
Abstract
Pseudomonas aeruginosa masters quorum sensing (QS) communication to coordinately regulate pathogenicity-associated group behaviors including the production of virulence factors and biofilm formation, which facilitate the invasion into the hosts, counteract host immune system, as well as promote the resistance/tolerance toward conventional antibiotics. Three main QS systems are employed by the pathogen, denoted as las (Gambello and Iglewski 1991; Passador et al. 1993), rhl (Ochsner et al. 1994; Ochsner and Reiser 1995), and pqs (Pesci et al. 1999). All the networks are hierarchically interconnected: las controls the other two systems; pqs positively regulates the rhl signaling, whereas rhl in turn puts a negative feedback upon pqs (Wilder et al. 2011; McGrath et al. 2004). Regarding the central role of QS for the infectious process, the interruption of these pathways by blocking the receptors or inhibiting the signal synthesis via small molecules is an attractive therapeutic strategy to attenuate the bacterial pathogenicity, thereby overcoming intractable P. aeruginosa infections (Rasmussen and Givskov 2006).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Quorum Sense
- Quorum Sense Inhibition
- Pseudomonas Quinolone Signal
- Virulence Factor Production
- Attractive Therapeutic Strategy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Pseudomonas aeruginosa masters quorum sensing (QS) communication to coordinately regulate pathogenicity-associated group behaviors including the production of virulence factors and biofilm formation, which facilitate the invasion into the hosts, counteract host immune system, as well as promote the resistance/tolerance toward conventional antibiotics. Three main QS systems are employed by the pathogen, denoted as las (Gambello and Iglewski 1991; Passador et al. 1993), rhl (Ochsner et al. 1994; Ochsner and Reiser 1995), and pqs (Pesci et al. 1999). All the networks are hierarchically interconnected: las controls the other two systems; pqs positively regulates the rhl signaling, whereas rhl in turn puts a negative feedback upon pqs (Wilder et al. 2011; McGrath et al. 2004). Regarding the central role of QS for the infectious process, the interruption of these pathways by blocking the receptors or inhibiting the signal synthesis via small molecules is an attractive therapeutic strategy to attenuate the bacterial pathogenicity, thereby overcoming intractable P. aeruginosa infections (Rasmussen and Givskov2006).
Synthetic QSIs Blocking Receptor Signaling
The interference with the QS receptors via QSIs is a promising approach to efficiently interrupt the communication networks, thereby decreasing the QS-controlled pathogenicity (Kalia 2013; Rasmussen and Givskov 2006). Generally, such QSIs are derived from the natural ligands of the target receptors, structurally unrelated natural products, or small molecules/fragments. These QSIs could be either pure antagonists or weak agonists, all of which effectively compete with the natural agonists preventing a sufficient stimulation of the receptors.
N-Acyl l-Homoserine Lactone (AHL) Receptors as Targets
In Gram-negative bacteria, N-acyl l-homoserine lactones (AHLs) are the most commonly used signal molecules for QS. The innate receptors of these autoinducers belong to the LuxR type which act as transcriptional regulators upon activation by their native agonists. In P. aeruginosa, the so-called las and rhl systems have been identified to be the key AHL-based QS systems. The native ligands of the involved cytoplasmic receptors LasR and RhlR are N-(3-oxo-dodecanoyl)-l-homoserine lactone (3-oxo-C12-HSL or OdDHL) and N-butanoyl-l-homoserine lactone (C4-HSL or BHL), respectively (Fig. 1a).

(a) Structure of N-acyl l-homoserine lactone-based autoinducers in P. aeruginosa. (b) A selection of reported substituents as head group replacements found in the literature (McInnis and Blackwell 2011a, b; Jog et al. 2006; Ishida et al. 2007; Smith et al. 2003; Kim et al. 2009; Persson et al. 2005; Morkunas et al. 2012; Hodgkinson et al. 2012). (c) A selection of substituents as tail section replacements found in the literature (Persson et al. 2005; O’Loughlin et al. 2013; Geske et al. 2005, 2007, 2008; Stacy et al. 2013; Amara et al. 2009). Abbreviations: 3-oxo-C 12 -HSL N-(3-oxo-dodecanoyl)-l-homoserine lactone, C 4 -HSL N-butanoyl-l-homoserine lactone
Notably, a higher rank in the regulatory hierarchy is accounted to the las system. An additional LuxR-type receptor – the “orphan” receptor QscR also responding to 3-oxo-C12-HSL – has been reported for this particular human pathogen (Chugani et al. 2001). However, the major part of scientific studies concentrates on the development of LasR antagonists to disrupt AHL signaling in P. aeruginosa. Desirable cellular effects of LasR- and/or RhlR-targeting QSIs would be the reduction of virulence factor production (e.g., elastase, hydrogen cyanide, pyocyanin, pyoverdine, rhamnolipids, or alkaline protease) as well as the attenuation of biofilm formation. Indeed, several groups have reported on the successful development of synthetic agents with promising in cellulo activities. The following section provides a detailed, yet not exhaustive, overview on the structural space covered by synthetic QSIs interfering with AHL signaling. In general, compounds addressing LasR and RhlR can be divided into two categories: (1) structural mimics of AHLs and (2) structurally unrelated substances.
AHL Mimics
Many nonnatural agonists and antagonists of LuxR-type receptors possess structural features very similar to the native ligands or are actually direct synthetic derivatives thereof (Fig. 1b, c).
Quite obviously, the AHL scaffold can be divided into two sections. The head group consists of the five-membered homoserine lactone moiety, while the tail region comprises a linear N-acyl residue of varying length. The amide-based linker between both segments facilitates modular approaches for straightforward synthesis and derivatization. Many reports in the literature dealing with synthetic AHL analogues focus on structural modifications in one of these two sections while leaving the other part of the molecule constant. Nevertheless, combinations of nonnatural head and tail modules have also been described.
