
Abstract
Proteolytic enzymes constitute 2–3 % of all known human genes and represent an important tool for the control of the biological functions of proteins. During gliomagenesis, the complex regulation of proteases in transformed and stromal cells is impaired as a result of several factors, and proteases critically contribute to the hallmarks of gliomas. Proteins regulated by proteases include components of the extracellular matrix, local mediators, cell surface receptors, ion channels and adhesion molecules, cytoskeletal proteins, components of the intracellular signaling cascades, and regulators of the cell cycle. Via the proteolytic modifications of these substrates and/or by non-proteolytic mechanisms, the extracellular as well as intracellular proteases contribute to the increased invasiveness of glioma cells, promote the self-renewal and proliferation of glioma stem-like cells, and facilitate tumor neovascularization. The role of proteases in glioma progression is therefore multifaceted and complex. Glioma-associated proteases represent attractive therapeutic targets and several approaches were proposed including the inhibition of the extracellular proteases involved in glioma invasiveness as well as the inhibition of the proteasome and γ-secretase. However, a more precise understanding of the pathogenetic role of proteases in individual glioma patients at different stages of the disease is necessary as indicated by the relatively low therapeutic efficacy of the protease inhibitors in the initial clinical trials. Identification of the key protease-dependent processes in individual glioma patients, the most effective modes of protease targeting, and optimal delivery schedules and routes seem to be crucial to improve the therapeutic outcomes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Proteolytic Enzymes: General Overview
With a total of 585 proteases listed in the degradome database (http://degradome.uniovi.es/dindex.html), proteases constitute approximately 2–3 % of all known human genes (Puente et al. 2003). Biochemically, proteases belong to hydrolases that cleave the covalent bonds linking amino acids in the polypeptide backbone releasing smaller protein fragments and/or individual amino acids (Hooper 2002), or removing posttranslationally attached proteins such as ubiquitin or SUMO (Clague et al. 2012; Hickey et al. 2012). Based on the nature of their active site and the mechanism of action, five main classes of proteases are distinguished, i.e., aspartic, cysteine, metallo, serine, and threonine, with additional classes found in lower organisms (http://merops.sanger.ac.uk/, Rawlings et al. 2012). The specificity of the peptide bond cleavage varies widely: some proteases are highly specific (e.g., blood coagulation proteases, caspases) whereas others (e.g., proteasome, cathepsins, proteinase K) cleave a wide variety of substrates at several positions.
Besides its role in protein digestion and catabolism, the proteolytic cleavage represents a very important and mostly irreversible mechanism of protein function regulation on the posttranslational level (Clague et al. 2012; Hickey et al. 2012; Turk et al. 2012a). Proteolysis can lead to
-
1.
Protein activation [e.g., zymogen activation, protein maturation by proprotein convertases such as furin (Seidah and Chretien 1999; Maret et al. 2012)],
-
2.
Protein inactivation [e.g., cleavage of a specific inhibitor of caspase-activated DNAse by caspase-3 during apoptosis (Enari et al. 1998)],
-
3.
Adjustment of the biological activity of the protein [e.g., chemokine processing by matrix metalloproteinases or aminopeptidases (Wolf et al. 2008)],
-
4.
Changes affecting the turnover of the protein (e.g., changed susceptibility to degradation after the removal of ubiquitin by deubiquitinating enzymes (Wilkinson 2009), or protein cleavage that enables further proteolytic processing).
Proteolysis thus determines the spatiotemporal bioavailability of proteins either quantitatively (e.g., degradation of structural or nutritive proteins) or qualitatively by highly specific (“limited”) proteolysis of particular peptide bonds, fine-tuning the biological activities of regulatory peptides (Konkoy and Davis 1996).
The activity of proteases can be controlled at several levels. In addition to the regulation of the expression level via transcriptional and posttranscriptional mechanisms and protein degradation, several proteases are synthesized in an inactive form (zymogen) that requires proteolytic cleavage in order to be converted to the active form. This activation is frequently accomplished by proteolysis in a multiprotein complex and represents a well-established regulatory step for, e.g., matrix metalloproteinases (MMPs) (Kessenbrock et al. 2010), plasminogen, urokinase-type plasminogen activator (uPA), and caspases (Donepudi and Grutter 2002). The activity of proteases is further regulated by their sequestration to specialized cellular compartments such as nucleus (Clague et al. 2012; Geng et al. 2012), endoplasmic reticulum, Golgi apparatus, endosomes, lysosomes, or secretory granules (Shen and Prywes 2004; Colbert et al. 2009; Lemberg 2011; Krzewski and Coligan 2012; Turk et al. 2012b; Bergbold and Lemberg 2013; Hattori and Tsujimoto 2013; Repnik et al. 2013; Seidah et al. 2013), mitochondria (Bulteau and Bayot 2011; Anand et al. 2013), and invadosomes (Brisson et al. 2012), where they meet with a stabilizing and optimal reaction microenvironment as well as with the target substrates. Besides this, proteases are controlled by a number of endogenous extracellular (secretory) and intracellular inhibitors. While α2-macroglobulin and α1-antitrypsin have the ability to inhibit a broad range of proteases, other endogenous inhibitors are more selective for individual proteases or protease classes, although there is some overlap in their specificities (Turk 2006; Mason and Joyce 2011). The serpins such as plasminogen activator inhibitors (PAI) 1 and 2 or α2-antiplasmin (Law et al. 2006) or the Kunitz-type inhibitors such as tissue factor pathway inhibitor (TFPI) predominantly inhibit serine proteases, whereas cystatins (Turk et al. 2008) and calpastatin inhibit the cysteine proteases cathepsins and calpains, respectively (Turk 2006; Turk et al. 2008; Cox 2009; Mason and Joyce 2011; Campbell and Davies 2012). Some intracellular serpins, such as serpinB3, serpinB4, and serpinB9, can function as cross-class inhibitors inhibiting serine as well as cysteine proteases (Law et al. 2006; Izuhara et al. 2008). The activity of MMPs, ADAMs (“A Disintegrin And Metalloproteinase”), and ADAMTS (“A Disintegrin And Metalloproteinase with Thrombospondin Motifs”) is regulated by the four members of the tissue inhibitor of matrix metalloproteinase (TIMP) family TIMP1-4, which form tight 1:1 complexes with the target proteases (Murphy 2011). TIMP1–4 differ somewhat in their ability to inhibit individual metalloproteases and in their expression pattern. TIMP1 has a restricted inhibitory potential as it does not inhibit the membrane-type MMPs (MT-MMPs); it is widely expressed extracranially, but its expression in the brain is confined to the regions with persistent neuronal plasticity such as the hippocampus, the olfactory bulb, and the cerebellum (Rivera et al. 2010; Murphy 2011). TIMP2 is constitutively expressed in many tissues and is the most abundantly expressed TIMP in the brain at least in the rat (Fager and Jaworski 2000). TIMP2 plays a dual role in regulating metalloproteases; besides being a protease inhibitor, TIMP2 is critically involved in the complex process of MMP2 activation. TIMP2 forms a trimolecular complex with MMP2 and MT1-MMP (MMP14) thereby enabling efficient activation of MMP2 by MMP14 on the cell surface (Strongin et al. 1995). These results of in vitro studies are further supported by the observation that TIMP2 knockout mice have impaired MMP2 activation, which can be rescued by exogenous TIMP2 (Caterina et al. 2000; Wang et al. 2000). TIMP3 is expressed in several tissues and its ablation in mice leads to emphysema-like damage of the lungs and increased apoptosis in the mammary gland suggesting its important role in regulating metalloproteases (Murphy 2011). Expression of the recently described TIMP4 is restricted to heart, kidney, pancreas, colon, testes, adipose tissue, and brain (Melendez-Zajgla et al. 2008). The data on TIMP4 function are limited, but the protein is suspected to have pro-tumorigenic activities (Melendez-Zajgla et al. 2008). It is important to stress that the majority of the endogenous protease inhibitors exhibits several physiological and pathological roles that are independent of their ability to inhibit the activity of proteases (Rivera et al. 2010), which must be considered when interpreting their effects on protease-mediated processes in the experimental models.
In addition to the protease genes, several proteins highly homologous to proteases but devoid of the proteolytic activity due to amino-acid substitution(s) in the active site are encoded in the human genome (Puente et al. 2003). For example, human ADAMs from the metzincin subgroup of the zinc protease superfamily comprise 19 members, of which approximately half are enzymatically inactive (Seals and Courtneidge 2003; Klein and Bischoff 2011). The inactive protease homologues may have important regulatory functions in sequestering inhibitors or protease substrates, or participate on protein–protein interactions that are unrelated to their “evolutionary original” proteolytic role. This latter aspect is also typical for several “bona fide” proteases, as they frequently contain non-protease domains that enable non-hydrolytic protein–protein interactions (Del Rosso et al. 2002; Rozanov et al. 2004; Mina-Osorio 2008; Sakamoto and Seiki 2009; Dufour et al. 2010; Redondo-Munoz et al. 2010). Thus, a number of proteases can serve as receptors (Mina-Osorio 2008; Klein and Bischoff 2011; Raj et al. 2013), signaling molecules (Rogove et al. 1999; Sower et al. 1999; Beffert et al. 2006; LaRusch et al. 2010; Strongin 2010), or modulator molecules directly interacting with and influencing other components of the signal transduction pathways (Sumitomo et al. 2000).
Proteases act intracellularly, in the extracellular space as well as in body fluids and secretions. The intracellular proteases are indispensable for proper protein maturation [e.g., proprotein convertases (Seidah and Chretien 1999)], contribute to the cytoskeletal remodeling [e.g., calpains (Franco and Huttenlocher 2005)] and regulation of transcription (Best et al. 2002; Chapman 2004), remove misfolded, damaged, or unneeded proteins (lysosomal proteases, proteasomes) (Das et al. 2012; Kaminskyy and Zhivotovsky 2012; Viry et al. 2014), and initiate and execute apoptosis (Pop and Salvesen 2009; Goldschneider and Mehlen 2010). Proteolysis also takes place within biological membranes (Lemberg 2011; Bergbold and Lemberg 2013). This unique mechanism mediated by, e.g., presenilins or rhomboids frequently leads to the release of effector peptides, often with a signaling or transcription factor activity, from transmembrane proteins such as Notch (Lemberg 2011). Proteases acting in the extracellular space are either secreted (e.g., uPA, MMPs 2 and 9, cathepsins, ADAMTS) or plasma membrane bound (e.g., MT1-MMP, ADAMs). Nevertheless, the secreted proteases can also associate with the cell surface by binding to specific membrane receptors as demonstrated for uPA, MMP2, MMP9, and procathepsin-B and their respective receptors uPAR, α-v-β-3 integrin, CD44, and annexin II (Brooks et al. 1996; Yu and Stamenkovic 1999; Mohamed and Sloane 2006; Eden et al. 2011). This binding enhances the proteolytic activity not only by the spatial concentration of the protease but frequently also decreases the effects of endogenous inhibitors.
The widespread distribution of proteases reflects their participation in almost every physiological process on the level of individual cells as well as on the level of the whole organism. Proteases exert control of cell behavior including its metabolism, signal sensing and transduction, proliferation and death, as well as participate among others in angiogenesis, blood clotting, and immune defense (see Lopez-Otin and Bond (2008) for review).
Proteases from various classes are expressed in the brain and fulfil numerous biological functions. The serine proteases neurotrypsin and neuropsin were originally identified due to their abundant expression in the neural tissue, but other more “traditional” serine proteases such as thrombin, plasmin, tPA, and trypsin 4 are also locally produced in the brain (see Wang et al. (2008b), Almonte and Sweatt (2011) and references therein). Similarly, metalloproteases such as MMP2, MMP9, several ADAMs, and ADAMTS4 and 5 are expressed in the central nervous system (for review see Yang et al. 2006; Rivera et al. 2010). Extracellular as well as intracellular brain proteases play an important role in neuronal signaling. Besides the processing, conversion, and inactivation of neuropeptides and growth factors such as pro-BDNF (brain-derived neurotrophic factor) (Hallberg et al. 2005; Almonte and Sweatt 2011), proteases trigger intracellular signaling cascades through “protease-activated receptors” (PAR) 1–4. These receptors are expressed on the surface of neurons, microglia, and astrocytes and their proteolytic cleavage exposes an amino-terminal part of the molecule that acts as a tethered intramolecular ligand for the same receptor and activates signal transduction through G proteins (see Noorbakhsh et al. (2003) for review). Ligand-gated ion channels may also be cleaved by proteases with subsequent changes in their turnover or signaling. For example, the intracellular calcium-activated cysteine proteases calpains cleave the NR2A subunit of the NMDA receptors (Guttmann et al. 2002) as well as degrade GRIP (glutamate receptor-interacting protein), a molecule important for the morphological and functional organization of the synapses (Lu et al. 2001). Likewise, the extracellular serine protease tPA is thought to be directly involved in glutaminergic transmission as it is induced by neuronal activity (Qian et al. 1993) and facilitates NMDA receptor-mediated signaling by cleaving the NR1 subunit (Nicole et al. 2001). Neuronal neurotransmitter receptors are also regulated by a spatiotemporally limited activation of caspases. In hippocampal neurons, NMDA receptor signaling causes caspase-3 activation by the mitochondrial pathway. The activated caspase-3 subsequently cleaves and inactivates the serine-threonine protein kinase Akt1, which blocks AMPA receptor internalization and inhibits long term depression (LTD) (Li et al. 2010b; Li and Sheng 2012). Similarly, caspase-1 was demonstrated to function as a negative regulator of the AMPA receptor signaling, resulting in the inhibition of long term potentiation (LTP) (Lu et al. 2006a). Other proteases with signaling roles in the brain include secretases that are indispensable for the Notch-1 signaling by liberating its C-terminal intracellular domain (Prox et al. 2012) and the ubiquitin-proteasome system, which impacts, e.g., on the Notch, Wnt, BMP, or Shh signal transduction pathways by regulating their intracellular effectors (Lehman 2009). The neuropsin-mediated cleavage of fibronectin (Almonte and Sweatt 2011) and of the L1 cell adhesion molecule (L1CAM, Shimizu et al. 1998) as well as the processing of beta-dystroglycan and N-cadherin by MMP9 (Dziembowska and Wlodarczyk 2012) are other examples of the remodeling of the extracellular matrix and the cleavage of cell adhesion molecules by which several proteases play an important role in LTP, synapse remodeling, and neuronal plasticity [for further details see, e.g., Tomimatsu et al. (2002), Reif et al. (2007), Stephan et al. (2008), Wright and Harding (2009), Shiosaka and Ishikawa (2011), Dziembowska and Wlodarczyk (2012), Li and Sheng (2012)].
In addition, proteases are essential factors during brain development. MMPs such as MMP3 and MMP9 contribute to neurite outgrowth, axon guidance, and neuronal migration (Vaillant et al. 1999, 2003; Van Hove et al. 2012). A similar role was proposed for the plasminogen activator-plasmin system (Seeds et al. 1997) and caspases (Williams et al. 2006; Westphal et al. 2010). ADAMs, most notably ADAM 10, 22, and 23, seem to have a complex role during central nervous system (CNS) development affecting the proliferation, neuronal migration, axon growth, and differentiation of neuronal progenitors, and their absence in mice results in serious CNS defects and early mortality (reviewed in Yang et al. 2006). Likewise, cathepsins were demonstrated to be indispensable for the proper development of the CNS. Combined deletion of the cysteine cathepsins B and L leads to neuronal loss with profound brain atrophy as well as early mortality in mice (Felbor et al. 2002). The deficiency of the aspartate protease cathepsin-D in mice was reported to cause disturbed myelin structure (Mutka et al. 2010), accumulation of ceroid, astrogliosis, and early mortality (Shevtsova et al. 2010) paralleling the phenotype of the congenital neuronal ceroid lipofuscinosis caused by cathepsin-D deficiency in humans (Siintola et al. 2006; Fritchie et al. 2009). The phenotype in cathepsin-K knockout mice is less severe and involves changes in hippocampal cytoarchitecture as well as learning and memory deficits (Dauth et al. 2011).
Imbalances in the proteolytic activities lead to several pathologies (Lopez-Otin and Bond 2008) including inflammatory, neurodegenerative, and malignant diseases affecting the CNS. Proteases were thus shown to participate on the pathogenesis of Alzheimer’s (Bernstein 2005) and Parkinson’s disease (Crocker et al. 2005) as well as the pathogenesis of ischemic (Morancho et al. 2010), traumatic (Knoblach and Faden 2005), inflammatory, and infectious (Kieseier and Bernal 2005) states.
The concept of the role of proteolytic enzymes in malignant diseases dates back to the half of the last century (Fischer 1946). At that time, proteases were viewed as effectors facilitating tumor cell dissemination by cleaving the protein components of the extracellular matrix. This is certainly the case for some of the proteases localized extracellularly or concentrated in the specialized regions of plasma membrane where their activity, together with the adhesion molecules, mediates interactions of the cancer cells with their surroundings (Stylli et al. 2008). However, experimental work in the ensuing years led to a more complex picture emphasizing the importance of proteases for several aspects of malignancy (reviewed in Nomura and Katunuma 2005; van Hinsbergh et al. 2006; Lopez-Otin and Matrisian 2007; Kessenbrock et al. 2010). For example, the membrane-bound MT1-MMP (MMP14) can mediate intracellular proteolysis of a centrosomal protein pericentrin-2, which leads to chromosomal instability and aneuploidy (Golubkov and Strongin 2007). Similar role at the early stages of tumor development was suggested for other proteases as well. Caspases, the initiators and executors of apoptotic cell death, are generally viewed as “guardians” protecting the organism from cancer. However, the activation of the DNA fragmentation factor by caspase-3 may lead to increased mutation frequency, genome instability, and chromosomal translocations when the apoptotic program is not completed (Aplan 2006; Lovric and Hawkins 2010). In addition, the caspase-3-mediated activation of prostaglandin E2 production and secretion by the tumor cells exposed to cytotoxic therapies can in fact enhance tumor repopulation by the surviving tumor cells (Huang et al. 2011) and thus eventually contribute to the cancer recurrence and progression. This together with other reports supports the notion that proteases in addition to tissue invasion and metastasis participate in cancer initiation, generation of genomic instability in tumor cells (Radisky et al. 2005), tumor angiogenesis, immune escape, dysregulation of apoptosis (Abraham et al. 2005), and cell proliferation, as well as on the infiltration of the tumors by immune cells (reviewed in McCawley and Matrisian 2001; Kessenbrock et al. 2010).
On the other hand, several proteases have rather anti-tumorigenic effects, e.g., by inactivating bioactive peptides, generating anti-angiogenic fragments from the extracellular matrix, inhibiting cell growth or modulating the inflammatory response (Lopez-Otin and Matrisian 2007). In other cases the net pro- or anti-oncogenic activity of a protease may critically depend on its cellular source as well as on other microenvironmental factors. For example, MMP9 may promote tumor growth and invasiveness as well as enhance angiogenesis by liberating VEGF (vascular endothelial growth factor) (Bergers et al. 2000), whereas the MMP9 mediated degradation of other extracellular matrix proteins such as type IV collagen or plasminogen produces potent suppressors of angiogenesis tumstatin and angiostatin, respectively (Hamano et al. 2003; Rege et al. 2005).
The role of proteases in cancer progression is therefore multifaceted and only partially elucidated. In this chapter we summarize the current knowledge of the expression of proteases in gliomas and discuss their regulation and potential mechanisms whereby they contribute to several aspects of glioma pathogenesis. Finally, the possible exploitation of proteases as therapeutic targets is also reviewed.
2 Dysregulation of Proteolytic Enzymes in Gliomas
Several studies (summarized in Table 12.1) addressed the expression and possible function of proteolytic enzymes in gliomas. Of these, the extracellularly localized proteases participating on glioma invasiveness and angiogenesis were most extensively studied and were the subject of a number of excellent reviews (Chintala et al. 1999; Fillmore et al. 2001; VanMeter et al. 2001; Binder and Berger 2002; Levicar et al. 2003b; Rao 2003; Lakka et al. 2005; Mentlein et al. 2012).
A very comprehensive view of the differential protease expression in gliomas can be obtained from the data in The Cancer Genome Atlas Research Network (2008). These data clearly show a consistent signature of up- or downregulated proteases and their homologues (Fig. 12.1a) corroborating the over- (e.g., MMP14, MMP2, MMP7, MMP9, tPA, uPA) or under-expression (e.g., kallikreins, ADAMTS8) observed in previous smaller studies. Importantly, it reveals the dysregulation of expression of numerous proteases that have been little or not at all explored in relation to glioma pathogenesis so far, making them candidates for future studies (e.g., CFI, CPVL, DPEP1, ENPEP, LTF, MEST, MAP1D, ubiquitin-specific proteases).

Dysregulation of protease expression in glioblastomas. (a) Hierarchical clustering of 528 protease gene expression profiles (rows) in 173 glioblastoma patients comprised in the Verhaak dataset (Verhaak et al. 2010, TCGA, Agilent platform). Data were normalized to the expression levels of healthy controls (median values of 10 control samples). The glioblastoma molecular subtype, as defined by Verhaak et al. (2010), is indicated at the top of the heatmap. Proteases displaying significant subtype-specific upregulation are indicated on the right side of the heatmap. (b) A subset of proteases from panel (a) that exert ≥2-fold upregulation of their median mRNA levels in a particular glioblastoma subtype compared to healthy controls and expression significantly higher (p ≤ 0.01) compared to the rest of the patients. Glioblastoma subtypes are indicated as in panel (a)
Several of the consistently upregulated proteases are implicated in the immune response (ADAMDEC1, C1RL, C1S, CFI, CPVL, LTF, PSMB8, PSMB9, SPPL2A) probably reflecting the proinflammatory state induced by gliomas and high infiltration by microglia/macrophages (Gabrusiewicz et al. 2011) and other immune cells. Similarly, proteases involved in the remodeling of the extracellular matrix and angiogenesis (ADAMTS5, MMP14, MMP2, MMP7, MMP9, tPA, uPA) are highly upregulated. The consistently downregulated proteases on the other hand comprise proteases or their homologues typical for the normally functioning neuronal tissue, i.e., involved in neurotransmission, ion channel regulation, and axonal growth (e.g., AMZ1, ASPA, DPP10, PCSK2, TRHDE, AGTPBP1, CPE, KLK8, PCSK1, ACY3, ASRGL1, DPP6, DPYSL2, DPYSL4, FOLH1, NELF, THOP1). In addition, several proteases with a proposed tumor suppressor function (e.g., ADAM11, PRSS3, ADAM23, ADAMTS8, CRMP1, KLK10) are downregulated.
Interestingly, a subset of proteases seems to be preferentially dysregulated in association with the individual glioblastoma molecular subtypes that were recently proposed by Verhaak et al. (2010) (Fig. 12.1b). Indeed, several proteases and protease inhibitors (e.g., ADAM12, caspases, cathepsins, uPA, cystatin A, serpins) are part of the Verhaak mesenchymal transcriptomic signature (Verhaak et al. 2010). In addition, a number of proteases are in fact upregulated solely or most markedly in this subtype (Fig. 12.1b), which might correspond to the higher overall necrosis with ensuing inflammatory infiltrate and expression of angiogenic genes in the mesenchymal subtype (Verhaak et al. 2010).
Albeit the transcriptomic TCGA data give very rich and complex information on the dysregulation of proteolytic systems based on over 500 glioblastoma cases, it is important to realize their limitations. As detailed above, the transcriptional regulation of protease expression represents only one possible level of control mechanisms, as proteolytic systems are often regulated on the posttranscriptional and posttranslational levels. In addition, although the TCGA data corroborate the heterogeneity of glioblastomas also with respect to the expression of proteases, they do not allow distinguishing the contribution of transformed and stromal cells and may therefore point to a changed cellular composition in different glioma subtypes rather than differential expression in the transformed cells. Irrespective of these limitations, they are a valuable source for selecting proteases for studies of glioma pathogenesis and identification of future therapeutic targets.
3 Signals and Mechanisms Affecting Expression and Activity of Proteases in Gliomas
The mechanisms leading to protease dysregulation in gliomas are complex and still only partially understood. However, several aberrant signaling pathways and mechanisms characteristic for gliomagenesis drive the tumor progression in part by affecting the proteolytic machinery (Fig. 12.2).

Examples of gliomagenesis relevant factors and events affecting the local proteolytic balance, and the biological consequences of dysregulated protease expression. PTEN phosphatase and tensin homolog, IDH isocitrate dehydrogenase, RTK receptor tyrosine kinase
3.1 Mechanisms Leading to Dysregulation of Protease Expression
The receptor tyrosine kinase (RTK)/Ras/PI3K signaling pathway is dysregulated in over 80 % of gliomas (Van Meir et al. 2010) and promotes cell proliferation, survival, and invasion. The most frequent alteration is EGFR gene amplification, overexpression, or the presence of a constitutively active mutant EGFRvIII variant; these are present in approximately half of the patients and are a characteristic phenotypic feature of the primary glioblastomas (Ohgaki and Kleihues 2007; Van Meir et al. 2010). It is worth mentioning that the active MMP9 was detected in 69 % of primary and just 14 % of secondary glioblastomas and its expression was strongly associated with the expression of EGFRvIII (Choe et al. 2002). Indeed, the EGFR signaling was demonstrated to increase the expression of several proteases in glioma cells in vitro including MMP9 (Kang et al. 2005; Zhao et al. 2010), MMP1 (Anand et al. 2011; Li et al. 2011), MMP2 (Lal et al. 2002; Park et al. 2006), MMP14 (Van Meter et al. 2004), and uPA (Amos et al. 2010). Besides that, EGFR signaling influences the subunit composition of the proteasome which was speculated to be associated with the radioresistance of glioma cells (Kim et al. 2008b). Protease expression can also be influenced by other alterations in the abovementioned RTK/Ras/PI3K signaling pathway such as PTEN (phosphatase and tensin homolog) deletion or Ras mutations, which occur in 37 % and 2 % of gliomas, respectively (Van Meir et al. 2010). PTEN inactivation in gliomas correlates with higher MMP9 expression (Comincini et al. 2009) and in a similar way as the activation of the downstream PI3K promotes the production of MMP2 and MMP9 in glioma cells in vitro (Koul et al. 2001; Kubiatowski et al. 2001; Park et al. 2002; Furukawa et al. 2006; Kwiatkowska et al. 2011). Similarly, constitutively active form of Ras was demonstrated to increase uPA expression and promote cell invasiveness in normal human astrocytes transformed by the introduction of human telomerase in combination with inactivation of p53/pRb by E6/E7 (Zhao et al. 2008b). The aberrant EGFR/Ras/PI3K pathway is also associated with the increased expression of tissue factor, a crucial cofactor for the initiation of the proteolytic blood clotting cascade (Rong et al. 2009; Magnus et al. 2010).
Glioblastoma microenvironment typically contains increased concentration of a number of cytokines including growth factors and chemokines, as well as local mediators such as adenosine or NO, which were demonstrated to influence the expression of proteases in glioma cells (Table 12.2).
Protease expression with the subsequent promotion of invasiveness is also affected by the interaction of glioma cells with the components of the extracellular matrix (ECM). Park et al. (2002) showed that binding of the hyaluronic acid to glioma cells activates the focal adhesion kinase (FAK)-ERK 1/2 signaling pathway and leads to increased MMP9 secretion. Similarly, tenascin C increases the invasiveness of glioma cells by stimulating the expression of MMP12 (Sarkar et al. 2006).
The Sp1 transcription factor promotes the expression of MMP2 (Qin et al. 1999; Guan et al. 2012), ADAM17 (Szalad et al. 2009), and cathepsin-B (Yan et al. 2000) in glioma cells and is highly expressed in the majority of gliomas. Similarly, the Ets-1 transcription factor (Oikawa and Yamada 2003) is overexpressed in glioblastomas (Kitange et al. 1999a) and mediates the transcription of MMP9 (Sahin et al. 2005), cathepsin-B (Yan et al. 2000) and uPA (Kitange et al. 1999b; Nakada et al. 1999b). Several MMP genes also contain the AP-1 element and their transcription may therefore be activated by Jun and Fos transcription factors (Westermarck and Kahari 1999) in response to a variety of signals from the extracellular milieu (Chakraborti et al. 2003).
