
Abstract
β-adrenoceptors are important in the regulation of heart function and have been studied extensively in recent decades. In vitro studies have shown downregulation of β-adrenoceptor density in heart failure and cardiac conditions that may lead to heart failure.
Novel methods have been developed to measure β-adrenoceptors in vivo with the use of positron emission tomography (PET). A PET study with the radioligand [11C]-CGP-12177 has shown promising results and measurements of β-adrenoceptor density with [11C]-CGP-12177 were shown to be reproducible and in agreement with in vitro studies. [11C]-CGP-12388 using a simpler method of radiochemical synthesis has been presented as an alternative. Also, transportable [18F]-labeled PET ligands are in development and applicable for more general use in PET centers lacking a cyclotron. Most PET studies with CGP radioligands were performed in the 1990s. The main limitation of [11C]-CGP-12177 and [11C]-CGP-12388, besides the troublesome production of the former, is the lack of subtype selectivity. Future perspectives may include the development of subtype-selective β-adrenergic receptor ligands to obtain more information about the pathophysiological role of the different subpopulations in vivo.
Using the full potential of PET, performance of regional measurements and longitudinal studies might add further knowledge to the pathophysiological role of the β-adrenoceptor in cardiac disease and the effect of interventions. This chapter will give an overview of the background of different β-adrenergic receptor types, their role in cardiac diseases, current PET imaging possibilities of the β-adrenergic receptor, and new developments in this field.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Positron Emission Tomography
- Human Heart
- Positron Emission Tomography Tracer
- Systolic Heart Failure
- Idiopathic Dilate Cardiomyopathy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
11.1 Introduction
Heart failure and arrhythmia are a major cause of mortality and morbidity (Bui et al. 2011; Heidenreich et al. 2011; Jhund et al. 2009; Rathi and Deedwania 2012). Cardiac sympathetic nervous system dysfunction is associated with heart failure and sudden cardiac death (Brunner-La Rocca et al. 2001). A common finding in heart failure and sudden cardiac death is a disturbed cardiac β-adrenergic receptor expression (Bristow 1984). Current status in the clinic is that patients are suboptimally selected for treatment, leading to over- and underdiagnosis of heart failure patients. Better selection of patients with high risk of fatal arrhythmias and risk of heart failure is needed for optimal targeted therapy. Also, patient selection for costly implantable cardioverter defibrillator (ICD) to prevent fatal arrhythmias should be improved. Finally, the prediction of the success rate of the costly cardiac synchronization therapy (CRT) in heart failure patients can be optimized by non-invasive imaging of the β-adrenoceptor density of the heart as the effect of CRT is related to the β-adrenoceptor density of the heart (Chakir et al. 2009).
PET is an accurate technique for non-invasive imaging of cardiac β-adrenergic receptor expression (Doze et al. 2002; Elsinga et al. 1998). The β-adrenoceptor plays an important role in the relation with heart failure development and arrhythmias and is therefore a potential therapeutic target for these pathologies (de Jong et al. 2005; Lefroy et al. 1993; Schafers et al. 1998; Wichter et al. 2000). A general feature of the failing human heart is a decrease in cardiac β-adrenoceptors that in most (but not all) cases is due to a selective decrease in β1-adrenoceptors leading to a shift in the β1-:β2-adrenoceptor ratio towards β2-adrenoceptors (Brodde et al. 2001). The PET tracers [11C]-CGP-12177 and [11C]-CGP-12388 are developed to image and quantify the cardiac β-adrenergic receptor density (Delforge et al. 1991; Doze et al. 2002; Elsinga et al. 1997). Due to the short half-life of carbon-11 (~20 min), these radiopharmaceuticals can only be used in PET centers with an on-site cyclotron. Another disadvantage is the nonselective binding of these PET tracers to different types of β-adrenergic receptors.
11.2 β-Adrenergic Receptors of the Heart
11.2.1 Background of β-Adrenergic Receptors in the Normal Heart
Four different β-adrenoceptor subtypes have been cloned so far and identified pharmacologically; they are designated β1-, β2-, β3-, and β4−adrenoceptors (Bylund et al. 1994, 1998; Kaumann and Molenaar 1997). It is generally accepted that, in the human heart, functional β1- and β2-adrenoceptors coexist. The expression of both receptors has been first demonstrated by radionuclide ligand binding studies and was subsequently confirmed by functional experiments (Bristow et al. 1990; Brodde et al. 1992a). The number of β-adrenoceptors is more or less evenly distributed in the right and left atrial and ventricular tissue; however, the proportion of β2-adrenoceptors is slightly higher in the atria (approximately 1/3 of the total β-adrenoceptors expression) than in the ventricles (about 20 % of the total β-adrenoceptors expression) (Brodde 1991; Steinfath et al. 1992a) and may be even higher (~50 %) in the atrioventricular conducting system (Elnatan et al. 1994). In the healthy human heart, the β1-adrenoceptor is the dominant subtype (β1 to β2 ratio = 3:1).
Both β1- and β2-adrenoceptors bind to adenylyl cyclase and cause increase of the intracellular amount of cAMP (Bristow et al. 1989; Brodde et al. 1984). In the human heart, adenylyl cyclase is preferentially activated by β2-adrenoceptor stimulation although β1-adrenoceptors predominate. This has been demonstrated in human right atrium (Brodde et al. 1984; Bruckner et al. 1984) and in human ventricular myocardium (Bristow et al. 1989; Kaumann et al. 1989). The mechanism underlying these different coupling efficiencies of human cardiac β1- and β2-adrenoceptors to adenylyl cyclase is not known. It can be explained by a general phenomenon that β2-adrenoceptors couple more efficiently to adenylyl cyclase than β1-adrenoceptors. In vitro experiments have convincingly shown that both β1- and β2-adrenoceptors can mediate positive inotropic effects of β-adrenoceptor agonists in isolated electrically driven atrial and ventricular preparations (Brodde et al. 2001). In right and left atria, β1- and β2-adrenoceptor stimulation can evoke maximum positive inotropic effects, while in right and left ventricles, only β1-adrenoceptor stimulation can evoke maximum positive inotropic effects and β2-adrenoceptor stimulation only submaximal positive inotropic effects (Kaumann et al. 1989; Motomura et al. 1990). High proportions of the β2 adrenoceptor are also found in the pacemaker and conduction regions, where they may be important in controlling heart rate and rhythm (Dzimiri 1999).
During the last few years, evidence has accumulated that, in addition to β1- and β2-adrenoceptors, a third or fourth (or both) β-adrenoceptor might exist in the human heart.
β3-adrenoceptor transcripts have been detected in the human heart (Pott et al. 2006). Stimulation of the β3-adrenoceptor produces a negative inotropic effect. The inhibition of contractility includes the inhibitory G protein, Gi/O, and results from the production of nitric oxide (NO) by the endothelial isoform of NO synthase (eNOS) and an increase in intracellular cGMP level (Gauthier et al. 1996, 1998). Compared to β1- and β2-adrenoceptor, the β3-adrenoceptor presents a relative in vitro and in vivo lack of desensitization following activation with agonists (Nantel et al. 1993). These features suggest that the expression of β3-adrenoceptor in heart may have pathophysiological significance.
