
Abstract
Static headspace as well as purge and trap are still widely in use for the analysis of volatile organic compounds. Since around 20 years, different types of microextraction techniques have been developed, which either require a strongly reduced amount of solvent or are completely solventless. In this chapter, we will focus on the solventless techniques, which can be combined with GC via injection or thermal desorption. In the first part a short overview of recent developments in solvent-free techniques such as headspace, purge and trap as well as microextraction techniques is given. The second part provides the theoretical background of the techniques with additional practical hints for their application.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Overview and Instrumental Set-Up
1.1 Scope
In the past, sample preparation often has been considered as a step of minor importance in the whole analytical process compared with separation and detection [1, 2]. In particular, capillary gas chromatography mass spectrometry (GC-MS) provides high sensitivity and selectivity in detection as well as high separation efficiency with plate numbers exceeding 100,000 and peak capacities >300. However, sample preparation remains as a bottleneck in the overall analytical procedure, because it is one of the most time-consuming steps and errors occurring in this primary step of the analytical process can hardly be corrected. Sample preparation prior to GC separation aims at the following goals: (1) improvement of the gas chromatographic behaviour of the analytes and analyte detectability, (2) separation of analytes from interfering matrix components and (3) enrichment of the target analytes. The first goal though has become less relevant in recent years because of better GC column quality and the use of liquid chromatography mass spectrometry for polar compounds, which often replaced previously established derivatisation procedures for many applications. The other points are still important when analytes in complex mixtures have to be determined and directives demanding identification and quantification at ever lower concentrations have to be fulfilled [2].
Classical sample preparation methods are liquid–liquid extraction (LLE) for liquid samples and Soxhlet extraction for solid samples. Both are simple and many protocols still rely on them [3, 4]. However, these methods typically need large volumes of sample and organic solvent (100–250 mL or even more), as well as repeated extractions for sufficient enrichment. A clean-up and further concentration by evaporation or distillation are often necessary. These methods can be laborious, time-consuming and prone to analyte losses. Furthermore, the used high-purity organic solvents are expensive, usually toxic and harmful to the environment and substantial quantities of solvent waste have to be handled. At the least, one should aim for a limited use of ozone layer destroying chlorinated solvents, e.g. chloroform and dichloromethane [1, 5, 6].
An alternative to LLE extraction is solid-phase extraction (SPE). In water analysis, SPE is nowadays the most widely used sample preparation technique for non-polar compounds [4]. It offers a wide field of applications and has been subject of detailed reviews [7, 8] and monographs [9]. In SPE, an aqueous sample passes through a solid sorbent bed, packed inside a disk or cartridge, in which the analytes are trapped on an immobilised phase and later re-extracted by organic solvents. Advantages of SPE are often lower (but not negligible) amounts of organic solvents, possibility for field sampling and automation of the sampling process [3, 4]. Miniaturised micro-SPE methods with the aim of sample and sorbent reduction have also been introduced [10, 11].
Evaluating the recent literature reveals that extraction, enrichment and matrix separation show major trends towards (1) miniaturisation [5], (2) online sample preparation, (3) automation [12, 13], (4) high sample throughput by lower processing times, (5) higher extraction yields [14] and better reproducibility, as well as (6) organic solvent reduction or exclusion [13, 15]. Nowadays, a variety of solventless enrichment and injection techniques for GC are available ranging from classical headspace injection to recently introduced sorptive enrichment formats. All of these will be discussed in this chapter with an emphasis on principles of the methods and similarities and differences among them that is often not directly obvious. A variety of acronyms for the different techniques have been established and can overwhelm potential users new to the topic. Therefore, an overview of acronyms is given in the glossary at the end of this article that includes also some techniques not dealt with specifically in this chapter. For details of individual methods we will refer to specialised monographs [4] and reviews. Although out of the scope of this chapter, we will mention miniaturised liquid extraction methods such as liquid-phase microextraction (LPME) [16], single drop microextraction (SDME) [17] and dispersive liquid–liquid microextraction (DLLME) [18]. All these methods employ organic solvents in the μL or nL range and reduce the solvent consumption tremendously.
1.2 Direct Headspace Analysis
The most prominent and widely used solventless injection technique is still direct headspace analysis [19, 20]. Headspace analysis is generally defined as an extraction of the vapour phase above a non-volatile liquid or solid phase [21]. The headspace approach is simple, very little expenditure on sample preparation is necessary and all modes of headspace analysis can easily be automated [20, 21].
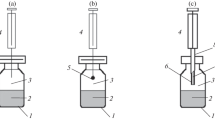
After the introduction of gas chromatography in the 1950s, the first application of static headspace (S-HS) with GC was reported in 1958 [21]. The simplest way to carry out static headspace analysis is to seal a sample in a gas-tight vial closed with a suitable septum, as it is shown in Fig. 11.1a. After sample equilibration, often at elevated temperatures, an aliquot of the headspace (generally 0.1–2.5 mL) is removed from the headspace with a gas-tight syringe and injected into the injection port of a gas chromatograph [20, 21].

(a) Typical static headspace sampling procedure. (b) Scheme of a pressure balanced static headspace system. In the left panel the pressurisation of the headspace sampling vial after equilibration is illustrated. Afterwards, the sample is introduced into the capillary column as shown in the right panel. (c) Scheme of a pressure/loop headspace system. In the first step pressurisation is carried out. This step is followed by a loop filling step and the subsequent transfer of the loop content into the GC column by the carrier gas stream
A major disadvantage is the often rather low sensitivity caused by low air–water partitioning constants (K aw) or vapour pressures of the target analytes. Another problem in S-HS is the internal pressure that is generated in the vial during thermostating and equilibration. It is the sum of the partial vapour pressures of all volatile sample compounds with the partial pressure of water being the main contributor. When using standard S-HS syringes, the pressurised headspace gas can expand through the open needle into the atmosphere, causing analyte loss during the transfer of the syringe to the injector. This problem can be avoided by using gas-tight syringes with valves, which can be closed during the transfer [20, 22]. However, in modern xyz-autosamplers that can be controlled very precisely reproducibility of headspace measurements without valve-closed syringes is also very good. An alternative option to avoid overpressure-related analyte losses is to replace the syringe by a heated transfer line, which is connected to the GC column. This approach was introduced by Perkin-Elmer as balanced pressure sampling system in 1968 [21]. Systems using sample loops are also common. The important steps of such a pressurised system are shown in Fig. 11.1b, c. In the first step, gas is pressed into the headspace of the sample vial to a set pressure level above the original pressure in the vial. By valve switching, the pressurised headspace is then temporarily connected directly to the GC column (Fig. 11.1b) or to a sample loop. The injected volume can be varied very precisely by varying the sampling time, or the exchange of the sample loop [21, 22]. A disadvantage of these systems is the need for a dedicated instrument since exchange of injection methods is not as easy as in syringe-based injection.
For an in-depth consideration we refer to excellent overview texts [23] and monographs on the theory and the fundamentals of headspace-GC analysis and instrument configurations [20, 21].
1.3 Sorbent-Based Techniques
1.3.1 Dynamic Headspace and Purge and Trap
To overcome the relatively low sensitivity of static headspace, exhaustive stripping methods were developed in the 1970s [21]. In principle, two modes can be distinguished. The first is dynamic headspace in which the headspace over a sample is continuously removed by a stream of an inert gas such as helium or nitrogen. This gas stream passes a sorbent trap in which the analytes are trapped. Due to the continuous removal of the target analytes from the headspace, no equilibrium can be established and the method will be exhaustive if the extraction time is long enough and if no breakthrough of the trapped analytes occurs. The second mode is purge and trap (P&T), where gas bubbles are purged trough the sample, stripping the analytes from the matrix and increasing the mass transfer to the headspace, again with subsequent trapping on a sorbent material (see Fig. 11.2).

Typical P&T system with sparger. (a) Analytes are purged by producing small gas bubbles with a glass frit. (b) Thermal desorption of the analytes from the sorbent packing material’s introduction into the GC injection port
In principle, the continuous extraction process could be carried out until all target analytes are completely removed from the sample and trapped as long as there is no breakthrough from the trap (exhaustive sampling). However, typically purge times of 10–15 min are used (see also Sect. 11.2.2.3).
The accumulation of water on the traps can cause problems during the subsequent chromatographic separation. A dry purge step, in which the trap is dried with a gas stream before thermal desorption, or a moisture control system, in which the water is condensed, can be used to avoid that. Typical dry purge times are between 1 and 4 min. The analyte traps are thermodesorbed after the extraction and the analytes are generally cryofocused to obtain small initial injection bands. If no cryofocusing is available a sorbent trap pre-heating step can be used to obtain a short desorption time and small injection bands. Typical desorption times are 2–4 min at desorption temperatures of 180–250 °C, depending on the trap material. Desorption flows are generally between 10 and 80 mL min−1. Different types of traps for various compound classes are available. Only few P&T methods have found their way into official procedures in Europe [24]. Contrary to this, several protocols of the Environmental Protection Agency (EPA) of the USA rely on P&T enrichment for the determination of volatiles in drinking, waste and hazardous wastewater [25, 26].
1.3.2 Thermal Desorption Tubes
Thermal desorption (TD) utilises tubes or GC liners, packed with sorbent materials, and is used mainly for airborne volatile analysis [2]. After sampling, the tubes are transferred to the GC and the analytes are thermodesorbed in a special thermal desorption unit or the GC injector. During desorption, the carrier gas flow through the tubes is in the opposite direction to the air flow during sampling. As in other analytical methods using thermal desorption in GC (like dynamic headspace and P&T), cryotrapping systems can be used to minimise the initial analyte band width [27].
TD is also a valuable alternative to headspace techniques for the isolation of volatile compounds from non-volatile solid, semi-solid and occasionally liquid matrices [2, 27–29]. For example, a sorbent packed liner was used as SPE extraction device; subsequent to the extraction, the liner was inserted directly into a programmable thermal vaporiser (PTV), where the residual water was removed through the split vent of the injector by evaporation in the carrier gas stream. The analytes stay trapped in the sorbent and are subsequently thermally desorbed onto the GC column for separation [30–32]. This is similar to a large volume injection (LVI) or direct aqueous injection (DAI) into a sorbent packed GC liner, combined with subsequent separation of water [33–38]. Gum-phase extraction (GPE) is very similar to SPE, but based on a packed bed of polydimethylsiloxane (PDMS) particles as trapping material [39, 40].
