
Abstract
In vitro screening for cardiovascular safety liabilities of novel drug candidates presents a challenge for the pharmaceutical industry. Such approaches rely on detecting pharmacologic effects on key components of complex integrated system early in drug discovery to define potential safety liabilities. Key to such studies are the concepts of hazard identification vs. risk assessment, drug specificity vs. selectivity, and an appreciation of the challenges faced when attempting to translate in vitro findings to preclinical in vivo as well as clinical effects. This chapter defines some key aspects of early safety pharmacology screening for cardiovascular liabilities, citing studies of two key depolarizing cardiac currents (fast sodium current and L-type calcium current) as examples linked to effects on cardiac conduction and repolarization.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cardiac conduction and repolarization
- Hazard identification
- Risk assessment
- Selectivity
- Sodium current
- Specificity
1 Introduction
An urgent need remains to reduce attrition in later stages of drug discovery. Aside from business considerations, attrition arises from two sources, namely, efficacy and safety concerns. For a drug to be successful, it must demonstrate efficacy along with acceptable safety. In the broadest contest, safety refers to overall drug effects, which include (a) potential on-target adverse effects (side effects consistent with a drug’s known mechanism of action), (b) off-target effects (side effects not related to a drug’s known or targeted mechanism of action), or (c) drug–drug interactions (metabolic interactions increasing the levels of the drug or concomitant medications). The balance between efficacy and safety forms the basis of the therapeutic index, defined as the ratio of the highest exposure to the drug that results in no toxicity to the exposure that produces the desired effectiveness (see a recent review by Muller and Milton 2012). Safety considerations can span across toxicologic studies (based more on form and structure) as well as functional studies (including more acute safety pharmacology studies).
The field of safety pharmacology employs the basic principles of pharmacology to provide data useful in evaluating risk/benefit assessments of evolving drug candidates (see Pugsley et al. 2008). In general, safety pharmacology studies evaluate functional endpoints, as compared to “classical” toxicology studies that focus more on morphological endpoints. More traditional safety pharmacology studies (see Bass et al. 2015) focus on acute in vivo studies ensuring safety of vital organ systems (“to identify undesirable pharmacodynamic properties of a substance that may have relevance to its human safety”) as outlined in the ICH S7A regulatory guidance (US FDA ICH S7A 2001). These studies typically evaluate later-stage discovery compounds (destined for first in human clinical studies) and performed under good laboratory practice (GLP) conditions to fulfill regulatory requirements. A newer area of safety pharmacology has since evolved whose goal is to identify potential hazards and risks of evolving drug candidates. The terms “Exploratory Safety Pharmacology” (Bass et al. 2009; Cavero 2009a, b) and “Frontloading” (Pugsley et al. 2008) have been used to describe these safety studies performed prior to or during lead selection. These studies, often performed in vitro, allow for reduced compound requirements (typically a few milligrams vs. hundreds of milligrams for in vivo studies) as well as reduced cost, animal usage, and more rapid turnaround times. Frontloading of early safety studies saves time and resources by removing compounds with potential liabilities early, informs project teams of potential toxicities, and guides further preclinical exploratory studies and clinical risk mitigation strategies with the goal of reducing late-stage attrition.
This chapter will present some perspectives on in vitro exploratory safety pharmacology studies, briefly discussing some strengths and important limitations of early safety screening. Some off-target cardiovascular safety assays will be cited, including binding assay studies, computational methods, and functional ion channel screening (emphasizing two prominent depolarizing cardiac currents, namely, fast sodium (INa) and calcium (ICa,L); key potassium currents are covered elsewhere in this volume). Emphasis within this chapter will focus on effects of small molecules, rather than biologics or biopharmaceuticals. For this later emerging class of therapeutics, adverse effects are more likely to be on-target-related and immune system-induced or arise with longer-term exposures (Kooijman et al. 2012; see Vargas et al. 2015) consistent with their greater specificity, more limited distribution, and longer half-lives (such as monoclonal antibodies) compared to small molecule therapeutics.
2 Consideration of Hazard Identification vs. Risk Assessment in Exploratory Safety Studies
It is informative to consider the early safety studies in drug discovery using the concepts of hazard identification and risk assessment. Borrowing definitions from the field of environmental risk, a hazard may be defined as a chemical’s intrinsic ability to cause harm or produce adverse effects. In contrast, risk is associated with the probability of harm or adverse effects will occur at select concentrations and scenarios. Bottled propane (as an example) represents a recognized hazard that has a low probability of causing harm when used properly; thus, the risk assessment depends on the amount of propane stored (concentration), conditions for storage, and use. Similarly, a novel drug candidate can be considered in regard to potential hazard and risk assessment.
In exploratory safety studies, hazard identification is often conducted during early selection of lead candidates. These studies serve to inform of potential risks and are used internally for decision-making, rank ordering of compounds, and subsequent lead optimization efforts. In general, early in vitro assays are conducted with simple preparations (transfected cells or membranes) that represent the most simplest of drug–receptor interactions (reflecting hazard identification). As these are reductionist approaches, they need not necessarily reflect functional effects on either cells, tissues, organs, or whole animals in normal or diseased states [representing more complete (and complex) risk assessment]. In contrast, later evaluations of risk assessment generally necessitate more complex assays (ranging from cell-based in vitro studies to ex vivo and organ studies). Such studies will subsequently include in vivo whole animal studies that form traditional safety pharmacology packages satisfying regulatory authorities. For example, an evolving compound may be shown to bind to beta-adrenergic receptors by ligand-displacement studies, thus identifying a potential hazard. Subsequent (follow-up) risk assessment requires further studies that could include functional in vitro studies involving evaluation of effects on heart rate and cardiac contractility (e.g., in isolated atrial preparations). Results from these efforts might indicate beta-receptor agonist effects (increased heart rate and cardiac contractility (depending on receptor–effector coupling and binding affinity)) or antagonist effects (decreased heart rate and contractility in the presence of beta-adrenergic stimulation). One critically important contribution of the safety pharmacologist is to recognize the strengths and limitations of early exploratory studies and their translation to more integrated preclinical (e.g., whole animal) and clinical studies.
