
Abstract
Vitamin D has emerged as a pleiotropic regulator of human physiology, and recent work has revealed that it has several roles in control of human immune system function. Vitamin D was originally characterized for its role in calcium homeostasis, and the active form, 1,25-dihydroxyvitamin D (1,25D), can be produced in the kidney by 1α-hydroxylation of circulating 25-hydroxyvitamin D catalyzed by the enzyme CYP27B1. Renal CYP27B1 expression is regulated by calcium regulatory inputs, and 1,25D produced in the kidney was thought to function largely as an endocrine hormone. However, it is now clear that CYP27B1 is expressed in numerous tissues, and that 1,25D acts at several sites in the body in an intracrine or paracrine manner. In particular, both CYP27B1 and the vitamin D receptor (VDR) are expressed in several cell types in the immune system, where CYP27B1 production is controlled by a number of immune-specific inputs. Recent research has opened several windows on the molecular mechanisms by which 1,25D signaling regulates both innate and adaptive immune responses in humans. Moreover, intervention trials are beginning to provide evidence that vitamin D supplementation can bolster clinical responses to infection. This review will discuss recent developments in our understanding of how immune signaling controls local vitamin D metabolism and how, in turn, the 1,25D-bound VDR modulates immune system function. A particular emphasis will be placed on the interplay between vitamin D signaling and signaling through different classes of pattern recognition receptors in the production of antimicrobial peptides during innate immune responses to microbial infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Vitamin D synthesis and sufficiency
Vitamin D is obtained from limited dietary sources or photochemical and thermal conversion of 7-dehydrocholesterol in skin [1]. ‘Vitamin D’ refers collectively to vitamin D3 (cholecalciferol), derived from metabolism of cholesterol, and related vitamin D2 (ergocalciferol), derived from the fungal steroid ergosterol. In animals, photochemical and thermal conversion of cutaneous 7-dehydrocholesterol occurs in the presence of solar ultraviolet B (~300–325 nm) radiation, which varies with time of the year, latitude, altitude and cloud cover. At 45° latitude (south of many of the major population centers of Europe) near sea level, UVB radiation intensity is insufficient for vitamin D synthesis for ~6 months of the year. This period, known as vitamin D winter, lengthens with increasing distance from the equator [1].
Dietary or cutaneous vitamin D must undergo two modifications to become biologically active. Hepatic hydroxylation catalyzed by CYP27A1, CYP2R1 and possibly other enzymes generates 25-hydroxyvitamin D (25D; refs. [2–5]), which has a half-life of several weeks and represents the major circulating metabolite and measure of vitamin D status. 25D is converted by 1α-hydroxylation catalyzed by CYP27B1 into the hormonally active 1,25-dihydroxyvitamin D (1,25D). The kidney is an important site of 1α-hydroxylation, which is stimulated by the calcium regulatory hormone parathyroid hormone (PTH). 1,25D released into the circulation from the kidney was long considered to be an important endocrine source of hormone. However, research in the last 10 years has revealed that CYP27B1 is widely expressed [6, 7], and, moreover, that 1,25D is produced locally in many tissues for function in an intracrine or paracrine manner. In a negative feedback loop, 1,25D strongly induces expression of the gene encoding CYP24, the enzyme that initiates catabolic degradation by catalyzing hydroxylation of 25D or 1,25D at the 24 position to produce biologically inactive metabolites.
The levels of circulating 25D consistent with vitamin sufficiency are a subject of considerable debate. Vitamin D deficiency and insufficiency are widely defined as circulating 25D levels of less than 20 ng/ml and 20–30 ng/ml, respectively (50 nM and 50–75 nM; refs. [3, 8–10]). Many researchers have long considered that circulating 25D concentrations of 30–32 ng/ml (75–80 nM; refs. [11–13]) or greater represent vitamin D sufficiency, as there is an inverse relationship between 25D levels and circulating PTH until 25D rises above 30 ng/ml, at which point PTH levels bottom-out. However, a recent Institute of Medicine (IOM) report defined insufficiency as circulating 25D levels of <20 ng/ml, but considered levels of 20–50 ng/ml as being ‘normal’, while >50 ng.ml (125 nM) was deemed ‘excessive’ [14]. Based on these definitions, the report concluded (controversially; [15, 16]) that much of the population was not vitamin D deficient or insufficient. Moreover, the report considered that, apart from bone health, there was insufficient experimental evidence for the importance of other physiological actions of vitamin D to inform guidelines for recommended dietary intake, due substantially to a lack of randomized, placebo-controlled clinical trials examining non-bone endpoints. However, as detailed below, studies in the last few years have shown that cells of the immune system are ‘wired’ to synthesize and respond to 1,25D upon detection of an infection. Importantly, clinical evidence from intervention trials for the protective effects of vitamin D sufficiency against infectious diseases is beginning to accumulate. Finally, while vitamin D intoxication does occur, it is rare and not observed until 25D levels of >150 ng/ml (375 nM; [3]). It is characterized by hypercalcemia, which if chronic leads to urinary calculi (renal or bladder stones) and renal failure.
