
Abstract
Atopic diseases are characterized by elevated levels of IgE and classically described as Th2 mediated. However, during the last years it has become clear that several other players also are important for the immune responses during atopic dermatitis. Among these are epithelial cells, innate lymphoid cells (ILCs), Th17 cells, and vitamin D. This chapter focuses on the role of these during the immune response to allergens and how the lack of filaggrin affects these mechanisms. Both human and mice studies are discussed because much of the knowledge about the effects of filaggrin on the immune system comes from studies using the Flaky tail mice (Flgft mice) lacking filaggrin.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Atopic Dermatitis
- Th17 Cell
- Experimental Autoimmune Encephalomyelitis
- Skin Inflammation
- Allergen Exposure
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
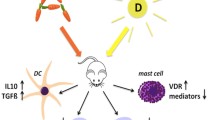
Atopic diseases are characterized by elevated levels of IgE and classically described as Th2 mediated. It is believed that following entry of an allergen into the skin, it is processed by local dendritic cells (DCs) that then migrate to the draining lymph node. Here, the DC presents the allergen for naïve CD4+ T cells, which subsequently differentiate into Th2 cells characterized by their production of IL-4, IL-5, and IL-13 (Fig. 5.1). The Th2 cells activate allergen-specific B cells and promote isotype switch to IgE. The allergen-specific IgE will bind and prime mast cells, which then can be activated following exposure to the allergen (see Fig. 5.1). During the last years it has become clear that several new players are involved in allergen-specific immune responses, among these the epithelial cells, innate lymphoid cells (ILCs), Th17 cells, and vitamin D. This chapter focuses on how filaggrin (or lack of) affects the immune response and vitamin D synthesis in the skin. Both human and mice studies are discussed as much of the knowledge about the effects of filaggrin on the immune system comes from studies using the Flaky tail mice (Flgft mice) lacking filaggrin [1–3] (Fig. 5.2).

Simple model for immune responses leading to AD. Skin exposure to allergens leads to activation of skin DC that migrates to the draining lymph nodes, where they present allergen for allergen-specific naïve CD4+ T cells. Due to the presence of IL-4, allergen-specific CD4+ T cells differentiate into Th2 cells. These subsequently activate allergen-specific B cells that differentiate into IgE-producing plasma cells. IgE bind to FcεR1 receptors on mast cells in the skin. Upon subsequent exposure to the allergen, IgE on the mast cells bind allergen and induce mast cell activation and thereby skin inflammation

Appearance of age-matched Flgft and WT mice (C57bl/6)
2 The T-Cell Response
Atopic dermatitis (AD) is a complex disease dependent on both genetic and environmental factors that induce a complex immune response. It is well known that T cells play a central role in the pathogenesis of AD [4, 5]. An AD mouse model showed that αβ T cells were required for skin inflammation, whereas γδ T cells and B cells were not required [5]. In addition, IL-4 expression was upregulated in inflamed skin and found to be produced by αβ T cells [5]. Interestingly, skin inflammation could be induced in mice lacking either B cells or CD40L indicating that IgE is not required for the development of skin inflammation [5].