The value of information gained from early exploratory safety pharmacology studies depends on the stage of drug discovery and available resources. If multiple drug candidates are under evaluation during early lead optimization, it might be prudent to eliminate the most conspicuous (potent) “offenders” based on rank ordering of results centered on hazard identification, thus saving resources for subsequent testing of remaining drug candidates. In contrast, if only a few drug candidates are available (e.g., after efficacy targets are achieved), then downstream studies with more complex, integrated systems focused on risk assessment would be warranted. The value of early hazard identification studies also depends on the extent (and timing) of subsequent risk assessment studies. A compound identified with a potential hazard (but with promising preclinical efficacy) might be pursued further should later risk assessments demonstrated limited liabilities at exposures where efficacy is anticipated. Clearly, care must be taken to avoid overinterpreting the extent of risk in order to avoid discarding promising compounds early in the drug pipeline that are in fact safe and would conceivably be eliminated during later safety testing. Finally, the combined results from hazard identification and subsequent risk assessment studies would both provide confidence in designing later risk mitigation strategies for compounds deemed worthy of further progression.
3 Specificity and Selectivity
One of the key strengths of early safety studies is the ability to identify potential off-target pharmacologic activity during the lead selection process. This process should occur concurrently with activities focused on identifying the best lead candidates. Broad profiling of activities, if conducted early, provides for the efficient attrition of multiple compounds and allows for more efficient medicinal chemistry efforts focused on lead optimization.
Two key concepts to consider regarding early compound profiling are specificity and selectivity. While different definitions may be found, I will refer to specificity (derived from Latin “species”) as describing a drug’s ability to exert a single effect by a single mechanism of action. In most cases, drugs are described based on their selectivity across multiple receptors, as most drugs are not specific and will interact with more than one receptor (especially at higher exposures). Single and multiple effects that may result from drug interactions with one receptor are referred to as “on-target” pharmacology. Indeed, on-target pharmacology may result in intended and adverse effects and often leads to understanding the physiology of affected systems. The antihistamine diphenhydramine provides such an example. As an antagonist at peripheral H1 receptors, this drug mitigates the effects of histamine release, thus providing relief from common allergies. However, as an antagonist at H1 receptors in the central nervous system, diphenhydramine produces drowsiness (an on-target effect sometimes used to benefit).
Effects shared by drugs with the same mechanism of action are referred to as “class effects” or “class actions.” Early safety pharmacology studies are useful in defining class effects related to potential safety issues and may prove critical in discerning the overall value of novel drug targets. A drug’s overall selectivity (that defines its “off-target” pharmacology) can be described based on differences in the dose–response curves for the multiple receptors or responses involved. It is preferable for a drug to exert its therapeutic actions (on-target effects) at lower exposures than for adverse or untoward effects elicited at off-target receptors, especially if the off-target effect is linked to a serious adverse effect (thus forming the basis for a therapeutic margin). However, when multiple receptors are involved in defining an overall effect, it is possible that both may define the overall safety profile. For example, it is postulated that the liabilities linked to hERG current block (proarrhythmia resulting from delayed ventricular repolarization due to reduced outward current) may be offset by concomitant block of L-type calcium current (which may act to shorten ventricular repolarization as a result of reduced inward current). These two opposing forces may balance, thus mitigating the potentially dangerous QT prolongation observed with hERG blockade alone (see Fermini and Fossa 2003 for discussion).
The weight-loss drug lorcaserin provides a recent example of a drug overcoming a selectivity-based safety liability. Lorcaserin was approved in 2012 as an adjunct to diet and exercise for chronic weight management in adult patients as well as patients overweight who also have at least one weight-related comorbidity. Lorcaserin is described as a selective 5HT2C serotonin receptor agonist that leads to GPCR-linked cellular excitatory activation that elicits accumulation of inositol phosphates and downstream activation of phospholipase C. Activation of the 5HT2C receptor has been implicated in feeding, and knockout mice have demonstrated hyperphagia and an obese phenotype. However, the 5HT2C receptor is one of at least 13 distinct 5-HT receptor subtypes cloned and characterized (see Hoyer et al. 2002). Heart valvulopathy, a significant adverse effect of an earlier weight-loss drug combination fenfluramine/dexfenfluramine, has been linked to off-target activation of 5HT-2B receptors in clinical and preclinical in vivo studies (Rothman et al. 2000; Elangbam et al. 2008; see Hutcheson et al. 2011 for a recent review). Selectivity of lorcaserin for the 5HT2C receptor was demonstrated to be 8–15× vs. the 5-HT2A receptor and 45–90× vs. the 5HT-2B receptor lending support for subsequent drug development and eventual approval of lorcaserin (FDA Lorcaserin Briefing Document 2010).
Broad profiling to characterize selectivity provides useful data required to prioritize candidates for further testing as well as guide follow-on studies to inform risk assessment. The extent of early profiling of compounds often depends on the resources available. Multiple contract research organizations offer in vitro screening services based on binding assays for receptors (GCPRs, nuclear receptors, kinases, etc.), enzymes, ion channels, and transporters; examples of such protocols can be found in such journals as Current Protocols in Pharmacology. Most early protocols rely on a competition assay model using radiolabeled ligands and either of three assay formats (filtration, scintillation proximity assay, and centrifugation); assays are typically optimized to provide fast, consistent, and reproducible results. One potential strategy is to initially screen compounds at a single high concentration (e.g., 1 or 10 μM), with follow-up studies on hits for IC50/Ki determinations when compounds display more than 50 % inhibition of control value or 50 % stimulation relative to control. Subsequent studies linking binding results to functional responses are required to effectively translate receptor studies and inform potential off-target effects, as both binding affinity and efficacy drive functional effects in the simplest of cellular systems (the next level of integration). Binding studies provide little information regarding specifics of drug–target interactions (e.g., agonism, competitive or noncompetitive antagonism, allosteric modulation, reverse agonists, desensitization, etc.) that may require further clarification.
