
Abstract
Paraneoplastic pemphigus (PNP) is a life-threatening autoimmune blistering disease associated with neoplasia, most commonly lymphoproliferative disorders. Clinical manifestations include severe oral ulceration and a polymorphous cutaneous eruption. Treatment includes systemic corticosteroids, rituximab and other immunosuppressive therapies. The response to treatment and prognosis in PNP is generally poor.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Points-
Paraneoplastic pemphigus (PNP) or paraneoplastic autoimmune multiorgan syndrome (PAMS) is a rare autoimmune blistering disease associated with neoplasia, most commonly lymphoproliferative disorders.
-
The pathogenesis of PNP is not fully understood. Multiple antibodies, mainly directed against proteins of the plakin family, are detected in the sera of patients with this disease. Antibodies to periplakin and envoplakin are strongly associated with PNP.
-
Clinical manifestations include severe oral ulceration and a polymorphous cutaneous eruption. Pulmonary disease consistent with bronchiolitis obliterans may occur and is often fatal.
-
Diagnosis is based on clinical, histopathological and immunological findings. Immunological investigations, including indirect immunofluorescence on rat bladder, ELISA, immunoblotting and immunoprecipitation, have high sensitivity and specificity for the diagnosis of PNP.
-
Treatment includes systemic corticosteroids, rituximab and other immunosuppressive therapies. The response to treatment and prognosis in PNP is generally poor.
Definition and Epidemiology
Paraneoplastic pemphigus (PNP) is a life-threatening autoimmune blistering disease associated with neoplasia, most commonly lymphoproliferative disorders. An alternative term for PNP, which has been increasingly used in the literature, is paraneoplastic autoimmune multiorgan syndrome (PAMS). The latter term reflects the diversity of the cutaneous manifestations and systemic involvement that may occur in this disorder. The incidence and prevalence of PNP are unknown, but it is a rare disease. Anhalt et al. first described PNP in 1990, and by 2011, over 450 cases had been reported in the literature (Czernik et al. 2011). PNP occurs worldwide and affects both sexes, with possible male predominance. The disorder can occur at any age but appears to be most common in the 45–70 years age group.
Basic Concepts of Pathogenesis
PNP is associated with a variety of neoplasms, both malignant and, less commonly, benign. Lymphoproliferative disorders account for the vast majority of PNP cases. The most frequently associated neoplasms are non-Hodgkin lymphoma (40 %), chronic lymphocytic leuchemia (CLL) (18–30 %) and Castleman’s disease (10–18 %), followed by carcinoma (9 %), thymoma (6 %), sarcoma (6 %) and Waldenström’s macroglobulinaemia (1 %). Monoclonal gammopathy and malignant melanoma each account for fewer than 1 % of cases (Kaplan et al. 2004). Castleman’s disease is the leading cause of PNP in children, and it has been reported with a higher frequency in Chinese patients. The most common non-haematological malignancy associated with PNP is carcinoma, with an increasing variety of adenocarcinomas and squamous cell carcinomas reported in the recent years (Webster et al. 2013). Sarcomas are less commonly implicated and include leiomyosarcoma and follicular dendritic cell sarcoma.
