
Abstract
Neurosurgical procedures, carried out routinely in health institutions, present postoperative complications that result from unavoidable brain injury inflicted by surgical maneuvers. These maneuvers, which include incisions, electrocauterization, and retraction, place brain tissue at the margins of the operative site at risk of injury. Brain edema is a major complication that develops subsequent to this surgically induced brain injury. In the present review, we will discuss type of injury as well as the animal model available to study it. In addition, we will discuss potential mediators, including vascular endothelial growth factor, metalloproteinases, and cyclooxygenases, which have been tested in in vivo experimental studies and have been shown to be potential targets for the development of clinical therapies for neuroprotection against brain edema.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
Introduction
The unique nature of brain tissue poses considerable challenges for neurosurgery. Under the rigid and tough protection of the skull and meninges, the brain is extremely vulnerable to the mechanical insults produced by neurosurgical maneuvers, such as direct incisions, electrocauterization, and retraction. Healthy tissues at the margins of the operative target are inevitably subjected to this surgical brain injury (SBI). Brain edema and hemorrhage are major complications that develop following SBI in neurosurgeries [3]. The blood–brain barrier (BBB), comprising the vascular endothelium, pericyte, and astrocytic processes, prevents the leakage of plasma proteins from the vascular bed into the brain tissue [25]. Vasogenic edema results from the passage of water along with the plasma proteins into the brain tissue because of damage to the capillary endothelium and the interendothelial tight junctions of the BBB, whereas cellular swelling of the injured brain cells results in cytotoxic edema. Both result in increased intracranial pressure, which may lead to further brain injury from cell death or hypoperfusion [3]. Currently, SBI is clinically addressed by nonspecific postoperative care; however, it has become possible to study potential therapies in the preclinical laboratory setting with the recent development of a rodent model for SBI. This review discusses the SBI rodent model and various molecules implicated in the pathogenesis of brain edema as well as treatments that have been applied to reduce brain edema in this model.
Surgical Brain Injury Animal Model
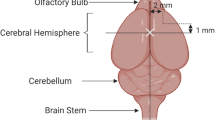
An in vivo rodent model was recently developed to study SBI pathophysiology as well as potential therapeutic targets [14, 16, 23]. This model was designed to mimic the injuries sustained from neurosurgical manipulation of brain tissue. The rodent brain is exposed through a small cranial window through which tissue resection is performed. The margins of the resection are designated in relation to the bregma, as shown in Fig. 1. The model provides consistently measureable brain edema using the brain water content (Fig. 2) in the perisurgical site at 24 h. Studies have shown that brain edema in the perisurgical site peaked 24 h after inducing SBI, was significantly higher up to day 3 post-injury, and had subsided by day 7; SBI was associated with neurobehavior deficits that had dissipated by day 7 [16, 23, 34]. Brain edema was assessed using brain water content measurement, Evans blue dye, and IgG extravasation, and by measuring the apparent diffusion coefficient (ADC) values (Table 1).

Partial right frontal lobectomy. Two incisions (dashed lines) are made leading away from the bregma (white X) along the sagittal and coronal planes, 2 mm lateral to sagittal and 1 mm rostral to coronal respectively

Twenty-four-hour brain water content. The figure shows marked edema in the right frontal region (bordering the surgical injury) of the brain compared with the sham control group 24 h after the surgical brain injury (SBI). The other brain regions did not differ statistically from each other after the injury. n = 6. p < 0.05 for * versus sham. Data are expressed as mean ± SEM. Statistical significance was verified using Student’s t test
Vascular Endothelial Growth Factor
Vascular endothelial growth factors (VEGFs) are a large family of proteins designated VEGF-A through VEGF-E and are expressed in the choroid plexus and neurons in the normal brain [24]. A well-known growth factor with angiogenic, mitogenic, and permeability-inducing effects, VEGF has also been shown to contribute to SBI-induced brain edema. VEGF-A has a potent hyperpermeability-inducing effect on the microvascular endothelium that is mediated through its receptor VEGF receptor-2 (VEGF-2, kdr) [24]. VEGF-2, a transmembrane tyrosine kinase present in the endothelium of the brain vessels, activates MAPK signaling [19]. An upregulation in VEGF has also been reported in models of TBI predominantly due to the infiltrating neutrophils [5]. VEGF administration and overexpression with viral vectors have both been associated with compromised integrity of the BBB in the rodent brain [29]. A potential mechanism by which VEGF-A might increase BBB permeability involves downregulating the expression of occludin, a tight junction protein; this would disrupt the organization of occludin and ZO-1, another junctional protein, leading to tight junction disassembly [25, 28]. Although VEGF may have potentially reparative actions during later phases after an injury, early inhibition of VEGF or its upstream mediator Src tyrosine kinase have been shown to reduce brain edema in various stroke models [16, 19, 24].