While established in vitro studies have traditionally focused on simpler subcellular assays, a growing number of whole cell-based screens are emerging. Such assays are likely useful for evaluating the integrated cellular response to a drug, representing multiple (and likely interdependent) effects on multiple cellular proteins and machinery, metabolic status, and other factors. It is the combination of these multiple factors that determines a drug’s overall efficacy or safety profile. Indeed, a drug may have different effects/efficacies [termed pluridimensional efficacy (Galandrin and Bouvier 2006)] dependent on the type of assay/assay system and conditions used [see Kenakin and Christopoulos (2012) for a recent review related to G protein-coupled receptors]. A potential complication of translation of in vitro cellular responses is provided by studies of seven transmembrane receptors that can form many conformations of the receptor, leading to behaviors where ligands can stabilize unique conformations that provide for selective activation of signaling pathways (termed “biased ligands”; see Kenakin 2011 for a review). G protein signaling vs. beta-arrestin recruitment to the parathyroid hormone receptor provides an example of a biased ligand (Gesty-Palmer et al. 2011). Thus, a specific assay to detect a ligand may report only a subset of efficacious compounds based on the assay conditions and measured endpoints. As compared to more simplified test systems, whole cell assays (e.g., using native cells or human-derived stem cells) may be less prone to demonstrate confounding bias as the integrated cellular response reflects the integrated response of multiple signaling pathways. The complexity inherent in cell-based screening approaches might be expected to differentiate subtle ligand effects (at the expense of making interpretation of cellular mechanisms and affected pathways more difficult).
A recent study by Loukine and colleagues (2012) described a computational approach to predict unintended “off-target”-based adverse drug reactions. After characterizing structural similarities of known ligand molecules for 73 biological targets, the authors searched for structural relationships of groups of ligands compared to test compounds. (This approach is somewhat different from other conventional methods that determine the strength of drug–receptor interactions based only on ligand and receptor structures.) Approximately half of the off-target predictions were true, with affinities of the new off-targets ranging from 1 nM to 30 μM. Of the 656 marketed drugs tested on the 73 “side-effect” targets, each drug modulated an average of seven safety targets, with more than 10 % acting on approximately half of the targets. Finally, based on the drug set tested, the 10 most promiscuous targets identified were (in descending order) Nav1.5 cardiac sodium channel, 5-HT2B serotonin receptor, 5-HT2A serotonin receptor, α2a adrenergic receptor, 5-HT1A serotonin receptor, α1A adrenergic receptor, M2 muscarinic receptor, hERG (IKr) potassium channel, H2 histamine receptor, and D4 dopamine receptor. With the exception of Nav1.5 and hERG (both ion channels), most promiscuous targets were G protein-coupled receptors, followed by transporters; enzymes, nuclear receptors, and ligand-gated ion channels were less promiscuous, and peptide-recognizing receptors were identified the least. This method provides an example of an early computational prediction of off-target interactions that can guide subsequent selection of appropriate interrogative in vitro and in vivo screening studies (should the compound be synthesized). However, given the high rate of false-positive findings with this approach, it should be considered as one component of a more complete early safety screening exercise.
Finally, it is informative to (re)evaluate exploratory safety pharmacology studies when a potential safety signal emerges from later preclinical studies (e.g., GLP-based assays to satisfy regulatory authorities) or subsequent clinical studies. Such reviews are informative regarding defining assay performance, which can include either the presence or absence of responses (concordance vs. discordance) as well as sensitivity of responses in preclinical vs. clinical studies (e.g., graphically using concentration–response relationship). Conversely, a preclinical signal not validated in clinical studies should be interrogated in order to establish potential mechanisms (e.g., species differences), as well as to inform on the level of translatable risk involved. Such studies are essential in evaluating the safety of subsequent backup candidates.
4 Exploratory Safety Pharmacology Studies with Cardiac Channels Involved in Cardiac Conduction
Screening of cardiac ion channels (beyond the expected regulatory hERG current screening) is assuming a more prominent role in early safety pharmacology studies. This greater emphasis arises from multiple sources, including familiarity with the prominent role of hERG screening (as detailed in the US FDA ICH S7B Guidance 2005), growing knowledge and characterization of inherited ion channelopathies linked to proarrhythmia, greater attention to observed ECG changes now evaluated as part of evolving thorough QT studies, counterscreening of cardiac channels based on homologies of noncardiac channel targets from various therapeutic areas, and a growing awareness of cardiotoxicity with oncology therapeutics. Enabling technologies for expressing ion channels in various systems and the growing number of automated patch clamp platforms for functional studies (see Möller and Witchel 2011) provide further impetus for such studies. Often subtle differences between different channel isoforms (of targets vs. non-targets, e.g., among the nine recognized sodium channel isoforms) necessitate counterscreening early to provide efficient compound differentiation in lead optimization. The following sections will discuss the basis for screening of two inwardly directed (depolarizing) cardiac ion currents that play a prominent role in defining conduction and the action potential plateau of ventricular myocardium, namely, sodium (INa or Nav1.5, encoded by the SCN5a gene), and calcium (ICa,L, or Cav1.2, encoded by the CaCNA1C gene). Despite the importance of these channels in maintaining normal cardiac rhythm, their evaluation is not specifically covered in current regulatory guidances. The reader is referred to Chap. 7 in this book for discussion of hERG screening.
5 Fast Sodium Current
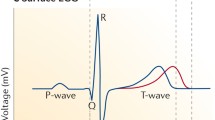
In working atrial and ventricular tissues, fast inward sodium current flowing through Nav1.5 channels is responsible for the action potential upstroke. These voltage-dependent channels rapidly activate (open) and inactivate (close) over the time course of a few msec, producing a strong and transient depolarizing inward current. The rapid current kinetics, along with large current density, provides for rapid, non-decremental conduction through working myocardium. As would be expected, block of this current results in slowed conduction, manifest as prolongation of the QRS duration (a measure of ventricular conduction) and prolongation of the PR interval (reflecting the combined effects on atrial conduction and conduction through specialized ventricular conduction pathways (Purkinje fibers) leading to working myocardium).
Reduction of fast inward sodium current is generally considered a risk factor for proarrhythmia. The Cardiac Arrhythmia Suppression Trial (CAST) demonstrated increased mortality in post-myocardial infarction patients when treated with either of the three local anesthetic-type antiarrhythmic agents (flecainide, encainide, and moricizine) to suppress ventricular premature depolarizations (Epstein et al. 1993; CAST investigators 1989; Echt et al. 1991). Sodium current block may also lead to negative inotropic effects due to reduced intracellular calcium concentrations affected by the cardiac Na+/Ca++ exchange pump (Ito et al. 1996), though this relationship is complex. Prolongation of the QRS duration in the presence of cardiovascular disease likely reflects pathophysiologic progression that is linked to proarrhythmia (see Nada et al. 2013 for a review). However, the relationship between QRS prolongation and proarrhythmia in patients without cardiac pathology is uncertain.