Intratumoral hypoxia, a typical feature of glioblastoma microenvironment affecting multiple signaling pathways, as well as mutations of IDH (isocitrate dehydrogenase) lead to the stabilization and thereby activation of the HIF1-α subunit of the transcription factor HIF (hypoxia inducible factor), which accelerates glioma progression in part through the upregulation of MMP2, MMP9, and ADAM17 (Brat et al. 2004; Fujiwara et al. 2007; Zheng et al. 2007; Fu et al. 2012).
Recently (see also Chap. 4) alterations in the expression of microRNAs (miRNAs) were described in glioblastomas (Moller et al. 2013) and the mechanisms by which miRNAs may influence the phenotype of glioma cells may also involve the regulation of proteases. miRNAs may directly target the protease mRNA, or exert an indirect effect by modulating the pathways regulating the expression of proteases or their inhibitors. In glioblastoma specimens, miR-211 expression is silenced by promotor hypermethylation and its expression level inversely correlates with MMP9 expression (Asuthkar et al. 2012). Similar negative correlation was also observed between MMP9 and miR-491-5p (Yan et al. 2011). Importantly, both miRNAs were demonstrated to downregulate MMP9 in glioma cells in vitro resulting in decreased invasiveness (Yan et al. 2011; Asuthkar et al. 2012). Other examples of the effects of miRNAs on the proteolytic balance include the negative regulation of MMP16 by miR-146b-5p (Xia et al. 2009; Li et al. 2013a), MMP3 by miR-152 (Zheng et al. 2013), and ADAM17 by miR-145 (Lu et al. 2013). The expression of MMP2 and MMP9 in glioma cells in vitro is further influenced by miR-7, which is downregulated in gliomas, and affects the proteases indirectly by downregulating FAK (Wu et al. 2011). In addition to these tumor suppressor miRNAs, protease targets were also demonstrated for the oncogenic miRNAs. miR-10b is overexpressed in gliomas (Moller et al. 2013) and induces glioma cell invasiveness by targeting the homeobox DNA-binding domain containing transcription factor HOXD10, a negative regulator of MMP14 and uPAR (Sun et al. 2011).
Interestingly, the expression of proteases involved in the remodeling of ECM was recently demonstrated to be induced by the therapeutic interventions used to treat gliomas, thereby somewhat paradoxically contributing to the tumor recurrence. Glioma cells exposed to ionizing radiation exhibit enhanced invasiveness caused by several mechanisms including the upregulation of proteases (Wild-Bode et al. 2001; Park et al. 2006; Badiga et al. 2011; Shankar et al. 2014). A rapid increase in the expression and activation of MMP2, MMP9, and MMP14 after sublethal irradiation was demonstrated in glioma cells in vitro and an MMP inhibitor o-phenantroline was able to reduce the increased invasiveness of the irradiated glioma cells (Wild-Bode et al. 2001). Similarly, treatment with a plasmid silencing the expression of MMP2 resulted in the reversal of the proinvasive effects of irradiation and increased the radiosensitivity of glioma cells (Badiga et al. 2011). The radiation-mediated enhancement of protease expression is probably p53 independent (Wild-Bode et al. 2001) and is suppressed in the presence of the active PTEN (Park et al. 2006). In the case of MMP2, radiation increases its transcription as a result of the activation of the Src and EGFR signaling with the ensuing activation of p38, PI3K, and Akt (Park et al. 2006). Bevacizumab, a monoclonal antibody targeting VEGF, was similarly shown to promote the invasiveness of glioma cells (Lucio-Eterovic et al. 2009; de Groot et al. 2010). Glioma cells exposed to bevacizumab upregulated MMP12, MMP9, MMP2, as well as plasminogen in vitro and a similar upregulation was observed in experimental tumors in mice treated with bevacizumab contributing to their more infiltrative growth (Lucio-Eterovic et al. 2009). This increase of MMP2 and MMP14 expression was also detected in glioma patients treated with bevacizumab (de Groot et al. 2010; Furuta et al. 2014) and the increase of MMP9 in the urine was in fact suggested as a marker for bevacizumab failure in glioma patients (Takano et al. 2010).
3.2 Mechanisms Leading to Dysregulation of Protease Activation
Besides the dysregulation of the protease gene expression, the increased activity of extracellular proteases is frequently a result of an inappropriate zymogen activation caused by the protease cofactor overexpression. The cell surface receptors urokinase-type plasminogen activator receptor (uPAR, CD87) (Yamamoto et al. 1994b) and tissue factor (TF, fIII) (Hamada et al. 1996), a crucial cofactor for the initiation of blood coagulation cascade, are both upregulated in gliomas. uPAR is a multifunctional glycosylphosphatidylinositol (GPI)-linked membrane protein that binds several extracellular ligands including uPA (Eden et al. 2011). The binding of uPA to uPAR not only promotes the activation of uPA leading to enhanced pericellular proteolysis but also directly regulates glioma cell adhesion and migration through uPAR (see Mohanam et al. (1999), Eden et al. (2011) and Sect. 12.4.1.3). TF is a transmembrane receptor and cofactor of the coagulation factor VIIa and its overexpression contributes to the procoagulative state in gliomas (Rong et al. 2006). Similarly to uPAR, TF promotes the malignant phenotype of glioma cells by activating intracellular signaling independent of its role in protease activation (Dutzmann et al. 2010; Gessler et al. 2010).
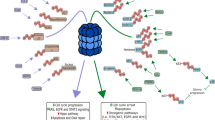
Zymogens may further be inappropriately activated as a result of the dysregulation of the local “proteolytic context” (reviewed in Mason and Joyce 2011). Interestingly, some aspects of the complex proteolytic systems resemble the basic features of the signaling cascades such as signal amplification, cross talk, and a purposeful adaptation. Within the broader, functionally interrelated proteolytic networks, the interactions of individual proteases are frequently reciprocal (Fig. 12.3). For example, uPA can both activate and be activated by plasmin and cathepsin-B (Mason and Joyce 2011) and some proteases facilitate the activity of other proteases by inactivating their inhibitors [e.g., MMPs can inactivate a range of serpins (Kessenbrock et al. 2010) and cathepsin-B cleaves some TIMPs and serpins (Mason and Joyce 2011)]. The glioma proteases cathepsin-B, uPA, and a number of the MMPs seem to occupy critical nodes within the complex proteolytic systems due to the broad range of their respective activators, the ability to activate numerous other proteases and to inactivate several protease inhibitors (Mason and Joyce 2011).

The proteolytic network of extracellular proteases implicated in glioma progression. Proteases from various classes interact in a multidirectional network through the proteolytic activation of zymogens (green lines) and inactivation (red lines) of endogenous protease inhibitors. Cysteine proteases in black, metalloproteases in red, serine proteases in green, aspartate proteases in blue; the protease inhibitors of individual protease classes are shown in the corresponding shades. MMP matrix metalloproteinase, uPA urokinase-type plasminogen activator, uPAR urokinase-type plasminogen activator receptor
Altered availability of the endogenous protease inhibitors may be another mechanism contributing to the dysregulation of proteolytic activity in gliomas, the best example being the imbalance between the cysteine cathepsins and cystatins that is thought to contribute to tumor invasion. In gliomas, cystatin-E/M downregulation was reported in 78 % of cases and the reduced expression correlated with its promoter hypermethylation (Qiu et al. 2008). Cystatin-C is also downregulated in glioblastomas compared to the low grade tumors and may be a predictive factor for patient survival (Nakabayashi et al. 2005). Further, Gole et al. (2012) showed that at the tumor margin the expression of cystatin-B is decreased, while cathepsin-B is, compared to the central part of the tumor, redistributed into the extracellular space. Such pathologic regulation of the availability of the cathepsin activity may lead to increased invasiveness (Gole et al. 2012). These results are supported by the findings that ectopic expression of the cathepsin-B inhibitor cystatin-C reduces in vitro glioma invasion as well as in vivo tumor formation (Konduri et al. 2002). Tissue factor pathway inhibitor-2 (TFPI-2) is a Kunitz-type serine protease inhibitor that inhibits a variety of serine proteases and is a potential tumor suppressor in several malignancies (Sierko et al. 2007). In gliomas, TFPI-2 is probably silenced by promoter hypermethylation (Konduri et al. 2003) and its expression is inversely correlated with tumor grade. Moreover, the protein was demonstrated to inhibit glioma cell invasion in vitro suggesting that its loss may be directly linked to glioma aggressiveness (Rao et al. 2001).
An opposite role is evident for the serpin plasminogen activator inhibitor 1 (PAI-1), the major inhibitor of the fibrinolytic system. Paradoxically, PAI-1 expression is associated with worse prognosis in several tumors (Van De Craen et al. 2012). In gliomas, a grade-dependent upregulation of PAI-1 is consistently reported with the most intense expression in the areas of vascular proliferation and perinecrotic areas (Rao et al. 1993a; Kono et al. 1994; Landau et al. 1994; Arai et al. 1998; Muracciole et al. 2002; Colin et al. 2009); in addition, high PAI-1 expression (Muracciole et al. 2002) and serum levels (Iwadate et al. 2008) are negative prognostic factors in glioma patients. This seeming paradox likely reflects the non-protease-mediated functions of PAI-1 (Van De Craen et al. 2012) that include its effects on cell adhesion and migration (Bryan et al. 2008; Paugh et al. 2008) and promotion of angiogenesis (Hjortland et al. 2004). The data for other endogenous protease inhibitors are equivocal. Changes in the expression of TIMPs were reported in gliomas by several groups. The most consistent data exist for the extracellular matrix-associated TIMP3, which is regarded as a tumor suppressor. TIMP3 promoter is hypermethylated in gliomas (Liu et al. 2010) and the hypermethylation correlates with the loss of TIMP3 expression in secondary glioblastomas; in addition, the region on chromosome 22 containing the TIMP3 gene is frequently deleted in a large proportion of secondary and some primary glioblastomas (Nakamura et al. 2005). Other mechanisms that may lead to TIMP3 downregulation involve its targeting by miR21 (Gabriely et al. 2008) and the frequent loss of the cell cycle regulator P14ARF encoded by the CDKN2A locus. P14ARF acts as a tumor suppressor via p53 stabilization but was recently shown to inhibit human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3 (Zerrouqi et al. 2012). TIMP3 inhibits invasion and promotes apoptosis in several tumor cells (Baker et al. 1999). However in a glioma model, the presence of TIMP3 did not significantly affect the antitumor efficacy of an oncolytic adenovirus (Lamfers et al. 2005). For TIMP1, 2, and 4 some studies indicate decreased expression in glioblastomas compared to normal brain (Nakagawa et al. 1994; Mohanam et al. 1995; Aaberg-Jessen et al. 2009) and inhibitory effects on glioma cells (Merzak et al. 1994; Nakano et al. 1995; Matsuzawa et al. 1996; Hoshi et al. 2000; Groft et al. 2001; Nakada et al. 2001; Takahashi et al. 2002; LuW et al. 2004), whereas others suggest that these TIMPs are produced in excess and may participate on the tumor progression (Nakagawa et al. 1995; Lampert et al. 1998; Kachra et al. 1999; Saxena et al. 1999; Pagenstecher et al. 2001; Lu et al. 2004; Blazquez et al. 2008a; Rorive et al. 2010; Crocker et al. 2011). These conflicting results may reflect not only the different methodologies and the heterogeneity of the studied tumors but also the ambiguous role of TIMPs in tumor pathogenesis. This is best illustrated by TIMP2 that promotes the activation of MMP2 [see Sect. 12.1 and Strongin et al. (1995)], may activate the promigratory signaling pathways by binding to MMP14 on the cell membrane (Sounni et al. 2010), and has anti-angiogenic effects independent of MMPs (see Stetler-Stevenson and Seo (2005) for review).
The production of the membrane-bound and secreted proteases in stromal cells, such as reactive astrocytes, microglia, endothelial, and infiltrating immune cells, is an additional factor, which increases the complexity of their biological impact in gliomas (Fig. 12.4) (Rivera et al. 2010). Because of their localization in the extracellular space, these stromal proteases may augment the proteolytic systems deployed by the glioma cells. Examples of this cooperation include the activation of glioma MMP2 by microglial MMP14, which promotes glioma growth (Markovic et al. 2009) or activation of MMP2 in reactive astrocytes by glioma cell-derived plasmin (Le et al. 2003). Interestingly, forced expression of MMP14 directly in glioma cells induced their death (Markovic et al. 2009) further strengthening the ordered and highly context-dependent role of proteases.

Examples of the extracellular proteases expressed by individual constituents of the glioma microenvironment. It is important to note that the list is non-exhaustive as the expression of several proteases was not yet analyzed in all cell types and the expression in individual cell types may dynamically change in response to microenvironmental stimuli. Cysteine proteases in black, metalloproteases in red, serine proteases in green, aspartate proteases in blue. MMP matrix metalloproteinase, uPA urokinase-type plasminogen activator, tPA tissue-type plasminogen activator, ECE endothelin converting enzyme, MT MMPs membrane type MMPs, GCP-II glutamate carboxypeptidase II
With the exception of the activation of the caspase cascade, the reports on the mechanisms regulating the expression and activity of intracellular proteases implicated in glioma pathogenesis are scarce. Hypermethylation of the caspase-8 promoter, which leads to the block of the extrinsic apoptotic pathway, was observed in over 50 % of glioblastomas by Skiriute et al. (2012) and was associated with shorter patient survival. Several mechanisms were proposed to regulate the activity of the cysteine proteases calpains, although their in vivo relevance mostly remains to be determined (see Franco and Huttenlocher (2005) for review). Elevation of the intracellular concentration of Ca2+, autocatalytic cleavage of the enzymes, and the interaction with phospholipids and the endogenous inhibitor calpastatin were demonstrated to influence the activity of calpains (Franco and Huttenlocher 2005). Interestingly, calpain-2 is phosphorylated by ERK after the activation of the EGFR signaling, which is essential for EGF-induced motility in fibroblasts (Glading et al. 2000, 2004), and may thus play an important role in the motility of glioma cells as well (see Sect. 12.4.1.5).
In conclusion, multiple proteases show differential expression in glioma tissue as a result of a number of different pathogenetic mechanisms eventually leading to the dysregulation of the proteolytic homeostasis in gliomas. In addition, the dysregulation of the proteolytic activity in the extracellular space can be triggered by various mechanisms originating from changes in the stromal as well as the malignant cells. The relative importance of the various mechanisms may change during glioma progression and in some cases the proteolytic homeostasis can be impaired even without an overt overexpression of the protease(s). Conversely, the overexpression of a protease by itself need not be sufficient to change the proteolytic balance in the presence of counteracting compensatory mechanisms in the tumor microenvironment or in tumor cells.
4 Protease-Mediated Protein Processing in Gliomas and Participation of Proteases on the Biological Hallmarks of Gliomas
Extracellularly localized and plasma membrane-bound proteases are important tools used both by glioma cells as well as by the stromal cells to shape the glioma microenvironment. Their substrates include structural protein components of the extracellular matrix (ECM), zymogens of other proteases, and regulatory molecules such as chemokines, cytokines, and cell surface receptors or plasma membrane-bound proteins that may be released into the extracellular space (Fig. 12.5). These proteases therefore frequently serve as direct (e.g., via protease-activated receptors) or indirect (e.g., by modifying or releasing signaling molecules) mediators of intercellular communication. The biological effects of their proteolytic activities are manifold; proteases contribute to the profound remodeling of the unique ECM of human brain observed in glioblastoma (Bellail et al. 2004) and promote glioma cell dissemination. Cleavage of the ECM not only removes the physical barriers to glioma dissemination but also releases growth factors sequestered in the ECM (e.g., VEFG, TGF-β) and produces fragments of ECM proteins with new biological activities (e.g., anti-angiogenic peptides) that influence glioma cell proliferation, resistance to apoptosis, and neovascularization. Finally, by shedding cell surface molecules responsible for the activation of the immune system such as NKG2D ligands (Eisele et al. 2006; Wolpert et al. 2014) and activating latent TGF-β (Huber et al. 1992; Leitlein et al. 2001), proteases aid glioma cells to escape from the immune surveillance.

Proteins regulated by plasma membrane-bound and extracellular proteases in the glioma microenvironment. CAR Coxsackievirus and Adenovirus Receptor, ECM extracellular matrix, HB-EGF heparin-binding EGF-like growth factor, HGF hepatocyte growth factor, PAR protease-activated receptors
In contrast with the extracellularly localized proteases, the role of the intracellular proteases is largely restricted to the expressing cells, possibly with the exception of some proteases present in the secretory pathway and exosomes or released into the extracellular space due to apoptotic or necrotic disintegration of the cells. The substrates of these proteases relevant for the progression of glioblastomas are less explored, but include the cytoskeletal and organellar proteins, components of the signal transduction pathways, proteins regulating and executing vesicular transport, gene expression and replication, autophagy, and apoptosis. Importantly, the proteasome together with the deubiquitinating enzymes (DUBs) are critical regulators of many cellular proteins implicated in glioma pathogenesis (Fig. 12.6).

Proteins regulated by intracellular proteases contributing to glioma pathogenesis. CAR Coxsackievirus and Adenovirus Receptor, CDK cyclin dependent kinases, FASN fatty acid synthase, IkB inhibitors of NFκB, p75NTR p75 neurotrophin receptor, DUBs deubiquitinating enzymes
4.1 Proteases and Glioma Invasion
The widespread infiltration of the surrounding brain tissue by glioma cells is a long recognized hallmark of gliomas (Bramwell 1888) and in gliomas with IDH mutation, individual invading glioma cells were demonstrated to be dispersed throughout the brain (Sahm et al. 2012). The locoregional treatments (i.e., surgical removal of the tumor with subsequent radiation therapy) therefore almost invariably fail and the tumor recurs usually within 2–3 cm of the original location.
Several reports establish a critical role of the extracellular and membrane-bound metalloproteases (especially MMP2, MMP9, MMP14, ADAMTS), the serine protease u-PA, and secreted lysosomal cysteine proteases including cathepsin-B in glioma invasiveness (reviewed in Binder and Berger 2002; Levicar et al. 2003b; Rao 2003; Mentlein et al. 2012). Indeed, MMP2 and MMP9 (Kim et al. 2011), ADAM17 (Chen et al. 2013a), as well as uPA and its receptor uPAR (Yamamoto et al. 1994a; Zhang et al. 2000; Colin et al. 2009) are abundantly expressed at the invasive edge. In addition, the urokinase-type plasminogen activator receptor (uPAR) and cathepsin-B are expressed by glioma cells infiltrating the surrounding brain tissue (Mikkelsen et al. 1995).
The activation of proteolytic systems seems to be sufficient as well as necessary for glioma cell invasiveness and involves proteases expressed by the malignant as well as stromal cells (Le et al. 2003). MMP14 is a key enzyme endowing glioma cells with the ability to spread and migrate on myelin (Paganetti et al. 1988; Amberger et al. 1994). Interestingly, even non-glioma cells such as fibroblasts could gain the capacity to migrate on an otherwise unpermissive myelin substrate as well as to invade the myelinated optic nerve fibers after the introduction of MMP14 (Belien et al. 1999). Similarly, introduction of ADAM17 into an astrocytic cell line caused upregulation of several invasion and angiogenesis-promoting genes and resulted in increased invasiveness and formation of high grade brain tumors upon intracranial implantation (Katakowski et al. 2009). MMP2 (gelatinase-A) is overexpressed in the majority of glioblastomas (Yamamoto et al. 1996; Kunishio et al. 2003; Hagemann et al. 2012) and a number of studies demonstrated that MMP2 may be one of the shared downstream components critical for the execution of the cell invasion program. MMP2 is produced as an inactive zymogen that is activated in the extracellular space by a complex mechanism that involves MMP14 and TIMP2 (see Kessenbrock et al. (2010) and references therein). Blocking MMP2 by RNAi (Kargiotis et al. 2008; Badiga et al. 2011), inhibitors (Noha et al. 2000; Nuti et al. 2011), or interference with the pathways that drive its expression (e.g., by modulating miRNA (Nan et al. 2010; Pan et al. 2012), the PI3K-Akt (Koul et al. 2001; Kubiatowski et al. 2001; Kwiatkowska et al. 2011; Jung et al. 2013) and other signaling pathways (Blazquez et al. 2008b; Kamino et al. 2011; Guan et al. 2012) decrease the invasiveness of glioma cells. Similar indispensable proinvasive role was demonstrated by targeting uPA and its receptors uPAR, MMP9, cathepsin-B (Kondraganti et al. 2000; Gondi et al. 2004a, b; Lakka et al. 2004) and cathepsin-L (Levicar et al. 2003b) with an additive effect when multiple proteases were targeted.
Recently, a number of other proteases including the ones localized predominantly intracellularly were also shown to contribute to glioma invasiveness. These include the metallo- [MMP1 (Gessler et al. 2011), MMP3 (Mercapide et al. 2003), MMP13 (Inoue et al. 2010), MMP15 (Zhang et al. 2005), MMP16 (Li et al. 2013a), MMP19 (Lettau et al. 2010), MMP26 (Deng et al. 2010), ADAMs 8 and 19 (Wildeboer et al. 2006), ADAMTS 4 and 5 (Held-Feindt et al. 2006)] as well as serine [(Miyata et al. 2007), FAP (Mentlein et al. 2011), PCSK6 (Delic et al. 2012), PCSK5A (Maret et al. 2012), furin (Mercapide et al. 2002; Wick et al. 2004), HGFA (Uchinokura et al. 2006)], and cysteine proteases [cathepsin-H (Sivaparvathi et al. 1996a), cathepsin-L (Sivaparvathi et al. 1996c; Levicar et al. 2003a), calpain-2 (Jang et al. 2010)].
The pathways and mechanisms by which proteases contribute to glioma invasion are diverse, strongly context-dependent, and in several cases highly redundant and overlapping.
4.1.1 Extracellular Matrix Cleavage
By cleaving the ECM, proteases remove the physical barriers hindering glioma invasion. Hyaluronic acid, tenascins, and the proteoglycans lecticans are major components of the brain ECM (Bellail et al. 2004). The fibrillar proteins typical for the ECM in other organs were previously thought to be largely restricted to the vicinity of blood vessels (collagen IV, fibronectin, laminin) or completely absent (collagen I), but there is increasing evidence that they are produced by glioma cells in situ (Paulus et al. 1994; Senner et al. 2008; Huijbers et al. 2010; Payne and Huang 2013; Serres et al. 2013). The brain-specific components and mesenchymal fibrillar proteins are subject to proteolytic degradation by several proteases. For example, the lecticans are cleaved by numerous ADAMTS, in particular the aggrecanases ADAMTS4 and ADAMTS5 (Held-Feindt et al. 2006) that are typically elevated in brain tumors, but contribution of several other MMPs including MMP19 is likely (Lettau et al. 2010). The broad range of extracellular components cleaved by MMPs (Yong et al. 2001; Hagemann et al. 2012) and aspartate and cysteine cathepsins, together with the profound dysregulation of their activities in brain tumors, makes these proteases most likely candidates involved in the degradation of ECM.
4.1.2 Modification of the Function of Cell Adhesion Molecules
CD44 is a major hyaluronic acid cell receptor on glioma cells and promotes their migration and invasion upon proteolytic cleavage. MMP9 binds to and cleaves CD44 (Chetty et al. 2012); in addition, CD44 can be cleaved by ADAM10 in response to CD44 ligation (Murai et al. 2004). The released extracellular fragment of CD44 was shown to directly promote glioma migration and invasion (Chetty et al. 2012), whereas the cell membrane-bound CD44 fragment is cleaved by γ-secretase. The liberated intracellular domain translocates to the cell nucleus where it acts as a transcription factor (Murakami et al. 2003) and may play a role in glioma cell adhesion (Chetty et al. 2012). Other examples of proteolytically processed cell adhesion molecules implicated in glioma migration are the ADAM 10 substrates N-cadherin (Kohutek et al. 2009) and L1CAM (CD171) (Yang et al. 2011).
4.1.3 Direct or Indirect Activation of the Migration-Promoting Signaling Pathways
Protease expression is associated with the activation of motility-promoting signaling cascades. Motility is decreased in glioma cells after the downregulation of uPA by RNAi in parallel with the disorganization of the cytoskeleton, downregulation of the small GTPase of the Rho-subfamily Cdc42, and decreased PI3K/Akt phosphorylation (Chandrasekar et al. 2003). The underlying mechanisms may include binding of uPA to uPAR, which converts the latter into a membrane-bound chemokine and triggers intracellular signaling (Eden et al. 2011). In addition, uPA may proteolytically activate HGF (Naldini et al. 1992), a cell motility enhancing growth factor (Moriyama et al. 1996). The tissue factor (TF) is robustly upregulated in glioma cells in response to hypoxia (Rong et al. 2009) and may trigger the local activation of the coagulation cascade proteases such as fVIIa or thrombin that subsequently initiate the promigratory signaling through the protease-activated receptors (PARs) (Gessler et al. 2010). Similarly to uPA, thrombin facilitates the maturation of HGF by activating the protease HGF activator (Uchinokura et al. 2006). Metalloproteases such as MMP14 were also demonstrated to promote promigratory MAPK signaling possibly through EGFR transactivation as well as by EGFR-independent mechanisms (Gingras et al. 2001; Langlois et al. 2007).
Zheng et al. (2007) further showed that ADAM17, a sheddase involved in the release of membrane-bound growth factors and cytokines, is implicated in the hypoxia-mediated increase in glioma invasiveness by activating the EGFR signaling pathway. Proteases such as MMP9 are also able to promote glioma cell migration by cleaving the IGFII-IGFBP complexes thereby releasing the cell motility enhancing IGF-II (Insulin-like growth factor-II, Rorive et al. (2008) and references therein). The cytokine TGF-β released from the extracellular matrix or from infiltrating microglia is another mediator that strongly promotes glioma invasiveness (Wesolowska et al. 2008; Ye et al. 2012). TGF-β is produced in a latent form and several proteases including plasmin, MMP2, thrombin, MMP14, and furin-like proteases are involved in its maturation (Leitlein et al. 2001; Jenkins 2008).
4.1.4 Removal of Migration Inhibitors and Proteolytic Modification of Extracellular Matrix-Promoting Glioma Migration
The brain ECM and myelin in particular are inhibitory to cellular migration (Caroni and Schwab 1988; Schwab and Caroni 1988). bNI-220, one of the most potent CNS myelin inhibitory proteins, was shown to be cleaved and inactivated by MMP14 (Belien et al. 1999), which promoted glioma cell invasiveness. Another inhibitory molecule is brevican (Yamada et al. 1997), a CNS-specific proteoglycan of the lectican family, that is upregulated in gliomas (Gary et al. 1998; Viapiano et al. 2005; Viapiano and Matthews 2006) and requires proteolysis for the promotion of glioma invasion. The studies by Viapiano et al. (2008) and Hu et al. (2008) demonstrate that the transfection of full length brevican that is resistant to the ADAMTS-mediated cleavage does not promote the malignant phenotype of glioma cells, whereas proteolytic cleavage or an N-terminal fragment of the brevican molecule promotes glioma cell adhesion, migration, and invasiveness as well as in vivo tumor growth (Hu et al. 2008; Viapiano et al. 2008). Although the downstream mechanisms remain to be established, the resulting N-terminal fragment of brevican seems to promote EGFR activation and expression of cell adhesion molecules and fibronectin (Hu et al. 2008). The EGFR activation may further promote glioma invasiveness by increasing protease expression (see Sect. 12.3). Yet another example of a secreted ECM molecule modified by proteolysis is the TGF-β-induced protein (betaig-h3). This protein has pleiotropic effects on cell–cell and cell–extracellular matrix interactions (Thapa et al. 2007) and influences adhesion and migration. In a study by Kim et al. (2012), betaig-h3 inhibited glioma cell invasion in vitro and its MMP9-mediated cleavage was suggested to be in part responsible for the proinvasive effects of MMP9 (Kim et al. 2012).
4.1.5 Cytoskeleton and Membrane Protrusion Remodeling
The dynamics of the actin fibers and the remodeling of the focal adhesions are critical determinants of cellular motility. Intracellular proteases such as the cysteine proteases of the calpain family importantly contribute to this process by the cleavage of the adhesion complex proteins (e.g., tallin, FAK, and paxillin) and of the actin regulators (e.g., cortactin and the small GTPase RhoA) (Carragher and Frame 2002; Franco and Huttenlocher 2005). In neuronal cells, EGF and brain-derived neurotrophic factor (BDNF) lead to calpain-2 phosphorylation via MAPK signaling (Zadran et al. 2010) and this posttranslational modification is necessary for the promigratory effects of EGF signaling (Glading et al. 2000, 2004; Cuevas et al. 2003). In gliomas, calpain-2 promotes cell migration after the binding of betaig-h3 (TGF-β-induced protein) to the α-5-β-1 integrin on the surface of glioma cells (Ma et al. 2012) and was shown to be required for glioma invasiveness by regulating the invadopodia dynamics and MMP2 secretion (Jang et al. 2010; Lal et al. 2012).
