
Abstract
The MYC proto-oncogene is downstream of many growth-related signal transduction pathways that senses the cellular environment, such that the presence of growth factors and nutrients stimulates MYC expression, which in turn produces a transcriptional factor that amplifies the expression of a broad spectrum of genes involving metabolism, cell cycle, and cell death regulation. Acute deregulation of MYC presumably leads to metabolic imbalances that trigger cell cycle checkpoints, such as p53 or ARF, rendering the cells non-proliferative or dead. During tumor development, however, loss of checkpoints unleashes the ability of deregulated MYC to drive nutrient (glucose and glutamine) uptake, lipogenesis, nucleotide and protein synthesis for ribosomal biogenesis, and cell cycle progression. Deregulation of MYC causes cells to be addicted to nutrients to support a constitutive program of growth. Herein, we review how MYC regulates metabolism and how MYC addiction could be exploited to develop therapies that are directed at metabolic pathways.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The field of cancer metabolism has grown rapidly over the last decade as more links between oncogenes and metabolism are uncovered 90 years after Otto Warburg made his seminal observations that many cancers consume vast amounts of glucose and convert it to lactate (Vander Heiden et al. 2009; Koppenol et al. 2011; Cantor and Sabatini 2012). To grow, proliferate, and survive, cancer cells exhibit different metabolic programs including the Warburg effect. However, the Warburg effect per se is insufficient to provide cancer cells with all the substrates for growth and survival. In the past decade, many genetic changes in cancers have been linked to the Warburg effect, glutaminolysis, carbohydrate, and fatty acid metabolism. Among the genetic alterations, oncogenic MYC stood out as a canonical oncogene that was first to be linked mechanistically to altered glucose metabolism, when it was discovered to transactivate LDHA, encoding lactate dehydrogenase A (Shim et al. 1997). MYC is now known to amplify the expression of thousands of genes, with the bulk involved in various aspects of cell metabolism including glycolysis, glutaminolysis, nucleotide, protein, and fatty acid metabolism (Dang 2012). MYC is also involved in mitochondrial and ribosome biogenesis. ln this chapter, we will provide an overview of key roles of MYC in regulating metabolism and how they contribute to cancer development and progression.
2 MYC and Cancer
Cell growth or the increase in cell mass required for cell proliferation involves the import of nutrients and their conversion to cell mass and ATP. MYC and the mTOR pathway are critical for cell growth. Discovery of the retroviral v-MYC oncogene (Duesberg and Vogt 1979) led to the identification of its cellular homologue termed c-MYC (Vennstrom et al. 1982). Herein, the human gene will be termed MYC (italicized) and the protein termed MYC. The normal proto-oncogene MYC is downstream of many growth factor signaling pathways, including receptor tyrosine kinase pathway, T cell receptor pathway, and WNT signaling pathways that regulated its expression through sensing extracellular cues (Dang 2012). The MYC proto-oncogene is a member of the MYC family, which includes MYCN and MYCL. While MYCN is commonly amplified in neuroblastoma, an aggressive childhood cancer, MYCL is only occasionally amplified in some human small cell lung cancer (Brodeur et al. 1984; Nau et al. 1985).
The MYC proto-oncogene is activated by chromosomal translocations in human Burkitt’s lymphoma (Dalla-Favera et al. 1982; Taub et al. 1982). The juxtaposition of MYC to one of three immunoglobulin enhancers via chromosomal translocations deregulates its expression. MYC is now found to be one of the most frequently amplified human oncogene among many different types of cancers, illustrating its central role in human cancer development (Beroukhim et al. 2010; Atlas 2012a, b; Cancer Genome Atlas Research N 2012; Cancer Genome Atlas Research N et al. 2013) (Fig. 5.1).

MYC alterations in human cancer and cancer cell lines. TCGA data are displayed through cBioPortal. Note that MYC is largely amplified across multiple human cancers
Furthermore, loss of upstream regulators such as APC in the WNT pathway can also lead to deregulated MYC gene expression in cancers (Beroukhim et al. 2010; He et al. 1998). In addition to its important role in cancer, MYC plays a pivotal role in maintaining the pluripotency of stem cells (Varlakhanova et al. 2010). Notably, MYC is also one of the four transcription factors that can induce pluripotency in human skin fibroblast (Takahashi and Yamanaka 2006). These observations suggest that MYC’s contribution to cellular dedifferentiation might be central to its neoplastic transforming activity.
Given its important role in growth, MYC expression is tightly regulated in non-transformed cells such that its acute overexpression results in activation of cell cycle checkpoints such as ARF and p53 (Dang 2012). Hence in normal cells, overexpressed MYC leads to cell growth arrest or apoptosis. Unlike their normal counterparts, many cancers with deregulated MYC lose these checkpoints such as loss of p53 in human Burkitt’s lymphoma (Schmitz et al. 2012, 2014).
The role of MYC in tumorigenesis has been extensively studied in a number of human cell lines and transgenic mouse models. While overexpression of MYC in cells that have lost checkpoints results in tumorigenesis, loss of MYC can trigger cell death in a Burkitt’s lymphoma model cell line with a tetracycline-regulated MYC transgene (Yustein et al. 2010). Activation of an MYC transgene specifically in the liver of a mouse model induced the formation of large liver tumors, which also exhibited the so-called oncogene addiction. Being addicted, these tumors regressed upon silencing of the MYC transgene (Felsher 2010). These findings suggest that MYC is important in tumor initiation and maintenance. Once these tumors are established, they can also be addicted to MYC, perhaps partially via the metabolic pathways that MYC regulates. Indeed, we hypothesize that deregulated MYC results in deregulated cell growth signaling that requires a commensurate constitutive source of bioenergetic nutrients, such that the MYC-transformed cell becomes addicted to glucose, glutamine, and other nutrients (Fig. 5.2). In this regard, we will review in this chapter the connections between MYC, cell growth, proliferation, and metabolism as they are related to cancer biology and therapy.

MYC proto-oncogene mediates a transcriptional program that provides nutrients for the cell and stimulates it to grow. In normal cells, it is surmised that growth factors and nutrients are both required for cell proliferation, such that nutrient deprivation will result in diminished (lowercase MYC) MYC expression leading to growth arrest
3 Function of MYC
MYC protein is composed of a transactivation domain at N-terminal and a helix-loop-helix leucine zipper domain at C-terminal for DNA binding (Baudino and Cleveland 2001). Upon induction, MYC heterodimerizes with its partner MAX, which is also a helix-loop-helix leucine zipper protein, to bind the consensus DNA sequence (CACGTG or E-box) or its variants and alter gene expression (Dang 2012).
