
Abstract
The clearance of viral infections is reliant on the coordination and balance of inflammatory factors necessary for viral destruction and immunoregulatory mechanisms necessary to prevent host pathology. In the case of persistent viral infections, immunoregulatory pathways prevent the immune response from clearing the virus, resulting in a long-term equilibrium between host and pathogen. Consequently, negative immune regulators are being considered as a therapeutic target to treat persistent and chronic viral infections. In this review, we will highlight the current understanding of the important negative immune regulator interleukin-10 (IL-10) in persistent viral infection. Though its main role for the host is to limit immune-mediated pathology, IL-10 is a multifunctional cytokine that differentially regulates a number of different hematopoietic cell types. IL-10 has been shown to play a role in a number of infectious diseases and many viral pathogens specifically exploit the IL-10 pathway to help evade host immunity. Recent advances have demonstrated that manipulation of IL-10 signaling during persistent viral infection can alter T cell responses in vivo and that this manipulation can lead to the clearance of persistent viral infection. Furthermore, there have been crucial advances in the understanding of factors that induce IL-10. We summarize lessons learned about IL-10 in model organisms and human persistent infections and conclude with the potential use of IL-10 to treat persistent viral infections.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Human Immunodeficiency Virus
- Human Immunodeficiency Virus Infection
- West Nile Virus
- High Viral Load
- Human Immunodeficiency Virus Patient
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Persistent viral infections are infections in which the host immune response is unable to clear the primary infection, resulting in a long-term equilibrium between the host and the virus. The equilibrium is the result of a number of factors that include host immune regulation, active suppression, immune evasion, and the level of viral replication. These factors are not static and can change over time depending on the actions of host immunity and the viral pathogen. As is apparent with HIV, the equilibrium can eventually be tipped in favor of the virus. Due to the disease severity of persistent infections, such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV), much research has been directed at elucidating the events that lead to persistent infection and how factors might be manipulated to tip the balance in favor of the host rather than the virus.
There are a variety of mechanisms to achieve persistence. Some viruses such as the herpes simplex viruses utilize latency in which the virus lies dormant within host cells to escape immunity resulting in low or no viral antigen except during periods of reactivation in which the virus reemerges. These reactivation periods are generally quickly controlled by the immune response. Other viral infections such as HIV, Hepatitis B virus (HBV), hepatitis C virus (HCV), and lymphocytic choriomeningitis virus (LCMV) rely on interfering with the function of the immune cells necessary for orchestrating anti-viral responses and clearing viral infection. These chronic infections are characterized by high levels of viral replication and high expression of negative immune regulators resulting in immune suppression of CD4 and CD8 T cell responses. Disruption in the generation of T cells results in delayed and/or failed clearance of virus and abrogation of T cell function contributes to the inability to control multiple persistent infections (Wherry 2011).
Murine infection with the enveloped RNA virus LCMV is a widely-used model to investigate T cell suppression during persistent/chronic viral infection. Developed in the 1980s, there are two well-characterized strains of LCMV used to compare and contrast immunologic factors unique to either acute or persistent viral infection, Armstrong 53b (ARM) and Clone 13 (Cl-13) (reviewed in (Dutko and Oldstone 1983; Oldstone and Campbell 2011)). These two strains differ by only three amino acids, but can result in very different disease outcomes. ARM induces a robust T cell response that clears the infection by 10 days post infection (Ahmed et al. 1984). Comparatively, Cl-13 replicates to much higher titers, inducing multiple host-based suppressive pathways, thereby generating a systemic persistent viremic infection that is not cleared from most tissues until 2–3 months later.
1.1 T Cell Immunity During Viral Infection
At the onset of a viral infection, the presence of virus is detected via innate immune receptors such as toll-like receptors which trigger an inflammatory response leading to the activation of the adaptive immune response. Antigen presenting cells such as dendritic cells (DCs) pick up the viral antigen and travel back to secondary lymphoid tissue (lymph nodes, spleen, peyer’s patches, etc.) where they present captured antigen on MHC I and MHCII to activate virus-specific CD8 and CD4 T cells. Clearance or control of viral infection is facilitated by effective anti-viral CD4+ and CD8+ T cell responses (Berger et al. 2000; Matloubian et al. 1994; Tishon et al. 1993, 1995). Naïve CD8 T cells mature into cytotoxic T cells [CTLs] that express inflammatory and antiviral cytokines and kill infected cells. Naïve CD4 T cells differentiate into one of several types of helper T cells that have a variety of functions that include expression of inflammatory cytokines, activation/expansion of CTLs, and regulating B cell class switching. The magnitude of the T cell response is controlled by the balance of factors that increase the activation and activity of immune cells (i.e., positive immune regulators) and factors that inhibit or decrease activity (i.e., negative immune regulators). In viral infections that become persistent, T cell responses are fairly normal at the initial stages of infection. In the acute phase of Cl-13 infection at 5 dpi, the numbers of LCMV-specific T cells and their cytokine production resemble those of ARM. Anti-viral effector CD4 and CD8 T cells produce IFNγ, TNFα, and IL-2 in response to antigen stimulation and CD8 T cells exhibit the ability to lyse infected cells. However, soon after, negative regulation triggers a hyporesponsive state in T cells termed “exhaustion” which was first described in the LCMV system (Zajac et al. 1998). The exhaustive state is characterized by an hierarchical diminishment of functional capacity that includes loss of proliferative ability and progresses to a decreased ability to produce key antiviral and immune stimulatory cytokines, and decreased cytolytic activity (Brooks et al. 2005; Wherry et al. 2003; Zajac et al. 1998). It is a state separate from anergy or tolerance with a distinct pattern of cellular and transcriptional expression. By 9 dpi in persistent Cl-13 infected mice, there are significantly fewer LCMV-specific T cells, illustrating physical deletion. Those anti-viral T cells that remain express fewer cytokines and have lost their cytolytic abilities, exhibiting severe hypofunctionality (Wherry 2011). The degeneration of anti-viral T cell function is compounded by the decreased ability of the immune system to mount de novo immune responses, thereby hampering the generation of new anti-viral T cells as well as disabling the host’s ability to fight secondary infections in most cases (Ahmed et al. 1984; Oldstone et al. 1988).
