
Abstract
Lignin is a complex heterogeneous aromatic polymer consisting of up to 30 % of plant material. Its aromatic structure suggests that it is a possible renewable source for aromatic chemicals. However, the natural complexity and high stability of lignin makes its depolymerization a highly challenging task. Many efforts have been directed toward a better understanding of the structure and composition of lignin in order to design more efficient and greener deconstruction paths. This chapter aims at providing an overview of key advances in the field of lignin depolymerization, with special emphasis on chemical catalysis, ionic liquids, and biocatalysis. The various technologies are discussed and critically evaluated in terms of potential for further industrial implementation. Research gaps that still need to be addressed and the most promising approaches are highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Introduction
Together with cellulose and hemicellulose, lignin is an important component of plant material constituting up to 30 % of the weight and 40 % of the fuel value of biomass [1]. Lignin is a complex three-dimensional amorphous polymer consisting of phenylpropanoid units of various types, which makes it the widest natural source of bio-based aromatic chemicals.
Despite this singularly attractive chemical property, only few commercial examples of lignin application exist besides its use as energy source [2]. Most of the lignin markets are limited to low value products, e.g., dispersing, binding, or stabilizing agents. Efforts have also been done to develop higher value applications of lignin in its macromolecular form, which includes using lignin in thermoplastics, phenolic and epoxy resins, polyurethane foams, or carbon fibers. These polymeric applications have been well summarized in several reviews and chapters [3, 4] and are not discussed herein.
In this chapter we focus on the production of value-added low molecular weight chemicals from lignin. Except for the production of vanillin and DMSO from lignin sulfonate by Borregaard in Norway [5, 6], most commercial lignin depolymerization routes are still in their infancy. Several reasons explain the limited commercial maturity of these reactions. Among them are: (i) the recalcitrance of lignin toward both chemical and biochemical attacks [7] and, (ii) the complexity of the reaction media produced which imply high separation costs. Today, the valorization of lignin into biochemicals represents a real challenge in terms of both sustainability and environmental protection [5, 8, 9]. The need to develop high-volume production of aromatics from lignin is an urgent matter considering the depletion of fossil resources. To be advantageous against the currently available production of petroleum-based aromatics, the new processes have to be efficient, economical, and environmentally friendly. The present chapter aims at summarizing new promising green approaches for the depolymerization of lignin.
10.2 Lignin Structure and Properties
Lignin is biosynthesized in plants by free-radical polymerization from three phenolic monomers, namely p-coumaryl alcohol, coniferyl alcohol, or sinapyl alcohol which correspond to the p-hydroxyphenyl (H), guaiacyl (G), or syringyl (S) units of lignin, respectively (Fig. 10.1). The proportion of each unit varies with the type of biomass, the part of the plant, the growing conditions, the geographical growing area, the extraction process, and even sometimes with the analytical methods used for characterization. Despite these heterogeneities, general trends have been drawn for various families of lignin. Softwood lignin consists almost exclusively of G units, while hardwood lignin contains a mixture of S and G units, and grass lignin contains minor amounts of H units in addition to G and S units [1]. The additional methoxyl groups on the aromatic rings of hardwood and grass lignins prevent formation of resistant 5-5′ (aryl aryl) linkages, and thus cause hardwood and grass lignins to form more linear and less condensed structures compared to softwood.

Lignin building blocks and their corresponding units
The phenylpropanoid units of lignin are bound together by ether bonds or C–C bonds which play a major role in determining the reactivity of lignin. In native lignin, ether bonds represent two thirds of the linkages or more, while the rest corresponds to C–C bonds [10]. A generic schematic representation of lignin is provided in Fig. 10.2 that includes the major functional groups and linkages known to arise in softwood, hardwood, or grass lignins. Major linkages occurring between the structural units of lignin are β-O-4 (β-aryl ether), 5-5′ (aryl aryl), β-5 (phenylcoumaran), α-O-4 (α-aryl ether), and 4-O-5 (diaryl ether) (Table 10.1), with the β-O-4 linkage being, by far, the most abundant. Other linkages, such as α-O-γ (aliphatic ether), β–β (resinol), and β-1 (spirodienone) can also be found in lignin [10–12]. The most abundant and “easy” to break β-O-4 linkage has attracted the most attention in attempts to depolymerize lignin.

Schematic representation of lignin (1: β-O-4; 2: 5-5′; 3: β-5/α-O-4; 4: α-O-4; 5: 4-O-5′; 6: β-1; 7: β–β; 8: spirodienone; 9: dibenzodioxocin)
The chemical structure of grass lignins is much less understood than that of wood lignins, likely due to their more complex structural features, their wider variability from species to species, and their more recent entry in the world of biorefineries as byproduct of cellulosic ethanol production [15–17]. Nevertheless, it has been recurrently observed that lignin in non-woody plants is naturally acetylated, coumarylated, and/or p-hydroxybenzoylated. Grass lignins are acylated by p-coumaric acid at the γ-position of lignin side chains, usually on the S units [15, 18]. Ferulic acid can also be present in smaller amounts with linkage occurring through either ether or ester bonds [19]. The content of ester groups in grass lignin is strongly affected by the pulping conditions, with acid or alkaline reagents acting as hydrolysis promoters.
Analyses of lignin chemical structure including its monolignol ratio, inter-unit linkages and functional groups have advanced considerably over the two last decades owing to the substantial progress of NMR technology. In particular, two-dimensional (2D) NMR correlation experiments, such as heteronuclear single quantum coherence (HSQC) spectroscopy, have contributed enormously to the elucidation of lignin subunits and lignin carbohydrate complexes [20, 21]. 2D NMR, however, has its limitations since it is not quantitative and it can lead to various overlapping signals. A complementary, more quantitative, approach was developed by Argyropoulos and his team, which consists of derivatizing all OH groups of lignin with an appropriate phosphorous reagent and analyzing quantitatively the phosphorylated lignin by 31P NMR [18, 22]. Since the pioneering work of Argyropoulos, quantitative 31P NMR has been widely used to quantify the aliphatic, phenolic, and carboxylic OH groups of numerous wood and grass lignins (Table 10.2). Knowledge of the content of various G, S, or H groups in lignin is a predeterminant of the type of target chemicals that can be obtained by depolymerization of a specific lignin.
While the original distribution of phenolic groups in lignin is mainly governed by the biomass, the absolute content of aliphatic, carboxylic, and in lesser extent phenolic OH groups is largely dependent on the process applied for its isolation. A full description of the production process, properties, and applications of various commercial lignins, including sulfite, kraft, soda, or organosolv lignin has been provided by Lora [2] and reader are referred to the preceding chapter of this book as well. Milled wood lignin obtained by milling in water/dioxane is the lignin that is the closest to native lignin [30]. It is generally rich in aliphatic OHs and poorer in phenolic and carboxylic OHs. Heated alkaline or acid processes such as soda and kraft processes or organosolv processes, respectively, induce a decrease in aliphatic OH groups, and an increase in either carboxylic or phenolic OH groups.
In summary, with its unique structure and chemical properties, lignin represents the only viable renewable source for the production of aromatic compounds. However, its high complexity and variability imply that lignin needs to be well characterized in order to target the desired chemicals from an appropriate source. In addition, lignin heterogeneity also implies that transformation processes need to be selective enough to limit the cost of product purification.
10.3 Chemical Catalysis for Depolymerization of Lignin
Lignin can be depolymerized using technologies including pyrolysis [31–33], homogenous acid/base-catalysis [12, 34–41], and thermal hydrogenolysis [32, 42, 43]. However due to harsh conditions used and a lack of selectivity, most of these techniques give rise to a large number of monomeric products in addition to substantial amounts of unwanted side products such as char and light gases. Such a broad product array requires dedicated separation efforts and a wide variety of catalytic reforming in order to upgrade all the products into useful chemicals. Chemical catalysis has thus been regarded as a key technology to tackle, in a more efficient and selective way, the chemical and biological resistance of lignin. Many catalytic pathways have been reported to depolymerise lignin or its model compounds, e.g., β-ether dimers and oligomers, under oxidative or reducing conditions, and several comprehensive reviews are available on the subject [7, 10, 11, 44].
10.3.1 Reductive Lignin Depolymerization
Most studies reported on reductive depolymerization of lignin have been focused on the production and upgrading of bio-oils and fuels [11] that fall beyond the scope of this chapter. Non-catalyzed thermochemical conversion of lignin into monomeric phenols has also been well documented, as reviewed by Roy and coworkers [42] and more recently by Pandey and Kim [32]. In this section, we focus on the production of phenols or arenes from lignin using catalytic reductive reactions as opposed to pure thermal processes.
Catalytic reduction of lignin or its model compounds normally occurs at temperatures within the range 100–400 °C and gives rise to aromatic compounds, such as phenols, benzene, toluene, xylene, and even alkanes if hydrogen upgrading is involved [10]. Reactions of a substrate with hydrogen, from H2 or from a hydrogen donor, can be of three main types: hydrogenation, i.e., saturation of C=O, C=C, or C≡C bonds; hydrogenolysis, i.e., cleavage of carbon–carbon or carbon–heteroatom bonds; or hydrodeoxygenation, i.e., removal of oxygen from oxygenated substrates. Hydrogenation generally occurs simultaneously with hydrogenolysis or hydrodeoxygenation, and is largely dependent on the catalyst system and reaction conditions. Since ether bonds constitute the most frequent linkages in lignin, many examples of lignin hydrogenolysis can also be considered as hydrodeoxygenation reactions. Table 10.3 gathers reported examples of hydrogenation and hydrodeoxygenation (or hydrogenolysis) applied to real lignins or whole biomass.
The first attempts to reductively depolymerize lignin have been conducted in the 1930s. Since then, many studies have been reported that consist of pressurized molecular hydrogen and a monometallic or bimetallic catalyst either in a solubilized or supported form (Table 10.3). The use of catalysts enhances hydrodeoxygenation by suppressing the formation of char and increasing the yield of oil [32]. Nickel (especially nickel-molybdenum) catalysts have been the most studied catalysts in lignin hydrotreatment. High partial hydrogen pressure (>1 MPa) and high temperature (often >300 °C) have both been shown to favor higher lignin conversion rates [51]. However, although conversion rates higher than 60 % have been repeatedly reported in lignin hydrotreatment (Table 10.3) the actual yields of individual phenols or arenes present in the resulting oils rarely exceed few tens of mg per g of lignin (<1 wt% of lignin). In addition, in most of these studies the stability and reusability of the catalyst have not been well documented.
A patent by Engel and Steigleder claimed that hydrocracking of Kraft lignin gave rise to high yields of phenols and cresols using a supported Ni-tungsten catalyst [47]. Nevertheless, the individual yields of phenolics were not provided, and the reaction was carried out in phenol, which constitutes a major environmental barrier to the industrial development of this process.
The use of fossil-fuel derived H2 for deoxygenation of lignin affects the carbon footprint of the processes. Finding greener sources of H2 could therefore contribute to reducing the negative environmental impact of lignin depolymerization. Tetralin has been used as a source of hydrogen in various hydrocracking processes but it is not renewable and it is often associated with phenol or cresol used as solvent [66]. A renewable source of hydrogen that has recently attracted growing attention for depolymerization of lignin into phenolics is formic acid, obtainable from cellulose. This first non-catalyzed method involves the treatment of lignin in a high pressure reactor using formic acid as active H2 donor and alcohol as solvent [67, 68]. Upon heating at 300 °C, formic acid decomposes into CO2 and H2, and the latter combines with oxygen from the methoxyl groups of lignin to form water, thus reducing the number of possible phenolic products. While promising, the method still requires optimization in terms of yields, product isolation, and solvent recovery. A second approach combines the use of formic acid with that of catalysts [7, 60, 62, 63]. Ni-based catalyst with formic acid was found to provide optimum depolymerization results as compared to noble metals including Rh, Ru, Pd, and Pt. The use of aluminium-substituted mesoporus material (Al-SBA-15) as support was advantageous due to its good (hydro) thermal stability, large surface area, and high acidity which, like for formic acid, helps quenching aromatic radicals. Importantly, Ni-catalysts supported on Al-SBA-15 were shown to be fairly stable and reusable under the investigated reaction conditions [62, 63]. Formic acid thus appears as a promising H2 replacement for the reductive depolymerization of lignin. In spite of these positive results, the separation of a complex product mixture and the utilization of an expensive formic acid reagent are key issues to be addressed in order to bring this technology to industrial applications [7].