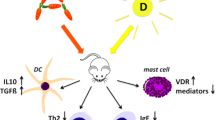
CD4+ T-cell differentiation is classically divided into Th1 and Th2 responses dominated by IFNγ, IL-2 and IL-4, IL-5 and IL-13, respectively (Fig. 5.3) [6, 7]. Recently, a new CD4+ T-cell subtype has been identified, namely, the Th17 cells (see below). A mouse model for AD-like skin inflammation was used to further investigate the role of Th1 and Th2 cells in AD. Impaired eosinophil recruitment to the skin was seen in mice lacking either IL-4 or IL-5, whereas IFNγ did not seem to be involved in eosinophil recruitment [8]. However, IFNγ seemed to be involved in the response by other mechanisms as reduced skin inflammation was seen in mice lacking either IL-5 or IFNγ [8]. In accordance with this, IL-4, IL-5, IL-13, IFNγ, and IL-12 are upregulated in the skin from AD patients compared to healthy skin [9, 10]. Interestingly, these cytokines appear to be involved at different stages of the disease. The initial inflammation seems to be driven by IL-4 and IL-13, as these are the dominating cytokines in acute lesional skin [9, 10]. In contrast, chronic inflammation seems to be maintained by IL-5, IL-12, and IFNγ [9, 10]. Increased eosinophil infiltration was seen in chronic compared to acute lesions, which correlates with the increased expression of IL-5 in chronic lesions [9]. Thus, the acute inflammation appears to be mediated by Th2 cells and their production of IL-4 and IL-13, whereas the chronic inflammation seems to involve both Th1 and Th2 cells.

Schematic representation of differentiation of ILC and naïve CD4+ T cells showing cytokines and transcription factor involved in the differentiation as well as effector cytokines produced by the cells
Exposure of the skin to protein allergens (ovalbumin (OVA), Dermatophagoides pteronyssinus (Derp1)), contact allergens, and irritants induces a more vigorous inflammatory response in Flgft mice than in control mice [1, 3]. The response to protein antigens seems to involve Th1, Th2, and Th17 cells, which correlates well with the findings in patients with AD (see below [1, 3, 9, 10]). Interestingly, only minor cytokine induction was seen following allergen exposure in control mice having an intact skin barrier [1, 3]. Thus, lack of filaggrin seems to increase the risk of developing allergen-specific T-cell responses mediated by Th1, Th2, and Th17 cells. To date very few studies have investigated how the lack of filaggrin affects T-cell responses in humans. However, by combining Derp1-specific tetramer staining together with IL-4 and filaggrin analysis, one study showed that individuals lacking filaggrin had an increased number of Derp1-specific IL-4-producing CD4+ positive cells in their blood compared to individual with wild-type filaggrin gene (FLG) mutation status [11]. Taken together, even though AD classically is described as mediated by Th2 cells, other effector CD4+ T cells seem to be important at different stages of the disease. Lack of filaggrin seems to increase the risk of developing allergen-specific T-cell responses, probably due to increased skin penetration of allergens that elicit a basic inflammatory response and thereby provides a reduced activation threshold for the T cells. In addition, a recent study shows that AD patients with FLG loss-of-function mutations had increased levels of IL-1α and IL-1β in the skin compared to both healthy controls and AD patients without FLG mutations [12]. Interestingly, the increased IL-1α and IL-1β correlated inversely with the “natural moisturizing factors” (NMFs) that correlated inversely with skin pH [12]. As IL-1 is a pro-inflammatory cytokine known to be involved in the initiation of the immune response in general (e.g., by inducing maturation and migration of DC), increased IL-1 levels are likely to lower the immune activation threshold within the skin of patients lacking filaggrin [13].