Opposite regulation has been described for β1-adrenoceptor and β3-adrenoceptor in the failing human heart (Moniotte et al. 2001). In addition to the classically observed β1-adrenoceptor downregulation (Bristow et al. 1982), an upregulation of β3-adrenoceptor was described (Moniotte et al. 2001). Despite increased β3-adrenoceptor expression, the negative inotropic effect was slightly reduced in failing heart tissue compared with responses observed in non-failing samples because of concurrent alterations in post-receptor coupling mechanisms, especially decreased eNOS expression. Nevertheless, the reduction in β3-adrenoceptor response is less than that obtained with β1-adrenoceptor stimulation.
In addition to the possible existence of cardiodepressant β3-adrenoceptors, Kaumann and colleagues had postulated the existence of a putative β4-adrenoceptor in the human heart that upon stimulation causes positive inotropic effects (Brodde and Michel 1999; Gauthier et al. 2000; Kaumann and Molenaar 1997). This receptor type, which had never been cloned and was primarily stimulated by CGP-12177, had properties clearly different from the β3-adrenoceptor. The β4-adrenoceptor interacts with nonconventional partial agonists, e.g., CGP-12177, that cause cardiostimulant effects at concentrations considerably higher than those that block β1- and β2,-adrenoceptors.
11.2.2 β-Adrenergic Receptor Expression in the Failing Heart
Chronic excessive sympathetic activation leads to substantial and pathologic downregulation of postsynaptic β-adrenergic receptors. The distribution of β1- and β2-adrenoceptors in the human heart can be inhomogeously altered in pathological situations such as heart failure or by pharmacological interventions. A general feature of the failing human heart is a decrease in cardiac β-adrenoceptors that in most cases is due to a selective decrease in β1-adrenoceptors leading to a shift in the β1-:β2-adrenoceptor ratio towards β2-adrenoceptors (Brodde et al. 2001). In patients with biventricular failure, β-adrenoceptors are downregulated in both right and the left ventricle (Pitschner et al. 1993). Interestingly, it appears that the decrease in β-adrenoceptors is more pronounced in ventricular tissue than in atrial tissue (Brodde et al. 1998). On the other hand, in patients with primary pulmonary hypertension who exhibit isolated right ventricular failure, β-adrenoceptors are chamber specifically downregulated only in right ventricles (Bristow et al. 1991). β3-adrenoceptors are overexpressed in heart failure and hypertension and could constitute a new therapeutic target (Moniotte et al. 2001).
Myocardial ischemia will result in upregulation of membrane-bound β-adrenoceptors (Maisel et al. 1985; Majmudar and Nahrendorf 2012). However, some studies also find downregulation of β-adrenoceptors in ischemic hearts (Rhee and Tyler 1985). Conflicting results may be explained by the fact that ongoing ischemia or hypertension proceeding to heart failure may cause downregulation of β-adrenoceptors, whereas short-term ischemia may cause upregulation. Diabetes may also be related to altered adrenergic receptor properties and density (Heyliger et al. 1982; Williams et al. 1983). Altered adrenergic receptor properties may underlie, at least in part, the chronotropic and inotropic abnormalities of cardiac performance that are associated with the diabetic state.
Also, the use of chemotherapy may influence the β-adrenoceptor density. Kenk and colleagues found that adriamycin-induced toxicity did not change presynaptic noradrenaline uptake but decreased β-adrenergic receptors in cardiac tissues (Kenk et al. 2010).
The transplanted human heart is a denervated organ; animal studies have shown that denervation can induce β-adrenoceptor sensitization (Brodde 1993). Whether this also occurs in the transplanted human heart is not completely understood at present. Assessment of β-adrenoceptor density over a long period after heart transplantation did not result in any upregulation (Brodde et al. 1991).
There was, however, a redistribution of β1- and β2-adrenoceptors with time after heart transplantation: β1-adrenoceptors decreased whereas β2-adrenoceptors increased (Brodde et al. 1991; Farrukh et al. 1993; Steinfath et al. 1992b). Finally, treatment of patients with β-adrenoceptor blockers can affect distribution of cardiac β1- and β2-adrenoceptors. Thus, chronic treatment of patients with coronary artery disease with β1-adrenoceptor blockers such as metoprolol, atenolol, or bisoprolol leads to a selective increase of cardiac β1-adrenoceptors (Brodde 1990). This indicates that chronic treatment with β1-adrenoceptors blockers sensitizes cardiac β2-adrenoceptors. A similar cardiac β2-adrenoceptor sensitizing effect of chronic β1-adrenoceptor blocker treatment has also been found in vivo, in patients with coronary artery disease (Hall et al. 1991) as well as in healthy volunteers (Hall et al. 1993). The mechanism of this cardiac β1-/β2-adrenoceptor crossover interaction is, however, not known. A previous study has demonstrated that carvedilol rather than metoprolol is the drug of choice for improving the hemodynamics and ventricular remodeling in the failing heart (Zhao et al. 2007). The blockade of β3-adrenoceptors may play a part in these beneficial effects on both left and right ventricles. In patients with heart failure, carvedilol is associated with a larger increase in left ventricular ejection fraction (LVEF) at rest, left ventricular stroke volume, and stroke work during exercise than metoprolol (Metra et al. 2000). Metoprolol diminishes left ventricular remodeling, but unlike carvedilol, it has no significant impact on right ventricular remodeling during chronic heart failure.
11.3 In Vitro Measurement of β-Adrenoceptor Density
The role of the β-adrenoceptor in the regulation of myocardial contraction has been extensively investigated, both in animal models and in human tissue. Assessment of β-adrenoceptor density in a membrane preparation became possible with the introduction of high-affinity, radiolabeled β-adrenergic antagonists, [3H]-DHA (Lefkowitz et al. 1974) and [125I]-IHYP (Aurbach et al. 1974). A disadvantage of these assays is the use of lipophilic radionuclide ligands, which leads to high nonspecific binding and binding to internalized receptors. With the introduction of [3H]-CGP-12177, a hydrophilic β-adrenergic receptor ligand, and the development of methods to measure cardiac β-adrenoceptors in isolated cells (Buxton and Brunton 1985) and tissue (Watson-Wright et al. 1989), it was a breakthrough to measure β-adrenoceptors at the surface of intact cells (Staehelin et al. 1983) in a physiological state. In vitro measurements in human cardiac tissue in the non-failing human heart have shown that β-adrenoceptor density varies between 70 and 100 fmol/mg protein (Brodde 1991). This variation may be due to the different circumstances in which tissues are obtained, different methods of transportation of tissues to the laboratory, different radionuclide ligands, and/or differences in the methodology of the measurements.