TD is not a new technique, but fully automated systems are only in use for about 10 years, because automation of the sample introduction is difficult [2]. In 2002, de Koning et al. designed a system which features a fully automated liner exchange, where a Focus XYZ sample preparation robot was combined with a modified PTV injector, whose head could be opened and closed automatically [41]. Two systems are commercially available by now, the ALEX™ (Automatic Liner EXchange) from Gerstel (Mülheim a.d. Ruhr, Germany) and the LINEX™ (LINer EXchanger) from ATAS GL (Veldhoven, The Netherlands).
1.4 Miniaturised Sorbent-Based Techniques
1.4.1 Classification and General Features
Microextraction methods are defined as techniques in which the volume of the extraction phase is very small compared to the sample volume [42]. In batch microextraction techniques typically only a small fraction of the analytes is extracted, in contrast to exhaustive methods [43]. In such cases, microextraction devices can be used as equilibrium sampling devices (ESDs) that have a negligible impact on the sample (see also below for SPME) [44].
Miniaturised extraction has several advantages: (1) The devices can be used directly in the field without long preparation and with little equipment, (2) it is often easier to implement miniaturised devices in already existing systems and a connection with GC is mostly straightforward, (3) miniaturised devices or techniques have lower operating costs and (4) the methods can be partly or fully automated, which leads to higher sample throughput and better reproducibility [45–47].
1.4.2 Coated and Packed Capillaries
Historically, the first microextraction methods used coated capillaries. The first applications using capillaries with polymer coatings such as polydimethylsiloxane (PDMS) as sorptive extraction phase have been shown already in the 1980s [48]. Organic substances were extracted from water and trapped in an open-tubular capillary column coated with cross-linked PDMS. After a drying step the analytes were analysed by thermodesorption (TD) and GC [49, 50]. This approach was named open-tubular trapping (OTT). The method was developed as an alternative to solid-phase extraction and the main advantage of the method was the easy removal of water by purging a short gas plug through the column [51]. Desorption of the trapped analytes is done either by thermal desorption or with a small amount of solvent. However, this approach suffers from complex instrumental set-ups and unfavourable sampling conditions such as high pressure drops for long traps and limited sample flows [52]. The same principle as in OTT, i.e. using internal capillary coatings, is also employed in in-tube solid-phase microextraction [53–55], in-tube extraction (ITE) and capillary microextraction (CME), which were developed later, mainly to overcome drawbacks of solid-phase microextraction (SPME) as discussed below.
1.4.3 Solid-Phase Microextraction
Solid-phase microextraction (SPME) was introduced in 1989 [56] and was commercialised in 1993. Initially, the principle was developed as a preconcentration method for spectroscopic remote sensing devices based on glass fibres, but later its high potential as a solventless extraction method for subsequent chromatographic analysis was realised. SPME is nowadays the most prominent and widely used microextraction method with hundreds of publications and just as many applications. Nevertheless, so far only few standardised procedures have been implemented (e.g. two German standard methods on pesticides and volatile organic compounds (VOC) in water [57, 58] and US EPA Method 8272 on polycyclic aromatic hydrocarbons (PAHs) in pore water [59]).
SPME combines several sample preparation steps such as sampling, enrichment and clean-up. A thin fused silica fibre coated with extraction phase (7–100 μm film thickness d f and typically 1 cm length) is utilised for the extraction of analytes from the sample. The fibre diameter is 0.1–0.2 mm. The extraction phase volume varies, depending on the fibre diameter, between 0.2 μL (0.1 mm, d f = 7 μm) and 1.6 μL (0.2 mm, d f = 100 μm). Commercially available SPME coatings are discussed in Sect. 11.3.2. The fragile fibre is encompassed in a special syringe holder for its protection during penetration of vial and GC injector septa. Fibres can be used in two application modes. One mode is direct immersion of the fibre into the sample (DI-SPME); the other one is analyte extraction from the headspace above the sample (see Fig. 11.3a). After the extraction, desorption is performed thermally in the GC injector.

(a) Headspace solid-phase microextraction (HS-SPME) and direct immersion solid-phase microextraction (DI-SPME) redrawn after ref. [46]. (b) High capacity headspace sorptive extraction (HSSE) using stir bars or coated glass rods in the sample headspace redrawn after refs. [52, 68]; (c) stir-bar sorptive extraction (SBSE) redrawn after ref. [69] and Dual twister after ref. [70]; (d) rod extraction redrawn after ref. [71]
Disadvantages of SPME are the often limited lifetimes of relatively expensive fibres and bleeding from thick-film coatings [60]. The limited lifetime has its origin in the technical construction of the SPME device itself. The most common practical problems facing SPME are mechanical damages of the coating due to scraping, needle bending and fibre rupture caused by the fragility of the fused silica support [61]. Several attempts to overcome these drawbacks of the initial SPME fibres have been introduced, such as the introduction of bendable metal alloy and StableFlex™ fibres [43].
In some cases, the limited sorption capacity due to the very low extractant phase volume (see further discussion below) may also be a problem.
The simplest and cheapest way to perform SPME is manual sampling, which is meaningful if the number of samples is small. For SPME sampling, a manual fibre holder as well as an injection support for SPME fibres is available. In case of manual SPME, it is important to bring the fibre in the same position in the injection port in every analysis to obtain reproducible results.
For a detailed discussion of SPME we refer the reader to numerous monographs [4, 62, 63] as well as reviews [5, 13, 42, 43, 64–67].
1.4.4 Stir-Bar Sorptive Extraction and Headspace Sorptive Extraction
Stir-bar sorptive extraction (SBSE) utilises a glass incorporated magnetic stir bar coated with an extraction phase as shown in Fig. 11.3c. The method was introduced by Baltussen and Sandra [72] and is based on the same principle as solid-phase microextraction, but exhibits a higher extraction phase volume and due to this a higher sorption capacity. It was commercialised under the brand name “Twister™” by Gerstel (Mülheim a.d. Ruhr, Germany). Four different PDMS-coated stir bars with coating volumes of 24, 63, 47 and 126 μL are commercially available to date [48]. Additionally, a polyacrylate-coated stir bar for extraction of more polar analytes became available recently [73]. Bicchi et al. introduced a dual twister, which consists of a short PDMS tube closed at both ends with two magnets (Fig. 11.3c, lower right). The inner tube volume is filled with an activated carbon packing material [70].
As shown in Fig. 11.3b, c, the stir bar is either placed directly into the sample or in case of high capacity headspace sorptive extraction (HSSE), in the headspace above the sample. In case of direct immersion, the stir bar is stirred for a fixed time with a fixed rotation velocity. Typical extraction times are between 30 and 60 min [39]. After extraction, the bar is removed from the sample solution with tweezers, dried with a lint-free tissue, and transferred into a glass thermal desorption tube. The tube is then introduced automatically into a thermal desorption unit. Desorbed compounds are cryofocused with liquid nitrogen in a programmed temperature vaporiser (PTV) [73, 74].
For a more thorough treatment of SBSE we refer to several recent reviews dealing with SBSE and its application [73–75].
Another format of HSSE was introduced by Tienpont et al. for extraction of volatiles from aqueous solutions as well as flavour aroma compounds [52]. As shown in Fig. 11.3b, headspace bars consist of a ca. 5 cm long glass rod with a PDMS tubing over the last centimetre. These rods are mounted in the screw caps of headspace vials or flasks. After extraction, the rods are put manually in a glass desorption tube for thermal desorption as it is used for SBSE extraction. Commercially available are headspace bars with 0.5 and 1 mm PDMS coating (Gerstel, Mülheim a.d. Ruhr, Germany).
1.4.5 Silicone Rod and Silicone Tube Extraction
One of the easiest and cheapest microextraction methods is to use silicone rods (SRs) or silicone tubes (STs) for extraction. Popp et al. employed such PDMS materials in the form of rods (see Fig. 11.3d) and tubes for the enrichment of organic compounds [71]. In terms of analyte extraction, the SRs and STs are very similar to SPME and SBSE but with the advantage of being inexpensive, flexible and robust [48]. PDMS rods or tubes are sold by the metre with diameters of 1 or 2 mm and can be cut to the length needed. The overall extraction procedure is the same as for SBSE. The sample bottles can be shaken by an overhead or a plate shaker. Thermal desorption can be carried out in TD units. SRs and STs with different sizes and phase volumes (8–635 μL) have been applied for the extraction of a large variety of organic micropollutants [48]. A disadvantage of SRs and STs can be the presence of interfering monomers or additives resulting from the polymer since the materials are not of analytical grade. Thus, a thorough pre-cleaning by solvents or heating might be necessary.
1.4.6 Coated and Packed Needles
Instead of capillaries discussed in Sect. 11.1.3.2, also internally coated or packed needles can be used for extraction. An advantage of using needles instead of capillaries is the generally easier automation using autosamplers. Methods that use coated needles are the capillary adsorption trap (INCAT) [76–78] and solid-phase dynamic extraction (SPDE), the latter of which is shown in Fig. 11.4a. SPDE was introduced in 1996, as a simplified method for solid-phase extraction from a sample, employing a syringe in which the inner surface of a cannula or needle was at least partly coated with a stationary phase, such that aspirating the sample into the needle results in sorption of the components of interest in the stationary phase. Aspiration of a solvent may be employed for re-extracting the analytes from the stationary phase for direct injection into a chromatographic instrument, or the components of interest may be introduced into a GC by thermal desorption in the hot injection port. Following SPME in terminology, this technique was named solid-phase dynamic extraction (SPDE) whereby the term “dynamic extraction” originates from the active pumping of the sample through the cannula for several times [79]. SPDE utilises a 2.5 mL headspace syringe with a removable needle that is coated with an immobilised extraction phase on the inside, similar to a fused silica GC column. For extraction, the needle is immersed in the headspace above the sample. The syringe plunger is then moved up and down several times for a dynamic extraction of the sample, and the analytes are sorbed by the internal coating. After several extraction cycles (aspirating and dispensing a preset gas volume), the analytes are thermally desorbed from the coating into the hot GC injector. To facilitate the analyte transport into the GC injector a certain desorption gas volume of an inert highly pure gas (nitrogen or helium) from the injector or a special gas station is aspirated into the syringe before injection.