PNP is an autoimmune disorder induced by an underlying neoplastic process. The pathogenesis of PNP is not entirely understood, but thought to involve both humoral and cell-mediated immune mechanisms. The humoral response is characterized by production of antibodies mainly directed against intracellular proteins of the plakin family, which are essential components of desmosomes and play a key role in epidermal cell adhesion. Antibodies directed against two of these proteins, periplakin and envoplakin, are the most characteristic of PNP, and their detection is a highly sensitive and specific laboratory feature of the disease. Other commonly associated antibodies include those to desmoplakins I and II, bullous pemphigoid antigen 1 (BPAG1), plectin and a recently identified alpha-2-macroglobulin-like-1 antigen (A2ML1). Desmoglein 3 and 1 (Dsg 3, 1) antibodies are present in some cases, and antibodies to desmocollins, laminin-332 and plakophilin-3 have also been reported (Yashiro et al. 2013). In addition to circulating autoantibodies and tissue-bound antibodies in skin and mucous membranes, deposits of autoantibodies have been found in many internal organs of the affected patients. The role of most antibodies in the pathogenesis of PNP is uncertain, but those to periplakin and envoplakin may be pathogenic. The mechanism by which neoplasms induce antibodies to plakins and other antigens is unknown. Proposed mechanisms include dysregulation of the immune system, cross-reactivity between tumour and epidermal antigens and epitope spreading (increased exposure of multiple antigens to the immune system, as a result of tissue injury). The latter theory may explain the variety of antibodies found in PNP.
There is growing evidence that cell-mediated immunity is involved in the pathophysiology of PNP. Apoptosis of keratinocytes, mediated by cytotoxic T-lymphocytes, along with an increased local production of interferon-γ (IFN-γ) and TNF-α, has been demonstrated in PNP (Billet et al. 2006). In addition, CD8+ cytotoxic T-lymphocytes, CD56+ natural killer cells and CD68 + monocytes/macrophages have been found at the dermal–epidermal junction of affected patients. These inflammatory infiltrates are similar to the ones present in other diseases characterized by cell-mediated cytotoxic reactions, such as erythema multiforme and lichen planus. Therefore, it is possible that lichenoid variants of PNP predominantly involve cellular immunity, whereas vesiculobullous eruptions are mainly mediated by humoral responses.
Clinical Presentation
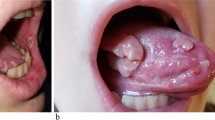
Approximately two-thirds of patients are known to have neoplasia at the time of onset of PNP. All PNP patients develop mucosal erosions, with severe, painful stomatitis being the most constant feature and the first manifestation of the disease in about 45 % of patients. The oral erosions are typically widespread, involving the tongue (particularly lateral borders), oropharynx and vermilion border of the lips. The erosions tend to appear more necrotic than those observed in pemphigus vulgaris (PV). Multiple other mucosal surfaces can be affected, including the conjunctiva, larynx, anogenital region and gastrointestinal tract.
The cutaneous involvement in PNP is characterized by polymorphous eruptions that significantly overlap with other dermatological diseases. The spectrum of cutaneous lesions includes vesicles and bullae, which may be flaccid or tense, erythematous/violaceous scaly papules and plaques, targetoid lesions and erosions. Based on the morphology and histological findings, five clinical variants of PNP have been described: pemphigus-like, bullous pemphigoid-like, lichen planus-like, graft-versus-host-disease (GVHD)-like and erythema multiforme-like. Pustules and vegetative lesions can occur, and a combination of various lesion types is often present in PNP patients. Involvement of the palms, soles and periungual skin is common, unlike PV in which it is rare. Extensive epidermal detachment, resembling toxic epidermal necrolysis (TEN), has also been described. Pulmonary involvement is increasingly recognized in PNP and manifests with a bronchiolitis obliterans-like disease. It is thought to occur in about 30–40 % of patients. Clinically it is characterized by dyspnoea in the presence of a relatively normal chest X-ray and an obstructive pattern on pulmonary function testing. High-resolution CT may demonstrate features of bronchiolitis obliterans, such as expiratory air trapping and bronchial wall thickening. The pulmonary disease is usually progressive and often leads to fatal respiratory failure. The mechanisms of respiratory involvement in PNP are not fully understood, but it has been shown that respiratory epithelial cells express the plakin proteins which are recognized by PNP antibodies, and their reactivity against pulmonary epithelial antigens may cause sloughing of epithelial cells with subsequent obstruction of small airways. In addition, T-cell-mediated cytotoxicity appears to play an important role in pulmonary disease.