The oncoprotein MYC has many binding targets, perhaps up to 15 % of genes (Fernandez et al. 2003; Cawley et al. 2004). Global mapping of MYC binding sites in the human genome using human Burkitt’s model lymphoma cells had demonstrated that approximately 3,000 genes are associated with MYC (Zeller et al. 2006). Among these 3,000 MYC-bound genes, 688 that were found to have altered are involved in protein synthesis and cell metabolism, suggesting that MYC activates metabolic reprogramming in cancer cells to fulfill the increased metabolic needs required for rapid growth. Subsequent global mapping of MYC binding sites has broadened the number of putative MYC targets, but expression analysis shows that many key metabolism genes are among the most upregulated genes.
Recent studies have suggested that MYC is a universal amplifier of gene expression through the release of RNA Pol II promoter pausing (Lin et al. 2012; Nie et al. 2012). However, MYC does not uniformly upregulate all genes to the same degree, suggesting that there is a hierarchy to global MYC regulation. That is, MYC regulates specific genes to different degrees depending on their specific cellular function. For example, MYC would not amplify tumor suppressor genes to the same extent as it would amplify the expression of growth-promoting genes in a cell stimulated to grow. Further, these observations could not explain MYC-mediated suppression. For instance, a few studies showed that MYC suppresses cyclin-dependent kinase inhibitors p15 and p21 by recruiting Miz-1 (Seoane et al. 2001, 2002; Staller et al. 2001). For cancer cells to proliferate, the balance between the expression of growth-promoting genes and growth-arresting genes needs to be maintained.
4 Role of MYC in Cell Growth and Proliferation
MYC consistently alters specific groups of genes that are involved in metabolism, protein biosynthesis, cell cycle regulation, angiogenesis, and apoptosis (Prendergast 1999; Nilsson and Cleveland 2003; Baudino et al. 2002). We will not discuss all of MYC’s target genes, but three functions relevant to cancer proliferation stand out: cell cycle, protein synthesis, and metabolism. While these roles of MYC have been studied separately, these processes are inextricably linked with metabolism, fueling MYC-driven cell growth and proliferation.
MYC has been shown to drive the cell cycle through E-box-dependent promoter regulation of cyclins D1 and D2, CDK4, and cyclin B1 (Fernandez et al. 2003; Bouchard et al. 2001; Menssen and Hermeking 2002; Hermeking et al. 2000). Via inhibiting Miz1, MYC can enhance the cell cycle through repression of CDK repressors p21 and p151NK4A (Wu et al. 2003; Seoane et al. 2001).
Cell cycle progression requires that cells attain a certain cell size before initiating DNA replication and the ensuing cell division to produce two daughter cells. In addition to accelerating the progression of the cell cycle, MYC also accelerates protein synthesis and increases cell size. Indeed, MYC overexpressing fibroblasts show protein synthesis that is about threefold higher than in MYC knockout fibroblasts (Mateyak et al. 1997). MYC plays a key role in regulating ribosome biogenesis (Kim et al. 2000; Schlosser et al. 2003; Poortinga et al. 2004). Studies have also shown that Drosophila mutants for these ribosomal protein genes have smaller body size which phenocopies a natural Drosophila MYC mutant fly, termed diminutive (Orian et al. 2003; Fernandez et al. 2003). Conversely, overexpressing MYC in Drosophila (which is called dMYC) increases their cell size, thus increasing the size of body parts (when dMYC is expressed in a tissue-specific fashion) (de la Cova et al. 2004; Moreno and Basler 2004; Secombe et al. 2004). Not only do dMYC-overexpressing cells grow bigger, but they competitively induce apoptosis on their surrounding cells which have lower levels of dMYC (de la Cova et al. 2004). In vertebrates, overexpression of MYC in the liver caused liver hypertrophy (Kim et al. 2000) and in B cells caused enlarged lymphocytes (Iritani et al. 2002). MYC is unique among transcription factors in that it can activate transcription mediated by all three RNA polymerases I, II, and III to drive ribosomal biogenesis and protein synthesis (Gomez-Roman et al. 2003). MYC binds to pol III-transcribed tRNA and 5S rRNA promoter genes which all play a major role in protein synthesis. MYC also directly stimulates rRNA transcription (Arabi et al. 2005; Grandori et al. 2005; Grewal et al. 2005). All these studies suggest several ways by which MYC increases cell size via ribosome biogenesis and protein synthesis. Taken together these studies show that MYC increases cell size and protein synthesis by driving ribosome synthesis, necessitating the increased uptake or synthesis of nucleotides and amino acids. Below we will discuss how MYC reprograms metabolism to work hand in hand with cell cycle and ribosome biogenesis to drive cancer cell growth and proliferation.
5 MYC and Metabolism
Cancer cells exhibit profound metabolic changes, promoting the synthesis of cellular building blocks to support cellular growth, proliferation, and survival (Ward and Thompson 2012). Cancer cells must produce sufficient lipids and phospholipids to build cellular and organelle membranes and sufficient nucleotides to replicate DNA, increase mRNA and build ribosomes, and acquire and produce sufficient amino acids to fuel protein synthesis.
The most noted change in cancer metabolism has been aerobic glycolysis or the Warburg effect, first noted by Otto Warburg in his landmark studies beginning in the 1920s (Koppenol et al. 2011; Warburg et al. 1927). In contrast to non-cancer cells where pyruvate derived from glucose via glycolysis enters the mitochondria and is oxidatively metabolized to maximize ATP production, proliferating cancer cells primarily convert pyruvate to lactate even in the presence of oxygen. Although aerobic glycolysis sacrifices ATP production per molecule of glucose, the increase of glucose flux in aerobic glycolysis provides the opportunity for cancer cells to maximize cellular building blocks via shunting of glycolytic intermediates into biosynthetic pathways. Glycolytic intermediates provide fuel for the pentose phosphate pathway, which provides ribose or nucleotide synthesis and NADPH for cellular reducing power; the serine biosynthesis pathway, which plays a critical role in nucleotide synthesis; and glycerol, which plays a key role in triglyceride metabolism (Ward and Thompson 2012).
In addition to glucose metabolism, cancer cells show additional metabolic changes. Many cancer cells become markedly dependent on glutamine for glutathione, protein, and nucleotide synthesis. Cancer cells often boost nucleotide metabolism, upregulating the synthesis of purine and pyrimidines to support DNA and RNA synthesis. Additionally, cancer cells reprogram how they synthesize and take up nonessential and essential amino acids.