Block of cardiac sodium current by small molecules is complex, as drug binding (and hence channel block) is modulated by the state of the channel (that affects drug affinity as well as drug access). Consideration of the dynamic interactions between drug (which may exist in charged and uncharged forms and may gain access to binding site(s) via different pathways) and various channel configurations (as the channel cycles through resting, open, and inactivated states with each heartbeat) is provided by the modulated receptor or guarded receptor hypothesis (Hondeghem and Katzung 1977, 1984; Hille 1977; Starmer and Courtney 1986). The extent of channel block is typically dependent on the rate of stimulation or electrical activity (use-dependent block) and the voltage prior to initiation of the upstroke (voltage-dependent block). Underlying these effects are time- and voltage-dependent modulations of the rates of drug association and dissociation. Thus, the extent of block is dependent on the rate and pattern of electrical activity, as well as the kinetics of drug-channel interactions These concepts are not new; using microelectrode techniques and measures of maximum upstroke velocity (Vmax), use-dependent block was first demonstrated with guinea pig papillary muscles treated with quinidine (Johnson and McKinnon 1957), and voltage-dependent block was described by Chen et al. (1975). While not necessarily sophisticated, techniques used in these early demonstrations still prove useful for characterizing local anesthetic effects of drugs in native cardiac preparations, with results more easily translatable to the intact heart. One of first demonstrations of voltage-dependent block using voltage-clamp techniques was shown by Bean et al. (1983) who demonstrated slowed sodium channel reactivation (recovery from block of cardiac sodium current) with lidocaine. In this study, potency of block at depolarized potentials (−65 mV approx. 10 μM) was significantly less than observed at hyperpolarized potentials (−120 mV, approx. 400 μM). Considering the multiple factors shown to modulate block of cardiac sodium current, one should recognize the limitation of a single IC50 value associated with one specific protocol to characterize block of cardiac sodium current.
In general, the kinetics of recovery from block determines the extent of block and defines three different subgroups of Class 1 cardiac drugs demonstrating local anesthetic effects within the Vaughan Williams classification (Vaughan Williams 1975, 1984; Nattel 1991). Thus, Class IA antiarrhythmic drugs (such as quinidine) slow the rate of rise of the action potential (and slow conduction) as a result of intermediate kinetics of association and dissociation from different states of the sodium channel. In contrast, Class 1B drugs (such as lidocaine) have little effect on the rate of depolarization or QRS duration due to rapid dissociation from sodium channels. Finally, Class IC agents (such as flecainide) markedly depress the rate of rise of the action potential and cause marked conduction slowing due to slow dissociation kinetics of drug from the channel (longer-lasting, cumulative block).
With the advent of automated patch clamp systems and heterologous expression systems, characterizing drug block of sodium current is easily accomplished (e.g., see Harmer et al. 2008; Penniman et al. 2010; Kirsch 2010). A more integrated assessment of local anesthetic effects that employs multiple approaches may provide more confidence regarding translation of early preclinical findings. A recent study demonstrated the ability to differentiate between “good” (lidocaine-like) and “bad” [flecainide-like (strong use-dependent block)] sodium channel-blocking activities using multiple in vitro approaches activities (block of human cardiac INa in transfected CHO cells), rabbit Purkinje fibers (measuring upstroke characteristics), arterially perfused left ventricular wedge preparations (measuring QRS/conduction velocity), and rabbit Langendorff hearts (Lu et al. 2010). Another recent study illustrated an integrated approach for the preclinical evaluation of evolving drug candidates on cardiac conduction (with experimental elements including patch clamp studies, QRS interval measures in isolated Langendorff preparations, and PR and QRS measures in dog or nonhuman primate; see Erdemli et al. 2012). The in vitro studies by Lu et al. also demonstrated the enhanced effect of flecainide with myocardial ischemia, confirming prior findings that flecainide results in more marked depression of conduction in ischemia/reperfused myocardium (Kou et al. 1987). This confirmation highlights the utility of benchmarking and mechanistic studies when assessing proarrhythmic risk of more potent sodium channel-blocking agents.
Translating the various preclinical findings characterizing sodium current block to clinical findings represents yet another challenge. A recent study by Harmer et al. (2011) explored the relationship between drug-induced block of cardiac sodium current and QRS duration effects described in clinical literature. Specifically, they compared safety margins for 98 compounds (defined as the ratio of IC50 values for hNAv1.5 block/free Cmax based on clinical exposures) vs. reported QRS prolongation. They reported that QRS prolongation occurred, on average, at free plasma levels 15-fold below the calculated safety margins. Similarly, (1) a recent study by Heath et al. (2011) demonstrated that free plasma concentrations of flecainide and mexiletine 6–30-fold below IC50 values for block of hNav1.5 were sufficient to prolong the QRS interval/duration by 10–20 % in preclinical and clinical studies, and (2) an abstract by Cordes et al. (2009) concluded that free plasma concentrations approx. 3–11-fold below the IC50 values for sodium current block were sufficient to produce QRS widening.
These above translational studies are somewhat surprising, considering the high current density of hNav1.5 in ventricular myocytes (and presumably high “depolarization reserve”; see Gintant et al. 2011). Indeed, computer simulations of cardiac propagation describe a nonlinear relationship between conduction and reduced maximal sodium current conductance, with a 50 % decrease in conductance resulting in only modest reduction of conduction velocity from 55 to 32 cm/s (Shaw and Rudy 1997). This study also suggested that extreme QRS prolongation is necessary for intraventricular conduction failure in normal hearts. The sensitivity of ventricular conduction to sodium current block may be related to the platforms and experimental conditions used to determine IC50 values for block [potential difference in properties of overexpressed sodium channels in HEK/CHO cells vs. native myocytes, experimental conditions (room temperature, low extracellular sodium conditions), voltage protocols used (often no consideration of voltage or rate dependence), and bath concentrations (with potential differences between achieved and nominal exposures)]. It is also possible that the retrospective analysis of clinical response may have been biased toward lower drug exposures. Despite these concerns, and based on present practices for evaluating drug effects on sodium current, minimal sodium current block appears to be sufficient to slow cardiac conduction in normal myocardium.
