
Abstract
Epstein–Barr virus (EBV) is an oncogenic human γ-herpes virus that causes various cancers such as Burkitt’s lymphoma, Hodgkin’s lymphoma, diffuse large B-cell lymphoma (DLBCL), gastric cancer, and nasopharyngeal carcinoma, one of the most common cancers in China.
miRNAs are small noncoding RNAs approximately 22 nucleotides long that posttranscriptionally regulate deadenylation, translation, and decay of their target messenger RNAs (mRNAs). The first viral miRNAs were discovered in human B-cells latently infected with EBV. EBV encodes at least 44 miRNAs. Forty of these miRNAs are transcribed from the BamH1 fragment A rightward transcript (BART) region. Several groups reported that EBV-encoded miRNAs target proapoptotic genes, preventing cells from entering the lytic phase. In this review, I discuss several recent findings centered on EBV-encoded miRNAs. In addition, I highlight the fact that small RNAs play important roles in inflammation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

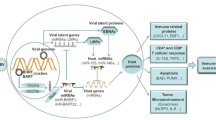
Keywords
1 Introduction
Epstein–Barr virus (EBV) was discovered by examination of electron micrographs of cells isolated from Burkitt’s lymphoma , a pediatric tumour common in sub-Saharan Africa, where its unusual geographic distribution indicated a viral etiology. EBV, which belongs to the γ-herpes virus family, has widespread distribution among humans and results in persistent asymptomatic infection of B cells. EBV usually results in clinically asymptomatic infections but may also cause infectious mononucleosis. In rare cases, EBV infection induces malignant transformation and development of cancers such as Burkitt’s lymphoma, gastric carcinoma, and nasopharyngeal carcinoma (NPC), one of the most common cancers in China (Fang et al. 2008). In addition, EBV causes several lymphoid malignancies, including acquired immunodeficiency syndrome (AIDS)-associated lymphoma, Hodgkin’s lymphoma , posttransplant lymphoma, age-associated B-cell lymphoma, and peripheral T- and NK-cell lymphomas (Parkin et al. 2005; Parkin 2006).
The primary site of EBV infection is the oropharyngeal cavity (Borza and Hutt-Fletcher 2002). EBV infection induces two distinct patterns of gene expression. During acute (lytic) EBV infection, the virus expresses all its genes sequentially. Linear, double-stranded viral genomes produced during the lytic state are packaged into virions. Shortly after the initial infection, EBV enters into a latent state and expresses only select ‘latent’ genes, which allows the virus to evade immune surveillance mechanisms of the host and establish a lifelong persistent infection (Ghosh et al. 2007). Serology analyses indicate that approximately 95 % adults worldwide are infected with EBV. After the primary infection, hosts remain lifelong carriers of the virus (Thorley-Lawson 2001).
2 EBV Micro-RNAs
2.1 Micro-RNAs
Micro-RNAs (miRNAs) are small, noncoding, single-stranded RNAs approximately 21–25 nucleotides in length. They posttranscriptionally regulate mRNA expression and are transcribed from the noncoding regions of genes in all multicellular organisms and in certain viruses (Wang et al. 2008; Chen and Rajewsky 2007). EBV was the first human virus found to encode miRNAs (Pfeffer et al. 2004). EBV encodes 44 miRNAs and a small RNA. EBV miRNAs are encoded by regions located within the BHRF1 and BamHI-A rightward transcript (BART) loci of the EBV genome. The BHRF1 cluster of miRNAs includes BHRF1-1, BHRF1-2, BHRF1-3, and BHRF1-4 (Chen and Rajewsky 2007; Pfeffer et al. 2004; Grundhoff et al. 2006). The remaining EBV miRNAs, except miR-BART2, are encoded by BART clusters 1 and 2. MiR-BART2 is encoded by a region outside the BART clusters (Chen and Rajewsky 2007; Pfeffer et al. 2004; Grundhoff et al. 2006; Griffiths-Jones et al. 2006). MiRNAs bind to the 3′ untranslated region (UTR) of mRNAs and interfere with their translation, thus downregulating protein expression. EBV miRNAs have been isolated from various EBV-associated carcinomas and lymphomas such as NPC, gastric carcinoma, diffuse large B-cell lymphoma, nasal T- and NK-cell lymphomas, and Hodgkin’s lymphoma (Pfeffer et al. 2004; do Kim et al. 2007). Viral miRNAs play vital roles in immunogenesis, survival and proliferation of host cells, differentiation, lymphomagenesis, and regulation of viral infection and latency (Table 19.1) (Pfeffer et al. 2004; Rana 2007; Xia et al. 2008; Barth et al. 2011; Lu et al. 2012).