Somewhat surprisingly, low levels of active caspase-3 likewise promote glioma migration and invasion probably by the processing of gelsolin (Gdynia et al. 2007), a protein involved in actin remodeling (Silacci et al. 2004).
4.2 Proteases and Glioma Cell Proliferation and Apoptosis
Results of recent studies provide evidence that multiple different proteolytic mechanisms contribute to the cell cycle deregulation in glioma cells. They include the proliferative signaling mediated by the presenilin-generated carboxyterminal fragment of the overexpressed Notch receptor (Stockhausen et al. 2010; Capaccione and Pine 2013), by increased activation or activity of the protease-activated receptors (Hayakawa et al. 2007; Gessler et al. 2010), by the upregulation of some ADAM family members possibly activating the EGFR-PI3K-AKT signaling pathway (Bulstrode et al. 2012; Zheng et al. 2012; Chen et al. 2013b), and by the reduced degradation of the overexpressed or mutated tyrosine kinase receptors such as EGFR and MET by the ubiquitin-proteasome system (Hede et al. 2013). The protein components of the cell cycle machinery as well as their regulators are targeted for degradation by the ubiquitin-proteasome system (Vlachostergios et al. 2012; Hede et al. 2013). However, several specific cysteine isopeptidases called deubiquitinating enzymes (DUBs), which remove the ubiquitin chains from the ubiquitinated proteins, reverse the function of ubiquitination as an integral component of the core cell cycle machinery and of the cell cycle check points (Clague et al. 2012; Fraile et al. 2012; Cox et al. 2013; Li et al. 2013a). Besides their involvement in the control of cell cycle progression, DUBs participate also in the regulation of the signaling pathways, DNA damage repair, and apoptosis (Panner et al. 2010; Ramakrishna et al. 2011; Clague et al. 2012; Eichhorn et al. 2012; Fraile et al. 2012). It was recently demonstrated that many DUBs are targets of reversible redox regulation (Kulathu et al. 2013; Lee et al. 2013) and thus their functions may be influenced by the inherent redox disturbances in cancer.
Defects in the initiation and execution of apoptosis represent a hallmark of malignant cells including transformed astrocytes and proteolytic enzymes are indispensable players in the apoptotic pathways. Although some glioma cell lines are sensitive to apoptosis induction by TRAIL (tumor necrosis factor-α-related apoptosis-inducing ligand) and anti-Fas agonistic antibodies, many others including those with the stem cell features show resistance against the extrinsic death receptor (DR)-mediated apoptosis (Xiao et al. 2002; Yang et al. 2007; Capper et al. 2009; Bellail et al. 2010; Tao et al. 2012). Despite the high frequency of CASP8 gene promoter, hypermethylation together with the absence of procaspase-8 protein expression in gliomas (Ashley et al. 2005; Martinez et al. 2007; Skiriute et al. 2012), reexpression of procaspase-8 in response to the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine treatment was not sufficient to restore TRAIL sensitivity in glioma cells. These results suggest the participation of additional factors responsible for the resistance to TRAIL (Capper et al. 2009). Indeed, other studies showed that the resistance of glioma cells against apoptosis induction by activated DRs is often caused by the combination of several factors including the downregulation of procaspase-8 and -10 expression and the inhibition of their activation at the death-inducing signaling complex (DISC) by c-FLIPL and c-FLIPS, RIP, and PED/PEA-15 proteins (Xiao et al. 2002; Yang et al. 2007; Bellail et al. 2010; Panner et al. 2010). There is evidence that glioma cells can be killed by the granzyme-B (GrB)/perforin apoptosis pathway triggered by tumor-infiltrating lymphocytes which establish immunological synapses with tumorigenic cells (Hishii et al. 1999; Barcia et al. 2009). The synapsing CD8+GrB+ T cells showed GrB polarization towards tumor cells, which displayed a pycnotic or fragmented nucleus and a high positivity of the cleaved caspase-3, together indicating induced entry of the tumor cells into the execution phase of apoptosis (Barcia et al. 2009). Despite the very low percentage of synapsing cytotoxic T lymphocytes in all examined glioma cases, suggesting a deficient immune response (Barcia et al. 2009), a recent immunohistochemical study revealed that the higher expression of cleaved caspase-3 in gliomas was associated with longer survival of surgically treated glioma patients (Kobayashi et al. 2007). Tumor microenvironment of gliomas frequently contains hypoxic and acidic regions. Since hypoxia and acidic stress induce overexpression of serpinB9 in glioma stem cells (Li et al. 2009; Hjelmeland et al. 2011), this serpin, being a physiological irreversible inhibitor of GrB (Rousalova and Krepela 2010), may protect them from the GrB/perforin-mediated immune killing. Compared to the normal brain cortex tissue and primary neurons, glioma cells and tumors are hypersensitive to the (cytochrome-c + dATP)-mediated induction of the apoptosome-driven activation of procaspase-9 and -3 (Johnson et al. 2007). The authors attributed the differential sensitivity of the apoptosome apparatus activation to high Apaf-1 mRNA and protein levels in the tumor tissue compared with low Apaf-1 levels in the normal adjacent brain tissue. These differences in Apaf-1 levels correlated with differences in the levels of the transcription factor E2F1, an activator of Apaf-1 transcription, which was overexpressed in gliomas and bound to Apaf-1 promoter specifically in the tumor tissue (Johnson et al. 2007). A systems medicine model was recently suggested to predict the susceptibility of gliomas to apoptosis execution via the apoptosome-dependent procaspase activation. Based on the Apaf-1, procaspase-9, procaspase-3, Smac, and XIAP protein expression levels, the mathematical model predicted the sensitivity of glioma cell lines to temozolomide in vitro. Even more importantly, higher sensitivity to apoptosis induction was predicted by the model for the patients with longer progression free survival (Murphy et al. 2013).
Taken together, the proteolytic balance represents a prominent homeostatic regulator affecting both the proliferation and apoptosis of glioma cells.
4.3 Proteases and Glioma Neovascularization, Formation of Necrotic Areas and Pseudopalisades
Glioblastomas rank among the most vascularized neoplasms and typically contain areas of microvascular proliferation and a large number of dysplastic vessels. In fact, endothelial cells in glioblastomas are responsible for a substantial proportion of the extracellular matrix degrading proteolytic activity within the glioma microenvironment. Using CNS-1 cells in a rat glioma model, Regina et al. demonstrated that endothelial cells were the dominant source of the MMP9 activity in the tumors (Regina et al. 2003). MMP2, MMP9 (Rao et al. 1996; Vince et al. 1999; Raithatha et al. 2000), cathepsin-B (Mikkelsen et al. 1995; Sivaparvathi et al. 1995; Wang et al. 2005), and uPA (Yamamoto et al. 1994a) are expressed by vascular structures in gliomas. Interestingly, whereas endothelial cells in the intracranial rat glioma tumors strongly upregulated MMP9, this upregulation was absent in the subcutaneous tumors, suggesting that complex microenvironmental signals are important for the induction of proteases in endothelial cells (Regina et al. 2003).
Mechanistically, membrane-bound and extracellular proteases are well-known regulators as well as executors of the process of glioma neovascularization (Lakka et al. 2005). The vascular structures are generated with the participation of local as well as bone marrow-derived cells by several mutually nonexclusive mechanisms including angiogenesis, vasculogenesis, vascular cooption, vascular mimicry, and transdifferentiation of glioma stem cells into vascular cell types (Weis and Cheresh 2011). During angiogenesis, the endothelial cell-produced matrix metalloproteinases, cysteine proteases such as cathepsin-B, and serine proteases such as uPA and plasmin mediate the degradation of the basal membrane, thus permitting the invasion of endothelial cells and sprouting of new vessels (Lakka et al. 2005). The proteolytic remodeling of extracellular matrix also leads to the release of proangiogenic growth factors. By releasing sequestered VEGF, MMP9 was shown to participate on the angiogenic switch in pancreatic cancer (Bergers et al. 2000) and a similar mechanism seems to operate in glioblastomas (Du et al. 2008). In an animal model, ablation of tumor cell derived and stromal MMP9 impeded vascular remodeling and recruitment of endothelial and pericyte progenitors (i.e., vasculogenesis) (Du et al. 2008). Interestingly, MMP9 on bone marrow-derived CD45+ tumor infiltrating cells was sufficient to induce angiogenesis by increasing the bioavailability of VEGF in this model (Du et al. 2008). The expression of MMP9 was also able to reverse the anti-angiogenic and growth inhibitory effect of SPARC overexpression in meduloblastoma cells (Bhoopathi et al. 2010). Thus the presence of a supercritical amount of MMP9 in the glioma microenvironment, most likely irrespective of its cellular source, seems to be necessary and sufficient to induce vascular remodeling and angiogenesis.
Another mode of glioma neovascularization involves the accumulation of glioma cells along preexisting vessels (vascular cooption) with an ensuing intravascular thrombosis, which leads to necrosis and hypoxia-induced angiogenesis (Rong et al. 2009). Proteases play various roles during this process. The enzymatic activity of matrix metalloproteinases contributes to the disruption of the endothelial cell barrier function (Ishihara et al. 2008; Rivera et al. 2010) and vessel destabilization. The serine proteases of the blood coagulation cascade thus gain access to the increased levels of tissue factor (TF) expressed by glioma cells, which initiates blood clotting and occlusion of the vessel (reviewed in Rong et al. 2009). Angiogenesis is activated by the resulting tissue hypoxia. In addition thrombin generated by the coagulation cascade stimulates endothelial cells by upregulating the α-v-β-3 integrin (Tsopanoglou et al. 2002), promotes the production of VEGF in glioma cells through PAR-2 signaling (Yamahata et al. 2002; Dutra-Oliveira et al. 2012), and leads to the activation of the proangiogenic HGF (Abounader and Laterra 2005; Uchinokura et al. 2006). The pathologic activation of the proteolytic coagulation cascade was also suggested to foster glioma stem cells in the perivascular niche (Garnier et al. 2010; Magnus et al. 2010).
Other proteases may be involved in glioma neovascularization by mechanisms not directly involving the degradation of the ECM. Endothelin converting enzyme (ECE) is expressed by glioma cells and glioma vasculature together with other components of the endothelin system (Egidy et al. 2000; Naidoo et al. 2005). ECE proteolytically converts a precursor protein into endothelin-1, which promotes vascularization due to its proproliferative and promigratory effects on endothelial cells (reviewed in Kaur et al. 2005). Clinical studies targeting ET-1 in glioblastoma are underway (Phuphanich et al. 2008) and an alternative approach could involve ECE inhibition (Berger et al. 2005). Glutamate carboxypeptidase II (prostate-specific membrane antigen, PSMA) is expressed in the neovasculature of several malignancies (Chang et al. 1999) including glioblastoma (Wernicke et al. 2011). In addition to being a maker of tumor neovascularization (Chang et al. 1999), PSMA seems to be functionally important as its absence or inhibition was demonstrated to prevent angiogenesis through the modification of integrin signaling in endothelial cells (Conway et al. 2006; Grant et al. 2012).
Proteases are driving the formation of the pseudopalisades, a typical morphological feature of glioblastomas. These highly cellular regions typically surround necrotic areas, are hypoxic, exhibit significant gelatinolytic activity (Brat et al. 2004), and abundant expression of cathepsin-B and PAI-1 (Colin et al. 2009). The increased cellularity of these structures is not due to the increased cell proliferation but rather reflects an increased migratory capacity of the glioma cells (Brat et al. 2004), which is promoted by proteases. Hypoxia, EGFR activation, and PTEN loss increase the expression of tissue factor in glioma cells (Rong et al. 2009) and the interaction of this protease cofactor with the coagulation fVIIa together with the activation of the protease-activated receptors (PARs) subsequently promotes their migration (reviewed in Dutzmann et al. 2010).
Noteworthy, proteases also exert anti-angiogenic effects (Rege et al. 2005; Lopez-Otin and Matrisian 2007; Ribatti 2009). ADAMTS1 and 8 were identified based on their sequence homology with the angioinhibitory thrombospondin-1 and similarly to thrombospondin-1 inhibit endothelial cell proliferation and angiogenesis (Vazquez et al. 1999), possibly by the sequestration of VEGF (Luque et al. 2003) or via the release of anti-angiogenic peptides from thrombospondin-1 and 2 (Lee et al. 2006). Interestingly, ADAMTS8 is downregulated in the majority of gliomas by a so far unknown mechanism (Dunn et al. 2006) and may therefore contribute to the excessive neovascularization of these tumors. A cell surface protease neprilysin (neutral endopeptidase 24.11, CD10) likewise inhibits angiogenesis by cleaving and inactivating the basic fibroblast growth factor (Goodman et al. 2006). Somewhat unexpectedly, anti-angiogenic effects are also observed in the case of proteases traditionally viewed as promoters of tumor progression. Several endogenous angiogenesis inhibitors such as endostatin, tumstatin, and angiostatin are proteolytic fragments produced from the ECM or plasminogen (Rege et al. 2005; Ribatti 2009). Indeed, MMP9 is crucial for the liberation of the C-terminal domain from collagen-IV producing tumstatin (Hamano et al. 2003) and the furin-mediated activation of MMP14 is crucial for the generation of vasculostatin from the membrane-bound brain-specific angiogenesis inhibitor 1 (BAI1) (Cork et al. 2012). Supporting the functional importance of these processes, the absence of MMP9 in mice leads to decreased levels of circulating tumstatin and increased growth of experimental tumors (Hamano et al. 2003). Similarly, the absence of MMP12 and MMP19 is associated with enhanced tumor growth owing to the promotion of angiogenesis (Houghton et al. 2006; Jost et al. 2006). Even more surprisingly, a proteolytically produced fragment of MMP2 corresponding to its hemopexin domain blocks glioma growth by inhibiting angiogenesis as well as proliferation and migration of glioma cells (Bello et al. 2001). This might explain the bewildering results of a recent study that showed a reduced in vivo growth of glioma cells overexpressing MMP2 and a more pronounced destabilization of the tumor vasculature together with higher tumor proliferation in its absence (Tremblay et al. 2011).
Proteases probably participate in the anti-angiogenic treatment-induced changes of the tumor vessels towards a more mature morphology and function. This vascular “normalization” (Goel et al. 2011) improves the perfusion and oxygenation of the tumors and is an important mechanism supporting the effectiveness of radiotherapy and cytotoxic therapies (Jain 2005; Sato 2011). In a mouse glioma model, VEGFR2 blockade promoted pericyte recruitment and a temporary restoration of the vascular morphology accompanied by the remodeling of the abnormally thick basal membrane. Collagenase IV enzymatic activity in the perivascular areas was significantly increased by the anti-VEGFR2 treatment, and the thinning of the basal membrane was prevented by a coadministration of a matrix metalloprotease inhibitor, further confirming the important role of the proteases in the process (Winkler et al. 2004).
In conclusion, the effects of proteases on angiogenesis are diverse as proteases may promote or inhibit the formation of new vessels depending on the stage of tumor development and the presence of their substrates in the tumor microenvironment.
5 Exploitation of Glioma-Associated Proteases as Possible Markers and Therapeutic Targets: Failures and Promises
Numerous studies examined the association between the expression of proteases and clinicopathologic data and expression of several proteases was even suggested to predict patient survival.
Patients with anaplastic astrocytomas with positive immunoreactivity for the cleaved caspase-3 in more than 10 % tumor cells had better survival rate in a study by Kobayashi et al. (2007). Similarly, higher immunohistochemical staining for carboxypeptidase-E was associated with longer survival (Horing et al. 2012). On the contrary, higher expression of ADAM8 (He et al. 2012a), CSN5 (Jab1) (He et al. 2012b), MMP13 (Wang et al. 2012), kallikrein 6 (Drucker et al. 2013), uPA (Bindal et al. 1994; Hsu et al. 1995), MMP1 (Zhang et al. 2011; Xu et al. 2013), and cathepsin-B (Colin et al. 2009) are negative prognostic factors in gliomas. However, these potentially clinically applicable results are mostly based on small scale studies or explorative microarray analyses and thus will require validation in larger patient cohorts before these candidate proteases can be accepted as reliable molecular markers in neurooncology (Jansen et al. 2010; Weller et al. 2013). Analysis of the TCGA data shows a possible prognostic significance of the proteases USP15 (Eichhorn et al. 2012) and YME1-like-1 (Bredel et al. 2009). These promising microarray-based results will need to be converted into standardized clinically applicable assays enabling the risk assessment for the individual glioma patient.
According to some authors, proteases may also be exploited for the visualization of the tumor tissue. Complete surgical resection of the tumor is an important factor influencing the prognosis of glioma patients and several methods such as microscopic surgery, intraoperative MRI, or 5-ALA fluorescence-guided resection are currently used to improve the identification and surgical removal of the tumor tissue (Black et al. 1997; Stummer et al. 2006). A recent work by Cutter et al. (2012) provides evidence that the differential expression of proteases such as cysteine cathepsins in the tumor tissue compared to normal brain may be utilized to differentiate tumor borders. The authors used fluorescently quenched activity-based probes that were constructed from suicide protease inhibitors. Proteolytic cleavage of such probes leads to the liberation of the quencher and the covalent attachment of the fluorochrome to the protease active site. Topical application of such probes rapidly visualized tumors in a preclinical glioma model as well as in the resected human glioma tissue (Cutter et al. 2012) suggesting its possible utility in guiding glioma resection.
Dysregulated proteolytic activity and often a direct role in glioma progression make proteases promising therapeutic targets. Several concepts may be envisioned for the therapeutic exploitation of glioma-associated proteases.
In addition to directly targeting the “prooncogenic” functions of glioma-associated proteases, the enzymatic activity of certain proteases may be used for the selective activation of a prodrug in the tumor tissue (tumor-activated prodrugs) (Atkinson et al. 2008). This approach relies on the highly elevated expression of the target protease in the tumor and its ability to efficiently convert the prodrug into a highly cytotoxic compound. The serine protease prostate-specific antigen (PSA) and the MMPs were studied in this context in several tumor types (Atkinson et al. 2008), but to the best of our knowledge, this approach has not been tested in gliomas. Similarly, the expression of proteases restricted to or highly upregulated in gliomas may be utilized as a means of targeted delivery of conventional chemo- or radiotherapy. GCP-II (PSMA, prostate-specific membrane antigen) is being tested as a therapeutic target in prostate cancer (Mullard 2013), but its expression in the vasculature of several tumors including gliomas make it a possible target in gliomas as well (Wernicke et al. 2011). Recently, the pronounced expression of uPA in gliomas was suggested for the selective targeting of the tumors by a recombinant uPA-directed oncolytic virus that markedly improved survival in a rodent glioma model (Hasegawa et al. 2010).
Downregulation of the target protease by RNA interference or the inhibition of the upstream signaling cascades driving protease expression are potential therapeutic approaches, the latter possibly being a contributing factor to the effects of therapies targeting receptor tyrosine kinases and the downstream signaling pathways (Lakka et al. 2000; Mao et al. 2012). Indeed, data from preclinical glioma models suggest that inhibition or genetic ablation of proteases or their respective critical activators in glioma cells leads to the induction of apoptosis (Gondi et al. 2009; Chetty et al. 2010), suppression of cell proliferation, invasiveness and inhibition of tumor-induced angiogenesis, as well as tumorigenicity in experimental animals (Rao 2003; Lakka et al. 2005; Badiga et al. 2011; Veeravalli et al. 2012; Kesanakurti et al. 2013).
Low-molecular-weight protease inhibitors have become an effective strategy in the treatment of cardiovascular and infectious diseases (reviewed in Turk 2006) and the hope is that they could also improve the outcomes in cancer patients.
Given the important role of MMPs in glioma invasiveness, MMP inhibitors were among the first protease inhibitors tested in glioblastomas. Four clinical trials testing the broad spectrum MMP inhibitor marimastat in combination with temozolomide or radiotherapy in patients with grade III and IV tumors were published. Overall, the therapeutic effects were small and the treatment was accompanied by musculoskeletal toxicity in a large proportion of patients (Groves et al. 2002, 2006; Larson et al. 2002; Levin et al. 2006). Possibly, more specific MMP inhibitors and/or optimized dosing and timing of administration may prove more efficacious in future studies, in particular considering the potential of MMP inhibitors to block the proinvasive effects of radiotherapy and anti-angiogenic treatments (Lucio-Eterovic et al. (2009) and Sect. 12.3.1).
More recently, proteasome inhibition was suggested as a potential therapeutic modality in gliomas. Proteasome is responsible for the proteolytic removal of regulatory proteins with growth inhibitory activity such as the CDK inhibitor p27 (Piva et al. 1999), the inhibitors of NFκB (IkB), and proteins p53, BID (Unterkircher et al. 2011), NOXA (Ohshima-Hosoyama et al. 2011), or activated caspases-8 and -3 (Kim et al. 2004) (see also Fig. 12.6). Proteasomal inhibition therefore induces the accumulation of these proteins; in addition, it enhances the expression of the apoptosis-inducing receptors such as Fas (Tani et al. 2001) or the TRAIL receptor DR5 (Hetschko et al. 2008) and triggers the endoplasmic reticulum stress (Kardosh et al. 2008). By these complex mechanisms proteasome inhibitors cause growth arrest and sensitization to proapoptotic stimuli as demonstrated in glioma cell lines (Kitagawa et al. 1999; Wagenknecht et al. 1999, 2000; Tani et al. 2001; Yin et al. 2005; Pedeboscq et al. 2008; Zanotto-Filho et al. 2012) and in patient-derived primary cells (Koschny et al. 2007). Proteasome inhibitors also enhanced the effectiveness of other chemotherapeutics (Ceruti et al. 2006), radiation (Ng et al. 2009), and TRAIL (La Ferla-Bruhl et al. 2007; Hetschko et al. 2008; Balyasnikova et al. 2011; Jane et al. 2011; Kahana et al. 2011; Seol 2011; Unterkircher et al. 2011) and had synergistic inhibitory effects on glioma stem-like cells in combination with the histonedeacetylase inhibitors (Asklund et al. 2012). In preclinical animal models, the antitumor activity was observed for lactacystin (Legnani et al. 2006; Wang et al. 2013a). However, studies using the new proteasome inhibitor salinosporamide A (Vlashi et al. 2010) or the most frequently utilized proteasome inhibitor approved for clinical application in other cancers bortezomib (Labussiere et al. 2008) failed at reducing the tumor growth in a xenotransplantation glioma model. Phase I clinical trials demonstrated safety of bortezomib in glioma patients (Kubicek et al. 2009; Phuphanich et al. 2010), but a phase II trial in patients with recurrent glioblastoma failed to show its efficacy in combination with vorinostat (Friday et al. 2012). Several other phase I and II studies of bortezomib in combination with other therapeutic modalities (Table 12.3) as well as testing of new proteasome inhibitors with improved in vivo efficacy (Roth et al. 2009) are underway (Vlachostergios et al. 2013). Another possibility to target the ubiquitin-proteosome system lies in the modulation of the deubiquinating enzymes (DUBs, reviewed in Hussain et al. (2009), Fraile et al. (2012)]. By cleaving the isopeptide bonds joining ubiquitin to other proteins, DUBs stabilize a number of proteins essential for the cell cycle regulation, chromatin remodeling, DNA damage repair, and signaling pathways including the membrane-bound receptors such as EGFR (Fraile et al. 2012; Liu et al. 2014). In the case of DUBs associated with the proteasome (i.e., UCHL5, USP14, and POH1), however, the inhibition leads to the block of proteasomal degradation of polyubiquitinated proteins. b-AP15, an inhibitor of the proteasome-associated DUBs, was recently demonstrated to have antitumor activity against a range of cancer cells with glioma cell lines of the NCI60 panel being particularly sensitive (D’Arcy et al. 2011). With few exceptions (see Table 12.1), the role of DUBs in glioma pathogenesis is nevertheless largely unknown and more research is needed before using DUB inhibitors or approaches aiming at restoring the activities of tumor-suppressor DUBs (Fraile et al. 2012) in glioma therapy.
Some protease inhibitors were found to have novel, unexpected antitumor activities. The aspartate protease inhibitors widely used in the treatment of HIV infection such as nelfinavir and ritonavir were demonstrated to inhibit glioma growth. The mechanisms are currently not fully elucidated but presumably involve the induction of endoplasmic reticulum stress, interference with the activity of Hsp90, and reduction of MMP expression (Laurent et al. 2004; Liuzzi et al. 2004; Pyrko et al. 2007) (see Kast et al. (2013) for review). Clinical studies in glioma patients were rapidly initiated (Table 12.3) as these drugs have a favorable safety profile and have already been in clinical use.
Inhibitors of the γ-secretase (GSIs) seem to be another promising class of drugs (see Golde et al. (2013) for review). Originally designed for the treatment of Alzheimer’s disease, GSIs are currently tested in several cancers including gliomas because of their inhibitory effect on the Notch signaling (Golde et al. 2013; Takebe et al. 2014). This pathway typically promotes the self-renewal and inhibits the differentiation of stem cells including glioma stem cells (Chen et al. 2010; Hu et al. 2011; Kristoffersen et al. 2013). Gliomas of the proneural subtype may be in particular sensitive to the inhibition of γ-secretase, as the Notch pathway seems to be typically activated in this subtype (Saito et al. 2014, #2290). In addition, the GSIs may also block the neurotrophin-induced glioma invasion (Wang et al. 2008a). In line with these results, GSIs inhibited glioma growth in preclinical models and enhanced the effects of temozolomide (Gilbert et al. 2010). Despite these promising results, there are several important areas that remain to be explored, such as possible off-target effects of GSIs on signal peptidase and the effects of GSI on angiogenesis and immune functions (Golde et al. 2013). For example, a recent report concluded that GSIs reduced the growth of xenotransplanted gliomas, but somewhat surprisingly enhanced the proliferation of endothelial cells in response to VEGF and increased the vascular density in the tumors (Zou et al. 2013). The vasculature was dysfunctional as the perfusion and oxygenation of the treated tumors was decreased similarly to observations in other cancers (see Liu et al. (2013) for review). In addition, γ-secretase was suggested to function as a tumor suppressor in squamous cell carcinoma (Li et al. 2007). Indeed, a prematurely halted clinical trial in patients with Alzheimer’s disease (Doody et al. 2013) reported increased incidence of skin cancer. Inhibition of γ-secretase may have negative effect on the proliferation and self-renewal of normal stem cells in several organs (van Es et al. 2005; Hu et al. 2011). Despite a phase I trial with a γ-secretase inhibitor MK-0752 in pediatric patients with recurrent CNS malignancies demonstrated good tolerability (Fouladi et al. 2011), the obvious complexity of the effects of GSIs on the tumor microenvironment still precludes the straightforward claim that γ-secretase represents a suitable target for antiglioma treatment and requires further studies with careful evaluation of its therapeutic efficacy and safety.
Other proteases were also suggested as potential targets in glioma treatment mostly based on preclinical data. Inhibition of the sheddase ADAM10 was demonstrated to block glioma growth by interfering with the abovementioned Notch signaling pathway (Floyd et al. 2012). In addition, the inhibition of ADAM10 may improve the ability of the immune system to eradicate glioma stem cell (Wolpert et al. 2014) and also block glioma cell migration and adhesion (Table 12.1, see Moss et al. (2008) for general review). Targeting of the extracellularly localized cathepsin-B by cell impermeable inhibitors (Withana et al. 2012) or its elimination by RNAi were also proposed, although at least in gliomas, the effects of downregulating cathepsin-B were most pronounced when combined with the targeting of other extracellular proteolytic systems (Gondi and Rao 2013). The proteolytic activation of the immunosuppressive TGF-β in gliomas may represent another example of a potential functional target of protease inhibitors (Huber et al. 1992; Leitlein et al. 2001).
Given the known high risk of thromboembolic events in patients with high grade gliomas and preclinical studies suggesting possible anticancer effects of low-molecular-weight heparin and direct thrombin inhibitors, inhibition of the blood coagulation proteases may seem beneficial in glioma patients (Ornstein et al. 2002; Hua et al. 2005a, b). The results from the so far performed clinical trials with low-molecular-weight heparins are however ambiguous (Perry et al. 2010), although some hints of potential benefit can be found (Robins et al. 2008; Zincircioglu et al. 2012). Given the observed increased risk of bleeding associated with their use (Perry et al. 2010) and the limited data on their clinical efficacy, the role of anticoagulation as an adjuvant antitumor therapy remains to be established (Perry 2010).