Advances in cardiac monitoring in early clinical trials and automated ECG analysis have made it easier to interrogate effects of drugs on ventricular conduction (QRS duration) in early clinical studies. Given that preclinical data suggests a potential clinical liability for QRS prolongation with a promising clinical candidate, it should be possible to de-risk the candidate by directly evaluating clinical responses early in clinical development (as in early clinical dose-ascending phase one tolerability studies). Such efforts require consideration of the power of clinical studies to reliably detect small changes in the QRS interval.
6 L-Type Calcium Current
In working ventricular myocardium, the predominant inward current that flows after the action potential upstroke is L-type calcium current (Cav1.2, encoded by the CaCNA1C gene). The cardiac L-type calcium current was so named because of its slow kinetics of current decay (“L for long-lasting”); it has also been referred to as the dihydropyridine receptor due to its sensitivity to this chemical series (see below). In cardiac membranes, this voltage-dependent channel is composed of an alpha 1c subunit, b2a subunit, and a2-delta subunit; the alpha 1c subunit consists of the voltage sensor and channel pore composed of 4 homologous motifs, each containing six transmembrane segments. The auxiliary subunits play important regulatory roles that may be overlooked when using heterologous expression systems. It is likely that various functionally distinct subpopulations of L-type calcium channels with regionally distinct functional properties and regulation exist [e.g., those at dyadic junctions vs. plasma membrane microdomains such as lipid rafts and caveolae (see review by Best and Kamp 2012)].
The three classes of calcium channel pore alpha1 subunits (Cav.1, Cav2, and Cav3) demonstrate marked differences in their regulation: the Cav1 family is regulated primarily by second messenger activated kinase pathway and protein phosphorylation, while the Cav2 family is regulated by direct binding of signaling proteins; less is known about regulation of the Cav3 family. A second, smaller, and more rapid calcium current [termed T-type calcium current (“T” for transient), CaV3.x] is also present in embryonic heart and specialized tissues of adult heart [nodal regions and specialized conduction pathways; the reader is referred to a recent review for more details (Ono and Iijima 2010)].
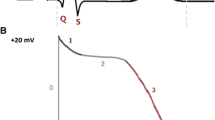
L-Type calcium current plays critical roles in modulating multiple cardiac functions. Thus, the influx of calcium ions is responsible for initiating muscle contraction and modulating contractility. From an electrophysiological perspective, L-type calcium current plays a prominent role in defining the configuration of the ventricular action potential. In contrast to fast inward sodium current, the relatively slow kinetics of activation delays the contribution of this current until inactivation of the fast inward sodium current and the early part of the action potential plateau. Under voltage-clamp conditions, ICa,L is activated at potentials positive to −40 mV, showing peak activation near +10 to +20 mV. This current also acts to sustain the action potential plateau at sufficiently positive potentials to enable activation of delayed rectifier current (hERG) that contributes to terminal repolarization (phase 3 of the action potential). ICa.L inactivation is complex, being dependent on both voltage and current [calcium-dependent inactivation (CDI); see Grandi et al. 2010; Tuckwell 2012 for reviews].
In regard to cardiac contractility and inotropy, the L-type calcium channel provides the transient influx of calcium ions responsible for calcium-induced calcium release (CICR) from sarcoplasmic reticulum stores, primarily through activation of the ryanodine receptors (RYR2) (see Bers 2002 for a review). As a consequence, intracellular calcium levels rise from low basal levels (<100 nM) to low micromolar levels with each action potential, leading to optimal binding of Ca++ to troponin C and induction of contraction. Thus, along with the sodium–calcium exchanger, L-type calcium currents figure prominently in modulating basal and transient levels of intracellular calcium to define intrinsic cardiac contractility and electrophysiology (see Banyasz et al. 2012). L-Type calcium current also demonstrates calcium-induced inactivation, a fact that highlights the integrated nature of cellular calcium handling and electromechanical coupling (an aspect of electrophysiological response often overlooked in studies employing channels expressed in heterologous systems). Finally, intracellular calcium levels likely also play signaling roles in the myocardium to influence more longer-term adaptation, such as hypertrophy; this area (important for longer-term chronic effects) is under active investigation.
L-Type calcium current plays a role in certain forms of arrhythmias. An early after depolarization (EAD) is a form of triggered electrical activity in which there is a slowing or reversal of repolarization during the plateau or early phase of action potential repolarization (see January and Riddle 1989; also Weiss et al. 2010). This premature depolarization results from a regenerative increase in net outward current, typically in the setting of slowed repolarization, indicative of reduced “repolarization reserve” (for a recent review, see Varró and Baczkó (2011)). In the voltage range of approximately −30–0 mV, there is overlap of steady-state activation and inactivation of ICa,L that results in the so-called “L-type window” current. Time- and voltage-dependent recovery from inactivation may allow a regenerative increase in ICa,L to elicit an EAD. If sufficiently strong (and properly coupled to surrounding myocardium), EADs may provide sufficient stimulus to produce a triggered beat (extrasystole) or run or triggered beats (potentially ventricular tachycardia). Block of L-type calcium current mitigates EAD activity. However, this effect is complex, in that other calcium-dependent currents (e.g., calcium current exchanger) may also be affected (see Banyasz et al. 2012). EADs represent an integrated cellular response that would not be detected in typical simpler (in vitro) ionic current studies. However, such activity has been reported in human pluripotent stem cell-derived cardiocytes [representing earlier in vitro studies not requiring animal tissues (Ma et al. 2011)]; such preparations may be amenable to earlier electrophysiological safety pharmacology studies derived from normal (Jonsson et al. 2012) or diseased sources (Liang et al. 2013).