One of the first papers in the early 1980s reported a decreased β-adrenoceptor density in the failing human heart using in vitro ligand binding to homogenized myocardial samples of hearts excised from cardiac transplant recipients (Bristow et al. 1982) (Table 11.1). They found reductions in β-adrenoceptor density of approximately 50 %. In the late 1980s, it was found that the severity of heart failure is related to the reduction of the β-adrenoceptor density and the responsiveness to agonists (Bohm et al. 1988). This downregulation of β-adrenoceptors has been explained by an enhanced sympathetic drive to the heart and hence endogenous downregulation by an elevated release of cardiac-derived noradrenaline (Ruffolo and Kopia 1986), leading to a loss of cardiac contractility (Brodde et al. 1992b). The reduction in receptor density in idiopathic dilated cardiomyopathy is selective for the β1-adrenoceptor subtype (Bristow et al. 1986; Brodde 1991) and is accompanied by a similar decrease in β1-adrenoceptor mRNA levels (Ihl-Vahl et al. 1996). This results in a physiological loss of receptors (Pitschner et al. 1993) and is correlated with the severity of heart failure (Engelhardt et al. 1996).
The levels of β2-adrenoceptor and β2-adrenoceptor mRNA remain unaffected but it is believed that these receptors become uncoupled (Brodde 1991). Patients with severe left ventricular dysfunction showed fewer β-adrenergic receptors in lymphocytes, as measured in radioligand binding assays (Colucci et al. 1981). However, although changes in lymphocyte β2-adrenoceptors are significantly correlated with changes in cardiac β2-adrenoceptors, they are not related to changes in cardiac β1-adrenoceptors, which predominate in all parts of the human heart. Furthermore, circulating lymphocytes are not exposed to the local environment of neuronally released catecholamines in the myocardial interstitium. The use of lymphocyte β2-adrenoceptors as a tool for predicting the status of cardiac β-adrenoceptors is therefore quite limited (Brodde et al. 1989), and thus cardiac tissue will be needed to evaluate cardiac β-adrenoceptor function. Abnormal sympathetic nervous system and β-adrenoceptor signaling is also associated with diabetes. Thackeray and colleagues used [3H]-CGP-12177 to examine altered β-adrenoceptor expression in diabetic rat hearts (Thackeray et al. 2011). Reduced cardiac [3H]-CGP-12177 binding in the presence of sustained hyperglycemia corresponded to a decrease in relative β-adrenoceptor expression. Their study indirectly supports the use of [11C]-CGP-12177 for assessment of cardiac dysfunction in diabetes, by evaluating the cardiac β-adrenoceptor density.
11.4 Non-invasive Imaging of Cardiac β-Adrenergic Receptors
11.4.1 PET Imaging and Density Measurement of Cardiac β-Adrenergic Receptors
Several postsynaptic receptor ligands have been labeled and proposed as PET tracers for cardiac quantification and imaging (Elsinga et al. 1998; Law et al. 2010; Tseng et al. 2001). However, the clinical use of receptor-targeted tracers has been limited to a few studies and still faces significant challenge. High specific binding, high affinity, and hydrophilicity, which avoids binding to internalized inactive receptors, lack of pharmacologic effects, and, finally, a simple and reliable tracer synthesis, are requirements that must be met for a widespread application of receptor ligands for cardiac PET. [11C]-CGP-12177, a hydrophilic nonselective β-adrenoreceptor antagonist, is still the most widely used tracer for adrenergic receptor imaging (Caldwell et al. 2008; Elsinga et al. 1998; Link et al. 2003; Naya et al. 2009) Synthesis of this tracer is not simple and requires [11C]-phosgene as a precursor, which has prevented a broader clinical application until now. CGP-12177 has high receptor affinity and fast plasma clearance, suggesting feasibility for clinical imaging. A graphical method, which adjusts for kinetics related to metabolites, has been established for quantification in humans (Delforge et al. 2002). This approach requires a dual-injection protocol with tracer doses of high and low specific activity (Fig. 11.1). β-Adrenergic receptor density (B max) measured by [11C]-CGP-12177 PET correlated well with in vitro measurements of myocardial samples in both healthy volunteers and patients with congestive cardiomyopathy (Delforge et al. 2002). [11C]-CGP-12388 is a non-subtype-selective β-adrenergic receptor antagonist and an isopropyl analog of CGP-12177. CGP-12388 can be labeled easier than CGP-12177 via a one-step procedure using 2-[11C]-acetone (Elsinga et al. 1994). It is equally hydrophilic compared to [11C]-CGP-12177 and the biodistribution and retention of CGP-12388 is reported to be similar to CGP-12177 (Doze et al. 2002). Both CGP ligands have been applied in the biologically active S-enantiomer and can be blocked by pindolol (Fig. 11.1).

PET images of a human volunteer acquired with [11C]-CGP-12388. Transaxial cross sections in the time frame 14–60 min postinjection are displayed. The upper row is the control study; the bottom row is the pindolol-blocked study (Elsinga et al. 2001)
[18F]-fluorocarazolol and [11C]-carazolol are non-subtype-selective, lipophilic radioligands with high affinity for β1- and β2-adrenoceptors. The use of fluorine-18 instead of carbon-11 has the advantages of higher specific activity and a longer half-life, which enables prolonged PET studies.
[11C]-carazolol has been evaluated by Berridge and coworkers in mice and pigs (Berridge et al. 1994). The pig heart was clearly visualized. Specific binding to β-adrenoceptors was demonstrated by injection of the bioactive [11C]-isomer (specific and nonspecific binding), followed by a second injection of the (R)-isomer (only nonspecific binding). [18F]-fluorocarazolol has been evaluated in several animal models and in humans. Specific binding to β-adrenoceptors of [18F]-fluorocarazolol was demonstrated: (1) by injection of the (S)-isomer and subsequent injection of the (R)-isomer, (2) by blocking experiments with various β-adrenoceptor agonists and antagonists (van Waarde et al. 1995), and (3) by saturation experiments (Doze et al. 1998). The in vivo binding of fluorocarazolol was found to be stereospecific (activity residing in the (S)-isomer). It could be blocked by drugs that bind to β1- and β2-adrenoceptors, and specific binding was in good agreement with β-adrenoceptor densities determined by in vitro assays. Metabolite analyses of [18F]-fluorocarazolol showed a rapid (<5 min) appearance of polar metabolites in plasma, while at 60 min postinjection, 92 and 82 % of the total radioactivity in lung and heart remained native [18F]-fluorocarazolol (van Waarde et al. 1995). In PET images of male Wistar rats, the lungs were clearly visible and pulmonary uptake of radioactivity was strongly decreased (>90 %) after pretreatment of the animals with propranolol. The heart could not be visualized. However, PET scans after i.v. injection of [18F]-fluorocarazolol in human volunteers clearly showed β-adrenoceptors in both lung and heart (Visser et al. 1997). Cardiac uptake of radioactivity was strongly inhibited after ingestion of pindolol (to 39 % of the control value at 60 min postinjection). These pilot studies in humans were performed with noncarrier-added [18F]-fluorocarazolol (∼1 nmol), after it had been shown that fluorocarazolol is not acutely toxic in rodents at doses >10,000-fold higher than were administered to volunteers. For quantification of receptor densities with compartment models, a dual-injection protocol is required involving the administration of a pharmacological dose of the radioligand (∼100 nmol). Such protocols can only be carried out after extensive toxicological screening of the experimental drug. Unfortunately, fluorocarazolol showed a positive Ames test (mutagenicity in bacterial strains) during such examination. Therefore, it was decided to terminate all human studies with [18F]-fluorocarazolol. In contrast to fluorocarazolol, the available toxicological data of carazolol show that the compound is nontoxic even at very high doses. Evaluation in humans should indicate the suitability of [11C]-carazolol as a radiopharmaceutical for clinical PET, although this PET ligand is lipophilic.