The expression of VEGF was increased at 24 h after SBI in the ipsilateral frontal region surrounding the surgical resection site [16]. The Src family is implicated in VEGF-dependent hyperpermeability [25] and has been shown to be involved in SBI-induced BBB disruption and subsequent brain edema in rodents [16]. An increase in expression of VEGF and p-ERK 1/2 as well as a corresponding decrease in the tight junction protein ZO-1 were reversed when rats were pretreated with PP1, an Src tyrosine kinase inhibitor, prior to inducing SBI [16]. In addition, VEGF was shown to play a possible role in the erythropoietin-induced increase in brain edema during the early phase in a SBI rodent model. Erythropoietin administration led to significantly increased brain water content in the perisurgical region at 24 h and was associated with increased expression of VEGF [23]. Thus, VEGF-A and its upstream Src tyrosine kinase present potential therapeutic targets for preserving the BBB and reducing brain edema following neurosurgical procedures.
VEGF-B, another member of the VEGF family expressed in the CNS, mediates its effects through the VEGF receptor-1 (VEGF-1, flt-1); VEGF-B is likely responsible for maintaining the BBB in a steady state [25]. Furthermore, when bound to the soluble extracellular portion of VEGFR-1, VEGF is inactive, sequestered, and unable to bind to VEGF receptors [24]. The role of VEGF-B and its receptors in brain edema development following SBI remains to be elucidated.
Matrix Metalloproteinases
Matrix metalloproteinases (MMPs), zinc-dependent endopeptidases involved in tissue remodeling and repair, have been implicated in the SBI-induced destruction of the extracellular matrix proteins of the neurovascular unit. The target substrates of MMPs include collagen IV, fibronectin, and laminin, all of which are critical to maintaining the integrity of the BBB [28]. MMP-2 (gelatinase A) and MMP-9 (gelatinase B) degrade the basal lamina and tight junction proteins of the BBB and promote BBB injury that leads to vasogenic edema during the acute stage in experimental models of brain injury and stroke [9, 28]. Upregulation of MMPs has been demonstrated following subarachnoid hemorrhage, cerebral ischemia, traumatic brain injury (TBI), and intracerebral hemorrhage, and has been shown to contribute to BBB disruption during the early stage of injury [8, 30, 31, 33]. Additionally, MMP-9 knockout mice had improved functional outcomes, lowered BBB permeability, and reduced lesion volume after transient focal cerebral ischemia and TBI [1, 32].
Similarly, the role of MMPs in mediating BBB disruption and brain edema after SBI was demonstrated by Yamaguchi and colleagues [34]. An upregulation of MMP-2 and MMP-9 was temporally associated with BBB disruption after SBI in rats. A significant increase in MMP activity, particularly that of MMP-9, compared with presurgery levels was detected at days 1–14 after SBI, with highest MMP activities observed at days 1 and 3 coinciding with peak values in brain edema; MMP inhibitor, reversed these effects, preserving the BBB integrity and reducing SBI-induced brain edema as early as 3 h post-injury [34]. Evidence of the role of MMPs in the development of brain edema in the early stages after stroke and in SBI models supports the potential use of MMP inhibition to prevent brain edema following neurosurgical procedures.