Some hERG-blocking drugs are not linked to proarrhythmia (e.g., verapamil and fluoxetine). For these drugs, it has been postulated that hERG block may be mitigated by L-type calcium channel block, reestablishing a “balance” between inward and outward currents and thereby reducing proarrhythmia liability (Fermini and Fossa 2003; Martin et al. 2004). Indeed, recent computer simulations suggest that consideration of drug effects on three cardiac currents (hERG, INa, and ICa,L) improved the predictive classification of 31 marketed drugs (based on the assessment of AP prolongation in model correlated with clinical risk of torsades de pointes arrhythmia (Mirams et al. 2011)). It is also possible that the mitigation of proarrhythmic risk (associated with delayed repolarization) by L-type calcium channel block results from effects on calcium handling/homeostasis originally perturbed by electrophysiological changes (prolongation) of the action potential. In favor of this notion, a recent study by Johnson et al. (2013) using canine ventricular myocytes suggested that late diastolic release of sarcoplasmic calcium during beta-adrenergic stimulation caused prolongation of the following action potential by reducing calcium-dependent inactivation of L-type calcium current. The resulting prolongation of the action potential leads to increased beat to beat variability of repolarization, a recognized hallmark of proarrhythmia. Future studies will be necessary to properly attribute the contributions of direct channel effects vs. intracellular calcium handling effects to mitigation of proarrhythmia by hERG-blocking drugs. Such endeavors are important to prevent unwarranted elimination of evolving drug candidates from further consideration based solely on early detection of hERG blockade.
Numerous studies have demonstrated that ICa,L is modulated by drugs and other factors, a fact not unexpected considering the multiple functional roles for this current. A voltage-clamp study demonstrating the dynamic modulation of direct block of ICa,L with verapamil by state-dependent changes in the channel is provided by Nawrath and Wegener (1997). Other indirect effects of pharmacologic agents are recognized, for example, that beta-adrenergic stimulation increases inotropy (in part) by phosphorylation of L-type calcium channels through cAMP-dependent protein kinase A signaling (McDonald et al. 1994) to increase current (without affecting single channel density) as part of an integrated response to increase cardiac inotropy. Hypoxia also inhibits basal L-type calcium current, consistent with modulation of current by redox status of the cardiac myocyte (Hool et al. 2005). The calcium-sensing protein calmodulin also acts as a transducer and targets L-type calcium channel [see Saucerman and Bers (2012) for a recent review]. The role which drugs may play in modulating known physiologic alterations of channel function is largely unstudied and would not be detected in most early (non-cell-based) safety screening assays.
Calcium channel blockers act to directly reduce L-type calcium current. When considered using schemes proposed for antiarrhythmic drugs, they are placed into the Vaughan Williams Class IV category. In general, calcium channel blockers are conveniently characterized within three structural chemical groups, with each binding to different binding sites on the channel: phenylalkylamines (e.g., verapamil) bind to the V binding site, benzothiazepines (e.g., diltiazem) bind to the D binding site, and dihydropyridines (e.g., nifedipine) bind to the N binding site. As this channel is present in the heart as well as smooth muscle layers of peripheral vasculature, calcium channel blockers may also dilate coronary arteries and peripheral arterioles in addition to reducing cardiac contractility (negative inotropic effect) and affecting conduction through the AV node (negative dromotropic effect). In general, dihydropyridines have minimal effect on cardiac conduction or heart rate, while verapamil and diltiazem are known for slowing of AV conduction and decreased SA nodal automaticity. In contrast to block of hERG and fast inward sodium current, translating the in vitro effects of calcium current reduction to either negative inotropic or chronotropic effects is more difficult due to the more integrated nature of these responses.
Despite the complex role that ICa,L plays in cardiac (and extracardiac) tissues, early functional screening (using voltage-clamp techniques) is valuable in the early detection of potential cardiovascular liabilities (hazard identification). However, the translation of macroscopic current data to inform on safety margins for clinical studies (risk assessment) requires an understanding of the role of ICa,L in more complex physiological systems. Such information may be provided by follow-up in vitro (tissues and organs) and in vivo (whole animal) electrophysiological studies that place early current studies in context. It should also be recognized that the knowledge of the regulation of normal complex physiological systems may be limited and less may be known for human disease states. Thus, care is necessary in attempting to translate such early signals to clinical effects, and attempts to overinterpret early safety margins should be avoided. While it is possible to monitor negative dromotropic effects (slowed PR conduction) of evolving drug candidates early in phase 1 clinical studies (e.g., via careful monitoring of PR interval changes on ECG linked to drug plasma concentrations), it is much more difficult to noninvasively assess acute drug effects on cardiac contractility.
7 Conclusions
One important role of the safety pharmacologist is to recognize the strengths and limitations of early exploratory studies in the setting of interpretation and translation to more integrated preclinical assays and final clinical studies. Such efforts will ultimately require comparing data across multiple assays with multiple compounds, ideally across multiple groups in the precompetitive space. For any early in vitro safety pharmacology assay, the ever-present challenge is to strike the appropriate balance between assay sensitivity (ability to detect true positive preclinical results) and assay specificity (ability to detect true negative preclinical results) in relation to “gold-standard” clinical observations. A recent study comparing results from the in vitro hERG functional current assay to clinical findings of QT prolongation in thorough QT studies (Gintant 2011) provides one approach to qualitatively assess the utility and performance of early safety assays based on defining receiver operator curves. However, it is likely that (even with assay optimization) overall performance of early safety assays will not be fully predictive due to the lack of systems complexity of such early assays relative to the integrated responses observed clinically (including cardiac repolarization, blood pressure, heart rate). Despite this limitation, even such “less than perfect” approaches will enhance the probability of success of advancing novel therapeutics and provide for the more efficient discovery and development of safe and effective drugs.