11.4.2 PET Imaging of β-Adrenergic Receptors in the Failing Heart
Several factors may induce changes of membrane-bound β-adrenergic receptor density. Major causes are (1) heart failure, (2) myocardial ischemia with or without diabetes, (3) hypertension, and (4) toxic damage. The first study measuring β-adrenoceptor density with [11C]-CGP-12177 PET in patients showed a decreased β-adrenoceptor density in vivo in a group of patients with heart failure due to idiopathic dilated cardiomyopathy (Merlet et al. 1993) (Table 11.2). The [11C]-CGP-12177 PET measurements correlated with β-adrenoceptor density in endomyocardial biopsy. Moreover, these in vivo measurements correlated with functional measurements of β-contractile responsiveness to intracoronary dobutamine infusion. These studies were followed by reports of the group of Camici concerning patients with hypertrophic cardiomyopathy in different phases of disease. Their first report using [11C]-CGP-12177 PET showed a slightly reduced β-adrenoceptor density in patients with primary hypertrophic cardiomyopathy with preserved left ventricular function (Lefroy et al. 1993). These results were in agreement with the hypothesis of an increased sympathetic activity in the heart, which is supported by an elevated myocardial noradrenaline content (Kawai et al. 1983; Tsukamoto et al. 2007) and cardiac spillover of noradrenaline (Brush et al. 1989) in patients with hypertrophic cardiomyopathy. A group with secondary hypertrophic cardiomyopathy due to hypertension and aortic stenosis without heart failure showed a comparable reduction in β-adrenoceptor with [11C]-CGP-12177 PET (Choudhury et al. 1996b). A study in a mixed group of patients with hypertrophic cardiomyopathy with and without signs of heart failure showed a lower β-adrenoceptor density in patients with signs of heart failure and a correlation between β-adrenoceptor density and ventricular function using [11C]-CGP-12177 PET (Choudhury et al. 1996a). From these studies it might be concluded that β-adrenoceptor downregulation precedes clinical heart failure and may be an early clinical marker of left ventricular dysfunction. A study of de Jong and colleagues investigated whether decreased myocardial β-adrenoceptor density in patients with idiopathic dilated cardiomyopathy (IDC) can be estimated using [11C]-CGP-12388 PET (de Jong et al. 2005). They concluded that [11C]-CGP-12388 PET is applicable for the measurement of myocardial β-adrenoceptor density in patients. A highly significant reduction in β-adrenoceptor density was found with a significant difference in β-adrenoceptor density (p < 0.005) between patients with IDC (B max 5.4 ± 1.3 pmol/g) and healthy controls (B max 8.4 ± 1.5 pmol/g). A prospective longitudinal study may yield further evidence to support this finding (de Jong et al. 2005).
Link and colleagues used [11C]-meta-hydroxyephedrine ([11C]-mHED) to image norepinephrine transporter function as an indicator of presynaptic function and [11C]-CGP-12177 to measure global and regional cell surface β-adrenoceptor density as an indicator of postsynaptic function in 19 normal subjects and 9 congestive heart failure patients (Link et al. 2003). Presynaptic, but not postsynaptic, function was significantly different between normals and congestive heart failure patients. Presynaptic function was well matched to postsynaptic function in the normal hearts but significantly different and poorly matched in the congestive heart failure patients studied.
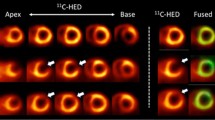
Caldwell and colleagues evaluated in 13 patients with ischemic congestive heart failure and 25 aged-matched healthy volunteers the presynaptic function with [11C]-mHED and the postsynaptic function with [11C]-CGP-12177 (Caldwell et al. 2008) (Fig. 11.2).

Short-axis PET images of [11C]-mHED (35- to 45 min sum) and [11C]-CGP-12177 (10- to 20 min sum from injection 1) showing left ventricular activity in a chronic congestive heart failure patient. Apical slices are at upper left and basal slices are at lower right of each panel. Arrows indicate extensive mismatch between [11C]-mHED and [11C]-CGP (Caldwell et al. 2008)
Myocardial blood flow was assessed with [15O]-water PET, but global and regional mean blood flow was not different between congestive heart failure and healthy subjects. They found reduced [11C]-mHED and [11C]-CGP-12177 activity in congestive heart failure patients compared with the healthy volunteers and also a mismatch (ratio B max of [11C]-CGP-12177 to [11C]-mHED uptake) between pre- and postsynaptic left ventricular sympathetic function in patients with severe congestive heart failure. After 1.5 year of follow-up, four individuals had an adverse outcome (congestive heart failure death, new or recurrent cardiac arrest or progressive congestive heart failure leading to transplantation). Three of the four patients had mismatch scores >3 times that of the healthy subjects or the congestive heart failure patients without an adverse outcome. Sympathetic signaling in such regions would be more dependent on circulating catecholamines, which are probably lower than those in a normally functioning myoneural junction (Bristow et al. 1992). This decrease could lead to β-adrenoceptor upregulation. However, in patients with dilated cardiomyopathy, [11C]-mHED PET is significantly correlated with the density but not the affinity of uptake-1 sites in the human heart, suggesting either loss of neurons or downregulation of uptake-1 in dilated cardiomyopathy (Ungerer et al. 1998).
After myocardial infarction, LV (left ventricle) remodeling is observed in non-infarcted LV myocardium. LV remodeling is closely associated with systolic heart failure. Myocardial dysfunction is related to the downregulation of cardiac postsynaptic β-adrenoceptors. A recent [11C]-CGP-12177 PET study found out that in the remote non-infarcted region in patients, β-adrenoceptor downregulation was observed, which was related to deterioration of local myocardial systolic function (Ohte et al. 2012).