An alternative solution to the complex downstream processing of mixtures of high boiling phenols is to proceed to the reduction of the resulting phenols into arenes or diarenes, expected to be more volatile and hence more easily valorizable using conventional refinery processes. Recently, Rinaldi and his group managed to deconstruct organosolv lignin by combining transfer hydrogenation (using isopropanol), hydrogenolysis with a Raney-Nickel catalyst, and an acidic beta-zeolite [69]. The proposed approach was conducted at relatively mild temperatures (150–240 °C) and preliminary results indicated the cleavage of ether linkages, demethoxylation of phenol units, and dehydroxylation of phenol intermediates resulting in 40 % conversion of lignin into oil mainly composed of arenes and alkanes. Jongerius et al. [70] also applied a two-step approach consisting of (i) lignin depolymerization over a 1 % Pt/Al2O3 catalyst at 225 °C in alkaline ethanol–water, and (ii) subsequent hydrodeoxygenation of the lignin-oil obtained using CoMo/Al2O3 and Mo2/Carbon nanofiber at 300 °C. Although encouraging, these complex two-step processes still require an increase of yields and selectivity before they could move to industrial scale.
In view of the difficulty to convert polymeric lignin into simple phenols in high yield, researchers have started to investigate the possible deconstruction of lignin directly from the lignocellulose matrix [64, 65, 71]. In a remarkable study, Song et al. designed a Ni-based magnetically separable catalyst able to depolymerise native birch wood lignin into monomeric phenols [64]. The proposed methodology conducted in methanol, ethanol, or ethylene glycol, at moderate temperatures (200 °C), and under argon (active H species were provided by the alcohol), achieved conversions of ca. 50 % and lignin was selectively cleaved into propylguaiacol and propylsyringol at >90 %. This is one of the first methodologies that allow converting lignin selectively, in green solvent, under relatively mild conditions, and with a catalyst that can be recovered and reused. Parsell et al. also reported a system applicable to whole lignocellulosics, i.e., a Zn/Pd/C catalyst that convert lignin in intact hardwood biomass directly into dihydroeugenol and 2,6-dimethoxy-4-propyl phenol, leaving behind the carbohydrates as a solid residue [65]. Interestingly, the system worked better for intact lignocellulose than for organosolv lignin and the leftover carbohydrate residue was easily hydrolysable by cellulases. Finally, Van den Bosch et al. reported a biorefinery process where a Ru/C catalyst was able to yield carbohydrate pulp and lignin oil (50 % of phenolic monomers, mainly 4-n-propylguaiacol and 4-n-propylsyringol) from hardwood with a nearly complete retention of cellulose in the pulp [72]. These “lignin-first” strategies demonstrate that deconstruction of lignin directly from the lignocellulosic matrix could be done more selectively and efficiently than from the isolated lignin. The new “lignin-first” approach could possibly constitute the basis of new industrial developments for lignin depolymerization.
10.3.2 Oxidative Lignin Depolymerization
In comparison to reductive depolymerization, oxidation of lignin-like material occurs at milder temperatures (20–200 °C) and gives rise to polyfunctionalized aromatic chemicals including aldehydes, ketones, and acids. Oxygen, air, hydrogen peroxide, or peracids are oxidative agents that have been most investigated for this purpose; nitrobenzene has also been used but mainly for analytical purposes. Catalysts used for lignin oxidation comprise organometallics, metal oxides, metal-free organics, acid/base, or metal salts [10].
Among the reported oxidation methods, many use simple lignin model compounds that lack key lignin structural features and properties; only few have been devoted to lignin itself. Oxidative processes applied to actual lignin are summarized in Table 10.4. When targeting aromatic fine chemicals, hydrogen peroxide is normally avoided to prevent ring opening and retain the aromatic features of products. Oxygen is almost always used as oxidative agent due to its abundance, low cost, and environmental friendliness. It is often coupled with alkaline conditions that are used to enhance lignin dissolution and to favor oxidation reactions of ether bonds at the Cα and Cβ positions, C–C bond cleavage in the side chains, quinone methide formation, and/or nucleophilic addition of hydroxide ion on quinone methide [34].
Vanillin (or syringaldehyde) is generally obtained as the major product of oxidation, depending on the lignin origin, but at individual yield that rarely exceeds 10 wt% (Table 10.4). Vanillate (syringate) or acetovanillone (acetosyringone) are also formed in significant amount. At higher pH, the formation of aldehydes is favored versus that of acids [86]. Compounds, such as guaiacol and syringol, lactones, dimers, or organic acids are also formed although their miniscule amounts are usually not provided in the literature.
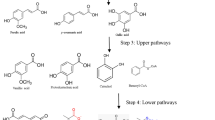
The main drawback of oxidative processes resides in the production of radicals that lead to partial re-polymerization and multiple reaction pathways associated to low selectivity [84]. Industrially, the only aromatic chemical that has ever been produced by oxidation of lignin is vanillin. Until the 1980s, vanillin was produced via alkaline oxidation of lignosulfonates, the byproduct resulting from sulfite pulping processes [5, 87]. However, with 160 kg of caustic liquids generated for each kg of vanillin produced, these processes raised environmental concerns and were largely abandoned in North America by the late 80’s. Today, Borregaard is the only company that produces vanillin from Norway spruce lignosulfonates. Although yields around 8 % have been reported in the literature for alkaline oxidation of lignin [78], Borregaard claimed a vanillin yield of 1 % [6], thus leaving plenty of room for the development of more efficient green processes that are more selective, economically viable, and bypass potentially harmful byproducts.
Kraft processes are currently the most widely used pulping processes in the world. Developing a depolymerization process that works well for Kraft lignin could thus be the key to success. New oxidation processes of Kraft lignin involving the use of polyoxometalates as reversible oxidants in combination with radical scavengers to prevent lignin fragments from repolymerizing have been reported [74, 75]. These new approaches that take place in alcohols and do not involve high concentrations of alkali processes are very promising in terms of environmental footprint. Another process was recently reported by Stahl and his group that allowed fairly high yields of syringyl compounds and their guaiacyl analogues, using a two-step process. This consists of (i) chemoselective aerobic organocatalytic oxidation of secondarybenzylic alcohols of lignin, and (ii) mild depolymerization of oxidized lignin in aqueous formic acid [84, 85] (Table 10.4). The metal-free process worked effectively for native hardwood (aspen) lignin. If applicable to Kraft lignins, and more specifically to the more condensed softwood Kraft lignins, it could open the door to a highly effective and selective system for the production of guaiacyl diketone and vanillin.
Oxidative depolymerization of lignin so far leads mainly to vanillin or syringaldehyde, along with many other phenolic compounds and organic acids. None of the metal-catalyzed lignin oxidation processes have been scaled up to an industrial level due in part to high amounts of alkali needed, low yields and selectivities, complex, and costly downstream treatments, as well as catalyst poisoning and reusability issues. Current processes lead to complex mixtures, which are economically and technically hard to fractionate into pure compounds [88]. Developing new non-alkaline chemical processes for oxidative lignin depolymerization as well as new technologies for vanillin/syringaldehyde isolation from the reaction medium therefore appears to be prerequisites to a successful industrial scale up of chemical production of vanillin or syringaldehyde from lignin. Alternatively, a way to make the new processes economically viable is to co-produce, along with vanillin, lignin fragments that are usable as macromonomers for the production of bio-based polymers. Such an approach was reported by Borges da Silva et al. where vanillin was co-produced with a polymer that is suitable for the production of polyurethane foams [86].
10.3.3 Summary
Although many homogeneous and heterogeneous catalysts have been investigated at the laboratory scale for lignin depolymerization, the industrial implementation of these catalysts is still hampered by the associated harsh conditions, low yields, and reusability issues. The usual products of lignin being either alcohols or aromatic compounds, solid catalysts remain very susceptible to surface saturation and deactivation by the coordinating products. New approaches aimed at depolymerizing lignin directly from lignocellulosic biomass appear to work under milder and greener conditions and might just be the way to go.
10.4 Photocatalysis
Photocatalysis has been used as a clean oxidative technology leading to the total mineralization of various organic pollutants including phenols and other aromatics [89, 90]. In particular, photocatalysis was proven to permit decolourization of paper mill effluents by completely degrading lignins and phenols into CO2 [91–98]. Titanium dioxide (TiO2) is generally considered to be the best photocatalyst for this reaction due to its high photosensitizing power, chemical stability, and commercial availability. In some cases, the photocatalytic activity of TiO2 can be enhanced by doping it with a noble metal such as platinum [95] or nonmetal ions such as sulfur or boron [99], or by adding ferrous iron in the medium [100]. Efforts have also been made to improve the separation of catalyst from the medium by immobilizing TiO2 on a support, such as sepiolite [97], carbon fibers [94], glass plates [99], or (CeO2, La2O3, or C) nanotubes [101]. Zinc oxide (ZnO) is another semiconductor material that was tested for lignin depolymerization and that appeared to be an even better photocatalyst than TiO2 for degradation of Kraft wheat straw lignin [102].
Since most of these photocatalytic systems were developed to decolorize pulping effluents, emphasis was usually put on the ability of the catalyst to decrease the color or chemical oxygen demand (COD) in the solution. Various parameters (pH, load of catalyst, temperature) effects were investigated but little effort was dedicated to the identification of lignin products. Ksibi et al. first identified phenolics by gas chromatography–mass spectrometry (GC-MS) when photodegrading soluble alfalfa lignin using a TiO2/UV photocatalytic technique under aerobic conditions [93]. When replacing air with H2O2, Kamwilaisak and Wright confirmed the formation of organic acids, namely acetic acid, malonic acid, and succinic acid, as main products of lignin TiO2-catalyzed photodegradation [103]. The photodegradation of phenolic compounds by TiO2 was suggested to occur via (i) ·OH radical attack on the phenyl rings producing catechol, resorcinol, and hydroquinone; (ii) phenyl ring opening to give malonic acid; (iii) degradation of malonic acid (and lignin side chains) to short-chain organic acids, such as maleic, oxalic, acetic, and formic acids; and (iv) release of CO2 by decarboxylation of formic acid. Interestingly, under certain conditions, succinic, and malonic acids were produced in significant amounts and photocatalysis appeared as a potential process to produce organic acids from lignin. The challenge for this reaction was to get a highly active photocatalyst that does not favor complete degradation of products into CO2. Keeping this in mind, Tonucci et al. designed a mild photocatalytic TiO2 system working under aerobic conditions, that showed low carbon consumption, good preservation of aromatic rings, and that greatly reduced mineralization [104]. The system was tested with lignosulfonates only. However, it would be interesting to see whether positive results can also be obtained with water-insoluble lignin.
10.5 Electrocatalysis
Electrochemical oxidation has also been considered as a potential alternative in lignin oxidative degradation. Various anodes ranging from simple ones (e.g., Pb, Ni [105, 106]) to more complex ones (Ti/Sb-SnO2, Ti/PbO2, Ti/Ta2O5-IrO2, Ti/SnO2-IrO2, Ti/RuO2-IrO2, and Ti/TiO2-IrO2) [107–109] have been used or designed for the purpose. Among the various IrO2-based electrodes tested, Ti/RuO2-IrO2 electrode exhibited the highest activity and stability [107]. Depending on the systems and the advancement of reaction, vanillin/vanillic acid or organic acids were identified as the main products. Vanillin and other low molecular weight phenolic intermediates can undergo condensation by phenolic dimerization or condensation of quinonic radicals [110]. Quinonic radicals can be decarbonylated and lead to cyclopentadienes; quinones can undergo ring opening to form various dicarboxylic acids that further evolve to simpler diacids, such as maleic or oxalic acid [110]. The production of phenolic compounds thus requires limiting secondary reactions. It can be enhanced by stopping the reaction at the early stage of the reaction [106] or by conducting continuous extraction to avoid further decomposition reactions of the primary intermediate products [111].