Noteworthy, the native lactone pentacycles of 3-oxo-C12-HSL and C4-HSL are easily hydrolyzed under physiological conditions. The resulting linear product is QS inactive (McInnis and Blackwell 2011b). Thus, replacing this moiety by stable bioisosters is a worthwhile endeavor providing access to nonnatural QS modulators with an in vivo half-life superior to the natural ligands. The range of possible substituents is broad and includes penta- and hexacyclic thiolactones, homo- and heteroaromatic residues, as well as saturated ring systems (Fig. 1b). Interestingly, modifications within the head group of AHL-receptor agonists enable to achieve the desirable functional inversion to yield promising synthetic antagonists. Structural differences between agonists and antagonists can be very subtle. For example, Suga and coworkers have shown that a compound possessing the natural 3-oxo-dodecanoyl tail and a nonnatural 2-aminocyclohexanoyl head group is a potent agonist and can be converted into an antagonist through head group replacement by a 2-aminocyclohexanone substituent (compound 1) (Jog et al. 2006). A similar effect can be observed for the usage of a 2-aminophenol analogue (2) (Smith et al. 2003). The latter derivative has the advantage that it has no stereoisomers which abolishes the need for stereo control and/or racemate separation during synthesis/purification. Moreover, it has been reported that the incorporation of aromatic head groups usually yields an antagonistic functional profile (Hodgkinson et al. 2012). In summary, nonnatural head groups usually contain a cyclic motif which can have diverse electronic and/or chemical properties as well as varied substitution patterns providing control over agonist/antagonist functionality of the desired compound without compromising affinity to the respective LuxR-type receptor.
Reported variations in the tail region of synthetic AHL mimics are also quite numerous (Fig. 1c). Indeed, the difference between the native AHL signal molecules in P. aeruginosa lies in the length and chemistry of this section mediating receptor selectivity. The absence of the β-keto motif in the RhlR-selective autoinducer (C4-HSL) inspired researchers to omit this structure also for the generation of LasR-addressing modulators. Interestingly, also short alkyl chains and even cyclic structures are accepted in this part of the molecule. Hence, substituted homo- and heteroaromatics with varying alkyl linker chains have been successfully incorporated into antagonists of LuxR-type AHL receptors in P. aeruginosa (Geske et al. 2008). Introduction of 1,2,3-triazole-based “click” linkers by Blackwell and coworkers allowed for additional synthetic modularity and combinatorial library generation (Stacy et al. 2013). However, antagonists possessing nonnatural tail groups that mimic the linear unbranched structure of the native signal molecules usually demonstrate higher potency than multi-cyclic or angled motifs (Geske et al. 2007). Incorporation of electrophilic reactive groups within such a linear tail section resulted in covalent QSI addressing LasR (e.g., compound 6) (Amara et al. 2009).
Combining favorable nonnatural head and tail groups with each other may yield rather unexpected results. Spring and coworkers have shown that many of these “chimeric” compounds are of low potency or essentially inactive (Hodgkinson et al. 2012). Hence, conservation of the native 3-oxo-dodecanoyl chain in antagonists with nonnatural head groups can be mandatory for strong QS inhibition. A selection of promising AHL-mimicking LasR and RhlR antagonists and their effects on P. aeruginosa is given in Fig. 2.

Structurally Unrelated AHL-Interfering QSI
The application of experimental screening methodologies using compound libraries led to the identification of structurally diverse LasR/RhlR antagonists (Wu et al. 2004; Musthafa et al. 2012; Muh et al. 2006a, b). A selection of respective compounds together with biological activities is given in Fig. 3. One structure showing very promising effects in vitro is referred to as C-30 (10). This compound is a synthetic derivative of a marine natural compound found in Delisea pulchra and has been investigated for in vivo efficacy in murine infection models. 10 was capable of attenuating QS-mediated virulence in mice and improved bacterial clearance from infected animal lungs (Wu et al. 2004). However, the molecular mode of action by which C-30 (10) interacts with its target receptor (LasR) is not yet fully elucidated.

Finally, it has to be stated that means by which the potency of such LuxR-type antagonists can be determined are various and range from recombinant reporter gene assays, over the direct quantification of receptor-regulated downstream products, to the investigation of effects on biofilm formation. Each of these biological evaluation methodologies is highly dependent on experimental parameters like used cell culture media, effective concentration of organic cosolvents like dimethyl sulfoxide, or, importantly, chosen P. aeruginosa strain. Hence, a comparison of QSI interfering with AHL signaling developed by separate working groups is difficult and in many cases not practical. Additionally, not in all cases it has been analyzed whether a cellular effect (e.g., reduction of virulence factor production) was mediated via antagonism of LasR, RhlR, or both. Indeed, a very strong dual antagonizing agent might prove ineffective in the cellular context as the las and the rhl systems can act reciprocally on the production of key virulence factors. Thus, detailed biological studies including in vivo experiments will be necessary to guide further developments in the field of AHL-interfering QSI in P. aeruginosa.
4-Hydroxy-2-Alkylquinoline (HAQ) Receptor as Target
PqsR is the receptor of the P. aeruginosa-specific pqs QS circuit and functions as a critical regulator that fine-tunes a large set of pathogenicity-associated genes that encode for virulence factors, such as pyocyanin, elastase B, and hydrogen cyanide. Pseudomonas quinolone signal (PQS) and 2-heptyl-4-hydroxyquinoline (HHQ) – the two most predominant members of the 2-alkyl-4-hydroxyquinoline (HAQ) family – are the agonists of the receptor (Fig. 4) and serve as the signal molecules of the network. While QSIs interfering with receptors of las and rhl systems are being intensively investigated, only a few compounds targeting PqsR (PqsR antagonists) have been reported.