2.2 Functions of EBV miRNAs
EBV miR-BART2 expression is low during latency. It prevents aberrant expression of BALF5 mRNA, which is essential for viral DNA replication during lytic infection (Barth et al. 2008), The sequence of miR-BART2 is perfectly complementary to the 3′ UTR of BALF5 mRNA. Thus, miR-BART2 inhibits viral DNA replication by degrading BALF5 mRNA. MiRNA-guided cleavage of mRNAs requires association with AGO2 (Meister et al. 2004), a member of the Argonaute family of proteins and a component of RNA-induced silencing complex (RISC). MiR-BART2 associates with AGO2 and guides the sequence-specific cleavage of BALF5 mRNA. MiR-BART2-guided cleavage of BALF5 mRNA substantially decreases after induction of the lytic cycle in EBV-infected cells (Barth et al. 2008). The amount of miR-BART2 is reduced during lytic infection, which in turn derepresses BALF5 expression (Barth et al. 2008). However, it is unclear whether miR-BART2-mediated regulation of viral replication is completely controlled by BALF5.
MiR-BART6, which is regulated by RNA editing, is another regulator of the shift from latent EBV infection to lytic infection (Iizasa et al. 2010). Editing of wild-type primary (pre)-miR-BART6 dramatically decreases loading of miR-BART6-5p onto RISC without affecting the processing of precursor or mature miRNAs (Iizasa et al. 2010). Editing of pre-miRNA might affect selection and loading of the guide strand onto RISC (Khvorova et al. 2003). MiR-BART6-5p silences DICER by binding to multiple target sites in the 3′ UTR of Dicer mRNA. In contrast, miR-BART6-3p is unable to perform this function (Iizasa et al. 2010).
MiR-BART5 promotes host cell survival by regulating p53-upregulated modulator of apoptosis (PUMA) (Choy et al. 2008). PUMA is an apoptotic protein belonging to the BH3-only group of the Bcl-2 family of proteins and is encoded by BBC3 (Han et al. 2001; Nakano and Vousden 2001; Yu et al. 2001). The 3′ UTR of BBC3 is perfectly complementary to miR-BART5, and binding of miR-BART5 to this region suppresses PUMA expression.
Abundant expression of miR-BART5 significantly downregulates PUMA expression in 60 % of NPC tissues (Choy et al. 2008). BART5 uses this mechanism to promote survival of NPC cells, EBV-infected gastric carcinoma cells, and EBV-infected epithelial cells (Choy et al. 2008). Therefore, miR-BART5 may be a good target for anticancer therapy in EBV-infected cancer cells.
LMP1 is a viral protein expressed during type III latent EBV infections (Hislop et al. 2007; Pagano et al. 2004). LMP1 promotes cell growth and B-cell transformation, resists serum deprivation-induced apoptosis, and induces phenotypic changes in epithelial cells. BART1 cluster miRNAs negatively regulate LMP1 expression and limit its inappropriately high levels, thereby preventing apoptosis induced by LMP1-mediated changes in UPR. BART1 cluster miRNAs, namely miR-BART16, miR-BART17-5p, and miR-BART1-5p, are recognized by target sites in the 3′ UTR of the mRNA expressing LMP1 (Lo et al. 2007). These miRNAs regulate LMP1 expression at the posttranscriptional level.
BHRF1 is a latent protein expressed in growth-transformed cells that contributes to virus-associated lymphomagenesis (Kelly et al. 2009). MiR-BHRF1 downregulates BHRF1 expression, modulates cell transformation (Seto et al. 2010), and promotes B-cell proliferation following EBV infection. EBV-infected B cells lacking miR-BHRF1 progress into the cell cycle less efficiently and eventually die through apoptosis (Seto et al. 2010). MiR-BHRF1 is constitutively expressed in LCLs (Seto et al. 2010). The proportion of G1/G0 cells increases whereas that of S-phase cells decreases in the absence of miR-BHRF1 (Seto et al. 2010), indicating its key role in controlling proliferation of latently-infected cells. EBV-mediated differentiation of resting B cells into active B cells requires time. MiR-BHRF1 acts at the stage of the EBV life cycle during which multiple EBV oncogenes are activated.