Although electrochemical oxidation is a potentially promising technology for lignin oxidation/modification, the high cost and the electrode fouling caused by condensation of intermediate products limit its application [107]. Electrocatalysts with high activity, long lifetime, and low cost have thus been the focus of the newest research for lignin electrooxidation [107]. Nevertheless, the yields of the products are still too low to make the overall process economically viable [10].
Various synergistic approaches aiming at improving the yield of depolymerization have been explored. An electrochemical approach combining anode oxidation and electrogenerated H2O2 oxidation has been recently developed for converting lignin into aromatic chemicals [109]. Using GC-MS for the analysis of reaction products, the authors confirmed that C–C and C–O–C bonds were cleaved synergistically by direct anodic oxidation and indirect H2O2 oxidation, and the macromolecules were gradually depolymerized into monomers and dimers. In another synergistic example, a combination of photoelectrocatalysis using a Ta2O5-IrO2 thin film as electrocatalyst, and TiO2 nanotube arrays as photocatalyst provided ca. 92 % lignin oxidation as compared to 66 % using a single electrochemical approach under similar conditions [112]. Finally, the electro-oxidative cleavage of alkali lignin was successfully conducted in a protic ionic liquid (IL), namely triethylammonium methanesulfonate, using a highly active Ru0.25V0.05Ti0.7O x anode [113]. Vanadium was identified as a crucial element in the electrode due to its ability to promote single electron transfer. The protic IL, with its ability for proton transfer from the acid to the base, provided a suitable medium for dissolution of lignin, ensured electrolysis at higher potentials and promoted the oxidative lignin cleavage mechanism. Interestingly, the product distribution was strongly affected by the applied potential. That higher potentials resulted in the formation of molecules of smaller molecular weights suggested the possibility of tuning the system (electrode and IL) toward specific selected products.
10.6 Ionic Liquids
Ionic liquids (ILs) are salts composed of large organic cations and inorganic or organic anions that exist as liquids at a relatively low temperature (<100 °C) [114, 115]. ILs tend to have good thermal and electrochemical stability, very low volatility, implying ease of recycling with virtually no VOCs (volatile organic compounds) release, and nonflammability, implying low or no risk of explosion [116–118]. The vast number of possible ion combinations in ILs results in a wide range of physical properties to choose from when selecting an IL for use as solvent or catalyst. The greener properties of ILs compared to conventional solvents associated with the extensive diversity of tunable properties made ILs the center of interest over the last few decades. Although most commercial developments involving ILs have been dedicated to petroleum-based reagents [119], new technologies are emerging where ILs are applied to pretreat, fractionate, or convert lignocellulosic materials.
10.6.1 Dissolution of Lignocellulosic Biomass in ILs
The transformation of lignocellulosic feedstocks has always been challenging due to their low solubility in almost any solvent [116]. Manufacturing of cellulose and its derivatives are normally conducted in environmentally undesirable media consisting of polar organic solvents mixed with charged compounds, e.g., dimethyl sulfoxide/tetrabutylammonium fluoride (DMSO/TBAF) or LiCl/dimethylacetamide (LiCl/DMAc) [116]. With their unique chemical inertness and solvation properties [120], ILs thus offer a promising greener alternative for the processing of biomass.
Interest in ILs for biomass dissolution started with the contribution of Rogers’ group who found that 1-butyl-3-methylimidazolium chloride, [BMIM][Cl], could dissolve cellulose at concentrations as high as 25 % and who then demonstrated that whole wood could be fully solubilized in 1-ethyl-3-methylimidazolium acetate, [EMIM][OAc] [121, 122]. A high number of studies reporting the processing of biomass in ILs then followed this pioneering work, with most studies being focused on cellulose dissolution and processing.
The easy solubilization of polysaccharides in ILs has been exploited for the development of pretreatment technologies aimed at increasing the enzymatic hydrolysis of cellulose [116, 118, 123, 124]. Indeed, it appears that cellulose obtained after reconstitution from IL solutions is less crystalline and therefore more accessible to cellulase than the original material [123].
Several reviews are now available on the combination of ILs with cellulose and lignocellulosic material that covered the dissolution mechanisms, the effect of ions on solubility, the constituent regeneration or reconstitution, and the use of ILs in pretreatment before cellulose hydrolysis [116–118, 125–129].
10.6.2 Dissolution of Lignin in ILs
Compared to the wide efforts dedicated to the understanding of dissolution of polysaccharides in ILs, much less is known about the potential of ILs to dissolve lignin. Several studies investigated the dissolution of whole biomass in ILs followed by the addition of antisolvent, e.g.,water or water/acetone, to precipitate cellulose-rich or lignin-rich fractions [122, 124, 130–132]. Solvents such as [BMIM][Cl] and 1-allyl-3-methylimidazolium chloride, [AMIM][Cl], gave the best results for both hardwood and softwood [123, 133]. [EMIM][OAc] also led to excellent results with complete dissolution of hardwood at 90 °C [122, 133] or softwood at >150 °C [131]. Significantly more rapid dissolution of biomass and more effective lignin removal were observed with increasing temperature, but higher temperatures were found to degrade the ILs and therefore decrease their recyclability [124, 131, 134].
Another approach makes use of ILs to allow dissolution of lignin while leaving behind the undissolved polysaccharide material. Excellent lignin extraction yields (>93 %) were achieved by Tan et al. using the aromatic alkylbenzenesulfonate (ABS) anion with BMIM cation [135]. Unfortunately, few drawbacks arose when using [BMIM][ABS] that include: (i) the relatively high extraction temperatures (190 °C), (ii) the necessity to pretreat the biomass with steam, (iii) a considerable loss of carbohydrate, and (iv) difficulties to recover the IL. Inspired by the initial results of Tan et al. [135], Pinkert et al. studied the dissolution of lignin using imidazolium acesulfamate ILs [136]. The authors’s choice was driven by three reasons: (i) the low cost and nontoxicity of potassium acesulfamate ([K][ace] is used as sugar substitute), (ii) previous studies suggested that large and bulky IL anions with delocalized charge do not dissolve cellulose, and (iii) the aromatic character of acesulfamate should allow a good interaction with lignin. Applying imidazolium [ace] to lignin dissolution from wood led to a lignin extraction yield of 43 % under gentle extraction conditions (T = 100 °C, t = 2 h), a steadiness of cellulose crystallinity, and a constancy of extraction yields over multiple sequential recycling of the IL (up to 6 runs). However, before any commercial plan could be realized for using [ace] IL to extract lignins from biomass some technical issues such as the sensitivity of the system to water present in biomass and the need to increase the recycling runs above 100 times, need to be addressed.
ILs were also used to dissolve isolated lignins. Different cations and anions screened for this purpose are summarized in Table 10.5. The role of cation consists in generating interactions between the IL and the biopolymer, whereas anion is primarily responsible for the initial disruption of the inter- and intramolecular hydrogen bonding that exist in the biopolymer. Imidazolium-based cations appeared to be efficient for the dissolution of lignin. Kilpelainen et al. suggested that π–π interactions exist between the imidazolium-based cations and the aromatic compounds of lignin [141], a hypothesis that was confirmed by the increased dissolution of softwood lignin when an allyl group replaced the butyl group of [BMIM][Cl] [123]. When dissolving softwood lignin with the cation [BMIM]+ and various anions, the solubility varies with the anions following the order: [OTf]− ~ [OMs]− ~ [MeSO4]− > > [OAc]− > [HCOO]− > [Cl]− ~ [Br]−> > [BF4]− ~ [PF6]−, suggesting that the strongly hydrogen-bonding anions such as [OTf]− or [OMs]− are efficient solvents for lignin, while the large non-coordinating anions, such as [BF4]− and [PF6]− are inefficient [118, 137, 138]. Chloride anion was used recently in combination with alkyl-diazabicyclo[5.4.0]undec-7-enium [DBUC n ] cation to dissolve softwood Kraft lignin but compared to imidazolium, these poorly unsaturated cations exhibited a modest dissolution power [140].
Recently, it was demonstrated that adding an appropriate amount of water to ILs (optimum ratio IL/H2O = 70/30) facilitated lignin dissolution, likely due to the increased mobility and diffusion constant of ions in the presence of water [138]. The addition of water promoted release of more ions from the IL stack, thus increasing the interaction probability between lignin and ILs.
10.6.3 Conversion of Lignin in ILs
A number of studies reporting the modification of lignin during IL biomass deconstruction have been published and recently reviewed by Brandt et al. [132]. Tan et al. reported that lignin extracted with [EMIM][ABS] had a lower molecular weight and a narrower polydispersity than one obtained by aqueous auto-catalyzed pretreatment [135]. Similarly, Kim et al. compared the characteristics of a lignin extracted from poplar wood using [EMIM][OAc] with the properties of milled wood lignin extracted from the same biomass and concluded that IL extraction led to a less polydisperse lignin of smaller molecular weight [130]. George et al. studied the impact of a range of ILs on several commercial lignins and demonstrated an intense effect of anion on the fragmentation mechanism and degree of depolymerization, while the cation did not play any significant role [142]. ILs containing alkyl sulfate anions appeared to have the greatest ability to fragment lignin and shorten polymer length. The order of molecular weight reduction was sulfate > lactate > acetate > chloride > phosphate. The anion was believed to undergo nucleophilic attack on the β-O-4 lignin linkages, thus reducing the average molecular weight of lignin.
Besides the observations on lignin chemical properties after IL treatment, ILs were also used as solvent for chemical transformation of lignin. Main research efforts addressing the conversion of lignin in ILs have been dedicated to either its acidolytic cleavage or catalytic oxidation. Moreover, most of the reported studies involved homogeneous or heterogeneous catalysis [118].
ILs can be used for lignin acidolytic depolymerization due to their ability to act as both an acidic catalyst and a solvent [7, 143]. For instance, 1-H-3-methylimidazolium chloride was shown to promote acidolytic cleavage of β-O-4 linkages in lignin over relatively mild temperatures (110–150 °C) [144]. The reaction was proposed to occur via protonation (or coordination) of the ether linkages, followed by attack of water (or any other nucleophile present in the system), similarly to the reactions taking place in conventional solvents.
ILs can also be used as solvent for the oxidative depolymerization of lignin, as reviewed recently [9]. A few good examples that use ILs as solvents for the production of aromatic chemicals from lignin are listed in Table 6 of Ref. [9]. Most of the oxidative reactions in ILs were applied to either organosolv or alkali lignin and were conducted using ILs based on phosphate or sulfonate anions due to (i) their stability against oxidation and, (ii) their ability to dissolve lignin. The oxidation of organosolv lignin in ILs in the presence of transition metals and molecular oxygen or air has been demonstrated by Stärk et al. [145], Weckhuysen and his team [146, 147], and Liu et al. [148]. Under optimal conditions, 66.3 % of the lignin could be converted into several monomeric units using Mn(NO3)2 in 1-ethyl-3-methylimidazolium trifluoromethanesulfonate [EMIM][CF3SO3] [145]. The predominant product was either dimethoxy-1,4-benzoquinone or syringaldehyde, depending upon the conditions applied. Interestingly, the potential antitumor agent, dimethoxy-1,4-benzoquinone, could be isolated in 11.5 wt% overall yield using a simple extraction/crystallization process [145]. Similarly, Alcell and soda lignins were oxidized under mild conditions (0.5 MPa O2, 80 °C) using CoCl2·6H2O and NaOH in 1-ethyl-3-methylimidazolium diethylphosphate [EMIM][Et2PO4]. The catalyst rapidly oxidized benzyl and other alcohol functionalities in lignin, but left phenolic functionality, and 5-5′, β-O-4 and phenylcoumaran linkages intact [146]. In situ ATR-FT-IR, Raman and UV-Vis spectroscopy allowed demonstrating that the reaction proceeded via the coordination of alcohol-containing substrates to cobalt, followed by formation of a Co-superoxo species [147]. Finally, in another very interesting study, under optimal conditions (2.5 MPa O2, 1.5 h, 175 °C), up to 100 % of the lignin could be converted into several monomeric units using CuSO4 in dimethyl phosphate-based ILs [148]. Using [MMIM][Me2PO4], a total yield of 29.7 % aldehydes (vanillin, syringaldehyde, p-hydroxybenzaldehyde) was obtained, which was significantly higher than the yields usually obtained in aqueous NaOH systems (see Table 10.4 for oxidation yields in aqueous systems). In contrast to the previous studies which did not address the recycling of IL, Liu et al. confirmed that the products and IL phase were easily separated by solvent extraction, allowing the recycling and reuse of the IL-CuSO4 phase up to six times without loss of efficiency [148].