The prognosis in PNP is poor, with mortality rates approaching 90 %. However, a recent study of 53 patients showed a 5-year survival rate of 38 %, suggesting that better outcomes may be possible (Leger et al. 2012). The main causes of death include sepsis, multiorgan failure, respiratory failure due to bronchiolitis obliterans and progression of an underlying malignancy. Erythema multiforme-like eruptions and keratinocyte necrosis have been shown to be predictors of fatal outcome. The prognosis is better in PNP associated with Castleman’s disease and thymoma, after resection of the tumours.
Diagnosis
The diagnosis of PNP is based on the combination of clinical and laboratory findings. There are no generally accepted diagnostic criteria, but the following mandatory criteria have been suggested: severe stomatitis, positive indirect immunofluorescence (IIF) on rat/monkey bladder epithelium, positive direct immunofluorescence (DIF) if negative IIF, detection of antibodies against envoplakin or periplakin and a concurrent neoplasm.
The laboratory tests used in PNP include histopathology, DIF and serological studies. The most common histopathological findings are suprabasal acantholysis, lichenoid interface dermatitis and keratinocyte necrosis. However, the histological features in PNP are not diagnostic as they are highly variable and may overlap with many other conditions.
DIF findings in PNP include epithelial intercellular and/or basement membrane zone (BMZ) IgG and/or C3. Combined intercellular and BMZ binding is unusual and should prompt suspicion about PNP. The sensitivity of DIF for PNP ranges from 41 to 79 %. IIF labelling of rat bladder epithelium has a high sensitivity and specificity for PNP of up to 86 and 99 %, respectively (Joly et al. 2000). IIF performed on rat bladder epithelium is positive exclusively in PNP, whereas IIF on monkey oesophagus detects antibodies in both PV and PNP. Enzyme-linked immunosorbent assay (ELISA) for circulating antibodies against envoplakin and periplakin has a high diagnostic accuracy in PNP, with a sensitivity and specificity around 80 and 96 %, respectively (Probst et al. 2009). Immunoblotting and immunoprecipitation are additional serological tests for the diagnosis of PNP. They detect antibodies directed to periplakin (190 kD), envoplakin and desmoplakin II (210 kD double band), desmoplakin I (250 kD), BPAG1 (230 kD), plectin (>400 kD), A2ML1 antigen (170 kD) and other proteins found in PNP. Immunoblotting and immunoprecipitation are highly sensitive specific tests, but their availability may be limited.
Once the diagnosis of PNP is established, evaluation for an underlying malignancy is necessary.
Differential Diagnosis
-
Oral mucositis due to chemotherapy and other causes of severe oral ulceration
-
Pemphigus (vulgaris, drug-induced)
-
Bullous pemphigoid and other autoimmune blistering disorders
-
Mucous membrane pemphigoid
-
Drug eruptions
-
Lichen planus
-
GVHD
-
Erythema multiforme
-
Stevens–Johnson syndrome and TEN
General Principles of Treatment
Treatment of PNP requires close collaboration between oncology, dermatology and respiratory physicians. Response to treatment in PNP is variable and generally poor. The cutaneous lesions are more responsive to therapy than the stomatitis, which is often refractory. Treatment of an underlying malignancy may be beneficial, but PNP commonly progresses despite controlling the neoplastic disease. In cases associated with thymoma and Castleman’s disease, complete tumour resection sometimes results in remission of PNP. Supportive measures and adequate analgesia play an essential role in the management of PNP. In terms of specific treatment, this is similar to that of PV both in terms of the drugs used and doses. Systemic corticosteroids (typically prednisolone 1 mg/kg/day) are usually the first-line therapy for patients with PNP. Pulsed intravenous steroids (e.g. methylprednisolone 500–1,000 mg on 1–5 consecutive days) can also be used as initial therapy. Prednisolone is slowly tapered over months and often combined with other immunosuppressive agents, such as azathioprine, ciclosporin, mycophenolate mofetil and cyclophosphamide (→see Pemphigus chapter). Due to the rarity of PNP, there is insufficient evidence to date to support any particular treatment modality or therapeutic regime. In case reports and small case series, the following doses of immunosuppressive agents have been used, alongside steroids: azathioprine 100 mg/day, ciclosporin 5–7 mg/kg/day and mycophenolate mofetil 1–2 g/day. Several treatment regimens have been described for cyclophosphamide, for instance, cyclophosphamide 500 mg intravenously on days 1–3 every 3 weeks with dexamethasone 100 mg intravenously at 3-week intervals (Frew and Murrell 2011). Rituximab, a monoclonal antibody against CD20, expressed on B-lymphocytes, has shown efficacy for PNP in case reports, albeit inconsistently (Frew and Murrell 2011). The main dosage schedule used is 375 mg/m2 intravenously once weekly for 4 weeks with (or without) subsequent doses at the intervals of several weeks to months. Alemtuzumab, a monoclonal antibody to CD52, expressed on T- and B-lymphocytes, has recently been reported to be effective in PNP associated with CLL. Other therapeutic options, such as plasmapheresis and intravenous immunoglobulin (e.g. 2 g/kg/cycle over 2–4 consecutive days, with cycles repeated every 4–6 weeks), have been used as adjuncts to immunosuppressive therapy.
Treatment should continue until disease control is achieved (as defined by the absence of new and healing of existing cutaneous and mucosal lesions), lack of efficacy is apparent or serious side effects occur. Careful monitoring and close liaison with oncologists are essential when using immunosuppressive therapy in the context of an underlying malignancy. A particular caution should be taken in the setting of an undiagnosed neoplasm, as empirical immunosuppression in such circumstances can be detrimental.
Abbreviations
- A2ML1:
-
Alpha-2-macroglobulin-like-1 antigen
- BMZ:
-
Basement membrane zone
- BPAG1:
-
Bullous pemphigoid antigen 1
- DIF:
-
Direct immunofluorescence
- Dsg:
-
Desmoglein
- ELISA:
-
Enzyme-linked immunosorbent assay
- IIF:
-
Indirect immunofluorescence
- PAMS:
-
Paraneoplastic autoimmune multiorgan syndrome
- PNP:
-
Paraneoplastic pemphigus
- TEN:
-
Toxic epidermal necrolysis
References
Billet SE, Grando SA, Pittelkow MR. Paraneoplastic autoimmune multiorgan syndrome: review of the literature and support for a cytotoxic role in pathogenesis. Autoimmunity. 2006;39(7):617.
Czernik A, Camilleri M, Pittelkow MR, Grando SA. Paraneoplastic autoimmune multiorgan syndrome: 20 years after. Int J Dermatol. 2011;50(8):905–14.
Frew JW, Murrell DF. Current management strategies in paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome). Dermatol Clin. 2011;29(4):607–12.
Joly P, Richard C, Gilbert D, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43(4):619.
Kaplan I, Hodak E, Ackerman L, et al. Neoplasms associated with paraneoplastic pemphigus: a review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol. 2004;40(6):553–62.
Leger S, Picard D, Ingen-Housz-Oro S, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol. 2012;148(10):1165.
Probst C, Schlumberger W, Stöcker W, et al. Development of ELISA for the specific determination of autoantibodies against envoplakin and periplakin in paraneoplastic pemphigus. Clin Chim Acta. 2009;410(1–2):13–8.
Webster G, Marques PM, Ortega Filho RC, et al. Paraneoplastic pemphigus: initial manifestation of lung cancer. Braz J Otorhinolaryngol. 2013;79(2):258.
Yashiro M, Nakano T, Taniguchi T, et al. IgA paraneoplastic pemphigus in angioimmunoblastic T-cell lymphoma with antibodies to desmocollin 1, type VII collagen and laminin 332. Acta Derm Venereol. 2013;4.