Discovery of PqsR antagonists following ligand-based and fragment-based approaches and the biological effects on the production of virulence factor pyocyanin in P. aeruginosa. Abbreviations: HHQ 2-heptyl-4-hydroxyquinoline, PQS Pseudomonas quinolone signal, IC 50 inhibitor concentration to achieve a half-maximal degree of inhibition
Following a ligand-based approach, two research groups have individually discovered potent PqsR antagonists. Hartmann and coworkers identified the first PqsR antagonists (e.g., compound 13, Fig. 4) by means of introducing strong electron-withdrawing groups into the 6-position of HHQ (Lu et al. 2012). Interestingly, such in vitro highly active antagonists reveal opposite functionality (agonistic activity) in P. aeruginosa culture, which is attributed to an unexpected functional inversion mediated by a bacterial enzyme PqsH. Overcoming the problem via further structural optimization resulted in the most potent PqsR antagonist to date (inhibitor concentration to achieve a half-maximal degree of inhibition (IC50) toward pyocyanin: 2 μM, compound 14), which demonstrated anti-virulence efficacy in vivo, thereby providing the first proof of concept for the PqsR-targeting therapeutic strategy (Lu et al. 2014). Meanwhile, Williams and coworkers reported a series of novel PqsR antagonists based on a quinazolinone (QZN) core mimicking the quinolone scaffold of the natural ligands (compound 15) (Ilangovan et al. 2013). The QZN compounds do not only strongly inhibit the pqs QS and production of pyocyanin but also attenuate the biofilm formation of P. aeruginosa. Importantly, this work provided the first co-crystal structures of the PqsR ligand-binding domain with either agonist or antagonist giving a deep insight into the ligand-receptor interactions (Ilangovan et al. 2013).
Application of fragment-based approaches is another promising way to discover potential PqsR antagonists. Based on the knowledge that the κ-opioid receptor agonist (±)-trans-U50488 stimulates the transcription of the PqsR-controlled operon, this compound was identified as a PqsR binder via biophysical methods (Klein et al. 2012). To simplify this compound into smaller molecules and modify it into potent antagonists, 106 fragments with key features derived from U50488 were screened and the best hit 16 was further optimized resulting in a hydroxamic acid 17 with both high activity and ligand efficiency. As expected, this antagonist significantly diminished the pyocyanin production. Similarly, attractive hits were discovered via fragment screening of a library collection composed of 720 small molecules (Zender et al. 2013). The most outstanding hit 18 having a 2-phenyl-1,3,4-thiadiazole core was subsequently transformed into potent PqsR antagonist 19, which successfully suppressed pqs QS activity as well as production of virulence factor pyocyanin. Particularly, it is worth to note that 19 is more drug-like than other known PqsR antagonists regarding physicochemical properties.
Overall, PqsR has been attracting attention and the recent contributions highlight QSIs antagonizing PqsR as promising anti-virulence compounds combating P. aeruginosa.
Synthetic QSIs Blocking Signal Molecule Biosynthesis
An alternative approach for interference with QS is inhibition of the signal molecule synthesis. Although reports applying this strategy are far less numerous than those about signal reception inhibition, evidence indicates that inhibition of signal synthesis is feasible and effective both in vitro and in vivo (LaSarre and Federle 2013). Usually, signal molecules are generated via a cascade of enzymes, which can be targeted individually for inhibition of signal molecule production.
AHL Biosynthesis
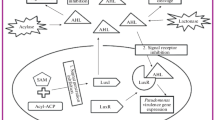
In P. aeruginosa, AHL signals C4-HSL and 3-oxo-C12-HSL are produced by the LuxI-type AHL synthases LasI and RhlI, respectively (Raychaudhuri et al. 2005; Parsek et al. 1999; Gould et al. 2004a). Both enzymes use l-S-adenosylmethionine as substrate. The second substrates, butanoyl-acyl carrier protein (ACP) for RhlI and 3-oxo-dodecanoyl-ACP for LasI, are likely derived from fatty acid biosynthesis. Thereby, FabI, a nicotinamide adenine dinucleotide-dependent enoyl-ACP reductase, reduces trans-2-enoyl-ACPs to the corresponding acyl-ACPs. FabB, a β-ketoacyl-ACP synthase I, condenses malonyl-ACP with an acyl-ACP resulting in the respective 3-oxo-acyl-ACP (Fig. 5a) (Hoang and Schweizer 1999). So far, only a limited number of reports on AHL biosynthesis inhibition have been published.

Current model for N-acyl l-homoserine lactone biosynthesis in P. aeruginosa (a) and representative QSIs (b). Abbreviations: AHL N-acyl l-homoserine lactone, ACP acyl carrier protein, NADH/NAD + reduced/oxidized form of nicotinamide adenine dinucleotide, SAM l-S-adenosylmethionine, MTA 5-methylthioadenosine, C 4 -HSL N-butanoyl-l-homoserine lactone, 3-oxo-C 12 -HSL N-(3-oxo-dodecanoyl)-l-homoserine lactone
Triclosan (20, Fig. 5b) was identified as potent inhibitor of FabI (IC50 of 0.2 μM) that could suppress the production of C4-HSL in vitro. However, as P. aeruginosa was resistant to triclosan due to active efflux, the validity of FabI-targeting QS inhibition could not be demonstrated yet (Hoang and Schweizer 1999).