EBV miR-BART7-3p enhances cell migration and invasion in vitro and cancer metastasis in vivo. EMT is characterized by the loss of epithelial markers and gain of mesenchymal features in NPC cells. Mechanistic studies indicate that EBV miR-BART7-3p targets PTEN , a major human tumour suppressor, and modulates PI3K/Akt/GSK-3β signalling, thus upregulating the expression and nuclear accumulation of Snail and β-catenin, which favor EMT (Lu et al. 2012; Cai et al. 2015).
3 EBV-Encoded Secretory miRNAs
3.1 Secretory miRNAs
Cellular and viral miRNAs control gene expression by repressing the translation of mRNAs into proteins, a process that is tightly regulated in healthy cells but that is deregulated in cancerous and virus-infected cells. Interestingly, miRNAs are not strictly intracellular; they are found in peripheral blood and are secreted into the culture media in small vesicles called exosomes (Kosaka et al. 2010). It has been suggested that exosome-associated miRNAs play important roles in intercellular communication (Kosaka et al. 2010); however, experimental evidence supporting this is not enough. Moreover, the dynamics and mechanism of miRNA secretion by exosomes are poorly understood. It is unclear whether miRNAs are secreted in physiologically relevant amounts and whether exogenous exosome-associated miRNAs can access the molecular machinery to undergo processing.
3.2 EBV-Encoded Secretory miRNAs
Pegtel et al. were the first to show that exosomes deliver viral miRNAs to noninfected cells (Pegtel et al. 2010). They used EBV B95.8-immortalised LCLs and showed that exosomes containing BHRF1 cluster miRNAs targeted CXCL11 mRNA in nearby uninfected cells. Furthermore, they showed that non-B cells obtained from EBV-infected patients with elevated viral loads contained EBV miRNAs, suggesting that exosomes transferred miRNAs to uninfected cells in vivo. These findings were confirmed by two studies reporting the release of exosomes from NPC cells. Gourzones et al. detected EBV miR-BART-containing exosomes in serum samples from patients with NPC and from mice xenografted with human NPC cells (Lu et al. 2012; Gourzones et al. 2010).
4 EBV miRNAs Regulate Inflammation and Immune Evasion
4.1 Inflammation and Oncogenesis
The potential of chronic inflammation to lead to oncogenesis has been established in malignancies. Chronic hepatitis caused by HCV can develop into hepatocarcinoma, autoimmune-mediated chronic colitis, colon cancer, Helicobacter pilori-induced chronic gastritis , and gastric cancer . These transitions require molecular and cellular interactions involving immune and nonimmune cells, cytokines , pathogens, and other factors (Grivennikov 2013; Shlomai et al. 2014).
Deficiency of proinflammatory cytokines such as TNF-alpha limits inflammation and suppresses oncogenesis in several models. Conversely, genetic deletion of immunosuppressive cytokines such as IL-10, and TGF-beta exacerbate inflammation and facilitate oncogenesis (Rickinson 2014). The mechanisms underlying inflammation-mediated regulation of oncogenesis have not been fully elucidated, but several studies are currently addressing this issue. Proinflammatory cytokines cause oxidative stress and production of reactive oxygen species (ROS) that induce DNA damage such as double strand breaks (DSB), and genomic instability (Lemercier 2015; Anuranjani 2014). Genomic instability can also be induced by active mutagenesis caused by activation-induced cytidine deaminase (AID). The minimal promoter of the gene encoding AID is controlled by NF-kB and gene expression is induced by several proinflammatory cytokines such as TNF-alpha (Okazaki et al. 2007). AID was originally discovered in the year 2000 as an essential component of the DNA modification step of class switch recombination (CSR) and somatic hypermutation (SHM) events that occur in B-cell immunoglobulin genes. AID expression is tightly regulated only in transient pre-B cells, in germinal center B cells (GC-B cells), and in activated mature B cells (Okazaki et al. 2007; Shimizu et al. 2012; Honjo et al. 2012). Interestingly, AID dysregulation induces SHM in genes other than immunogloblins. In B-cell lymphoma , MYC, BCL6, PIM1, and numerous other genes were found to be massively mutated (Kotani et al. 2005). Moreover, aberrant AID expression outside B cells has been reported to be involved in oncogenesis associated with cancers where inflammation has been linked to oncogenesis, such as gastric cancer , hepatocarcinoma, and others (Okazaki et al. 2007).