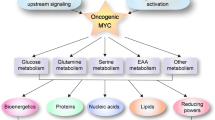
One of the great advances in our understanding of cancer metabolism over the last 15 years is relationship between the recurrent genetic changes observed in cancer and the metabolic phenotypes of the resultant cancers. The p53 protein, one of the most frequently mutated or lost genes in cancer, has been shown to be a key regulator of glucose, glutamine, and amino acid metabolism (Bensaad et al. 2006; Hu et al. 2010; Jiang et al. 2011; Maddocks et al. 2013). The mammalian/mechanistic target of rapamycin (mTOR) complex 1, which serves as both a metabolic sensor and regulator, is recurrently activated in cancers via constitutive activation of growth factor/PI3K/AKT signaling pathways via activating mutations or loss of inhibitors such as PTEN, LKB1, or tuberous sclerosis complex proteins (Laplante and Sabatini 2012; Willems et al. 2012; Fresno Vara et al. 2004; Rodon et al. 2013; Atlas 2012a, b; Song et al. 2012; Sanchez-Cespedes 2011; Luo et al. 2010). Common KRAS mutations have been reported to activate glucose, glutamine, and nucleotide metabolism (Son et al. 2013; Gaglio et al. 2011; Ying et al. 2012). However, the oncogene that has perhaps the best studied role in metabolism is MYC (Dang 2012). Expression of MYC induces profound metabolic reprogramming in cancer, controlling glucose, glutamine, nucleotide, lipid, and amino acid metabolism (Fig. 5.3).

MYC stimulates genes involved in glycolysis, glutaminolysis, lipid, and amino acid synthesis as well as mitochondrial and ribosomal biogenesis. Glucose and glutamine are depicted to be transported and catabolized through glycolysis and the mitochondrion to produce ATP and the building blocks for nucleotide, lipid, and protein synthesis. The mitochondrion is depicted as a central biosynthetic organelle in addition to its function in oxidative phosphorylation. FA fatty acid, MCT1 monocarboxylate transporter 1
5.1 MYC, the Warburg Effect, and Mitochondria
Aerobic glycolysis or the Warburg effect relies on increased uptake and retention of glucose and increased glycolysis to convert this glucose to pyruvate and, consequently, to lactate. In cancer cells, MYC controls a transcriptional program to promote the Warburg effect. Glycolysis requires the uptake of glucose into the cell by glucose transporters. MYC can directly upregulate the glucose transporter GLUT1 (Osthus et al. 2000). After uptake by transporters, glucose is phosphorylated by hexokinases and becomes trapped in the cell. Hexokinase II has been shown to be overexpressed in cancer and can be induced by MYC (Kim et al. 2004). MYC also shows almost uniform upregulation of glycolytic genes, including phosphoglucose isomerase, phosphofructokinase, glyceraldehyde-3-phosphate dehydrogenase, phosphoglycerate kinase, and enolase, binding to the promoters of these genes in upregulating them (Osthus et al. 2000; Kim et al. 2004). By upregulating almost the entire pathway, MYC is capable of enhancing the Warburg effect.
In certain experimental systems, MYC can induce the expression of PKM2 as well as influencing the splicing of the PKM transcript to PKM2 by upregulating the RNA binding splicing proteins HNRNPA1 and HNRNPA2 (David et al. 2010). PKM2 is an important enzyme in cancer metabolism, which differs from its alternative splice form PKM1 in its ability to promote the Warburg effect, regulate pentose phosphate pathway flux, regulate serine biosynthesis, and bind phosphotyrosines (Ye et al. 2012; Anastasiou et al. 2011; Christofk et al. 2008; Chaneton et al. 2012). Upon phosphorylation by ERK, PKM2 but not PKM1 appears to translocate to the nucleus and phosphorylates H3 tyrosine 11 at the MYC promoter, enhancing MYC expression (Yang et al. 2012).
Aerobic glycolysis requires the glycolytic cofactor NAD+ for the activity of GAPDH. Cancer cells can regenerate NAD+ from NADH by converting pyruvate to lactate via the enzyme lactate dehydrogenase. MYC directly upregulates the lactate dehydrogenase A (LDHA), promoting the conversion of pyruvate to lactate (Shim et al. 1997). Inhibiting LDHA genetically or pharmacologically diminishes the growth of MYC-dependent cancer cell lines (Le et al. 2010). The buildup of lactate can be toxic to cells, so cancer cells undergoing aerobic glycolysis need to excrete lactate. MYC upregulates the monocarboxylate transporter 1 (MCT1/SLC16A1), which transports lactate out of the cells. Inhibiting lactate export via MCT1 inhibition can result in cell death in MYC-dependent cells (Doherty et al. 2014).
Mitochondria, being the powerhouses of cellular metabolism, are also affected by MYC. Genes involved in mitochondrial biogenesis are upregulated by MYC in both mammals and Drosophila (Orian et al. 2003; O’Connell et al. 2003; Morrish et al. 2003; Wonsey et al. 2002; Li et al. 2005). MYC also targets ferritin, IRP1, IRP2, and transferrin receptor (TFRC1) which are all genes involved in iron metabolism largely involving the mitochondrion (O’Connell et al. 2003; Bowen et al. 2002; Wu et al. 1999; O’Donnell et al. 2006). In addition to iron metabolism, nucleotide synthesis genes are also upregulated by MYC, including carbamoyl phosphate synthetase, aspartate transcarbamoylase, dihydroorotase (CAD), and ornithine decarboxylase (ODC) (Bello-Fernandez et al. 1993; Miltenberger et al. 1995; Liu et al. 2008). Specifically, DHODH, which is a target of MYC, requires a functional mitochondrial electron transport chain for its catalytic conversion of orotate to dihydroorotate in nucleotide synthesis. Hence many pathways influenced by MYC require the function of mitochondria to support cell growth and metabolism.
Although MYC plays an important role in inducing glycolysis and mitochondrial function, tumor cells are often hypoxic due to the imperfect neo-vasculature found in solid tumors. The hypoxia-inducible factor (HIF1) inhibits pyruvate conversion to acetyl-CoA, by shunting it to lactate via activation of lactate dehydrogenase A (LDHA) and pyruvate dehydrogenase kinase (PDK1) that inhibits pyruvate dehydrogenase (PDH). This activation of LDHA, and suppression of PDH by HIF1, stops glucose from supplying carbons to the TCA cycle (Kim et al. 2006). Surprisingly, glutamine’s involvement in the TCA cycle persists under hypoxia (Le et al. 2012).
5.2 MYC and Glutamine Metabolism
Glutamine is another major bioenergetic source for tumor cells, especially as a source of nitrogen and carbon for nucleotide and amino acid synthesis. Glutamine is imported to the cells through glutamine transporter (e.g., ASCT2). Glutamine is then converted to glutamate by glutaminase. Glutamate can be further metabolized to α-ketoglutarate (αKG) through glutamine dehydrogenase, glutamine pyruvate transaminase (GPT), or glutamine oxaloacetate transaminase (GOT) to enter TCA cycle and be catabolized by the mitochondria.
