is a phosphaturic hormone produced and secreted by bone cells of the osteoblastic lineage, predominantly late osteoblasts and osteocytes, and was initially identified as the mediator of the human disorder autosomal dominant hypophosphatemic rickets (ADHR) [44]. FGF23 is synthesized as a 32 kDa, 251 amino acid protein, with a signal (leader) sequence of 24 amino acids, an NH2-terminal FGF homology domain of 155 amino acids, and a unique sequence in its carboxyl (COOH)-domain of 72 amino acids [45]. The molecule can be cleaved between Arg179 and Ser180 by a subtilisin-like proprotein convertase, yet to be identified, to generate NH2-terminal and COOH-terminal fragments; the entire sequence of the secreted 25–251 amino acid protein appears to be necessary for its biological action. O-glycosylation within the 162–228 region apparently reduces the susceptibility of the protein to proteolysis [46], and it is possible that the COOH-terminal fragment may compete with intact FGF23 for binding to its receptor complex and function as a competitive inhibitor [47]. ADHR patients carry missense mutations at the proteolytic cleavage site of FGF23 (176RXXR179), which confers resistance to inactivation by proteolytic cleavage [48]. As a result, ADHR patients exhibit increased blood levels of intact FGF23 and Pi-wasting phenotypes.
2.3.1 Regulation of FGF23 Production
2.3.1.1 Local Regulators of FGF23 Production
Local regulators produced in osteoblast/osteocytes appear to be important in modulating the release of FGF23. Mutations in the gene encoding the membrane protein PHEX (phosphate-regulating neutral endopeptidase with homology to endopeptidase on the X chromosome) [49–51] and in the gene encoding the SIBLING (small integrin-binding ligand interacting glycoproteins) protein DMP‑1 (dentin matrix protein-1) [52, 53] increase FGF23 expression and induce renal Pi wasting in mice and humans. In one apparent mechanism [54], DMP-1 binds to the osteocyte through its integrin-binding domains and to PHEX through its ASARM domain [acidic serine aspartate-rich matrix extracellular phosphoglycoprotein (MEPE)-associated motif]. Binding of DMP‑1 to PHEX inhibits production of active FGF‑23, while disruption of either DMP‑1 or PHEX releases this inhibition, and active FGF23 production is increased. Local relative levels of the mineralization regulators pyrophosphate and phosphate appear to be important modulators [55, 56] and disruption of bone mineralization may release low molecular weight FGFs from the bone matrix which may activate the osteocyte FGFR and stimulate transcription of the FGF‑23 gene [57]. Finally reduced iron and tissue hypoxemia have also been reported to stimulate FGF23 release [58].
2.3.1.2 Systemic Regulators
Extremely high FGF23 levels are observed in primary deficiency of the Klotho protein (genetic deletion or mutational hypomorph) [59, 60] and the much more common secondary Klotho deficiency in chronic kidney disease (CKD) [61, 62]. However, there are no in vitro data to date to support a direct effect of Klotho, either locally produced or circulating, on FGF23 production.
A critical regulator of FGF23 release is 1,25(OH)2D, acting via the VDR [63] in part by directly upregulating gene expression [64] and in part by inhibiting PHEX and by inducing expression of the gene encoding ecto-nucleotide pyrophosphatase/phosphodiesterase (Enpp1), which could alter local levels of pyrophosphate and phosphate [65]. 1,25(OH)2D may also increase the levels of the hypoxia-inducible transcription factor, HIF1A, which might mediate the effects of tissue hypoxemia on FGF23 release [66]. PTH has also been reported to stimulate synthesis and secretion of FGF23 through activation of the PTHR1 on osteocytes/osteoblasts [67, 68].
Increased Pi in the diet and increases in serum Pi can both increase FGF23 release by osteocytic cells; however, the mechanism is still unclear. Nevertheless, this observation has led to the concept that the skeleton therefore serves as a sensor of Pi levels in a manner analogous to the function of the parathyroid glands as a Ca++ sensor.