4.2 EBV-Related Cancer and Inflammation
EBV-related cancers are usually accompanied by severe inflammation. EBV-positive DLBCL of the elderly and EBV-positive Hodgkin’s lymphoma show massive infiltration of tumours with lymphocytes , eosinophils, stromal cells, and fibroblasts . NKT lymphomas have poor prognoses and are characterised by severe inflammation. These observations strongly suggest that inflammation is involved in EBV oncogenesis (Lu et al. 2012). This hypothesis is supported by a study showing that in EBV-associated lymphoma , only tumour cells without bystander cells fail to be engrafted into immunodeficient mice. This observation suggests that bystander cells support tumour cell survival. EBV infection has been shown to activate NFkB, which as previously mentioned induces AID expression in mature B cells (Okazaki et al. 2007). However, whether aberrant AID expression during EBV infections is involved in EBV oncogenesis remains to be elucidated. Intriguingly, certain EBV-related cancers present with coinfection with malaria or HIV. For example, endemic Burkitt’s lymphoma is common in sub-Saharan Africa, which also happens to be the endemic area for malaria. Several lines of evidence, including epidemiological studies, strongly indicate that malaria induces Burkitt’s lymphoma through a mechanism that involves T-cell dysfunction, or other direct cofactors. Similarly, HIV infection has been reported to play certain roles in EBV-induced lymphomagenesis. The fact that T-cell maintenance in the HAART era is not associated with decreased incidence of EBV-related lymphomas such as Hodgkin’s lymphoma , suggests that HIV infection and not the degree of immunocompromise, plays a role in these diseases (Rickinson 2014).
4.3 EBV miRNAs Regulate Inflammation and Immune Evasion
Several EBV miRNAs have been reported to be involved in inflammation and immune evasion. Major histocompatibility complex class I polypeptide-related sequence B (MICB) is a ligand of the NKG2D type II receptor, a stress-induced immune molecule (Bahram et al. 1994; Groh et al. 1996). Both B cells and endothelial cells, which are the targets of EBV, express MICB. Binding of MICB activates NK, CD8+αβ, and CD8+γδ T cells (Suarez-Alvarez et al. 2009). MICB expression on the cell surface is upregulated in response to various insults such as viral infection, tumour formation, heat shock, and DNA damage. Therefore, EBV downregulates MICB expression to decrease immune detection by NK cells . Previous studies have shown that downregulated MICB expression decreases the lysis of infected cells by NK cells (Stern-Ginossar et al. 2007). The 3′ UTR of MICB mRNA has potential binding sites for EBV miR-BART2-5p (Nachmani et al. 2009). EBV downregulates MICB expression by employing miR-BART2-5p, thus decreasing NK-cell–mediated lysis to prevent detection by immune cells.
MiR-BART1-1 is expressed from the 5′ UTR and miR-BART1-2 and miR-BART1-3 are expressed from the 3′ UTR of BHRF1 in EBV-infected cells (Xia et al. 2008). MiR-BART1-3 expression is markedly elevated in EBV-infected type III latent cells (Xia et al. 2008). In addition, miR-BART1-3 has been detected in cells isolated from EBV-positive primary effusion lymphoma and AIDS-associated diffuse large B-cell lymphoma (Xia et al. 2008). BHRF1 cluster miRNAs are characteristically detected in EBV-infected type III latent cells (Xing and Kieff 2007). EBV miR-BHRF1-3 regulates host immunity by downregulating interferon (IFN) -inducible T-cell–attracting chemokine (I-TAC, also known as CXCL11). CXCL11 belongs to the CXC family of chemokines, and its expression is strongly induced by both IFN-β and IFN-γ (Rani et al. 1996). CXCL11 promotes cell-mediated immunity by attracting activated T cells. The 3′ UTR of CXCL11 mRNA shows 100 % complementarity to miR-BART1-3 and therefore is a target of miR-BART1-3. MiR-BART1-3 inversely regulates CXCL11 expression whereas the antisense sequence of miR-BART1-3 has an opposite effect (Pfeffer et al. 2004). MiR-BART1-3 significantly reduces CXCL11 expression at both the mRNA and protein levels (Xia et al. 2008). Thus, because cellular chemokines are the targets of viral miRNAs, EBV may regulate antigen processing and presentation and downregulate CTL cytokine networks through this mechanism.