Furthermore, noradrenaline uptake-1 mechanism and β-adrenoceptor density are reduced in the myocardium of patients with chronic LV dysfunction and evidence of hibernating myocardium (John et al. 2007). The increased sympathetic activity to the heart in these patients is a generalized rather than regional phenomenon which is likely to contributing to the remodeling process of the whole left ventricle rather than playing a causative role in hibernating myocardium.
In patients with syndrome X (Rosen et al. 1996) and asthma (Qing et al. 1997b), i.e., patients with normal left ventricular function, myocardial β-adrenoceptor density was found to be equal to that in normal volunteers, which is in agreement with the general hypothesis that β-adrenoceptor downregulation is only associated with heart failure. Interestingly, myocardial β-adrenoceptor downregulation was also observed in patients with arrhythmogenic right ventricular cardiomyopathy (Schafers et al. 1998). Although these patients have no heart failure, some evidence suggests that their local synaptic catecholamine levels are increased, which apparently causes downregulation of β-adrenoceptor similar to that in patients with heart failure (Wichter et al. 2000). A pharmacological intervention study has been performed in healthy volunteers. This study showed downregulation of pulmonary (Hayes et al. 1996) as well as myocardial β-adrenoceptors (Qing et al. 1997a) after 2 weeks of treatment with a β2-adrenoceptor agonist (albuterol). Naya and colleagues examined if [11C]-CGP-12177 PET could predict improvement of cardiac function by beta-blocker carvedilol treatment in patients with IDC (Naya et al. 2009). They found that myocardial β-adrenoceptor density is more tightly related to improvement of LVEF due to carvedilol than is cardiac contractile reserve as assessed by dobutamine stress echocardiography in patients with IDC. Patients with decreased myocardial β-adrenoceptor have higher resting adrenergic drive, as reflected by plasma norepinephrine, and may receive greater benefit from being treated by anti-adrenergic drugs.
11.5 New Developments
So far, production methods of β-receptor PET ligands were very complex, hampering their widespread use. Because of the potential clinical importance of cardiac β-adrenergic receptor imaging with PET, radiopharmaceuticals should be developed for PET sites without proper production facilities. To this end new radiopharmaceuticals need to be developed which are labeled with [18F] instead of [11C], as [18F] has a longer half-life (110 min) and can be transported to sites within a range of 4 h transport time, which is routinely done on a commercial basis for [18F]-FDG. Beside the disadvantage of the short half-life of carbon-11, CGP-derivatives are non-subtype-selective β-adrenergic receptor ligands. A more selective β-adrenergic receptor ligand characterized with fast plasma clearance and with a high affinity is needed, and β-adrenergic receptor 1 subtype will be the optimal choice in heart studies.
A [18F]-labeled β1-adrenoceptor PET ligand with these optimal properties as mentioned before is needed. Law and colleagues developed and applied a fluoroethoxy derivative of the β1-adrenoceptor antagonist ICI 89406, labeled with fluorine-18 [(S)-[18F]-FICI] (Law et al. 2010) in an animal study. Although in vitro membrane studies showed that (S)-FICI had high affinity and selectivity for β1- adrenoceptors, this study in mice and rats failed to demonstrate high specific binding of (S)-[18F]-F-ICI to myocardial β1-adrenoceptor.
Novel [18F]-fluorination techniques, such as click chemistry, new lead molecules can be synthesized that showed high affinity for β-adrenoceptors. In this click reaction, the bio-orthogonal functional groups alkyne and azide react to form triazoles.
The “click reaction” catalyzed by Cu(I) is a well-established method for rapid and highly efficient synthesis of 1,4-disubstituted triazoles from a wide variety of substrates. Using this method to prepare a β-adrenoceptor ligand, the hydroxyl propylamine moiety (crucial for binding to β-adrenoceptors) can partially be maintained and [18F] is introduced as a novel moiety, hopefully not causing mutagenicity of the carazolol derivatives. A lead compound being a [18F]-fluorinated analog of carazolol, [18F]-FPTC, was produced by a click reaction between a PEGylated [18F]-alkyne and an azidoalcohol derivative of 4-hydroxycarbazol.
A number of studies, either in animals or in human patients, have demonstrated that functionally active autoantibodies targeting the human β1-adrenergic receptor (anti-b1AR-abs) may play an important role in the development and clinical course of progressive cardiac dilatation and failure and increase the risk of developing malignant arrhythmia (Iwata et al. 2001a, b; Magnusson et al. 1994). The presence of these autoantibodies is associated with a markedly worse prognosis in patients with dilated cardiomyopathy (DCM) and ischemic heart disease.
The disadvantage of these anti-b1AR-antibodies is the interaction with the [18F]-labeled β1-adrenoceptor PET ligands which may cause interference with the PET tracer binding to β1-adrenergic receptors.
Future perspectives may include the development of [18F]-labeled subtype-selective β-adrenoceptor ligands to obtain more information about the pathophysiological role of the different subpopulations in vivo. Subtype-selective ligands are being developed for the β1-adrenoceptor as well as the β2-adrenoceptor, but thus far no suitable ligands have been produced and evaluated in clinical studies.
PET has been shown to be a promising technique for the investigation of the role of β-adrenoceptors in cardiac diseases. So far most studies have focused on their role in patients with systolic heart failure (i.e., with a reduced LVEF). However, it is currently unknown whether β-receptor density plays also a role in the development of heart failure and specifically in the development of heart failure with a preserved ejection fraction. The lifetime risk for developing heart failure is 20 % (Lloyd-Jones et al. 2002). Due to the ageing population, the incidence and prevalence of heart failure will increase, not only systolic heart failure but even more heart failure with a preserved ejection fraction (Brouwers et al. 2013). Several studies have identified risk factors for new onset of heart failure, including age, the presence of hypertension, and a history of ischemic heart disease. However, so far the role of β-adrenoreceptor in this is unknown and warrants further investigation.
11.6 Conclusions
The development of new methods to measure β-adrenoceptor in vivo might help us to further understand β-adrenoceptor function and provide additional prognostic information and assist in clinical decisions about therapeutic interventions. New perspectives will lie in the development and application of [18F]-labeled subtype-selective ligands and in using the full potential of PET to perform regional and longitudinal studies.