In-needle and in-tube techniques with internal coatings and packing materials as extraction phase. (a) Solid-phase dynamic extraction (SPDE), (b) in-tube extraction (ITEX) and (c) needle trap (NT)
Removeable SPDE needles with PDMS (polydimethylsiloxane), PDMS/AC (polydimethylsiloxane + 10 % active charcoal), CT-5 (5 % diphenyl/95 % dimethylpolysiloxane), Carbowax (polyethylene glycol, PEG), CT-1701 (14 % cyanopropyl/86 % dimethylpolysiloxane), OV-225 (25 % phenyl-/25 % cyanopropylmethylsilicone/50 % polysiloxane) and custom-made coatings are commercially available by Chromtech GmbH (Idstein, Germany). The film thickness of the coatings inside the needle is 50 μm. Two needle lengths are available (56 and 74 mm). The longer one is used in combination with a Peltier cooling device for extraction phase cooling to obtain higher extraction yields (due to the exothermic extraction process). With a commercially available apparatus, a maximum temperature difference of 40 °C is possible, so that at room temperature an extraction phase temperature of −15 °C can be obtained [80]. SPDE needles possess around 4–6 times larger extraction phase volumes than a 100-μm SPME fibre [81, 82]. However, it turned out that SPDE needs relatively long extraction times compared to SPME, because every dispensing step causes a significant loss of retained analytes, leading to a rather low extraction yield. This loss can be up to ~50 %. To overcome this problem, van Durme et al. developed an accelerated solid-phase dynamic extraction (ASPDE), in which only aspiration of a volume into a bigger syringe is carried out. Thus the system is used in terms of breakthrough sampling similar to D-HS and maximum loading capacity is reached in much shorter times [83]. Another possibility to reduce the loss of analyte during syringe dispensing is to use a high dispensing volume flow and a low aspirating flow (see also discussion in Sect. 11.2.2.2).
In a second class of in-needle extraction techniques packing materials (particulate material or bundles of polymeric fibres) are used as extraction phases. With packings, an increase in the sorbent mass and surface can be achieved and principally all adsorptive materials and particulate partitioning materials, such as PDMS, can be filled in such needles.
A needle concentrator device was developed by Kubinek et al. [84]. The method utilised a glass syringe connected to a 9 cm long stainless-steel needle, filled with Porapak Q and aluminium oxide. The syringe needle is immersed directly into the sample which is then aspirated into the syringe. The analytes are adsorbed by passing the Porapak Q and the aluminium oxide bed. The device is then flushed with air to remove residual water. Afterwards, analytes are thermally desorbed into the hot injector. Aluminium oxide acts as water reservoir, which evaporates and transports the analytes out of the needle with the vapour stream. A similar thermal desorption, without aluminium oxide is used for the needle trap device.
In-tube extraction (ITEX™) was commercialised in 2006 by CTC Analytics AG (Zwingen, Switzerland) [85]. It features a sorbent bed volume of 160 μL and so far is the only fully automated device with needle packing. It features a sorbent packed needle surrounded by an external heater, mounted on a gas-tight syringe with a side port. Enrichment is carried out similar to SPDE by aspirating and dispensing the syringe several times, pumping the sample headspace through the sorbent bed.
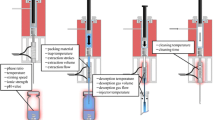
However, a loss of analytes during the dispensing steps is not as pronounced as in SPDE, because in general stronger interactions between the sorbates and the packed sorbent occur. The analytes are thermodesorbed into the GC injector with desorption gas, preferentially aspirated inert carrier gas from the injector. Although in principle a portion of the sample headspace could be used as well, this may lead to rapid deterioration of oxygen-sensitive packings and problems with residual humidity may result. Thus, we do not recommend the latter approach. After the syringe is withdrawn from the injector, the plunger is moved above the side port and the heated trap is flushed with inert gas (N2, He) several times to avoid carryover. A schematic depiction of the steps in the ITEX procedure is given in Fig. 11.5.

Steps of the ITEX procedure from left to right: Sample conditioning through heating and stirring/shaking, adsorption by dynamic headspace extraction, thermodesorption from the heated trap, trap cleaning by a stream of helium or nitrogen. Graphic reprinted with permission from [86]
The initial ITEX system required a special autosampler head, which hampered the quick exchange of analytical methods. In 2009, this drawback has been overcome with the release of a new ITEX 2 system, which can be mounted on any PAL-type autosampler without modification.
A similar packed needle trap device (NT) was developed concurrently by Wang and Pawlizyn [87]. The needle trap, shown in Fig. 11.4c, is either filled with Carboxen 1000 or with a mixed packing of PDMS, DVB and Carboxen particles. NTs with packing materials between 60 and 80 mesh (DVB, Carboxen, Carbosieve, etc.) are supplied by PAS Technology (Magdala, Germany). For sampling, the needle can either be exposed to the sample headspace for passive [88] sampling, or active sampling can be performed by a common syringe or syringe pump [89, 90]. After sampling, a small amount of extraction air is left in the syringe and used for desorption in the hot GC injector.
A fibre packed needle is an extraction device composed of a needle, packed longitudinally with a bundle of Zylon™ or Technora™ fibres and was developed by Saito et al. for GC measurements of volatile aromatic and organic compounds in aqueous solutions [91]. The needle is filled with a special technique on a 20 mm section with fibres, containing approximately 830 single filaments (12 μm o.d.) which are surface coated with polymeric materials.
An advantage of all in-tube techniques is that they can easily be automated by xyz-autosampler systems. This is also mandatory for quantitative work when pre-equilibrium sampling is performed, since it is important to keep extraction parameters like temperatures, extraction time, stirring speed and the flows during sampling and desorbing constant in all measurements. A disadvantage of all these techniques (as for the capillaries discussed in Sect. 11.1.3.2) is that particles are able to block the needles and tubes; therefore samples have to be clean, or headspace extraction has to be used.
1.5 Injection, Thermal Desorption, Cryofocusing
In case of static headspace injection and in-needle extraction techniques, it is suitable to use needles with a side port hole, because the septum will not be cut out by the thicker needle tips. It also prevents the liner from being contaminated by septum particles, which can lead to chromatographic interferences, peak tailing and carryover. A plugging of the needle by septum material is also diminished. Normal injection volumes in S-HS vary between 0.1 and 1 mL. Particularly gaseous samples are injected by split injections. Splitting prevents peak broadening and peak splitting, but leads to a lower sensitivity. The used split ratios are generally small and have the effect that the injected gas volume is transferred faster into the GC capillary. To obtain a small initial injection band of the introduced analytes and sharp peaks, cryofocusing should be applied [92]. A sharpening of the injection band can also be accomplished by using a thick-film analytical column or a short thick-film retention gap.
SPME fibres are compatible with all heated injector ports, whereby the needle exposure depth has to be adjusted to place the fibre in the hot injector zone. Therefore, it is advantageous to measure the actual injector temperature profile. Additionally, low-volume SPME liner (splitless liner with 0.75–0.8 mm ID) is available.
In the case of SPDE, the temperature profile of the injector should be equal over the whole injector body, because the coating is spread over the length of the needle, which is about five times longer than an SPME fibre. Other methods, like dynamic headspace, P&T or ITEX, use external heaters and are independent of the injector temperature profile.
For a fast desorption from the fibre, which leads to sharper peaks, narrow bore GC liners with ~0.75 mm internal diameter are optimal. Such special SPME narrow bore liners are available for most types of injectors. The fibre has to be exposed completely from its surrounding supporting sheath; otherwise peak splitting can occur. To prevent septum particles in the injector during SPME analysis, a septum-free injection system with Merlin microseals™ can be used (only for 23 gauge needles).
As for static headspace injection, a thick-film analytical column or a short thick-film retention gap can be applied to sharpen the initial injection band.
2 Theoretical Background
2.1 Classification According to Extraction Mode
In Fig. 11.6, the basic principles of all extraction modes frequently used in solvent-free extraction techniques and the corresponding relevant method parameters are summarised. Generally, one can distinguish batch and dynamic sampling approaches. Although in batch methods sample mixing by stirring or agitation is carried out, the term dynamic is limited here to systems in which the sample is penetrating or passing a sorbent material by active pumping or sucking. The batch sampling approaches are shown in Fig. 11.6a–c, where one can distinguish two-phase and three-phase systems.

Basic principles of extraction modes in solvent-free extraction techniques and relevant method parameters. Condensed phases are shown as hatched areas; the extraction phase is shaded in grey
In Fig. 11.6a, a two-phase system without an additional extraction phase is shown. This kind of system represents the typical static headspace sampling. In Fig. 11.6b the extraction phase is directly in contact with the sample phase. Systems with direct immersion sampling such as DI-SPME, DI-SBSE, SR and ST extraction are representatives. Figure 11.6c represents a three-phase system with an extraction phase placed in the headspace. Methods such as HS-SPME and HSSE fall in this category.
Figure 11.6d, e represents dynamic extraction methods in which the sample flows in one direction through the extraction device and passes an extraction phase coating or a packing. This type of extraction device arrangement can be found in micro-SPE, OTT, CME and in-tube SPME. Dynamic sampling, such as carried out by HS-SPDE, ITEX and NT, is described in Fig. 11.6 (panels f–h). Finally, Fig. 11.6, panels i and j describe purge and trap and dynamic headspace methods, respectively.
2.2 Thermodynamics
2.2.1 Batch Extraction
The most important factor governing the thermodynamics and thus the equilibrium partitioning in a static system such as shown in Fig. 11.6a–c is the distribution constant K es which is defined as
where c s is the concentration of analyte in the sample in mol L−1 and c e is the concentration of analyte in the extractant (extraction phase) in mol L−1; n s, n e are the amounts of the analyte remaining in the sample and the amount of analyte in the extractant phase in mol, respectively. V e is the volume of the extractant and V s is the volume of sample in L, respectively. β is the phase ratio of the static extraction system and is defined as V s/V e. Note that definitions of distribution constants and phase ratios are not consistent in the literature and might be used reciprocally, in particular in dealing with static headspace. In S-HS, the phase ratio β is defined as the sample volume to headspace volume ratio V s/V h. In case of S-HS no extraction phase is available; here only the partitioning of the analyte between the headspace and the sample phase, represented by K hs, occurs, whereby the concentration in the headspace c hs can be calculated from the initial concentration of the sample c 0 by
The sensitivity depends only on the phase ratio and the partitioning constant K hs. In case of aqueous solutions, K hs is identical to the air–water partitioning constant K aw. In Fig. 11.7 the effect of phase ratio and temperature is shown exemplarily based on calculated c hs according to Eq. (11.2). Temperature-dependent K aw values required as input parameters are tabulated in the literature or can be calculated by van’t Hoff-type equations, using compound-specific curve fitting parameters [93–95]. It is clear from the figure that the concentration in the headspace and thus the sensitivity directly correlate with K aw of a compound. Furthermore, if K aw is high, β becomes significant and a higher phase ratio further increases sensitivity up to β = 2. Beyond this ratio there is no substantial effect of β on sensitivity any more. Thus, for such compounds the headspace volume should be kept below one-third of the vial volume to obtain highest extraction yields. If K aw is low as for ethanol, β becomes less significant.