Cancer cells are addicted to glutamine, which was documented few decades ago, for the following reasons (Reitzer et al. 1979). First, glutamine helps to feed TCA cycle and results in a truncated TCA cycle, which allows acetyl-CoA from glycolytic pathway to be used for de novo fatty acid synthesis and cholesterol synthesis instead of making citrate with oxaloacetate. Fatty acids and cholesterols are important for building new cell membrane. Other intermediates of TCA cycle can also be used for biosynthesis under the constant supply of αKG from glutamine. The role of glutamine to replenish the intermediates in TCA cycle is far more critical in cancer cells than normal cells due to increased biosynthesis. Second, glutamine and its derivatives glutamate and aspartate are source for nucleotide synthesis, which is also increased in proliferated cancer cells.
MYC drives glutamine metabolism by targeting a number of genes that are involved in the pathway. For instance, MYC directly binds to the promoter region of two high-affinity glutamine importers ASCT2 and SN2 and upregulated their mRNA expression to increase glutamine import (Wise et al. 2008). To also increase flux from glutamine to glutamate, MYC activates glutaminase (GLS) both transcriptionally and posttranscriptionally (Wise et al. 2008; Gao et al. 2009). In both P493 Burkitt’s lymphoma and PC3 prostate cancer cell line, MYC activation increases GLS protein at a much higher level compared to mRNA expression, suggesting that MYC regulates mitochondrial GLS indirectly (Gao et al. 2009). Further investigation showed that MYC suppresses miR-23a and miR-23b, resulting in a derepressed GLS protein translation from miR-23a/b (Gao et al. 2009). As a result, elevated GLS facilitates glutamine to enter TCA cycle in the mitochondria.
MYC-overexpressed cells often exhibit addiction to glutamine. High level of MYC appears to prime fibroblast to a glutamine-dependent state (Yuneva et al. 2007). Upon glutamine deprivation, cells underwent apoptosis. This observation, however, can be rescued by oxaloacetate and pyruvate, suggesting TCA cycle intermediate depletion leads to apoptosis (Yuneva et al. 2007). Similar observations were made in glioma cells. Using small-interference RNA (siRNA) targeting MYC helps glioma cells to develop resistance to glutamine deprivation (Wise et al. 2008).
MYC-dependent glutaminolysis is observed to be a critical alternative energy source pathway in nutrient-limiting environment, specifically under glucose and oxygen deprivation conditions (Le et al. 2012). C13-labeled glutamine was used to track the flux of glutamine metabolism in P493 B cell in the presence and absence of glucose. Interestingly, under glucose-deprived condition, much higher levels of these labeled isotopologues of TCA cycle intermediates, such as fumarate, malate, and aspartate, were found compared to glucose-abundant condition (Le et al. 2012). This observation suggests that while glucose is still a preferable carbon source of TCA cycle, glutamine can virtually replace glucose in the absence of glucose. Nevertheless, under glucose-abundant condition, glutamine is used for glutathione synthesis to reduce oxidative stress (Le et al. 2012). Flux analysis using C13, N15 glutamine as a tracer in another study has documented that glutamine can also contribute to proline biosynthesis (Liu et al. 2012). A recent study using hyperpolarized C13-pyruvate magnetic resonance spectroscopic imaging (MRSI) to visualize tumor formation and regression in an MYC-inducible mouse hepatocellular carcinoma cancer model has observed that glutamine is converted to αKG through increased flux from pyruvate to alanine in premalignant stage (Hu et al. 2011).
A tissue-specific relationship and MYC and glutamine metabolism have been demonstrated in MYC-driven liver cancer versus lung cancer (Yuneva et al. 2012). Differential expression of glutamine synthetase determines the direction of glutamine metabolism (Yuneva et al. 2012). For instance, with low glutamine synthetase expression, MYC-induced liver tumors exhibit increased glutamine catabolism (Yuneva et al. 2012). In contrast, glutamine accumulation found in MYC-induced lung cancers is possibly associated with elevated glutamine synthetase (Yuneva et al. 2012). As tissue origins of tumors can dictate the expression of metabolic pathways, the fate of glutamine can be varied based on the tissue of origin even on the same oncogenic background.
Given that MYC-induced tumors rely on glutamine metabolism for cell growth and survival, targeting glutaminase appears to be a feasible way to treat MYC-overexpressed cancer. In fact, BPTES, a glutaminase inhibitor, has been shown to effectively slow down tumor growth in P493 B cells as well as MYC-induced tumor xenografts (Le et al. 2012; Wang et al. 2010).
5.3 MYC and Amino Acid Transporters and Synthesis
MYC plays a key role in the acquisition and uptake of the amino acids required for cell growth. MYC stimulates the uptake and catabolism of glutamine by upregulating glutamine transporters and the enyzme glutaminase. As discussed above, MYC can drive the synthesis of serine and glycine from glucose via upregulation of key enzymes. Additionally, MYC-driven increase in glutamine metabolism can promote the synthesis of alanine and aspartate, as glutamine-derived glutamate is used to transaminate pyruvate to produce alanine and oxaloacetate to produce aspartate. MYC also promotes the synthesis of proline from glutamine by upregulating proline synthesis genes and indirectly downregulating genes that degrade proline (Liu et al. 2012).
Leucine plays a key role in cancer metabolism through its key role in the regulation of mTORC1 activity (Nicklin et al. 2009). Glutamine can be exchanged through the L-type amino acid transporter (LAT1 composed of SLC7A5 and SLC3A2) for leucine, with glutamine uptake being the rate-limiting step (Nicklin et al. 2009). MYC promote the uptake of glutamine through the regulation of SLC1A5 and then stimulate its exchange for leucine by upregulating SLC7A5 (Gao et al. 2009; Hayashi et al. 2012). While MYC activation has been shown to stimulate leucine uptake (Murphy et al. 2013), the crosstalk between SLC1A5, SLC7A5, MYC, and mTOR remains to be fully elucidated (Sinclair et al. 2013).
5.4 Fatty Acid Metabolism
Glucose first enters the cell via glucose transporters and is retained intracellularly once it is phosphorylated by hexokinases. This six-carbon phosphate then is phosphorylated again and split into two three-carbon structures that can then be converted to glycerol. This glycerol can either be used for lipogenesis or to make pyruvate. In the case of its transformation to pyruvate (the process of glycolysis), the pyruvate is then transaminated to alanine with glutamate derived from glutamine as the nitrogen donor or converted to acetyl-CoA in the Krebs cycle. Acetyl-CoA is then transformed into citrate and oxaloacetate, completing the Krebs cycle, generating ATP, carbon dioxide, and other carbon substrates for other pathways. For example, citrate could be exported into the cytoplasm and converted to acetyl-CoA by ATP citrate lyase (ACLY) for lipogenesis.