Interference with C4-HSL production by inhibiting the synthase RhlI with substrate analogues, reaction intermediates, and final products was investigated by Greenberg and coworkers (Parsek et al. 1999). Inter alia, the substrate analogue l-S-adenosylcysteine (21, Fig. 5b) and the reaction product 5′-methylthioadenosine (22, Fig. 5b) strongly inhibited RhlI activity in vitro at micromolar concentrations (Parsek et al. 1999). However, these homologues have not been evaluated in cellulo as they are likely to affect the central pathways of amino acid and fatty acid metabolism (Scutera et al. 2014).
Although the crystal structure of LasI has been elucidated (Gould et al. 2004a, b), no inhibitor targeting the synthase in P. aeruginosa has been described so far. However, recent advances in inhibiting LuxI synthases in other bacterial species might pave the way for the development of such inhibitors in P. aeruginosa (Chung et al. 2011; Christensen et al. 2013).
HAQ Biosynthesis
The pqs QS system, which is unique to P. aeruginosa, makes use of HAQs as signal molecules, among which PQS and its precursor HHQ play a major role (Xiao et al. 2006). Their biosynthesis requires the enzymes PqsA-D and PqsH (Fig. 6a). Thereby, PqsA acts as a ligase catalyzing the formation of anthraniloyl coenzyme A (CoA) from anthranilate, adenosine triphosphate, and CoA (Coleman et al. 2008). PqsD, a β-ketoacyl-ACP synthase III (FabH)-type condensing enzyme, has been shown to catalyze the condensation reaction between anthraniloyl-CoA and β-ketodecanoic acid to give HHQ in vitro (Pistorius et al. 2011; Steinbach et al. 2013). However, recent studies revealed that in the cellular context of P. aeruginosa, PqsD more likely employs anthraniloyl-CoA and malonyl-CoA as substrates to form 3-(o-aminophenyl)-3-keto-propionic acid. This reactive intermediate is then condensated with octanoyl to yield HHQ by a PqsB/PqsC complex (Dulcey et al. 2013). As the exact mechanism of action of PqsB/PqsC still remains elusive, there have been no attempts so far to develop QSIs targeting PqsB/PqsC. Finally, HHQ is converted into PQS by the monooxygenase PqsH (Schertzer et al. 2010). However, PqsH has not been considered as suitable target as a pqsH mutant displayed wild-type virulence in mice (Xiao et al. 2006). Thus, most efforts have been put into the development of small molecule inhibitors blocking PqsA and PqsD.


Current model for 4-hydroxy-2-alkylquinoline biosynthesis in P. aeruginosa (a) and representative QSIs (b). Abbreviations: HAQ 4-hydroxy-2-alkylquinoline, CoA coenzyme A, ATP adenosine triphosphate, AMP adenosine monophosphate, PP i pyrophosphate; II, HHQ 2-heptyl-4-hydroxyquinoline, NADH/NAD + reduced/oxidized form of nicotinamide adenine dinucleotide, PQS Pseudomonas quinolone signal
The first reported inhibitor of PQS production in P. aeruginosa was methyl anthranilate (23, Fig. 6b), an analogue of the PqsA substrate anthranilate that was able to decrease the levels of PQS-dependent virulence factor elastase. However, concentrations in the millimolar range were necessary to see a pronounced inhibitory effect on PQS formation (Calfee et al. 2001). Although it could be excluded that methyl anthranilate was a substrate or inhibitor of PqsA (Coleman et al. 2008), the exact mechanism of action remained unknown.
Rahme and coworkers aimed at developing more potent substrate analogues based on the anthranilate structure (Lesic et al. 2007). Introduction of electron-withdrawing halogen atoms into the phenyl ring should restrict formation of an activated carbonyl. Indeed, these derivatives strongly inhibited HHQ and PQS formation (Lesic et al. 2007). For instance, 6-fluoroanthranilic acid (24, Fig. 6b) exhibited an IC50 of 109 μM regarding inhibition of PQS synthesis (Maurer et al. 2013). Excitingly, these compounds were shown to have therapeutic benefits in vivo, where they increased survival and limited systemic dissemination of P. aeruginosa in a thermal injury mouse model (Lesic et al. 2007). These effects were concluded to be likely due to PqsA inhibition as anthranilic acid accumulated in cultures grown in the presence of the compounds.
Pesci and coworkers purified PqsA for the first time and developed an in vitro assay for identifying substrates and inhibitors of PqsA (Coleman et al. 2008). For example, anthranilate derivatives bearing chloro- and fluoro-substituents in 4- to 6-position of the benzene ring, such as compound 24 (K m of 11 μM), were found to be substrates of PqsA. In contrast, 3-chloroanthranilic (25, Fig. 6b) acid was identified as an inhibitor of PqsA (K i of 12.9 μM). In general, the extent of inhibition of PQS synthesis achievable with both substrates and inhibitors did not correlate with respective K m or K i values. Finally, it remained to be elucidated whether the inhibition of PQS production by the substrates was due to competition with anthranilate or to the inhibition of downstream enzymes by formed CoA thioesters (Coleman et al. 2008).
In 2011, Müller and coworkers developed an in vitro enzyme assay with purified PqsD using anthraniloyl-CoA and β-ketodecanoic acid as substrates and detecting the product HHQ (Pistorius et al. 2011). As a starting point for the identification of PqsD inhibitors, known inhibitors of FabH, a structural and functional homologue of PqsD, were tested. Indeed, compounds 26(IC50 of 65 μM) and 27 (IC50 of 35 μM) could be identified as the first inhibitors of PqsD (Fig. 6b). However, their activity was only moderate and they were not tested in cellular assays as they were expected to exhibit antibiotic activity (Pistorius et al. 2011).