Further Reading
Anan T, Shimizu F, Hatano Y, et al. Paraneoplastic pemphigus associated with corneal perforation and cutaneous alternariosis: a case report and review of cases treated with rituximab. J Dermatol. 2011;38(11):1084.
Anhalt GJ. Paraneoplastic pemphigus. J Investig Dermatol Symp Proc. 2004;9:29–33.
Anhalt GJ, Kim SC, Stanley JR, et al. Paraneoplastic pemphigus. An autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990;323(25):1729–35.
Bech R, Baumgartner-Nielsen J, Peterslund NA, et al. Alemtuzumab is effective against severe chronic lymphocytic leukaemia-associated paraneoplastic pemphigus. Br J Dermatol. 2013;169(2):469–72.
Cho JH, Kim NJ, Ko SM, et al. A case report of paraneoplastic pemphigus associated with esophageal squamous cell carcinoma. Cancer Res Treat. 2013;45(1):70–3.
Kimyai-Asadi A, Jih MH. Paraneoplastic pemphigus. Int J Dermatol. 2001;40:367–72.
Lambert J, Bracke S, van Roy F, et al. Serum plakophilin-3 autoreactivity in paraneoplastic pemphigus. Br J Dermatol. 2010;163(3):630–2.
Li J, Bu DF, Huang YC, Zhu XJ. Role of autoantibodies against the linker subdomains of envoplakin and periplakin in the pathogenesis of paraneoplastic pemphigus. Chin Med J. 2009;122(5):486–95.
Miida H, Kazama T, Inomata N, et al. Severe gastrointestinal involvement in paraneoplastic pemphigus. Eur J Dermatol. 2006;16(4):420–2.
Mimouni D, Anhalt GJ, Lazarova Z, et al. Paraneoplastic pemphigus in children and adolescents. Br J Dermatol. 2002;147:725–32.
Nguyen VT, Ndoye A, Bassler KD, et al. Classification, clinical manifestations, and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome: a reappraisal of paraneoplastic pemphigus. Arch Dermatol. 2001;137:193–206.
Nousari HC, Deterding R, Wojtczack H, et al. The mechanism of respiratory failure in paraneoplastic pemphigus. N Engl J Med. 1999;340(18):1406.
Numata S, Teye K, Tsuruta D, et al. Anti-alpha-2-macroglobulin-like-1 autoantibodies are detected frequently and may be pathogenic in paraneoplastic pemphigus. J Invest Dermatol. 2013;133(7):1785–93.
Sehgal VN, Srivastava G. Paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome. Int J Dermatol. 2009;48(2):162–9.
Wang J, Zhu X, Li R, et al. Paraneoplastic pemphigus associated with Castleman tumor: a commonly reported subtype of paraneoplastic pemphigus in China. Arch Dermatol. 2005;141:1285–93.
Yamada T, Nakamura S, Demitsu T, et al. Paraneoplastic pemphigus mimicking toxic epidermal necrolysis associated with B-cell lymphoma. J Dermatol. 2013;40(4):286–8.
Zhang J, Qiao QL, Chen XX, et al. Improved outcomes after complete resection of underlying tumors for patients with paraneoplastic pemphigus: a single-center experience of 22 cases. J Cancer Res Clin Oncol. 2011;137(2):229–34.
Zimmermann J, Bahmer F, Rose C, et al. Clinical and immunopathological spectrum of paraneoplastic pemphigus. J Dtsch Dermatol Ges. 2010;8(8):598–606.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Golberg, O., Harman, K.E. (2015). Paraneoplastic Pemphigus. In: Katsambas, A.D., Lotti, T.M., Dessinioti, C., D’Erme, A.M. (eds) European Handbook of Dermatological Treatments. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-45139-7_73
Download citation
DOI: https://doi.org/10.1007/978-3-662-45139-7_73
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-45138-0
Online ISBN: 978-3-662-45139-7
eBook Packages: MedicineMedicine (R0)