10.6.4 Summary
Recent studies involving lignin and ILs demonstrate that ILs can be used (i) to extract selectively lignin from lignocellulosic biomass, and (ii) to oxidatively depolymerise lignin more selectively than in aqueous media. The possible control of selectivity by changing the experimental conditions (IL nature, reaction temperature, catalyst loading, extracting solvent, etc.) opens the door to new ILs-based processes for the production of value-added chemicals from lignin [9]. Although the conversion and depolymerization of lignin can be achieved in ILs, and even controlled by changing the structure of the IL or conditions, bulk separation of the products from ILs remains a formidable challenge that needs to be overcome [118].
10.7 Biocatalysis
The high selectivity and efficiency of enzymatic catalysts, the mild operating conditions required, the broad range of substrates, and the ability of some enzymes to react under adverse conditions (e.g., high temperatures, extreme pH values) are all advantageous properties when considering the application of biocatalysis for industrial processes [149]. Given the lack of selectivity observed when subjecting lignin to chemical treatments, one could see biocatalysis as a suitable means to depolymerise lignin in a selective, cleaner, and more environmental friendly way. However to date, no biocatalytic system has been shown to be efficient enough to access aromatics or non-aromatics from lignin in yields that could be commercialized.
In a desire to combine both an easier access of enzymes to cellulose for biofuel production and the valorization of lignin into value-added chemicals, there has been recently a renewed interest in the microbial breakdown of lignin [150]. While microbial degradation of lignin had been widely studied in white-rot and brown-rot fungi [151–154], emphasis has recently been placed on lignin-degrading bacteria which offer more opportunities for protein expression and genetic manipulation [155–159]. The present section is focused on recent progress in ligninolytic green biotechnology—either as microbes or enzymes—for the production of aromatic and non-aromatic chemicals.
10.7.1 Microbial Lignin Degradation
Lignin was reported to be biodegraded only under aerobic conditions [151]. The initial steps of lignin biodegradation consist in introducing new functional groups into its macromolecular structure by oxidative enzymes, which render lignin more susceptible towards its subsequent degradation by other enzymes [149]. Studies on the microbial degradation of lignin have focused primarily on white-rot and brown-rot fungi, with Phanerochaete chrysosporium being by far the most studied of all white-rot fungi [151, 152, 160]. White-rot fungi are much more active lignin-degrader than brown-rot fungi. Although white-rot fungi (basidiomycetes) do not use lignin as a carbon source for their growth, they have developed nonspecific methods for the degradation of lignin [161]. The initial depolymerization of lignin is thought to be promoted by extracellular oxidative enzymes (oxidoreductases) whereas subsequent transformation of smaller molecular weight lignin fragments is assumed to occur intracellularly [154]. The extracellular enzymes involved in lignin depolymerization are lignin peroxidases (LiP), manganese peroxidases (MnP), versatile peroxidases (VP) and phenol oxidases, also known as laccases.
Despite the extensive study of fungal lignin degradation since the mid-1980s, there is still no commercial biocatalytic process for lignin depolymerization, in part due to the practical challenges of fungal protein expression and fungal genetic manipulation [157]. In the last decade, efforts have thus been focused toward the breakdown of lignin by bacteria as indicated by the recent outbreak of reports on the subject [157, 159]. Various bacterial strains identified to have activity for lignin breakdown have been isolated from soils [158, 162–164] or from guts of termites [165, 166]. Most of these strains fall within three classes: actinomycetes, α-proteobacteria, and γ-proteobacteria [157]. Their activity is often less than that of white-rot fungus P. chrysosporium, but comparable to other lignin-degrading fungi [153]. Although the metabolic pathway of lignin-degrading bacteria is much less understood than that of white-rot and brown-rot fungi, there are indications that bacteria use similar types of extracellular enzymes found in fungi, i.e., peroxidases and laccases [157]. The next section provides more details on the enzymatic pathways involved in lignin degradation and transformation.
10.7.2 Enzymatic Pathways
10.7.2.1 Lignin Peroxidases (LiP)
LiP were the first lignolytic enzymes to be isolated from P. chrysosporium in the mid 80’s. LiP are characterized by their high redox potential (~1.2 V), low pH optima and molecular mass varying from 37 to 50 kDa for different white-rot fungus strains [161]. Relatively non-specific to their substrates, LiP have been known to oxidize phenolic and non-phenolic aromatic substrates by abstracting one electron via a mechanism involving cation radicals. Its catalytic cycle is similar to other peroxidase enzymes, in which the resting state of the enzyme contains ferric heme, which reacts with H2O2 to form a compound I oxo-ferryl intermediate (two-electron oxidized form), and subsequently a compound II intermediate (one-electron oxidized form). Veratryl alcohol (VA), produced by ligninolytic fungi, has been proposed to act as a redox mediator, or electron shuttle. LiP differ from other peroxidases by the fact that the heme environment provides high redox potential to the oxo-ferryl complex, which allows oxidation of unusually high potential sites, such as aromatic rings [150, 159]. LiP oxidize the substrates in multistep electron transfers by forming intermediate radicals such as phenoxy radicals and VA radical cations. Phenoxy radicals can undergo nonenzymatic reactions such as repolymerization on the lignin polymer and/or Cα–Cβ breakdown, yielding p-quinones [161]. LiP are active on a wide range of aromatic compounds, such as VA, methoxybenzenes, and nonphenolic β-O-4 linked arylglycerol β-aryl ethers with a redox potential up to 1.4 V in the presence of H2O2 [167].
The main drawback in using purified LiP for oxidative depolymerization of lignin is the repolymerization reaction that occurs when the small phenolic products are not removed or consumed [168, 169]. The preference of LiP for phenolic lignin units favors the coupling of phenoxy radicals and increases the propensity to polymerize rather than depolymerise lignin samples under in vitro conditions [167]. A second drawback comes from the difficulties associated with the fast decay of H2O2, indispensable for LiP (and MnP) catalysis, and the inactivation of LiP (and MnP) for certain amounts of H2O2 [170].
10.7.2.2 Manganese Peroxidase (MnP)
MnP is a heme-containing glycoprotein of ~40–60 kDa, optimum pH of 4–7, and temperature of 40–60 °C, which was also first discovered in P. chrysosporium. It is produced and secreted by almost all basidiomycetes, including white-rot and various soil colonizing fungi [167]. Like LiP, MnP requires H2O2 as an oxidant, but in contrast to LiP, it also requires the presence of Mn2+ and chelators (organic acids) such as oxalate or malate. MnP catalyses the oxidation of Mn2+ to Mn3+, which in turn can oxidize a large number of phenolic substrates. Complexed Mn3+ is widely accepted as a diffusible oxidant, able to oxidize secondary substrates at a distance away from the active site of MnP [150]. The Mn3+-chelate complex oxidizes phenolic compounds (such as 2,6-dimethoxyphenol, guaiacol, 4-methoxyphenol and phenolic lignin residues), but is inactive on VA or nonphenolic substrates [167]. It is also small enough to diffuse into lignin or analogous structures, which are not necessarily available to the enzymes [161].
The drawbacks associated to the broader usage of LiP, i.e., repolymerization and inconvenient use of H2O2 apply to MnP.
10.7.2.3 Versatile Peroxidases (VP)
VP are glycoproteins which act in a bifunctional way by sharing typical features of the MnP and LiP fungal peroxidase families. Like MnP they oxidize Mn (II), and similar to LiP they oxidize both phenolic and non-phenolic aromatic compounds, including VA, methoxybenzenes, and lignin model compounds [167].
10.7.2.4 Laccases
Laccases (or benzenediol:oxygen oxidoreductase) are glycosylated blue multi-copper oxidoreductases, produced by many fungi, bacteria, plants, and insect. A comprehensive review on the ability of laccases to breakdown lignin is available elsewhere [171].
In plant, laccases are thought to participate in lignin biosynthesis, while in fungi, they are suspected to contribute to lignin depolymerization, though this last role is still unclear and controversial [150]. Laccases subtract one electron from phenolic–OH groups thus forming phenoxy radicals, which undergo polymerization via radical coupling. As a consequence, the polymerizing or depolymerizing effect of laccase treatment on native or technical lignin is ambiguous [171].
Laccase contains four copper ions of three different types: one type 1 (T1) copper, which gives laccase its characteristic blue color; one type 2 (T2) and two type 3 (T3) copper ions forming a trinuclear cluster. T1 is the site where substrate oxidation takes place, whereas O2 reduction occurs at the trinuclear cluster site [171].
Laccase alone is not able to break down nonphenolic units of lignin [171]. Indeed, laccases have low redox potentials (0.5–0.8 V) that restrict their action to the oxidation of the phenolic lignin fragments [167]. The restriction of laccase action to phenolic subunits can, however, be overcome by use of a small molecular weight mediator that acts as electron shuttle [172]. Mediators are believed to expand the reach of laccase thanks to their comparatively smaller size that helps them diffuse into the plant cell wall or polymeric structures. In addition, they also broaden the range of oxidizable substrates including non-phenolic units by having higher redox potential than laccase. Natural compounds such as 4-hydroxybenzylic alcohol, p-cinnamic acid, sinapic acid, or syringaldehyde can act as laccase mediators. Synthetic compounds such as 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS), 1-hydroxybenzotriazole (HBT), 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), or violuric acid can also be co-added with laccases to increase the activity of the latter [171].
In contrast to ligninolytic enzymes LiP, MnP, and VP, phenol oxidases such as laccases use O2 as the final electron acceptor rather than H2O2, which could be a great advantage for industrial applications. The main drawbacks, however, in using laccases for oxidative transformation of lignin are the non-fully elucidated action mode of laccases on lignin, especially the ability of laccase to polymerise and depolymerise lignin, and the low extracellular production of laccases in basidiomycete fungi.
10.7.2.5 Other Enzymes
The above ligninolytic enzymes are characterized by their technically unresolved lack of specificity and their high predisposition to provoke the repolymerization of previously released monolignols [173]. Bacterial enzymes may be superior to their fungal counterparts with regard to specificity, thermostability, halotolerance, and mediator dependency [161]. New types of enzymes are therefore being investigated to favor depolymerization of lignin into monomers. Various β-O-4 aryl ether cleaving enzymes or enzyme systems have been isolated from microbes including fungus 2BW-1 [174] and bacteria Sphingobium paucimobilis SYK-6 and Novosphingobium sp. [173, 175–178], and biochemically characterized. Biphenyl bond cleavage enzyme systems involving demethylation on one ring, followed by dioxygenase-catalyzed ring opening of the resulting catechol, and C–C hydrolase at the benzylic ketone site have also been reported [150]. A tetrahydrofolate-dependent O-demethylase gene was also isolated from S. paucimobilis SYK-6 and found to convert syringate into 3-O-methylgallate [179]. A review recently published by Bugg and Rahmanpour describes recent developments in the understanding of bacterial enzymes for lignin breakdown [180]. The enzymes covered in this review include dye-decolorizing peroxidases (DyP), bacterial laccases, and beta-etherase enzymes. While the knowledge of microbial lignin degradation pathways is still incomplete, the use of pathway engineering methods to construct genetically modified microbes to convert lignin to renewable chemicals (e.g., vanillin, adipic acid) via fermentation seems a possible way to go [180].