The development of exhaustion directly contributes to impaired control of viral infection, consequently, motivating extensive examination of T cell exhaustion. Transcriptional profiling comparing functionally active CTLs versus “exhausted” CTLs has uncovered differences in expression of a variety of genes for inhibitory receptors, transcription factors, metabolic pathways, chemotaxins, and migration factors (Wherry et al. 2007). Although many factors were found to be associated with T cell exhaustion, only two factors have been shown to have therapeutic impact when either genetically deleted or neutralized alone in vivo: programmed death-1 (PD-1) and interleukin-10 (IL-10). PD-1 is a member of the CD28 receptor family expressed on activated T cells and negatively regulates these cells by binding its cognate ligands PD-L1 and PD-L2 found on the surface of other cell types including DCs and non-hematopoietic cells (Sharpe et al. 2007). High levels of PD-1 are found on the surface of exhausted CD4+ and CD8+ cells during HIV (Day et al. 2006; Trautmann et al. 2006), HBV (Peng et al. 2008), HCV (Urbani et al. 2006), and persistent LCMV infection (Barber et al. 2006). Ex vivo blockade results in resurrection of T cell function (Nakamoto et al. 2009; Trautmann et al. 2006; Tzeng et al. 2012; Urbani et al. 2006) and in vivo blockade in persistent LCMV leads to early clearance (Barber et al. 2006).
IL-10 is also a potent regulator of T cell exhaustion which we will focus on for the remainder of this review. IL-10 was first described in 1989 as a factor secreted by Th2 cells to inhibit cytokine secretion by Th1 cells (Fiorentino et al. 1989) highlighting IL-10’s main role as an inflammatory cytokine. Since this first description, study of this cytokine has illuminated a variety of functions (Moore et al. 2001) revealing pleiotropic effects on different cell populations and with diverse suppressive roles during persistent viral infection (Ouyang et al. 2011; Wilson and Brooks 2011).
2 Establishing IL-10 as a Cause of Viral Persistence
2.1 IL-10 in LCMV
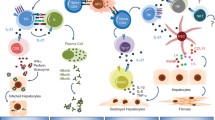
The first description of IL-10 as a major contributing factor to viral persistence came in 2005 from studies of LCMV (Brooks et al. 2006; Ejrnaes et al. 2006). The work stemmed from observations of the differential patterns of IL-10 expression during acute versus persistent infection. Serum IL-10 levels peak at equivalent levels in both ARM and CL-13 infected mice (Brooks et al. 2008b) and then quickly decrease by 2 and 3 days post-infection (Fig. 1). IL-10 levels in ARM continue to decrease over the next 12 days, while in Cl-13 infection these levels reverse to increase over the same period. The kinetics of IL-10 increase correlate with viremia (Brooks et al. 2008b; Wilson et al. 2012) and is concomitant with T cell exhaustion. Accordingly, blockade of this increase in IL-10 prevented T cell exhaustion, maintaining robust T cell responses, as measured by the number of anti-viral CD4 and CD8 T cells and their ability to produce IFN-γ, IL-2, and TNF-α. This augmented response resulted in early clearance of CL-13 infection (Brooks et al. 2006), establishing IL-10 as a major contributor to the decrease in virus-specific T cells and their function. Even after T cell exhaustion is established, blockade of IL-10 is able to rescue T cell activity and instigate early viral clearance Deletion of the il10 gene has a similar effect, with il10-/- mice displaying greater numbers of anti-viral T cells as well as higher cytokine production compared to their wild-type counterparts (Brooks et al. 2006). These data established that IL-10 is necessary to both instill and maintain the immune suppressive state during persistent viral infection.

IL-10 and T cell responses during acute versus persistent infection. IL-10 is initially by infection with both acute and persistent strains of LCMV. However, levels of IL-10 continue to increase in persistent viral infection, concomitantly with viral load. IL-10 supresses T cell responses and is a major contributor to T cell exhaustion in which T cells lose the capacity to secrete multiple cytokines and CTLs lose the capacity to lyse infected cells
Establishment of the role of IL-10 in LCMV persistence brought about renewed interest in the interactions of IL-10 during HIV, HCV, and HBV infections. Because it is difficult to perform tightly controlled experiments in humans, it has only been possible, thus far, to establish correlations between IL-10 and human viral infections. However, IL-10 is elicited during these infections as IL-10 correlates positively with viral load in HIV (Brockman et al. 2009; Orsilles et al. 2006), HBV (Peppa et al. 2010), and HCV (Akcam et al. 2012; Claassen et al. 2012; Reiser et al. 1997; Sofian et al. 2012).
2.2 IL-10 in HIV
The role of IL-10 during HIV infection is complex and multi-faceted. Although it is difficult to quantify the contribution of IL-10 to T cell exhaustion during HIV infection, ex vivo studies suggest that IL-10 is a major contributing factor. IL-10 expression positively correlates with HIV viremia (Brockman et al. 2009; Orsilles et al. 2006) and ex vivo blockade of IL-10 enhances activity of both CD4 and CD8 T cells isolated from HIV patients as measured by proliferation and expression of IFN-γ, TNF-α, and IL-2 (Bento et al. 2009; Brockman et al. 2009; Landay et al. 1996; Porichis et al. 2013; Yang et al. 2009). Notably, IL-10 blockade was effective for HIV patients with uncontrolled viremia, but not for those with highly controlled viremia (elite controllers or those on antiretroviral treatment) (Brockman et al. 2009), suggesting that HIV progression may be associated with IL-10-mediated immune suppression. However, this may be only during the pre-AIDS phase of infection, as IL-10 blockade was less successful at enhancing T cell responses in those with AIDS (<200 CD4 T cells/ul) (Landay et al. 1996).