The pathway for fatty acid synthesis takes place in the cytoplasm. Citrate from the Krebs cycle in mitochondria is first released into the cytoplasm and converted into acetyl-CoA by ATP citrate lyase (ACLY). The production of malonyl-CoA by acetyl-CoA carboxylase (ACACA) is the first committed step of fatty acid synthesis. Acetyl-CoA carboxylase (ACACA) is the major site of regulation of fatty acid synthesis. Fatty acid synthase (FASN) then converts the malonyl-CoA into a 16-carbon palmitate. Tracing glucose in MYC-induced cells has been shown to be incorporated into increased acetyl-CoA production and in turn increased palmitate synthesis, consistent with the metabolic pathway of glucose conversion to acetyl-CoA and then in turn to palmitate (Morrish et al. 2010). Though this shows the involvement of MYC in lipid metabolism, this also has epigenetic implications, because the traced acetyl-CoA is shown to be incorporated into the acetylation of H4K16.
This upregulation of the lipogenesis pathway by MYC was found in Burkitt’s lymphoma, hepatocellular carcinoma, and osteocarcinoma cell lines. However, not all cancers induce lipogenesis. Many prostate cancers oxidize lipids to make ATP (Tennakoon et al. 2013). Others have also shown that pharmaceutical inhibition of lipid oxidation in N- MYC-amplified neuroblastoma cells leads to cell cycle arrest, apoptosis, and neuronal differentiation (Zirath et al. 2013). This differentiation is accompanied with lipid accumulation. Gene expression analysis shows inhibition of MYC’s correlation with decreased expression of oxidative phosphorylation and fatty acid oxidation genes (Zirath et al. 2013). These observations suggest that cancer metabolic networks depend on the specific cell type, the driving oncogene(s), and the tumor microenvironment.
5.5 MYC and Nucleotide Biosynthesis
MYC drives glucose and glutamine metabolism to provide carbon sources for biosynthesis and continually generate ATP to support tumor cell growth and survival. However, for cancer cells to proliferate (increase cell numbers), sufficient supply of nucleotides is equally crucial. Global mapping of MYC target genes using ChIP-PET has indicated that many genes that are involved in nucleotide synthesis pathway are direct MYC targets (Zeller et al. 2006). For instance, the enzymes that are involved in dNTP metabolism, such as inosine monophosphate dehydrogenase (IMPDH), thymidylate synthase (TS), and phosphoribosyl pyrophosphate synthetase 2 (PRPS2), were found to be induced by MYC (Mannava et al. 2008; Liu et al. 2008). Targeting IMPDH by its specific inhibitor mycophenolic acid (MPA) results in apoptosis and S phase arrest in P493 B cells, which can be rescued by exogenous guanosine (Liu et al. 2008).
Despite direct regulation of nucleotide synthesis pathway, MYC also promotes the channeling of glycolytic intermediates to make amino acids that are required for nucleotide synthesis, such as serine and glycine (Vazquez et al. 2011). Glycolytic intermediate 3-phosphoglycerate is oxidized by phosphoglycerate dehydrogenase (PHGDH) and then converted to serine in a series of reactions that are catalyzed by phosphoserine aminotransferase 1 (PSAT1) and phosphoserine phosphatase (PSPH). Further, serine hydroxymethyltransferase (SHMT) can convert serine to glycine while simultaneously converting tetrahydrofolate to 5,10 methylenetetrahydrofolate. Several studies have shown PHGDH, and PSPH can be induced by MYC (Vazquez et al. 2011). In addition, both mitochondrial and cytoplasmic SHMTs that were documented are direct MYC targets (Nikiforov et al. 2002). However, decreased tumor burden was not found when breed Burkitt’s lymphoma mouse model expresses human MYC transgenes with inactivated alleles of PHGDH or SHMT, suggesting that target genes that are involved in serine and glycine pathway individually may not be sufficient (Nilsson et al. 2012).
5.6 MYC and Oncometabolites
While altered metabolism in cancers is often thought of as downstream of oncogenes, recent studies have shown that metabolites themselves can contribute to tumor formation most likely through alterations in the cancer epigenome. Mutations in TCA cycle enzymes can lead to the accumulation of succinate and fumarate, which are believed to promote cancer through alteration of epigenetic state and reactive oxygen species (Letouze et al. 2013; Sullivan et al. 2013). As MYC can increase the amount of glutamine entering the TCA cycle, it is possible that the MYC activation could stimulate succinate and fumarate accumulation in the TCA cycle mutant cell lines.
The most studied oncometabolite to date is 2-hydroxyglutarate. Isocitrate dehydrogenase (IDH) catalyzes the formation of alpha-ketoglutarate from isocitrate in the TCA cycle. Mutants IDH1 and IDH2, which are recurrently mutated in several types of cancer including leukemia and gliomas, produce 2-hydroxyglutarate. 2-Hyrdroxyglutarate can inhibit alpha-ketoglutarate-dependent histone demethylases (Chowdhury et al. 2011; Lu et al. 2012) and DNA demethylase Tet2, causing epigenetic changes which inhibit cellular differentiation (Figueroa et al. 2010). However, recent studies suggest that mutant IDH is not the only source of 2-hydroxyglutarate in cancer. A recent study showed the accumulation of 2-hydroxyglutarate in triple negative breast cancers lacking an IDH mutation (Terunuma et al. 2014). These high 2-hydroxyglutarate breast cancers, which exhibited a hypermethylation, showed strong overexpression of MYC and had an MYC expression signature. Knockdown of MYC in these breast cancer cell lines decreased levels of 2-hydroxyglutarate. MYC increased the metabolism of glutamine, which was the source of carbons used to produce the 2-hydroxyglutatarate. While this study suggests that MYC may upregulate 2-hydroxyglutarate, it is not yet known whether this applies to tissues beyond breast cancer.
6 MYC-Driven Metabolism and Cancer Therapy
Although MYC is an intriguing therapeutic target, for example, via bromodomain inhibitors, its downstream transcriptional targets and altered metabolism offer additional therapeutic opportunities (Loven et al. 2013; Delmore et al. 2011). Increased understanding of the reliance of cancer on altered metabolism and the mutations that underlie metabolic reprogramming have driven interest in using anti-metabolism therapies to treat cancer. However, once these therapies reach the clinic, it will be challenging to predict which tumors respond to which therapies. Although MYC status will likely not be sufficient to predict therapeutic response to antimetabolic therapies in all cases due to tissue- and tumor-specific effects, MYC-driven metabolic reprogramming provides intriguing therapeutic targets. MYC drives nucleotide metabolism, which is the target of some of the oldest and most successful chemotherapies. Blocking glucose metabolism by inhibiting glucose uptake and glycolysis is challenging due to the reliance of the vast majority of non-cancer cells on glucose. Targeting aerobic glycolysis through inhibition of lactate dehydrogenase or monocarboxylate transporter-dependent lactate export is a potentially viable target; the lack of high-quality inhibitors and the dependence of rapidly growing non-cancer cells on aerobic glycolysis present challenges in targeting aerobic glycolysis (Doherty et al. 2014; Le et al. 2010). Glutamine metabolism was first tried in the clinic using nonspecific amino acid analogues which alter the activity of a large variety of enzymes, leading to off-target effect of lack of efficacy (Rajagopalan and DeBerardinis 2011; Shapiro et al. 1979). However, the identification of allosteric inhibitors of glutaminase have opened the door to a less toxic inhibition of glutamine metabolism (Robinson et al. 2007; Le et al. 2012). A glutaminase inhibitor began clinical trials in early 2014 (Gross et al. 2014).