References
Banyasz T, Horvath B, Jian Z, Izu LT, Chen-Izu Y (2012) Profile of L-type Ca(2+) current and Na(+)/Ca(2+) exchange current during cardiac action potential in ventricular myocytes. Heart Rhythm 9(1):134–142
Bass AS, Cartwright ME, Mahon C, Morrison R, Snyder R, McNamara P, Bradley P, Zhou YY, Hunter J (2009) Exploratory drug safety: a discovery strategy to reduce attrition in development. J Pharmacol Toxicol Methods 60(1):69–78
Bass AS, Hombo T, Kasai C, Kinter LB, Valentin J-P (2015) A historical view and vision into the future of the field of safety pharmacology. In: Pugsley MK, Curtis MJ (eds) Principles of safety pharmacology, Handbook of experimental pharmacology. Springer, Berlin
Bean BP, Cohen CJ, Tsien RW (1983) Lidocaine block of cardiac sodium channels. J Gen Physiol 81(5):613–642
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415(6868):198–205
Best JM, Kamp TJ (2012) Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol 52(2):376–387
FDA Briefing Document (2010) Lorcaserin advisory committee meeting, Sept 6, 2010
Cavero I (2009a) Safety pharmacology society: 8th annual meeting. Expert Opin Drug Saf 8(2):237–247
Cavero I (2009b) Exploratory safety pharmacology: a new safety paradigm to de-risk drug candidates prior to selection for regulatory science investigations. Expert Opin Drug Saf 8(6):627–647
Chen CM, Gettes LS, Katzung BG (1975) Effect of lidocaine and quinidine on steady-state characteristics and recovery kinetics of (dV/dt)max in guinea pig ventricular myocardium. Circ Res 37(1):20–29
Cordes J, Li C, Dugas J, Austin-LaFrance R, Lightbown I, Engwall M et al (2009) Translation between in vitro inhibition of the cardiac Nav1.5 channel and preclinical and clinical QRS widening. J Pharm Tox Methods 60:221 (abstract)
Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL et al (1991) Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The cardiac arrhythmia suppression trial. N Engl J Med 324(12):781–788
Elangbam CS, Job LE, Zadrozny LM, Barton JC, Yoon LW, Gates LD, Slocum N (2008) 5-hydroxytryptamine (5HT)-induced valvulopathy: compositional valvular alterations are associated with 5HT2B receptor and 5HT transporter transcript changes in Sprague-Dawley rats. Exp Toxicol Pathol 60(4–5):253–262
Epstein AE, Hallstrom AP, Rogers WJ, Liebson PR, Seals AA, Anderson JL, Cohen JD, Capone RJ, Wyse DG (1993) Mortality following ventricular arrhythmia suppression by encainide, flecainide, and moricizine after myocardial infarction. The original design concept of the cardiac arrhythmia suppression trial (CAST). JAMA 270(20):2451–2455
Erdemli G, Kim AM, Ju H, Springer C, Penland RC, Hoffmann PK (2012) Cardiac safety implications of hNav1.5 Blockade and a framework for pre-clinical evaluation. Front Pharmacol 3:6. doi:10.3389/fphar.2012.00006 (Epub 2012 Jan 26)
Fermini B, Fossa AA (2003) The impact of drug-induced QT interval prolongation on drug discovery and development. Nat Rev Drug Discov 2(6):439–447
Galandrin S, Bouvier M (2006) Distinct signaling profiles of beta1 and beta2 adrenergic receptor ligands toward adenylyl cyclase and mitogen-activated protein kinase reveals the pluridimensionality of efficacy. Mol Pharmacol 70(5):1575–1584
Gesty-Palmer D, Luttrell LM (2011) Refining efficacy: exploiting functional selectivity for drug discovery. Adv Pharmacol 62:79–107
Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ (2006) Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem 281(16):10856–10864
Gintant G (2011) An evaluation of hERG current assay performance: translating preclinical safety studies to clinical QT prolongation. Pharmacol Ther 129(2):109–119
Gintant GA, Gallacher DJ, Pugsley MK (2011) The ‘overly-sensitive’ heart: sodium channel block and QRS interval prolongation. Br J Pharmacol 164(2):254–259
Grandi E, Morotti S, Ginsburg KS, Severi S, Bers DM (2010) Interplay of voltage and Ca-dependent inactivation of L-type Ca current. Prog Biophys Mol Biol 103(1):44–50
Harmer AR, Abi-Gerges N, Easter A, Woods A, Lawrence CL, Small BG, Valentin JP, Pollard CE (2008) Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorks. J Pharmacol Toxicol Methods 57(1):30–41
Harmer AR, Valentin JP, Pollard CE (2011) On the relationship between block of the cardiac Na+ channel and drug-induced prolongation of the QRS complex. Br J Pharmacol 164(2):260–273
Heath BM, Cui Y, Worton S, Lawton B, Ward G, Ballini E, Doe CP, Ellis C, Patel BA, McMahon NC (2011) Translation of flecainide- and mexiletine-induced cardiac sodium channel inhibition and ventricular conduction slowing from nonclinical models to clinical. J Pharmacol Toxicol Methods 63(3):258–268
Hille B (1977) Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor interaction. J Gen Physiol 69:487–515
Hondeghem LM, Katzung BG (1977) Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta 472:373–398
Hondeghem LM, Katzung BG (1984) Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annu Rev Pharmacol Toxicol 24:387–423
Hool LC, Di Maria CA, Viola HM, Arthur PG (2005) Role of NAD(P)H oxidase in the regulation of cardiac L-type Ca2+ channel function during acute hypoxia. Cardiovasc Res 67(4):624–635
Hoyer D, Hannon JP, Martin GR (2002) Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav 71(4):533–554
Hutcheson JD, Setola V, Roth BL, Merryman WD (2011) Serotonin receptors and heart valve disease–it was meant 2B. Pharmacol Ther 132(2):146–157
Investigators CAST (1989) Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators. N Engl J Med 321(6):406–412
Ito K, Nagafuchi K, Taga A, Yorikane R, Koike H (1996) Possible involvement of altered Na+ -Ca2+ exchange in negative inotropic effects of class I antiarrhythmic drugs on rabbit and rat ventricles. J Cardiovasc Pharmacol 27(3):355–361
January CT, Riddle JM (1989) Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res 64(5):977–990
Johnson EA, McKinnon MG (1957) The differential effect of quinidine and pyrilamine on the myocardial action potential at various rates of stimulation. J Pharmacol Exp Ther 120(4):460–468
Johnson DM, Heijman J, Bode EF, Greensmith DJ, van der Linde H, Abi-Gerges N, Eisner DA, Trafford AW, Volders PG (2013) Diastolic spontaneous calcium release from the sarcoplasmic reticulum increases beat-to-beat variability of repolarization in canine ventricular myocytes after β-adrenergic stimulation. Circ Res 112(2):246–256