FGF23 is also regulated by serum Ca++ [69–71], as initially suggested by several lines of evidence. Thus, serum Ca++ levels are independently associated with FGF23 levels in dialysis patients, in transplant recipients, and in patients with primary hyperparathyroidism [3]. In patients with severe secondary and tertiary (persistent) hyperparathyroidism referred for parathyroidectomy, postoperative changes of FGF23 are limited and related to changes of Ca++ [72]. In patients with acute untreated hypoparathyroidism occurring after thyroidectomy [73], FGF23 levels are initially reduced when patients are hyperphosphatemic but still hypocalcemic. In contrast, in patients with chronic hypoparathryoidism and hyperphosphatemia who are normocalcemic on calcium and calcitriol treatment, serum FGF-23 levels are elevated [74]. Consequently treatment of hypocalcemia in patients with chronic hypoparathyroidism may raise FGF23 which will promote phosphaturia, but the serum levels of phosphorus concentrations may not completely normalize, possibly because the concerted phosphaturic actions of both PTH and FGF23 may be required.
In vitamin D receptor-null mice, dietary calcium supplementation significantly increases serum calcium levels, FGF23 messenger RNA abundance, and circulating FGF23 levels. In PTH-null mice that are hypocalcemic and hyperphosphatemic, FGF23 levels are reduced [75]. In wild-type mice and PTH-null mice, acute elevation of either serum Ca++ or Pi by intraperitoneal injection increased serum FGF23 levels. However, increases in serum Pi by chronic exposure to a high dietary Pi load were accompanied by severe hypocalcemia, which appeared to blunt stimulation of FGF23 release. Calcium-mediated increases in serum FGF23 required a threshold of at least normal serum Pi levels. Similarly, Pi-elicited increases in FGF23 were markedly blunted if serum Ca++ was less than normal. The best correlation between Ca++ and Pi and serum FGF23 was found between FGF23 and the Ca++ × Pi product.
2.3.2 Actions of FGF23
In addition to being regulated by serum Ca++ and Pi levels, FGF23 in turn acts to modulates serum Ca++ and Pi levels, thus ensuring that the Ca++ × Pi product remains within a physiological range. FGF23 is therefore not only a phosphoregulatory hormone but a dual calciophosphoregulatory hormone (Figs. 8.1 and 8.2).

Model of hormonal regulation of Pi (and Ca++) homeostasis. In the presence of normal renal function, increased dietary phosphorus (Pi) (1) facilitated by 1,25(OH)2D action on the intestine (2) may increase serum Pi levels. Increased Pi levels can directly or indirectly increase PTH secretion (3) leading to increased renal Pi excretion (4), but also to increased renal Ca++ retention, increased 1,25(OH)2D production (5), and increased release of Ca++ and Pi from the bone (6). The increased serum Pi in the presence of a threshold level of serum Ca++ can now increase FGF23 secretion from the bone (7), as well as PTH (8), and 1,25(OH)2D (8) which may also each increase FGF23 production. Secreted FGF23 can decrease 1,25(OH)2D (9) thus reducing its capacity to further enhance intestinal Pi absorption and can also inhibit PTH (10) thus reducing stimulation of renal production of 1,25(OH)2D, Pi mobilization from the bone, and enhancement of Pi excretion. Serum Pi per se may also reduce 1,25(OH)2D production (11). FGF23 can inhibit proximal tubular NaPi2a and 2c (12) and produce phosphaturia (13) in place of the suppressed PTH, thereby reducing the Pi (and Ca++ × Pi product). With prolonged elevation of FGF23 and suppression of 1,25(OH)2D, secondary hyperparathyroidism may occur to prevent significant hypocalcemia