Abbreviations
- CRT:
-
Cardiac synchronization therapy
- DCM:
-
Dilated cardiomyopathy
- eNOS:
-
Endothelial isoform of NO synthase
- ICD:
-
Implantable cardioverter defibrillator
- IDC:
-
Idiopathic dilated cardiomyopathy
- LV:
-
Left ventricle
- LVEF:
-
Left ventricular ejection fraction
- NO:
-
Nitric oxide
- PET:
-
Positron emission tomography
References
Aurbach GD, Fedak SA, Woodard CJ et al (1974) Beta-adrenergic receptor: stereospecific interaction of iodinated beta-blocking agent with high affinity site. Science 186:1223–1224
Berridge MS, Nelson AD, Zheng L et al (1994) Specific beta-adrenergic receptor binding of carazolol measured with PET. J Nucl Med 35:1665–1676
Bohm M, Beuckelmann D, Brown L et al (1988) Reduction of beta-adrenoceptor density and evaluation of positive inotropic responses in isolated, diseased human myocardium. Eur Heart J 9:844–852
Bristow MR (1984) Myocardial beta-adrenergic receptor downregulation in heart failure. Int J Cardiol 5:648–652
Bristow MR, Ginsburg R, Minobe W et al (1982) Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med 307:205–211
Bristow MR, Ginsburg R, Umans V et al (1986) Beta 1- and beta 2-adrenergic-receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective beta 1-receptor down-regulation in heart failure. Circ Res 59:297–309
Bristow MR, Hershberger RE, Port JD et al (1989) Beta 1- and beta 2-adrenergic receptor-mediated adenylate cyclase stimulation in nonfailing and failing human ventricular myocardium. Mol Pharmacol 35:295–303
Bristow MR, Hershberger RE, Port JD et al (1990) Beta-adrenergic pathways in nonfailing and failing human ventricular myocardium. Circulation 82:I12–I25
Bristow MR, Anderson FL, Port JD et al (1991) Differences in beta-adrenergic neuroeffector mechanisms in ischemic versus idiopathic dilated cardiomyopathy. Circulation 84:1024–1039
Bristow MR, Minobe W, Rasmussen R et al (1992) Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest 89:803–815
Brodde OE (1990) Beta- and alpha-adrenoceptor-agonists and -antagonists in chronic heart failure. Basic Res Cardiol 85(Suppl 1):57–66
Brodde OE (1991) Beta 1- and beta 2-adrenoceptors in the human heart: properties, function, and alterations in chronic heart failure. Pharmacol Rev 43:203–242
Brodde OE (1993) Beta-adrenoceptors in cardiac disease. Pharmacol Ther 60:405–430
Brodde OE, Michel MC (1999) Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev 51:651–690
Brodde OE, O’Hara N, Zerkowski HR et al (1984) Human cardiac beta-adrenoceptors: both beta 1- and beta 2-adrenoceptors are functionally coupled to the adenylate cyclase in right atrium. J Cardiovasc Pharmacol 6:1184–1191
Brodde OE, Michel MC, Gordon EP et al (1989) Beta-adrenoceptor regulation in the human heart: can it be monitored in circulating lymphocytes? Eur Heart J 10(Suppl B):2–10
Brodde OE, Khamssi M, Zerkowski HR (1991) Beta-adrenoceptors in the transplanted human heart: unaltered beta-adrenoceptor density, but increased proportion of beta 2-adrenoceptors with increasing posttransplant time. Naunyn Schmiedebergs Arch Pharmacol 344:430–436
Brodde OE, Broede A, Daul A et al (1992a) Receptor systems in the non-failing human heart. Basic Res Cardiol 87(Suppl 1):1–14
Brodde OE, Hillemann S, Kunde K et al (1992b) Receptor systems affecting force of contraction in the human heart and their alterations in chronic heart failure. J Heart Lung Transplant 11:S164–S174
Brodde OE, Vogelsang M, Broede A et al (1998) Diminished responsiveness of Gs-coupled receptors in severely failing human hearts: no difference in dilated versus ischemic cardiomyopathy. J Cardiovasc Pharmacol 31:585–594
Brodde OE, Bruck H, Leineweber K et al (2001) Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res Cardiol 96:528–538
Brouwers FP, de Boer RA, van der Harst P et al (2013) Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J 34:1424–1431
Bruckner R, Meyer W, Mugge A et al (1984) Alpha-adrenoceptor-mediated positive inotropic effect of phenylephrine in isolated human ventricular myocardium. Eur J Pharmacol 99:345–347
Brunner-La Rocca HP, Esler MD, Jennings GL et al (2001) Effect of cardiac sympathetic nervous activity on mode of death in congestive heart failure. Eur Heart J 22:1136–1143
Brush JE Jr, Eisenhofer G, Garty M et al (1989) Cardiac norepinephrine kinetics in hypertrophic cardiomyopathy. Circulation 79:836–844
Bui AL, Horwich TB, Fonarow GC (2011) Epidemiology and risk profile of heart failure. Nat Rev Cardiol 8:30–41
Buxton IL, Brunton LL (1985) Direct analysis of beta-adrenergic receptor subtypes on intact adult ventricular myocytes of the rat. Circ Res 56:126–132
Bylund DB, Eikenberg DC, Hieble JP et al (1994) International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 46:121–136
Bylund DB, Bond RA, Clarke DE, Eikenburg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman PB, Milinoff PB, Ruffolo RR, Strosberg AD, Trendelenburg UG (1998) Adrenoceptors. In: Bylund DB, Bond RA, Clarke DE, Eikenburg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman PB, Milinoff PB, Ruffolo RR, Strosberg AD, Trendelenburg UG (eds) The IUPHAR compendium of receptor characterization and classification. IUPHAR Media, London, pp 58–74
Caldwell JH, Link JM, Levy WC et al (2008) Evidence for pre- to postsynaptic mismatch of the cardiac sympathetic nervous system in ischemic congestive heart failure. J Nucl Med 49:234–241
Chakir K, Daya SK, Aiba T et al (2009) Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation 119:1231–1240
Choudhury L, Guzzetti S, Lefroy DC et al (1996a) Myocardial beta adrenoceptors and left ventricular function in hypertrophic cardiomyopathy. Heart 75:50–54
Choudhury L, Rosen SD, Lefroy DC et al (1996b) Myocardial beta adrenoceptor density in primary and secondary left ventricular hypertrophy. Eur Heart J 17:1703–1709
Colucci WS, Alexander RW, Williams GH et al (1981) Decreased lymphocyte beta-adrenergic-receptor density in patients with heart failure and tolerance to the beta-adrenergic agonist pirbuterol. N Engl J Med 305:185–190
de Jong RM, Willemsen AT, Slart RH et al (2005) Myocardial beta-adrenoceptor downregulation in idiopathic dilated cardiomyopathy measured in vivo with PET using the new radioligand (S)-[11C]CGP12388. Eur J Nucl Med Mol Imaging 32:443–447
Delforge J, Syrota A, Lancon JP et al (1991) Cardiac beta-adrenergic receptor density measured in vivo using PET, CGP 12177, and a new graphical method. J Nucl Med 32:739–748