3 The IL-23/TH17 Axis in AD
Following the discovery that IL-23, and not IL-12, was required for the induction of experimental autoimmune encephalomyelitis (EAE), the mouse model of multiple sclerosis, intensive work was carried out to characterize the effector CD4+ T cells responding to IL-23, which eventually lead to identification of IL-17-producing CD4+ T cells (Th17) in 2005 [14–16]. It is now known that TGF-β and IL-6 are required for initiation of Th17 differentiation, whereas IL-23 is required for the stabilization of the Th17 cells (see Fig. 5.3) [7]. Th17 cells are involved in the pathogenesis of a variety of autoimmune and inflammatory diseases such as EAE, inflammatory bowel diseases, and psoriasis [17]. The primary cytokines produced by Th17 cells are IL-17 and IL-22, both of which stimulate epithelial cells to produce a variety of inflammatory cytokines (e.g., IL-1β, IL-6, TNFα), chemokines (e.g., CXCL8), and antimicrobial peptides (e.g., β-defensin, cathelicidin) [17]. Th17 cells most likely play a role in AD as an increased percentage of Th17 cells are found in the blood and lesional skin of AD patients [18, 19]. Interestingly, Th17 cells seem to serve as an initial cytokine source as they are more prevalent in acute than chronic lesions [18, 19]. Furthermore, Th17 cells are likely associated with the severity of AD as a direct correlation between severity of the inflammation and the percentage of Th17 cells in the blood has been found [18]. The role of Th17 cells in the immune response to protein allergen has been further investigated by using an OVA sensitization mouse model. Here mice were exposed to OVA either epicutaneously (EC) or intraperitoneally (IP) [20]. It was shown that EC OVA sensitization induced both a local and a systemic Th17 response, whereas IP OVA sensitization did not [20]. In contrast, the production of IL-4 and IFNγ following OVA sensitization seemed to be independent on the sensitization route [20]. The Th17 response also appears to drive airway inflammation as neutrophil influx and bronchial hyperactivity induced by OVA inhalation in EC-sensitized mice could be reversed by IL-17 blockade [20]. The reason why EC sensitization, in contrast to IP sensitization, leads to Th17 responses might be explained by the ability of skin-derived DC to produce IL-23, a feature that is not observed in splenic DC [20]. In this model, mice were tape-stripped before OVA exposure of the skin, a procedure that is known to induce disruption of the skin barrier. As EC sensitization with allergens seems to play an important role in allergen sensitization of patients with AD, and as patients lacking filaggrin have an increased risk of developing asthma [21, 22], it was suggested that allergen exposure of skin lacking filaggrin leads to a Th17 response, which upon later allergen exposure of the airways induces a Th17-dependent airway inflammation [20]. In agreement with this, three studies on Flgft mice have shown an increased IL-17 production in Flgft mice compared to control mice [1–3]. The increased IL-17 production was found both in the skin at steady state and in OVA-specific CD4+ T cells after EC OVA sensitization [1, 3]. Interestingly, a similar Th17 response was seen in Flgft mice and control mice following IP sensitization with OVA [3]. Taken together, allergen exposure of the skin seems to favor a Th17 response, and lack of filaggrin increases the risk of developing allergen-specific Th17 responses that again increases the risk of developing severe AD and asthma (Fig. 5.4).

Model for the role of IL-17 in initiation of AD
Even though Th17 cells were first described as the IL-17-producing cells, it is now clear that several other types of cells can produce IL-17, i.e., CD8+ T cells, γδ T cells, and ILC [3, 17, 23]. During the last years much focus has been on ILC. The ILC are characterized by lack of expression of markers associated with T cells, B cells, DCs, macrophages, and granulocytes [23]. Interestingly, it seems that ILCs can be subdivided based on transcription factors and cytokine production in a way similar to the CD4+ T effector cells (see Fig. 5.3). Thus, ILC1 are the innate analogs to Th1 cells, ILC2 are the innate analogs to Th2 cells, and, finally, LTi and ILC3 are the innate analogs to Th17 cells [7, 23]. A recent study indicated that cells other than Th17 cells might be responsible of the increased level of IL-17 found in the skin of Flgft mice at steady state [24]. In this study, Flgft mice were crossed to RAG2-deficient mice lacking both T and B cells [24]. Lesional skin inflammation characterized by fur loss, erythematous scaly skin, and periocular swelling was seen in 88 % of Flgft mice after 32 weeks. In contrast, no sign of skin inflammation was seen in mice lacking both filaggrin and RAG2, indicating that the adaptive immune response is required for the skin inflammation in Flgft mice [24]. However, increased levels of both IL-17A and IL-22 were found in the skin of RAG2−/− Flgft mice compared to mice only lacking RAG2, indicating that LTi and/or ILC3 might be involved in the increased level of IL-17A found in Flgft mice at steady state [24]. Thus, it is likely that ILC subtypes are involved in the inflammatory response induced by the lack of filaggrin; however, this needs further investigations (see Fig. 5.4).