In addition to modulating T cell function, IL-10 may negatively regulate the size of important immune cell populations. CD4 T cells from HIV-infected individuals are highly sensitive to apoptosis (Clerici et al. 1994; Estaquier et al. 1995) and this sensitivity can be decreased by blocking IL-10, suggesting that IL-10 contributes to the depletion of this cell population. Not only does IL-10 control cell death in the T cell compartment, but also in the DC compartment. DCs produce IL-10 during HIV infection and this production targets them for lysis by NK cells (Alter et al. 2010). Hence, not only does IL-10 debilitate the anti-viral T cell response, but also enables the virus to directly target cells necessary to generate new anti-viral T cells.
The role of IL-10 in HIV extends beyond the suppression of immune cells. IL-10 has been observed to both induce and block viral replication depending on the cell type examined (Finnegan et al. 1996; Leghmari et al. 2008; Takeshita et al. 1995; Weissman et al. 1995). In infected macrophages/monocyte cell lines, exposure to IL-10 in the presence of TNFα induces viral replication (Finnegan et al. 1996; Weissman et al. 1995). Conversely, IL-10 was shown to block viral replication in ex vivo stimulated CD4 T cells (Bento et al. 2009). The variability of these observations may stem in part from the differential modulation of cellular receptors such as HIV co-receptors CCR5 and CXCR4, which are affected by IL-10 signaling (Kwon and Kaufmann 2010). Overall, it is clear that the role of IL-0 in HIV is not simple and is likely dependent on the cell-type.
2.3 IL-10 in HBV and HCV
The involvement of IL-10 in chronic HBV and HCV infections is controversial and has been difficult to study due to the shortage of small animal models. In chronic HBV-infected individuals, high serum IL-10 levels correlate with higher viral loads (Peppa et al. 2010). In the liver, the main site of HBV infection, IL-10 likely suppresses the activity of natural killer (NK) cells, which have a prominent population in this tissue (Maini and Peppa 2013). The suppression of NK activity, including their ability to produce IFN-γ, which is important to T cell activation (Maini and Peppa 2013), occurs in a manner that is inversely and temporally correlated with IL-10 levels (Dunn et al. 2009) and blockade of IL-10/IL-10R ex vivo can restore IFN-γ secretion by NK cells (Peppa et al. 2010). Thus, these data suggest that IL-10 regulates the behavior of NK cells in the liver.
Though whether IL-10 is a factor in suppression of anti-HCV responses is still under investigation, it is likely that there is some degree of contribution. IL-10 levels of HCV-infected patients are higher than those of uninfected individuals (Claassen et al. 2012; Reiser et al. 1997; Sofian et al. 2012). Ex vivo blockade of IL-10 is followed by improved function of exhausted HCV-specific T cells (Ludewig et al. 2012; Rigopoulou et al. 2005). Interestingly, long-term administration of IL-10 to individuals with HCV-related liver disease led to an increase in viral burden. The increase in viral titers was accompanied by the arrest of liver damage as demonstrated by a reduction in hepatic inflammation and fibrosis (Nelson et al. 2003), demonstrating that anti-HCV immunity is susceptible to control by HCV in the chronic phase. Further support for the role of IL-10 in chronic HCV infection is derived from studies examining genetic polymorphisms that control the level of host IL-10 secretion. Although the data is highly variable, meta-analyses have demonstrated a correlation between susceptibility to chronic HCV and specific single nucleotide polymorphisms in the il10 promoter, especially the IL-10–1082A/G polymorphism (Sun et al. 2013; Zhang et al. 2010). More research is necessary to establish the precise role of IL-10 in HBV and HCV infection and how this pathway can be manipulated to benefit the host.
3 Cellular Sources of IL-10 and Its Target(s)
A number of different lymphocyte populations are capable of producing IL-10 (Moore et al. 2001), however, the source(s) of IL-10 are likely dependent on the specific virus and the site, route, and cellular tropism of the particular infection as reflected by the differing profiles of IL-10 expression during different infection. For example, among acute viral infections, effector T cells in the lung secrete high levels of the cytokine during influenza infection (Sun et al. 2009), while splenic CD4 T cells are a major source of IL-10 during infection with west nile virus (Bai et al. 2009). During Cl-13 infection, one of the main producers of IL-10 during Cl-13 infection are the CD8α- subset of DCs (Ng and Oldstone 2012; Wilson et al. 2012). Comparatively, only minimal expression of IL-10 by DCs has been observed in ARM-infected mice (Brooks et al. 2006; Wilson et al. 2012). DC-specific deletion of il10 results in a significant but not complete decrease in serum IL-10 levels (Ng and Oldstone 2012), suggesting that DCs are a major source of IL-10 but that other cell-types contribute as well (Ng and Oldstone 2012; Richter et al. 2013). A small percentage of B cells as well as CD4 T cells demonstrate IL-10 expression, while expression is negligable in CD8 T cells. Differing results have been observed with macrophages. However, it is interesting to note that antigen-presenting cells that express IL-10 also express high levels of other suppressive factors, including PD-L1 and indoleamine 2,3 dioxygenase (IDO) (Ng and Oldstone 2012; Wilson et al. 2012). Thus, when T cells interact with these cells, there is the delivery of multiple suppressive signals.