Dependency of headspace concentrations on phase ratio β and temperature. Values calculate using Eq. (11.2) for three different analytes (dichloromethane, ethyl acetate, ethanol) at an initial analyte concentration of 100 μmol L−1
Raising the temperature increases K aw and thus the analyte concentration in the headspace as is evident from the calculated headspace concentrations shown in Fig. 11.7a, b. In general, compounds with a higher affinity to the aqueous phase show a stronger variation of the partitioning constant with changing temperature (compare K aw values in Fig. 11.7a, b).
In the static two-phase system depicted in Fig. 11.6b, in which a sample is in direct contact with a solid or liquid extraction phase, the amount of analyte extracted at equilibrium n e is given by the following mass balance:
With increasing sample volume V s, the amount of analyte extracted increases until V s is significantly larger than the factor K es V e, and n e does not increase further. With large sample volumes V s, small K es and small V e, the amount of analytes extracted is insignificant to the amount of analytes in the sample. In microextraction methods, V e is generally orders of magnitude smaller than V s. In such cases, the following simplification of Eq. (11.3) is frequently used although this is only valid for rather small K es:
For example, with typical values for V s (10 mL) and V e (1 μL), K es needs to be smaller than 103 to neglect the term K es V e compared with V s. However, for many compounds and coatings, higher K es values are found.
Nevertheless, the very high phase ratio β typical in static two-phase extractions such as SPME may lead to a negligible depletion of total analyte mass in the sample and thus results in minimum disturbance of the investigated system. It therefore can be used to measure freely dissolved concentrations instead of total concentrations. These aspects make such methods very useful for probing biological and living systems, investigating multiphase distribution equilibria, and repeated sampling from the same system to perform process studies [44, 96, 97].
In general, the recovery R or extraction yield for a two-phase batch system can be calculated by
where n 0 is the initial amount of analyte in the sample. Higher recoveries in particular for compounds with low K es values can be obtained by using higher extraction phase volumes and resulting lower phase ratios. In Fig. 11.8 this is shown exemplarily with the comparison of recoveries in SPME, SPDE and SBSE depending on K PDMS/water. Chloroform is only extracted by SBSE significantly, whereas for pyrene even SPME yields a recovery of >50 %; thus in the latter case, depletion in the sample is not negligible any more.

Recovery for various extraction techniques under equilibrium conditions, calculated for an aqueous sample volume of 10 mL and the indicated PDMS volume. Chloroform, ethylbenzene and pyrene are indicated as exemplary target compounds with log K PDMS/water of 1.7, 2.7 and 4.3, respectively
A change in temperature from T 0 to T influences the K es value according to:
where ΔH is the change in molar enthalpy of the phase transfer process and R is the gas constant. Partitioning into the fibre is an exothermic process (ΔH > 0). An increase of temperature decreases the extraction phase/sample distribution and thus yields lower recovery and sensitivity.
The last batch system that has to be considered here is a three-phase system in which the extraction phase is in the headspace above a sample (see Fig. 11.6c). As already mentioned, this kind of system can be found in HS-SPME and HSSE. The amount of analyte extracted n e at equilibrium can be expressed by extension of the mass balance equation (11.3) by the “headspace capacity term” K hs V h in the denominator, which leads to Eq. (11.7):
Fundamentally, the situation is similar to static headspace. The headspace sensitivity is controlled by the phase ratio V s/V h, the headspace/sample partitioning constant K hs and the extraction phase/headspace distribution constant K eh.
Although an increase in temperature increases the headspace to sample partitioning constant, the sample vessel temperature should not be too high. As pointed out for direct immersion the enthalpy of sorption in general is negative and thus K eh decreases if the headspace temperature rises. To minimise this sensitivity drop, extraction phase cooling devices were developed, to exploit the so-called “coldfinger effect”. For HS-SPDE a dedicated Peltier needle cooler is available [80]. The influence of the extraction phase cooling on the extraction phase–headspace distribution constant can be expressed by
where K eh is the distribution constant of a compound between the cold extraction phase, which is at temperature T e and the hot headspace at temperature T h. C p is the constant pressure heat capacity of the compound and \( {K}_{\mathrm{e}\mathrm{h}\left({T}_{\mathrm{e}}\right)} \) is the extraction phase–headspace distribution constant, when both extraction phase and headspace have the temperature T e. A disadvantage of exploiting the coldfinger effect to increase extraction yields is a potential interference by water condensation in case of aqueous samples.
As a rule of thumb, if no cooling device is available, the sample temperature for headspace extractions in three-phase systems should be between 40 and 60 °C.
Equation (11.7) for static three-phase systems can be generalised to describe multiphase distribution equilibria as sketched in Fig. 11.9.

Equilibrium extraction in a multiphase system
In any multiphase system, the concentration in the extraction phase under equilibrium conditions is given by
where K i V i is the analyte capacity in phase i.
2.2.2 Flow Through Systems
When using sorbent tubes or traps (see Fig. 11.6, panels d and e, i and j), the breakthrough curve of an analyte is important. At the beginning of the sampling, the analyte in the sample or an air phase in contact with the sample is sorbed on the sorbent and the concentration in the outflow is zero. In case there is a constant inflow concentration c in over time, eventually the sorption capacity for the compound is reached, and the concentration of the compound in the outflow c out rises and finally reaches the concentration of the inflow. This elution profile is called the breakthrough curve and it is typically assumed that this curve can be described as the integral of a Gaussian peak.
The most important parameters describing flow through extraction in the volume domain are (1) the specific retention volume, (2) the specific breakthrough volume (sometimes also called safe sample volume V safe) and (3) the complete sample elution volume V elution. The latter is relevant for desorption of the compounds during injection. The former two parameters relevant for sampling/extraction are sketched in Fig. 11.10.

Breakthrough curve. Parameters describing breakthrough in elution profiles in the frontal (upper panel) and pulse injection (lower panel) technique. V M: dead volume, t M: dead time (time a nonretained marker needs to pass the sorbent bed)
The specific retention volume is also called 50 % breakthrough volume BTV50 % and is given in mL g−1 of sorbent. BTV50 % indicates that the outflow concentration reaches 50 % of the inflow concentration when this volume has passed the sorbent. Although shapes of elution profiles may differ, BTV50 % is characteristic for a given compound and a specific sorbent at a given temperature [98]. It equals the partition constant between the sample and the sorbent, K es. BTV50 % can be determined by either a frontal or an elution (pulse injection) technique. In the former, it is equivalent to the inflection point in the elution profile and in the latter to its net retention volume [99].
For Gaussian shaped breakthrough curves the specific breakthrough volume or V safe can be approximated as
Independent of the specific elution profile less than 5 % of the analytes are lost by breakthrough when limiting sampling to this volume.
Assuming no sorption hysteresis, the breakthrough curve also allows to estimate the gas volume needed for a complete elution of the analyte. As a rule of thumb, this volume V elution is given as
However, for short extraction tubes with sorbent beds or coatings that provide only a small number of theoretical plates (i.e. less than 2–5), deviations from the Gaussian modelled breakthrough curve may occur and Eq. (11.10) is not valid anymore. For this case, alternative equations to describe elution profiles have been proposed by Lövkvist and Jönsson that allow the calculation of the breakthrough volume depending on plate number and accepted breakthrough level in % [100].
2.2.3 Salting Out
The addition of salt can increase the partitioning coefficient K hs or K es, which can lead to higher extraction yields and thus higher sensitivities. The effect is also called “salting out effect”. In preparative organic chemistry this effect is often used to enhance the extraction yield of a compound during LLE. Addition of salt to water results in dissociation of the salt to ions forced by strong ion–dipole interactions. Hydration shells form around the dissociated ions, which are variable in thickness depending on their hydration enthalpies. The formation of hydration shells binds this water which is not any longer available for solvation of organic compounds in the sample. As a consequence, a decrease of “free water” volume and therefore an increase of initial compound concentration in the remaining “free water” should result [101]. The effect of salt addition on extraction yields and the air–water partitioning constant can be described quantitatively by the “salting out” or “Setschenow constants” K s. It is possible to determine the Setschenow constants according to Eq. (11.12) using measurements at various salt concentrations [102]:
where γ w is the activity coefficient of the compound in pure water, γ w,salt is the activity coefficient in the saline aqueous solution and [salt]total is the total molar salt concentration. Note that K s values are salt specific. Common salts used for salting out are NaCl, KCl, Na2CO3 and Na2SO4. Using the calculated K s the modified K hs or K es, K salths or K saltes , can be calculated by Eq. (11.13) (identical for K es):
The equation has to be used with care, because at high salt concentrations (>15 % (w/w)) considerable deviations from the behaviour described by Eq. (11.12) can occur.
The addition of salt can also be used to minimise differences in matrix composition of samples that otherwise may influence extraction yields to a different extent. If salt is used in sample preparation, it should be tempered to remove possible organic residues. Note that the addition of salt also leads to changes in the sample volume. A disadvantage of salt addition in the case of direct contact between extraction phase and sample is the formation of a salt crust on the extraction phase material during thermal desorption that can lead to damage and diminished extraction phase lifetime. Furthermore, there is still conflicting evidence with regard to the influence and extent of salting out for partitioning between two condensed phases. Depending on the extraction phase and the target analyte, even a “salting in effect” has been observed in a few cases. Surely, the use of salt as a means to influence phase partitioning requires further systematic investigations.
2.2.4 pH
For ionisable species a transformation into the undissociated form will increase the extraction phase-sample partitioning constant K es in direct immersion techniques and the air–water partitioning constant K aw in headspace techniques.
To quantify the influence of a certain pH value on K es the following equation can be used:
K undiss.es and K a are the extraction phase-sample partitioning constant and the dissociation constant of the undissociated compound, respectively [103]. Highest extraction yields can be achieved when pH values for acid analysis are adjusted at minimum two units lower than the pK a value of the corresponding undissociated acid [62] and for base analysis two units higher than the pK a value of the corresponding protonated base. The same discussion applies to headspace systems replacing K es by K aw.
One needs to keep in mind though that very low (<2) or very high pH values (>11) can lead to a fast destruction of the extraction phase in direct immersion techniques, in particular for SPME coatings [62].