10.7.3 Small Chemicals Obtained from Lignin Using Biocatalysis
Oxidative breakdown of spruce wood lignin by P. chrysosporium was found to lead to 28 low molecular weight products, 10 of which were aromatic carboxylic acids obtained by Cα–Cβ cleavage, and 13 others were acyclic 2,4-hexadiene-1,6-dioic acids (e.g., muconic acid) resulting from oxidative ring cleavage [181]. Many similar phenolic compounds have been detected when treating lignin with bacterial lignin degraders such as S. paucimobilis or Bacillus sp. [150, 157]. However, the natural microbial systems tend to produce the aromatic chemicals as intermediates which are further degraded into smaller molecules, and even down to CO2 in the case of P. chrysosporium.
Recently, with the aim to produce vanillin in larger amounts and higher selectivity compared to chemical catalysis, a targeted pathway engineering strategy was applied to Rhodococcus jostii strain RHA1 [182]. When grown on minimal medium containing 2.5 % wheat straw lignocellulose and 0.05 % glucose, the strain in which the vanillin dehydrogenase gene had been deleted was found to accumulate vanillin with yields of up to 96 mg/L after 144 h, together with smaller amounts of ferulic acid and 4-hydroxybenzaldehyde. This pioneering work established that vanillin could be produced from lignin using a predictive gene deletion and demonstrated that lignin breakdown pathways could in principle be engineered for the production of aromatics.
Pathway engineering has also been used in Pseudomonas putida KT2440 to accumulate cis,cis-muconic acid from degradation of aromatic ring, via blockage of the protocatechuate cleavage pathway, and rerouting via catechol cleavage [183]. Using an alkaline pretreated liquor, muconic acid was obtained at a yield of 0.7 g/L after 24 h and subsequently reduced into adipic acid by Pd/C hydrogenation [183]. Interestingly, muconic acid could be obtained at high purity (>97 %) after a two-step purification. Given these two recent examples, metabolic engineering and synthetic biology seem to offer a tangible way for obtaining reasonable yields of chemicals from lignin via the reconstructed catabolic pathways.
10.7.4 Scale-up of Enzyme Production
Despite the potential industrial use of fungal peroxidases, the actual application of these enzymes in industrial processes is hampered by (i) the limited availability of the proteins in the natural hosts, and (ii) their rather low stability [170]. Efforts have been made to produce LiP and MnP recombinant proteins, using either homologous or heterologous host systems but those led to limited success [170]. Given the lack of efficient microbial expression systems for peroxidases, a significant number of studies have been carried out to express laccases genes in various fungal hosts. In spite of the active secretion of recombinant laccase in several heterologous systems, the levels of recombinant laccase were too low for industrial purposes [184]. The lack of efficient microbial expression systems together with the large amount of enzymes requirement constitute a serious bottleneck in the industrial application of fungal enzymes.
Until recently, the enzymology of bacterial lignin degradation was not well understood [180]. However, genome sequences of few lignin degraders, e.g., R. jostii RHA1 and P. putida KT2440, have recently become available. A better understanding of the metabolic pathway and their gene regulation should facilitate the goals of synthetic biology to address the bottlenecks encountered with fungal enzymes and hence engineer new routes for large scale production of lignin-derived chemicals from renewable feedstocks.
10.7.5 Summary
A wide variety of lignin-degrading fungi and bacteria have been identified, among which the white-rot fungi are the most active and the only ones that can completely break down lignin to CO2 and H2O. Aromatic carboxylic acids and aldehydes obtained by Cα–Cβ cleavage as well as acyclic 2,4-hexadiene-1,6-dioic acids resulting from ring cleavage are the products that have been the most often observed, usually as intermediates. The main extracellular enzymes participating in lignin degradation are the LiP, MnP, and laccases. Attempts to produce these enzymes using either homologous or heterologous host systems led to limited success. Efforts are currently centered on the elucidation of the enzymology of bacterial lignin breakdown. Metabolic engineering has been successfully applied recently to lignin degraders, allowing the production of chemicals like vanillin or muconic acid from lignin at concentration of 0.096 and 0.7 g/L, respectively. These promising results are encouraging signs for a great future for the biological depolymerization of lignin.
10.8 Computational Approaches
At first sight, the natural structural complexity of lignin seems rather incompatible with first-principles, computational approaches, which are limited with regard to the size and complexity of molecules they can be used for. Significant computational effort has been dedicated to understanding lignin model compounds reactivity. Few research groups have used β-O-4 lignin models to investigate homolytic bond dissociation energies [185–188], kinetic parameters [189, 190], and free-energy pathways occurring under various reaction conditions [191–193]. However, computational modeling of the heterogeneous lignin using the information gathered with only dimers would not have been representative.
In a recent computational screening approach, Kulik and coworkers have developed a chemical discovery technique to identify the chemically relevant putative fragments in eight of the known polymeric linkages of lignin [194]. From the identified cleavage pathways and resulting fragments the authors concluded that (i) ether bonds cleave readily in lignin polymers; (ii) biphenyl linkages are more recalcitrant; (iii) spirodienone linkages can fragment into many possible products with a high frequency of cleavage; and (iv) breaking an “easy” to break ether bond could also result in the spontaneous cleavage of harder to break biphenyl-like bonds [194]. While the two first observations had been previously made by others, the latter two were newly revealed by this theoretical approach. After the pioneering work conducted by Kulik’s team with lignin oligomers consisting of up to six monomers, one hopes to see environmentally friendly theoretical approaches unraveling more mysteries of lignin in the near future.
10.9 Conclusions and Future Outlook
Currently, all aromatic chemicals are produced from petroleum-based sources. Lignin is a complex heterogeneous aromatic polymer consisting of up to 30 % of plant material. Its aromatic structure suggests that it is a possible renewable source for aromatic chemicals. However, the natural complexity and high stability of lignin makes its depolymerization a highly challenging task. Despite a strong renewed interest for lignin depolymerization over the last decade, a very limited number of lignin reactions, e.g., production of vanillin from lignosulfonates, have been actually scaled up and commercialized.
On a general point of view, the majority of efforts dedicated to lignin conversion have been done using simple model compounds that lack key lignin structural features and properties. Efforts are therefore necessary to confirm the applicability of promising processes to actual lignins which themselves offer another level of challenge by being largely a heterogenous and poorly soluble material.
This chapter presents recent advances and challenges to be addressed in the field of lignin depolymerization, with emphasis on chemical catalysis, ILs, or microbial and enzymatic approaches.
Although many homogeneous and heterogeneous catalysts have been investigated at the laboratory scale for lignin depolymerization, the industrial implementation of these catalysts was hampered by the harsh conditions, low selectivity, low yields, and issues with the reusability of catalysts. Efforts have still to be done to (i) increase the activity and selectivity of chemical catalysts; (ii) increase the tolerance of catalysts to impurities, especially sulfur or water; (iii) increase the separability and reusability of catalysts; and (iv) develop new technologies able to isolate phenolic compounds efficiently and economically. The promising results (50 % depolymerization, >90 % selectivity) recently obtained using reductive catalyzed depolymerization of lignin directly from lignocellulosic biomass opened the door to a new approach for lignin depolymerization, i.e., the “lignin-first” processes. By involving milder and greener conditions than the depolymerization of isolated lignin, this new approach gives rise to more environmentally friendly reactions that are more selective and therefore less costly in product recovery. Though all “lignin-first” experiments have been conducted at the lab-scale so far, they are encouraging to demonstrate the technology on a larger scale, and more efforts should be pursued towards the scale-up of “lignin-first” biorefinery processes. Under oxidative conditions, continuous separation of products is an effective way to increase the reaction yields by limiting radical condensation of products. Dedicating more effort to the engineering needs of the oxidative depolymerization of lignin should allow producing aromatic aldehyde and acids in higher yields and higher purity.
ILs are seen as another new green option for lignin conversion. Indeed, the use of ILs is becoming more and more accepted in the chemical industry and recent studies involving lignin and ILs demonstrate that ILs can be applied (i) to extract selectively lignin from lignocellulosic biomass and (ii) to oxidatively depolymerise lignin more selectively than in aqueous media. Despite the tremendous progress done in the last few years on lignin depolymerization in ILs, bulk separation of the products from ILs remains a major challenge that researchers should continue dedicating their efforts to.
Finally, lignin can also be deconstructed biologically. Regardless of the strong activity of fungal peroxidases for lignin depolymerization, the actual application of these enzymes in industrial processes is yet to take place. Efforts are now being pursued to elucidate the enzymology of bacterial lignin breakdown, and metabolic engineering is underway to construct genetically modified microbes that can convert lignin into renewable chemicals. The recent case studies of vanillin production (0.096 g/L) from lignin or muconic acid (0.7 g/L) from an alkaline pretreated liquor blaze the trail for a potentially great future for the biological depolymerization of lignins.