In relation to human infections, IL-10 is up-regulated by multiple cell types. During HIV infection, IL-10-producing cells include CD4 T cells, DCs, B cells, and natural killer (NK) cells (Brockman et al. 2009; Leghmari et al. 2008). Although this expression is likely due to host-programmed upregulation, HIV also may actively induce expression of IL-10. In vitro studies have demonstrated that monocytic cell lines and primary patient cells incubated with HIV proteins Tat, gp120, and nef upregulate expression of IL-10 (Blazevic et al. 1996; Borghi et al. 1995; Brigino et al. 1997; Contreras et al. 2004; Gee et al. 2007; Gupta et al. 2008; Masood et al. 1994; Schols and De Clercq 1996). In HBV infection, T cells and monocytes isolated from the blood of chronic HBV patients secrete IL-10 upon stimulation with HBV antigen (Hyodo et al. 2004) and increased levels of IL-10 are also elicited from the peripheral leukocytes of HCV-infected individuals when exposed to the HCV core antigen (Barrett et al. 2008). While research has identified some sources of IL-10, further research is necessary to determine whether/how the identity of the IL-10 producing cell has impact on determining the specific downstream effects of IL-10.
Much has been elucidated regarding the effects of IL-10 in T cell function, yet the exact mechanism by which IL-10 elicits T cell exhaustion is not known as IL-10 can signal on multiple cell-types. Many lymphocytes express the receptor for IL-10 (IL-10R), including CD4 and CD8 T cells (Moore et al. 2001). Hence, there are several possibilities for the targets of IL-10 signaling. The first is signaling directly on T cells. In CL-13 infection, since DCs express IL-10, T cells that come into contact should be directly exposed to the cytokine. Surprisingly, specific deletion of IL-10R on T cells does not alleviate T cell exhaustion or lead to accelerated clearance of the virus (Ng and Oldstone, unpublished observation), suggesting that IL-10 signaling on T cells is not the route by which IL-10 elicits exhaustion. Comparatively, during acute infection with ARM, IL-10 signals directly on anti-viral CD4 T cells to limit the size of the effector and memory population (Brooks et al. 2010). Ex vivo blockade of IL-10R improves T cell function in cells from HIV- and HCV- infected patients (Brockman et al. 2009; Ludewig et al. 2012; Rigopoulou et al. 2005), however, these experiments have all been performed on PBMC, and thus, do not provide evidence on the target cell.
A second possibility for the mechanism of IL-10-mediated exhaustion is that IL-10 may suppress T cell function indirectly via antigen-presenting cells such as DCs. IL-10 signaling is known to inhibit maturation and expression of MHC I, MHC II, and stimulatory molecules CD80 and CD86 on monocytes and DCs (Chan et al. 2006; Koppelman et al. 1997) which in turn would affect their ability to fully activate T cells. DCs of CL-13 infected mice display a reduction in these molecules as well as a reduced capacity to activate T cells (Sevilla et al. 2000, 2004). However, the concept of IL-10 suppression of stimulatory molecules on DCs is not supported by current data as deletion of IL-10R on DCs does not alter their expression of stimulatory molecules during Cl-13 infection (Ng and Oldstone 2012). Thirdly, it is possible that IL-10 signaling on multiple cell-types is responsible for the observed exhaustion and that only by blocking signaling on all of these cells can T cell function be rescued. Indeed, cell-specific deletion of IL-10 signaling does not affect T cell exhaustion (unpublished observation), while IL-10R knockout mice and mice treated with anti-IL-10R antibody do not undergo T cell exhaustion and clear Cl-13 infection early (Brooks et al. 2006). Whether multiple cell types or an as yet-to-be determined single cell-type are the target(s) of IL-10 remains to be determined.
4 Triggers of IL-10 and T Cell Exhaustion
The precise molecular events that induce IL-10 and cause T cell exhaustion are still being examined. Data, thus far, suggests at least two factors. The first factor is viral antigen. In multiple infections, IL-10 expression and magnitude of expression is correlated with viral load (Fig. 1) both longitudinally within an infected organism and across patient groups. Furthermore, prolonged exposure to high viral antigen load leads to T cell exhaustion (Fahey and Brooks 2010; Mueller and Ahmed 2009; Richter et al. 2012; Wherry 2011). LCMV studies examining CD8 T cell exhaustion have observed that increasing the number of antigen-presenting cells results in decreased T cell function, suggesting that the amount of antigen that the T cell encounters regulates functionality. The initiation of T cell exhaustion is not simply linked to the level of viremia, but to the amount and length of antigen presentation that is important in determining exhaustion. In combination with the amount of encountered antigen, the antigen-presenting cell may also play a role. Recognition of antigen on non-hematopoietic cells promotes T cell exhaustion. This is consistent with the observation that non-hematopoietic stromal cells in lymphoid tissue upregulate PD-L1 during Cl-13 infection, which prevents immunopathology (Mueller et al. 2007, 2010). Interestingly, these cells are implicated in regulation of peripheral tolerance and T cell activation.