5 Concluding Remarks
EBV-associated cancers are generally difficult to cure. Despite extensive studies on well-known concepts and methods, the molecular mechanisms through which EBV induces tumourigenesis and eludes immune surveillance remain unclear. Recent studies using mouse models of EBV-mediated lymphoproliferative diseases have shown that EBV infection of B cells is necessary but not sufficient to induce tumourigenesis because all peripheral mononuclear cells are needed to generate tumours in these mice (Kuppers 2009). Immune cells are also indispensable for EBV-induced tumourigenesis. However, the detailed roles of inflammation in EBV-induced lymphomagenesis and the relationship between these cells and EBV-infected cells with respect to tumourigenesis remain unclear. Moreover, the mechanisms underlying drug resistance, which results in poor prognoses of EBV-related tumours, have not yet been elucidated. Therefore, it is important to study the biology of EBV-associated tumours from a new perspective such as that provided by investigations focused on EBV miRNAs.
References
Anuranjani BM (2014) Concerted action of Nrf2-ARE pathway, MRN complex, HMGB1 and inflammatory cytokines - implication in modification of radiation damage. Redox Biol 2:832–846
Bahram S, Bresnahan M, Geraghty DE, Spies T (1994) A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci U S A 91(14):6259–6263
Barth S, Pfuhl T, Mamiani A et al (2008) Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res 36(2):666–675
Barth S, Meister G, Grasser FA (2011) EBV-encoded miRNAs. Biochim Biophys Acta 1809(11–12):631–640
Borza CM, Hutt-Fletcher LM (2002) Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8(6):594–599
Cai LM, Lyu XM, Luo WR, Cui XF, Ye YF, Yuan CC, Peng QX, Wu DH, Liu TF, Wang E, Marincola FM, Yao KT, Fang WY, Cai HB, Li X (2015) EBV-miR-BART7-3p promotes the EMT and metastasis of nasopharyngeal carcinoma cells by suppressing the tumor suppressor PTEN. Oncogene 34(17):2156–66
Chen K, Rajewsky N (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8(2):93–103
Choy EY, Siu KL, Kok KH et al (2008) An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med 205(11):2551–2560
do Kim N, Chae HS, Oh ST et al (2007) Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J Virol 81(2):1033–1036
Fang W, Li X, Jiang Q et al (2008) Transcriptional patterns, biomarkers and pathways characterizing nasopharyngeal carcinoma of Southern China. J Transl Med 6:32
Ghosh SK, Forman LW, Akinsheye I, Perrine SP, Faller DV (2007) Short, discontinuous exposure to butyrate effectively sensitizes latently EBV-infected lymphoma cells to nucleoside analogue antiviral agents. Blood Cells Mol Dis 38(1):57–65
Gourzones C, Gelin A, Bombik I et al (2010) Extra-cellular release and blood diffusion of BART viral micro-RNAs produced by EBV-infected nasopharyngeal carcinoma cells. Virol J 7:271
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34(Database issue):D140–144
Grivennikov SI (2013) Inflammation and colorectal cancer: colitis-associated neoplasia. Semin Immunopathol 35(2):229–244
Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T (1996) Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci U S A 93(22):12445–12450
Grundhoff A, Sullivan CS, Ganem D (2006) A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 12(5):733–750
Han J, Flemington C, Houghton AB et al (2001) Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc Natl Acad Sci U S A 98(20):11318–11323
Hislop AD, Taylor GS, Sauce D, Rickinson AB (2007) Cellular responses to viral infection in humans: lessons from Epstein-Barr virus. Annu Rev Immunol 25:587–617
Honjo T, Kobayashi M, Begum N, Kotani A, Sabouri S, Nagaoka H (2012) The AID dilemma: infection, or cancer? Adv Cancer Res 113:1–44
Iizasa H, Wulff BE, Alla NR et al (2010) Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J Biol Chem 285(43):33358–33370
Kelly GL, Long HM, Stylianou J et al (2009) An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog 5(3):e1000341
Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115(2):209–216
Kosaka N, Iguchi H, Ochiya T (2010) Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci 101(10):2087–2092
Kotani A, Okazaki IM, Muramatsu M et al (2005) A target selection of somatic hypermutations is regulated similarly between T and B cells upon activation-induced cytidine deaminase expression. Proc Natl Acad Sci U S A 102(12):4506–4511
Kuppers R (2009) Molecular biology of Hodgkin lymphoma. Hematol Am Soc Hematol Educ Progr:1491–496
Lemercier C (2015) When our genome is targeted by pathogenic bacteria. Cell Mol Life Sci 72(14):2665–2676
Lo AK, To KF, Lo KW et al (2007) Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A 104(41):16164–16169
Lu J, Chanda B, Kotani A (2012) EBV encoded miRNAs in EBV related malignancy. Hematol Sci Pract 409–425.
Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T (2004) Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 15(2):185–197
Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O (2009) Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5(4):376–385
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7(3):683–694
Okazaki IM, Kotani A, Honjo T (2007) Role of AID in tumorigenesis. Adv Immunol 94:245–273
Pagano JS, Blaser M, Buendia MA et al (2004) Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol 14(6):453–471
Parkin DM (2006) The global health burden of infection-associated cancers in the year 2002. Int J Cancer 118(12):3030–3044
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55(2):74–108
Pegtel DM, Cosmopoulos K, Thorley-Lawson DA et al (2010) Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107(14):6328–6333
Pfeffer S, Zavolan M, Grasser FA et al (2004) Identification of virus-encoded microRNAs. Science 304(5671):734–736
Rana TM (2007) Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol 8(1):23–36
Rani MR, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM (1996) Characterization of beta-R1, a gene that is selectively induced by interferon beta (IFN-beta) compared with IFN-alpha. J Biol Chem 271(37):22878–22884
Rickinson AB (2014) Co-infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol 26:99–115
Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W (2010) Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog 6(8):e1001063
Shimizu T, Marusawa H, Endo Y, Chiba T (2012) Inflammation-mediated genomic instability: roles of activation-induced cytidine deaminase in carcinogenesis. Cancer Sci 103(7):1201–1206
Shlomai A, de Jong YP, Rice CM (2014) Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin Cancer Biol 26:78–88
Stern-Ginossar N, Elefant N, Zimmermann A et al (2007) Host immune system gene targeting by a viral miRNA. Science 317(5836):376–381
Suarez-Alvarez B, Lopez-Vazquez A, Baltar JM, Ortega F, Lopez-Larrea C (2009) Potential role of NKG2D and its ligands in organ transplantation: new target for immunointervention. Am J Transplant 9(2):251–257
Thorley-Lawson DA (2001) Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1(1):75–82
Wang X, Gu J, Zhang MQ, Li Y (2008) Identification of phylogenetically conserved microRNA cis-regulatory elements across 12 Drosophila species. Bioinformatics 24(2):165–171
Xia T, O’Hara A, Araujo I et al (2008) EBV microRNAs in primary lymphomas and targeting of CXCL-11 by ebv-mir-BHRF1-3. Cancer Res 68(5):1436–1442
Xing L, Kieff E (2007) Epstein-Barr virus BHRF1 micro- and stable RNAs during latency III and after induction of replication. J Virol 81(18):9967–9975
Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7(3):673–682
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Kotani, A. (2016). Roles of Epstein–Barr Virus Micro RNAs in Epstein–Barr Virus-Associated Malignancies. In: Miyasaka, M., Takatsu, K. (eds) Chronic Inflammation. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56068-5_19
Download citation
DOI: https://doi.org/10.1007/978-4-431-56068-5_19
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56066-1
Online ISBN: 978-4-431-56068-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)