Despite the wealth of encouraging data in the preclinical studies, protease inhibitors were so far largely ineffective in the majority of the clinical trials in cancer patients (Coussens et al. 2002), including patients with gliomas (Groves et al. 2002, 2006; Larson et al. 2002; Levin et al. 2006; Ahluwalia et al. 2011). There are a number of causes that may have contributed to these failures:
-
1.
Malignancies frequently “hijack” physiologically occurring mechanisms including the deployment of proteases; many “on-target” side effects should therefore be anticipated due to the interference with their physiological functions (see Sect. 12.1). This risk is illustrated by the musculoskeletal side effects observed in the clinical studies using MMP inhibitors (Coussens et al. 2002). In addition, several proteases act as tumor suppressors or produce anti-angiogenic mediators (see Sect. 12.4.3, Bello et al. (2001), Rege et al. (2005), Lopez-Otin and Matrisian (2007), Cork et al. (2012)] and their inhibition may thus facilitate tumorigenesis.
-
2.
The complexity, functional overlap, and redundancy of the proteolytic cascades in gliomas suggest that targeting of several different types of proteases may be necessary to achieve the desired biological effects. This not only complicates the design of low molecular weight inhibitors capable of targeting several biochemically distinct protease classes but also brings about an increased risk of “on-target” side effects.
-
3.
Incomplete understanding of the role of proteases in tumor progression and spatiotemporal and interindividual differences in the dependence of the tumors on the protease-mediated events. The variability of protease expression (Fig. 12.1, Jaworski et al. 2010) and their differing functional role at various stages of tumor progression requires careful characterization and selection of appropriate patients for clinical trials as well as for subsequent use in clinical practice.
-
4.
Limitations of current preclinical glioma models in determining the clinical efficacy of anti-protease therapies. The in vitro systems and long term propagated glioma cell lines grown in serum-containing media have numerous limitations (Schulte et al. 2011) and may produce misleading results. For example, an adenoviral-mediated overexpression of PAI-1 in a glioma cell line reduced their invasiveness through Matrigel, but not in brain tissue (Hjortland et al. 2003). Similarly, several studies with the proteasome inhibitor bortezomib in glioma cell lines indicated its growth inhibitory effects, which was not recapitulated in more complex in vivo models or so far completed clinical studies (Friday et al. 2012; Vlachostergios et al. 2013). Reliable preclinical models recapitulating the heterogeneity of tumor cells (Snuderl et al. 2011), enabling the assessment of the effects of therapeutic interventions on the stromal cells (Jones and Holland 2012), as well as the complex host-tumor pro-tumorigenic and anti-tumorigenic interactions are needed.
-
5.
Absence of appropriate biomarkers of achieving target protease inhibition in the clinical setting. Identification of the biologically active dose is necessary before performing large phase III trials. Furthermore, the design of clinical trials represents another challenge as the effect of agents that do not directly kill cancer cells cannot be measured by reduction of tumor size (Coussens et al. 2002).
-
6.
Last but not least: some of the compounds targeting proteases were rushed into clinical trials at an unprecedented pace and in several cases in suboptimal settings. Enormous need to improve the outcome of otherwise untreatable diseases, but also patent issues and competition may have contributed to such decisions in the past, but these factors are unfortunately likely to play a role in the future trials as well (Coussens et al. 2002).
Conclusion
Proteases are important regulators of the biological functions of proteins under both physiological and pathological conditions. Their dysregulation in gliomas is a consequence of complex changes in the glioma microenvironment and involves transformed glioma cells as well as various cell types present in the tumor stroma. Despite some common patterns, glioblastomas are, similarly to other parameters, heterogeneous also in terms of protease expression and activity. Proteases participate on almost every aspect of gliomagenesis acting both as regulators and executors and may therefore represent promising therapeutic targets. However, the role of individual proteases at various stages of the disease progression is usually complex, highly context dependent, and redundant. This might have contributed to the failures of the initial attempts on therapeutic targeting of proteases in gliomas as well as in other cancers. Designing effective and safe anti-glioma strategies exploiting proteases will therefore require a more in-depth understanding of their physiological and pathophysiological roles. Equally important will be the identification of critical protease-mediated processes in individual glioma subtypes and careful selection of subgroups of glioma patients most likely to benefit from therapies interfering with the functions of proteases.
Abbreviations
- ADAM:
-
A disintegrin and metalloproteinase
- ADAMTS:
-
A disintegrin and metalloproteinase with thrombospondin motifs
- bFGF:
-
Basic fibroblast growth factor
- BDNF:
-
Brain-derived neurotrophic factor
- BMP:
-
Bone morphogenic protein
- CAM:
-
Cell adhesion molecule
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- CXCL12:
-
Chemokine (C-X-C Motif) Ligand 12 (SDF stromal cell-derived factor)
- CXCL16:
-
Chemokine (C-X-C Motif) Ligand 16
- DISC:
-
Death-inducing signaling complex
- ECE:
-
Endothelin converting enzyme
- ECM:
-
Extracellular matrix
- EGFR:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular signal-regulated kinase
- FAK:
-
Focal adhesion kinase
- GCP-II:
-
Glutamate carboxypeptidase-II (N-acetyl-l-aspartyl-l-glutamate peptidase I, NAALADase I, PSMA, prostate-specific membrane antigen)
- GrB:
-
Granzyme-B
- HB-EGF:
-
Heparin-binding EGF-like growth factor
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia inducible factor
- IDH:
-
Isocitrate dehydrogenase
- IFN:
-
Interferon
- IGF:
-
Insulin-like growth factor
- MAPK:
-
Mitogen-activated protein kinase
- miRNA:
-
Micro RNA
- MMP:
-
Matrix metalloproteinase
- MT-MMP:
-
Membrane-type MMP
- PAI:
-
Plasminogen activator inhibitor
- PAR:
-
Protease-activated receptor
- PI3K:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- PTEN:
-
Phosphatase and tensin homolog
- RNAi:
-
RNA interference
- RTK:
-
Receptor tyrosine kinase
- Shh:
-
Sonic hedgehog
- SUMO:
-
Small ubiquitin-like modifier
- TF:
-
Tissue factor (fIII)
- TFPI:
-
Tissue factor pathway inhibitor
- TGF:
-
Transforming growth factor
- TIMP:
-
Tissue inhibitor of matrix metalloproteinase
- TNF:
-
Tumor necrosis factor
- tPA:
-
Tissue-type plasminogen activator
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- uPA:
-
Urokinase-type plasminogen activator
- uPAR:
-
Urokinase-type plasminogen activator receptor
- USP:
-
Ubiquitin-specific protease
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Aaberg-Jessen C, Christensen K, Offenberg H, Bartels A, Dreehsen T, Hansen S, Schroder HD, Brunner N, Kristensen BW (2009) Low expression of tissue inhibitor of metalloproteinases-1 (TIMP-1) in glioblastoma predicts longer patient survival. J Neurooncol 95(1):117–128
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 7(4):436–451
Abraham R, Schafer J, Rothe M, Bange J, Knyazev P, Ullrich A (2005) Identification of MMP-15 as an anti-apoptotic factor in cancer cells. J Biol Chem 280(40):34123–34132
Ahluwalia MS, Patton C, Stevens G, Tekautz T, Angelov L, Vogelbaum MA, Weil RJ, Chao S, Elson P, Suh JH, Barnett GH, Peereboom DM (2011) Phase II trial of ritonavir/lopinavir in patients with progressive or recurrent high-grade gliomas. J Neurooncol 102(2):317–321
Almonte AG, Sweatt JD (2011) Serine proteases, serine protease inhibitors, and protease-activated receptors: roles in synaptic function and behavior. Brain Res 1407:107–122
Amberger VR, Paganetti PA, Seulberger H, Eldering JA, Schwab ME (1994) Characterization of a membrane-bound metalloendoprotease of rat C6 glioblastoma cells. Cancer Res 54(15):4017–4025
Amos S, Redpath GT, Dipierro CG, Carpenter JE, Hussaini IM (2010) Epidermal growth factor receptor-mediated regulation of urokinase plasminogen activator expression and glioblastoma invasion via C-SRC/MAPK/AP-1 signaling pathways. J Neuropathol Exp Neurol 69(6):582–592
Anand M, Van Meter TE, Fillmore HL (2011) Epidermal growth factor induces matrix metalloproteinase-1 (MMP-1) expression and invasion in glioma cell lines via the MAPK pathway. J Neurooncol 104(3):679–687
Anand R, Langer T, Baker MJ (2013) Proteolytic control of mitochondrial function and morphogenesis. Biochim Biophys Acta 1833(1):195–204
Annabi B, Laflamme C, Sina A, Lachambre MP, Beliveau R (2009) A MT1-MMP/NF-kappaB signaling axis as a checkpoint controller of COX-2 expression in CD133+ U87 glioblastoma cells. J Neuroinflammation 6:8
Aplan PD (2006) Causes of oncogenic chromosomal translocation. Trends Genet 22(1):46–55
Arai Y, Kubota T, Nakagawa T, Kabuto M, Sato K, Kobayashi H (1998) Production of urokinase-type plasminogen activator (u-PA) and plasminogen activator inhibitor-1 (PAI-1) in human brain tumours. Acta Neurochir 140(4):377–386
Ariza A, Fernandez LA, Inagami T, Kim JH, Manuelidis EE (1988) Renin in glioblastoma multiforme and its role in neovascularization. Am J Clin Pathol 90(4):437–441
Ashley DM, Riffkin CD, Muscat AM, Knight MJ, Kaye AH, Novak U, Hawkins CJ (2005) Caspase 8 is absent or low in many ex vivo gliomas. Cancer 104(7):1487–1496
Asklund T, Kvarnbrink S, Holmlund C, Wibom C, Bergenheim T, Henriksson R, Hedman H (2012) Synergistic killing of glioblastoma stem-like cells by bortezomib and HDAC inhibitors. Anticancer Res 32(7):2407–2413
Asuthkar S, Velpula KK, Chetty C, Gorantla B, Rao JS (2012) Epigenetic regulation of miRNA-211 by MMP-9 governs glioma cell apoptosis, chemosensitivity and radiosensitivity. Oncotarget 3(11):1439–1454
Atkinson JM, Siller CS, Gill JH (2008) Tumour endoproteases: the cutting edge of cancer drug delivery? Br J Pharmacol 153(7):1344–1352
Backes DM, Siddiq A, Cox DG, Calboli FC, Gaziano JM, Ma J, Stampfer M, Hunter DJ, Camargo CA, Michaud DS (2013) Single-nucleotide polymorphisms of allergy-related genes and risk of adult glioma. J Neurooncol 113(2):229–238
Badiga AV, Chetty C, Kesanakurti D, Are D, Gujrati M, Klopfenstein JD, Dinh DH, Rao JS (2011) MMP-2 siRNA inhibits radiation-enhanced invasiveness in glioma cells. PLoS One 6(6):e20614
Baker AH, George SJ, Zaltsman AB, Murphy G, Newby AC (1999) Inhibition of invasion and induction of apoptotic cell death of cancer cell lines by overexpression of TIMP-3. Br J Cancer 79(9–10):1347–1355
Balyasnikova IV, Ferguson SD, Han Y, Liu F, Lesniak MS (2011) Therapeutic effect of neural stem cells expressing TRAIL and bortezomib in mice with glioma xenografts. Cancer Lett 310(2):148–159
Barcia C Jr, Gomez A, Gallego-Sanchez JM, Perez-Valles A, Castro MG, Lowenstein PR, Barcia C Sr, Herrero MT (2009) Infiltrating CTLs in human glioblastoma establish immunological synapses with tumorigenic cells. Am J Pathol 175(2):786–798
Barrow J, Adamowicz-Brice M, Cartmill M, MacArthur D, Lowe J, Robson K, Brundler MA, Walker DA, Coyle B, Grundy R (2011) Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol 13(2):212–222
Beffert U, Durudas A, Weeber EJ, Stolt PC, Giehl KM, Sweatt JD, Hammer RE, Herz J (2006) Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J Neurosci 26(7):2041–2052
Belien AT, Paganetti PA, Schwab ME (1999) Membrane-type 1 matrix metalloprotease (MT1-MMP) enables invasive migration of glioma cells in central nervous system white matter. J Cell Biol 144(2):373–384
Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG (2004) Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol 36(6):1046–1069
Bellail AC, Tse MC, Song JH, Phuphanich S, Olson JJ, Sun SY, Hao C (2010) DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J Cell Mol Med 14(6A):1303–1317
Bello L, Lucini V, Carrabba G, Giussani C, Machluf M, Pluderi M, Nikas D, Zhang J, Tomei G, Villani RM, Carroll RS, Bikfalvi A, Black PM (2001) Simultaneous inhibition of glioma angiogenesis, cell proliferation, and invasion by a naturally occurring fragment of human metalloproteinase-2. Cancer Res 61(24):8730–8736
Bergbold N, Lemberg MK (2013) Emerging role of rhomboid family proteins in mammalian biology and disease. Biochim Biophys Acta 1828(12):2840–2848
Berger Y, Dehmlow H, Blum-Kaelin D, Kitas EA, Loffler BM, Aebi JD, Juillerat-Jeanneret L (2005) Endothelin-converting enzyme-1 inhibition and growth of human glioblastoma cells. J Med Chem 48(2):483–498
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2(10):737–744
Bernstein H-G (2005) Proteases and Alzheimer’s disease: present knowledge and emerging concepts of therapy. In: Lendeckel U, Hooper N (eds) Proteases in the brain, vol 3. Springer, New York, NY, pp 1–23
Best JL, Ganiatsas S, Agarwal S, Changou A, Salomoni P, Shirihai O, Meluh PB, Pandolfi PP, Zon LI (2002) SUMO-1 protease-1 regulates gene transcription through PML. Mol Cell 10(4):843–855
Bethke L, Sullivan K, Webb E, Murray A, Schoemaker M, Auvinen A, Kiuru A, Salminen T, Johansen C, Christensen HC, Muir K, McKinney P, Hepworth S, Dimitropoulou P, Lophatananon A, Feychting M, Lonn S, Ahlbom A, Malmer B, Henriksson R, Swerdlow A, Houlston R (2008) The common D302H variant of CASP8 is associated with risk of glioma. Cancer Epidemiol Biomarkers Prev 17(4):987–989
Bhoopathi P, Chetty C, Gujrati M, Dinh DH, Rao JS, Lakka SS (2010) The role of MMP-9 in the anti-angiogenic effect of secreted protein acidic and rich in cysteine. Br J Cancer 102(3):530–540
Bindal AK, Hammoud M, Shi WM, Wu SZ, Sawaya R, Rao JS (1994) Prognostic significance of proteolytic enzymes in human brain tumors. J Neurooncol 22(2):101–110
Binder DK, Berger MS (2002) Proteases and the biology of glioma invasion. J Neurooncol 56(2):149–158
Black PM, Moriarty T, Alexander E III, Stieg P, Woodard EJ, Gleason PL, Martin CH, Kikinis R, Schwartz RB, Jolesz FA (1997) Development and implementation of intraoperative magnetic resonance imaging and its neurosurgical applications. Neurosurgery 41(4):831–842, discussion 842–835
Blazquez C, Carracedo A, Salazar M, Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G, Guzman M (2008a) Down-regulation of tissue inhibitor of metalloproteinases-1 in gliomas: a new marker of cannabinoid antitumoral activity? Neuropharmacology 54(1):235–243
Blazquez C, Salazar M, Carracedo A, Lorente M, Egia A, Gonzalez-Feria L, Haro A, Velasco G, Guzman M (2008b) Cannabinoids inhibit glioma cell invasion by down-regulating matrix metalloproteinase-2 expression. Cancer Res 68(6):1945–1952
Bodey B, Bodey B Jr, Siegel SE, Kaiser HE (2000) Matrix metalloproteinase expression in childhood astrocytomas. Anticancer Res 20(5A):3287–3292
Bodey B, Bodey V, Siegel SE, Nasir A, Coppola D, Hakam A, Kaiser HE (2004) Immunocytochemical detection of members of the caspase cascade of apoptosis in high-grade astrocytomas. In Vivo 18(5):593–602
Bramwell B (1888) Intracranial tumours. Pentland, Edinburgh
Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG (2004) Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res 64(3):920–927
Bredel M, Scholtens DM, Harsh GR, Bredel C, Chandler JP, Renfrow JJ, Yadav AK, Vogel H, Scheck AC, Tibshirani R, Sikic BI (2009) A network model of a cooperative genetic landscape in brain tumors. JAMA 302(3):261–275
Brisson L, Reshkin SJ, Gore J, Roger S (2012) pH regulators in invadosomal functioning: proton delivery for matrix tasting. Eur J Cell Biol 91(11–12):847–860
Brooks PC, Stromblad S, Sanders LC, von Schalscha TL, Aimes RT, Stetler-Stevenson WG, Quigley JP, Cheresh DA (1996) Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85(5):683–693
Bryan L, Paugh BS, Kapitonov D, Wilczynska KM, Alvarez SM, Singh SK, Milstien S, Spiegel S, Kordula T (2008) Sphingosine-1-phosphate and interleukin-1 independently regulate plasminogen activator inhibitor-1 and urokinase-type plasminogen activator receptor expression in glioblastoma cells: implications for invasiveness. Mol Cancer Res 6(9):1469–1477
Bulstrode H, Jones LM, Siney EJ, Sampson JM, Ludwig A, Gray WP, Willaime-Morawek S (2012) A-Disintegrin and Metalloprotease (ADAM) 10 and 17 promote self-renewal of brain tumor sphere forming cells. Cancer Lett 326(1):79–87
Bulteau AL, Bayot A (2011) Mitochondrial proteases and cancer. Biochim Biophys Acta 1807(6):595–601
Busek P, Stremenova J, Sromova L, Hilser M, Balaziova E, Kosek D, Trylcova J, Strnad H, Krepela E, Sedo A (2012) Dipeptidyl peptidase-IV inhibits glioma cell growth independent of its enzymatic activity. Int J Biochem Cell Biol 44(5):738–747
Caccamo DV, Keohane ME, McKeever PE (1994) Plasminogen activators and inhibitors in gliomas: an immunohistochemical study. Mod Pathol 7(1):99–104
Campbell RL, Davies PL (2012) Structure-function relationships in calpains. Biochem J 447(3):335–351
Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068
Capaccione KM, Pine SR (2013) The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 34(7):1420–1430
Capper D, Gaiser T, Hartmann C, Habel A, Mueller W, Herold-Mende C, von Deimling A, Siegelin MD (2009) Stem-cell-like glioma cells are resistant to TRAIL/Apo2L and exhibit down-regulation of caspase-8 by promoter methylation. Acta Neuropathol 117(4):445–456
Caroni P, Schwab ME (1988) Two membrane protein fractions from rat central myelin with inhibitory properties for neurite growth and fibroblast spreading. J Cell Biol 106(4):1281–1288
Carragher NO, Frame MC (2002) Calpain: a role in cell transformation and migration. Int J Biochem Cell Biol 34(12):1539–1543
Caterina JJ, Yamada S, Caterina NC, Longenecker G, Holmback K, Shi J, Yermovsky AE, Engler JA, Birkedal-Hansen H (2000) Inactivating mutation of the mouse tissue inhibitor of metalloproteinases-2(Timp-2) gene alters proMMP-2 activation. J Biol Chem 275(34):26416–26422
Ceruti S, Mazzola A, Abbracchio MP (2006) Proteasome inhibitors potentiate etoposide-induced cell death in human astrocytoma cells bearing a mutated p53 isoform. J Pharmacol Exp Ther 319(3):1424–1434
Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T (2003) Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 253(1–2):269–285
Chandrasekar N, Mohanam S, Gujrati M, Olivero WC, Dinh DH, Rao JS (2003) Downregulation of uPA inhibits migration and PI3k/Akt signaling in glioblastoma cells. Oncogene 22(3):392–400
Chang SS, Reuter VE, Heston WD, Bander NH, Grauer LS, Gaudin PB (1999) Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res 59(13):3192–3198
Chapman HA (2004) Cathepsins as transcriptional activators? Dev Cell 6(5):610–611
Chen J, Kesari S, Rooney C, Strack PR, Chen J, Shen H, Wu L, Griffin JD (2010) Inhibition of notch signaling blocks growth of glioblastoma cell lines and tumor neurospheres. Genes Cancer 1(8):822–835
Chen X, Chen L, Chen J, Hu W, Gao H, Xie B, Wang X, Yin Z, Li S, Wang X (2013a) ADAM17 promotes U87 glioblastoma stem cell migration and invasion. Brain Res 1538:151–158
Chen X, Chen L, Zhang R, Yi Y, Ma Y, Yan K, Jiang X, Wang X (2013b) ADAM17 regulates self-renewal and differentiation of U87 glioblastoma stem cells. Neurosci Lett 537:44–49
Chetty C, Lakka SS, Bhoopathi P, Gondi CS, Veeravalli KK, Fassett D, Klopfenstein JD, Dinh DH, Gujrati M, Rao JS (2010) Urokinase plasminogen activator receptor and/or matrix metalloproteinase-9 inhibition induces apoptosis signaling through lipid rafts in glioblastoma xenograft cells. Mol Cancer Ther 9(9):2605–2617
Chetty C, Vanamala SK, Gondi CS, Dinh DH, Gujrati M, Rao JS (2012) MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell Signal 24(2):549–559
Chintala SK, Tonn JC, Rao JS (1999) Matrix metalloproteinases and their biological function in human gliomas. Int J Dev Neurosci 17(5–6):495–502
Choe G, Park JK, Jouben-Steele L, Kremen TJ, Liau LM, Vinters HV, Cloughesy TF, Mischel PS (2002) Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin Cancer Res 8(9):2894–2901
Choi GC, Li J, Wang Y, Li L, Zhong L, Ma B, Su X, Ying J, Xiang T, Rha SY, Yu J, Sung JJ, Tsao SW, Chan AT, Tao Q (2014) The metalloprotease ADAMTS8 displays antitumor properties through antagonizing EGFR-MEK-ERK signaling and is silenced in carcinomas by CpG methylation. Mol Cancer Res 12(2):228–238
Chu Q, Orr BA, Semenkow S, Bar EE, Eberhart CG (2013) Prolonged inhibition of glioblastoma xenograft initiation and clonogenic growth following in vivo Notch blockade. Clin Cancer Res 19(12):3224–3233
Clague MJ, Coulson JM, Urbe S (2012) Cellular functions of the DUBs. J Cell Sci 125(Pt 2):277–286
Clark EB, Jovov B, Rooj AK, Fuller CM, Benos DJ (2010) Proteolytic cleavage of human acid-sensing ion channel 1 by the serine protease matriptase. J Biol Chem 285(35):27130–27143
Colbert JD, Matthews SP, Miller G, Watts C (2009) Diverse regulatory roles for lysosomal proteases in the immune response. Eur J Immunol 39(11):2955–2965
Colin C, Voutsinos-Porche B, Nanni I, Fina F, Metellus P, Intagliata D, Baeza N, Bouvier C, Delfino C, Loundou A, Chinot O, Lah T, Kos J, Martin PM, Ouafik L, Figarella-Branger D (2009) High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta Neuropathol 118(6):745–754
Comincini S, Paolillo M, Barbieri G, Palumbo S, Sbalchiero E, Azzalin A, Russo MA, Schinelli S (2009) Gene expression analysis of an EGFR indirectly related pathway identified PTEN and MMP9 as reliable diagnostic markers for human glial tumor specimens. J Biomed Biotechnol 2009:924565
Conway RE, Petrovic N, Li Z, Heston W, Wu D, Shapiro LH (2006) Prostate-specific membrane antigen regulates angiogenesis by modulating integrin signal transduction. Mol Cell Biol 26(14):5310–5324
Cork SM, Kaur B, Devi NS, Cooper L, Saltz JH, Sandberg EM, Kaluz S, Van Meir EG (2012) A proprotein convertase/MMP-14 proteolytic cascade releases a novel 40 kDa vasculostatin from tumor suppressor BAI1. Oncogene 31(50):5144–5152
Coussens LM, Fingleton B, Matrisian LM (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295(5564):2387–2392
Cox JL (2009) Cystatins and cancer. Front Biosci (Landmark Ed) 14:463–474
Cox JL, Wilder PJ, Gilmore JM, Wuebben EL, Washburn MP, Rizzino A (2013) The SOX2-interactome in brain cancer cells identifies the requirement of MSI2 and USP9X for the growth of brain tumor cells. PLoS One 8(5):e62857
Crocker S, Smith P, Park D (2005) Calpain proteolysis and the etiology of Parkinson’s disease: an emerging hypothesis. In: Lendeckel U, Hooper N (eds) Proteases in the brain, vol 3. Springer, New York, NY, pp 25–61
Crocker M, Ashley S, Giddings I, Petrik V, Hardcastle A, Aherne W, Pearson A, Bell BA, Zacharoulis S, Papadopoulos MC (2011) Serum angiogenic profile of patients with glioblastoma identifies distinct tumor subtypes and shows that TIMP-1 is a prognostic factor. Neuro Oncol 13(1):99–108
Cuevas BD, Abell AN, Witowsky JA, Yujiri T, Johnson NL, Kesavan K, Ware M, Jones PL, Weed SA, DeBiasi RL, Oka Y, Tyler KL, Johnson GL (2003) MEKK1 regulates calpain-dependent proteolysis of focal adhesion proteins for rear-end detachment of migrating fibroblasts. EMBO J 22(13):3346–3355
Cutter JL, Cohen NT, Wang J, Sloan AE, Cohen AR, Panneerselvam A, Schluchter M, Blum G, Bogyo M, Basilion JP (2012) Topical application of activity-based probes for visualization of brain tumor tissue. PLoS One 7(3):e33060
D’Abaco GM, Ng K, Paradiso L, Godde NJ, Kaye A, Novak U (2006) ADAM22, expressed in normal brain but not in high-grade gliomas, inhibits cellular proliferation via the disintegrin domain. Neurosurgery 58(1):179–186, discussion 179-186
D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S (2011) Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 17(12):1636–1640
Das G, Shravage BV, Baehrecke EH (2012) Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 4(6). doi:10.1101/cshperspect.a008813
Dauth S, Sirbulescu RF, Jordans S, Rehders M, Avena L, Oswald J, Lerchl A, Saftig P, Brix K (2011) Cathepsin K deficiency in mice induces structural and metabolic changes in the central nervous system that are associated with learning and memory deficits. BMC Neurosci 12:74
de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 12(3):233–242
Del Rosso M, Fibbi G, Schmitt M (2002) Non-enzymatic activities of proteases: from scepticism to reality. Biol Chem 383(1):1–4
Delic S, Lottmann N, Jetschke K, Reifenberger G, Riemenschneider MJ (2012) Identification and functional validation of CDH11, PCSK6 and SH3GL3 as novel glioma invasion-associated candidate genes. Neuropathol Appl Neurobiol 38(2):201–212
Demchik LL, Sameni M, Nelson K, Mikkelsen T, Sloane BF (1999) Cathepsin B and glioma invasion. Int J Dev Neurosci 17(5–6):483–494
Deng Y, Li W, Li Y, Yang H, Xu H, Liang S, Zhang L, Li Y (2010) Expression of Matrix Metalloproteinase-26 promotes human glioma U251 cell invasion in vitro and in vivo. Oncol Rep 23(1):69–78