References
Holladay J, White JF, Bozell JJ, Johnson D (2007) Top value-added chemicals from biomass. Vol. II: results of screening for potential candidates from biorefinery lignin; Report PNNL-16983. U.S. Department of Commerce: Springfield, VA, pp 1–79
Lora J (2008) Industrial commercial lignins: Sources, properties and application. In: Belgacem MN, Gandini A (eds) Monomers, polymers and composites from renewable resources. Elsevier, Amsterdam, pp 225–241
Gandini A, Belgacem MN (2008) Lignins as components of macromolecular materials. In: Belgacem MN, Gandini A (eds) Monomers, polymers and composites from renewable resources. Elsevier, Amsterdam, pp 243–271
Doherty WOS, Mousavioun P, Fellows CW (2011) Value-adding to cellulosic ethanol: lignin polymers. Ind Crop Prod 33:259–276
Hocking MB (1997) Vanillin: synthetic flavoring from spent sulfite liquor. J Chem Educ 74:1055–1059
Borregaard http://www.borregaard.com/Business-Areas/Other-Businesses/Borregaard-Ingredients/ (14 Dec 2015)
Xu C, Arancon RAD, Labidi J, Luque R (2014) Lignin depolymerisation strategies: towards valuable chemicals and fuels. Chem Soc Rev 43:7485–7500
Ragauskas AJ, Beckham GT, Biddy MJ, Chandra R, Chen F, Davis MF, Davison BH, Dixon RA, Gilna P, Keller M, Langan P, Naskar AK, Saddler JN, Tschaplinski TJ, Tuskan GA, Wyman CE (2014) Lignin valorization: improving lignin processing in the biorefinery. Science 344:1246843
Chatel G, Rogers RD (2014) Review: oxidation of lignin using ionic liquids-an innovative strategy to produce renewable chemicals. ACS Sustain Chem Eng 2:322–339
Li C, Zhao X, Wang A, Huber GW, Zhang T (2015) Catalytic transformation of lignin for the production of chemicals and fuels. Chem Rev 115:11559–11624
Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM (2010) The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev 110:3552–3599
Rodrigues Pinto PC, da Silva EAB, Rodrigues AE (2011) Insights into oxidative conversion of lignin to high-added-value phenolic aldehydes. Ind Eng Chem Res 50:741–748
Chakar FS, Ragauskas AJ (2004) Review of current and future softwood kraft lignin process chemistry. Ind Crop Prod 20:131–141
Capanema EA, Balakshin MY, Kadla JF (2005) Quantitative characterization of a hardwood milled wood lignin by nuclear magnetic resonance spectroscopy. J Agric Food Chem 53:9639–9649
Buranov AU, Mazza G (2008) Lignin in straw of herbaceous crops. Ind Crop Prod 28:237–259
Zhang AP, Lu FC, Sun RC, Ralph J (2009) Ferulate-coniferyl alcohol cross-coupled products formed by radical coupling reactions. Planta 229:1099–1108
Monteil-Rivera F, Phuong M, Ye M, Halasz A, Hawari J (2013) Isolation and characterization of herbaceous lignins for applications in biomaterials. Ind Crop Prod 41:356–364
Crestini C, Argyropoulos DS (1997) Structural analysis of wheat straw lignin by quantitative P-31 and 2D NMR spectroscopy. The occurrence of ester bonds and alpha-O-4 substructures. J Agric Food Chem 45:1212–1219
Scalbert A, Monties B, Lallemand J-Y, Guittet E, Rolando C (1985) Ether linkage between phenolic acids and lignin fractions from wheat straw. Phytochemistry 24:1359–1362
Ralph J, Marita J, Ralph S, Hatfield RD, Lu F, Ede RM, Peng J, Quideau S, Helm R, Grabber J, Kim H, Jimenez-Monteon G, Zhang Y, Jung H-JG, Landucci L, MacKay J, Sederoff R, Chapple C, Boudet A (1999) Solution-state NMR of lignins. In: Argyropoulos DS (ed) Advances in lignocellulosics characterization. Tappi Press, Atlanta, pp 55–108
Pu Y, Cao S, Ragauskas AJ (2011) Application of quantitative 31P NMR in biomass lignin and biofuel precursors characterization. Energy Environ Sci 4:3154–3166
Argyropoulos DS (1994) Quantitative phosphorus-31 NMR analysis of lignins, a new tool for the lignin chemist. J Wood Chem Technol 14:45–63
Cateto CA, Barreiro MF, Rodrigues AE, Brochier-Salon MC, Thielemans W, Belgacem MN (2008) Lignins as macromonomers for polyurethane synthesis: a comparative study on hydroxyl group determination. J Appl Polym Sci 109:3008–3017
Monteil-Rivera F, Paquet L (2015) Solvent-free catalyst-free microwave-assisted acylation of lignin. Ind Crop Prod 65:446–453
Yang Q, Wu SB, Lou R, Lv GJ (2011) Structural characterization of lignin from wheat straw. Wood Sci Technol 45:419–431
Hu G, Cateto C, Pu Y, Samuel R, Ragauskas AJ (2012) Structural characterization of switchgrass lignin after ethanol organosolv pretreatment. Energy Fuels 26:740–745
Hattalli S, Benaboura A, Ham-Pichavant F, Nourmamode A, Castellan A (2002) Adding value to Alfa grass (Stipa tenacissima L.) soda lignin as phenolic resins. 1. Lignin characterization. Polym Degrad Stab 75:259–264
Ahvazi B, Wojciechowicz O, Ton-That T-M, Hawari J (2011) Preparation of lignopolyols from wheat straw soda lignin. J Agric Food Chem 59:10505–10516
Moghaddam L, Zhang Z, Wellard RM, Bartley JP, Hara IMO, Doherty WOS (2014) Characterisation of lignins isolated from sugarcane bagasse pretreated with acidified ethylene glycol and ionic liquids. Biomass Bioenergy 70:498–512
Fengel D, Wegener G (1984) Chemical composition and analysis of wood. In: Fengel D, Wegener G (eds) Wood chemistry ultrastructure reactions. Walter de Gruyter, Berlin, pp 26–65
Jae J, Tompsett GA, Lin YC, Carlson TR, Shen J, Zhang T, Yang B, Wyman CE, Conner WC, Huber GW (2010) Depolymerization of lignocellulosic biomass to fuel precursors: maximizing carbon efficiency by combining hydrolysis with pyrolysis. Energy Environ Sci 3:358–365
Pandey MP, Kim CS (2011) Lignin depolymerization and conversion: a review of thermochemical methods. Chem Eng Technol 34:29–41
Wang K, Kim KH, Brown RC (2014) Catalytic pyrolysis of individual components of lignocellulosic biomass. Green Chem 16:727–735
Tarabanko VE, Petukhov DV, Selyutin GE (2004) New mechanism for the catalytic oxidation of lignin to vanillin. Kinet Catal 45:569–577
Wong Z, Chen K, Li J (2010) Formation of vanillin and syringaldehyde in an oxygen delignification process. Bioresoure 5:1509–1516
Araújo JDP, Grande CA, Rodrigues AE (2010) Vanillin production from lignin oxidation in a batch reactor. Chem Eng Res Des 88:1024–1032
Roberts VM, Stein V, Reiner T, Lemonidou A, Li X, Lercher JA (2011) Towards quantitative catalytic lignin depolymerization. Chem Eur J 17:5939–5948
Beauchet R, Monteil-Rivera F, Lavoie JM (2012) Conversion of lignin to aromatic-based chemicals (L-chems) and biofuels (L-fuels). Bioresour Technol 121:328–334
Gasson JR, Forchheim D, Sutter T, Hornung U, Kruse A, Barth T (2012) Modeling the lignin degradation kinetics in an ethanol/formic acid solvolysis approach. Part 1. Kinetic model development. Ind Eng Chem Res 51:10595–10606
Forchheim D, Gasson JR, Hornung U, Kruse A, Barth T (2012) Modeling the lignin degradation kinetics in an ethanol/formic acid solvolysis approach. Part 2. Validation and transfer to variable conditions. Ind Eng Chem Res 51:15053–15063
Toledano A, Serrano L, Labidi J (2012) Organosolv lignin depolymerisation with different base catalysts. J Chem Technol Biotechnol 87:1593–1599
Amen-Chen C, Pakdel H, Roy C (2001) Production of monomeric phenols by thermochemical conversion of biomass: a review. Bioresour Technol 79:277–299
Zakzeski J, Jongerius AL, Bruijnincx PCA, Weckhuysen BM (2012) Catalytic lignin valorization process for the production of aromatic chemicals and hydrogen. ChemSusChem 5:1602–1609
Bozell J (2014) Approaches to the selective catalytic conversion of lignin: a grand challenge for biorefinery development. In: Nicholas KM (ed) Selective catalysis for renewable feedstocks and chemicals. Springer, Berlin, pp 229–255
Harris EE, D’Ianni J, Adkins H (1938) Reaction of hardwood lignin with hydrogen. J Am Chem Soc 60:1467–1470
Kashima K, Maeda Y, Oshima M (1964) Method for liquefying lignin. Canadian Patent 700210
Engel DJ, Steigleder KZ (1987) Hydrocracking process for liquefaction of lignin. U.S. Patent 4,647,704
Urban P, Engel DJ (1988) Process for liquefaction of lignin U.S. Patent 4,731,491
Ratcliff MA, Johnson DK, Posey FL, Chum HL (1988) Hydrodeoxygenation of lignins and model compounds—scientific note. Appl Biochem Biotechnol 17:151–160
Meier D, Berns J, Faix O, Balfanz U, Baldauf W (1994) Hydrocracking of organocell lignin for phenol production. Biomass Bioenergy 7:99–105
Meier D, Ante R, Faix O (1992) Catalytic hydropyrolysis of lignin: influence of reaction conditions on the formation and composition of liquid products. Bioresour Technol 40:171–177
Oasmaa A, Alen R, Meier D (1993) Catalytic hydrotreatment of some technical lignins. Bioresour Technol 45:189–194
Oasmaa A, Johansson A (1993) Catalytic hydrotreating of lignin with water-soluble molybdenum catalyst. Energy Fuels 7:426–429
Torr KM, van de Pas DJ, Cazeils E, Suckling ID (2011) Mild hydrogenolysis of in-situ and isolated Pinus radiate lignins. Bioresour Technol 102:7608–7611
Horácek J, Homola F, Kubicková I, Kubicka D (2012) Lignin to liquids over sulfide catalysts. Catal Today 179:191–198
Song Q, Wang F, Xu J (2012) Hydrogenolysis of lignosulfonate into phenols over heterogeneous nickel catalysts. Chem Commun 48:7019–7021
Zhang JG, Asakura H, van Rijn J, Yang J, Duchesne P, Zhang B, Chen X, Zhang P, Saeys M, Yan N (2014) Highly efficient, NiAu-catalyzed hydrogenolysis of lignin into phenolic chemicals. Green Chem 16:2432–2437
Zhang JG, Teo J, Chen X, Asakura H, Tanaka T, Teramura K, Yan N (2014) A series of NiM (M = Ru, Rh, and Pd) bimetallic catalysts for effective lignin hydrogenolysis in water. ACS Catal 4:1574–1583
Ma XL, Tian Y, Hao WY, Ma R, Li YD (2014) Production of phenols from catalytic conversion of lignin over a tungsten phosphide catalyst. Appl Catal A 481:64–70
Xu W, Miller SJ, Agrawal PK, Jones CW (2012) Depolymerization and hydrodeoxygenation of switchgrass lignin with formic acid. ChemSusChem 5:667–675
Barta K, Warner GR, Beach ES, Anastas PT (2014) Depolymerization of organosolv lignin to aromatic compounds over Cu-doped porous metal oxides. Green Chem 16:191–196
Toledano A, Serrano L, Balu AM, Luque R, Pineda A, Labidi J (2013) Fractionation of organosolv lignin from olive tree clippings and its valorization to simple phenolic compounds. ChemSusChem 6:529–536
Toledano A, Serrano L, Pineda A, Romero AA, Luque R, Labidi J (2014) Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: catalyst screening. Appl Catal B Environ 145:43–55
Song Q, Wang F, Cai J, Wang Y, Zhang J, Yua W, Xu J (2013) Lignin depolymerization (LDP) in alcohol over nickel-based catalysts via a fragmentation–hydrogenolysis process. Energy Environ Sci 6:994–1007
Parsell T, Yohe S, Degenstein J, Jarrell T, Klein I, Gencer E, Hewetson B, Hurt M, Kim JI, ChoudhariH Saha B, Meilan R, Mosier N, Ribeiro F, Delgass WN, Chapple C, KenttamaaHI Agrawal R, Abu-Omar MM (2015) A synergistic biorefinery based on catalytic conversion of lignin prior to cellulose starting from lignocellulosic biomass. Green Chem 17:1492–1499