Taken together, IL-17 is most likely produced by both Th17 cells and ILC in the inflammatory response observed in AD. Lack of filaggrin leads to an impaired barrier function and thereby probably to danger signals that activate skin DC to produce IL-23. IL-23 subsequently stimulates IL-17 production from ILC and Th17 cells. IL-17 in the skin stimulates keratinocytes to produce pro-inflammatory cytokines and chemokines that eventually lead to increased skin inflammation (see Fig. 5.4).
4 The Role of TSLP in the Response
Keratinocytes constitute the majority of the cells in the epidermis and were originally described mechanistically as the cells that form the physical barrier between the environment and the body. However, it has become clear that keratinocytes also play an important immune-modulating role due to their ability to produce a variety of cytokines (e.g., IL-1β, IL-23, TNFα, and IL-10 in response to pathogens, stress, and other environmental triggers) [25–28]. Cytokines produced by keratinocytes can modify the activation and differentiation of skin DC. Thymic stromal lymphopoietin (TSLP) mainly produced by keratinocytes, fibroblasts, and stromal cells can induce a Th2 response [29]. TSLP is highly expressed in the epidermis of patients with AD [30]. Stimulation of CD11c+ DC with TLSP in vitro leads to activation and differentiation of DC that promote Th2 differentiation (Fig. 5.5) [30]. By use of a transgenic mouse model, where TSLP specifically can be induced in the keratinocytes, it was shown that mice developed spontaneous AD characterized by skin inflammation, increased number of skin-homing Th2 cells, and elevated levels of serum IgE 2–3 weeks after TSLP induction [31]. Interestingly, induction of TSLP in TCRβΚΟ mice lacking all CD4+ and CD8+ αβ T cells still lead to skin inflammation, suggesting that T cells are not necessary for the induction of the allergic response [31]. It was suggested that the response could be induced by TSLP acting directly on activated macrophages, eosinophils, mast cells, and other myeloid effector cells [31]. However, ILC2 cells could also be involved. TSLP can induce cytokine production by ILC2 in the skin independent of IL-33 and IL-25 [32]. An increased frequency of ILC2 was found in lesional skin from patients with AD [32]. Furthermore, AD-like skin inflammation could be significantly reduced either by depleting ILC or by using TSLP receptor KO mice [32]. This indicates that TSLP might play an important role in the induction of AD by stimulating ILC2 to produce IL-5 and IL-13. In agreement with studies in human AD patients, TSLP was found to be more expressed in skin from Flgft mice than in control mice [33]. Furthermore, it was found that the expression and activity of the endogenous proteases kallikrein 5, 7, and 14, which activate TSLP production in keratinocytes, were higher in skin from Flgft mice compared to control mice in steady state [33]. It can, therefore, be suggested that the increased activity of the endogenous proteases caused by the lack of filaggrin leads to increased production of TSLP via the protease-activated receptor-2 in keratinocytes and that this plays an important role in the induction of both ILC2 and Th2 cells.

Model for the involvement of TSLP in the immune response during AD
5 Vitamin D, Filaggrin, and Immune Responses
Several studies have demonstrated that vitamin D regulates keratinocyte growth and differentiation and affects immune responses [34–39]. The major source of vitamin D for most humans is 7-dehydrocholesterol (7-DHC) in the plasma membrane of keratinocyte [40, 41]. The first stage of vitamin D synthesis depends on the UVB (280–320 nm)-mediated photoconversion of 7-DHC to previtamin D3 in the skin. Once formed, previtamin D3 is rapidly converted to vitamin D3 that diffuses to the blood circulation, where it is bound to the vitamin D-binding protein (DBP). Vitamin D3 is subsequently metabolized in the liver to 25-hydroxyvitamin D3 (25(OH)D3) and then in the kidney to its biologically active form 1,25-dihydroxyvitamin D3 (1,25(OH)2D3) [40, 42, 43]. 1,25(OH)2D3 is classically considered to function as a endocrine regulator of calcium homeostasis. However, the understanding of vitamin D metabolism and physiological function has evolved dramatically in recent years. Vitamin D is now recognized as a pleiotropic regulator of human physiology with emerging roles in several tissues including the immune system and the skin [44].