Model of hormonal regulation of Ca++ (and Pi) homeostasis. Increased intestinal absorption of Ca++ (1) facilitated by increased 1,25(OH)2D (2) or increased bone resorption (3) or both may result in increased serum calcium (Ca++) (and Pi). Serum Ca++, by stimulating the renal CaSR (4), can enhance Ca++ excretion (5). Serum Ca++, acting via the CaSR in the parathyroid gland, can inhibit PTH secretion (6). Decreased PTH results in reduced mobilization of skeletal Ca++ (and Pi) (7), reduced renal production of 1,25(OH)2D (8), and reduced renal Ca++ retention (9) but also in reduced renal Pi clearance. Elevated serum Ca++ in the presence of high normal or elevated serum Pi can then increase FGF23 secretion (10) which can further inhibit PTH secretion (11) and kidney-derived 1,25(OH)2D (12) and normalize the Ca++ × Pi product
FGF23 combines with an FGFR, as well as with Klotho, an obligate coreceptor, in order to transmit the signal of FGF23 to target organs [76]. Thus, Klotho protein forms constitutive binary complexes with FGFR1c, FGFR3c, and FGFR4 which increase the affinity of these FGFRs selectively to FGF23 [77] and produce a heterotrimeric complex which is required for FGF23 to activate downstream signaling molecules, including FGFR substrate-2α and mitogen-activated protein kinases (MAPKs) such as extracellular signal-regulated kinases (ERK1/2). Klotho is a type I membrane protein but may also be expressed, due to alternative RNA splicing, as a secreted form that lacks the transmembrane and intracellular domains; it may therefore also act as a humoral factor [78] and may also have β-glucuronidase activity [79]. A unique structural feature of the endocrine FGFs, including FGF23, is their lack of a heparin-binding domain that is conserved in all paracrine/autocrine FGFs [80]. This heparin-binding domain binds to heparan sulfate (HS) in the extracellular matrix, thereby imposing some restriction to the secretion of non-endocrine FGFs and increasing their local concentration to support their paracrine/autocrine mode of action; in addition, the HS-binding domain is essential for FGFR activation, forming a complex of HS, FGF, and FGFR [81]. Absence of the heparin-binding domain in endocrine FGFs may facilitate their release from sites of production and Klotho proteins substitute for HS in enhancing receptor binding by endocrine FGFs. Although FGFRs are quite ubiquitous, Klotho expression is relatively restricted and may confer tissue specificity for FGF23 action [42].
The major target for FGF23 is the kidney, where it acts to promote phosphate excretion and to decrease production and increase clearance of 1,25(OH)2D. FGF23 suppresses Pi reabsorption [28] by inhibiting NaPi2a and NaPi2c on the apical brush border membrane of proximal tubular cells [82]. Although all the FGF23 actions seem to occur in the proximal tubule, Klotho expression is higher in the distal tubules [83]. Because proximal tubules also express Klotho, albeit in lower quantities [84], FGF23 may signal directly in proximal tubules to regulate their function with a small number of FGFR–Klotho complexes. Alternatively FGF23 may act on distal convoluted tubules where Klotho is most abundantly expressed and initiate release of a paracrine factor(s) that acts on adjacent proximal tubules. It is currently unclear but unlikely that circulating Klotho can serve as a coreceptor for FGFR.
FGF23 also lowers blood levels of 1,25(OH)2D by downregulating the expression of the CYP27B1 gene [31] and by upregulating gene expression of 24-hydroxylase (CYP24A1), which converts 1,25(OH)2D to inactive metabolites [85]. Thus, FGF23 suppresses synthesis and promotes degradation of the active hormonal form of vitamin D. By diminishing circulating 1,25(OH)2D levels, FGF23 can therefore also indirectly reduce vitamin D-stimulated Pi (and Ca++) absorption in the intestine (Figs. 8.1 and 8.2).