The necessity of a high viral antigen load to trigger of T cell exhaustion emphasizes several points about virus-host interactions: (1) For a viral infection to become persistent, the virus must evade a functional T cell response long enough to replicate to levels that will trigger T cell exhaustion; (2) To prevent T cell exhaustion, the host must control viral replication to sufficiently low levels. The result of the virus-host interaction is likely determined very early on in infection and involves a family of cytokines until recently only perceived as anti-viral. Based on very recent work, the mechanism for how viral antigen triggers T cell exhaustion may involve the induction of type I interferons (IFN), such as IFNα and IFNβ. Two studies have elucidated a role for type I IFN in instituting viral persistence during LCMV infection (Teijaro et al. 2013; Wilson et al. 2013). Infection with both ARM and CL-13 initiates the production of IFNα in the first 24 h (Teijaro et al. 2013; Wilson et al. 2013). However, CL-13 infection produces significantly more IFNα and IFNβ, whereas ARM produces less IFNα and no measureable IFNβ, suggesting an association between persistent viruses and IFN-I signaling. Blockade of type I IFN significantly impacts a number of factors, including expression of negative immune regulators. At 1, 5, and 9 days post-infection, there is lower expression of IL-10 (Fig. 2) and PD-1 ligand despite significantly higher viral loads (Teijaro et al. 2013; Wilson et al. 2013). At 9 days post-infection when T cells are typically exhausted, type I IFN blockade results in significantly greater numbers of cytokine-producing anti-viral CD4 T cells despite significantly higher viral load and greater numbers of antigen presenting cells; CD8 T cells not only are initiated at a slower rate, but also exhibit a similar trend at later timepoints (Ng, Teijaro and Oldstone, unpublished observation). Surprisingly, these cells do not exhibit the characteristic signs of exhaustion despite prolonged exposure to high viral antigen loads. Ultimately, the contribution of these factors culminates in early clearance by around 40 versus 60 days post-infection. In essence, type I IFN is a master regulator, controlling expression of negative immune regulators and initiation of T cell exhaustion. Type I IFN blockade decouples the link between antigen exposure and T cell exhaustion suggesting that it is the ability of high viral load to elicit type I IFN that ultimately results in T cell exhaustion.

Type I interferon (IFN) is a determinant of IL-10 expression and viral persistence. High expression levels of type I IFN predict high expression of IL-10. Early blockade of type I IFN signaling by an anti-IFN α/β receptor antibody results in the induction of significantly lower levels of serum IL-10 in the early phase of infection (day 1 and 5) and once persistence is established (day 9) compared to isotype control mice. This suggests that IL-10 expression is controlled by type I IFN. Concomitantly, treated mice exhibit greater numbers of anti-viral CD4 T cells. Treated mice clear Cl-13 infection at an accelerated rate in a CD4 T cell-dependent manner
5 Il-10 as a Therapeutic
Because IL-10 is a dominant suppressive pathway in the regulation of T cell exhaustion, it presents an interesting potential therapeutic target for persistent viral infections that are characterized by dampened T cell responses. Although the IL-10 pathway is an attractive target, manipulation of IL-10 must be balanced carefully to enhance anti-viral responses and with the need to minimize host tissue damage. T cell exhaustion severely hinders viral clearance; however, this strategy is likely a host-derived mechanism to prevent life-threatening immunopathology. Inhibition of the IL-10 pathway during LCMV not only enhances viral clearance, but also decreases survival. Importantly, the pleiotropic effect of IL-10 on different cell-types must also be considered as the function of various immune cells may be up- or down-regulated simultaneously. However, in proof of principle this strategy is viable. As discussed earlier, neutralization of IL-10 after the onset of T cell exhaustion, rescues CD4 and CD8 T cell responses and viral control. Human in vivo studies have yet to be performed, however, in vitro blockade of IL-10 is followed by improved function of exhausted T cells in HIV (Landay et al. 1996; Yang et al. 2009), HBV, and HCV. This treatment could be further improved upon by utilizing therapeutic vaccination. The combination of IL-10 blockade to relieve the immune suppressive environment with an LCMV-specific DNA vaccine to stimulate immunity enhances both anti-LCMV CD4 and CD8 T cell responses leading to significant viral control that exceeds IL-10 blockade alone (Brooks et al. 2008b). This strategy is not only of interest for amplifying anti-viral responses, but also applicable for enhancing vaccine responses against other pathogens. HIV- and HCV-infected individuals have suboptimal immunological responses to vaccines such as influenza vaccination or HBV vaccination (De Sousa dos Santos et al. 2004; Malaspina et al. 2005; Moorman et al. 2011). Using blocking IL-10 signaling would likely improve the responses to vaccination resulting in better protection against secondary infection.
Another therapeutic consideration is the targeting of several inhibitory pathways to restore T cell function, since exhausted T cells express multiple inhibitory receptors. As discussed earlier, the PD-1 pathway is another dominant suppressive pathway that limits T cell function during persistent infections. PD-1 functions independently of the IL-10 pathway as evidenced by unchanged expression when the other pathway is manipulated. The simultaneous blockade of both pathways creates a synergistic effect that leads to faster clearance of persistent LCMV infection than the blockade of one pathway alone (Brooks et al. 2008a). Furthermore, blockade of either inhibitory receptor T-cell immunoglobulin domain and mucin domain 3 (Tim-3) or LAG-3, though ineffective alone (Blackburn et al. 2009; Jin et al. 2010; Leitner et al. 2013), have been shown to be effective at restoring T cell function when either are paired with PD-1 (Blackburn et al. 2009; Jin et al. 2010). Thus, it may be possible to utilize all four pathways to alleviate T cell exhaustion depending on the extent of hypofunctionality and potential immune pathology. Only further investigation of anti-IL-10 treatment will determine if it is a viable strategy to decrease T cell exhaustion in human chronic infections.
6 Conclusion
Collectively, the data presented in this review show that IL-10, though elicited early during most inflammatory events, is a prominent factor in persistent viral infections. In spite of the major advances in our understanding of IL-10 as a key suppressive pathway in persistent infection, there is still much left to be elucidated. First, it is important to understand how IL-10 is elicited from specific cellular subsets, because this likely determines the downstream effects of IL-10. Second, resolving the specific cellular mechanism by which IL-10 signals T cell exhaustion is crucial for attaining a complete understanding of this pathway and how it can be manipulated for therapeutic benefit. Lastly, the clinical contribution of IL-10 must be better quantitated to better define the potential impact of any manipulations of the IL-10 pathway, although this may prove difficult due to the relative lack of animal models in HIV, HBV, and HCV. Hopefully, these questions will be answered quickly to define the potential of this cytokine in the treatment of persistent viral infections.