Deryugina EI, Soroceanu L, Strongin AY (2002) Up-regulation of vascular endothelial growth factor by membrane-type 1 matrix metalloproteinase stimulates human glioma xenograft growth and angiogenesis. Cancer Res 62(2):580–588
Donepudi M, Grutter MG (2002) Structure and zymogen activation of caspases. Biophys Chem 101–102:145–153
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, C. Alzheimer’s Disease Cooperative Study Steering, Siemers E, Sethuraman G, Mohs R, G. Semagacestat Study (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369(4):341–350
Drucker KL, Paulsen AR, Giannini C, Decker PA, Blaber SI, Blaber M, Uhm JH, O’Neill BP, Jenkins RB, Scarisbrick IA (2013) Clinical significance and novel mechanism of action of kallikrein 6 in glioblastoma. Neuro Oncol 15(3):305–318
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, Vandenberg S, Johnson RS, Werb Z, Bergers G (2008) HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 13(3):206–220
Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J (2010) Role of matrix metalloproteinase-9 dimers in cell migration: design of inhibitory peptides. J Biol Chem 285(46):35944–35956
Dunn JR, Reed JE, du Plessis DG, Shaw EJ, Reeves P, Gee AL, Warnke P, Walker C (2006) Expression of ADAMTS-8, a secreted protease with antiangiogenic properties, is downregulated in brain tumours. Br J Cancer 94(8):1186–1193
Dutra-Oliveira A, Monteiro RQ, Mariano-Oliveira A (2012) Protease-activated receptor-2 (PAR2) mediates VEGF production through the ERK1/2 pathway in human glioblastoma cell lines. Biochem Biophys Res Commun 421(2):221–227
Dutzmann S, Gessler F, Harter PN, Gerlach R, Mittelbronn M, Seifert V, Kogel D (2010) The pro-migratory and pro-invasive role of the procoagulant tissue factor in malignant gliomas. Cell Adh Migr 4(4):515–522
Dziembowska M, Wlodarczyk J (2012) MMP9: a novel function in synaptic plasticity. Int J Biochem Cell Biol 44(5):709–713
Dziembowska M, Danilkiewicz M, Wesolowska A, Zupanska A, Chouaib S, Kaminska B (2007) Cross-talk between Smad and p38 MAPK signalling in transforming growth factor beta signal transduction in human glioblastoma cells. Biochem Biophys Res Commun 354(4):1101–1106
Eden G, Archinti M, Furlan F, Murphy R, Degryse B (2011) The urokinase receptor interactome. Curr Pharm Des 17(19):1874–1889
Egidy G, Eberl LP, Valdenaire O, Irmler M, Majdi R, Diserens AC, Fontana A, Janzer RC, Pinet F, Juillerat-Jeanneret L (2000) The endothelin system in human glioblastoma. Lab Invest 80(11):1681–1689
Eichhorn PJ, Rodon L, Gonzalez-Junca A, Dirac A, Gili M, Martinez-Saez E, Aura C, Barba I, Peg V, Prat A, Cuartas I, Jimenez J, Garcia-Dorado D, Sahuquillo J, Bernards R, Baselga J, Seoane J (2012) USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat Med 18(3):429–435
Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, Weller M, Friese MA (2006) TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 129(Pt 9):2416–2425
Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391(6662):43–50
Endo S, Yokosawa H, Ishii S (1989) Involvement of endopeptidase-24.11 in degradation of substance P by glioma cells. Neuropeptides 14(3):177–184
Esteve PO, Tremblay P, Houde M, St-Pierre Y, Mandeville R (1998) In vitro expression of MMP-2 and MMP-9 in glioma cells following exposure to inflammatory mediators. Biochim Biophys Acta 1403(1):85–96
Esteve PO, Chicoine E, Robledo O, Aoudjit F, Descoteaux A, Potworowski EF, St-Pierre Y (2002) Protein kinase C-zeta regulates transcription of the matrix metalloproteinase-9 gene induced by IL-1 and TNF-alpha in glioma cells via NF-kappa B. J Biol Chem 277(38):35150–35155
Fager N, Jaworski DM (2000) Differential spatial distribution and temporal regulation of tissue inhibitor of metalloproteinase mRNA expression during rat central nervous system development. Mech Dev 98(1–2):105–109
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL, Eberhart CG (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28(1):5–16
Felbor U, Kessler B, Mothes W, Goebel HH, Ploegh HL, Bronson RT, Olsen BR (2002) Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A 99(12):7883–7888
Fillmore HL, VanMeter TE, Broaddus WC (2001) Membrane-type matrix metalloproteinases (MT-MMPs): expression and function during glioma invasion. J Neurooncol 53(2):187–202
Fischer A (1946) Mechanism of the proteolytic activity of malignant tissue cells. Nature 157:442
Fischer U, Leidinger P, Keller A, Folarin A, Ketter R, Graf N, Lenhof HP, Meese E (2010) Amplicons on chromosome 12q13-21 in glioblastoma recurrences. Int J Cancer 126(11):2594–2602
Flannery T, Gibson D, Mirakhur M, McQuaid S, Greenan C, Trimble A, Walker B, McCormick D, Johnston PG (2003) The clinical significance of cathepsin S expression in human astrocytomas. Am J Pathol 163(1):175–182
Flannery T, McQuaid S, McGoohan C, McConnell RS, McGregor G, Mirakhur M, Hamilton P, Diamond J, Cran G, Walker B, Scott C, Martin L, Ellison D, Patel C, Nicholson C, Mendelow D, McCormick D, Johnston PG (2006) Cathepsin S expression: an independent prognostic factor in glioblastoma tumours–a pilot study. Int J Cancer 119(4):854–860
Floyd DH, Kefas B, Seleverstov O, Mykhaylyk O, Dominguez C, Comeau L, Plank C, Purow B (2012) Alpha-secretase inhibition reduces human glioblastoma stem cell growth in vitro and in vivo by inhibiting Notch. Neuro Oncol 14(10):1215–1226
Formolo CA, Williams R, Gordish-Dressman H, MacDonald TJ, Lee NH, Hathout Y (2011) Secretome signature of invasive glioblastoma multiforme. J Proteome Res 10(7):3149–3159
Forsyth PA, Wong H, Laing TD, Rewcastle NB, Morris DG, Muzik H, Leco KJ, Johnston RN, Brasher PM, Sutherland G, Edwards DR (1999) Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br J Cancer 79(11–12):1828–1835
Forsyth PA, Krishna N, Lawn S, Valadez JG, Qu X, Fenstermacher DA, Fournier M, Potthast L, Chinnaiyan P, Gibney GT, Zeinieh M, Barker PA, Carter BD, Cooper MK, Kenchappa RS (2014) p75 neurotrophin receptor cleavage by alpha- and gamma-secretases is required for neurotrophin mediated proliferation of brain tumor initiating cells. J Biol Chem 289(12):8067–8085
Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar A, Demuth T, Kun LE, Boyett JM, Gilbertson RJ (2011) Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 29(26):3529–3534
Fraile JM, Quesada V, Rodriguez D, Freije JM, Lopez-Otin C (2012) Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 31(19):2373–2388
Franco SJ, Huttenlocher A (2005) Regulating cell migration: calpains make the cut. J Cell Sci 118(Pt 17):3829–3838
Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E, Jaeckle K, Galanis E (2012) Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol 14(2):215–221
Friedberg MH, Glantz MJ, Klempner MS, Cole BF, Perides G (1998) Specific matrix metalloproteinase profiles in the cerebrospinal fluid correlated with the presence of malignant astrocytomas, brain metastases, and carcinomatous meningitis. Cancer 82(5):923–930
Fritchie K, Siintola E, Armao D, Lehesjoki AE, Marino T, Powell C, Tennison M, Booker JM, Koch S, Partanen S, Suzuki K, Tyynela J, Thorne LB (2009) Novel mutation and the first prenatal screening of cathepsin D deficiency (CLN10). Acta Neuropathol 117(2):201–208
Fu Y, Zheng Y, Li K, Huang R, Zheng S, An N, Liang A (2012) Mutations in isocitrate dehydrogenase 2 accelerate glioma cell migration via matrix metalloproteinase-2 and 9. Biotechnol Lett 34(3):441–446
Fujiwara S, Nakagawa K, Harada H, Nagato S, Furukawa K, Teraoka M, Seno T, Oka K, Iwata S, Ohnishi T (2007) Silencing hypoxia-inducible factor-1alpha inhibits cell migration and invasion under hypoxic environment in malignant gliomas. Int J Oncol 30(4):793–802
Fukuda ME, Iwadate Y, Machida T, Hiwasa T, Nimura Y, Nagai Y, Takiguchi M, Tanzawa H, Yamaura A, Seki N (2005) Cathepsin D is a potential serum marker for poor prognosis in glioma patients. Cancer Res 65(12):5190–5194
Fukushima S, Kato S, Maeda M, Shigemori M (2008) Caspase-9 pathway activation by inhibiting endogenous fibroblast growth factor signaling in human glioma cells. Int J Oncol 32(2):467–473
Furukawa K, Kumon Y, Harada H, Kohno S, Nagato S, Teraoka M, Fujiwara S, Nakagawa K, Hamada K, Ohnishi T (2006) PTEN gene transfer suppresses the invasive potential of human malignant gliomas by regulating cell invasion-related molecules. Int J Oncol 29(1):73–81
Furuta T, Nakada M, Misaki K, Sato Y, Hayashi Y, Nakanuma Y, Hamada JI (2014) Molecular analysis of a recurrent glioblastoma treated with bevacizumab. Brain Tumor Pathol 31(1):32–39
Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, Krichevsky AM (2008) MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol 28(17):5369–5380
Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B (2011) Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS One 6(8):e23902
Gallo M, Ho J, Coutinho FJ, Vanner R, Lee L, Head R, Ling EK, Clarke ID, Dirks PB (2013) A tumorigenic MLL-homeobox network in human glioblastoma stem cells. Cancer Res 73(1):417–427
Garnier D, Milsom C, Magnus N, Meehan B, Weitz J, Yu J, Rak J (2010) Role of the tissue factor pathway in the biology of tumor initiating cells. Thromb Res 125(Suppl 2):S44–S50
Gary SC, Kelly GM, Hockfield S (1998) BEHAB/brevican: a brain-specific lectican implicated in gliomas and glial cell motility. Curr Opin Neurobiol 8(5):576–581
Gdynia G, Grund K, Eckert A, Bock BC, Funke B, Macher-Goeppinger S, Sieber S, Herold-Mende C, Wiestler B, Wiestler OD, Roth W (2007) Basal caspase activity promotes migration and invasiveness in glioblastoma cells. Mol Cancer Res 5(12):1232–1240
Geng F, Wenzel S, Tansey WP (2012) Ubiquitin and proteasomes in transcription. Annu Rev Biochem 81:177–201
Gessi S, Sacchetto V, Fogli E, Merighi S, Varani K, Baraldi PG, Tabrizi MA, Leung E, Maclennan S, Borea PA (2010) Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem Pharmacol 79(10):1483–1495
Gessler F, Voss V, Dutzmann S, Seifert V, Gerlach R, Kogel D (2010) Inhibition of tissue factor/protease-activated receptor-2 signaling limits proliferation, migration and invasion of malignant glioma cells. Neuroscience 165(4):1312–1322
Gessler F, Voss V, Seifert V, Gerlach R, Kogel D (2011) Knockdown of TFPI-2 promotes migration and invasion of glioma cells. Neurosci Lett 497(1):49–54
Gilbert CA, Daou MC, Moser RP, Ross AH (2010) Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res 70(17):6870–6879
Gingras D, Bousquet-Gagnon N, Langlois S, Lachambre MP, Annabi B, Beliveau R (2001) Activation of the extracellular signal-regulated protein kinase (ERK) cascade by membrane-type-1 matrix metalloproteinase (MT1-MMP). FEBS Lett 507(2):231–236
Girgis AH, Bui A, White NM, Yousef GM (2012) Integrated genomic characterization of the kallikrein gene locus in cancer. Anticancer Res 32(3):957–963
Glading A, Chang P, Lauffenburger DA, Wells A (2000) Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem 275(4):2390–2398
Glading A, Bodnar RJ, Reynolds IJ, Shiraha H, Satish L, Potter DA, Blair HC, Wells A (2004) Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol 24(6):2499–2512
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91(3):1071–1121
Goh KY, Poon WS, Chan DT, Ip CP (2005) Tissue plasminogen activator expression in meningiomas and glioblastomas. Clin Neurol Neurosurg 107(4):296–300
Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L (2013) gamma-Secretase inhibitors and modulators. Biochim Biophys Acta 1828(12):2898–2907
Goldschneider D, Mehlen P (2010) Dependence receptors: a new paradigm in cell signaling and cancer therapy. Oncogene 29(13):1865–1882
Gole B, Duran Alonso MB, Dolenc V, Lah T (2009) Post-translational regulation of cathepsin B, but not of other cysteine cathepsins, contributes to increased glioblastoma cell invasiveness in vitro. Pathol Oncol Res 15(4):711–723
Gole B, Huszthy PC, Popovic M, Jeruc J, Ardebili YS, Bjerkvig R, Lah TT (2012) The regulation of cysteine cathepsins and cystatins in human gliomas. Int J Cancer 131(8):1779–1789
Golubkov VS, Strongin AY (2007) Proteolysis-driven oncogenesis. Cell Cycle 6(2):147–150
Gondi CS, Rao JS (2013) Cathepsin B as a cancer target. Expert Opin Ther Targets 17(3):281–291
Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS (2004a) Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biol 1(2):165–176
Gondi CS, Lakka SS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Tung CH, Weissleder R, Rao JS (2004b) Adenovirus-mediated expression of antisense urokinase plasminogen activator receptor and antisense cathepsin B inhibits tumor growth, invasion, and angiogenesis in gliomas. Cancer Res 64(12):4069–4077
Gondi CS, Talluri L, Dinh DH, Gujrati M, Rao JS (2009) RNAi-mediated downregulation of MMP-2 activates the extrinsic apoptotic pathway in human glioma xenograft cells. Int J Oncol 35(4):851–859
Goodman OB Jr, Febbraio M, Simantov R, Zheng R, Shen R, Silverstein RL, Nanus DM (2006) Neprilysin inhibits angiogenesis via proteolysis of fibroblast growth factor-2. J Biol Chem 281(44):33597–33605
Grant CL, Caromile LA, Ho V, Durrani K, Rahman MM, Claffey KP, Fong GH, Shapiro LH (2012) Prostate specific membrane antigen (PSMA) regulates angiogenesis independently of VEGF during ocular neovascularization. PLoS One 7(7):e41285
Groft LL, Muzik H, Rewcastle NB, Johnston RN, Knauper V, Lafleur MA, Forsyth PA, Edwards DR (2001) Differential expression and localization of TIMP-1 and TIMP-4 in human gliomas. Br J Cancer 85(1):55–63
Groves MD, Puduvalli VK, Hess KR, Jaeckle KA, Peterson P, Yung WK, Levin VA (2002) Phase II trial of temozolomide plus the matrix metalloproteinase inhibitor, marimastat, in recurrent and progressive glioblastoma multiforme. J Clin Oncol 20(5):1383–1388
Groves MD, Puduvalli VK, Conrad CA, Gilbert MR, Yung WK, Jaeckle K, Liu V, Hess KR, Aldape KD, Levin VA (2006) Phase II trial of temozolomide plus marimastat for recurrent anaplastic gliomas: a relationship among efficacy, joint toxicity and anticonvulsant status. J Neurooncol 80(1):83–90
Grunda JM, Nabors LB, Palmer CA, Chhieng DC, Steg A, Mikkelsen T, Diasio RB, Zhang K, Allison D, Grizzle WE, Wang W, Gillespie GY, Johnson MR (2006) Increased expression of thymidylate synthetase (TS), ubiquitin specific protease 10 (USP10) and survivin is associated with poor survival in glioblastoma multiforme (GBM). J Neurooncol 80(3):261–274
Guan H, Cai J, Zhang N, Wu J, Yuan J, Li J, Li M (2012) Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int J Cancer 130(3):593–601
Guo P, Imanishi Y, Cackowski FC, Jarzynka MJ, Tao HQ, Nishikawa R, Hirose T, Hu B, Cheng SY (2005) Up-regulation of angiopoietin-2, matrix metalloprotease-2, membrane type 1 metalloprotease, and laminin 5 gamma 2 correlates with the invasiveness of human glioma. Am J Pathol 166(3):877–890
Guttmann RP, Sokol S, Baker DL, Simpkins KL, Dong Y, Lynch DR (2002) Proteolysis of the N-methyl-d-aspartate receptor by calpain in situ. J Pharmacol Exp Ther 302(3):1023–1030
Hagemann C, Anacker J, Ernestus RI, Vince GH (2012) A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J Clin Oncol 3(5):67–79
Hallberg M, Grevès P, Nyberg F (2005) Neuropeptide processing. In: Lendeckel U, Hooper N (eds) Proteases in the brain, vol 3. Springer, New York, NY, pp 203–234
Hamada K, Kuratsu J, Saitoh Y, Takeshima H, Nishi T, Ushio Y (1996) Expression of tissue factor correlates with grade of malignancy in human glioma. Cancer 77(9):1877–1883
Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R (2003) Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 3(6):589–601
Hamasuna R, Kataoka H, Moriyama T, Itoh H, Seiki M, Koono M (1999) Regulation of matrix metalloproteinase-2 (MMP-2) by hepatocyte growth factor/scatter factor (HGF/SF) in human glioma cells: HGF/SF enhances MMP-2 expression and activation accompanying up-regulation of membrane type-1 MMP. Int J Cancer 82(2):274–281
Hamasuna R, Kataoka H, Meng JY, Itoh H, Moriyama T, Wakisaka S, Koono M (2001) Reduced expression of hepatocyte growth factor activator inhibitor type-2/placental bikunin (HAI-2/PB) in human glioblastomas: implication for anti-invasive role of HAI-2/PB in glioblastoma cells. Int J Cancer 93(3):339–345
Hasegawa Y, Kinoh H, Iwadate Y, Onimaru M, Ueda Y, Harada Y, Saito S, Furuya A, Saegusa T, Morodomi Y, Hasegawa M, Saito S, Aoki I, Saeki N, Yonemitsu Y (2010) Urokinase-targeted fusion by oncolytic Sendai virus eradicates orthotopic glioblastomas by pronounced synergy with interferon-beta gene. Mol Ther 18(10):1778–1786
Hattori A, Tsujimoto M (2013) Endoplasmic reticulum aminopeptidases: biochemistry, physiology and pathology. J Biochem 154(3):219–228
Hayakawa Y, Kurimoto M, Nagai S, Kurosaki K, Tsuboi Y, Hamada H, Hayashi N, Endo S (2007) Thrombin-induced cell proliferation and platelet-derived growth factor-AB release from A172 human glioblastoma cells. J Thromb Haemost 5(11):2219–2226
He S, Ding L, Cao Y, Li G, Deng J, Tu Y, Wang B (2012a) Overexpression of a disintegrin and metalloprotease 8 in human gliomas is implicated in tumor progression and prognosis. Med Oncol 29(3):2032–2037
He SM, Zhao ZW, Wang Y, Zhao JP, Wang L, Hou F, Gao GD (2012b) Potential role of Jun activation domain-binding protein 1 and phosphorylated p27 expression in prognosis of glioma. Brain Tumor Pathol 29(1):3–9
Hede SM, Savov V, Weishaupt H, Sangfelt O, Swartling FJ (2013) Oncoprotein stabilization in brain tumors. Oncogene. doi:10.1038/onc.2013.445
Held-Feindt J, Paredes EB, Blomer U, Seidenbecher C, Stark AM, Mehdorn HM, Mentlein R (2006) Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int J Cancer 118(1):55–61
Hetschko H, Voss V, Seifert V, Prehn JH, Kogel D (2008) Upregulation of DR5 by proteasome inhibitors potently sensitizes glioma cells to TRAIL-induced apoptosis. FEBS J 275(8):1925–1936
Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13(12):755–766
Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ (1998) Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell 95(3):365–377
Hishii M, Kurnick JT, Ramirez-Montagut T, Pandolfi F (1999) Studies of the mechanism of cytolysis by tumour-infiltrating lymphocytes. Clin Exp Immunol 116(3):388–394
Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN (2011) Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ 18(5):829–840
Hjortland GO, Bjornland K, Pettersen S, Garman-Vik SS, Emilsen E, Nesland JM, Fodstad O, Engebraaten O (2003) Modulation of glioma cell invasion and motility by adenoviral gene transfer of PAI-1. Clin Exp Metastasis 20(4):301–309
Hjortland GO, Lillehammer T, Somme S, Wang JB, Halvorsen T, Juell S, Hirschberg H, Fodstad O, Engebraaten O (2004) Plasminogen activator inhibitor-1 increases the expression of VEGF in human glioma cells. Exp Cell Res 294(1):130–139
Hooper NM (2002) Proteases: a primer. Essays Biochem 38:1–8
Horing E, Harter PN, Seznec J, Schittenhelm J, Buhring HJ, Bhattacharyya S, von Hattingen E, Zachskorn C, Mittelbronn M, Naumann U (2012) The “go or grow” potential of gliomas is linked to the neuropeptide processing enzyme carboxypeptidase E and mediated by metabolic stress. Acta Neuropathol 124(1):83–97
Hoshi M, Harada A, Kawase T, Uyemura K, Yazaki T (2000) Antitumoral effects of defective herpes simplex virus-mediated transfer of tissue inhibitor of metalloproteinases-2 gene in malignant glioma U87 in vitro: consequences for anti-cancer gene therapy. Cancer Gene Ther 7(5):799–805
Houghton AM, Grisolano JL, Baumann ML, Kobayashi DK, Hautamaki RD, Nehring LC, Cornelius LA, Shapiro SD (2006) Macrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastases. Cancer Res 66(12):6149–6155
Houri N, Huang KC, Nalbantoglu J (2013) The Coxsackievirus and Adenovirus Receptor (CAR) undergoes ectodomain shedding and regulated intramembrane proteolysis (RIP). PLoS One 8(8):e73296
Hsu DW, Efird JT, Hedley-Whyte ET (1995) Prognostic role of urokinase-type plasminogen activator in human gliomas. Am J Pathol 147(1):114–123
Hu B, Kong LL, Matthews RT, Viapiano MS (2008) The proteoglycan brevican binds to fibronectin after proteolytic cleavage and promotes glioma cell motility. J Biol Chem 283(36):24848–24859
Hu YY, Zheng MH, Cheng G, Li L, Liang L, Gao F, Wei YN, Fu LA, Han H (2011) Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC Cancer 11:82
Hua Y, Tang L, Keep RF, Schallert T, Fewel ME, Muraszko KM, Hoff JT, Xi G (2005a) The role of thrombin in gliomas. J Thromb Haemost 3(9):1917–1923
Hua Y, Tang LL, Fewel ME, Keep RF, Schallert T, Muraszko KM, Hoff JT, Xi GH (2005b) Systemic use of argatroban reduces tumor mass, attenuates neurological deficits and prolongs survival time in rat glioma models. Acta Neurochir Suppl 95:403–406
Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY (2011) Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med 17(7):860–866
Huber D, Philipp J, Fontana A (1992) Protease inhibitors interfere with the transforming growth factor-beta-dependent but not the transforming growth factor-beta-independent pathway of tumor cell-mediated immunosuppression. J Immunol 148(1):277–284
Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C, Isacke CM (2010) A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PLoS One 5(3):e9808
Hussain S, Zhang Y, Galardy PJ (2009) DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 8(11):1688–1697
Inoue A, Takahashi H, Harada H, Kohno S, Ohue S, Kobayashi K, Yano H, Tanaka J, Ohnishi T (2010) Cancer stem-like cells of glioblastoma characteristically express MMP-13 and display highly invasive activity. Int J Oncol 37(5):1121–1131
Ishihara H, Kubota H, Lindberg RL, Leppert D, Gloor SM, Errede M, Virgintino D, Fontana A, Yonekawa Y, Frei K (2008) Endothelial cell barrier impairment induced by glioblastomas and transforming growth factor beta2 involves matrix metalloproteinases and tight junction proteins. J Neuropathol Exp Neurol 67(5):435–448
Iwadate Y, Hayama M, Adachi A, Matsutani T, Nagai Y, Hiwasa T, Saeki N (2008) High serum level of plasminogen activator inhibitor-1 predicts histological grade of intracerebral gliomas. Anticancer Res 28(1B):415–418
Iwatsuki K, Kumara E, Yoshimine T, Nakagawa H, Sato M, Hayakawa T (2000) Elastase expression by infiltrating neutrophils in gliomas. Neurol Res 22(5):465–468
Izuhara K, Ohta S, Kanaji S, Shiraishi H, Arima K (2008) Recent progress in understanding the diversity of the human ov-serpin/clade B serpin family. Cell Mol Life Sci 65(16):2541–2553
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307(5706):58–62
Jane EP, Premkumar DR, Pollack IF (2011) Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther 10(1):198–208
Jang HS, Lal S, Greenwood JA (2010) Calpain 2 is required for glioblastoma cell invasion: regulation of matrix metalloproteinase 2. Neurochem Res 35(11):1796–1804
Jansen M, Yip S, Louis DN (2010) Molecular pathology in adult gliomas: diagnostic, prognostic, and predictive markers. Lancet Neurol 9(7):717–726
Jaworski DM, Stradecki HM, Penar PL, Pendlebury WW, Pennington CJ, Edwards DR, Broaddus WC, Fillmore FL (2010) Degradome profiling fails to identify a unique protease signature in primary malignant brain tumors. Neuro Oncol 12(Suppl 4):iv1–iv7
Jenkins G (2008) The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol 40(6–7):1068–1078
Jin X, Jin X, Sohn YW, Yin J, Kim SH, Joshi K, Nam DH, Nakano I, Kim H (2013) Blockade of EGFR signaling promotes glioma stem-like cell invasiveness by abolishing ID3-mediated inhibition of p27(KIP1) and MMP3 expression. Cancer Lett 328(2):235–242
Johnson CE, Huang YY, Parrish AB, Smith MI, Vaughn AE, Zhang Q, Wright KM, Van Dyke T, Wechsler-Reya RJ, Kornbluth S, Deshmukh M (2007) Differential Apaf-1 levels allow cytochrome c to induce apoptosis in brain tumors but not in normal neural tissues. Proc Natl Acad Sci U S A 104(52):20820–20825
Jones TS, Holland EC (2012) Standard of care therapy for malignant glioma and its effect on tumor and stromal cells. Oncogene 31(16):1995–2006
Jost M, Folgueras AR, Frerart F, Pendas AM, Blacher S, Houard X, Berndt S, Munaut C, Cataldo D, Alvarez J, Melen-Lamalle L, Foidart JM, Lopez-Otin C, Noel A (2006) Earlier onset of tumoral angiogenesis in matrix metalloproteinase-19-deficient mice. Cancer Res 66(10):5234–5241
Juillerat-Jeanneret L, Celerier J, Chapuis Bernasconi C, Nguyen G, Wostl W, Maerki HP, Janzer RC, Corvol P, Gasc JM (2004) Renin and angiotensinogen expression and functions in growth and apoptosis of human glioblastoma. Br J Cancer 90(5):1059–1068
Jung CH, Kim EM, Park JK, Hwang SG, Moon SK, Kim WJ, Um HD (2013) Bmal1 suppresses cancer cell invasion by blocking the phosphoinositide 3-kinase-Akt-MMP-2 signaling pathway. Oncol Rep 29(6):2109–2113
Kachra Z, Beaulieu E, Delbecchi L, Mousseau N, Berthelet F, Moumdjian R, Del Maestro R, Beliveau R (1999) Expression of matrix metalloproteinases and their inhibitors in human brain tumors. Clin Exp Metastasis 17(7):555–566
Kahana S, Finniss S, Cazacu S, Xiang C, Lee HK, Brodie S, Goldstein RS, Roitman V, Slavin S, Mikkelsen T, Brodie C (2011) Proteasome inhibitors sensitize glioma cells and glioma stem cells to TRAIL-induced apoptosis by PKCepsilon-dependent downregulation of AKT and XIAP expressions. Cell Signal 23(8):1348–1357