In addition, FGF23 acts directly on the parathyroid gland FGFRs (likely via FGFR1 and FGFR3) and Klotho [26, 27] in an ERK1/2-dependent manner [86, 87] to suppress PTH synthesis and secretion [86, 87]; FGF23 may also increase the levels of parathyroid CaSR and VDR to indirectly inhibit PTH gene expression, secretion, and cell proliferation via Ca++ and 1,25(OH)2D respectively [88]. However, in most clinical and pathological situations associated with chronically increased circulating FGF23 concentrations, secondary hyperparathyroidism is present. In mice overexpressing FGF23 [89], secondary hyperparathyroidism also tends to occur, suggesting that the effects of FGF23 on Pi, Ca++, and 1,25(OH)D metabolism, which act to stimulate PTH production, may overcome any direct inhibitory effects of FGF23 on PTH release. Downregulation of FGFRs and Klotho in the parathyroids that reduces sensitivity to FGF23 signaling has been reported as the underlying cause of FGF23 resistance in some [90, 91], but not all studies [92] of secondary hyperparathyroidism in uremia. In Hyp mice which have a loss of Phex function with resultant increased FGF23 and which are phenocopies of X-linked hypophosphatemic rickets (XLH) in man, secondary hyperparathyroidism occurs and deletion of the gene encoding PTH results in early lethality due to hypocalcemia [93]. Hyperparathyroidism, therefore, is an integral component in the pathophysiology of Hyp, and likely XLH, despite excess circulating FGF23 and may serve as a compensatory mechanism to prevent severe hypocalcemia in mice and perhaps in patients afflicted with the disorder.
3 Endocrine Regulation of Pi and Ca++ Metabolism
When renal function is normal, in the presence of an increased dietary load of Pi, serum Pi levels may increase, enhanced by 1,25(OH)2D action on the intestine. This increased serum Pi may directly [94] or indirectly (by reducing serum Ca++ levels) increase PTH secretion and facilitate renal Pi excretion. However, PTH may also mobilize Pi (and Ca++) from the bone and enhance 1,25(OH)2D production with resultant increased intestinal absorption of Pi (and Ca++). In the presence of a threshold level of serum Ca++, Pi can then increase FGF23 secretion from the bone. 1,25(OH)2D per se and PTH may also increase skeletal production of FGF23. Secreted FGF23 can then inhibit 1,25(OH)2D production, thus reducing its capacity to further enhance intestinal Pi absorption, and can also inhibit PTH thus reducing its capacity to mobilize Pi from the bone, as well as to stimulate renal production of 1,25(OH)2D and to promote Pi excretion. Pi per se may also inhibit 1,25(OH)2D production. FGF23 can inhibit proximal tubular NaPi2a and 2c in the kidney and can produce phosphaturia in place of the suppressed PTH, restoring the serum Pi (and Ca++ x Pi product) to normal. With prolonged elevation of FGF23 and prolonged suppression of 1,25(OH)2D, secondary hyperparathyroidism may occur to prevent significant hypocalcemia (Fig. 8.1).
Increased serum Ca++ may arise from increased intestinal absorption of Ca++, (with Pi) or from increased bone resorption (with Pi) or from both mechanisms. The increased serum Ca++ per se by stimulating the renal CaSR can enhance renal Ca++ excretion. The increased serum Ca++ acting via the CaSR in the parathyroid gland can inhibit PTH secretion. Decreased PTH results in reduced bone resorption and concomitant mobilization of skeletal Ca++ (and Pi), decreased renal Ca++ retention, and reduced production of 1,25(OH)2D in the kidney, but also in increased Pi retention. Elevated serum Ca++ in the presence of high normal or elevated serum Pi can also increase FGF23 secretion which can further inhibit PTH secretion, can inhibit kidney-derived 1,25(OH)2D, and replace the action of PTH in promoting phosphaturia (Fig. 8.2), thereby normalizing the Ca++ × Pi product.
These complex interrelationships underlie the exquisite controls that have evolved to maintain serum Pi and Ca++ levels within a defined range when one of these ions is dysregulated but also to ensure an appropriate ratio by maintaining a normal Ca++ × Pi product.