References
Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB (1984) Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med 160:521–540
Akcam FZ, Tigli A, Kaya O, Ciris M, Vural H (2012) Cytokine levels and histopathology in chronic hepatitis B and chronic hepatitis C. J Interf Cytokine Res: Off J Int Soc Interf Cytokine Res 32:570–574
Alter G, Kavanagh D, Rihn S, Luteijn R, Brooks D, Oldstone M, van Lunzen J, Altfeld M (2010) IL-10 induces aberrant deletion of dendritic cells by natural killer cells in the context of HIV infection. J Clin Invest 120:1905–1913
Bai F, Town T, Qian F, Wang P, Kamanaka M, Connolly TM, Gate D, Montgomery RR, Flavell RA, Fikrig E (2009) IL-10 signaling blockade controls murine West Nile virus infection. PLoS Pathog 5:e1000610
Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439:682–687
Barrett L, Gallant M, Howley C, Bowmer MI, Hirsch G, Peltekian K, Grant M (2008) Enhanced IL-10 production in response to hepatitis C virus proteins by peripheral blood mononuclear cells from human immunodeficiency virus-monoinfected individuals. BMC Immunol 9:28
Bento CA, Hygino J, Andrade RM, Saramago CS, Silva RG, Silva AA, Linhares UC, Brindeiro R, Tanuri A, Rosenzwajg M et al (2009) IL-10-secreting T cells from HIV-infected pregnant women downregulate HIV-1 replication: effect enhanced by antiretroviral treatment. AIDS 23:9–18
Berger DP, Homann D, Oldstone MB (2000) Defining parameters for successful immunocytotherapy of persistent viral infection. Virology 266:257–263
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ (2009) Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol 10:29–37
Blazevic V, Heino M, Lagerstedt A, Ranki A, Krohn KJ (1996) Interleukin-10 gene expression induced by HIV-1 Tat and Rev in the cells of HIV-1 infected individuals. J Acquir Immune Defic Syndr Hum Retrovirol 13:208–214
Borghi P, Fantuzzi L, Varano B, Gessani S, Puddu P, Conti L, Capobianchi MR, Ameglio F, Belardelli F (1995) Induction of interleukin-10 by human immunodeficiency virus type 1 and its gp120 protein in human monocytes/macrophages. J Virol 69:1284–1287
Brigino E, Haraguchi S, Koutsonikolis A, Cianciolo GJ, Owens U, Good RA, Day NK (1997) Interleukin 10 is induced by recombinant HIV-1 Nef protein involving the calcium/calmodulin-dependent phosphodiesterase signal transduction pathway. Proc Natl Acad Sci U S A 94:3178–3182
Brockman MA, Kwon DS, Tighe DP, Pavlik DF, Rosato PC, Sela J, Porichis F, Le Gall S, Waring MT, Moss K et al (2009) IL-10 is up-regulated in multiple cell types during viremic HIV infection and reversibly inhibits virus-specific T cells. Blood 114:346–356
Brooks DG, Teyton L, Oldstone MB, McGavern DB (2005) Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol 79:10514–10527
Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB (2006) Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12:1301–1309
Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB (2008a) IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A 105:20428–20433
Brooks DG, Lee AM, Elsaesser H, McGavern DB, Oldstone MB (2008b) IL-10 blockade facilitates DNA vaccine-induced T cell responses and enhances clearance of persistent virus infection. J Exp Med 205:533–541
Brooks DG, Walsh KB, Elsaesser H, Oldstone MB (2010) IL-10 directly suppresses CD4 but not CD8 T cell effector and memory responses following acute viral infection. Proc Natl Acad Sci U S A 107:3018–3023
Chan A, Baird M, Mercer AA, Fleming SB (2006) Maturation and function of human dendritic cells are inhibited by orf virus-encoded interleukin-10. J Gen Virol 87:3177–3181
Claassen MA, de Knegt RJ, Turgut D, Groothuismink ZM, Janssen HL, Boonstra A (2012) Negative regulation of hepatitis C virus specific immunity is highly heterogeneous and modulated by pegylated interferon-alpha/ribavirin therapy. PLoS ONE 7:e49389
Clerici M, Sarin A, Coffman RL, Wynn TA, Blatt SP, Hendrix CW, Wolf SF, Shearer GM, Henkart PA (1994) Type 1/type 2 cytokine modulation of T-cell programmed cell death as a model for human immunodeficiency virus pathogenesis. Proc Natl Acad Sci U S A 91:11811–11815
Contreras X, Bennasser Y, Bahraoui E (2004) IL-10 production induced by HIV-1 Tat stimulation of human monocytes is dependent on the activation of PKC beta(II) and delta isozymes. Microbes Infect/Institut Pasteur 6:1182–1190
Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C et al (2006) PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354
De Sousa dos Santos S, Lopes MH, Simonsen V, Caiaffa Filho HH (2004) Haemophilus influenzae type b immunization in adults infected with the human immunodeficiency virus. AIDS Res Hum Retroviruses 20:493–496
Dunn C, Peppa D, Khanna P, Nebbia G, Jones M, Brendish N, Lascar RM, Brown D, Gilson RJ, Tedder RJ et al (2009) Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 137:1289–1300
Dutko FJ, Oldstone MB (1983) Genomic and biological variation among commonly used lymphocytic choriomeningitis virus strains. J Gen Virol 64(Pt 8):1689–1698
Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, von Herrath MG (2006) Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med 203:2461–2472
Estaquier J, Idziorek T, Zou W, Emilie D, Farber CM, Bourez JM, Ameisen JC (1995) T helper type 1/T helper type 2 cytokines and T cell death: preventive effect of interleukin 12 on activation-induced and CD95 (FAS/APO-1)-mediated apoptosis of CD4+ T cells from human immunodeficiency virus-infected persons. J Exp Med 182:1759–1767