Hartmann and coworkers initiated several rational design projects for the development of potent, selective, and non-bactericidal PqsD inhibitors. In the course of these studies, three classes of PqsD inhibitors have been identified following a design approach based on known inhibitors of FabH, an experimental screening, and a ligand-based strategy. For each class, a series of compounds was synthesized and evaluated for its inhibitory potency in an in vitro PqsD assay. Therefrom, structure-activity relationships were derived. Furthermore, molecular docking based on the crystal structure of PqsD (Bera et al. 2009), biochemical assays, and biophysical methods including surface plasmon resonance spectroscopy (Henn et al. 2012) were applied to characterize the compounds regarding binding site, binding mode, or molecular interactions with the target. Based on that knowledge, structural optimizations were performed that led to potent PqsD inhibitors with IC50 values in the single-digit micromolar to submicromolar range (Storz et al. 2012; Weidel et al. 2013; Sahner et al. 2013; Hinsberger et al. 2014; Storz et al. 2013).
The design approach based on known FabH inhibitors (Pistorius et al. 2011) resulted in two subclasses of compounds with 2-benzamidobenzoic acid core structure. The 3′-sulfonamide-substituted series was found to reversibly bind to the substrate access channel within PqsD. The most potent inhibitor 28 (Fig. 6b) exhibited an IC50 value of 1.2 μM (Weidel et al. 2013). The 3′-phenoxy/4′-phenyl-substituted 2-benzamidobenzoic acids, originally reported as inhibitors of bacterial RNA polymerase (RNAP), were systematically optimized regarding their activity and selectivity profile. The most promising compound 29 (Fig. 6b) strongly inhibited PqsD (IC50 of 6.2 μM) while not affecting RNAP (Hinsberger et al. 2014).
From the class of 5-aryl-ureidothiophene-2-carboxylic acids identified by experimental screening, compounds 30 (IC50 of 0.5 μM) and 31 (IC50 of 2 μM) turned out to be the most potent (Fig. 6b). As the latter binds covalently to the active site, it carries the potential of strong biological effects (Sahner et al. 2013).
The ligand-guided design strategy led to a potent class of PqsD inhibitors, the nitrophenyl-methanols, which showed time-dependent inhibitory activity, tight-binding behavior, and active site binding (Storz et al. 2013). The most promising member of this series, compound 32 (Fig. 6b), exhibited high potency (IC50 of 3.2 μM) and high ligand efficiency (0.39). Applied at micromolar concentrations, this compound strongly inhibited HHQ and PQS synthesis as well as biofilm formation in P. aeruginosa without affecting growth. Thus, by the use of 32, it was shown for the first time that inhibition of signal molecule synthesis is feasible with a PqsD inhibitor and that PqsD is a valid anti-biofilm target (Storz et al. 2012).
Conclusion
Numerous highly active blockers of QS receptors as well as inhibitors of QS signal synthases were identified using diverse design strategies during the last decade. Among the developed compounds, several showed anti-infective activities. However, studies providing detailed in vivo data are rare. Nevertheless, the recent scientific achievements emphasize the potential applicability of QSIs as a weapon to treat the recalcitrant infectious diseases caused by P. aeruginosa.
Opinion
Numerous scientific contributions demonstrate that inhibition of QS can be regarded as a promising strategy against P. aeruginosa infections, and the discovery of small molecules targeting QS is rapidly progressing. However, there are obstacles that hinder the successful translation of such QSIs into real anti-infective drugs. First, the methodologies used for biological evaluation of QSIs are various and the results are highly dependent on experimental conditions, especially on the chosen P. aeruginosa strain. This makes a comparison of QSIs developed by different working groups difficult or even impossible. Second, the molecular target of QSIs discovered by cellular screening approaches is often unknown, which hampers a directed optimization of their activity, selectivity, and pharmacokinetic profile. Third, QSIs derived from target-based drug discovery approaches often fail to exhibit cellular activity. Development of such QSIs overcoming the Gram-negative cell wall and escaping the widespread efflux pumps remains a very challenging task. Fourth, despite exciting in vitro and in cellulo activities achieved with QSIs, an in-depth evaluation of such inhibitors in advanced animal models or even clinical trials is still to be performed. Such studies are mainly hampered by lack of drug-like molecules. Many QSIs, especially those derived from natural products, suffer from chemical instability, exhibit toxic effects, or possess inappropriate pharmacokinetic properties. Thus, in our opinion, current QS research should focus on development of drug-like molecules applicable for in-depth in vivo studies providing the proof of concept for QS inhibition-based treatment of P. aeruginosa infections.