Conclusion
Initial studies on mineral ion regulation focussed on the Ca++-regulating hormones PTH and 1,25(OH)2D, but these were also known to have profound effects on renal and intestinal handling of Pi. Analysis of genetic diseases of renal Pi wasting identified FGF23, as a primary hormonal regulator of Pi homeostasis. Subsequently FGF23 was found to participate in complex feedback loops with the classic Ca++-regulating hormones, and all three hormones are now known to share interacting functions on regulating both Ca++ and Pi homeostasis.
References
Christakos S (2012) Recent advances in our understanding of 1,25-dihydroxyvitamin D(3) regulation of intestinal calcium absorption. Arch Biochem Biophys 523:73–76
Forster IC, Hernando N, Biber J, Murer H (2006) Proximal tubular handling of phosphate: A molecular perspective. Kidney Int 70:1548–1559
Brown EM (2013) Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract Res Clin Endocrinol Metab 27:333–343
Demay MB, Kiernan MS, DeLuca HF, Kronenberg HM (1992) Sequences in the human parathyroid hormone gene that bind the 1,25-dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A 89:8097–8101
Kremer R, Bolivar I, Goltzman D, Hendy GN (1989) Influence of calcium and 1,25-dihydroxycholecalciferol on proliferation and proto-oncogene expression in primary cultures of bovine parathyroid cells. Endocrinology 125:935–941
Panda DK, Miao D, Bolivar I et al (2004) Inactivation of the 25-hydroxyvitamin D 1alpha-hydroxylase and vitamin D receptor demonstrates independent and interdependent effects of calcium and vitamin D on skeletal and mineral homeostasis. J Biol Chem 279:16754–16766
Tregear GW, Van Rietschoten J, Greene E et al (1973) Bovine parathyroid hormone: minimum chain length of synthetic peptide required for biological activity. Endocrinology 93:1349–1353
Goltzman D, Peytremann A, Callahan E et al (1975) Analysis of the requirements for parathyroid hormone action in renal membranes with the use of inhibiting analogues. J Biol Chem 250:3199–3203
Jüppner H, Abou-Samra AB, Freeman M et al (1991) A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 254:1024
Abou-Samra AB, Jüppner H, Force T et al (1992) Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A 89:2732–2736
Lambers TT, Bindels RJ, Hoenderop JG (2006) Coordinated control of renal Ca2+ handling. Kidney Int 69:650–654
van Abel M, Hoenderop JG, van der Kemp AW et al (2005) Coordinated control of renal Ca(2+) transport proteins by parathyroid hormone. Kidney Int 68:1708–1721
Cha SK, Wu T, Huang CL (2008) Protein kinase C inhibits caveolae-mediated endocytosis of TRPV5. Am J Physiol Renal Physiol 294:F1212–F1221
Topala CN, Schoeber JP, Searchfield LE et al (2009) Activation of the Ca(2+)-sensing receptor stimulates the activity of the epithelial Ca(2+) channel TRPV5. Cell Calcium 45:331–339
Custer M, Lotscher M, Biber J et al (1994) Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Physiol 266:F767–F774
Bacic D, Lehir M, Biber J et al (2006) The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int 69:495–503
Segawa H, Yamanaka S, Onitsuka A et al (2007) Parathyroid hormone-dependent endocytosis of renal type IIc Na-Pi cotransporter. Am J Physiol Renal Physiol 292:F395–F403
Traebert M, Volkl H, Biber J et al (2000) Luminal and contraluminal action of 1–34 and 3–34 PTH peptides on renal type IIa Na-P(i) cotransporter. Am J Physiol Renal Physiol 278:F792–F798
Brenza HL, Kimmel-Jehan C, Jehan F et al (1998) Parathyroid hormone activation of the 25-hydroxyvitamin D3-1alpha-hydroxylase gene promoter. Proc Natl Acad Sci U S A 95:1387–1391