2.3 Kinetics in Solvent-Free Extraction
2.3.1 Batch Systems
When the enrichment is carried out by direct immersion of the extraction phase into the sample, mixing by stirring, shaking and other methods can be used to promote diffusion of analytes from the sample into the extractant, by minimising the boundary layer around the phase. Two different ways to agitate a sample within a vial for SPME extraction are shown. In (a) the agitation by an external agitator is shown. Here the vial is agitated in cycles and a rotation of the fibre in the sample is induced. In (b) the sample agitation by stirring with a magnetic bar is shown. In such cases it is better to place the SPME fibre not directly in the middle of the vial because the rotational speed of the sample is higher between vial centre and wall; thus equilibrium will be attained faster. This, however, only affects the time required for equilibration; it does not affect the position of equilibrium itself or other properties of the static process (see Fig. 11.11a). In two-phase batch systems a boundary or Prandtl layer of the thickness δ as shown in Fig. 11.11a is formed. Outside this boundary layer, it is assumed that the analyte flux to the extraction phase is convection controlled. In contrast, the flux through the boundary layer is only controlled by diffusion of the analyte. The layer thickness depends on the rate of convection, which can be increased by increasing sample agitation. When the extraction kinetics are only controlled by diffusion through the boundary layer at the contact area to the extraction phase, which is assumed for thin extraction phases (d e < 200 μm) and partition constants K es > 100 [4], the time t e to reach 95 % of the equilibrium concentration in the extractant phase can be estimated by Eq. (11.15):

Influence of sample agitation on the extraction time
where B geo is a geometric factor that depends on the geometry of the extraction phase (e.g. 3 for a cylindrical geometry), d e is the thickness of the extraction phase and D s is the diffusion coefficient of the analyte in the sample.
In case of thicker extraction phases and smaller extractant-sample partition constants (K es < 100), the mass transfer within the extraction phase controls the extraction rate and the diffusion coefficient of the analyte in the extraction phase D e becomes dominant. In these cases, Eq. (11.16) can be used to estimate the time in which 95 % of the equilibrium amount in the extractant phase can be reached:
In case of headspace extraction in a three-phase system such as shown in Fig. 11.6c, the extraction time depends on three diffusion coefficients in the different phases. The diffusion coefficient D h of a compound in the headspace is three orders of magnitude higher than that of the same compound in the aqueous sample phase D s and five to six orders of magnitude than that in the extraction phase D e; thus, it is never the limiting factor in kinetics of the extraction process.
The headspace volume can have an enormous influence on the time to reach equilibrium in the system. If K hs is high and the headspace volume is large compared with the sample volume, there is no need to re-establish equilibrium between the sample and the gas phase during sampling. Under these conditions, the extraction can be very fast and can even be independent from sample agitation or stirring [62]. However, as was pointed out in the thermodynamic considerations on batch extractions, the sensitivity of a headspace method can be increased by increasing the phase ratio β; thus a small headspace volume is preferred for maximum sensitivity. Since K eh always will be orders of magnitude higher than K aw this also applies to three-phase systems. When the sample phase is large compared with the headspace volume, equilibrium between sample and headspace may have to be re-established during sampling. In this case, the extraction time increases, because the diffusion coefficient of the analyte in the sample is low (compared to that in the headspace), slowing the mass transfer to the headspace and hence the extraction process. Thus, the choice of the phase ratio β needs to be balanced in terms of sensitivity versus extraction time considerations.
2.3.2 Flow Through in Capillaries and Needles
For homogeneous samples, being liquids or gases, flowing in one direction through a tube, such as a fused silica capillary column in OTT or an in-tube SPME device, the extraction process can be described by chromatographic theory. In this geometry of the extraction device, with the extraction phase coated on the inner wall, the concentration time profile in longitudinal (x-direction) can be adopted from the dispersion of a concentration front [4]:
where u s is the linear velocity of the sample through the tube and V v is the void volume of the tubing containing the extraction phase. σ is the mean square root dispersion of the front, which is defined as
where H is the height equivalent to theoretical plate in chromatographic systems. Equation (11.18) states that the analyte front migrates through the tube with a velocity proportional to the linear velocity of the sample and is inversely related to the partition constant.
When using approaches in which aspiration, as well as dispensing through a needle or tube takes place, every dispensing step can lead to a significant loss of retained analytes, which can result in rather long sampling times to reach equilibrium [83]. This can be overcome by using only aspiration and much faster dispensing than aspirating flows in case of aspiration and dispensing sampling.
2.3.3 Purge and Trap
By using a stripping instrument such as presented in Fig. 11.2a and depicted schematically in 7i, bubbles of air or an inert gas are produced near the bottom of a vessel and then rise to the surface of the sample solution or slurry. In order to minimise extraction times, the exiting gas should have achieved equilibrium with the liquid sample. Therefore, the velocity of the rising bubbles should be sufficiently small and the well mixed sample column sufficiently high to establish air–water equilibrium. Furthermore, the bubbles need to be small, to achieve a large interface area, but also large enough, that adsorption at the air–water interface can be neglected. The theoretically extracted amount for different purge times can be calculated by using the K aw in the following exponential equation:
where G is the purge volume per unit time, V s is the volume of the sample ands c 0(t) and c e(t) are the initial concentration and the concentration in water at time t, respectively. Resulting extraction profiles are shown in Fig. 11.12 where the effect of different K aw on extraction time is clearly visible.

Extraction profiles in P&T systems: Calculated extraction efficiency vs. purge time profile for four compounds with different air–water partitioning constants. The purge gas flow is 40 mL min−1 and the sample volume 25 mL. Vinyl chloride (VC) has the highest air water partitioning constant and bromoform the lowest
3 Choice of Extraction Phases and Sorption Materials
3.1 Interactions Between Analyte and Extractant Phase
The efficiency of an extraction process depends on the distribution constant between the extraction phase and the sample K es. This is the characteristic parameter that describes the properties of an extraction material and the affinity of the analyte towards it [46]. Therefore, the selection of an appropriate coating or extraction phase for a given set of analytes is of high importance. Fundamentally, one can distinguish between absorptive and adsorptive extraction materials. In case of liquid coatings, partitioning into the extraction phase, with additional solvation of the molecules by the liquid coating, takes place. Analyte molecules can diffuse within the whole liquid coating during the extraction time. These phases obtain high sorption capacities. In contrast, solid sorbents have a defined crystalline structure, and due to very small diffusion coefficients in solids, in an adequate timescale, only adsorption on the sorbent surface takes place. Because only a limited number of adsorption sites are available on surfaces, a competition of analytes for free sites can occur at higher concentrations. This causes displacement of analytes (competition effects) with lower affinity to the sorption phase [62, 104]. Absorptive interactions are typically much weaker compared to adsorption on active surfaces. Therefore, analyte desorption can be performed under softer conditions, such as lower temperature and shorter desorption times, which prevents degradation of thermolabile analytes. On the other hand, although adsorptive materials need higher desorption temperatures, they can also withstand higher temperatures than absorption phases without thermal degradation.
The sorption affinity of an organic compound towards a liquid polymer extraction material depends on the nature of prevalent interaction forces. These forces can principally be divided into three groups. The first group contains apolar compounds such as alkanes and PCBs that interact only by van der Waals interactions. The second group contains monopolar compounds, such as ethers, carbonyl compounds and chlorinated hydrocarbons, which interact by van der Waals interactions and as either H-acceptors or H-donors. The third group contains bipolar compounds, which are able to interact by van der Waals interactions and as well as H-donors and as H-acceptors. Compounds that can be assigned to the last group are, e.g. alcohols, phenols, carboxylic acids, amines, etc. The knowledge of these interactions can be utilised in the choice of an appropriate sorbent material for a certain type of organic compounds. Although it cannot replace experimental comparison, it can help to limit the number of sorbents to be evaluated for a given application.
3.2 Solid Sorbent Materials
A vast number of adsorbent materials are commercially available. They have been used for the analysis of VOCs, particularly in air analysis, in combination with direct thermal desorption, dynamic headspace or purge and trap. In-tube extraction needles, as well as needle traps, can be filled with such materials, which results in a huge variety of different sorptive materials for different applications. Multi-stage adsorbent traps consisting of a combination of layered sorbents further increase the number of available extractants and are often suited for analysis of target analytes with widely differing physico-chemical properties. The sorbents used for sorptive enrichment in combination with thermal desorption should meet the following criteria [99]:
-
Complete enrichment of the analytes of interest
-
Complete and fast desorption of the analytes
-
Homogenous and inert surface to avoid artefact formation, irreversible adsorption and catalytic effects during sampling, if applicable storage of the loaded adsorbent tubes, and desorption
-
Low affinity to water to avoid displacement and hydrolysis reactions and to minimise deterioration of the gas chromatographic analysis
-
Low adsorption capacity for other inorganic constituents of air such as N2O, SO2, CO2 and ozone.
-
High inertness against reactive species such as ozone
-
High mechanical and thermal stability
-
Multiple usability for a variety of compounds
Basically, such sorbents can be classified in three groups: (1) inorganic materials, (2) carbon-based adsorbents and (3) organic polymers. The first group comprises materials such as silica gel, zeolites or alumina which have been more and more replaced by organic sorbents [99].
In Table 11.1 an overview of the most frequently used organic sorption materials is presented.
Carbon-based adsorbents can be sub-classified into activated carbon, carbon molecular sieves and graphitised carbon blacks. Activated carbons are micro-porous carbon materials with a broad pore size distribution and high specific surface areas (800–1,500 m2 g−1). Activated carbons have a chemically heterogeneous surface with mineral admixtures and several functional groups, such as hydroxyl, carbonyl and carboxylic functions. Enrichment is, therefore, caused by non-specific and specific H-acceptor or H-donor interactions. Due to this, polar analytes, for example alcohols, could be irreversibly adsorbed and even thermal desorption may be insufficient to remove such compounds. Owing to its functional groups, water is strongly retained on activated carbon than on other carbon adsorbent materials with specific retention volumes that may exceed 1,400 mL g−1 [105]. Although activated carbons are thermally stable materials and thermal desorption is possible the maximum applicable temperature is lower than for the other carbon-based materials.
Carbon molecular sieves are mainly produced by pyrolysis of organic polymers, e.g. poly(vinylidene chloride), poly(vinyl chloride) and sulphonated polymers. Their main representatives are Carboxen, Carbosphere, Carbosieve and Ambersorb materials. As molecular sieves, they have a well-defined micro-porous structure with a sharp pore size distribution and a high specific surface area. Adsorption to these materials is mainly based on non-specific van der Waals interactions. Although carbon molecular sieves belong to the non-polar adsorbents they adsorb substantial amounts of water, which could be explained by a condensation of water in the micropores. The carbon molecular sieves are distributed as spherical particles and high temperature stability makes them ideal for thermal desorption. As shown in Table 11.1, carbon molecular sieves are superior for enrichment of small molecules in the range of C2–C5. Because of this property, they are used in mixed-bed adsorbents behind weaker sorbents to prevent breakthrough of such small molecular compounds. Strongly sorbing compounds are then retained on the weaker sorbent and thus can be easier thermally desorbed, whereas the C2–C5 compounds will not be fully retained by the weak sorbent but be trapped by the molecular sieve and will be thermally desorbed at moderate temperatures.