Cyclooxygenase-2
Various inflammatory mediators have been implicated in BBB disruption and brain edema after stroke and brain injury [25, 27]. The role of cyclooxygenases, enzymes that catalyze the conversion of arachidonic acid to prostaglandins and thromboxanes [6], in mediating brain edema development has been extensively studied. The upregulation of cyclooxygenase-2 (COX-2), expressed in various cell types including neurons, astrocytes, endothelial cells, macrophages, and microglia in the CNS, has been demonstrated following focal and global cerebral ischemia, neonatal hypoxia–ischemia, and intracerebral hemorrhage [4, 6, 10, 11]. Selective inhibition of COX-2 provided protection after ICH by reducing prostaglandin E2 production, thereby decreasing inflammation, brain edema, and cell death, which translated into improved functional recovery [6]. Further, the role of COX-2 in mediating preconditioning-induced protection has been suggested. COX-2 has been shown to mediate ischemic preconditioning in vitro [18]. Studies from our laboratory have determined COX-2 to be a potential mediator of hyperbaric oxygen preconditioning (HBO-PC) in SBI and global ischemia rodent models; animals preconditioned for 1 h daily for 5 days with HBO prior to inducing injury had significantly improved neurological function and brain edema [4, 17].
Cyclooxygenase-2 has been shown to play a part in SBI pathophysiology as well. An increase in COX-2 expression was detected 24 h after SBI in mice, and HBO-PC significantly attenuated the increase in COX-2 possibly through suppression of HIF-1α, the upstream regulator of COX-2 [17, 26]. The study showed that HBO-PC increased COX-2 level twofold in comparison to the four-fold increase by the SBI, which suggested that protection conferred by HBO-PC might have involved brain preconditioning by increasing COX-2 to subinjurious levels. Furthermore, the protective effects of HBO-PC were reversed in the presence of selective COX-2 inhibitor. These studies demonstrate HBO-PC or COX-2 inhibition to be promising therapies in attenuating brain edema following neurosurgical procedures.
Conclusion
The SBI rodent model mimics injuries caused by neurosurgical procedures and produces postoperative brain edema. It allows for the study of the cellular signaling pathways and the identification of key molecular targets for neuroprotective pretreatment before neurosurgical intervention. To date, VEGF, MMPs, and COXs have been revealed to be potential targets for therapy. As clinically applicable therapeutic intervention for SBI is likely to result in significant benefits for patients and healthcare organizations, further preclinical and clinical studies are necessary to explore the applicability of these targets.
References
Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, Fini ME, Lo EH (2001) Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci 21:7724–7732
Bravo TP, Matchett GA, Jadhav V, Martin RD, Jourdain A, Colohan A, Zhang JH, Tang J (2008) Role of histamine in brain protection in surgical brain injury in mice. Brain Res 1205:100–107
Bruder N, Ravussin P (1999) Recovery from anesthesia and postoperative extubation of neurosurgical patients: a review. J Neurosurg Anesthesiol 11:282–293
Cheng O, Ostrowski RP, Wu B, Liu W, Chen C, Zhang JH (2011) Cyclooxygenase-2 mediates hyperbaric oxygen preconditioning in the rat model of transient global cerebral ischemia. Stroke 42:484–490
Chodobski A, Chung I, Kozniewska E, Ivanenko T, Chang W, Harrington JF, Duncan JA, Szymdynger-Chodobska J (2003) Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience 122:853–867
Chu K, Jeong SW, Jung KH, Han SY, Lee ST, Kim M, Roh JK (2004) Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. J Cereb Blood Flow Metab 24:926–933
Di F, Yan-Ting G, Hui L, Tao T, Zai-Hua X, Xue-Ying S, Hong-Li X, Yun-Jie W (2008) Role of aminoguanidine in brain protection in surgical brain injury in rat. Neurosci Lett 448:204–207
Ding JY, Kreipke CW, Schafer P, Schafer S, Speirs SL, Rafols JA (2009) Synapse loss regulated by matrix metalloproteinases in traumatic brain injury is associated with hypoxia inducible factor-1 alpha expression. Brain Res 1268:125–134
Donkin JJ, Vink R (2010) Mechanisms of cerebral edema in traumatic brain injury: therapeutic developments. Curr Opin Neurol 23:293–299