Rouleau MF, Mitchell J, Goltzman D (1990) Characterization of the major parathyroid hormone target cell in the endosteal metaphysis of rat long bones. J Bone Miner Res 5:1043–1053
Miao D, He B, Karaplis AC, Goltzman D (2002) Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest 109:1173–1182
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423:337–342
Silva BC, Costa AG, Cusano NE et al (2011) Catabolic and anabolic actions of parathyroid hormone on the skeleton. J Endocrinol Invest 34:801–810
MacLaughlin JA, Anderson RR, Holick MF (1982) Spectral character of sunlight modulates photosynthesis of previtamin D3 and its photoisomers in human skin. Science 216:1001–1003
Dastani Z, Berger C, Langsetmo L et al (2014) In healthy adults, biological activity of vitamin D, as assessed by serum PTH, is largely independent of DBP concentrations. J Bone Miner Res 29:494–499
Zhu JG, Ochalek JT, Kaufmann M et al (2013) CYP2R1 is a major, but not exclusive, contributor to 25-hydroxyvitamin D production in vivo. Proc Natl Acad Sci U S A 110:15650–15655
Jones G, Strugnell SA, DeLuca HF (1998) Current understanding of the molecular actions of vitamin D. Physiol Rev 78:1193–1231
Murayama A, Takeyama K, Kitanaka S et al (1999) Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology 140:2224–2231
Liu P, Stenger S, Li H et al (2006) Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311:1770–1773
St-Arnaud R (2010) CYP24A1-deficient mice as a tool to uncover a biological activity for vitamin D metabolites hydroxylated at position 24. J Steroid Biochem Mol Biol 121:254–256
Shimada T, Hasegawa H, Yamazaki Y et al (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19:429–435
Pike JW, Meyer MB (2014) Fundamentals of vitamin D hormone-regulated gene expression. J Steroid Biochem Mol Biol 144PA:5–11. pii: S0960-0760(13)00234-3. doi:10.1016/j.jsbmb.2013.11.004. PMID: 24239506
Meyer MB, Watanuki M, Kim S et al (2006) The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25-dihydroxyvitamin D3 in intestinal cells. Mol Endocrinol 20:1447–1461
Fleet JC, Wood RJ (1994) Identification of calbindin D-9 k mRNA and its regulation by 1,25-dihydroxyvitamin D3 in Caco-2 cells. Arch Biochem Biophys 308:171–174
Christakos S, Dhawan P, Ajibade D et al (2010) Mechanisms involved in vitamin D mediated intestinal calcium absorption and in non-classical actions of vitamin D. J Steroid Biochem Mol Biol 121:183–187
Haussler MR, Whitfield GK, Kaneko I et al (2013) Molecular mechanisms of vitamin D action. Calcif Tissue Int 92:77–98
Suda T, Takahashi N, Martin TJ (1992) Modulation of osteoclast differentiation. Endocr Rev 3:66–80
Miao D, He B, Lanske B et al (2004) Skeletal abnormalities in Pth-null mice are influenced by dietary calcium. Endocrinology 145:2046–2053
Tanaka H, Seino Y (2004) Direct action of 1,25-dihydroxyvitamin D on bone: VDRKO bone shows excessive bone formation in normal mineral condition. J Steroid Biochem Mol Biol 89–90:343–345
Kim S, Yamazaki M, Zella LA et al (2006) Activation of receptor activator of NF-kappaB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol 26:6469–6486
Eswarakumar VP, Lax I, Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16:139–149
Mohammadi M, Olsen SK, Ibrahimi OA (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 16:107–137
Beenken A, Mohammadi M (2012) The structural biology of the FGF19 subfamily. Adv Exp Med Biol 728:1–24
White KE, Evans WE, O’Riordan JLH et al (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26:345–348
Yamashita T (2005) Structural and biochemical properties of fibroblast growth factor 23. Ther Apher Dial 9:313–318