Kamino M, Kishida M, Kibe T, Ikoma K, Iijima M, Hirano H, Tokudome M, Chen L, Koriyama C, Yamada K, Arita K, Kishida S (2011) Wnt-5a signaling is correlated with infiltrative activity in human glioma by inducing cellular migration and MMP-2. Cancer Sci 102(3):540–548
Kaminskyy V, Zhivotovsky B (2012) Proteases in autophagy. Biochim Biophys Acta 1824(1):44–50
Kanakis D, Lendeckel U, Theodosiou P, Dobrowolny H, Mawrin C, Keilhoff G, Bukowska A, Dietzmann K, Bogerts B, Bernstein HG (2013) ADAM 12: a putative marker of oligodendrogliomas? Dis Markers 34(2):81–91
Kang CS, Pu PY, Li YH, Zhang ZY, Qiu MZ, Huang Q, Wang GX (2005) An in vitro study on the suppressive effect of glioma cell growth induced by plasmid-based small interference RNA (siRNA) targeting human epidermal growth factor receptor. J Neurooncol 74(3):267–273
Kardosh A, Golden EB, Pyrko P, Uddin J, Hofman FM, Chen TC, Louie SG, Petasis NA, Schonthal AH (2008) Aggravated endoplasmic reticulum stress as a basis for enhanced glioblastoma cell killing by bortezomib in combination with celecoxib or its non-coxib analogue, 2,5-dimethyl-celecoxib. Cancer Res 68(3):843–851
Kargiotis O, Chetty C, Gondi CS, Tsung AJ, Dinh DH, Gujrati M, Lakka SS, Kyritsis AP, Rao JS (2008) Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene 27(35):4830–4840
Kast RE, Boockvar JA, Bruning A, Cappello F, Chang WW, Cvek B, Dou QP, Duenas-Gonzalez A, Efferth T, Focosi D, Ghaffari SH, Karpel-Massler G, Ketola K, Khoshnevisan A, Keizman D, Magne N, Marosi C, McDonald K, Munoz M, Paranjpe A, Pourgholami MH, Sardi I, Sella A, Srivenugopal KS, Tuccori M, Wang W, Wirtz CR, Halatsch ME (2013) A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 4(4):502–530
Kasza A, Koj A (2002) Cytokines regulate plasminogen activation system in astrocytoma cells. J Physiol Pharmacol 53(1):95–104
Katakowski M, Jiang F, Zheng X, Gutierrez JA, Szalad A, Chopp M (2009) Tumorigenicity of cortical astrocyte cell line induced by the protease ADAM17. Cancer Sci 100(9):1597–1604
Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG (2005) Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol 7(2):134–153
Keerthivasan S, Keerthivasan G, Mittal S, Chauhan SS (2007) Transcriptional upregulation of human cathepsin L by VEGF in glioblastoma cells. Gene 399(2):129–136
Kegel L, Aunin E, Meijer D, Bermingham JR (2013) LGI proteins in the nervous system. ASN Neuro 5(3):167–181
Kenig S, Alonso MB, Mueller MM, Lah TT (2010) Glioblastoma and endothelial cells cross-talk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett 289(1):53–61
Kesanakurti D, Chetty C, Dinh DH, Gujrati M, Rao JS (2013) Role of MMP-2 in the regulation of IL-6/Stat3 survival signaling via interaction with alpha5beta1 integrin in glioma. Oncogene 32(3):327–340
Kessenbrock K, Plaks V, Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141(1):52–67
Kieseier B, Bernal F (2005) Proteases in CNS infection and inflammation. In: Lendeckel U, Hooper N (eds) Proteases in the brain, vol 3. Springer, New York, NY, pp 109–137
Kim S, Choi K, Kwon D, Benveniste EN, Choi C (2004) Ubiquitin-proteasome pathway as a primary defender against TRAIL-mediated cell death. Cell Mol Life Sci 61(9):1075–1081
Kim JH, Choi C, Benveniste EN, Kwon D (2008a) TRAIL induces MMP-9 expression via ERK activation in human astrocytoma cells. Biochem Biophys Res Commun 377(1):195–199
Kim K, Brush JM, Watson PA, Cacalano NA, Iwamoto KS, McBride WH (2008b) Epidermal growth factor receptor vIII expression in U87 glioblastoma cells alters their proteasome composition, function, and response to irradiation. Mol Cancer Res 6(3):426–434
Kim CS, Jung S, Jung TY, Jang WY, Sun HS, Ryu HH (2011) Characterization of invading glioma cells using molecular analysis of leading-edge tissue. J Korean Neurosurg Soc 50(3):157–165
Kim YH, Kwon HJ, Kim DS (2012) Matrix metalloproteinase 9 (MMP-9)-dependent processing of betaig-h3 protein regulates cell migration, invasion, and adhesion. J Biol Chem 287(46):38957–38969
Kinder DH, Berger MS, Mueller BA, Silber JR (1993) Urokinase plasminogen-activator is elevated in human astrocytic gliomas relative to normal adjacent brain. Oncol Res 5(10–11):409–414
Kitagawa H, Tani E, Ikemoto H, Ozaki I, Nakano A, Omura S (1999) Proteasome inhibitors induce mitochondria-independent apoptosis in human glioma cells. FEBS Lett 443(2):181–186
Kitange G, Kishikawa M, Nakayama T, Naito S, Iseki M, Shibata S (1999a) Expression of the Ets-1 proto-oncogene correlates with malignant potential in human astrocytic tumors. Mod Pathol 12(6):618–626
Kitange G, Shibata S, Tokunaga Y, Yagi N, Yasunaga A, Kishikawa M, Naito S (1999b) Ets-1 transcription factor-mediated urokinase-type plasminogen activator expression and invasion in glioma cells stimulated by serum and basic fibroblast growth factors. Lab Invest 79(4):407–416
Klein T, Bischoff R (2011) Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. J Proteome Res 10(1):17–33
Knoblach S, Faden A (2005) Proteases in traumatic brain injury. In: Lendeckel U, Hooper N (eds) Proteases in the brain, vol 3. Springer, New York, NY, pp 79–108
Kobayashi T, Masumoto J, Tada T, Nomiyama T, Hongo K, Nakayama J (2007) Prognostic significance of the immunohistochemical staining of cleaved caspase-3, an activated form of caspase-3, in gliomas. Clin Cancer Res 13(13):3868–3874
Kodama T, Ikeda E, Okada A, Ohtsuka T, Shimoda M, Shiomi T, Yoshida K, Nakada M, Ohuchi E, Okada Y (2004) ADAM12 is selectively overexpressed in human glioblastomas and is associated with glioblastoma cell proliferation and shedding of heparin-binding epidermal growth factor. Am J Pathol 165(5):1743–1753
Kohutek ZA, diPierro CG, Redpath GT, Hussaini IM (2009) ADAM-10-mediated N-cadherin cleavage is protein kinase C-alpha dependent and promotes glioblastoma cell migration. J Neurosci 29(14):4605–4615
Komatsu K, Nakanishi Y, Nemoto N, Hori T, Sawada T, Kobayashi M (2004) Expression and quantitative analysis of matrix metalloproteinase-2 and -9 in human gliomas. Brain Tumor Pathol 21(3):105–112
Kondraganti S, Mohanam S, Chintala SK, Kin Y, Jasti SL, Nirmala C, Lakka SS, Adachi Y, Kyritsis AP, Ali-Osman F, Sawaya R, Fuller GN, Rao JS (2000) Selective suppression of matrix metalloproteinase-9 in human glioblastoma cells by antisense gene transfer impairs glioblastoma cell invasion. Cancer Res 60(24):6851–6855
Konduri S, Lakka SS, Tasiou A, Yanamandra N, Gondi CS, Dinh DH, Olivero WC, Gujrati M, Rao JS (2001) Elevated levels of cathepsin B in human glioblastoma cell lines. Int J Oncol 19(3):519–524
Konduri SD, Yanamandra N, Siddique K, Joseph A, Dinh DH, Olivero WC, Gujrati M, Kouraklis G, Swaroop A, Kyritsis AP, Rao JS (2002) Modulation of cystatin C expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene 21(57):8705–8712
Konduri SD, Srivenugopal KS, Yanamandra N, Dinh DH, Olivero WC, Gujrati M, Foster DC, Kisiel W, Ali-Osman F, Kondraganti S, Lakka SS, Rao JS (2003) Promoter methylation and silencing of the tissue factor pathway inhibitor-2 (TFPI-2), a gene encoding an inhibitor of matrix metalloproteinases in human glioma cells. Oncogene 22(29):4509–4516
Konkoy CS, Davis TP (1996) Ectoenzymes as sites of peptide regulation. Trends Pharmacol Sci 17(8):288–294
Kono S, Rao JS, Bruner JM, Sawaya R (1994) Immunohistochemical localization of plasminogen-activator inhibitor type-1 in human brain-tumors. J Neuropathol Exp Neurol 53(3):256–262
Koschny R, Holland H, Sykora J, Haas TL, Sprick MR, Ganten TM, Krupp W, Bauer M, Ahnert P, Meixensberger J, Walczak H (2007) Bortezomib sensitizes primary human astrocytoma cells of WHO grades I to IV for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Clin Cancer Res 13(11):3403–3412
Koul D, Parthasarathy R, Shen R, Davies MA, Jasser SA, Chintala SK, Rao JS, Sun Y, Benvenisite EN, Liu TJ, Yung WK (2001) Suppression of matrix metalloproteinase-2 gene expression and invasion in human glioma cells by MMAC/PTEN. Oncogene 20(46):6669–6678
Kristoffersen K, Villingshoj M, Poulsen HS, Stockhausen MT (2013) Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biol Ther 14(7):625–637
Krona A, Aman P, Orndal C, Josefsson A (2007) Oncostatin M-induced genes in human astrocytomas. Int J Oncol 31(6):1457–1463
Krzewski K, Coligan JE (2012) Human NK cell lytic granules and regulation of their exocytosis. Front Immunol 3:335
Kubiatowski T, Jang T, Lachyankar MB, Salmonsen R, Nabi RR, Quesenberry PJ, Litofsky NS, Ross AH, Recht LD (2001) Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. J Neurosurg 95(3):480–488
Kubicek GJ, Werner-Wasik M, Machtay M, Mallon G, Myers T, Ramirez M, Andrews D, Curran WJ Jr, Dicker AP (2009) Phase I trial using proteasome inhibitor bortezomib and concurrent temozolomide and radiotherapy for central nervous system malignancies. Int J Radiat Oncol Biol Phys 74(2):433–439
Kulathu Y, Garcia FJ, Mevissen TE, Busch M, Arnaudo N, Carroll KS, Barford D, Komander D (2013) Regulation of A20 and other OTU deubiquitinases by reversible oxidation. Nat Commun 4:1569
Kunishio K, Okada M, Matsumoto Y, Nagao S (2003) Matrix metalloproteinase-2 and -9 expression in astrocytic tumors. Brain Tumor Pathol 20(2):39–45
Kwiatkowska A, Kijewska M, Lipko M, Hibner U, Kaminska B (2011) Downregulation of Akt and FAK phosphorylation reduces invasion of glioblastoma cells by impairment of MT1-MMP shuttling to lamellipodia and downregulates MMPs expression. Biochim Biophys Acta 1813(5):655–667
La Ferla-Bruhl K, Westhoff MA, Karl S, Kasperczyk H, Zwacka RM, Debatin KM, Fulda S (2007) NF-kappaB-independent sensitization of glioblastoma cells for TRAIL-induced apoptosis by proteasome inhibition. Oncogene 26(4):571–582
Labussiere M, Pinel S, Delfortrie S, Plenat F, Chastagner P (2008) Proteasome inhibition by bortezomib does not translate into efficacy on two malignant glioma xenografts. Oncol Rep 20(5):1283–1287
Lagadec C, Vlashi E, Frohnen P, Alhiyari Y, Chan M, Pajonk F (2014) The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem Cells 32(1):135–144
Lakka SS, Jasti SL, Kyritsis AP, Yung WK, Ali-Osman F, Nicolson GL, Rao JS (2000) Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by the extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clin Exp Metastasis 18(3):245–252
Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, Rao JS (2004) Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 23(27):4681–4689
Lakka SS, Gondi CS, Rao JS (2005) Proteases and glioma angiogenesis. Brain Pathol 15(4):327–341
Lal A, Glazer CA, Martinson HM, Friedman HS, Archer GE, Sampson JH, Riggins GJ (2002) Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res 62(12):3335–3339
Lal S, La Du J, Tanguay RL, Greenwood JA (2012) Calpain 2 is required for the invasion of glioblastoma cells in the zebrafish brain microenvironment. J Neurosci Res 90(4):769–781
Lamfers ML, Gianni D, Tung CH, Idema S, Schagen FH, Carette JE, Quax PH, Van Beusechem VW, Vandertop WP, Dirven CM, Chiocca EA, Gerritsen WR (2005) Tissue inhibitor of metalloproteinase-3 expression from an oncolytic adenovirus inhibits matrix metalloproteinase activity in vivo without affecting antitumor efficacy in malignant glioma. Cancer Res 65(20):9398–9405
Lampert K, Machein U, Machein MR, Conca W, Peter HH, Volk B (1998) Expression of matrix metalloproteinases and their tissue inhibitors in human brain tumors. Am J Pathol 153(2):429–437
Landau BJ, Kwaan HC, Verrusio EN, Brem SS (1994) Elevated levels of urokinase-type plasminogen activator and plasminogen activator inhibitor type-1 in malignant human brain tumors. Cancer Res 54(4):1105–1108
Langlois S, Nyalendo C, Di Tomasso G, Labrecque L, Roghi C, Murphy G, Gingras D, Beliveau R (2007) Membrane-type 1 matrix metalloproteinase stimulates cell migration through epidermal growth factor receptor transactivation. Mol Cancer Res 5(6):569–583
Larson DA, Prados M, Lamborn KR, Smith V, Sneed PK, Chang S, Nicholas KM, Wara WM, Devriendt D, Kunwar S, Berger M, McDermott MW (2002) Phase II study of high central dose gamma knife radiosurgery and marimastat in patients with recurrent malignant glioma. Int J Radiat Oncol Biol Phys 54(5):1397–1404
LaRusch GA, Mahdi F, Shariat-Madar Z, Adams G, Sitrin RG, Zhang WM, McCrae KR, Schmaier AH (2010) Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood 115(24):5111–5120
Laurent M, Martinerie C, Thibout H, Hoffman MP, Verrecchia F, Le Bouc Y, Mauviel A, Kleinman HK (2003) NOVH increases MMP3 expression and cell migration in glioblastoma cells via a PDGFR-alpha-dependent mechanism. FASEB J 17(13):1919–1921
Laurent N, de Bouard S, Guillamo JS, Christov C, Zini R, Jouault H, Andre P, Lotteau V, Peschanski M (2004) Effects of the proteasome inhibitor ritonavir on glioma growth in vitro and in vivo. Mol Cancer Ther 3(2):129–136
Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, Rosado CJ, Langendorf CG, Pike RN, Bird PI, Whisstock JC (2006) An overview of the serpin superfamily. Genome Biol 7(5):216
Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, Rewcastle B, Yong VW (2003) Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci 23(10):4034–4043
Lee NV, Sato M, Annis DS, Loo JA, Wu L, Mosher DF, Iruela-Arispe ML (2006) ADAMTS1 mediates the release of antiangiogenic polypeptides from TSP1 and 2. EMBO J 25(22):5270–5283
Lee JG, Baek K, Soetandyo N, Ye Y (2013) Reversible inactivation of deubiquitinases by reactive oxygen species in vitro and in cells. Nat Commun 4:1568
Legnani FG, Pradilla G, Thai QA, Fiorindi A, Recinos PF, Tyler BM, Gaini SM, DiMeco F, Brem H, Olivi A (2006) Lactacystin exhibits potent anti-tumor activity in an animal model of malignant glioma when administered via controlled-release polymers. J Neurooncol 77(3):225–232
Lehman NL (2009) The ubiquitin proteasome system in neuropathology. Acta Neuropathol 118(3):329–347
Leitlein J, Aulwurm S, Waltereit R, Naumann U, Wagenknecht B, Garten W, Weller M, Platten M (2001) Processing of immunosuppressive pro-TGF-beta 1,2 by human glioblastoma cells involves cytoplasmic and secreted furin-like proteases. J Immunol 166(12):7238–7243
Lemberg MK (2011) Intramembrane proteolysis in regulated protein trafficking. Traffic 12(9):1109–1118
Lettau I, Hattermann K, Held-Feindt J, Brauer R, Sedlacek R, Mentlein R (2010) Matrix metalloproteinase-19 is highly expressed in astroglial tumors and promotes invasion of glioma cells. J Neuropathol Exp Neurol 69(3):215–223
Levicar N, Strojnik T, Kos J, Dewey RA, Pilkington GJ, Lah TT (2002) Lysosomal enzymes, cathepsins in brain tumour invasion. J Neurooncol 58(1):21–32
Levicar N, Dewey RA, Daley E, Bates TE, Davies D, Kos J, Pilkington GJ, Lah TT (2003a) Selective suppression of cathepsin L by antisense cDNA impairs human brain tumor cell invasion in vitro and promotes apoptosis. Cancer Gene Ther 10(2):141–151
Levicar N, Nuttall RK, Lah TT (2003b) Proteases in brain tumour progression. Acta Neurochir (Wien) 145(9):825–838
Levin VA, Phuphanich S, Yung WK, Forsyth PA, Maestro RD, Perry JR, Fuller GN, Baillet M (2006) Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol 78(3):295–302
Li Z, Sheng M (2012) Caspases in synaptic plasticity. Mol Brain 5:15
Li T, Wen H, Brayton C, Das P, Smithson LA, Fauq A, Fan X, Crain BJ, Price DL, Golde TE, Eberhart CG, Wong PC (2007) Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma-secretase. J Biol Chem 282(44):32264–32273
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB, Rich JN (2009) Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15(6):501–513
Li R, Li G, Deng L, Liu Q, Dai J, Shen J, Zhang J (2010a) IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol Rep 23(6):1553–1559
Li Z, Jo J, Jia JM, Lo SC,Whitcomb DJ, Jiao S, Cho K, Sheng M (2010b) Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 141(5):859–871
Li M, Mukasa A, Inda MM, Zhang J, Chin L, Cavenee W, Furnari F (2011) Guanylate binding protein 1 is a novel effector of EGFR-driven invasion in glioblastoma. J Exp Med 208(13):2657–2673
Li Y, Wang Y, Yu L, Sun C, Cheng D, Yu S, Wang Q, Yan Y, Kang C, Jin S, An T, Shi C, Xu J, Wei C, Liu J, Sun J, Wen Y, Zhao S, Kong Y (2013a) miR-146b-5p inhibits glioma migration and invasion by targeting MMP16. Cancer Lett 339(2):260–269
Li ZH, Yu Y, Du C, Fu H, Wang J, Tian Y (2013b) RNA interference-mediated USP22 gene silencing promotes human brain glioma apoptosis and induces cell cycle arrest. Oncol Lett 5(4):1290–1294
Liu BL, Cheng JX, Zhang W, Zhang X, Wang R, Lin H, Huo JL, Cheng H (2010) Quantitative detection of multiple gene promoter hypermethylation in tumor tissue, serum, and cerebrospinal fluid predicts prognosis of malignant gliomas. Neuro Oncol 12(6):540–548
Liu B, Wang L, Shen LL, Shen MZ, Guo XD, Wang T, Liang QC, Wang C, Zheng J, Li Y, Jia LT, Zhang H, Gao GD (2012) RNAi-mediated inhibition of presenilin 2 inhibits glioma cell growth and invasion and is involved in the regulation of Nrg1/ErbB signaling. Neuro Oncol 14(8):994–1006
Liu Z, Zanata SM, Kim J, Peterson MA, Di Vizio D, Chirieac LR, Pyne S, Agostini M, Freeman MR, Loda M (2013) The ubiquitin-specific protease USP2a prevents endocytosis-mediated EGFR degradation. Oncogene 32(13):1660–1669
Liu Z, Fan F, Wang A, Zheng S, Lu Y (2014) Dll4-Notch signaling in regulation of tumor angiogenesis. J Cancer Res Clin Oncol 140(4):525–536
Liuzzi GM, Mastroianni CM, Latronico T, Mengoni F, Fasano A, Lichtner M, Vullo V, Riccio P (2004) Anti-HIV drugs decrease the expression of matrix metalloproteinases in astrocytes and microglia. Brain 127(Pt 2):398–407
Lopez-Otin C, Bond JS (2008) Proteases: multifunctional enzymes in life and disease. J Biol Chem 283(45):30433–30437
Lopez-Otin C, Matrisian LM (2007) Emerging roles of proteases in tumour suppression. Nat Rev Cancer 7(10):800–808
Lovric MM, Hawkins CJ (2010) TRAIL treatment provokes mutations in surviving cells. Oncogene 29(36):5048–5060
Lu X, Wyszynski M, Sheng M, Baudry M (2001) Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: implications for synaptic plasticity. J Neurochem 77(6):1553–1560
Lu KV, Jong KA, Rajasekaran AK, Cloughesy TF, Mischel PS (2004) Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Lab Invest 84(1):8–20
LuW, ZX, Hong B, Liu J, Yue Z (2004) Suppression of invasion in human U87 glioma cells by adenovirus-mediated co-transfer of TIMP-2 and PTEN gene. Cancer Lett 214(2):205–213
Lu C, Wang Y, Furukawa K, Fu W, Ouyang X, Mattson MP (2006a) Evidence that caspase-1 is a negative regulator of AMPA receptor-mediated long-term potentiation at hippocampal synapses. J Neurochem 97(4):1104–1110
Lu Z, Wang Y, Zhang Q, Zhang X, Wang S, Xie H, Li Y, Jiao B, Zhang J (2006b) Association between the functional polymorphism in the matrix metalloproteinase-7 promoter and susceptibility to adult astrocytoma. Brain Res 1118(1):6–12
Lu DY, Leung YM, Cheung CW, Chen YR, Wong KL (2010) Glial cell line-derived neurotrophic factor induces cell migration and matrix metalloproteinase-13 expression in glioma cells. Biochem Pharmacol 80(8):1201–1209
Lu Y, Chopp M, Zheng X, Katakowski M, Buller B, Jiang F (2013) MiR-145 reduces ADAM17 expression and inhibits in vitro migration and invasion of glioma cells. Oncol Rep 29(1):67–72
Lucio-Eterovic AK, Piao Y, de Groot JF (2009) Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res 15(14):4589–4599
Ludwig A, Schulte A, Schnack C, Hundhausen C, Reiss K, Brodway N, Held-Feindt J, Mentlein R (2005) Enhanced expression and shedding of the transmembrane chemokine CXCL16 by reactive astrocytes and glioma cells. J Neurochem 93(5):1293–1303
Luque A, Carpizo DR, Iruela-Arispe ML (2003) ADAMTS1/METH1 inhibits endothelial cell proliferation by direct binding and sequestration of VEGF165. J Biol Chem 278(26):23656–23665
Ma J, Cui W, He SM, Duan YH, Heng LJ, Wang L, Gao GD (2012) Human U87 astrocytoma cell invasion induced by interaction of betaig-h3 with integrin alpha5beta1 involves calpain-2. PLoS One 7(5):e37297
Magnus N, Garnier D, Rak J (2010) Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 116(5):815–818
Mao H, Lebrun DG, Yang J, Zhu VF, Li M (2012) Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest 30(1):48–56
Mares V, Stremenova J, Lisa V, Kozakova H, Marek J, Syrucek M, Soula O, Sedo A (2012) Compartment- and malignance-dependent up-regulation of gamma-glutamyltranspeptidase and dipetidylpeptidase-IV activity in human brain gliomas. Histol Histopathol 27(7):931–940
Maret D, Sadr MS, Sadr ES, Colman DR, Del Maestro RF, Seidah NG (2012) Opposite roles of furin and PC5A in N-cadherin processing. Neoplasia 14(10):880–892
Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kalin R, van Rooijen N, Holmbeck K, Heppner FL, Kiwit J, Matyash V, Lehnardt S, Kaminska B, Glass R, Kettenmann H (2009) Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A 106(30):12530–12535
Martinez R, Setien F, Voelter C, Casado S, Quesada MP, Schackert G, Esteller M (2007) CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis 28(6):1264–1268
Mason SD, Joyce JA (2011) Proteolytic networks in cancer. Trends Cell Biol 21(4):228–237
Matsuzawa K, Fukuyama K, Hubbard SL, Dirks PB, Rutka JT (1996) Transfection of an invasive human astrocytoma cell line with a TIMP-1 cDNA: modulation of astrocytoma invasive potential. J Neuropathol Exp Neurol 55(1):88–96
Matthews RT, Gary SC, Zerillo C, Pratta M, Solomon K, Arner EC, Hockfield S (2000) Brain-enriched hyaluronan binding (BEHAB)/brevican cleavage in a glioma cell line is mediated by a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family member. J Biol Chem 275(30):22695–22703
McCawley LJ, Matrisian LM (2001) Matrix metalloproteinases: they’re not just for matrix anymore! Curr Opin Cell Biol 13(5):534–540
McCormick D (1993) Secretion of cathepsin B by human gliomas in vitro. Neuropathol Appl Neurobiol 19(2):146–151
McCready J, Broaddus WC, Sykes V, Fillmore HL (2005) Association of a single nucleotide polymorphism in the matrix metalloproteinase-1 promoter with glioblastoma. Int J Cancer 117(5):781–785
McGillicuddy LT, Fromm JA, Hollstein PE, Kubek S, Beroukhim R, De Raedt T, Johnson BW, Williams SM, Nghiemphu P, Liau LM, Cloughesy TF, Mischel PS, Parret A, Seiler J, Moldenhauer G, Scheffzek K, Stemmer-Rachamimov AO, Sawyers CL, Brennan C, Messiaen L, Mellinghoff IK, Cichowski K (2009) Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 16(1):44–54
Medeiros Mdos S, Balmforth AJ, Vaughan PF, Turner AJ (1991) Hydrolysis of atrial and brain natriuretic peptides by the human astrocytoma clone D384 and the neuroblastoma line SH-SY5Y. Neuroendocrinology 54(3):295–302
Mehling M, Simon P, Mittelbronn M, Meyermann R, Ferrone S, Weller M, Wiendl H (2007) WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism? Acta Neuropathol 114(2):111–119
Melendez-Zajgla J, Del Pozo L, Ceballos G, Maldonado V (2008) Tissue inhibitor of metalloproteinases-4. The road less traveled. Mol Cancer 7:85
Mentlein R, Hattermann K, Hemion C, Jungbluth A, Held-Feindt J (2011) Expression and role of the cell surface protease seprase/fibroblast activation protein-alpha (FAP-alpha) in astroglial tumors. Biol Chem 392(3):199–207
Mentlein R, Hattermann K, Held-Feindt J (2012) Lost in disruption: role of proteases in glioma invasion and progression. Biochim Biophys Acta 1825(2):178–185
Mercapide J, Lopez De Cicco R, Bassi DE, Castresana JS, Thomas G, Klein-Szanto AJ (2002) Inhibition of furin-mediated processing results in suppression of astrocytoma cell growth and invasiveness. Clin Cancer Res 8(6):1740–1746
Mercapide J, Lopez De Cicco R, Castresana JS, Klein-Szanto AJ (2003) Stromelysin-1/matrix metalloproteinase-3 (MMP-3) expression accounts for invasive properties of human astrocytoma cell lines. Int J Cancer 106(5):676–682
Merzak A, Parker C, Koochekpour S, Sherbet GV, Pilkington GJ (1994) Overexpression of the 18A2/mts1 gene and down-regulation of the TIMP-2 gene in invasive human glioma cell lines in vitro. Neuropathol Appl Neurobiol 20(6):614–619
Miake H, Tsuchiya K, Nakamura A, Ikeda K, Levesque L, Fraser PE, St-George Hyslop PH, Mizusawa H, Uchihara T (1999) Glial expression of presenilin epitopes in human brain with cerebral infarction and in astrocytoma. Acta Neuropathol 98(4):337–340
Mikkelsen T, Yan PS, Ho KL, Sameni M, Sloane BF, Rosenblum ML (1995) Immunolocalization of cathepsin B in human glioma: implications for tumor invasion and angiogenesis. J Neurosurg 83(2):285–290
Mina-Osorio P (2008) The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med 14(8):361–371
Miyata S, Fukushima T, Kohama K, Tanaka H, Takeshima H, Kataoka H (2007) Roles of Kunitz domains in the anti-invasive effect of hepatocyte growth factor activator inhibitor type 1 in human glioblastoma cells. Hum Cell 20(4):100–106
Miyazaki K, Umenishi F, Funahashi K, Koshikawa N, Yasumitsu H, Umeda M (1992) Activation of TIMP-2/progelatinase A complex by stromelysin. Biochem Biophys Res Commun 185(3):852–859
Mohamed MM, Sloane BF (2006) Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer 6(10):764–775
Mohanam S, Wang SW, Rayford A, Yamamoto M, Sawaya R, Nakajima M, Liotta LA, Nicolson GL, Stetler-Stevenson WG, Rao JS (1995) Expression of tissue inhibitors of metalloproteinases: negative regulators of human glioblastoma invasion in vivo. Clin Exp Metastasis 13(1):57–62