Vuori A, Bredenberg JB-S (1988) Liquefaction of Kraft lignin: 1. Primary reactions under mild thermolysis conditions. Holzforshung 42:155–161
Kleinert M, Barth T (2008) Phenols from lignin. Chem Eng Technol 31:736–745
Kleinert M, Barth T (2008) Towards a lignincellulosic biorefinery: direct one-step conversion of lignin to hydrogen-enriched biofuel. Energy Fuels 22:1371–1379
Wang X, Rinaldi R (2013) A route for lignin and bio-oil conversion: dehydroxylation of phenols into arenes by catalytic tandem reactions. Angew Chem Int Ed 52:11499–11503
Jongerius AL, Bruijnincx PCA, Weckhuysen BM (2013) Liquid-phase reforming and hydrodeoxygenation as a two-step route to aromatics from lignin. Green Chem 15:3049–3056
Yan N, Zhao C, Dyson PJ, Wang C, L-t Liu, Kou Y (2008) Selective degradation of wood lignin over noble-metal catalysts in a two-step process. ChemSusChem 1:626–629
Van den Bosch S, Schutyser W, Vanholme R, Driessen T, Koelewijn S-K, Renders T, De Meester B, Huijgen WJJ, Dehaen W, Courtin CM, Lagrain B, Boerjan W, Sels BF (2015) Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps. Energy Environ Sci 8:1748–1763
Crestini C, Pro P, Neri V, Saladino R (2005) Methyltrioxorhenium: a new catalyst for the activation of hydrogen peroxide to the oxidation of lignin and lignin model compounds. Bioorg Med Chem 13:2569–2578
Voitl T, von Rohr PR (2008) Oxidation of lignin using aqueous polyoxometalates in the presence of alcohols. ChemSusChem 1:763–769
Voitl T, von Rohr PR (2010) Demonstration of a process for the conversion of Kraft lignin into vanillin and methyl vanillate by acidic oxidation in aqueous methanol. Ind Eng Chem Res 49:520–525
Xiang Q, Lee YY (2001) Production of oxychemicals from precipitated hardwood lignin. Appl Biochem Biotechnol 91–93:71–80
Wu G, Heitz M (1995) Catalytic mechanism of Cu2+ and Fe3+ in alkaline O2 oxidation of lignin. J Wood Chem Technol 15:189–202
Bjørsvik H-R, Minisci F (1999) Fine chemicals from lignosulfonates. 1. Synthesis of vanillin by oxidation of lignosulfonates. Org Proc Res Dev 3:330–340
Azarpira A, Ralph J, Lu FC (2014) Catalytic alkaline oxidation of lignin and its model compounds: a pathway to aromatic biochemicals. BioEnergy Res 7:78–86
Villar JC, Caperos A, Garcia-Ochoa F (2001) Oxidation of hardwood kraft-lignin to phenolic derivatives with oxygen as oxidant. Wood Sci Technol 35:245–255
Sales FG, Maranhão LCA, Lima Filho NM, Abreu CAM (2007) Experimental evaluation and continuous catalytic process for fine aldehyde production from lignin. Chem Eng Sci 62:5386–5391
Deng HB, Lin L, Sun Y, Pang CS, Zhuang JP, Ouyang PK, Li ZJ, Liu SJ (2008) Perovskite-type oxide LaMnO3: an efficient and recyclable heterogeneous catalyst for the wet aerobic oxidation of lignin to aromatic aldehydes. Catal Lett 126:106–111
Zhou XF (2014) Selective oxidation of Kraft lignin over zeolite-encapsulated Co(II) [H4]salen and [H2]salen complexes. J Appl Polym Sci 131:40809(1-9)
Rahimi A, Azarpira A, Kim H, Ralph J, Stahl SS (2013) Chemoselective metal-free aerobic oxidation in lignin. J Am Chem Soc 135:6415–6418
Rahimi A, Ulbrich A, Coon JJ, Stahl SS (2014) Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 515:249–252
Borges da Silva EA, Zabkova M, Araújo JD, Cateto CA, Belgacem MF, Barreiro MN, Rodrigues AE (2009) An integrated process to produce vanillin and lignin-based polyurethanes from Kraft lignin. Chem Eng Res Des 87:1276–1292
Wong JT (2012) Technological, commercial, organizational, and social uncertainties of a novel process for vanillin production from lignin. MBA Project, Simon Fraser University
Fache M, Boutevin B, Caillol S (2016) Vanillin production from lignin and its use as a renewable chemical. ACS Sustain Chem Eng 4:35–46
Fox MA, Dulay MT (1993) Heterogeneous photocatalysis. Chem Rev 93:341–357
Lathasree S, Nageswara R, Sivasankar B, Sadasivam V, Rengaraj K (2004) Heterogeneous photocatalytic mineralization of phenols in aqueous solutions. J Mol Catal A Chem 223:101–105
Kobayakawa K, Sato Y, Nakamura S, Fujishima A (1989) Photodecomposition of kraft lignin catalyzed by TiO2. Bull Chem Soc Jpn 62:3433–3436
Tanaka K, Calanag CR, Hisanaga T (1999) Photocatalyzed degradation of lignin on TiO2. J Mol Catal A Chem 138:287–294
Ksibi M, Ben Amor S, Cherif S, Elaloui E, Houas A, Elaloui M (2003) Photodegradation of lignin from black liquor using a UV/TiO2 system. J Photochem Photobiol A 154:211–218
Yuan RS, Guan RB, Liu P, Zheng JT (2007) Photocatalytic treatment of wastewater from paper mill by TiO2 loaded on activated carbon fibers. Colloids Surf A 293:80–86
Ma YS, Chang CN, Chiang YP, Sung HF, Chao AC (2008) Photocatalytic degradation of lignin using Pt/TiO2 as the catalyst. Chemosphere 71:998–1004
Ugurlu M, Karaoglu MH (2009) Removal of AOX, total nitrogen and chlorinated lignin from bleached Kraft mill effluents by UV oxidation in the presence of hydrogen peroxide utilizing TiO2 as photocatalyst. Environ Sci Pollut Res 16:265–273
Ugurlu M, Karaoglu MH (2011) TiO2 supported on sepiolite: preparation, structural and thermal characterization and catalytic behaviour in photocatalytic treatment of phenol and lignin from olive mill wastewater. Chem Eng J 166:859–867
Ugurlu M, Karaoglu MH, Vaizogullar AI (2013) Decolourization and removal of phenol compounds from olive mill wastewater byO2/UV/NaBO2H2O2-SH2O. Fresenius Environ Bull 22:752–763
Portjanskaja E, Stepanova K, Klauson D, Preis S (2009) The influence of titanium dioxide modifications on photocatalytic oxidation of lignin and humic acids. Catal Today 144:26–30
Portjanskaja E, Preis S (2007) Aqueous photocatalytic oxidation of lignin: the influence of mineral admixtures. Int J Photoenergy 76730:1–7
Rangel R, Mercado GJL, Bartolo-Perez P, Garcia R (2012) Nanostructured-[CeO2, La2O3, C]/TiO2catalysts for lignin photodegradation. Sci Adv Mater 4:573–578
Kansal SK, Singh M, Sud D (2008) Studies on TiO2/ZnO photocatalysed degradation of lignin. J Hazard Mater 153:412–417
Kamwilaisak K, Wright PC (2012) Investigating laccase and titanium dioxide for lignin degradation. Energy Fuels 26:2400–2406
Tonucci L, Coccia F, Bressan M, Alessandro N (2012) Mild photocatalysed and catalysed green oxidation of lignin: A useful pathway to low-molecular-weight derivatives. Waste Biomass Valorization 3:165–174
Bailey A, Brooks HM (1946) Electrolytic oxidation of lignin. J Am Chem Soc 68:445–446
Moodley B, Mulholland DA, Brookes HC (2011) The electrooxidation of lignin in Sappi Saiccor dissolving pulp mill effluent. Water SA 37:33–40
Tolba R, Tian M, Wen J, Jiang Z-H, Chen A (2010) Electrochemical oxidation of lignin at IrO 2-based oxide electrodes. J Electroanal Chem 649:9–15
Shao D, Liang JD, Cui XM, Xu H, Yan W (2014) Electrochemical oxidation of lignin by two typical electrodes: Ti/Sb-SnO2 and Ti/PbO2. Chem Eng J 244:288–295
Zhu HB, Chen YM, Qin TF, Wang L, Tang Y, SunYZ Wan PY (2014) Lignin depolymerization via an integrated approach of anode oxidation and electro-generated H2O2 oxidation. RSC Adv 4:6232–6238
Constant S, Robitzer M, Quignard F, Di Renzo F (2012) Vanillin oligomerization as a model of side reactions in lignin fragmentation. Catal Today 189:123–128
Parpot P, Bettencourt AP, Carvalho AM, Belgsir EM (2000) Biomass conversion: attempted electrooxidation of lignin for vanillin production. J Appl Electrochem 30:727–731
Tian M, Wen J, MacDonald D, Asmussen RM, Chen A (2010) A novel approach for lignin modification and degradation. Electrochem Commun 12:527–530
Reichert E, Wintringer R, Volmer DA, Hempelmann R (2012) Electro-catalytic oxidative cleavage of lignin in a protic ionic liquid. Phys Chem Chem Phys 14:5214–5221
Wasserscheid P, Stark A (eds) (2010) Handbook of green chemistry: green solvents, vol 6—ionic liquids, Wiley-VCH
Peleteiro S, Rivas S, Alonso JL, Santos V, Parajo JC (2015) Utilization of ionic liquids in lignocellulose biorefineries as agents for separation, derivatization, fractionation, or pretreatment. J Agric Food Chem 63:8093–8102
Olivier-Bourbigou H, Magna L, Morvan D (2010) Ionic liquids and catalysis: Recent progress from knowledge to applications. Appl Catal A 373:1–56
Stark A (2011) Ionic liquids in the biorefinery: a critical assessment of their potential. Energy Environ Sci 4:19–32
Hossain MM, Aldous L (2012) Ionic liquids for lignin processing: dissolution, isolation, and conversion. Aust J Chem 65:1465–1477
Plechkova NV, Seddon KR (2008) Applications of ionic liquids in the chemical industry. Chem Soc Rev 37:123–150
Parvulescu VI, Hardacre C (2007) Catalysis in ionic liquids. Chem Rev 107:2615–2665
Swatloski RP, Spear SK, Holbrey JD, Rogers RD (2002) Dissolution of cellulose with ionic liquids. J Am Chem Soc 124:4974–4975
Sun N, Rahman M, Qin Y, Maxim ML, Rodriguez H, Rogers RD (2009) Complete dissolution and partial delignification of wood in the ionic liquid 1-ethyl-3-methylimidazolium acetate. Green Chem 11:646–655
Lee SH, Doherty TV, Linhardt JS (2009) Ionic liquid-mediated selective extraction of lignin from wood leading to enhanced enzymatic cellulose hydrolysis. Biotechnol Bioeng 102:1368–1376
Fu D, Mazza G, Tamaki Y (2010) Lignin extraction from straw by ionic liquids and enzymatic hydrolysis of the cellulosic residues. J Agric Food Chem 58:2915–2922
Zhu S, Wu Y, Chen Q, Yu Z, Wang C, Jin S, Ding Y, Wu G (2006) Dissolution of cellulose with ionic liquids and its application: a mini-review. Green Chem 8:325–327
Pinkert A, Marsh KN, Pang SS, Staiger MP (2009) Ionic liquids and their interaction with cellulose. Chem Rev 109:6712–6728
Maki-Arvela P, Anugwom I, Virtanen P, Sjoholm R, Mikkola JP (2010) Dissolution of lignocellulosic materials and its constituents using ionic liquids—a review. Ind Crop Prod 32:175–201
Mora-Pale M, Meli L, Doherty TV, Linhardt RJ, Dordick JS (2011) Room temperature ionic liquids as emerging solvents for the pretreatment of lignocellulosic biomass. Biotechnol Bioeng 108:1229–1245
Wang H, Gurau G, Rogers RD (2012) Ionic liquid processing of cellulose. Chem Soc Rev 41:1519–1537
Kim J-Y, Shin E-J, Eoma I-Y, Wonb K, Kim YH, Choi D, Choi IG, Choi JW (2011) Structural features of lignin macromolecules extracted with ionic liquid from poplar wood. Bioresour Technol 102:9020–9025
Li W, Sun N, Stoner B, Jiang X, Lu X, Rogers RD (2011) Rapid dissolution of lignocellulosic biomass in ionic liquids using temperatures above the glass transition of lignin. Green Chem 13:2038–2047
Brandt A, Grasvik J, Hallett JP, Welton T (2013) Deconstruction of lignocellulosic biomass with ionic liquids. Green Chem 15:550–583
Zavrel M, Bross D, Funke M, Buchs J, Spiess AC (2009) High-throughput screening for ionic liquids dissolving (ligno-)cellulose. Bioresour Technol 100:2580–2587
Tan SSY, MacFarlane DR (2009) Ionic liquids in biomass processing. Top Curr Chem 290:311–339