Delforge J, Mesangeau D, Dolle F et al (2002) In vivo quantification and parametric images of the cardiac beta-adrenergic receptor density. J Nucl Med 43:215–226
Doze P, van Waarde A, Elsinga PH et al (1998) Validation of S-1′-[18F]fluorocarazolol for in vivo imaging and quantification of cerebral beta-adrenoceptors. Eur J Pharmacol 353:215–226
Doze P, Elsinga PH, van Waarde A et al (2002) Quantification of beta-adrenoceptor density in the human heart with (S)-[11C]CGP 12388 and a tracer kinetic model. Eur J Nucl Med Mol Imaging 29:295–304
Dzimiri N (1999) Regulation of beta-adrenoceptor signaling in cardiac function and disease. Pharmacol Rev 51:465–501
Elnatan J, Molenaar P, Rosenfeldt FL et al (1994) Autoradiographic localization and quantitation of beta 1- and beta 2-adrenoceptors in the human atrioventricular conducting system: a comparison of patients with idiopathic dilated cardiomyopathy and ischemic heart disease. J Mol Cell Cardiol 26:313–323
Elsinga PH, van Waarde A, Visser GM et al (1994) Synthesis and preliminary evaluation of (R, S)-1-[2-((carbamoyl-4-hydroxy)phenoxy)-ethylamino]-3-[4-(1-[11C]-met hyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol ([11C]CGP 20712A) as a selective beta 1-adrenoceptor ligand for PET. Nucl Med Biol 21:211–217
Elsinga PH, van Waarde A, Jaeggi KA et al (1997) Synthesis and evaluation of (S)-4-(3-(2′-[11C]isopropylamino)-2-hydroxypropoxy) -2H-benzimidazol -2-one ((S)-[11C]CGP 12388) and (S)-4-(3-((1′-[18F]-fluoroisopropyl)amino)-2-hydroxypropoxy) -2H- benzimidazol-2-one ((S)-[18F]fluoro-CGP 12388) for visualization of beta-adrenoceptors with positron emission tomography. J Med Chem 40:3829–3835
Elsinga PH, van Waarde A, Visser TJ et al (1998) Visualization of beta-adrenoceptors using PET. Clin Positron Imaging 1:81–94
Elsinga PH, Doze P, van Waarde A et al (2001) Imaging of beta-adrenoceptors in the human thorax using (S)-[(11)C]CGP12388 and positron emission tomography. Eur J Pharmacol 433:173–176
Engelhardt S, Bohm M, Erdmann E et al (1996) Analysis of beta-adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: progressive reduction of beta 1-adrenergic receptor mRNA in heart failure. J Am Coll Cardiol 27:146–154
Farrukh HM, White M, Port JD et al (1993) Up-regulation of beta 2-adrenergic receptors in previously transplanted, denervated nonfailing human hearts. J Am Coll Cardiol 22:1902–1908
Gauthier C, Tavernier G, Charpentier F et al (1996) Functional beta3-adrenoceptor in the human heart. J Clin Invest 98:556–562
Gauthier C, Leblais V, Kobzik L et al (1998) The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest 102:1377–1384
Gauthier C, Langin D, Balligand JL (2000) Beta3-adrenoceptors in the cardiovascular system. Trends Pharmacol Sci 21:426–431
Hall JA, Petch MC, Brown MJ (1991) In vivo demonstration of cardiac beta 2-adrenoreceptor sensitization by beta 1-antagonist treatment. Circ Res 69:959–964
Hall JA, Ferro A, Dickerson JE et al (1993) Beta adrenoreceptor subtype cross regulation in the human heart. Br Heart J 69:332–337
Hayes MJ, Qing F, Rhodes CG et al (1996) In vivo quantification of human pulmonary beta-adrenoceptors: effect of beta-agonist therapy. Am J Respir Crit Care Med 154:1277–1283
Heidenreich PA, Trogdon JG, Khavjou OA et al (2011) Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 123:933–944
Heyliger CE, Pierce GN, Singal PK et al (1982) Cardiac alpha- and beta-adrenergic receptor alterations in diabetic cardiomyopathy. Basic Res Cardiol 77:610–618
Ihl-Vahl R, Eschenhagen T, Kubler W et al (1996) Differential regulation of mRNA specific for beta 1- and beta 2-adrenergic receptors in human failing hearts. Evaluation of the absolute cardiac mRNA levels by two independent methods. J Mol Cell Cardiol 28:1–10
Iwata M, Yoshikawa T, Baba A et al (2001a) Autoantibodies against the second extracellular loop of beta1-adrenergic receptors predict ventricular tachycardia and sudden death in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 37:418–424
Iwata M, Yoshikawa T, Baba A et al (2001b) Autoimmunity against the second extracellular loop of beta(1)-adrenergic receptors induces beta-adrenergic receptor desensitization and myocardial hypertrophy in vivo. Circ Res 88:578–586
Jhund PS, Macintyre K, Simpson CR et al (2009) Long-term trends in first hospitalization for heart failure and subsequent survival between 1986 and 2003: a population study of 5.1 million people. Circulation 119:515–523
John AS, Mongillo M, Depre C et al (2007) Pre- and post-synaptic sympathetic function in human hibernating myocardium. Eur J Nucl Med Mol Imaging 34:1973–1980
Kaumann AJ, Molenaar P (1997) Modulation of human cardiac function through 4 beta-adrenoceptor populations. Naunyn Schmiedebergs Arch Pharmacol 355:667–681
Kaumann AJ, Hall JA, Murray KJ et al (1989) A comparison of the effects of adrenaline and noradrenaline on human heart: the role of beta 1- and beta 2-adrenoceptors in the stimulation of adenylate cyclase and contractile force. Eur Heart J 10(Suppl B):29–37
Kawai C, Yui Y, Hoshino T et al (1983) Myocardial catecholamines in hypertrophic and dilated (congestive) cardiomyopathy: a biopsy study. J Am Coll Cardiol 2:834–840
Kenk M, Thackeray JT, Thorn SL et al (2010) Alterations of pre- and postsynaptic noradrenergic signaling in a rat model of adriamycin-induced cardiotoxicity. J Nucl Cardiol 17:254–263
Law MP, Wagner S, Kopka K et al (2010) Preclinical evaluation of an 18F-labelled beta1-adrenoceptor selective radioligand based on ICI 89,406. Nucl Med Biol 37:517–526
Lefkowitz RJ, Mukherjee C, Coverstone M et al (1974) Stereospecific (3H)(minus)-alprenolol binding sites, beta-adrenergic receptors and adenylate cyclase. Biochem Biophys Res Commun 60:703–709
Lefroy DC, De Silva R, Choudhury L et al (1993) Diffuse reduction of myocardial beta-adrenoceptors in hypertrophic cardiomyopathy: a study with positron emission tomography. J Am Coll Cardiol 22:1653–1660
Link JM, Stratton JR, Levy W et al (2003) PET measures of pre- and post-synaptic cardiac beta adrenergic function. Nucl Med Biol 30:795–803
Lloyd-Jones DM, Larson MG, Leip EP et al (2002) Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation 106:3068–3072
Magnusson Y, Wallukat G, Waagstein F et al (1994) Autoimmunity in idiopathic dilated cardiomyopathy. Characterization of antibodies against the beta 1-adrenoceptor with positive chronotropic effect. Circulation 89:2760–2767