Fahey LM, Brooks DG (2010) Opposing positive and negative regulation of T cell activity during viral persistence. Curr Opin Immunol 22:348–354
Finnegan A, Roebuck KA, Nakai BE, Gu DS, Rabbi MF, Song S, Landay AL (1996) IL-10 cooperates with TNF-alpha to activate HIV-1 from latently and acutely infected cells of monocyte/macrophage lineage. J Immunol 156:841–851
Fiorentino DF, Bond MW, Mosmann TR (1989) Two types of mouse T helper cell. IV. Th2 clones secrete a factor that inhibits cytokine production by Th1 clones. J Exp Med 170:2081–2095
Gee K, Angel JB, Mishra S, Blahoianu MA, Kumar A (2007) IL-10 regulation by HIV-Tat in primary human monocytic cells: involvement of calmodulin/calmodulin-dependent protein kinase-activated p38 MAPK and Sp-1 and CREB-1 transcription factors. J Immunol 178:798–807
Gupta S, Boppana R, Mishra GC, Saha B, Mitra D (2008) HIV-1 Tat suppresses gp120-specific T cell response in IL-10-dependent manner. J Immunol 180:79–88
Hyodo N, Nakamura I, Imawari M (2004) Hepatitis B core antigen stimulates interleukin-10 secretion by both T cells and monocytes from peripheral blood of patients with chronic hepatitis B virus infection. Clin Exp Immunol 135:462–466
Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, Freeman GJ, Kuchroo VK, Ahmed R (2010) Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 107:14733–14738
Koppelman B, Neefjes JJ, de Vries JE, de Waal Malefyt R (1997) Interleukin-10 down-regulates MHC class II alphabeta peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity 7:861–871
Kwon DS, Kaufmann DE (2010) Protective and detrimental roles of IL-10 in HIV pathogenesis. Eur Cytokine Netw 21:208–214
Landay AL, Clerici M, Hashemi F, Kessler H, Berzofsky JA, Shearer GM (1996) In vitro restoration of T cell immune function in human immunodeficiency virus-positive persons: effects of interleukin (IL)-12 and anti-IL-10. J Infect Dis 173:1085–1091
Leghmari K, Bennasser Y, Bahraoui E (2008) HIV-1 Tat protein induces IL-10 production in monocytes by classical and alternative NF-kappa B pathways. Eur J Cell Biol 87:947–962
Leitner J, Rieger A, Pickl WF, Zlabinger G, Grabmeier-Pfistershammer K, Steinberger P (2013) TIM-3 does not act as a receptor for Galectin-9. PLoS Pathog 9:e1003253
Ludewig B, Stein JV, Sharpe J, Cervantes-Barragan L, Thiel V, Bocharov G (2012) A global “imaging” view on systems approaches in immunology. Eur J Immunol 42:3116–3125
Maini MK, Peppa D (2013) NK cells: a double-edged sword in chronic hepatitis B virus infection. Front Immunol 4:57
Malaspina A, Moir S, Orsega SM, Vasquez J, Miller NJ, Donoghue ET, Kottilil S, Gezmu M, Follmann D, Vodeiko GM et al (2005) Compromised B cell responses to influenza vaccination in HIV-infected individuals. J Infect Dis 191:1442–1450
Masood R, Lunardi-Iskandar Y, Moudgil T, Zhang Y, Law RE, Huang CL, Puri RK, Levine AM, Gill PS (1994) IL-10 inhibits HIV-1 replication and is induced by tat. Biochem Biophy Res Commun 202:374–383
Matloubian M, Concepcion RJ, Ahmed R (1994) CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68:8056–8063
Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A (2001) Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19:683–765
Moorman JP, Zhang CL, Ni L, Ma CJ, Zhang Y, Wu XY, Thayer P, Islam TM, Borthwick T, Yao ZQ (2011) Impaired hepatitis B vaccine responses during chronic hepatitis C infection: involvement of the PD-1 pathway in regulating CD4(+) T cell responses. Vaccine 29:3169–3176
Mueller SN, Ahmed R (2009) High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 106:8623–8628
Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, Larsen CP, Ahmed R (2007) Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci U S A 104:15430–15435
Mueller SN, Vanguri VK, Ha SJ, West EE, Keir ME, Glickman JN, Sharpe AH, Ahmed R (2010) PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J Clin Invest 120:2508–2515
Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, Gostick E, Price DA, Freeman GJ, Wherry EJ et al (2009) Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog 5:e1000313
Nelson DR, Tu Z, Soldevila-Pico C, Abdelmalek M, Zhu H, Xu YL, Cabrera R, Liu C, Davis GL (2003) Long-term interleukin 10 therapy in chronic hepatitis C patients has a proviral and anti-inflammatory effect. Hepatology 38:859–868
Ng CT, Oldstone MB (2012) Infected CD8alpha-dendritic cells are the predominant source of IL-10 during establishment of persistent viral infection. Proc Natl Acad Sci U S A 109:14116–14121
Oldstone MB, Campbell KP (2011) Decoding arenavirus pathogenesis: essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology 411:170–179
Oldstone MB, Salvato M, Tishon A, Lewicki H (1988) Virus-lymphocyte interactions. III. Biologic parameters of a virus variant that fails to generate CTL and establishes persistent infection in immunocompetent hosts. Virology 164:507–516
Orsilles MA, Pieri E, Cooke P, Caula C (2006) IL-2 and IL-10 serum levels in HIV-1-infected patients with or without active antiretroviral therapy. APMIS : acta pathologica, microbiologica, et immunologica Scandinavica 114:55–60
Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG (2011) Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 29:71–109
Peng G, Li S, Wu W, Tan X, Chen Y, Chen Z (2008) PD-1 upregulation is associated with HBV-specific T cell dysfunction in chronic hepatitis B patients. Mol Immunol 45:963–970
Peppa D, Micco L, Javaid A, Kennedy PT, Schurich A, Dunn C, Pallant C, Ellis G, Khanna P, Dusheiko G et al (2010) Blockade of immunosuppressive cytokines restores NK cell antiviral function in chronic hepatitis B virus infection. PLoS Pathog 6:e1001227