Mohanam S, Gladson CL, Rao CN, Rao JS (1999) Biological significance of the expression of urokinase-type plasminogen activator receptors (uPARs) in brain tumors. Front Biosci 4:D178–D187
Mohanam S, Jasti SL, Kondraganti SR, Chandrasekar N, Lakka SS, Kin Y, Fuller GN, Yung AW, Kyritsis AP, Dinh DH, Olivero WC, Gujrati M, Ali-Osman F, Rao JS (2001) Down-regulation of cathepsin B expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene 20(28):3665–3673
Mohanan V, Temburni MK, Kappes JC, Galileo DS (2013) L1CAM stimulates glioma cell motility and proliferation through the fibroblast growth factor receptor. Clin Exp Metastasis 30(4):507–520
Moller HG, Rasmussen AP, Andersen HH, Johnsen KB, Henriksen M, Duroux M (2013) A systematic review of microRNA in glioblastoma multiforme: micro-modulators in the mesenchymal mode of migration and invasion. Mol Neurobiol 47(1):131–144
Monod L, Diserens AC, Jongeneel CV, Carrel S, Ronco P, Verroust P, de Tribolet N (1989) Human glioma cell lines expressing the common acute lymphoblastic leukemia antigen (cALLa) have neutral endopeptidase activity. Int J Cancer 44(5):948–951
Monod L, Hamou MF, Ronco P, Verroust P, de Tribolet N (1992) Expression of cALLa/NEP on gliomas: a possible marker of malignancy. Acta Neurochir (Wien) 114(1–2):3–7
Morancho A, Rosell A, Garcia-Bonilla L, Montaner J (2010) Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann NY Acad Sci 1207:123–133
Mori T, Abe T, Wakabayashi Y, Hikawa T, Matsuo K, Yamada Y, Kuwano M, Hori S (2000) Up-regulation of urokinase-type plasminogen activator and its receptor correlates with enhanced invasion activity of human glioma cells mediated by transforming growth factor-alpha or basic fibroblast growth factor. J Neurooncol 46(2):115–123
Moriyama T, Kataoka H, Seguchi K, Tsubouchi H, Koono M (1996) Effects of hepatocyte growth factor (HGF) on human glioma cells in vitro: HGF acts as a motility factor in glioma cells. Int J Cancer 66(5):678–685
Moriyama T, Kataoka H, Hamasuna R, Yoshida E, Sameshima T, Iseda T, Yokogami K, Nakano S, Koono M, Wakisaka S (1999) Simultaneous up-regulation of urokinase-type plasminogen activator (uPA) and uPA receptor by hepatocyte growth factor/scatter factor in human glioma cells. Clin Exp Metastasis 17(10):873–879
Moss ML, Stoeck A, Yan W, Dempsey PJ (2008) ADAM10 as a target for anti-cancer therapy. Curr Pharm Biotechnol 9(1):2–8
Mullard A (2013) Maturing antibody-drug conjugate pipeline hits 30. Nat Rev Drug Discov 12(5):329–332
Munaut C, Noel A, Hougrand O, Foidart JM, Boniver J, Deprez M (2003) Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1-MMP, MMP-2 and MMP-9 in human glioblastomas. Int J Cancer 106(6):848–855
Muracciole X, Romain S, Dufour H, Palmari J, Chinot O, Ouafik L, Grisoli F, Branger DF, Martin PM (2002) PAI-1 and EGFR expression in adult glioma tumors: toward a molecular prognostic classification. Int J Radiat Oncol Biol Phys 52(3):592–598
Murai T, Miyazaki Y, Nishinakamura H, Sugahara KN, Miyauchi T, Sako Y, Yanagida T, Miyasaka M (2004) Engagement of CD44 promotes Rac activation and CD44 cleavage during tumor cell migration. J Biol Chem 279(6):4541–4550
Murakami D, Okamoto I, Nagano O, Kawano Y, Tomita T, Iwatsubo T, De Strooper B, Yumoto E, Saya H (2003) Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 22(10):1511–1516
Murphy G (2011) Tissue inhibitors of metalloproteinases. Genome Biol 12(11):233
Murphy AC, Weyhenmeyer B, Schmid J, Kilbride SM, Rehm M, Huber HJ, Senft C, Weissenberger J, Seifert V, Dunst M, Mittelbronn M, Kogel D, Prehn JH, Murphy BM (2013) Activation of executioner caspases is a predictor of progression-free survival in glioblastoma patients: a systems medicine approach. Cell Death Dis 4:e629
Mutka AL, Haapanen A, Kakela R, Lindfors M, Wright AK, Inkinen T, Hermansson M, Rokka A, Corthals G, Jauhiainen M, Gillingwater TH, Ikonen E, Tyynela J (2010) Murine cathepsin D deficiency is associated with dysmyelination/myelin disruption and accumulation of cholesteryl esters in the brain. J Neurochem 112(1):193–203
Naidoo V, Naidoo S, Mahabeer R, Raidoo DM (2005) Localization of the endothelin system in human diffuse astrocytomas. Cancer 104(5):1049–1057
Nakabayashi H, Hara M, Shimuzu K (2005) Clinicopathologic significance of cystatin C expression in gliomas. Hum Pathol 36(9):1008–1015
Nakada M, Nakamura H, Ikeda E, Fujimoto N, Yamashita J, Sato H, Seiki M, Okada Y (1999a) Expression and tissue localization of membrane-type 1, 2, and 3 matrix metalloproteinases in human astrocytic tumors. Am J Pathol 154(2):417–428
Nakada M, Yamashita J, Okada Y, Sato H (1999b) Ets-1 positively regulates expression of urokinase-type plasminogen activator (uPA) and invasiveness of astrocytic tumors. J Neuropathol Exp Neurol 58(4):329–334
Nakada M, Kita D, Futami K, Yamashita J, Fujimoto N, Sato H, Okada Y (2001) Roles of membrane type 1 matrix metalloproteinase and tissue inhibitor of metalloproteinases 2 in invasion and dissemination of human malignant glioma. J Neurosurg 94(3):464–473
Nakagawa T, Kubota T, Kabuto M, Sato K, Kawano H, Hayakawa T, Okada Y (1994) Production of matrix metalloproteinases and tissue inhibitor of metalloproteinases-1 by human brain tumors. J Neurosurg 81(1):69–77
Nakagawa T, Kubota T, Kabuto M, Sato K, Arai Y, Kodera T (1995) Production of tissue inhibitor of metalloproteinases-1 (TIMP-1) by human astrocytic tumors. Neurol Med Chir (Tokyo) 35(10):728–731
Nakamura M, Ishida E, Shimada K, Kishi M, Nakase H, Sakaki T, Konishi N (2005) Frequent LOH on 22q12.3 and TIMP-3 inactivation occur in the progression to secondary glioblastomas. Lab Invest 85(2):165–175
Nakano A, Tani E, Miyazaki K, Furuyama J, Matsumoto T (1993) Expressions of matrilysin and stromelysin in human glioma cells. Biochem Biophys Res Commun 192(3):999–1003
Nakano A, Tani E, Miyazaki K, Yamamoto Y, Furuyama J (1995) Matrix metalloproteinases and tissue inhibitors of metalloproteinases in human gliomas. J Neurosurg 83(2):298–307
Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio PM (1992) Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J 11(13):4825–4833
Nan Y, Han L, Zhang A, Wang G, Jia Z, Yang Y, Yue X, Pu P, Zhong Y, Kang C (2010) MiRNA-451 plays a role as tumor suppressor in human glioma cells. Brain Res 1359:14–21
Ng K, Nitta M, Hu L, Kesari S, Kung A, D’Andrea A, Chen CC (2009) A small interference RNA screen revealed proteasome inhibition as strategy for glioblastoma therapy. Clin Neurosurg 56:107–118
Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A (2001) The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 7(1):59–64
Noha M, Yoshida D, Watanabe K, Teramoto A (2000) Suppression of cell invasion on human malignant glioma cell lines by a novel matrix-metalloproteinase inhibitor SI-27: in vitro study. J Neurooncol 48(3):217–223
Nomura T, Katunuma N (2005) Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J Med Invest 52(1–2):1–9
Noorbakhsh F, Vergnolle N, Hollenberg MD, Power C (2003) Proteinase-activated receptors in the nervous system. Nat Rev Neurosci 4(12):981–990
Nuti E, Casalini F, Santamaria S, Gabelloni P, Bendinelli S, Da Pozzo E, Costa B, Marinelli L, La Pietra V, Novellino E, Margarida Bernardo M, Fridman R, Da Settimo F, Martini C, Rossello A (2011) Synthesis and biological evaluation in U87MG glioma cells of (ethynylthiophene)sulfonamido-based hydroxamates as matrix metalloproteinase inhibitors. Eur J Med Chem 46(7):2617–2629
Ogiichi T, Hirashima Y, Nakamura S, Endo S, Kurimoto M, Takaku A (2000) Tissue factor and cancer procoagulant expressed by glioma cells participate in their thrombin-mediated proliferation. J Neurooncol 46(1):1–9
Ohgaki H, Kleihues P (2007) Genetic pathways to primary and secondary glioblastoma. Am J Pathol 170(5):1445–1453
Ohshima-Hosoyama S, Davare MA, Hosoyama T, Nelon LD, Keller C (2011) Bortezomib stabilizes NOXA and triggers ROS-associated apoptosis in medulloblastoma. J Neurooncol 105(3):475–483
Oikawa T, Yamada T (2003) Molecular biology of the Ets family of transcription factors. Gene 303:11–34
Ornstein DL, Meehan KR, Zacharski LR (2002) The coagulation system as a target for the treatment of human gliomas. Semin Thromb Hemost 28(1):19–28
Paganetti PA, Caroni P, Schwab ME (1988) Glioblastoma infiltration into central nervous system tissue in vitro: involvement of a metalloprotease. J Cell Biol 107(6 Pt 1):2281–2291
Pagenstecher A, Wussler EM, Opdenakker G, Volk B, Campbell IL (2001) Distinct expression patterns and levels of enzymatic activity of matrix metalloproteinases and their inhibitors in primary brain tumors. J Neuropathol Exp Neurol 60(6):598–612
Pan SJ, Zhan SK, Pei BG, Sun QF, Bian LG, Sun BM (2012) MicroRNA-149 inhibits proliferation and invasion of glioma cells via blockade of AKT1 signaling. Int J Immunopathol Pharmacol 25(4):871–881
Panner A, Crane CA, Weng C, Feletti A, Fang S, Parsa AT, Pieper RO (2010) Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer Res 70(12):5046–5053
Park MJ, Kim MS, Park IC, Kang HS, Yoo H, Park SH, Rhee CH, Hong SI, Lee SH (2002) PTEN suppresses hyaluronic acid-induced matrix metalloproteinase-9 expression in U87MG glioblastoma cells through focal adhesion kinase dephosphorylation. Cancer Res 62(21):6318–6322
Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, Park IC, Rhee CH, Hong SI (2006) Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res 66(17):8511–8519
Paugh BS, Paugh SW, Bryan L, Kapitonov D, Wilczynska KM, Gopalan SM, Rokita H, Milstien S, Spiegel S, Kordula T (2008) EGF regulates plasminogen activator inhibitor-1 (PAI-1) by a pathway involving c-Src, PKCdelta, and sphingosine kinase 1 in glioblastoma cells. FASEB J 22(2):455–465
Paulus W, Huettner C, Tonn JC (1994) Collagens, integrins and the mesenchymal drift in glioblastomas: a comparison of biopsy specimens, spheroid and early monolayer cultures. Int J Cancer 58(6):841–846
Payne LS, Huang PH (2013) The pathobiology of collagens in glioma. Mol Cancer Res 11(10):1129–1140
Pedeboscq S, L’Azou B, Passagne I, De Giorgi F, Ichas F, Pometan JP, Cambar J (2008) Cytotoxic and apoptotic effects of bortezomib and gefitinib compared to alkylating agents on human glioblastoma cells. J Exp Ther Oncol 7(2):99–111
Perry JR (2010) Anticoagulation of malignant glioma patients in the era of novel antiangiogenic agents. Curr Opin Neurol 23(6):592–596
Perry JR, Julian JA, Laperriere NJ, Geerts W, Agnelli G, Rogers LR, Malkin MG, Sawaya R, Baker R, Falanga A, Parpia S, Finch T, Levine MN (2010) PRODIGE: a randomized placebo-controlled trial of dalteparin low-molecular-weight heparin thromboprophylaxis in patients with newly diagnosed malignant glioma. J Thromb Haemost 8(9):1959–1965
Phuphanich S, Carson KA, Grossman SA, Lesser G, Olson J, Mikkelsen T, Desideri S, Fisher JD, C. N. S. C. New Approaches to Brain Tumor Therapy (2008) Phase I safety study of escalating doses of atrasentan in adults with recurrent malignant glioma. Neuro Oncol 10(4):617–623
Phuphanich S, Supko JG, Carson KA, Grossman SA, Burt Nabors L, Mikkelsen T, Lesser G, Rosenfeld S, Desideri S, Olson JJ (2010) Phase 1 clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol 100(1):95–103
Piccinini M, Rinaudo MT, Anselmino A, Ramondetti C, Buccinna B, Fiano V, Ghimenti C, Schiffer D (2005) Characterization of the 20S proteasome in human glioblastomas. Anticancer Res 25(5):3203–3210
Piva R, Cancelli I, Cavalla P, Bortolotto S, Dominguez J, Draetta GF, Schiffer D (1999) Proteasome-dependent degradation of p27/kip1 in gliomas. J Neuropathol Exp Neurol 58(7):691–696
Pop C, Salvesen GS (2009) Human caspases: activation, specificity, and regulation. J Biol Chem 284(33):21777–21781
Prezas P, Scorilas A, Yfanti C, Viktorov P, Agnanti N, Diamandis E, Talieri M (2006) The role of human tissue kallikreins 7 and 8 in intracranial malignancies. Biol Chem 387(12):1607–1612
Prox J, Rittger A, Saftig P (2012) Physiological functions of the amyloid precursor protein secretases ADAM10, BACE1, and presenilin. Exp Brain Res 217(3–4):331–341
Puente XS, Sanchez LM, Overall CM, Lopez-Otin C (2003) Human and mouse proteases: a comparative genomic approach. Nat Rev Genet 4(7):544–558
Pullen NA, Fillmore HL (2010) Induction of matrix metalloproteinase-1 and glioma cell motility by nitric oxide. J Neurooncol 96(2):201–209
Pullen NA, Anand M, Cooper PS, Fillmore HL (2012) Matrix metalloproteinase-1 expression enhances tumorigenicity as well as tumor-related angiogenesis and is inversely associated with TIMP-4 expression in a model of glioblastoma. J Neurooncol 106(3):461–471
Pyrko P, Kardosh A, Wang W, Xiong W, Schonthal AH, Chen TC (2007) HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res 67(22):10920–10928
Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D (1993) Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature 361(6411):453–457
Qin H, Moellinger JD, Wells A, Windsor LJ, Sun Y, Benveniste EN (1998) Transcriptional suppression of matrix metalloproteinase-2 gene expression in human astroglioma cells by TNF-alpha and IFN-gamma. J Immunol 161(12):6664–6673
Qin H, Sun Y, Benveniste EN (1999) The transcription factors Sp1, Sp3, and AP-2 are required for constitutive matrix metalloproteinase-2 gene expression in astroglioma cells. J Biol Chem 274(41):29130–29137
Qiu J, Ai L, Ramachandran C, Yao B, Gopalakrishnan S, Fields CR, Delmas AL, Dyer LM, Melnick SJ, Yachnis AT, Schwartz PH, Fine HA, Brown KD, Robertson KD (2008) Invasion suppressor cystatin E/M (CST6): high-level cell type-specific expression in normal brain and epigenetic silencing in gliomas. Lab Invest 88(9):910–925
Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ (2005) Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 436(7047):123–127
Raithatha SA, Muzik H, Muzik H, Rewcastle NB, Johnston RN, Edwards DR, Forsyth PA (2000) Localization of gelatinase-A and gelatinase-B mRNA and protein in human gliomas. Neuro Oncol 2(3):145–150
Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, Muth D, Demmers JA, Zaki A, Fouchier RA, Thiel V, Drosten C, Rottier PJ, Osterhaus AD, Bosch BJ, Haagmans BL (2013) Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495(7440):251–254
Ramakrishna S, Suresh B, Baek KH (2011) The role of deubiquitinating enzymes in apoptosis. Cell Mol Life Sci 68(1):15–26
Rao JS (2003) Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer 3(7):489–501
Rao JS, Rayford A, Morantz RA, Festoff BW, Sawaya R (1993a) Increased levels of plasminogen-activator inhibitor-1 (Pai-1) in human brain-tumors. J Neurooncol 17(3):215–221
Rao JS, Steck PA, Mohanam S, Stetler-Stevenson WG, Liotta LA, Sawaya R (1993b) Elevated levels of M(r) 92,000 type IV collagenase in human brain tumors. Cancer Res 53(10 Suppl):2208–2211
Rao JS, Yamamoto M, Mohaman S, Gokaslan ZL, Fuller GN, Stetler-Stevenson WG, Rao VH, Liotta LA, Nicolson GL, Sawaya RE (1996) Expression and localization of 92 kDa type IV collagenase/gelatinase B (MMP-9) in human gliomas. Clin Exp Metastasis 14(1):12–18
Rao CN, Lakka SS, Kin Y, Konduri SD, Fuller GN, Mohanam S, Rao JS (2001) Expression of tissue factor pathway inhibitor 2 inversely correlates during the progression of human gliomas. Clin Cancer Res 7(3):570–576
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40(Database issue):D343–D350
Ray SK, Patel SJ, Welsh CT, Wilford GG, Hogan EL, Banik NL (2002) Molecular evidence of apoptotic death in malignant brain tumors including glioblastoma multiforme: upregulation of calpain and caspase-3. J Neurosci Res 69(2):197–206
Redondo-Munoz J, Ugarte-Berzal E, Terol MJ, Van den Steen PE, Hernandez del Cerro M, Roderfeld M, Roeb E, Opdenakker G, Garcia-Marco JA, Garcia-Pardo A (2010) Matrix metalloproteinase-9 promotes chronic lymphocytic leukemia b cell survival through its hemopexin domain. Cancer Cell 17(2):160–172
Rege TA, Fears CY, Gladson CL (2005) Endogenous inhibitors of angiogenesis in malignant gliomas: nature’s antiangiogenic therapy. Neuro Oncol 7(2):106–121
Regina A, Demeule M, Berube A, Moumdjian R, Berthelet F, Beliveau R (2003) Differences in multidrug resistance phenotype and matrix metalloproteinases activity between endothelial cells from normal brain and glioma. J Neurochem 84(2):316–324
Reif R, Sales S, Hettwer S, Dreier B, Gisler C, Wolfel J, Luscher D, Zurlinden A, Stephan A, Ahmed S, Baici A, Ledermann B, Kunz B, Sonderegger P (2007) Specific cleavage of agrin by neurotrypsin, a synaptic protease linked to mental retardation. FASEB J 21(13):3468–3478
Rempel SA, Rosenblum ML, Mikkelsen T, Yan PS, Ellis KD, Golembieski WA, Sameni M, Rozhin J, Ziegler G, Sloane BF (1994) Cathepsin B expression and localization in glioma progression and invasion. Cancer Res 54(23):6027–6031
Repnik U, Cesen MH, Turk B (2013) The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol 5(1):a008755
Rettig WJ, Chesa PG, Beresford HR, Feickert HJ, Jennings MT, Cohen J, Oettgen HF, Old LJ (1986) Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer Res 46(12 Pt 1):6406–6412
Ribatti D (2009) Endogenous inhibitors of angiogenesis: a historical review. Leuk Res 33(5):638–644
Rivera S, Khrestchatisky M, Kaczmarek L, Rosenberg GA, Jaworski DM (2010) Metzincin proteases and their inhibitors: foes or friends in nervous system physiology? J Neurosci 30(46):15337–15357
Robins HI, O’Neill A, Gilbert M, Olsen M, Sapiente R, Berkey B, Mehta M (2008) Effect of dalteparin and radiation on survival and thromboembolic events in glioblastoma multiforme: a phase II ECOG trial. Cancer Chemother Pharmacol 62(2):227–233
Robson DK, Ironside JW, Reid WA, Bogue PR (1990) Immunolocalization of cathepsin D in the human central nervous system and central nervous system neoplasms. Neuropathol Appl Neurobiol 16(1):39–44
Rogove AD, Siao C, Keyt B, Strickland S, Tsirka SE (1999) Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci 112(Pt 22):4007–4016
Rome C, Arsaut J, Taris C, Couillaud F, Loiseau H (2007) MMP-7 (matrilysin) expression in human brain tumors. Mol Carcinog 46(6):446–452
Rong Y, Durden DL, Van Meir EG, Brat DJ (2006) ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 65(6):529–539
Rong Y, Brat DJ (2009) Vaso-occlusive mechanisms that initiate hypoxia and necrosis in glioblastoma: the role of thrombosis and tissue factor. In: Teicher BA (ed) CNS cancer. Humana, Totowa, NJ, pp 507–528 (This is a book chapter- please see http://springerlink.bibliotecabuap.elogim.com/chapter/10.1007%2F978-1-60327-553-8_22)
Rorive S, Berton A, D’Haene N, Takacs CN, Debeir O, Decaestecker C, Salmon I (2008) Matrix metalloproteinase-9 interplays with the IGFBP2-IGFII complex to promote cell growth and motility in astrocytomas. Glia 56(15):1679–1690
Rorive S, Lopez XM, Maris C, Trepant AL, Sauvage S, Sadeghi N, Roland I, Decaestecker C, Salmon I (2010) TIMP-4 and CD63: new prognostic biomarkers in human astrocytomas. Mod Pathol 23(10):1418–1428
Roth P, Kissel M, Herrmann C, Eisele G, Leban J, Weller M, Schmidt F (2009) SC68896, a novel small molecule proteasome inhibitor, exerts antiglioma activity in vitro and in vivo. Clin Cancer Res 15(21):6609–6618
Rousalova I, Krepela E (2010) Granzyme B-induced apoptosis in cancer cells and its regulation (review). Int J Oncol 37(6):1361–1378
Rozanov DV, Sikora S, Godzik A, Postnova TI, Golubkov V, Savinov A, Tomlinson S, Strongin AY (2004) Non-proteolytic, receptor/ligand interactions associate cellular membrane type-1 matrix metalloproteinase with the complement component C1q. J Biol Chem 279(48):50321–50328
Ruano Y, Mollejo M, Camacho FI, Rodriguez de Lope A, Fiano C, Ribalta T, Martinez P, Hernandez-Moneo JL, Melendez B (2008) Identification of survival-related genes of the phosphatidylinositol 3'-kinase signaling pathway in glioblastoma multiforme. Cancer 112(7):1575–1584
Sahin A, Velten M, Pietsch T, Knuefermann P, Okuducu AF, Hahne JC, Wernert N (2005) Inactivation of Ets 1 transcription factor by a specific decoy strategy reduces rat C6 glioma cell proliferation and mmp-9 expression. Int J Mol Med 15(5):771–776
Sahm F, Capper D, Jeibmann A, Habel A, Paulus W, Troost D, von Deimling A (2012) Addressing diffuse glioma as a systemic brain disease with single-cell analysis. Arch Neurol 69(4):523–526
Saito N, Fu J, Zheng S, Yao J, Wang S, Liu DD, Yuan Y, Sulman EP, Lang FF, Colman H, Verhaak RG, Yung WK, Koul D (2014) A high Notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells 32(1):301–312. doi:10.1002/stem.1528
Sakamoto T, Seiki M (2009) Cytoplasmic tail of MT1-MMP regulates macrophage motility independently from its protease activity. Genes Cells 14(5):617–626
Salmaggi A, Croci D, Prina P, Cajola L, Pollo B, Marras CE, Ciusani E, Silvani A, Boiardi A, Sciacca FL (2006) Production and post-surgical modification of VEGF, tPA and PAI-1 in patients with glioma. Cancer Biol Ther 5(2):204–209
Sarkar S, Nuttall RK, Liu S, Edwards DR, Yong VW (2006) Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res 66(24):11771–11780
Sato Y (2011) Persistent vascular normalization as an alternative goal of anti-angiogenic cancer therapy. Cancer Sci 102(7):1253–1256
Sawaya R, Ramo OJ, Shi ML, Mandybur G (1991) Biological significance of tissue plasminogen activator content in brain tumors. J Neurosurg 74(3):480–486
Sawaya RE, Yamamoto M, Gokaslan ZL, Wang SW, Mohanam S, Fuller GN, McCutcheon IE, Stetler-Stevenson WG, Nicolson GL, Rao JS (1996) Expression and localization of 72 kDa type IV collagenase (MMP-2) in human malignant gliomas in vivo. Clin Exp Metastasis 14(1):35–42
Saxena A, Shriml LM, Dean M, Ali IU (1999) Comparative molecular genetic profiles of anaplastic astrocytomas/glioblastomas multiforme and their subsequent recurrences. Oncogene 18(6):1385–1390
Schafer C, Fels C, Brucke M, Holzhausen HJ, Bahn H, Wellman M, Visvikis A, Fischer P, Rainov NG (2001) Gamma-glutamyl transferase expression in higher-grade astrocytic glioma. Acta Oncol 40(4):529–535
Schulte A, Gunther HS, Phillips HS, Kemming D, Martens T, Kharbanda S, Soriano RH, Modrusan Z, Zapf S, Westphal M, Lamszus K (2011) A distinct subset of glioma cell lines with stem cell-like properties reflects the transcriptional phenotype of glioblastomas and overexpresses CXCR4 as therapeutic target. Glia 59(4):590–602
Schulz I, Zeitschel U, Rudolph T, Ruiz-Carrillo D, Rahfeld JU, Gerhartz B, Bigl V, Demuth HU, Rossner S (2005) Subcellular localization suggests novel functions for prolyl endopeptidase in protein secretion. J Neurochem 94(4):970–979
Schwab ME, Caroni P (1988) Oligodendrocytes and CNS myelin are nonpermissive substrates for neurite growth and fibroblast spreading in vitro. J Neurosci 8(7):2381–2393
Schwartzbaum J, Ahlbom A, Malmer B, Lonn S, Brookes AJ, Doss H, Debinski W, Henriksson R, Feychting M (2005) Polymorphisms associated with asthma are inversely related to glioblastoma multiforme. Cancer Res 65(14):6459–6465
Scrideli CA, Carlotti CG Jr, Okamoto OK, Andrade VS, Cortez MA, Motta FJ, Lucio-Eterovic AK, Neder L, Rosemberg S, Oba-Shinjo SM, Marie SK, Tone LG (2008) Gene expression profile analysis of primary glioblastomas and non-neoplastic brain tissue: identification of potential target genes by oligonucleotide microarray and real-time quantitative PCR. J Neurooncol 88(3):281–291
Seals DF, Courtneidge SA (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev 17(1):7–30
Seeds NW, Siconolfi LB, Haffke SP (1997) Neuronal extracellular proteases facilitate cell migration, axonal growth, and pathfinding. Cell Tissue Res 290(2):367–370
Seidah NG, Chretien M (1999) Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res 848(1–2):45–62
Seidah NG, Sadr MS, Chretien M, Mbikay M (2013) The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J Biol Chem 288(30):21473–21481
Senner V, Ratzinger S, Mertsch S, Grassel S, Paulus W (2008) Collagen XVI expression is upregulated in glioblastomas and promotes tumor cell adhesion. FEBS Lett 582(23–24):3293–3300
Seol DW (2011) p53-Independent up-regulation of a TRAIL receptor DR5 by proteasome inhibitors: a mechanism for proteasome inhibitor-enhanced TRAIL-induced apoptosis. Biochem Biophys Res Commun 416(1–2):222–225
Serres E, Debarbieux F, Stanchi F, Maggiorella L, Grall D, Turchi L, Burel-Vandenbos F, Figarella-Branger D, Virolle T, Rougon G, Van Obberghen-Schilling E (2013) Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene. doi:10.1038/onc.2013.305
Shankar A, Kumar S, Iskander A, Varma NR, Janic B, Decarvalho A, Mikkelsen T, Frank JA, Ali MM, Knight RA, Brown S, Arbab AS (2014) Subcurative radiation significantly increases proliferation, invasion, and migration of primary GBM in vivo. Chin J Cancer 33(3):148–158
Shen J, Prywes R (2004) Dependence of site-2 protease cleavage of ATF6 on prior site-1 protease digestion is determined by the size of the luminal domain of ATF6. J Biol Chem 279(41):43046–43051
Shevtsova Z, Garrido M, Weishaupt J, Saftig P, Bahr M, Luhder F, Kugler S (2010) CNS-expressed cathepsin D prevents lymphopenia in a murine model of congenital neuronal ceroid lipofuscinosis. Am J Pathol 177(1):271–279
Shimizu C, Yoshida S, Shibata M, Kato K, Momota Y, Matsumoto K, Shiosaka T, Midorikawa R, Kamachi T, Kawabe A, Shiosaka S (1998) Characterization of recombinant and brain neuropsin, a plasticity-related serine protease. J Biol Chem 273(18):11189–11196