The biological actions of 1,25(OH)2D3 are mediated by the vitamin D receptor (VDR) that belongs to the nuclear hormone receptor superfamily [45, 46]. Interaction of 1,25(OH)2D3 with VDR induces heterodimerization with the retinoid X receptor (RXR) and translocation of 1,25(OH)2D3-VDR/RXR complexes into the nucleus [44, 47, 48]. The 1,25(OH)2D3-VDR/RXR complexes bind to specific DNA sequences called vitamin D response elements (VDREs) in target genes, and dependent on the recruited co-regulators either augment or inhibit transcription of the target gene [48–50]. Both keratinocytes and various cells of the immune system express VDR, especially after their activation [38, 51–54].
The normal range of the 1,25(OH)2D3 concentration in serum is 50–175 pM, whereas the concentration of the precursor 25(OH)D3 is approximately 1,000-fold higher (50–160 nM). The conversion of 25(OH)D3 to 1,25(OH)2D3 is mediated by the 1-α hydroxylase CYP27B1 [55]. This conversion was at first believed exclusively to take place in the kidneys; however, it is now clear that CYP27B1 is expressed in various cell types including keratinocytes, macrophages, and activated T cells [56–58], and evidence is rapidly accumulating that local CYP27B1-catalyzed production of 1,25(OH)2D3 is critical for its physiological actions [37, 59]. In this context, the keratinocytes are the only cell type where the complete enzymatic machinery for the synthesis of 1,25(OH)2D3 from 7-DHC has been shown [41, 60–62]. Thus, it can be assumed that UVB-induced production of vitamin D3 in the skin might result in formation of substantial amounts of local 1,25(OH)2D3, which regulate keratinocyte growth and differentiation and affect the local immune response.
The keratohyalin granules in the stratum granulosum of the epidermis consist primarily of pro-filaggrin polymers [63] that are proteolytically cleaved into filaggrin monomers. Monomeric filaggrin binds to keratin to form tight bundles facilitating the collapse and flattening of the cells in the stratum corneum [64]. Subsequently, filaggrin is fully degraded to its constituents amino acids dominated by glutamine, arginine, and histidine [65]. Histidine is a substrate for histidase that is highly expressed in the stratum granulosum [66]. Histidase converts histidine to urocanic acid (UCA) in the upper layers of the epidermis. UCA has been suggested to be an important UV photoprotectant as it has a high extinction coefficient in the wavelength range from 260 to 310 nm [67, 68], and it was for several years used as a component of commercial sunscreens [69]. All the prevalent FLG mutations are either nonsense or frameshift mutations that result in loss of filaggrin production in the epidermis [70]. Because of the lower levels of filaggrin, individuals with FLG mutations have reduced levels of epidermal UCA and thereby reduced UCA-mediated absorption of UVB. This should, in theory, lead to a higher photoconversion of 7-DHC to previtamin D3 and thereby higher levels of 25(OH)D3 and 1,25(OH)2D3. This hypothesis is supported by in vitro experiments demonstrating that knockdown of filaggrin increased UVB sensitivity [71], by in vivo experiments demonstrating that mice with a mutated histidase gene have reduced levels of UCA in the skin and show increased sensitivity to UVB radiation [66], and finally by five general population studies that showed that FLG mutation carriers have 10 % higher mean serum 25(OH)D3 levels than controls [72]. How FLG mutations influence the local concentration of 1,25(OH)2D3 is not known, but it could well be assumed to augment the concentration and thereby have an impact on immune responses. 1,25(OH)2D3 also stimulates keratinocytes and macrophages to increased production of the antimicrobial peptide cathelicidin, which might be of benefit for AD patients [73, 74].