Maisel AS, Motulsky HJ, Insel PA (1985) Externalization of beta-adrenergic receptors promoted by myocardial ischemia. Science 230:183–186
Majmudar MD, Nahrendorf M (2012) Cardiovascular molecular imaging: the road ahead. J Nucl Med 53:673–676
Merlet P, Delforge J, Syrota A et al (1993) Positron emission tomography with 11C CGP-12177 to assess beta-adrenergic receptor concentration in idiopathic dilated cardiomyopathy. Circulation 87:1169–1178
Metra M, Giubbini R, Nodari S et al (2000) Differential effects of beta-blockers in patients with heart failure: a prospective, randomized, double-blind comparison of the long-term effects of metoprolol versus carvedilol. Circulation 102:546–551
Moniotte S, Kobzik L, Feron O et al (2001) Upregulation of beta(3)-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation 103:1649–1655
Motomura S, Deighton NM, Zerkowski HR et al (1990) Chronic beta 1-adrenoceptor antagonist treatment sensitizes beta 2-adrenoceptors, but desensitizes M2-muscarinic receptors in the human right atrium. Br J Pharmacol 101:363–369
Nantel F, Bonin H, Emorine LJ et al (1993) The human beta 3-adrenergic receptor is resistant to short term agonist-promoted desensitization. Mol Pharmacol 43:548–555
Naya M, Tsukamoto T, Morita K et al (2009) Myocardial beta-adrenergic receptor density assessed by 11C-CGP12177 PET predicts improvement of cardiac function after carvedilol treatment in patients with idiopathic dilated cardiomyopathy. J Nucl Med 50:220–225
Ohte N, Narita H, Iida A et al (2012) Cardiac beta-adrenergic receptor density and myocardial systolic function in the remote noninfarcted region after prior myocardial infarction with left ventricular remodeling. Eur J Nucl Med Mol Imaging 39:1246–1253
Pitschner HF, Droege A, Mitze M et al (1993) Down-regulated beta-adrenoceptors in severely failing human ventricles: uniform regional distribution, but no increased internalization. Basic Res Cardiol 88:179–191
Pott C, Brixius K, Bloch W et al (2006) Beta3-adrenergic stimulation in the human heart: signal transduction, functional implications and therapeutic perspectives. Pharmazie 61:255–260
Qing F, Rahman SU, Hayes MJ et al (1997a) Effect of long-term beta2-agonist dosing on human cardiac beta-adrenoceptor expression in vivo: comparison with changes in lung and mononuclear leukocyte beta-receptors. J Nucl Cardiol 4:532–538
Qing F, Rahman SU, Rhodes CG et al (1997b) Pulmonary and cardiac beta-adrenoceptor density in vivo in asthmatic subjects. Am J Respir Crit Care Med 155:1130–1134
Rathi S, Deedwania PC (2012) The epidemiology and pathophysiology of heart failure. Med Clin North Am 96:881–890
Rhee HM, Tyler L (1985) Myocardial ischemic injury and beta-adrenergic receptors in perfused working rabbit hearts. Adv Exp Med Biol 191:281–288
Rosen SD, Boyd H, Rhodes CG et al (1996) Myocardial beta-adrenoceptor density and plasma catecholamines in syndrome X. Am J Cardiol 78:37–42
Ruffolo RR Jr, Kopia GA (1986) Importance of receptor regulation in the pathophysiology and therapy of congestive heart failure. Am J Med 80:67–72
Schafers M, Lerch H, Wichter T et al (1998) Cardiac sympathetic innervation in patients with idiopathic right ventricular outflow tract tachycardia. J Am Coll Cardiol 32:181–186
Staehelin M, Simons P, Jaeggi K et al (1983) CGP-12177. A hydrophilic beta-adrenergic receptor radioligand reveals high affinity binding of agonists to intact cells. J Biol Chem 258:3496–3502
Steinfath M, Lavicky J, Schmitz W et al (1992a) Regional distribution of beta 1- and beta 2-adrenoceptors in the failing and nonfailing human heart. Eur J Clin Pharmacol 42:607–611
Steinfath M, von der Leyen H, Hecht A et al (1992b) Decrease in beta 1- and increase in beta 2-adrenoceptors in long-term follow-up after orthotopic cardiac transplantation. J Mol Cell Cardiol 24:1189–1198
Thackeray JT, Parsa-Nezhad M, Kenk M et al (2011) Reduced CGP12177 binding to cardiac beta-adrenoceptors in hyperglycemic high-fat-diet-fed, streptozotocin-induced diabetic rats. Nucl Med Biol 38:1059–1066
Tseng H, Link JM, Stratton JR et al (2001) Cardiac receptor physiology and its application to clinical imaging: present and future. J Nucl Cardiol 8:390–409
Tsukamoto T, Morita K, Naya M et al (2007) Decreased myocardial beta-adrenergic receptor density in relation to increased sympathetic tone in patients with nonischemic cardiomyopathy. J Nucl Med 48:1777–1782
Ungerer M, Hartmann F, Karoglan M et al (1998) Regional in vivo and in vitro characterization of autonomic innervation in cardiomyopathic human heart. Circulation 97:174–180
van Waarde A, Elsinga PH, Brodde OE et al (1995) Myocardial and pulmonary uptake of S-1′-[18F]fluorocarazolol in intact rats reflects radioligand binding to beta-adrenoceptors. Eur J Pharmacol 272:159–168
Visser TJ, van Waarde A, van der Mark TW et al (1997) Characterization of pulmonary and myocardial beta-adrenoceptors with S-1′-[fluorine-18]fluorocarazolol. J Nucl Med 38:169–174
Watson-Wright WM, Armour JA, Johnstone DE et al (1989) Myocardial slice: a physiological approach to beta-adrenergic ([3H] CGP-12177) receptor binding in hamster and guinea pig heart. J Pharmacol Methods 22:37–47
Wichter T, Schafers M, Rhodes CG et al (2000) Abnormalities of cardiac sympathetic innervation in arrhythmogenic right ventricular cardiomyopathy: quantitative assessment of presynaptic norepinephrine reuptake and postsynaptic beta-adrenergic receptor density with positron emission tomography. Circulation 101:1552–1558
Williams RS, Schaible TF, Scheuer J et al (1983) Effects of experimental diabetes on adrenergic and cholinergic receptors of rat myocardium. Diabetes 32:881–886
Zhao Q, Wu TG, Jiang ZF et al (2007) Effect of beta-blockers on beta3-adrenoceptor expression in chronic heart failure. Cardiovasc Drugs Ther 21:85–90
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Slart, R.H.J.A., van der Meer, P., Tio, R.A., van Veldhuisen, D.J., Elsinga, P.H. (2015). PET Imaging of Myocardial β-Adrenoceptors. In: Slart, R., Tio, R., Elsinga, P., Schwaiger, M. (eds) Autonomic Innervation of the Heart. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-45074-1_11
Download citation
DOI: https://doi.org/10.1007/978-3-662-45074-1_11
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-45073-4
Online ISBN: 978-3-662-45074-1
eBook Packages: MedicineMedicine (R0)