Abbreviations
- ACP:
-
Acyl carrier protein
- AHL:
-
N-acyl homoserine lactone
- AMP:
-
Adenosine monophosphate
- ATP:
-
Adenosine triphosphate
- C4-HSL:
-
N-butanoyl-l-homoserine lactone
- CoA:
-
Coenzyme A
- HAQ:
-
4-hydroxy-2-alkylquinoline
- HHQ:
-
2-heptyl-4-hydroxyquinoline
- IC50 :
-
Inhibitor concentration to achieve a half-maximal degree of inhibition
- MTA:
-
5-methylthioadenosine
- NADH/ NAD+ :
-
Reduced/oxidized form of nicotinamide adenine dinucleotide
- 3-oxo-C12 -HSL:
-
N-(3-oxo-dodecanoyl)-l-homoserine lactone
- PPi :
-
Pyrophosphate
- PQS:
-
Pseudomonas quinolone signal
- QS:
-
Quorum sensing
- QSI:
-
Quorum sensing inhibitor
- QZN:
-
Quinazolinone
- RNAP:
-
RNA polymerase
- SAM:
-
l-S-adenosylmethionine
References
Amara N, Mashiach R, Amar D, Krief P, Spieser SAH, Bottomley MJ, Aharoni A, Meijler MM (2009) Covalent inhibition of bacterial quorum sensing. J Am Chem Soc 131(30):10610–10619. doi:10.1021/Ja903292v
Bera AK, Atanasova V, Robinson H, Eisenstein E, Coleman JP, Pesci EC, Parsons JF (2009) Structure of PqsD, a pseudomonas quinolone signal biosynthetic enzyme, in complex with anthranilate. Biochemistry 48(36):8644–8655. doi:10.1021/Bi9009055
Calfee MW, Coleman JP, Pesci EC (2001) Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc Natl Acad Sci USA 98(20):11633–11637. doi:10.1073/pnas.201328498
Christensen QH, Grove TL, Booker SJ, Greenberg EP (2013) A high-throughput screen for quorum-sensing inhibitors that target acyl-homoserine lactone synthases. Proc Natl Acad Sci USA 110(34):13815–13820. doi:10.1073/pnas.1313098110
Chugani SA, Whiteley M, Lee KM, D'Argenio D, Manoil C, Greenberg EP (2001) QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc Natl Acad Sci USA 98(5):2752–2757. doi:10.1073/pnas.051624298
Chung J, Goo E, Yu S, Choi O, Lee J, Kim J, Kim H et al (2011) Small-molecule inhibitor binding to an N-acyl-homoserine lactone synthase. Proc Natl Acad Sci USA 108(29):12089–12094. doi:10.1073/pnas.1103165108
Coleman JP, Hudson LL, McKnight SL, Farrow JM, Calfee MW, Lindsey CA, Pesci EC (2008) Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme a ligase. J Bacteriol 190(4):1247–1255. doi:10.1128/Jb.01140-07
Dulcey CE, Dekimpe V, Fauvelle DA, Milot S, Groleau MC, Doucet N, Rahme LG, Lepine F, Deziel E (2013) The end of an old hypothesis: the pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem Biol 20(12):1481–1491. doi:10.1016/j.chembiol.2013.09.021
Gambello MJ, Iglewski BH (1991) Cloning and characterization of the Pseudomonas-aeruginosa lasr gene, a transcriptional activator of elastase expression. J Bacteriol 173(9):3000–3009
Geske GD, Wezeman RJ, Siegel AP, Blackwell HE (2005) Small molecule inhibitors of bacterial quorum sensing and biofilm formation. J Am Chem Soc 127(37):12762–12763. doi:10.1021/Ja0530321
Geske GD, O'Neill JC, Miller DM, Mattmann ME, Blackwell HE (2007) Modulation of bacterial quorum sensing with synthetic ligands: systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanisms of action. J Am Chem Soc 129(44):13613–13625. doi:10.1021/Ja074135h
Geske GD, Mattmann ME, Blackwell HE (2008) Evaluation of a focused library of N-aryl L-homoserine lactones reveals a new set of potent quorum sensing modulators. Bioorg Med Chem Lett 18(22):5978–5981. doi:10.1016/j.bmcl.2008.07.089
Gould TA, Schweizer HP, Churchill MEA (2004a) Structure of the Pseudomonas aeruginosa acyl-homoserinelactone synthase LasI. Mol Microbiol 53(4):1135–1146. doi:10.1111/j.1365-2958.2004.04211.x
Gould TA, Watson WT, Choi KH, Schweizer HP, Churchi MEA (2004b) Crystallization of Pseudomonas aeruginosa AHL synthase Lasl using beta-turn crystal engineering. Acta Crystallogr D Biol Crystallogr 60:518–520. doi:10.1107/S0907444903028300
Henn C, Boettcher S, Steinbach A, Hartmann RW (2012) Catalytic enzyme activity on a biosensor chip: combination of surface plasmon resonance and mass spectrometry. Anal Biochem 428(1):28–30. doi:10.1016/J.Ab.2012.05.024
Hentzer M, Wu H, Andersen JB, Riedel K, Rasmussen TB, Bagge N, Kumar N et al (2003) Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. Embo J 22(15):3803–3815. doi:10.1093/Emboj/Cdg366
Hinsberger S, de Jong JC, Groh M, Haupenthal J, Hartmann RW (2014) Benzamidobenzoic acids as potent PqsD inhibitors for the treatment of Pseudomonas aeruginosa infections. Eur J Med Chem 76:343–351. doi: 10.1016/j.ejmech.2014.02.014 S0223-5234(14)00134-2 [pii]
Hoang TT, Schweizer HP (1999) Characterization of Pseudomonas aeruginosa Enoyl-Acyl carrier protein reductase (FabI): a target for the antimicrobial triclosan and its role in acylated homoserine lactone synthesis. J Bacteriol 181(17):5489–5497
Hodgkinson JT, Galloway WRJD, Wright M, Mati IK, Nicholson RL, Welch M, Spring DR (2012) Design, synthesis and biological evaluation of non-natural modulators of quorum sensing in Pseudomonas aeruginosa. Org Biomol Chem 10(30):6032–6044. doi:10.1039/C2ob25198a