References
Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NEJ, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41:602–8.
Moniaga CS, Egawa G, Kawasaki H, Hara-Chikuma M, Honda T, Tanizaki H, et al. Flaky tail mouse denotes human atopic dermatitis in the steady state and by topical application with Dermatophagoides pteronyssinus extract. Am J Pathol. 2010;176:2385–93.
Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol. 2009;124(3):485–93, 493.e1.
Mihm Jr MC, Soter NA, Dvorak HF, Austen KF. The structure of normal skin and the morphology of atopic eczema. J Invest Dermatol. 1976;67:305–12.
Woodward AL, Spergel JM, Alenius H, Mizoguchi E, Bhan AK, Castigli E, et al. An obligate role for T-cell receptor alphabeta+ T cells but not T-cell receptor gammadelta+ T cells, B cells, or CD40/CD40L interactions in a mouse model of atopic dermatitis. J Allergy Clin Immunol. 2001;107:359–66.
Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57.
Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–55.
Spergel JM, Mizoguchi E, Oettgen H, Bhan AK, Geha RS. Roles of TH1 and TH2 cytokines in a murine model of allergic dermatitis. J Clin Invest. 1999;103:1103–11.
Hamid Q, Boguniewicz M, Leung DY. Differential in situ cytokine gene expression in acute versus chronic atopic dermatitis. J Clin Invest. 1994;94:870–6.
Hamid Q, Naseer T, Minshall EM, Song YL, Boguniewicz M, Leung DY. In vivo expression of IL-12 and IL-13 in atopic dermatitis. J Allergy Clin Immunol. 1996;98:225–31.
McPherson T, Sherman VJ, Aslam A, Crack L, Chan H, Lloyd-Lavery A, et al. Filaggrin null mutations associate with increased frequencies of allergen-specific CD4+ T-helper 2 cells in patients with atopic eczema. Br J Dermatol. 2010;163:544–9.
Kezic S, O’Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. 2012;129:1031–9.
Shornick LP, Bisarya AK, Chaplin DD. IL-1beta is essential for langerhans cell activation and antigen delivery to the lymph nodes during contact sensitization: evidence for a dermal source of IL-1beta. Cell Immunol. 2001;211:105–12.
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8.
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32.
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–41.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517.
Koga C, Kabashima K, Shiraishi N, Kobayashi M, Tokura Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J Invest Dermatol. 2008;128:2625–30.
Toda M, Leung DY, Molet S, Boguniewicz M, Taha R, Christodoulopoulos P, et al. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J Allergy Clin Immunol. 2003;111:875–81.
He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci U S A. 2007;104:15817–22.
O’Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2009;124:R2–6.
Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6.
Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells – how did we miss them? Nat Rev Immunol. 2013;13:75–87.
Leisten S, Oyoshi MK, Galand C, Hornick JL, Gurish MF, Geha RS. Development of skin lesions in filaggrin-deficient mice is dependent on adaptive immunity. J Allergy Clin Immunol. 2013;131:1247–50.
Enk AH, Katz SI. Identification and induction of keratinocyte-derived IL-10. J Immunol. 1992;149:92–5.
Enk AH, Katz SI. Early molecular events in the induction phase of contact sensitivity. Proc Natl Acad Sci U S A. 1992;89:1398–402.
Larsen JM, Bonefeld CM, Poulsen SS, Geisler C, Skov L. IL-23 and T(H)17-mediated inflammation in human allergic contact dermatitis. J Allergy Clin Immunol. 2009;123:486–92.
Schleimer RP, Kato A, Kern R, Kuperman D, Avila PC. Epithelium: at the interface of innate and adaptive immune responses. J Allergy Clin Immunol. 2007;120:1279–84.
Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol. 2012;130:845–52.
Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Lu YJ, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–80.
Yoo J, Omori M, Gyarmati D, Zhou B, Aye T, Brewer A, et al. Spontaneous atopic dermatitis in mice expressing an inducible thymic stromal lymphopoietin transgene specifically in the skin. J Exp Med. 2005;202:541–9.
Kim BS, Siracusa MC, Saenz SA, Noti M, Monticelli LA, Sonnenberg GF, et al. TSLP elicits IL-33-independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5:170ra16.
Moniaga CS, Jeong SK, Egawa G, Nakajima S, Hara-Chikuma M, Jeon JE, et al. Protease activity enhances production of thymic stromal lymphopoietin and basophil accumulation in flaky tail mice. Am J Pathol. 2013;182:841–51.
Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc Natl Acad Sci U S A. 2007;104:9428–33.
Svendsen ML, Daneels G, Geysen J, Binderup L, Kragballe K. Proliferation and differentiation of cultured human keratinocytes is modulated by 1,25(OH)2D3 and synthetic vitamin D3 analogues in a cell density-, calcium- and serum-dependent manner. Pharmacol Toxicol. 1997;80:49–56.
Rid R, Wagner M, Maier CJ, Hundsberger H, Hintner H, Bauer JW, et al. Deciphering the calcitriol-induced transcriptomic response in keratinocytes: presentation of novel target genes. J Mol Endocrinol. 2013;50:131–49.
Hewison M. Vitamin D and immune function: autocrine, paracrine or endocrine? Scand J Clin Lab Invest Suppl. 2012;243:92–102.
Nagy L, Szanto A, Szatmari I, Szeles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92:739–89.
White JH. Vitamin D metabolism and signaling in the immune system. Rev Endocr Metab Disord. 2012;13:21–9.
Holick MF. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am J Clin Nutr. 2004;80:1678S–88.
Lehmann B, Meurer M. Vitamin D metabolism. Dermatol Ther. 2010;23:2–12.
Holick MF, MacLaughlin JA, Clark MB, Holick SA, Potts Jr JT, Anderson RR, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210:203–5.
Webb AR, Holick MF. The role of sunlight in the cutaneous production of vitamin D3. Annu Rev Nutr. 1988;8:375–99.
Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26:662–87.
Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, et al. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci U S A. 1988;85:3294–8.
Moore DD, Kato S, Xie W, Mangelsdorf DJ, Schmidt DR, Xiao R, et al. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor. Pharmacol Rev. 2006;58:742–59.
Prufer K, Racz A, Lin GC, Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem. 2000;275:41114–23.
Haussler MR, Whitfield GK, Kaneko I, Haussler CA, Hsieh D, Hsieh JC, et al. Molecular mechanisms of vitamin D action. Calcif Tissue Int. 2013;92:77–98.
Kerner SA, Scott RA, Pike JW. Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proc Natl Acad Sci U S A. 1989;86:4455–9.
Pike JW, Meyer MB, Bishop KA. Regulation of target gene expression by the vitamin D receptor - an update on mechanisms. Rev Endocr Metab Disord. 2012;13:45–55.
Hong SP, Kim MJ, Jung MY, Jeon H, Goo J, Ahn SK, et al. Biopositive effects of low-dose UVB on epidermis: coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J Invest Dermatol. 2008;128:2880–7.
Demetriou SK, Ona-Vu K, Teichert AE, Cleaver JE, Bikle DD, Oh DH. Vitamin D receptor mediates DNA repair and is UV inducible in intact epidermis but not in cultured keratinocytes. J Invest Dermatol. 2012;132:2097–100.
von Essen MR, Kongsbak M, Schjerling P, Olgaard K, Odum N, Geisler C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol. 2010;11:344–9.