Conclusion
MYC has been studied over the years as a master oncogenic regulator, especially because it regulates many genes that are crucial for cancer cell growth and proliferation. Among many of its activities, three major functions of MYC were discussed: cell cycle, protein synthesis, and metabolism. In concert with MYC’s ability to induce cell proliferation, MYC also upregulates many cellular metabolic pathways that are involved in nutrient import and macromolecular biosynthesis. In essence, MYC is an amplifier of gene expression that coordinates the import of nutrients and the bioenergetics demands of replicating a cell, shunting nutrients into cell biomass. Glucose, glutamine, and lipid metabolic pathways are regulated by MYC in various cells to support the increased demand for energy and raw building blocks of proliferating cells. Normal proliferating cells depend on similar metabolic pathways; however, normal MYC expression is dependent on external cues and growth factors, such that altered metabolism induced by MYC is dependent on these external cues. Normal MYC expression is attenuated by the absence of growth factors or nutrients. By contrast, cancer cells with deregulated MYC expression that no longer requires external cues are forced to undergo cell growth independent of nutrient sensing. In this regard, MYC-dependent cancer cells are addicted to nutrients, such as glucose and glutamine. Insights into these metabolic pathways and how MYC regulates them allow for the identification of new therapeutic targets and provide the hope that new therapies might emerge in the clinic for different types of cancers.
References
Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334(6060):1278–1283. doi:10.1126/science.1211485
Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, Larsson LG, Wright AP (2005) c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol 7(3):303–310. doi:10.1038/ncb1225
Atlas TCG (2012a) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487(7407):330–337. doi:10.1038/nature11252
Atlas TCG (2012b) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70. doi:10.1038/nature11412
Baudino TA, Cleveland JL (2001) The Max network gone mad. Mol Cell Biol 21(3):691–702
Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16(19):2530–2543. doi:10.1101/gad.1024602
Bello-Fernandez C, Packham G, Cleveland JL (1993) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci USA 90(16):7804–7808
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126(1):107–120. doi:10.1016/j.cell.2006.05.036
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, Reich M, Winckler W, Lawrence MS, Weir BA, Tanaka KE, Chiang DY, Bass AJ, Loo A, Hoffman C, Prensner J, Liefeld T, Gao Q, Yecies D, Signoretti S, Maher E, Kaye FJ, Sasaki H, Tepper JE, Fletcher JA, Tabernero J, Baselga J, Tsao MS, Demichelis F, Rubin MA, Janne PA, Daly MJ, Nucera C, Levine RL, Ebert BL, Gabriel S, Rustgi AK, Antonescu CR, Ladanyi M, Letai A, Garraway LA, Loda M, Beer DG, True LD, Okamoto A, Pomeroy SL, Singer S, Golub TR, Lander ES, Getz G, Sellers WR, Meyerson M (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463(7283):899–905. doi:10.1038/nature08822
Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B (2001) Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev 15(16):2042–2047. doi:10.1101/gad.907901
Bowen H, Biggs TE, Baker ST, Phillips E, Perry VH, Mann DA, Barton CH (2002) c-Myc represses the murine Nramp1 promoter. Biochem Soc Trans 30(4):774–777, doi:10.1042/
Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM (1984) Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224(4653):1121–1124
Cancer Genome Atlas Research N (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489(7417):519–525. doi:10.1038/nature11404
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM (2013) The cancer genome Atlas Pan-cancer analysis project. Nat Genet 45(10):1113–1120. doi:10.1038/ng.2764
Cantor JR, Sabatini DM (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov 2(10):881–898. doi:10.1158/2159-8290.CD-12-0345
Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116(4):499–509
Chaneton B, Hillmann P, Zheng L, Martin AC, Maddocks OD, Chokkathukalam A, Coyle JE, Jankevics A, Holding FP, Vousden KH, Frezza C, O’Reilly M, Gottlieb E (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491(7424):458–462. doi:10.1038/nature11540
Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep 12(5):463–469. doi:10.1038/embor.2011.43
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452(7184):181–186. doi:10.1038/nature06667
Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM (1982) Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA 79(24):7824–7827
Dang CV (2012) MYC on the path to cancer. Cell 149(1):22–35. doi:10.1016/j.cell.2012.03.003
David CJ, Chen M, Assanah M, Canoll P, Manley JL (2010) HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463(7279):364–368. doi:10.1038/nature08697
de la Cova C, Abril M, Bellosta P, Gallant P, Johnston LA (2004) Drosophila myc regulates organ size by inducing cell competition. Cell 117(1):107–116
Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, Chesi M, Schinzel AC, McKeown MR, Heffernan TP, Vakoc CR, Bergsagel PL, Ghobrial IM, Richardson PG, Young RA, Hahn WC, Anderson KC, Kung AL, Bradner JE, Mitsiades CS (2011) BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146(6):904–917. doi:10.1016/j.cell.2011.08.017
Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, Tortosa M, Genau HM, Rounbehler RJ, Lu Y, Dang CV, Kumar KG, Butler AA, Bannister TD, Hooper AT, Unsal-Kacmaz K, Roush WR, Cleveland JL (2014) Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res 74(3):908–920. doi:10.1158/0008-5472.CAN-13-2034
Duesberg PH, Vogt PK (1979) Avian acute leukemia viruses MC29 and MH2 share specific RNA sequences: evidence for a second class of transforming genes. Proc Natl Acad Sci USA 76(4):1633–1637
Felsher DW (2010) MYC inactivation elicits oncogene addiction through both tumor cell-intrinsic and host-dependent mechanisms. Genes Cancer 1(6):597–604. doi:10.1177/1947601910377798
Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c-Myc protein. Genes Dev 17(9):1115–1129. doi:10.1101/gad.1067003
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, Melnick A (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18(6):553–567. doi:10.1016/j.ccr.2010.11.015
Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M (2004) PI3K/Akt signalling pathway and cancer. Cancer Treat Rev 30(2):193–204. doi:10.1016/j.ctrv.2003.07.007
Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F (2011) Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7:523. doi:10.1038/msb.2011.56
Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV (2009) c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458(7239):762–765. doi:10.1038/nature07823
Gomez-Roman N, Grandori C, Eisenman RN, White RJ (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421(6920):290–294. doi:10.1038/nature01327
Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ (2005) c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol 7(3):311–318. doi:10.1038/ncb1224
Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA (2005) Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol 7(3):295–302. doi:10.1038/ncb1223
Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, Mackinnon AL, Parlati F, Rodriguez ML, Shwonek PJ, Sjogren EB, Stanton TF, Wang T, Yang J, Zhao F, Bennett MK (2014) Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 13:890–901. doi:10.1158/1535-7163.MCT-13-0870
Hayashi K, Jutabha P, Endou H, Anzai N (2012) c-Myc is crucial for the expression of LAT1 in MIA Paca-2 human pancreatic cancer cells. Oncol Rep 28(3):862–866. doi:10.3892/or.2012.1878
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281(5382):1509–1512
Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O’Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Eick D, Vogelstein B, Kinzler KW (2000) Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci USA 97(5):2229–2234. doi:10.1073/pnas.050586197
Hu S, Balakrishnan A, Bok RA, Anderton B, Larson PE, Nelson SJ, Kurhanewicz J, Vigneron DB, Goga A (2011) 13C-pyruvate imaging reveals alterations in glycolysis that precede c-Myc-induced tumor formation and regression. Cell Metab 14(1):131–142. doi:10.1016/j.cmet.2011.04.012
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z (2010) Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA 107(16):7455–7460. doi:10.1073/pnas.1001006107
Iritani BM, Delrow J, Grandori C, Gomez I, Klacking M, Carlos LS, Eisenman RN (2002) Modulation of T-lymphocyte development, growth and cell size by the Myc antagonist and transcriptional repressor Mad1. EMBO J 21(18):4820–4830
Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X (2011) p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 13(3):310–316. doi:10.1038/ncb2172
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3(3):177–185. doi:10.1016/j.cmet.2006.02.002
Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, Dang CV (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 24(13):5923–5936. doi:10.1128/MCB.24.13.5923-5936.2004
Kim S, Li Q, Dang CV, Lee LA (2000) Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci USA 97(21):11198–11202. doi:10.1073/pnas.200372597
Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer 11(5):325–337. doi:10.1038/nrc3038
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149(2):274–293. doi:10.1016/j.cell.2012.03.017
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 107(5):2037–2042. doi:10.1073/pnas.0914433107
Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, Dang CV (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15(1):110–121. doi:10.1016/j.cmet.2011.12.009
Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo AP, Favier J (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23(6):739–752. doi:10.1016/j.ccr.2013.04.018
Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV (2005) Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25(14):6225–6234. doi:10.1128/MCB.25.14.6225-6234.2005
Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA (2012) Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151(1):56–67. doi:10.1016/j.cell.2012.08.026
Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM (2012) Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci USA 109(23):8983–8988. doi:10.1073/pnas.1203244109
Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, Dang CV (2008) Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 3(7):e2722. doi:10.1371/journal.pone.0002722
Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153(2):320–334. doi:10.1016/j.cell.2013.03.036
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483(7390):474–478. doi:10.1038/nature10860
Luo Z, Zang M, Guo W (2010) AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol 6(3):457–470. doi:10.2217/fon.09.174
Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, Vousden KH (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493(7433):542–546. doi:10.1038/nature11743
Mannava S, Grachtchouk V, Wheeler LJ, Im M, Zhuang D, Slavina EG, Mathews CK, Shewach DS, Nikiforov MA (2008) Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 7(15):2392–2400
Mateyak MK, Obaya AJ, Adachi S, Sedivy JM (1997) Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 8(10):1039–1048
Menssen A, Hermeking H (2002) Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci USA 99(9):6274–6279. doi:10.1073/pnas.082005599
Miltenberger RJ, Sukow KA, Farnham PJ (1995) An E-box-mediated increase in cad transcription at the G1/S-phase boundary is suppressed by inhibitory c-Myc mutants. Mol Cell Biol 15(5):2527–2535
Moreno E, Basler K (2004) dMyc transforms cells into super-competitors. Cell 117(1):117–129
Morrish F, Giedt C, Hockenbery D (2003) c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev 17(2):240–255. doi:10.1101/gad.1032503
Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, Hockenbery D (2010) Myc-dependent mitochondrial generation of acetyl-CoA contributes to fatty acid biosynthesis and histone acetylation during cell cycle entry. J Biol Chem 285(47):36267–36274. doi:10.1074/jbc.M110.141606
Murphy TA, Dang CV, Young JD (2013) Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab Eng 15:206–217. doi:10.1016/j.ymben.2012.07.008
Nau MM, Brooks BJ, Battey J, Sausville E, Gazdar AF, Kirsch IR, McBride OW, Bertness V, Hollis GF, Minna JD (1985) L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 318(6041):69–73
Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, Myer VE, MacKeigan JP, Porter JA, Wang YK, Cantley LC, Finan PM, Murphy LO (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136(3):521–534. doi:10.1016/j.cell.2008.11.044
Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, Zhao K, Levens D (2012) c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151(1):68–79. doi:10.1016/j.cell.2012.08.033
Nikiforov MA, Chandriani S, O’Connell B, Petrenko O, Kotenko I, Beavis A, Sedivy JM, Cole MD (2002) A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol Cell Biol 22(16):5793–5800
Nilsson JA, Cleveland JL (2003) Myc pathways provoking cell suicide and cancer. Oncogene 22(56):9007–9021. doi:10.1038/sj.onc.1207261
Nilsson LM, Forshell TZ, Rimpi S, Kreutzer C, Pretsch W, Bornkamm GW, Nilsson JA (2012) Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet 8(3):e1002573. doi:10.1371/journal.pgen.1002573
O’Connell BC, Cheung AF, Simkevich CP, Tam W, Ren X, Mateyak MK, Sedivy JM (2003) A large scale genetic analysis of c-Myc-regulated gene expression patterns. J Biol Chem 278(14):12563–12573. doi:10.1074/jbc.M210462200
O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, Dang CV (2006) Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol 26(6):2373–2386. doi:10.1128/MCB.26.6.2373-2386.2006
Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, Pierce S, Edgar BA, Parkhurst SM, Eisenman RN (2003) Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev 17(9):1101–1114. doi:10.1101/gad.1066903
Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV (2000) Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 275(29):21797–21800. doi:10.1074/jbc.C000023200
Poortinga G, Hannan KM, Snelling H, Walkley CR, Jenkins A, Sharkey K, Wall M, Brandenburger Y, Palatsides M, Pearson RB, McArthur GA, Hannan RD (2004) MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J 23(16):3325–3335. doi:10.1038/sj.emboj.7600335
Prendergast GC (1999) Mechanisms of apoptosis by c-Myc. Oncogene 18(19):2967–2987. doi:10.1038/sj.onc.1202727
Rajagopalan KN, DeBerardinis RJ (2011) Role of glutamine in cancer: therapeutic and imaging implications. J Nucl Med 52(7):1005–1008. doi:10.2967/jnumed.110.084244