Jonsson MK, Vos MA, Mirams GR, Duker G, Sartipy P, de Boer TP, van Veen TA (2012) Application of human stem cell-derived cardiomyocytes in safety pharmacology requires caution beyond hERG. J Mol Cell Cardiol 52(5):998–1008
Kenakin T (2011) Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 336(2):296–302
Kenakin T, Christopoulos A (2012) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 12(3):205–216
Kirsch G (2010) Mechanistic ion channel screening for drug discovery. Drug Discov Dev 13(9):28–30
Kooijman M, van Meer PJ, Moors EH, Schellekens H (2012) Thirty years of preclinical safety evaluation of biopharmaceuticals: did scientific progress lead to appropriate regulatory guidance? Expert Opin Drug Saf 11(5):797–801
Kou WH, Nelson SD, Lynch JJ, Montgomery DG, DiCarlo L, Lucchesi BR (1987) Effect of flecainide acetate on prevention of electrical induction of ventricular tachycardia and occurrence of ischemic ventricular fibrillation during the early postmyocardial infarction period: evaluation in a conscious canine model of sudden death. J Am Coll Cardiol 9(2):359–365
Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Nguyen PK, Wang PJ, Bers DM, Robbins RC, Wu JC (2013) Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease specific patterns of cardiotoxicity. Circulation (in press)
Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Côté S, Shoichet BK (2012) Urban L Large-scale prediction and testing of drug activity on side-effect targets. Nature 486(7403):361–367. doi:10.1038/nature11159
Lu HR, Rohrbacher J, Vlaminckx E, Van Ammel K, Yan GX, Gallacher DJ (2010) Predicting drug-induced slowing of conduction and pro-arrhythmia: identifying the ‘bad’ sodium current blockers. Br J Pharmacol 160(1):60–76
Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT (2011) High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am J Physiol Heart Circ Physiol 301(5):H2006–H2017
Martin RL, McDermott JS, Salmen HJ, Palmatier J, Cox BF, Gintant GA (2004) The utility of hERG and repolarization assays in evaluating delayed cardiac repolarization: influence of multi-channel block. J Cardiovasc Pharmacol 43(3):369–379
McDonald TF, Pelzer S, Trautwein W, Pelzer DJ (1994) Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev 74(2):365–507
Mirams GR, Cui Y, Sher A, Fink M, Cooper J, Heath BM, McMahon NC, Gavaghan DJ, Noble D (2011) Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc Res 91(1):53–61
Möller C, Witchel H (2011) Automated electrophysiology makes the pace for cardiac ion channel safety screening. Front Pharmacol 2:73
Muller PY, Milton MN (2012) The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov 11(10):751–761
Nada A, Gintant GA, Kleiman R, Gutstein DE, Gottfridsson C, Michelson EL, Strnadova C, Killeen M, Geiger MJ, Fiszman ML, Koplowitz LP, Carlson GF, Rodriguez I, Sager PT (2013) The evaluation and management of drug effects on cardiac conduction (PR and QRS intervals) in clinical development. Am Heart J 165(4):489–500. doi:10.1016/j.ahj.2013.01.011, Epub 2013 Feb 21
Nattel S (1991) Antiarrhythmic drug classifications. A critical appraisal of their history, present status, and clinical relevance. Drugs 41(5):672–701
Nawrath H, Wegener JW (1997) Kinetics and state-dependent effects of verapamil on cardiac L-type calcium channels. Naunyn Schmiedebergs Arch Pharmacol 355(1):79–86
Ono K, Iijima T (2010) Cardiac T-type Ca(2+) channels in the heart. J Mol Cell Cardiol 48(1):65–70
Penniman JR, Kim DC, Salata JJ, Imredy JP (2010) Assessing use-dependent inhibition of the cardiac Na(+/−) current (I(Na)) in the PatchXpress automated patch clamp. J Pharmacol Toxicol Methods 62(2):107–118
Pugsley MK, Authier S, Curtis MJ (2008) Principles of safety pharmacology. Br J Pharmacol 154(7):1382–1399
Rothman RB, Baumann MH, Savage JE, Rauser L, McBride A, Hufeisen SJ, Roth BL (2000) Evidence for possible involvement of 5-HT(2B) receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102(23):2836–2841
Saucerman JJ, Bers DM (2012) Calmodulin binding proteins provide domains of local Ca2+ signaling in cardiac myocytes. J Mol Cell Cardiol 52(2):312–316
Shaw RM, Rudy Y (1997) Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res 81(5):727–741
Starmer CF, Courtney KR (1986) Modeling ion channel blockade at guarded binding sites: application to tertiary drugs. Am J Physiol 251(4 Pt 2):H848–H856
Tuckwell HC (2012) Quantitative aspects of L-type Ca2+ currents. Prog Neurobiol 96(1):1–31
U.S. Food & Drug Administration (2001) ICH S7A safety pharmacology studies for human pharmaceuticals. Guidance for industry. Fed Reg 66:36791–36792
U.S. Food & Drug Administration (2005) Guidance for industry: ICH S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. Fed Reg 70:61133–61134
Vargas HM, Amouzadeh HR, Engwall MJ (2015) Safety pharmacology evaluation of biopharmaceuticals. In: Pugsley MK, Curtis MJ (eds) Principles of safety pharmacology, Handbook of experimental pharmacology. Springer, Berlin
Varró A, Baczkó I (2011) Cardiac ventricular repolarization reserve: a principle for understanding drug-related proarrhythmic risk. Br J Pharmacol 164(1):14–36
Vaughan Williams EM (1975) Classification of antidysrhythmic drugs. Pharmacol Ther B 1(1):115–138 (Review)
Vaughan Williams EM (1984) A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol 24(4):129–147 (Review)
Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z (2010) Early after depolarizations and cardiac arrhythmias. Heart Rhythm 7(12):1891–1899
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Gintant, G. (2015). In Vitro Early Safety Pharmacology Screening: Perspectives Related to Cardiovascular Safety. In: Pugsley, M., Curtis, M. (eds) Principles of Safety Pharmacology. Handbook of Experimental Pharmacology, vol 229. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-46943-9_2
Download citation
DOI: https://doi.org/10.1007/978-3-662-46943-9_2
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46942-2
Online ISBN: 978-3-662-46943-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)