Ilangovan A, Fletcher M, Rampioni G, Pustelny C, Rumbaugh K, Heeb S, Camara M et al (2013) Structural basis for native agonist and synthetic inhibitor recognition by the Pseudomonas aeruginosa quorum sensing regulator PqsR (MvfR). Plos Pathog 9(7). doi: ARTN e1003508 doi:10.1371/journal.ppat.1003508
Ishida T, Ikeda T, Takiguchi N, Kuroda A, Ohtake H, Kato J (2007) Inhibition of quorum sensing in Pseudomonas aeruginosa by N-acyl cyclopentylamides. Appl Environ Microbiol 73(10):3183–3188. doi:10.1128/Aem.02233-06
Jog GJ, Igarashi J, Suga H (2006) Stereoisomers of P-aeruginosa autoinducer analog to probe the regulator binding site. Chem Biol 13(2):123–128. doi:10.1016/j.chembiol.2005.12.013
Kalia VC (2013) Quorum sensing inhibitors: an overview. Biotechnol Adv 31(2):224–245. doi:10.1016/j.biotechadv.2012.10.004
Kim C, Kim J, Park HY, Lee JH, Park HJ, Kim CK, Yoon J (2009) Structural understanding of quorum-sensing inhibitors by molecular modeling study in Pseudomonas aeruginosa. Appl Microbiol Biotechnol 83(6):1095–1103. doi:10.1007/s00253-009-1954-3
Klein T, Henn C, de Jong JC, Zimmer C, Kirsch B, Maurer CK, Pistorius D, Muller R, Steinbach A, Hartmann RW (2012) Identification of small-molecule antagonists of the Pseudomonas aeruginosa transcriptional regulator PqsR: biophysically guided hit discovery and optimization. ACS Chem Biol 7(9):1496–1501. doi:10.1021/Cb300208g
LaSarre B, Federle MJ (2013) Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 77(1):73–111. doi:10.1128/Mmbr.00046-12
Lesic B, Lepine F, Deziel E, Zhang JW, Zhang QH, Padfield K, Castonguay MH et al (2007) Inhibitors of pathogen intercellular signals as selective anti-infective compounds. Plos Pathog 3(9):1229–1239. doi:ARTN e126. doi 10.1371/journal.ppat.0030126
Lu CB, Kirsch B, Zimmer C, de Jong JC, Henn C, Maurer CK, Musken M, Haussler S, Steinbach A, Hartmann RW (2012) Discovery of antagonists of PqsR, a key player in 2-alkyl-4-quinolone-dependent quorum sensing in Pseudomonas aeruginosa. Chem Biol 19(3):381–390. doi:10.1016/j.chembiol.2012.01.015
Lu CB, Maurer CK, Kirsch B, Steinbach A, Hartmann RW (2014) Overcoming the unexpected functional inversion of a PqsR antagonist in Pseudomonas aeruginosa: an in vivo potent antivirulence agent targeting pqs quorum sensing. Angew Chem Int Ed 53(4):1109–1112. doi:10.1002/anie.201307547
Maurer CK, Steinbach A, Hartmann RW (2013) Development and validation of a UHPLC-MS/MS procedure for quantification of the Pseudomonas Quinolone Signal in bacterial culture after acetylation for characterization of new quorum sensing inhibitors. J Pharm Biomed Anal 86:127–134. doi:10.1016/j.jpba.2013.07.047
McGrath S, Wade DS, Pesci EC (2004) Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS). FEMS Microbiol Lett 230(1):27–34. doi:10.1016/S0378-1097(03)00849-8
McInnis CE, Blackwell HE (2011a) Design, synthesis, and biological evaluation of abiotic, non-lactone modulators of LuxR-type quorum sensing. Bioorg Med Chem 19(16):4812–4819. doi:10.1016/j.bmc.2011.06.072
McInnis CE, Blackwell HE (2011b) Thiolactone modulators of quorum sensing revealed through library design and screening. Bioorg Med Chem 19(16):4820–4828. doi:10.1016/j.bmc.2011.06.071
Morkunas B, Galloway WRJD, Wright M, Ibbeson BM, Hodgkinson JT, O’Connell KMG, Bartolucci N, Della Valle M, Welch M, Spring DR (2012) Inhibition of the production of the Pseudomonas aeruginosa virulence factor pyocyanin in wild-type cells by quorum sensing autoinducer-mimics. Org Biomol Chem 10(42):8452–8464. doi:10.1039/C2ob26501j
Muh U, Hare BJ, Duerkop BA, Schuster M, Hanzelka BL, Heim R, Olson ER, Greenberg EP (2006a) A structurally unrelated mimic of a Pseudomonas aeruginosa acyl-homoserine lactone quorum-sensing signal. Proc Natl Acad Sci USA 103(45):16948–16952. doi:10.1073/pnas.0608348103
Muh U, Schuster M, Heim R, Singh A, Olson ER, Greenberg EP (2006b) Novel Pseudomonas aeruginosa quorum-sensing inhibitors identified in an ultra-high-throughput screen. Antimicrob Agents Chemother 50(11):3674–3679. doi:10.1128/Aac.00665-06
Musthafa KS, Balamurugan K, Pandian SK, Ravi AV (2012) 2,5-Piperazinedione inhibits quorum sensing-dependent factor production in Pseudomonas aeruginosa PAO1. J Basic Microbiol 52(6):679–686. doi:10.1002/jobm.201100292
O’Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL (2013) A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A 110(44):17981–17986. doi:10.1073/pnas.1316981110
Ochsner UA, Reiser J (1995) Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas-Aeruginosa. Proc Natl Acad Sci USA 92(14):6424–6428. doi:10.1073/pnas.92.14.6424
Ochsner UA, Fiechter A, Reiser J (1994) Isolation, characterization, and expression in Escherichia-Coli of the Pseudomonas-aeruginosa Rhlab genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J Biol Chem 269(31):19787–19795
Parsek MR, Val DL, Hanzelka BL, Cronan JE, Greenberg EP (1999) Acyl homoserine-lactone quorum-sensing signal generation. Proc Natl Acad Sci USA 96(8):4360–4365. doi:10.1073/pnas.96.8.4360
Passador L, Cook JM, Gambello MJ, Rust L, Iglewski BH (1993) Expression of Pseudomonas-Aeruginosa virulence genes requires cell-to-cell communication. Science 260(5111):1127–1130. doi:10.1126/science.8493556