Yasmin N, Konradi S, Eisenwort G, Schichl YM, Seyerl M, Bauer T, et al. beta-catenin promotes the differentiation of epidermal langerhans dendritic cells. J Invest Dermatol. 2013;133:1250–9.
Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. 2004;29:664–73.
Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, et al. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. 2001;86:888–94.
Hewison M, Burke F, Evans KN, Lammas DA, Sansom DM, Liu P, et al. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J Steroid Biochem Mol Biol. 2007;103:316–21.
Adams JS, Hewison M. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch Biochem Biophys. 2012;523:95–102.
White JH. Regulation of intracrine production of 1,25-dihydroxyvitamin D and its role in innate immune defense against infection. Arch Biochem Biophys. 2012;523:58–63.
Lehmann B, Rudolph T, Pietzsch J, Meurer M. Conversion of vitamin D3 to 1alpha,25-dihydroxyvitamin D3 in human skin equivalents. Exp Dermatol. 2000;9:97–103.
Lehmann B, Sauter W, Knuschke P, Dressler S, Meurer M. Demonstration of UVB-induced synthesis of 1 alpha,25-dihydroxyvitamin D3 (calcitriol) in human skin by microdialysis. Arch Dermatol Res. 2003;295:24–8.
Lehmann B, Schattiger K, Meurer M. Conversion of vitamin D3 to hormonally active 1alpha,25-dihydroxyvitamin D3 in cultured keratinocytes: relevance to cell growth and differentiation. J Steroid Biochem Mol Biol. 2010;121:322–3.
Sybert VP, Dale BA, Holbrook KA. Ichthyosis vulgaris: identification of a defect in synthesis of filaggrin correlated with an absence of keratohyaline granules. J Invest Dermatol. 1985;84:191–4.
Manabe M, Sanchez M, Sun TT, Dale BA. Interaction of filaggrin with keratin filaments during advanced stages of normal human epidermal differentiation and in ichthyosis vulgaris. Differentiation. 1991;48:43–50.
Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–40.
Barresi C, Stremnitzer C, Mlitz V, Kezic S, Kammeyer A, Ghannadan M, et al. Increased sensitivity of histidinemic mice to UVB radiation suggests a crucial role of endogenous urocanic acid in photoprotection. J Invest Dermatol. 2011;131:188–94.
Zenisek A, Kral JA, Hais IM. Sun-screening effect of urocanic acid. Biochim Biophys Acta. 1955;18:589–91.
Tabachnick J. Urocanic acid, the major acid-soluble, ultraviolet-absorbing compound in guinea pig epidermis. Arch Biochem Biophys. 1957;70:295–8.
Gibbs NK, Tye J, Norval M. Recent advances in urocanic acid photochemistry, photobiology and photoimmunology. Photochem Photobiol Sci. 2008;7:655–67.
Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–4.
Mildner M, Jin J, Eckhart L, Kezic S, Gruber F, Barresi C, et al. Knockdown of filaggrin impairs diffusion barrier function and increases UV sensitivity in a human skin model. J Invest Dermatol. 2010;130:2286–94.
Thyssen JP, Thuesen B, Huth C, Standl M, Carson CG, Heinrich J, et al. Skin barrier abnormality caused by filaggrin (FLG) mutations is associated with increased serum 25-hydroxyvitamin D concentrations. J Allergy Clin Immunol. 2012;130:1204–7.
Heine G, Hoefer N, Franke A, Nothling U, Schumann RR, Hamann L, et al. Association of vitamin D receptor gene polymorphisms with severe atopic dermatitis in adults. Br J Dermatol. 2013;168:855–8.
Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117:803–11.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Bonefeld, C.M., Nielsen, M.M., Geisler, C. (2014). Immune Activity and Vitamin D. In: Thyssen, J., Maibach, H. (eds) Filaggrin. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-54379-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-642-54379-1_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-54378-4
Online ISBN: 978-3-642-54379-1
eBook Packages: MedicineMedicine (R0)