Kato K, Jeanneau C, Tarp MA et al (2006) Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O‑glycosylation. J Biol Chem 281:18370–18377
Goetz R, Nakada Y, Hu MC et al (2010) Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc Natl Acad Sci U S A 107:407–412. PubMed: 19966287
Bai XY, Miao D, Goltzman D, Karaplis AC (2003) The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem 278(11):9843–9849
Francis F, Hennig S, Korn B et al (1995) A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 11:130–136
Yamazaki Y, Okazaki R, Shibata M et al (2002) Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab 87:4957–4960
Weber TJ, Liu S, Quarles LD (2003) Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res 18:1227–1234
Feng JQ, Ward LM, Liu S et al (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet 38:1310–1315
Turan S, Aydin C, Bereket A, Akcay T, Güran T, Yaralioglu BA, Bastepe M, Jüppner H (2010) Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia. Bone 46:402–409
Rowe PS (2012) The chicken or the egg: PHEX, FGF23 and SIBLINGs unscrambled. Cell Biochem Funct 30:355–375
Huitema LF, Apschner A, Logister I et al (2012) Entpd5 is essential for skeletal mineralization and regulates phosphate homeostasis in zebrafish. Proc Natl Acad Sci U S A 109:21372–21377
Mackenzie NC, Zhu D, Milne EM et al (2012) Altered bone development and an increase in FGF‑23 expression in Enpp1−/− mice. PLoS One 7(2012)
Wohrle S, Bonny O, Beluch N et al (2011) FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res 26:2486–2497
Wolf M, Koch TA, Bregman DB (2013) Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res 28:1793–1803
Kuro-o M, Matsumura Y, Aizawa H et al (1997) Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51
Ichikawa S, Imel EA, Kreiter ML et al (2007) A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest 117:2684–2691
Hu MC, Shi M, Zhang J et al (2011) Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22:124–136
Shimamura Y, Hamada K, Inoue K et al (2012) Serum levels of soluble secreted α-Klotho are decreased in the early stages of chronic kidney disease, making it a probable novel biomarker for early diagnosis. Clin Exp Nephrol 16:722–729
Saito H, Maeda A, Ohtomo S et al (2005) Circulating FGF‑23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem 280:2543–2549
Kolek OI, Hines ER, Jones MD et al (2005) 1{alpha},25-Dihydroxyvitamin D3 upregulates FGF23gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol 289(6):G1036–G1042
Turner AG, Hanrath MA, Morris HA et al (2014) The local production of 1,25(OH)2D3 promotes osteoblast and osteocyte maturation. J Steroid Biochem Mol Biol 144:114–118. doi:10.1016/j.jsbmb.2013.10.003. pii: S0960-0760(13)00196-9
Ormsby RT, Findlay DM, Kogawa M et al (2014) Analysis of vitamin D metabolism gene expression in human bone: Evidence for autocrine control of bone remodelling. J Steroid Biochem Mol Biol 144:110–113. doi:10.1016/j.jsbmb.2013.09.016. pii: S0960-0760(13)00190-8
Rhee Y, Bivi N, Farrow E et al (2011) Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 49:636–643
Lavi-Moshayoff V, Wasserman G, Meir T et al (2010) PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol 299:F882–F889
Lopez I, Rodriguez-Ortiz ME, Almaden Y et al (2011) Direct and indirect effects of parathyroid hormone on circulating levels of fibroblast growth factor 23 in vivo. Kidney Int 80:475–482
Rodriguez-Ortiz ME, Lopez I, Munoz-Castaneda JR et al (2012) Calcium deficiency reduces circulating levels of FGF23. J Am Soc Nephrol 23:1190–1197
Haussler MR, Whitfield GK, Kaneko I et al (2011) The role of vitamin D in the FGF23, klotho, and phosphate bone-kidney endocrine axis. Rev Endocr Metab Disord 13:57–69