Graphitised carbon blacks are produced from petroleum or natural gas soot, in an inert atmosphere at temperatures of about 2,700–3,000 °C. They are available under the names Carbotrap, Carbopack and Carbograph. With a higher degree of graphitisation, the specific surface area of the material, which varies between 5 and 260 m2 g−1, decreases. They are supplied as fine-grained powders which tend to form agglomerates and are of low mechanical stability. Graphitised carbon blacks for analytical purposes are non-polar adsorbents with a physically and chemically homogeneous surface and adsorption takes place on the basal planes of the graphite crystallites by non-specific interactions, whereby the shape and degree of polarisation determine the adsorption strength. Due to their high hydrophobicity, graphitised carbon blacks can be used for the sampling of VOCs in extremely humidic atmosphere.
The third group are porous organic polymers that belong to a large group of adsorbents with different surface areas and polarities. Porous organic polymers such as Chromosorb 101, 102, 106, Porapack P, HayeSep P and XAD-2 are all polystyrene-based materials. A major drawback of these adsorbents is their limited temperature stability which makes them less compatible with thermal desorption. Chromosorb 106 is the most frequently used sorbent out of this group but has the disadvantage that it is not applicable for trace analysis due to high background levels. For VOC analysis, Tenax, a material based on poly-(2,6-diphenyl-p-phenylene oxide), is the most important porous polymer. Nowadays, the high-purity version TenaxTA has replaced Tenax because of lower polymer bleeding that reduces background levels. A mixture of TenaxTA and graphitised carbon black called TenaxGR is also available, combining the advantages of both materials. Tenax is a very hydrophobic material, which is characterised by a high thermal stability. It has a low specific surface area (30 m2 g−1) and it is not suitable for sampling of highly volatile organics with carbon numbers lower than four.
To facilitate the choice of an adsorbent material, one can utilise a data collection on breakthrough volumes of about 200 compounds for different technical sorbents, as a function of temperature that is available on the WWW [106].
3.3 Liquid Polymer Extraction Phases
Liquid polymer extraction phases are mainly applied as coated extraction phases, such as used in SPME, SBSE and SPDE, or as bulk phase material in silicone rod, silicone tube or gum-phase extraction. As already mentioned, partitioning is the main extraction mechanism, and due to the weaker interactions compared to adsorption, mild thermal desorption conditions can be used. Therefore thermally induced decomposition is minimised.
The most widely used absorption material for non-polar compounds is polydimethylsiloxane (PDMS). It is inert and shows no irreversible adsorption and catalytic reactions. Water is not retained by PDMS, but the degradation products of PDMS can interfere with target analytes if non-specific detectors (e.g. FID, TCD, etc.) are used. With a mass selective detector, characteristic PDMS fragments can easily be identified and excluded. PDMS can withstand temperatures up to 300 °C.
Polyacrylate (PA) is suitable for more polar semi-volatile compounds, such as phenols and aliphatic amines. At room temperature, PA is a low-density solid polymer, which allows analyte diffusion into the coating.
Polyethylene glycol (PEG, Wax, Carbowax) is also a solid polymer at room temperature and can be used for polar compounds such as alcohols. Polyethylene glycol can dissolve substantial amounts of water and can form stable trihydrates [107], which changes the polarity of the extraction phase and lowers the extraction ability. PEG is not useable for high desorption temperatures, even in inert atmospheres, as pyrolysis at temperatures above 150 °C has been observed [108, 109]. A random scission of the main chain occurs already at temperatures around 80 °C in air [110].
Mixed phase coatings, with embedded adsorbent particles in the liquid extraction phase such as Carboxen PDMS (CAR/PDMS) have been designed for extraction of small, low molecular weight analytes at trace levels. The high porosity of Carboxen 1006 enables the fibre to strongly retain small analytes combined with high analyte capacity. A selection of the available extraction phases for SPME is given in Table 11.2.
Finally, a large number of laboratory-made extraction phases have been described in the literature. Some of these approaches use sol–gel reactions to modify the extraction phases to resist high pH values, water and/or higher desorption temperatures and to provide stability against frictional ripping during the extraction, especially when liquid samples are extracted directly. The number of these approaches is huge and would exceed the scope of this chapter; therefore we refer to more specialised literature [111].
3.4 Abraham Solvation Parameter Model
The Abraham solvation parameter model is one of the most useful approaches for the analysis and prediction of partitioning and sorption coefficients and thus can be used as a tool in the selection of appropriate extraction phases. The model tries to capture differences of all relevant molecular interactions of an analyte in two phases that determine the partitioning between these phases. To that end, five compound descriptors are used that are combined with complementary system descriptors specific for each pair of phases in order to predict the log K es. Such combinations are called linear solvation energy relationships (LSER) or polyparameter linear free energy relationships (pp-LFER). Abraham and co-workers have developed and refined the model most frequently used today. They set up two different equations for partitioning between two condensed phases [Eq. (11.20)] and between a gas phase and a condensed phase [Eq. (11.21)]:
Recently, it was shown by Goss [112] that equally good predictions of partition constants can be achieved by a unified pp-LFER of the form:
The capital letters in these equations are the compound descriptors; the small letters represent the complementary system descriptors. E is the excess molar refractivity in (cm3 mol−1)/10, which can be derived from the solute refractive index. S is the solute’s dipolarity/polarisability descriptor. A and B are measures of the solute’s hydrogen-bond acidity and hydrogen-bond basicity, respectively. V is the McGowan volume of the solute in (cm3 mol−1)/100 and L is the logarithm of the hexadecane–air partition constant at 298 K. The system descriptors quantify the differences in the abilities of the two phases to interact via these interactions with the solute. In this context they describe the sorbent or coating properties in contact with either a gaseous or an aqueous phase. Note that system descriptors are totally different for a sorbent in contact with air or water since the differences in the interactions in the two phases are captured. In order to integrate temperature dependence into this approach, one can establish pp-LFERs for different temperatures treating the system descriptors as temperature dependent and the compound descriptors as temperature independent [98].
Generally, system descriptors exhibit a steady trend with temperature, which facilitates safe inter- and extrapolation to other temperatures. A disadvantage of this method is that a complete calibration data set for every temperature is required. Sprunger et al. suggested a method for combining experimental partitioning coefficients from different temperatures into a single pp-LFER regression that contains temperature as a separate parameter and provided system descriptors for log K PDMS/water and K PDMS/headspace based on experimental partitioning data for 169 compounds [113].
Schneider and Goss combined the approach of Sprunger et al. in combination with the modified Abraham equation by Goss, whereby a smaller set of experimental calibration compounds is used than would be needed for calibration for different temperatures. Moreover, Schneider and Goss provided sorbent descriptors for the materials Tenax TA, Chromosorb 106, Porapak N and Carbopack F commonly used in trapping analytes from the gas phase [98]. They investigated 200 compounds at temperatures between 40 and 250 °C for these four sorbents. Since the 50 % breakthrough volume BTV50 % equals K es the pp-LFER model can also be used to predict breakthrough volumes. A calculator for the determination of BTV50 % is provided online by Schneider and Goss at free of charge [114]. Recently, this group has also established a pp-LFER for partitioning between the frequently used SPME coating polyacrylate and water at 25 °C derived from the literature data. In addition, to mimic biologically relevant situations, water was replaced by phosphate-buffered saline solution and temperature raised to 37 °C. However, it was found that neither the small temperature increase nor the salt addition significantly changed the resulting partition constants [115]. The comparison of the original Abraham pp-LFER and the unified Goss pp-LFER resulted in equally good fits and very similar system descriptors.
Table 11.3 comprises pp-LFERs published to date for sorbents of interest in solventless extraction methods. The direct comparison of PDMS–water and PA–water descriptors shows particularly strong differences in the a parameter. This is due to the strong H-bond accepting properties of PA that are nearly equal to water, whereas PDMS is a much weaker H-bond acceptor. As a consequence, H-bond donating organic compounds such as all bipolar compounds and H-bond acidic halocarbons are extracted to a much higher extent in PA than in PDMS. In contrast, apolar and monopolar H-acceptor compounds (carbonyl compounds, ethers) are extracted equally well with both phases. A second difference in system descriptors is the less negative s value for PA. This leads to higher extraction yields using PA for compounds with large S values such as PAHs and is a good example of the failure of the traditional “like-dissolves-like” concept, since this concept would suggest a higher recovery for non-polar PAHs by the non-polar PDMS than by the more polar PA.
4 Conclusions and Outlook
Automated solventless extraction techniques have matured over the past decades and are nowadays available for routine GC-based analysis of almost all typical volatile and semi-volatile compounds. In the coming years, such methods surely will also be included to a larger extent in standardisation efforts, thereby replacing more and more outdated liquid–liquid extraction methods. Although researchers constantly develop new formats of microextraction techniques many of these are just minor adaptations of already existing approaches. Fundamental new developments are not on the horizon. More research needs are in the further development of predictive tools for selection of appropriate methods, extraction materials and operational parameters, design of experiment approaches to minimise remaining experimental efforts and integration of such tools in software platforms to facilitate a rapid optimisation of analytical methods that is currently still dominated by mere trial-and-error approaches.