Shiosaka S, Ishikawa Y (2011) Neuropsin–a possible modulator of synaptic plasticity. J Chem Neuroanat 42(1):24–29
Sierko E, Wojtukiewicz MZ, Kisiel W (2007) The role of tissue factor pathway inhibitor-2 in cancer biology. Semin Thromb Hemost 33(7):653–659
Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 129(Pt 6):1438–1445
Silacci P, Mazzolai L, Gauci C, Stergiopulos N, Yin HL, Hayoz D (2004) Gelsolin superfamily proteins: key regulators of cellular functions. Cell Mol Life Sci 61(19–20):2614–2623
Sivaparvathi M, Sawaya R, Wang SW, Rayford A, Yamamoto M, Liotta LA, Nicolson GL, Rao JS (1995) Overexpression and localization of cathepsin B during the progression of human gliomas. Clin Exp Metastasis 13(1):49–56
Sivaparvathi M, Sawaya R, Gokaslan ZL, Chintala SK, Rao JS (1996a) Expression and the role of cathepsin H in human glioma progression and invasion. Cancer Lett 104(1):121–126
Sivaparvathi M, Sawaya R, Chintala SK, Go Y, Gokaslan ZL, Rao JS (1996b) Expression of cathepsin D during the progression of human gliomas. Neurosci Lett 208(3):171–174
Sivaparvathi M, Yamamoto M, Nicolson GL, Gokaslan ZL, Fuller GN, Liotta LA, Sawaya R, Rao JS (1996c) Expression and immunohistochemical localization of cathepsin L during the progression of human gliomas. Clin Exp Metastasis 14(1):27–34
Skiriute D, Vaitkiene P, Saferis V, Asmoniene V, Skauminas K, Deltuva VP, Tamasauskas A (2012) MGMT, GATA6, CD81, DR4, and CASP8 gene promoter methylation in glioblastoma. BMC Cancer 12:218
Snuderl M, Fazlollahi L, Le LP, Nitta M, Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD, Betensky RA, Louis DN, Iafrate AJ (2011) Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 20(6):810–817
Sounni NE, Rozanov DV, Remacle AG, Golubkov VS, Noel A, Strongin AY (2010) Timp-2 binding with cellular MT1-MMP stimulates invasion-promoting MEK/ERK signaling in cancer cells. Int J Cancer 126(5):1067–1078
Sower LE, Payne DA, Meyers R, Carney DH (1999) Thrombin peptide, TP508, induces differential gene expression in fibroblasts through a nonproteolytic activation pathway. Exp Cell Res 247(2):422–431
Stephan A, Mateos JM, Kozlov SV, Cinelli P, Kistler AD, Hettwer S, Rulicke T, Streit P, Kunz B, Sonderegger P (2008) Neurotrypsin cleaves agrin locally at the synapse. FASEB J 22(6):1861–1873
Stetler-Stevenson WG, Seo DW (2005) TIMP-2: an endogenous inhibitor of angiogenesis. Trends Mol Med 11(3):97–103
Stockhausen MT, Kristoffersen K, Poulsen HS (2010) The functional role of Notch signaling in human gliomas. Neuro Oncol 12(2):199–211
Stojic J, Hagemann C, Haas S, Herbold C, Kuhnel S, Gerngras S, Roggendorf W, Roosen K, Vince GH (2008) Expression of matrix metalloproteinases MMP-1, MMP-11 and MMP-19 is correlated with the WHO-grading of human malignant gliomas. Neurosci Res 60(1):40–49
Stremenova J, Krepela E, Mares V, Trim J, Dbaly V, Marek J, Vanickova Z, Lisa V, Yea C, Sedo A (2007) Expression and enzymatic activity of dipeptidyl peptidase-IV in human astrocytic tumours are associated with tumour grade. Int J Oncol 31(4):785–792
Strojnik T, Zajc I, Bervar A, Zidanik B, Golouh R, Kos J, Dolenc V, Lah T (2000) Cathepsin B and its inhibitor stefin A in brain tumors. Pflugers Arch 439(3 Suppl):R122–R123
Strojnik T, Kavalar R, Trinkaus M, Lah TT (2005) Cathepsin L in glioma progression: comparison with cathepsin B. Cancer Detect Prev 29(5):448–455
Strojnik T, Kavalar R, Zajc I, Diamandis EP, Oikonomopoulou K, Lah TT (2009) Prognostic impact of CD68 and kallikrein 6 in human glioma. Anticancer Res 29(8):3269–3279
Strongin AY (2010) Proteolytic and non-proteolytic roles of membrane type-1 matrix metalloproteinase in malignancy. Biochim Biophys Acta 1803(1):133–141
Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI (1995) Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 270(10):5331–5338
Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ, A. L.-G. S. Group (2006) Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol 7(5):392–401
Stylli SS, Kaye AH, Lock P (2008) Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci 15(7):725–737
Sumitomo M, Shen R, Walburg M, Dai J, Geng Y, Navarro D, Boileau G, Papandreou CN, Giancotti FG, Knudsen B, Nanus DM (2000) Neutral endopeptidase inhibits prostate cancer cell migration by blocking focal adhesion kinase signaling. J Clin Invest 106(11):1399–1407
Sun L, Yan W, Wang Y, Sun G, Luo H, Zhang J, Wang X, You Y, Yang Z, Liu N (2011) MicroRNA-10b induces glioma cell invasion by modulating MMP-14 and uPAR expression via HOXD10. Brain Res 1389:9–18
Sun C, Wang Q, Zhou H, Yu S, Simard AR, Kang C, Li Y, Kong Y, An T, Wen Y, Shi F, Hao J (2013) Antisense MMP-9 RNA inhibits malignant glioma cell growth in vitro and in vivo. Neurosci Bull 29(1):83–93
Szalad A, Katakowski M, Zheng X, Jiang F, Chopp M (2009) Transcription factor Sp1 induces ADAM17 and contributes to tumor cell invasiveness under hypoxia. J Exp Clin Cancer Res 28:129
Takahashi M, Fukami S, Iwata N, Inoue K, Itohara S, Itoh H, Haraoka J, Saido T (2002) In vivo glioma growth requires host-derived matrix metalloproteinase 2 for maintenance of angioarchitecture. Pharmacol Res 46(2):155–163
Takano S, Mashiko R, Osuka S, Ishikawa E, Ohneda O, Matsumura A (2010) Detection of failure of bevacizumab treatment for malignant glioma based on urinary matrix metalloproteinase activity. Brain Tumor Pathol 27(2):89–94
Takebe N, Nguyen D, Yang SX (2014) Targeting Notch signaling pathway in cancer: clinical development advances and challenges. Pharmacol Ther 141(2):140–149
Talieri M, Zoma M, Devetzi M, Scorilas A, Ardavanis A (2012) Kallikrein-related peptidase 6 (KLK6)gene expression in intracranial tumors. Tumour Biol 33(5):1375–1383
Tani E, Kitagawa H, Ikemoto H, Matsumoto T (2001) Proteasome inhibitors induce Fas-mediated apoptosis by c-Myc accumulation and subsequent induction of FasL message in human glioma cells. FEBS Lett 504(1–2):53–58
Tao J, Qiu B, Zhang D, Wang Y (2012) Expression levels of Fas/Fas-L mRNA in human brain glioma stem cells. Mol Med Rep 5(5):1202–1206
Tao BB, He H, Shi XH, Wang CL, Li WQ, Li B, Dong Y, Hu GH, Hou LJ, Luo C, Chen JX, Chen HR, Yu YH, Sun QF, Lu YC (2013) Up-regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J Clin Neurosci 20(5):717–720
Thapa N, Lee BH, Kim IS (2007) TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-beta. Int J Biochem Cell Biol 39(12):2183–2194
Thorns V, Walter GF, Thorns C (2003) Expression of MMP-2, MMP-7, MMP-9, MMP-10 and MMP-11 in human astrocytic and oligodendroglial gliomas. Anticancer Res 23(5A):3937–3944
Tirapelli LF, Bolini PH, Tirapelli DP, Peria FM, Becker AN, Saggioro FP, Carlotti CG Jr (2010) Caspase-3 and Bcl-2 expression in glioblastoma: an immunohistochemical study. Arq Neuropsiquiatr 68(4):603–607
Tomimatsu Y, Idemoto S, Moriguchi S, Watanabe S, Nakanishi H (2002) Proteases involved in long-term potentiation. Life Sci 72(4–5):355–361
Tremblay P, Beaudet MJ, Tremblay E, Rueda N, Thomas T, Vallieres L (2011) Matrix metalloproteinase 2 attenuates brain tumour growth, while promoting macrophage recruitment and vascular repair. J Pathol 224(2):222–233
Tsatas D, Kaye AH (2003) The role of the plasminogen activation cascade in glioma cell invasion: a review. J Clin Neurosci 10(2):139–145
Tsopanoglou NE, Andriopoulou P, Maragoudakis ME (2002) On the mechanism of thrombin-induced angiogenesis: involvement of alphavbeta3-integrin. Am J Physiol Cell Physiol 283(5):C1501–C1510
Turk B (2006) Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov 5(9):785–799
Turk V, Stoka V, Turk D (2008) Cystatins: biochemical and structural properties, and medical relevance. Front Biosci 13:5406–5420
Turk B, Turk D, Turk V (2012a) Protease signalling: the cutting edge. EMBO J 31(7):1630–1643
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D (2012b) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824(1):68–88
Uchinokura S, Miyata S, Fukushima T, Itoh H, Nakano S, Wakisaka S, Kataoka H (2006) Role of hepatocyte growth factor activator (HGF activator) in invasive growth of human glioblastoma cells in vivo. Int J Cancer 118(3):583–592
Unterkircher T, Cristofanon S, Vellanki SH, Nonnenmacher L, Karpel-Massler G, Wirtz CR, Debatin KM, Fulda S (2011) Bortezomib primes glioblastoma, including glioblastoma stem cells, for TRAIL by increasing tBid stability and mitochondrial apoptosis. Clin Cancer Res 17(12):4019–4030
Vaillant C, Didier-Bazes M, Hutter A, Belin MF, Thomasset N (1999) Spatiotemporal expression patterns of metalloproteinases and their inhibitors in the postnatal developing rat cerebellum. J Neurosci 19(12):4994–5004
Vaillant C, Meissirel C, Mutin M, Belin MF, Lund LR, Thomasset N (2003) MMP-9 deficiency affects axonal outgrowth, migration, and apoptosis in the developing cerebellum. Mol Cell Neurosci 24(2):395–408
Van De Craen B, Declerck PJ, Gils A (2012) The biochemistry, physiology and pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb Res 130(4):576–585
van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, Clevers H (2005) Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435(7044):959–963
van Hinsbergh VW, Engelse MA, Quax PH (2006) Pericellular proteases in angiogenesis and vasculogenesis. Arterioscler Thromb Vasc Biol 26(4):716–728
Van Hove I, Lemmens K, Van de Velde S, Verslegers M, Moons L (2012) Matrix metalloproteinase-3 in the central nervous system: a look on the bright side. J Neurochem 123(2):203–216
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ (2010) Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60(3):166–193
Van Meter TE, Broaddus WC, Rooprai HK, Pilkington GJ, Fillmore HL (2004) Induction of membrane-type-1 matrix metalloproteinase by epidermal growth factor-mediated signaling in gliomas. Neuro Oncol 6(3):188–199
VanMeter TE, Rooprai HK, Kibble MM, Fillmore HL, Broaddus WC, Pilkington GJ (2001) The role of matrix metalloproteinase genes in glioma invasion: co-dependent and interactive proteolysis. J Neurooncol 53(2):213–235
Vazquez F, Hastings G, Ortega MA, Lane TF, Oikemus S, Lombardo M, Iruela-Arispe ML (1999) METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem 274(33):23349–23357
Veeravalli KK, Ponnala S, Chetty C, Tsung AJ, Gujrati M, Rao JS (2012) Integrin alpha9beta1-mediated cell migration in glioblastoma via SSAT and Kir4.2 potassium channel pathway. Cell Signal 24(1):272–281
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, Getz G, Perou CM, Hayes DN (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1):98–110
Viapiano MS, Matthews RT (2006) From barriers to bridges: chondroitin sulfate proteoglycans in neuropathology. Trends Mol Med 12(10):488–496
Viapiano MS, Bi WL, Piepmeier J, Hockfield S, Matthews RT (2005) Novel tumor-specific isoforms of BEHAB/brevican identified in human malignant gliomas. Cancer Res 65(15):6726–6733
Viapiano MS, Hockfield S, Matthews RT (2008) BEHAB/brevican requires ADAMTS-mediated proteolytic cleavage to promote glioma invasion. J Neurooncol 88(3):261–272
Vince GH, Wagner S, Pietsch T, Klein R, Goldbrunner RH, Roosen K, Tonn JC (1999) Heterogeneous regional expression patterns of matrix metalloproteinases in human malignant gliomas. Int J Dev Neurosci 17(5–6):437–445
Viry E, Baginska J, Berchem G, Noman MZ, Medves S, Chouaib S, Janji B (2014) Autophagic degradation of GZMB/granzyme B: a new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 10(1):173–175
Vlachostergios PJ, Voutsadakis IA, Papandreou CN (2012) The ubiquitin-proteasome system in glioma cell cycle control. Cell Div 7(1):18
Vlachostergios PJ, Voutsadakis IA, Papandreou CN (2013) Mechanisms of proteasome inhibitor-induced cytotoxicity in malignant glioma. Cell Biol Toxicol 29(4):199–211
Vlashi E, Kim K, Lagadec C, Donna LD, McDonald JT, Eghbali M, Sayre JW, Stefani E, McBride W, Pajonk F (2009) In vivo imaging, tracking, and targeting of cancer stem cells. J Natl Cancer Inst 101(5):350–359
Vlashi E, Mattes M, Lagadec C, Donna LD, Phillips TM, Nikolay P, McBride WH, Pajonk F (2010) Differential effects of the proteasome inhibitor NPI-0052 against glioma cells. Transl Oncol 3(1):50–55
Wagenknecht B, Hermisson M, Eitel K, Weller M (1999) Proteasome inhibitors induce p53/p21-independent apoptosis in human glioma cells. Cell Physiol Biochem 9(3):117–125
Wagenknecht B, Hermisson M, Groscurth P, Liston P, Krammer PH, Weller M (2000) Proteasome inhibitor-induced apoptosis of glioma cells involves the processing of multiple caspases and cytochrome c release. J Neurochem 75(6):2288–2297
Waltereit R, Weller M (2002) The role of caspases 9 and 9-short (9S) in death ligand- and drug-induced apoptosis in human astrocytoma cells. Brain Res Mol Brain Res 106(1–2):42–49
Wang Z, Juttermann R, Soloway PD (2000) TIMP-2 is required for efficient activation of proMMP-2 in vivo. J Biol Chem 275(34):26411–26415
Wang M, Tang J, Liu S, Yoshida D, Teramoto A (2005) Expression of cathepsin B and microvascular density increases with higher grade of astrocytomas. J Neurooncol 71(1):3–7
Wang L, Rahn JJ, Lun X, Sun B, Kelly JJ, Weiss S, Robbins SM, Forsyth PA, Senger DL (2008a) Gamma-secretase represents a therapeutic target for the treatment of invasive glioma mediated by the p75 neurotrophin receptor. PLoS Biol 6(11):e289
Wang Y, Luo W, Reiser G (2008b) Trypsin and trypsin-like proteases in the brain: proteolysis and cellular functions. Cell Mol Life Sci 65(2):237–252
Wang J, Li Y, Wang J, Li C, Yu K, Wang Q (2012) Increased expression of matrix metalloproteinase-13 in glioma is associated with poor overall survival of patients. Med Oncol 29(4):2432–2437
Wang H, Zhang S, Zhong J, Zhang J, Luo Y, Pengfei G (2013a) The proteasome inhibitor lactacystin exerts its therapeutic effects on glioma via apoptosis: an in vitro and in vivo study. J Int Med Res 41(1):72–81
Wang L, Yuan J, Tu Y, Mao X, He S, Fu G, Zong J, Zhang Y (2013b) Co-expression of MMP-14 and MMP-19 predicts poor survival in human glioma. Clin Transl Oncol 15(2):139–145
Warich M, von Bossanyi P, Dietzmann K (1995) Expression of cathepsin D in human astrocytic neoplasias. Gen Diagn Pathol 141(2):93–96
Wei X, Lv T, Chen D, Guan J (2014) Lentiviral vector mediated delivery of RHBDD1 shRNA down regulated the proliferation of human glioblastoma cells. Technol Cancer Res Treat 13(1):87–93
Weis SM, Cheresh DA (2011) Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med 17(11):1359–1370
Weller M, Pfister SM, Wick W, Hegi ME, Reifenberger G, Stupp R (2013) Molecular neuro-oncology in clinical practice: a new horizon. Lancet Oncol 14(9):e370–e379
Wernicke AG, Edgar MA, Lavi E, Liu H, Salerno P, Bander NH, Gutin PH (2011) Prostate-specific membrane antigen as a potential novel vascular target for treatment of glioblastoma multiforme. Arch Pathol Lab Med 135(11):1486–1489
Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, Franciszkiewicz K, Chouaib S, Kaminska B (2008) Microglia-derived TGF-beta as an important regulator of glioblastoma invasion–an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 27(7):918–930
Westermarck J, Kahari VM (1999) Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J 13(8):781–792
Westphal D, Sytnyk V, Schachner M, Leshchyns’ka I (2010) Clustering of the neural cell adhesion molecule (NCAM) at the neuronal cell surface induces caspase-8- and -3-dependent changes of the spectrin meshwork required for NCAM-mediated neurite outgrowth. J Biol Chem 285(53):42046–42057
Wick W, Wild-Bode C, Frank B, Weller M (2004) BCL-2-induced glioma cell invasiveness depends on furin-like proteases. J Neurochem 91(6):1275–1283
Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W (2001) Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res 61(6):2744–2750
Wildeboer D, Naus S, Amy Sang QX, Bartsch JW, Pagenstecher A (2006) Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J Neuropathol Exp Neurol 65(5):516–527
Wilkinson KD (2009) DUBs at a glance. J Cell Sci 122(Pt 14):2325–2329
Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, Truman JW (2006) Local caspase activity directs engulfment of dendrites during pruning. Nat Neurosci 9(10):1234–1236
Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK (2004) Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 6(6):553–563
Withana NP, Blum G, Sameni M, Slaney C, Anbalagan A, Olive MB, Bidwell BN, Edgington L, Wang L, Moin K, Sloane BF, Anderson RL, Bogyo MS, Parker BS (2012) Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res 72(5):1199–1209
Wolf M, Albrecht S, Marki C (2008) Proteolytic processing of chemokines: implications in physiological and pathological conditions. Int J Biochem Cell Biol 40(6–7):1185–1198
Wolpert F, Tritschler I, Steinle A, Weller M, Eisele G (2014) A disintegrin and metalloproteinases 10 and 17 modulate the immunogenicity of glioblastoma-initiating cells. Neuro Oncol 16(3):382–391
Wright JW, Harding JW (2009) Contributions of matrix metalloproteinases to neural plasticity, habituation, associative learning and drug addiction. Neural Plast 2009:579382
Wu DG, Wang YY, Fan LG, Luo H, Han B, Sun LH, Wang XF, Zhang JX, Cao L, Wang XR, You YP, Liu N (2011) MicroRNA-7 regulates glioblastoma cell invasion via targeting focal adhesion kinase expression. Chin Med J (Engl) 124(17):2616–2621
Xia H, Qi Y, Ng SS, Chen X, Li D, Chen S, Ge R, Jiang S, Li G, Chen Y, He ML, Kung HF, Lai L, Lin MC (2009) microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res 1269:158–165
Xiao C, Yang BF, Asadi N, Beguinot F, Hao C (2002) Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J Biol Chem 277(28):25020–25025
Xie H, Xue YX, Liu LB, Wang P, Liu YH, Ying HQ (2011) Expressions of matrix metalloproteinase-7 and matrix metalloproteinase-14 associated with the activation of extracellular signal-regulated kinase1/2 in human brain gliomas of different pathological grades. Med Oncol 28(Suppl 1):S433–S438
Xu Y, Zhong Z, Yuan J, Zhang Z, Wei Q, Song W, Chen H (2013) Collaborative overexpression of matrix metalloproteinase-1 and vascular endothelial growth factor-C predicts adverse prognosis in patients with gliomas. Cancer Epidemiol 37(5):697–702
Yamada H, Fredette B, Shitara K, Hagihara K, Miura R, Ranscht B, Stallcup WB, Yamaguchi Y (1997) The brain chondroitin sulfate proteoglycan brevican associates with astrocytes ensheathing cerebellar glomeruli and inhibits neurite outgrowth from granule neurons. J Neurosci 17(20):7784–7795
Yamaguchi Y, Shao Z, Sharif S, Du XY, Myles T, Merchant M, Harsh G, Glantz M, Recht L, Morser J, Leung LL (2013) Thrombin-cleaved fragments of osteopontin are overexpressed in malignant glial tumors and provide a molecular niche with survival advantage. J Biol Chem 288(5):3097–3111
Yamahata H, Takeshima H, Kuratsu J, Sarker KP, Tanioka K, Wakimaru N, Nakata M, Kitajima I, Maruyama I (2002) The role of thrombin in the neo-vascularization of malignant gliomas: an intrinsic modulator for the up-regulation of vascular endothelial growth factor. Int J Oncol 20(5):921–928
Yamamoto M, Sawaya R, Mohanam S, Bindal AK, Bruner JM, Oka K, Rao VH, Tomonaga M, Nicolson GL, Rao JS (1994a) Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Res 54(14):3656–3661
Yamamoto M, Sawaya R, Mohanam S, Rao VH, Bruner JM, Nicolson GL, Rao JS (1994b) Expression and localization of urokinase-type plasminogen activator receptor in human gliomas. Cancer Res 54(18):5016–5020
Yamamoto M, Mohanam S, Sawaya R, Fuller GN, Seiki M, Sato H, Gokaslan ZL, Liotta LA, Nicolson GL, Rao JS (1996) Differential expression of membrane-type matrix metalloproteinase and its correlation with gelatinase A activation in human malignant brain tumors in vivo and in vitro. Cancer Res 56(2):384–392
Yan S, Berquin IM, Troen BR, Sloane BF (2000) Transcription of human cathepsin B is mediated by Sp1 and Ets family factors in glioma. DNA Cell Biol 19(2):79–91
Yan W, Zhang W, Sun L, Liu Y, You G, Wang Y, Kang C, You Y, Jiang T (2011) Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res 1411:108–115
Yanamandra N, Gumidyala KV, Waldron KG, Gujrati M, Olivero WC, Dinh DH, Rao JS, Mohanam S (2004) Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene 23(12):2224–2230
Yang P, Baker KA, Hagg T (2006) The ADAMs family: coordinators of nervous system development, plasticity and repair. Prog Neurobiol 79(2):73–94
Yang BF, Xiao C, Li H, Yang SJ (2007) Resistance to Fas-mediated apoptosis in malignant tumours is rescued by KN-93 and cisplatin via downregulation of c-FLIP expression and phosphorylation. Clin Exp Pharmacol Physiol 34(12):1245–1251
Yang M, Li Y, Chilukuri K, Brady OA, Boulos MI, Kappes JC, Galileo DS (2011) L1 stimulation of human glioma cell motility correlates with FAK activation. J Neurooncol 105(1):27–44
Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, Xiao HL, Wang B, Yi L, Wang QL, Jiang XF, Yang L, Zhang P, Qian C, Cui YH, Zhang X, Bian XW (2012) Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-beta1 signaling pathway. J Immunol 189(1):444–453
Yeh WL, Lu DY, Lee MJ, Fu WM (2009) Leptin induces migration and invasion of glioma cells through MMP-13 production. Glia 57(4):454–464
Yin D, Zhou H, Kumagai T, Liu G, Ong JM, Black KL, Koeffler HP (2005) Proteasome inhibitor PS-341 causes cell growth arrest and apoptosis in human glioblastoma multiforme (GBM). Oncogene 24(3):344–354
Yong VW, Power C, Forsyth P, Edwards DR (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2(7):502–511
Yu Q, Stamenkovic I (1999) Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev 13(1):35–48
Zadran S, Jourdi H, Rostamiani K, Qin Q, Bi X, Baudry M (2010) Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J Neurosci 30(3):1086–1095
Zanotto-Filho A, Braganhol E, Battastini AM, Moreira JC (2012) Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFkappaB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Invest New Drugs 30(6):2252–2262
Zerrouqi A, Pyrzynska B, Febbraio M, Brat DJ, Van Meir EG (2012) P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. J Clin Invest 122(4):1283–1295
Zhang X, Fei Z, Bu X, Zhen H, Zhang Z, Gu J, Chen Y (2000) Expression and significance of urokinase type plasminogen activator gene in human brain gliomas. J Surg Oncol 74(2):90–94
Zhang J, Sarkar S, Yong VW (2005) The chemokine stromal cell derived factor-1 (CXCL12) promotes glioma invasiveness through MT2-matrix metalloproteinase. Carcinogenesis 26(12):2069–2077
Zhang Y, Zhan H, Xu W, Yuan Z, Lu P, Zhan L, Li Q (2011) Upregulation of matrix metalloproteinase-1 and proteinase-activated receptor-1 promotes the progression of human gliomas. Pathol Res Pract 207(1):24–29
Zhao Y, Lyons CE Jr, Xiao A, Templeton DJ, Sang QA, Brew K, Hussaini IM (2008a) Urokinase directly activates matrix metalloproteinases-9: a potential role in glioblastoma invasion. Biochem Biophys Res Commun 369(4):1215–1220
Zhao Y, Xiao A, Dipierro CG, Abdel-Fattah R, Amos S, Redpath GT, Carpenter JE, Pieper RO, Hussaini IM (2008b) H-Ras increases urokinase expression and cell invasion in genetically modified human astrocytes through Ras/Raf/MEK signaling pathway. Glia 56(8):917–924
Zhao Y, Xiao A, diPierro CG, Carpenter JE, Abdel-Fattah R, Redpath GT, Lopes MB, Hussaini IM (2010) An extensive invasive intracranial human glioblastoma xenograft model: role of high level matrix metalloproteinase 9. Am J Pathol 176(6):3032–3049
Zheng X, Jiang F, Katakowski M, Kalkanis SN, Hong X, Zhang X, Zhang ZG, Yang H, Chopp M (2007) Inhibition of ADAM17 reduces hypoxia-induced brain tumor cell invasiveness. Cancer Sci 98(5):674–684
Zheng X, Jiang F, Katakowski M, Lu Y, Chopp M (2012) ADAM17 promotes glioma cell malignant phenotype. Mol Carcinog 51(2):150–164
Zheng X, Chopp M, Lu Y, Buller B, Jiang F (2013) MiR-15b and miR-152 reduce glioma cell invasion and angiogenesis via NRP-2 and MMP-3. Cancer Lett 329(2):146–154
Zhou Y, Zhang J, Liu Q, Bell R, Muruve DA, Forsyth P, Arcellana-Panlilio M, Robbins S, Yong VW (2005) The chemokine GRO-alpha (CXCL1) confers increased tumorigenicity to glioma cells. Carcinogenesis 26(12):2058–2068
Zincircioglu SB, Kaplan MA, Isikdogan A, Cil T, Karadayi B, Dirier A, Kucukoner M, Inal A, Yildiz I, Aggil F, Donmez O, Urakci Z, Pekkolay Z, Firat U (2012) Contribution of low-molecular weight heparin addition to concomitant chemoradiotherapy in the treatment of glioblastoma multiforme. J BUON 17(1):124–127
Zou Y, Cao Y, Yue Z, Liu J (2013) Gamma-secretase inhibitor DAPT suppresses glioblastoma growth via uncoupling of tumor vessel density from vessel function. Clin Exp Med 13(4):271–278
Acknowledgment
This work was supported by grants IGA 12237-5/2011, PRVOUK-P27/LF1/1, UNCE 204013 and by the project “BIOCEV—Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University” (CZ.1.05/1.1.00/02.0109), from the European Regional Development Fund.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Wien
About this chapter
Cite this chapter
Busek, P., Prevorovsky, M., Krepela, E., Sedo, A. (2014). Glioma-Associated Proteases. In: Sedo, A., Mentlein, R. (eds) Glioma Cell Biology. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1431-5_12
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1431-5_12
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1430-8
Online ISBN: 978-3-7091-1431-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)