Persson T, Hansen TH, Rasmussen TB, Skinderso ME, Givskov M, Nielsen J (2005) Rational design and synthesis of new quorum-sensing inhibitors derived from acylated homoserine lactones and natural products from garlic. Org Biomol Chem 3(2):253–262. doi:10.1039/B415761c
Pesci EC, Milbank JB, Pearson JP, McKnight S, Kende AS, Greenberg EP, Iglewski BH (1999) Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96(20):11229–11234
Pistorius D, Ullrich A, Lucas S, Hartmann RW, Kazmaier U, Muller R (2011) Biosynthesis of 2-Alkyl-4(1H)-quinolones in Pseudomonas aeruginosa: potential for therapeutic interference with pathogenicity. Chembiochem 12(6):850–853. doi:10.1002/cbic.201100014
Rasmussen TB, Givskov M (2006) Quorum-sensing inhibitors as anti-pathogenic drugs. Int J Med Microbiol 296(2–3):149–161. doi:10.1016/j.ijmm.2006.02.005
Raychaudhuri A, Jerga A, Tipton PA (2005) Chemical mechanism and substrate specificity of RhlI, an acylhomoserine lactone synthase from Pseudomonas aeruginosa. Biochemistry 44(8):2974–2981. doi:10.1021/Bi048005m
Sahner JH, Brengel C, Storz MP, Groh M, Plaza A, Muller R, Hartmann RW (2013) Combining in silico and biophysical methods for the development of Pseudomonas aeruginosa quorum sensing inhibitors: an alternative approach for structure-based drug design. J Med Chem 56(21):8656–8664. doi:10.1021/Jm401102e
Schertzer JW, Brown SA, Whiteley M (2010) Oxygen levels rapidly modulate Pseudomonas aeruginosa social behaviours via substrate limitation of PqsH. Mol Microbiol 77(6):1527–1538. doi:10.1111/j.1365-2958.2010.07303.x
Scutera S, Zucca M, Savoia D (2014) Novel approaches for the design and discovery of quorum-sensing inhibitors. Expert Opin Drug Discov 9(4):353–366. doi:10.1517/17460441.2014.894974
Smith KM, Bu YG, Suga H (2003) Library screening for synthetic agonists and antagonists of a Pseudomonas aeruginosa autoinducer. Chem Biol 10(6):563–571. doi:10.1016/S1074-5521(03)00107-8
Stacy DM, Le Quement ST, Hansen CL, Clausen JW, Tolker-Nielsen T, Brummond JW, Givskov M, Nielsen TE, Blackwell HE (2013) Synthesis and biological evaluation of triazole-containing N-acyl homoserine lactones as quorum sensing modulators. Org Biomol Chem 11(6):938–954. doi:10.1039/C2ob27155a
Steinbach A, Maurer CK, Weidel E, Henn C, Brengel C, Hartmann RW, Negri M (2013) Molecular basis of HHQ biosynthesis: molecular dynamics simulations, enzyme kinetic and surface plasmon resonance studies. BMC Biophys 6. doi:Unsp 10. doi:10.1186/2046-1682-6-10
Storz MP, Maurer CK, Zimmer C, Wagner N, Brengel C, de Jong JC, Lucas S et al (2012) Validation of PqsD as an anti-biofilm target in Pseudomonas aeruginosa by development of small-molecule inhibitors. J Am Chem Soc 134(39):16143–16146. doi:10.1021/Ja3072397
Storz MP, Brengel C, Weidel E, Hoffmann M, Hollemeyer K, Steinbach A, Muller R, Empting M, Hartmann RW (2013) Biochemical and biophysical analysis of a Chiral PqsD inhibitor revealing tight-binding behavior and enantiomers with contrary thermodynamic signatures. ACS Chem Biol 8(12):2794–2801. doi:10.1021/Cb400530d
Weidel E, de Jong JC, Brengel C, Storz MP, Braunshausen A, Negri M, Plaza A, Steinbach A, Muller R, Hartmann RW (2013) Structure optimization of 2-benzamidobenzoic acids as PqsD inhibitors for Pseudomonas aeruginosa infections and elucidation of binding mode by SPR, STD NMR, and molecular docking. J Med Chem 56(15):6146–6155. doi:10.1021/Jm4006302
Wilder CN, Diggle SP, Schuster M (2011) Cooperation and cheating in Pseudomonas aeruginosa: the roles of the las, rhl and pqs quorum-sensing systems. ISME J 5(8):1332–1343. doi:10.1038/ismej.2011.13
Wu H, Song Z, Hentzer M, Andersen JB, Molin S, Givskov M, Hoiby N (2004) Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J Antimicrob Chemother 53(6):1054–1061. doi:10.1093/Jac/Dkh223
Xiao GP, Deziel E, He JX, Lepine F, Lesic B, Castonguay MH, Milot S, Tampakaki AP, Stachel SE, Rahme LG (2006) MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol Microbiol 62(6):1689–1699. doi:10.1111/j.1365-2958.2006.05462.x
Zender M, Klein T, Henn C, Kirsch B, Maurer CK, Kail D, Ritter C, Dolezal O, Steinbach A, Hartmann RW (2013) Discovery and biophysical characterization of 2-amino-oxadiazoles as novel antagonists of PqsR, an important regulator of Pseudomonas aeruginosa virulence. J Med Chem 56(17):6761–6774. doi:10.1021/Jm400830r
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer India
About this chapter
Cite this chapter
Maurer, C.K., Lu, C., Empting, M., Hartmann, R.W. (2015). Synthetic Quorum Sensing Inhibitors (QSIs) Blocking Receptor Signaling or Signal Molecule Biosynthesis in Pseudomonas aeruginosa . In: Kalia, V. (eds) Quorum Sensing vs Quorum Quenching: A Battle with No End in Sight. Springer, New Delhi. https://doi.org/10.1007/978-81-322-1982-8_24
Download citation
DOI: https://doi.org/10.1007/978-81-322-1982-8_24
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-1981-1
Online ISBN: 978-81-322-1982-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)