Sato T, Tominaga Y, Ueki T et al (2004) Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am J Kidney Dis 44:481–487
Yamashita H, Yamazaki Y, Hasegawa H et al (2007) Fibroblast growth factor-23 (FGF23) in patients with transient hypoparathyroidism: its important role in serum phosphate regulation. Endocr J 54(3):465–470
Gupta A, Winer K, Econs MJ et al (2004) FGF-23 is elevated by chronic hyperphosphatemia. J Clin Endocrinol Metab 89:4489–4492
Quinn SJ, Thomsen AR, Pang JL et al (2013) Interactions between calcium and phosphorus in the regulation of the production of fibroblast growth factor 23 in vivo. Am J Physiol Endocrinol Metab 304(3):E310–E320
Juppner H, Wolf M (2012) αKlotho: FGF23 coreceptor and FGF23-regulating hormone. J Clin Invest 122:4336–4339
Kurosu H, Ogawa Y, Miyoshi M et al (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123
Kurosu H, Yamamoto M, Clark JD et al (2005) Suppression of aging in mice by the hormone Klotho. Science 309:1829–1833
Tohyama O, Imura A, Iwano A et al (2004) Klotho is a novel β-glucuronidase capable of hydrolyzing steroid β-glucuronides. J Biol Chem 279:9777–9784
Goetz R, Beenken A, Ibrahimi OA et al (2007) Molecular insights into the Klotho dependent, endocrine mode of action of FGF19 subfamily members. Mol Cell Biol 27:3417–3428
Urakawa I, Yamazaki Y, Shimada T et al (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–774
Murer H, Hernando N, Forster I, Biber J (2003) Regulation of Na/Pi transporter in the proximal tubule. Annu Rev Physiol 65:531–542
Farrow EG, Davis SI, Summers LJ, White KE (2009) Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol 20:955–960
Hu MC, Shi M, Zhang J et al (2010) Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24:3438–3450
Shimada T, Kakitani M, Yamazaki Y et al (2004) Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113(4):561–568
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V et al (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117:4003–4008
Krajisnik T, Bjorklund P, Marsell R et al (2007) Fibroblast growth factor-23 regulates parathyroid hormone and 1α-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol 195:125–131
Canalejo R, Canalejo A, Martinez-Moreno JM et al (2010) FGF23 fails to inhibit uremic parathyroid glands. J Am Soc Nephrol 21:1125–1135
Bai X, Miao D, Li J et al (2004) Transgenic mice overexpressing human fibroblast growth factor 23 (R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology 145(11):5269–5279
Galitzer H, Ben Dov IZ, Silver J, Naveh-Many T (2010) Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int 77:211–218
Komaba H, Goto S, Fujii H et al (2010) Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients. Kidney Int 77:232–238
Hofman-Bang J, Martuseviciene G, Santini MA et al (2010) Increased parathyroid expression of klotho in uremic rats. Kidney Int 78:1119–1127
Bai X, Miao D, Goltzman D, Karaplis AC (2007) Early lethality in Hyp mice with targeted deletion of Pth gene. Endocrinology 148(10):497
Slatopolsky E, Finch J, Denda M et al (1996) Phosphorus restriction prevents parathyroid gland growth-high phosphorus directly stimulates PTH secretion in vitro. J Clin Invest 97:2534–2540
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Italia
About this chapter
Cite this chapter
Goltzman, D., Karaplis, A.C. (2015). The PTH/Vitamin D/FGF23 Axis. In: Brandi, M., Brown, E. (eds) Hypoparathyroidism. Springer, Milano. https://doi.org/10.1007/978-88-470-5376-2_8
Download citation
DOI: https://doi.org/10.1007/978-88-470-5376-2_8
Published:
Publisher Name: Springer, Milano
Print ISBN: 978-88-470-5375-5
Online ISBN: 978-88-470-5376-2
eBook Packages: MedicineMedicine (R0)