Tan SSY, MacFarlane DR, Upfal J, Edye LA, Doherty WOS, Patti AF, Pringle JM, Scott JL (2009) Extraction of lignin from lignocellulose at atmospheric pressure using alkylbenzenesulfonate ionic liquid. Green Chem 11:339–345
Pinkert A, Goeke DF, Marsh KN, Pang S (2011) Extracting wood lignin without dissolving or degrading cellulose: investigations on the use of food additive-derived ionic liquids. Green Chem 13:3124–3136
Pu Y, Jiang N, Ragauskas AJ (2007) Ionic liquid as a green solvent for lignin. J Wood Chem Technol 27:23–33
Wang Y, Wei L, Li K, Ma Y, Ma N, Ding S, Wang L, Zhao D, Yan B, Wan W, Zhang Q, Wang X, Wang J, Li H (2014) Lignin dissolution in dialkylimidazolium-based ionic liquid–water mixtures. Bioresour Technol 170:499–505
Lateef H, Grimes S, Kewcharoenwong P, Feinberg B (2009) Separation and recovery of cellulose and lignin using ionic liquids: a process for recovery from paper-based waste. J Chem Technol Biotechnol 84:1818–1827
Diop A, Bouazza AH, Daneault C, Montplaisir D (2013) New ionic liquid for the dissolution of lignin. Bioresoure 8:4270–4282
Kilpelainen I, Xie H, King A, Granstrom M, Heikkinen S, Argyropoulos DS (2007) Dissolution of wood in ionic liquids. J Agric Food Chem 55:9142–9148
George A, Tran K, Morgan TJ, Benke PI, Berrueco C, Lorente E, Wu BC, Keasling JD, Simmons BA, Holmes BM (2011) The effect of ionic liquid cation and anion combinations on the macromolecular structure of lignins. Green Chem 13:3375–3385
Li B, Filpponen I, Argyropoulos DS (2010) Acidolysis of wood in ionic liquids. Ind Eng Chem Res 49:3126–3136
Cox BJ, Ekerdt JG (2012) Depolymerization of oak wood lignin under mild conditions using the acidic ionic liquid 1-H-3-methylimidazolium chloride as both solvent and catalyst. Bioresour Technol 118:584–588
Stärk K, Taccardi N, Bösmann A, Wasserscheid P (2010) Oxidative depolymerization of lignin in ionic liquids. ChemSusChem 3:719–723
Zakzeski J, Jongerius AL, Weckhuysen BM (2010) Transition metal catalyzed oxidation of Alcell lignin, soda lignin, and lignin model compounds in ionic liquids. Green Chem 12:1225–1236
Zakzeski J, Bruijnincx PCA, Weckhuysen BM (2011) In situ spectroscopic investigation of the cobalt-catalyzed oxidation of lignin model compounds in ionic liquids. Green Chem 13:671–680
Liu S, Shi ZL, Li L, Yu ST, Xie C, Song Z (2013) Process of lignin oxidation in an ionic liquid coupled with separation. RSC Adv 3:5789–5793
Sena-Martins G, Almeida-Vara E, Duarte JC (2008) Eco-friendly new products from enzymatically modified industrial lignins. Ind Crop Prod 27:189–195
Bugg TDH, Ahmad M, Hardiman EM, Rahmanpour R (2011) Pathways for degradation of lignin in bacteria and fungi. Nat Prod Rep 28:1883–1896
Kirk TK, Farrell RL (1987) Enzymatic ‘‘combustion’’: the microbial degradation of lignin. Annu Rev Microbiol 41:465–505
Ten Have R, Teunissen PJM (2001) Oxidative mechanisms involved in lignin degradation by white-rot fungi. Chem Rev 101:3397–3413
Ahmad M, Taylor CR, Pink D, Burton K, Eastwood D, Bending GD, Bugg TDH (2010) Development of novel assays for lignin degradation: comparative analysis of bacterial and fungal lignin degraders. Mol BioSyst 6:815–821
Hatakka A, Hammel KE (2010) Fungal biodegradation of lignocelluloses. In: The Mycota. Industrial Applications, 10, pp 319–340
Vicuna R (1988) Bacterial degradation of lignin. Enzyme Microb Technol 10:646–655
Zimmermann W (1990) Degradation of lignin by bacteria. J Biotechnol 13:119–130
Bugg TDH, Ahmad M, Hardiman EM, Singh R (2011) The emerging role for bacteria in lignin degradation and bio-product formation. Curr Opin Biotechnol 22:394–400
Bandounas L, Wierckx NJP, de Winde JH, Ruijssenaars HJ (2011) Isolation and characterization of novel bacterial strains exhibiting ligninolytic potential. BMC Biotechnol 11:94
Brown ME, Chang MCY (2014) Exploring bacterial lignin degradation. Curr Opin Chem Biol 19:1–7
Dey S, Maiti TK, Bhattacharyya BC (1994) Production of some extracellular enzymes by a lignin peroxidase-producing brown rot fungus, Polyporus ostreiformis, and its comparative abilities for lignin degradation and dye decolorization. Appl Environ Microbiol 60:4216–4218
Paliwal R, Rawat AP, Rawat M, Rai JPN (2012) Bioligninolysis: recent updates for biotechnological solution. Appl Biochem Biotechnol 167:1865–1889
DeAngelis KM, Allgaier M, Chavarria Y, Fortney JL, Hugenholtz P, Simmons B, Sublette K, Silver WL, Hazen TC (2011) Characterization of trapped lignin-degrading microbes in tropical forest soil. PLoS ONE 6:e19306
Taylor CR, Hardiman EM, Ahmad M, Sainsbury PD, Norris PR, Bugg TDH (2012) Isolation of bacterial strains able to metabolize lignin from screening of environmental samples. J Appl Microbiol 113:521–530
Huang X-F, Santhanam N, Badri DV, Hunter WJ, Manter DK, Decker SR, Vivanco JM, Reardon KF (2013) Isolation and characterization of lignin-degrading bacteria from rainforest soils. Biotechnol Bioeng 110:1616–1626
Pasti MB, Pometto AL, Nuti MP, Crawford DL (1990) Lignin-solubilizing ability of actinomycetes isolated from termite (Termitidae) gut. Appl Environ Microbiol 56:2213–2218
Kato K, Kosaki S, Sakuranaga M (1998) Degradation of lignin compounds by bacteria from termite guts. Biotechnol Lett 20:459–462
Pollegioni L, Tonin F, Rosini E (2015) Lignin-degrading enzymes. FEBS J 282:1190–1213
Haemmerli SD, Leisola MSA, Fiechter A (1986) Polymerisation of lignins by ligninases from Phanerochaete chrysosporium. FEMS Microbiol Lett 35:33–36
Hammel KE, Jensen KA, Mozuch MD, Landucci LL, Tien M, Pease EA (1993) Ligninolysis by a purified lignin peroxidase. J Biol Chem 268:12274–12281
Conesa A, Punt PJ, van den Hondel CAMJJ (2002) Fungal peroxidases: molecular aspects and applications. J Biotechnol 93:143–158
Munk L, Sitarz AK, Kalyani DC, Mikkelsen JD, Meyer AS (2015) Can laccases catalyze bond cleavage in lignin? Biotechnol Adv 33:13–24
Rochefort D, Leech D, Bourbonnais R (2004) Electron transfer mediator systems for bleaching of paper pulp. Green Chem 6:14–24
Reiter J, Strittmatter H, Wiemann LO, Schieder D, Sieber V (2013) Enzymatic cleavage of lignin beta-O-4-aryl ether bonds via net internal hydrogen transfer. Green Chem 15:1373–1381
Otsuka Y, Sonoki T, Ikeda S, Kajita S, Nakamura M, Katayama Y (2003) Detection and characterization of a novel extracellular fungal enzyme that catalyzes the specific and hydrolytic cleavage of lignin guaiacyl glycerol beta-aryl ether linkages. Eur J Biochem 270:2353–2362
Masai E, Ichimura A, Sato Y, Miyauchi K, Katayama Y, Fukuda M (2003) Roles of the enantioselective glutathione S-transferases in cleavage of β-aryl ether. J Bacteriol 185:1768–1775
Allocati N, Federici L, Masulli M, DiIlio C (2009) Glutathione transferases in bacteria. FEBS J 276:58–75
Gall DL, Kim H, Lu F, Donohoe TJ, Noguera DR, Ralph J (2014) Stereochemical features of glutathione-dependent enzymes in the Sphingobium sp. strain SYK-6 β-aryl etherase pathway. J Biol Chem 289:8656–8667
Picart P, Müller C, Mottweiler J, Wiermans L, Bolm C, Dominguez de Maria P, Schallmey A (2014) From gene towards selective biomass valorization: bacterial β-etherases with catalytic activity on lignin-like polymers. ChemSusChem 7:3164–3171
Masai E, Sasaki M, Minakawa Y, Abe T, Sonoki T, Miyauchi K, Katayama Y, Fukuda M (2004) A novel tetrahydrofolate-dependent O-demethylase gene is essential for growth of Sphingomonas paucimobilis SYK-6 with syringate. J Bacteriol 186:2757–2765
Bugg TDH, Rahmanpour R (2015) Enzymatic conversion of lignin into renewable chemicals. Curr Opin Chem Biol 29:10–17
Chen CL, Chang HM, Kirk TK (1983) Carboxylic acids produced through oxidative cleavage of aromatic rings during degradation of lignin in spruce wood by Phanerochaete chrysosporium. J Wood Chem Technol 3:35–57
Sainsbury PD, Hardiman EM, Ahmad M, Otani H, Seghezzi N, Eltis LD, Bugg TDH (2013) Breaking down lignin to high-value chemicals: the conversion of lignocellulose to vanillin in a gene deletion mutant of Rhodococcus jostii RHA1. ACS Chem Biol 8:2151–2156
Vardon DR, Franden MA, Johnson CW, Karp EM, Guarneri MT, Linger JG, Salm MJ, Strathman TJ, Beckham GT (2015) Adipic acid production from lignin. Energy Environ Sci 8:617–628
Kiiskinen LL, Saloheimo M (2004) Molecular cloning and expression in Saccharomyces cerevisiae of a laccase gene from the ascomycete Melanocarpus albomyces. Appl Environ Microbiol 70:137–144
Kim S, Chmely SC, Nimos MR, Bomble YJ, Foust TD, Paton RS, Beckham GT (2011) Computational study of bond dissociation enthalpies for a large range of native and modified lignins. J Phys Chem Lett 2:2846–2852
Parthasarathi R, Romero RA, Redondo A, Gnanakaran S (2011) Theoretical study of the remarkably diverse linkages in lignin. J Phys Chem Lett 2:2660–2666
Beste A, Buchanan AC III (2009) Computational study of bond dissociation enthalpies for lignin model compounds. Substituent effects in phenethyl phenyl ethers. J Org Chem 74:2837–2841
Younker JM, Beste A, Buchanan AC III (2011) Computational study of bond dissociation enthalpies for substituted β-O-4 lignin model compounds. ChemPhysChem 12:3556–3565
Beste A, Buchanan AC III (2011) Kinetic analysis of the phenyl-shift reaction in β-O-4 lignin model compounds: a computational study. J Org Chem 76:2195–2203
Beste A, Buchanan AC III (2012) Role of carbon–carbon phenyl migration in the pyrolysis mechanism of β-O-4 lignin model compounds: phenethyl phenyl ether and α-hydroxy phenethyl phenyl ether. J Phys Chem A 116:12242–12248
Beste A, Buchanan AC III, Harrison RJ (2008) Computational prediction of α/β selectivities in the pyrolysis of oxygen-substituted phenethyl phenyl ethers. J Phys Chem A 112:4982–4988
Beste A, Buchanan AC III (2012) Kinetic simulation of the thermal degradation of phenethyl phenyl ether, a model compound for the β-O-4 linkage in lignin. Chem Phys Lett 550:19–24
Beste A, Buchanan AC III (2013) Computational investigation of the pyrolysis product selectivity for α-hydroxy phenethyl phenyl ether and phenethyl phenyl ether: analysis of substituent effects and reactant conformer selection. J Phys Chem A 117:3235–3242
Mar BD, Qi HW, Liu F, Kulik HJ (2015) Ab initio screening approach for the discovery of lignin polymer breaking pathways. J Phys Chem A 119:6551–6562
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer-Verlag GmbH Germany
About this chapter
Cite this chapter
Monteil-Rivera, F. (2016). Green Processes for Lignin Conversion. In: C.K. Lau, P. (eds) Quality Living Through Chemurgy and Green Chemistry. Green Chemistry and Sustainable Technology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-53704-6_10
Download citation
DOI: https://doi.org/10.1007/978-3-662-53704-6_10
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-53702-2
Online ISBN: 978-3-662-53704-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)