References
Smith RM (2003) Before the injection – modern methods of sample preparation for separation techniques. J Chromatogr A 1000(1–2):3–27
de Koning S, Janssen HG, Brinkman UAT (2009) Modern methods of sample preparation for GC analysis. Chromatographia 69:S33–S78. doi:10.1365/s10337-008-0937-3
Jönsson JA, Mathiasson L (2000) Membrane-based techniques for sample enrichment. J Chromatogr A 902:205–225
Pawliszyn J (2002) Sampling and sample preparation for field and laboratory : fundamentals and new directions in sample preparation. Elsevier, Amsterdam
Pawliszyn J (2003) Sample preparation: Quo Vadis? Anal Chem 75(11):2543–2558
Noble D (1993) Here today, gone tomorrow halogenated solvents in analytical chemistry. Anal Chem 65(15):693–695A
Liska I (2000) Fifty years of solid-phase extraction in water analysis – historical development and overview. J Chromatogr A 885(1–2):3–16
Hennion M-C (1999) Solid-phase extraction: method development, sorbents, and coupling with liquid chromatography. J Chromatogr A 856:3–54
Fritz JS (1999) Solid-phase extraction. Wiley, New York
Saito Y, Jinno K (2002) On-line coupling of miniaturized solid-phase extraction and microcolumn liquid-phase separations. Anal Bioanal Chem 373(6):325–331
Saito Y, Jinno K (2003) Miniaturized sample preparation combined with liquid phase separations. J Chromatogr A 1000(1–2):53–67
O’Reilly J, Wang O, Setkova L, Hutchinson JP, Chen Y, Lord HL, Linton CM, Pawliszyn J (2005) Automation of solid-phase microextraction. J Sep Sci 28(15):2010–2022
David F, Van Hoeck E, Sandra P (2007) Towards automated, miniaturized and solvent-free sample preparation methods. Anal Bioanal Chem 387(1):141–144
Baltussen E, Cramers CA, Sandra PJF (2002) Sorptive sample preparation – a review. Anal Bioanal Chem 373(1–2):3–22
Nerin C, Salafranca J, Aznar M, Batlle R (2009) Critical review on recent developments in solventless techniques for extraction of analytes. Anal Bioanal Chem 393(3):809–833
He Y, Lee HK (1997) Liquid-phase microextraction in a single drop of organic solvent by using a conventional microsyringe. Anal Chem 69(22):4634–4640
Liu H, Dasgupta PK (1996) Analytical chemistry in a drop. Solvent extraction in a microdrop. Anal Chem 68(11):1817–1821
Rezaee M, Assadi Y, Milani Hosseini M-R, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid-liquid microextraction. J Chromatogr A 1116(1–2):1–9
Kolb B (1980) Applied headspace gas chromatography. Heyden, London
Kolb B, Ettre LS (2006) Static headspace-gas chromatography : theory and practice, 2nd edn. Wiley, Hoboken, NJ
Snow NH, Slack GC (2002) Head-space analysis in modern gas chromatography. TrAC Trends Anal Chem 21(9–10):608–617
Kolb B, Ettre LS (1991) Theory and practice of multiphase extraction. Chromatographia 32(11–12):505–513
Voice TC, Kolb B (1993) Static and dynamic headspace analysis of volatile organic compounds in soils. Environ Sci Technol 27(4):709–713
Wasserbeschaffenheit-Gaschromatographische Bestimmung einer Anzahl monocyclischer aromatischer Kohlenwasserstoffe, Naphthalin und einiger chlorierter Substanzen mittels Purge und Trap-Anreicherung und thermischer Desorption (ISO 15680:2003); Deutsche Fassung EN ISO 15680:2003 (DIN-EN ISO Method 15680: 2004)
Voice TC, Kolb B (1994) Comparison of European and American techniques for the analysis of volatile organic-compounds in environmental matrices. J Chromatogr Sci 32(8):306–311
Martínez E, Lacorte S, Llobet I, Viana P, Barceló D (2002) Multicomponent analysis of volatile organic compounds in water by automated purge and trap coupled to gas chromatography-mass spectrometry. J Chromatogr A 959(1–2):181–190
Woolfenden E (2010) Sorbent-based sampling methods for volatile and semi-volatile organic compounds in air Part 1: Sorbent-based air monitoring options. J Chromatogr A 1217(16):2674–2684
Demeestere K, Dewulf J, De Witte B, Van Langenhove H (2007) Sample preparation for the analysis of volatile organic compounds in air and water matrices. J Chromatogr A 1153(1–2):130–144
Woolfenden E (2010) Sorbent-based sampling methods for volatile and semi-volatile organic compounds in air Part 2: Sorbent selection and other aspects of optimizing air monitoring methods. J Chromatogr A 1217:2685–2694
Vreuls JJ, De Jong GJ, Ghijsen RT, Brinkman UAT (1993) On-line solid-phase extraction/thermal desorption for introduction of large volumes of aqueous samples into a capillary gas chromatograph: Part 2. J Microcolumn Sep 5(4):317–324
Vreuls JJ, Louter AJH, Brinkman UAT (1999) On-line combination of aqueous-sample preparation and capillary gas chromatography. J Chromatogr A 856(1–2):279–314
Mol HGJ, Janssen HGM, Cramers CA, Brinkman UAT (1993) Use of a temperature-programmed injector with a packed liner for direct water analysis and online reversed-phase GC-LC. J High Resolut Chromatogr 16(8):459–463
Teske J, Efer J, Engewald W (1997) Large-volume PTV injection: new results on direct injection of water samples in GC analysis. Chromatographia 46(11–12):580–586
Teske J, Efer J, Engewald W (1998) Large-volume PTV injection: comparison of direct water injection and in-vial extraction for GC analysis of triazines. Chromatographia 47(1–2):35–41
Müller RK, Grosse J, Thieme D, Lang R, Teske J, Trauer H (1999) Introduction to the application of capillary gas chromatography of performance-enhancing drugs in doping control. J Chromatogr A 843(1–2):275–285
Engewald W, Teske J, Efer J (1999) Programmed temperature vaporisers-based large volume injection in capillary gas chromatography. J Chromatogr A 842(1–2):143–161
Engewald W, Teske J, Efer J (1999) Programmed temperature vaporiser-based injection in capillary gas chromatography. J Chromatogr A 856(1–2):259–278
Teske J, Engewald W (2002) Methods for, and applications of, large-volume injection in capillary gas chromatography. TrAC Trends Anal Chem 21(9–10):584–593
Baltussen E, David F, Sandra P, Janssen HG, Cramers C (1999) Equilibrium sorptive enrichment on poly(dimethylsiloxane) particles for trace analysis of volatile compounds in gaseous samples. Anal Chem 71(22):5193–5198
Baltussen E, David F, Sandra P, Janssen HG, Cramers CA (1998) Sorption tubes packed with polydimethylsiloxane: a new and promising technique for the preconcentration of volatiles and semi-volatiles from air and gaseous samples. HRC J High Resolut Chromatogr 21(6):332–340
de Koning JA, Blokker P, Jungel P, Alkema G, Brinkman UAT (2002) Automated liner exchange – a novel approach in direct thermal desorption – gas chromatography. Chromatographia 56(3–4):185–190
Lord HL, Pawliszyn J (1998) Recent advances in solid-phase microextraction. LC GC Mag of Separ Sci 41–46
Quintana JB, Rodríguez I (2006) Strategies for the microextraction of polar organic contaminants in water samples. Anal Bioanal Chem 384:1447–1461
Mayer P, Tolls J, Hermens L, Mackay D (2003) Equilibrium sampling devices. Environ Sci Technol 37(9):184A–191A
Arthur CL, Killam LM, Buchholz KD, Pawliszyn J, Berg JR (1992) Automation and optimization of solid-phase microextraction. Anal Chem 64:1960–1966
Lord H, Pawliszyn J (2000) Evolution of solid-phase microextraction technology. J Chromatogr A 885(1–2):153–193
David F, Sandra P (2007) Stir bar sorptive extraction for trace analysis. J Chromatogr A 1152(1–2):54–69
van Pinxteren M, Paschke A, Popp P (2010) Silicone rod and silicone tube sorptive extraction. J Chromatogr A 1217(16):2589–2598
Grob K, Habich A (1985) Headspace gas analysis: the role and the design of concentration traps specifically suitable for capillary gas chromatography. J Chromatogr A 321:45–58
Burger BV, Munro Z (1986) Headspace gas analysis: quantitative trapping and thermal desorption of volatiles using fused-silica open tubular capillary traps. J Chromatogr A 370:449–464
Mol HGJ, Janssen H-GM, Cramers CA, Vreuls JJ, Brinkman UAT (1995) Trace level analysis of micropollutants in aqueous samples using gas chromatography with on-line sample enrichment and large volume injection. J Chromatogr A 703(1–2):277–307
Tienpont B, David F, Bicchi C, Sandra P (2000) High capacity headspace sorptive extraction. J Microcolumn Sep 12(11):577–584
Kataoka H (2002) Automated sample preparation using in-tube solid-phase microextraction and its application – a review. Anal Bioanal Chem 373(1–2):31–45
Nardi L (2002) Coupled in-tube SPME-HRGC: a complementary SPME technique to analyze aqueous samples by GC. Am Lab 34(1):30–37
Nardi L (2003) Guidelines for capillary extraction-capillary gas chromatography: preparation of extractors and analysis of aromatic compounds in water. J Chromatogr A 1017(1–2):1–15
Belardi RP, Pawliszyn JB (1989) The application of chemically modified fused silica fibers in the extraction of organics from water matrix samples and their rapid transfer to capillary columns. Water Pollut Res J Can 24(1):179–191
Anonymous (2006) German standard methods for the examination of water, waste water and sludge – Jointly determinable substances (group F) – Part 34: Determination of selected plant treatment agents, biocides and break-down products; Method using gas chromatography (GC-MS) after solid-phase micro extraction (SPME) (F 34), vol 38407-34. Beuth, Berlin
Anonymous (2009) German standard methods for the examination of water, waste water and sludge – Jointly determinable substances (group F) – Part 41: Determination of selected easily volatile organic compounds in water – Method using gas chromatography (GC-MS) after solid-phase micro extraction (SPME), vol 38407-41. Beuth, Berlin
Anonymous (2007) Parent and alkyl polycyclic aromatics in sediment pore water by solid-phase microextraction and gas chromatography/mass spectrometry in selected ion monitoring mode, vol Method 8272. Washington, DC
Wang H, Liu W, Guan Y (2004) In-tube solid-phase microextraction and on-line coupling with high-resolution GC. LC-GC Eur 17(3):144–151
Bigham S, Medlar J, Kabir A, Shende C, Alli A, Malik A (2002) Sol-gel capillary microextraction. Anal Chem 74(4):752–761
Pawliszyn J (1997) Solid phase microextraction: theory and practice. Wiley-VCH, New York
Pawliszyn J (1999) Applications of solid phase microextraction. Royal Society of Chemistry, Cambridge
Eisert R, Levsen K (1996) Solid-phase microextraction coupled to gas chromatography: a new method for the analysis of organics in water. J Chromatogr A 733(1–2):143–157
Alpendurada MF (2000) Solid-phase microextraction: a promising technique for sample preparation in environmental analysis. J Chromatogr A 889(1–2):3–14