Reitzer LJ, Wice BM, Kennell D (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 254(8):2669–2676
Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK, Hansen JC, Curthoys NP (2007) Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 406(3):407–414. doi:10.1042/BJ20070039
Rodon J, Dienstmann R, Serra V, Tabernero J (2013) Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol 10(3):143–153. doi:10.1038/nrclinonc.2013.10
Sanchez-Cespedes M (2011) The role of LKB1 in lung cancer. Familial Cancer 10(3):447–453. doi:10.1007/s10689-011-9443-0
Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D (2003) A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res 31(21):6148–6156
Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM (2014) Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb Perspect Med 4 (2):321–333. doi:10.1101/cshperspect.a014282
Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, Liu X, Powell J, Yang Y, Xu W, Zhao H, Kohlhammer H, Rosenwald A, Kluin P, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Ogwang MD, Reynolds SJ, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Pittaluga S, Wilson W, Waldmann TA, Rowe M, Mbulaiteye SM, Rickinson AB, Staudt LM (2012) Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490(7418):116–120. doi:10.1038/nature11378
Secombe J, Pierce SB, Eisenman RN (2004) Myc: a weapon of mass destruction. Cell 117(2):153–156
Seoane J, Le HV, Massague J (2002) Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 419(6908):729–734. doi:10.1038/nature01119
Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J (2001) TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol 3(4):400–408. doi:10.1038/35070086
Shapiro RA, Clark VM, Curthoys NP (1979) Inactivation of rat renal phosphate-dependent glutaminase with 6-diazo-5-oxo-L-norleucine. Evidence for interaction at the glutamine binding site. J Biol Chem 254(8):2835–2838
Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV (1997) c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A 94(13):6658–6663
Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA (2013) Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14(5):500–508. doi:10.1038/ni.2556
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443):101–105. doi:10.1038/nature12040
Song MS, Salmena L, Pandolfi PP (2012) The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 13(5):283–296. doi:10.1038/nrm3330
Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M (2001) Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol 3(4):392–399. doi:10.1038/35070076
Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 51(2):236–248. doi:10.1016/j.molcel.2013.05.003
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. doi:10.1016/j.cell.2006.07.024
Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, Aaronson S, Leder P (1982) Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A 79(24):7837–7841
Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, Zhang A, Xia X, Ilkayeva OR, Xin L, Ittmann MM, Rick FG, Schally AV, Frigo DE (2013) Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. doi:10.1038/onc.2013.463
Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, Karoly ED, Chan K, Samanta S, Prieto D, Hsu TY, Kurley SJ, Putluri V, Sonavane R, Edelman DC, Wulff J, Starks AM, Yang Y, Kittles RA, Yfantis HG, Lee DH, Ioffe OB, Schiff R, Stephens RM, Meltzer PS, Veenstra TD, Westbrook TF, Sreekumar A, Ambs S (2014) MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest 124(1):398–412. doi:10.1172/JCI71180
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324(5930):1029–1033. doi:10.1126/science.1160809
Varlakhanova NV, Cotterman RF, deVries WN, Morgan J, Donahue LR, Murray S, Knowles BB, Knoepfler PS (2010) myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation 80(1):9–19. doi:10.1016/j.diff.2010.05.001
Vazquez A, Markert EK, Oltvai ZN (2011) Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PLoS One 6(11):e25881. doi:10.1371/journal.pone.0025881
Vennstrom B, Sheiness D, Zabielski J, Bishop JM (1982) Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol 42(3):773–779
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA (2010) Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18(3):207–219. doi:10.1016/j.ccr.2010.08.009
Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J Gen Physiol 8(6):519–530
Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21(3):297–308. doi:10.1016/j.ccr.2012.02.014
Willems L, Tamburini J, Chapuis N, Lacombe C, Mayeux P, Bouscary D (2012) PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr Oncol Rep 14(2):129–138. doi:10.1007/s11912-012-0227-y
Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105(48):18782–18787. doi:10.1073/pnas.0810199105
Wonsey DR, Zeller KI, Dang CV (2002) The c-Myc target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc Natl Acad Sci U S A 99(10):6649–6654. doi:10.1073/pnas.102523299
Wu KJ, Polack A, Dalla-Favera R (1999) Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 283(5402):676–679
Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG (2003) Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 22(3):351–360. doi:10.1038/sj.onc.1206145
Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z (2012) PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150(4):685–696. doi:10.1016/j.cell.2012.07.018
Ye J, Mancuso A, Tong X, Ward PS, Fan J, Rabinowitz JD, Thompson CB (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci U S A 109(18):6904–6909. doi:10.1073/pnas.1204176109
Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA (2012) Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149(3):656–670. doi:10.1016/j.cell.2012.01.058
Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y (2007) Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 178(1):93–105. doi:10.1083/jcb.200703099
Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, Chen X, Bishop JM (2012) The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15(2):157–170. doi:10.1016/j.cmet.2011.12.015
Yustein JT, Liu YC, Gao P, Jie C, Le A, Vuica-Ross M, Chng WJ, Eberhart CG, Bergsagel PL, Dang CV (2010) Induction of ectopic Myc target gene JAG2 augments hypoxic growth and tumorigenesis in a human B-cell model. Proc Natl Acad Sci U S A 107(8):3534–3539. doi:10.1073/pnas.0901230107
Zeller KI, Zhao X, Lee CW, Chiu KP, Yao F, Yustein JT, Ooi HS, Orlov YL, Shahab A, Yong HC, Fu Y, Weng Z, Kuznetsov VA, Sung WK, Ruan Y, Dang CV, Wei CL (2006) Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc Natl Acad Sci U S A 103(47):17834–17839. doi:10.1073/pnas.0604129103
Zirath H, Frenzel A, Oliynyk G, Segerstrom L, Westermark UK, Larsson K, Munksgaard Persson M, Hultenby K, Lehtio J, Einvik C, Pahlman S, Kogner P, Jakobsson PJ, Henriksson MA (2013) MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci U S A 110(25):10258–10263. doi:10.1073/pnas.1222404110
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Wien
About this chapter
Cite this chapter
Gouw, A.M., Hsieh, A.L., Stine, Z.E., Dang, C.V. (2015). MYC Regulation of Metabolism and Cancer. In: Mazurek, S., Shoshan, M. (eds) Tumor Cell Metabolism. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1824-5_5
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1824-5_5
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1823-8
Online ISBN: 978-3-7091-1824-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)