Fathali N, Ostrowski RP, Lekic T, Jadhav V, Tong W, Tang J, Zhang JH (2010) Cyclooxygenase-2 inhibition provides lasting protection against neonatal hypoxic-ischemic brain injury. Crit Care Med 38:572–578
Graham SH, Hickey RW (2003) Cyclooxygenases in central nervous system diseases: a special role for cyclooxygenase 2 in neuronal cell death. Arch Neurol 60:628–630
Hao W, Wu X, Xu R (2009) The molecular mechanism of aminoguanidine-mediated reduction on the brain edema after surgical brain injury in rats. Brain Res 1282:156–161
Hyong A, Jadhav V, Lee S, Tong W, Rowe J, Zhang JH, Tang J (2008) Rosiglitazone, a PPAR gamma agonist, attenuates inflammation after surgical brain injury in rodents. Brain Res 1215:218–224
Jadhav V, Zhang JH (2007) Surgical brain injury: prevention is better than cure. Front Biosci 13:3793–3797
Jadhav V, Matchett G, Hsu FPK, Zhang JH (2007) Inhibition of Src tyrosine kinase and effect on outcomes in a new in vivo model of surgically induced brain injury. J Neurosurg 106:680–686
Jadhav V, Solaroglu I, Obenaus A, Zhang JH (2007) Neuroprotection against surgically induced brain injury. Surg Neurol 67:15–20
Jadhav V, Ostrowski RP, Tong W, Matus B, Jesunathadas R, Zhang JH (2009) Cyclo-oxygenase-2 mediates hyperbaric oxygen preconditioning-induced neuroprotection in the mouse model of surgical brain injury. Stroke 40:3139–3142
Kim E, Raval AP, Defazio RA, Perez-Pinzon MA (2007) Ischemic preconditioning via epsilon protein kinase C activation requires cyclooxygenase-2 activation in vitro. Neuroscience 145:931–941
Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH (2004) Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 24:916–925
Lee S, Jadhav V, Ayer R, Rojas H, Hyong A, Lekic T, Stier G, Martin R, Zhang JH (2008) The antioxidant effects of melatonin in surgical brain injury in rats. Acta Neurochir Suppl 102:367–371
Lee S, Jadhav V, Lekic T, Hyong A, Allard M, Stier G, Martin R, Zhang J (2008) Simvastatin treatment in surgically induced brain injury in rats. Acta Neurochir Suppl 102:401–404
Lo W, Bravo T, Jadhav V, Titova E, Zhang JH, Tang H (2007) NADPH oxidase inhibition improves neurological outcomes in surgically-induced brain injury. Neurosci Lett 414:228–232
Matchett G, Hahn J, Obeanus A, Zhang JH (2006) Surgically induced brain injury in rats: the effect of erythropoietin. J Neurosci Methods 158:234–241
Merrill MJ, Oldfield EH (2005) A reassessment of vascular endothelial growth factor in central nervous system pathology. J Neurosurg 103:853–868
Nag S, Manias JL, Stewart DJ (2009) Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol 118:197–217
Ostrowski RP, Colohan AR, Zhang JH (2006) Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res 4:399–414
Ostrowski RP, Colohan ART, Zhang JH (2005) Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab 25:554–571
Petty MA, Lo EH (2002) Junctional complexes of the blood–brain barrier: permeability changes in neuroinflammation. Prog Neurobiol 68:311–323
Proescholdt MA, Heiss JD, Walbridge S, Muhlhauser J, Capogrossi MC, Oldfield EH, Merrill MJ (1999) Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J Neuropathol Exp Neurol 58:613–627
Rosenberg GA, Yang Y (2007) Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg Focus 22(5):E4
Sehba FA, Mostafa G, Knopman J, Friedrich V Jr, Bederson JB (2004) Acute alterations in microvascular basal lamina after subarachnoid hemorrhage. J Neurosurg 101:633–640
Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH (2000) Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci 20:7037–7042
Wu H, Zhang Z, Zhao R, Li H, Song Y, Qi J, Wang J (2010) Time course of upregulation of inflammatory mediators in the hemorrhagic brain in rats: correlation with brain edema. Neurochem Int 57:248–253
Yamaguchi M, Jadhav V, Obeanus A, Colohan A, Zhang JH (2007) Matrix metalloproteinase inhibition attenuates brain edema in an in vivo model of surgically-induced brain injury. Neurosurgery 61:1067–1076
Conflict of Interest
We declare that we have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this paper
Cite this paper
Sherchan, P., Kim, C.H., Zhang, J.H. (2013). Surgical Brain Injury and Edema Prevention. In: Katayama, Y., Maeda, T., Kuroiwa, T. (eds) Brain Edema XV. Acta Neurochirurgica Supplement, vol 118. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1434-6_23
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1434-6_23
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1433-9
Online ISBN: 978-3-7091-1434-6
eBook Packages: MedicineMedicine (R0)