Risticevic S, Niri VH, Vuckovic D, Pawliszyn J (2009) Recent developments in solid-phase microextraction. Anal Bioanal Chem 393(3):781–795
Spietelun A, Kloskowski A, Chrzanowski W, Ni J (2013) Understanding solid-phase microextraction: key factors influencing the extraction process and trends in improving the technique. Chem Rev 113:1667–1685
Bicchi C, Iori C, Rubiolo P, Sandra P (2002) Headspace sorptive extraction (HSSE), stir bar sorptive extraction (SBSE), and solid phase microextraction (SPME) applied to the analysis of roasted Arabica coffee and coffee brew. J Agric Food Chem 50(3):449–459
Kataoka H (2005) Recent advances in solid-phase microextraction and related techniques for pharmaceutical and biomedical analysis. Curr Pharm Anal 1:65–84
Bicchi C, Cordero C, Liberto E, Rubiolo P, Sgorbini B, David F, Sandra P (2005) Dual-phase twisters: a new approach to headspace sorptive extraction and stir bar sorptive extraction. J Chromatogr A 1094(1–2):9–16
Montero L, Popp P, Paschke A, Pawliszyn J (2004) Polydimethylsiloxane rod extraction, a novel technique for the determination of organic micropollutants in water samples by thermal desorption-capillary gas chromatography-mass spectrometry. J Chromatogr A 1025(1):17–26
Baltussen E, Sandra P, David F, Cramers C (1999) Stir bar sorptive extraction (SBSE), a novel extraction technique for aqueous samples: theory and principles. J Microcolumn Sep 11(10):737–747
Prieto A, Basauri O, Rodil R, Usobiaga A, Fernandez LA, Etxebarria N, Zuloaga O (2010) Stir-bar sorptive extraction: a view on method optimisation, novel applications, limitations and potential solutions. J Chromatogr A 1217(16):2642–2666
Lancas FM, Queiroz MEC, Grossi P, Olivares IRB (2009) Recent developments and applications of stir bar sorptive extraction. J Sep Sci 32(5–6):813–824
Sanchez-Rojas F, Bosch-Ojeda C, Cano-Pavon JM (2009) A review of stir bar sorptive extraction. Chromatographia 69:S79–S94
McComb ME, Oleschuk RD, Giller E, Gesser HD (1997) Microextraction of volatile organic compounds using the inside needle capillary adsorption trap (INCAT) device. Talanta 44:2137–2143
Shojania S, McComb ME, Perreault H, Gesser HD, Chow A, Oleschuk RD (1999) Qualitative analysis of complex mixtures of VOCs using the inside needle capillary adsorption trap. Can J Chem 77(11):1716–1727
Shojania S, Oleschuk RD, McComb ME, Gesser HD, Chow A (1999) The active and passive sampling of benzene, toluene, ethyl benzene and xylenes compounds using the inside needle capillary adsorption trap device. Talanta 50(1):193–205
Lachenmeier DW, Kroener L, Musshoff F, Madea B (2003) Application of tandem mass spectrometry combined with gas chromatography and headspace solid-phase dynamic extraction for the determination of drugs of abuse in hair samples. Rapid Commun Mass Spectrom 17(5):472–478
Sieg K, Fries E, Püttmann W (2008) Analysis of benzene, toluene, ethylbenzene, xylenes and n-aldehydes in melted snow water via solid-phase dynamic extraction combined with gas chromatography/mass spectrometry. J Chromatogr A 1178(1–2):178–186
Musshoff F, Lachenmeier DW, Kroener L, Madea B (2002) Automated headspace solid-phase dynamic extraction for the determination of amphetamines and synthetic designer drugs in hair samples. J Chromatogr A 958(1–2):231–238
Bicchi C, Cordero C, Liberto E, Rubiolo P, Sgorbini B (2004) Automated headspace solid-phase dynamic extraction to analyse the volatile fraction of food matrices. J Chromatogr A 1024(1–2):217–226
Van Durme J, Demeestere K, Dewulf J, Ronsse F, Braeckman L, Pieters J, Van Langenhove H (2007) Accelerated solid-phase dynamic extraction of toluene from air. J Chromatogr A 1175(2):145–153
Kubinec R, Berezkin VG, Gorova R, Addova G, Mracnova H, Sojak L (2004) Needle concentrator for gas chromatographic determination of BTEX in aqueous samples. J Chromatogr B Analyt Technol Biomed Life Sci 800(1–2):295–301
Jochmann MA, Yuan X, Schilling B, Schmidt TC (2008) In-tube extraction for enrichment of volatile organic hydrocarbons from aqueous samples. J Chromatogr A 1179(2):96–105
Laaks J, Jochmann MA, Schilling B, Schmidt TC (2010) In-tube extraction of volatile organic compounds from aqueous samples: an economical alternative to purge and trap enrichment. Anal Chem 82(18):7641–7648
Wang AP, Fang F, Pawliszyn J (2005) Sampling and determination of volatile organic compounds with needle trap devices. J Chromatogr A 1072(1):127–135
Gong Y, Eom IY, Lou DW, Hein D, Pawliszyn J (2008) Development and application of a needle trap device for time-weighted average diffusive sampling. Anal Chem 80(19):7275–7282
Mieth M, Kischkel S, Schubert JK, Hein D, Miekisch W (2009) Multibed needle trap devices for on site sampling and preconcentration of volatile breath biomarkers. Anal Chem 81(14):5851–5857
Mieth M, Schubert JK, Groger T, Sabel B, Kischkel S, Fuchs P, Hein D, Zimmermann R, Miekisch W (2010) Automated needle trap heart-cut GC/MS and needle trap comprehensive two-dimensional GC/TOF-MS for breath gas analysis in the clinical environment. Anal Chem 82(6):2541–2551
Saito Y, Nakao Y, Imaizumi M, Takeichi T, Kiso Y, Jinno K (2000) Fiber-in-tube solid-phase microextraction: a fibrous rigid-rod heterocyclic polymer as the extraction medium. Fresenius J Anal Chem 368(7):641–643
Kolb B (1999) Headspace sampling with capillary columns. J Chromatogr A 842(1–2):163–205
Staudinger J, Roberts PV (1996) A critical review of Henry’s law constants for environmental applications. Crit Rev Environ Sci Technol 26(3):205–297
Staudinger J, Roberts PV (2001) A critical compilation of Henry’s law constant temperature dependence relations for organic compounds in dilute aqueous solutions. Chemosphere 44(4):561–576
Robbins GA, Wang S, Stuart JD (1993) Using the static headspace method to determine Henry’s law constants. Anal Chem 65(21):3113–3118
Heringa MB, Hermens JLM (2003) Measurement of free concentrations using negligible depletion-solid phase microextraction (nd-SPME). TrAC Trends Anal Chem 22(10):575–587
Ramos EU, Meijer SN, Vaes WHJ, Verhaar HJM, Hermens JLM (1998) Using solid-phase microextraction to determine partition coefficients to humic acids and bioavailable concentrations of hydrophobic chemicals. Environ Sci Technol 32(21):3430–3435
Schneider M, Goss KU (2009) Systematic investigation of the sorption properties of Tenax TA, Chromosorb 106, Porapak N, and Carbopak F. Anal Chem 81(8):3017–3021
Dettmer K, Engewald W (2002) Adsorbent materials commonly used in air analysis for adsorptive enrichment and thermal desorption of volatile organic compounds. Anal Bioanal Chem 373(6):490–500
Lovkvist P, Jonsson JA (1987) Capacity of sampling and preconcentration columns with a low number of theoretical plates. Anal Chem 59(6):818–821
Rabe S, Krings U, Berger RG (2003) Initial dynamic flavour release from sodium chloride solutions. Eur Food Res Technol 218(1):32–39
Schwarzenbach RP, Gschwend PM, Imboden DM (2003) Environmental organic chemistry, 2nd edn. Wiley, New York
Lou DW, Lee X, Pawliszyn J (2008) Extraction of formic and acetic acids from aqueous solution by dynamic headspace-needle trap extraction. Temperature and pH optimization. J Chromatogr A 1201(2):228–234
Zwank L, Berg M, Schmidt TC, Haderlein SB (2003) Compound-specific carbon isotope analysis of volatile organic compounds in the low-microgram per liter range. Anal Chem 75(20):5575–5583
Dettmer K, Engewald W (2003) Ambient air analysis of volatile organic compounds using adsorptive enrichment. Chromatographia 57:S339–S347
Graham NB, Zulfiqar M, Nwachuku NE, Rashid A (1990) Interaction of poly(ethylene oxide) with solvents: 4. Interaction of water with poly(ethylene oxide) crosslinked hydrogels. Polymer 31(5):909–916
Lattimer RP (2000) Mass spectral analysis of low-temperature pyrolysis products from poly(ethylene glycol). J Anal Appl Pyrolysis 56(1):61–78
Voorhees KJ, Baugh SF, Stevenson DN (1994) An investigation of the thermal-degradation of poly(ethylene glycol). J Anal Appl Pyrolysis 30(1):47–57
Han S, Kim C, Kwon D (1995) Thermal degradation of poly(ethyleneglycol). Polym Degrad Stab 47(2):203–208
Dietz C, Sanz J, Camara C (2006) Recent developments in solid-phase microextraction coatings and related techniques. J Chromatogr A 1103(2):183–192
Goss KU (2005) Comment on “free energy of transfer of a solute and its relation to the partition constant” – Reply. J Phys Chem B 109(37):17770–17770
Sprunger L, Proctor A, Acree WE, Abraham MH (2007) Characterization of the sorption of gaseous and organic solutes onto polydimethyl siloxane solid-phase microextraction surfaces using the Abraham model. J Chromatogr A 1175(2):162–173
Endo S, Droge STJ, Goss K-U (2011) Polyparameter linear free energy models for polyacrylate fiber-water partition coefficients to evaluate the efficiency of solid-phase microextraction. Anal Chem 83:1394–1400
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Glossary of Acronyms in Extraction and Injection
Glossary of Acronyms in Extraction and Injection
- ASPDE:
-
Accelerated solid-phase dynamic extraction
- CME:
-
Capillary microextraction
- DAI:
-
Direct aqueous injection
- DI:
-
Direct immersion
- DTD:
-
Direct thermal desorption
- ESD:
-
Equilibrium sampling device
- GPE:
-
Gum-phase extraction
- HS:
-
Headspace
- HSSE:
-
Headspace sorptive extraction
- INCAT:
-
Inside needle capillary adsorption trap
- In-tube SPME/ITSPME:
-
In-tube solid-phase microextraction
- ITE:
-
In-tube extraction
- ITEX:
-
In-tube extraction device
- LLE:
-
Liquid–liquid extraction
- LVI:
-
Large volume injection
- Micro-SPE:
-
Micro solid-phase extraction
- NT:
-
Needle trap
- OTME:
-
Open-tubular microextraction
- OTT:
-
Open-tubular trapping
- P&T:
-
Purge and trap
- SBSE:
-
Stir-bar sorptive extraction
- S-HS:
-
Static headspace
- SPDE:
-
Solid-phase dynamic extraction
- SPE:
-
Solid-phase extraction
- SPME:
-
Solid-phase microextraction
- SR:
-
Silicone rod
- ST:
-
Silicone tube
- TD:
-
Thermal desorption
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Jochmann, M.A., Laaks, J., Schmidt, T.C. (2014). Solvent-Free Extraction and Injection Techniques. In: Dettmer-Wilde, K., Engewald, W. (eds) Practical Gas Chromatography. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-54640-2_11
Download citation
DOI: https://doi.org/10.1007/978-3-642-54640-2_11
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-54639-6
Online ISBN: 978-3-642-54640-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)