Porichis F, Hart MG, Zupkosky J, Barblu L, Kwon DS, McMullen A, Brennan T, Ahmed R, Freeman GJ, Kavanagh DG et al (2013) Differential impact of PD-1 and/or IL-10 blockade on HIV-1-specific CD4 T cell and antigen-presenting cell functions. J Virol 88(5):2508–2518
Reiser M, Marousis CG, Nelson DR, Lauer G, Gonzalez-Peralta RP, Davis GL, Lau JY (1997) Serum interleukin 4 and interleukin 10 levels in patients with chronic hepatitis C virus infection. J Hepatol 26:471–478
Richter K, Brocker T, Oxenius A (2012) Antigen amount dictates CD8+ T-cell exhaustion during chronic viral infection irrespective of the type of antigen presenting cell. Eur J Immunol 42:2290–2304
Richter K, Perriard G, Behrendt R, Schwendener RA, Sexl V, Dunn R, Kamanaka M, Flavell RA, Roers A, Oxenius A (2013) Macrophage and T cell produced IL-10 promotes viral chronicity. PLoS Pathog 9:e1003735
Rigopoulou EI, Abbott WG, Haigh P, Naoumov NV (2005) Blocking of interleukin-10 receptor—a novel approach to stimulate T-helper cell type 1 responses to hepatitis C virus. Clin Immunol 117:57–64
Schols D, De Clercq E (1996) Human immunodeficiency virus type 1 gp120 induces anergy in human peripheral blood lymphocytes by inducing interleukin-10 production. J Virol 70:4953–4960
Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, de La Torre JC, Oldstone MB (2000) Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med 192:1249–1260
Sevilla N, McGavern DB, Teng C, Kunz S, Oldstone MB (2004) Viral targeting of hematopoietic progenitors and inhibition of DC maturation as a dual strategy for immune subversion. J Clin Invest 113:737–745
Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ (2007) The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 8:239–245
Sofian M, Aghakhani A, Farazi AA, Banifazl M, Eslamifar A, Rashidi N, Khadem Sadegh A, Ramezani A (2012) Serum profile of T helper 1 and T helper 2 cytokines in hepatitis C virus infected patients. Hepat Mon 12:e6156
Sun J, Madan R, Karp CL, Braciale TJ (2009) Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med 15:277–284
Sun, X.R., Wu, J., Shi, K.Q., and Tang, K.F. (2013). Relationship between IL-10 gene −1082A/G and −592C/A polymorphisms and the risk of hepatitis C infection: a meta-analysis. J Viral Hepat n/a-n/a
Takeshita S, Breen EC, Ivashchenko M, Nishanian PG, Kishimoto T, Vredevoe DL, Martinez-Maza O (1995) Induction of IL-6 and IL-10 production by recombinant HIV-1 envelope glycoprotein 41 (gp41) in the THP-1 human monocytic cell line. Cell Immunol 165:234–242
Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB (2013) Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211
Tishon A, Eddleston M, de la Torre JC, Oldstone MB (1993) Cytotoxic T lymphocytes cleanse viral gene products from individually infected neurons and lymphocytes in mice persistently infected with lymphocytic choriomeningitis virus. Virology 197:463–467
Tishon A, Lewicki H, Rall G, Von Herrath M, Oldstone MB (1995) An essential role for type 1 interferon-gamma in terminating persistent viral infection. Virology 212:244–250
Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS et al (2006) Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med 12:1198–1202
Tzeng HT, Tsai HF, Liao HJ, Lin YJ, Chen L, Chen PJ, Hsu PN (2012) PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS ONE 7:e39179
Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C (2006) PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol 80:11398–11403
Weissman D, Poli G, Fauci AS (1995) IL-10 synergizes with multiple cytokines in enhancing HIV production in cells of monocytic lineage. J Acquir Immune Defic Syndr Hum Retrovirol 9:442–449
Wherry EJ (2011) T cell exhaustion. Nat Immunol 12:492–499
Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R (2003) Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77:4911–4927
Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684
Wilson EB, Brooks DG (2011) The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol 350:39–65
Wilson EB, Kidani Y, Elsaesser H, Barnard J, Raff L, Karp CL, Bensinger S, Brooks DG (2012) Emergence of distinct multiarmed immunoregulatory antigen-presenting cells during persistent viral infection. Cell Host Microbe 11:481–491
Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG (2013) Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207
Yang H, Guimaraes-Walker A, Hibbs S, Dong T, Stacey A, Borrow P, Hanke T, Davenport MP, McMichael A, Dorrell L (2009) Interleukin-10 responses to therapeutic vaccination during highly active antiretroviral therapy and after analytical therapy interruption. AIDS 23:2226–2230
Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R (1998) Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med 188:2205–2213
Zhang LZ, Zhang TC, Pan FM, Zhang ZH, Li X (2010) Interleukin-10 gene polymorphisms in association with susceptibility to chronic hepatitis C virus infection: a meta-analysis study. Arch Virol 155:1839–1842
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Ng, C.T., Oldstone, M.B.A. (2014). IL-10: Achieving Balance During Persistent Viral Infection. In: Fillatreau, S., O'Garra, A. (eds) Interleukin-10 in Health and Disease. Current Topics in Microbiology and Immunology, vol 380. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-43492-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-662-43492-5_6
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-43491-8
Online ISBN: 978-3-662-43492-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)