2 The vitamin D receptor
1,25D binds and activates the vitamin D receptor (VDR), a member of the superfamily of nuclear receptors, which are ligand-activated transcription factors [17, 18]. Much of vitamin D physiology can be explained by the VDR functioning as a regulator of gene transcription, and expression profiling studies, particularly with microarrays, have been a valuable source of insights, particularly into novel aspects of vitamin D physiology [19–21]. The VDR is composed of a highly conserved DNA binding domain, and an α-helical ligand binding domain [18, 22]. In the presence of 1,25D, the VDR heterodimerizes with retinoid X receptors (RXRs), which are required for recognition of cognate vitamin D response elements (VDREs), located in the regulatory regions of 1,25D target genes.
VDREs are composed of direct repeats of PuG(G/T)TCA motifs separated by 3 bp (DR3) or everted repeats with 6 bp spacing (ER6; refs. [18, 23–25]). ER8 motifs are also recognized by the VDR and related retinoic acid receptors, and contribute to regulation by 1,25D of the gene encoding the cytokine interleukin 10, for example [26, 27]. DNA-bound VDR/RXRs sequentially recruit complexes of so-called coregulatory proteins, which stimulate histone modifications, chromatin remodeling, and RNA polymerase II binding necessary for transcriptional initiation [28, 29]. While numerous VDREs have been identified in relatively promoter proximal locations [20], recent work has provided evidence that the DNA-bound VDR can function at distances as great as 75 kb to regulate adjacent target gene transcription [30, 31]. The VDR can also repress transcription in the presence of 1,25D. VDR/RXR heterodimers can displace DNA-bound nuclear factor of activated T cells (NF-AT), thus repressing cytokine gene expression [32]. We recently, found that the ligand-bound VDR repressed transcription of the gene encoding cyclin D2 via its interaction with FoxO transcription factors [33].
3 Overview of vitamin D action in the immune system
Links between vitamin D or solar therapy and treatment of infectious diseases go back millennia. Hippocrates, the father of medicine, apparently used heliotherapy, or exposure to sunlight, to treat phthisis (tuberculosis; TB) [34]. Sun exposure re-emerged as a popular therapy for treatment of TB in pre-antibiotic era, dating from the mid 19th century with the spread of sanitoria. In addition, Niels Finsen won the 1903 Nobel Prize for the demonstration that UV light could treat cutaneous TB (lupus vulgaris). Cod-liver oil, an excellent source of vitamin D (and vitamin A), first described for treatment of chronic rheumatism in the 18th century, was used as early as 1849 to treat TB, and scrofula, a cervical tuberculosis lympadenopathy arising from infection of lymph nodes of the neck by M. tuberculosis (M.tb.) or related mycobacteria [35–37]. Finally, studies in the 1980’s showed that 1,25D inhibits the growth of M.tb. in cultured human macrophages [38], providing the first evidence for direct stimulation by 1,25D of innate immune responses to M.tb. infection.
A number of clinical studies have found associations between either vitamin D deficiency or specific restriction fragment length polymorphisms in the VDR gene, and susceptibility to TB or other infectious diseases, particularly respiratory tract infections of bacterial or viral origin (reviewed in [39]). Two recent intervention trials are notable additions in this regard. Martineau and colleagues investigated the effect of high-dose vitamin D3 on time to sputum culture conversion in patients undergoing antibiotic therapy for pulmonary TB [40]. Four doses of 2.5 mg (100,000 IU) of vitamin D3 (62 patients) or placebo (64 patients) were administered at 14-day intervals. Vitamin D therapy markedly raised mean serum 25D concentrations at 56 day (101.4 nM in the intervention group vs. 22.8 nM in the placebo group). Therapy did not have a statistically significant effect on the time to sputum culture conversion in the study population as a whole 36 day (vitamin D) vs. 43.5 d (placebo) (adjusted hazard ratio 1·39, 95% CI 0·90–2·16; p = 0.14), but it did significantly reduce the time to sputum culture conversion in participants with the VDR tt genotype (8·09, 95% CI 1·36–48·01; p = 0·02) [40]. The authors concluded that vitamin D therapy should benefit at least subset of vitamin D-deficient patients with pulmonary TB and recommended further studies to investigate the apparently selective response in patients with the tt VDR polymorphism [40].
A recently published randomized double-blind placebo-controlled trial provided strong evidence that vitamin D supplementation would be of benefit in prevention of seasonal influenza A infections [41]. School children in Japan were supplemented with 1,200 IU of vitamin D3 or placebo from December to March. 10.8% of the treatment group (18/167) came down with influenza, whereas 18.6% (31/167) of the placebo group became ill (relative risk, 0.58; 95% CI: 0.34, 0.99; P = 0.04). The reduction was even more prominent in children who had not been taking vitamin D supplements prior to the trial [41]. These results clearly suggest that vitamin D supplementation may be effective in preventing influenza, particularly in vitamin D-deficient children. It will be important to investigate the molecular mechanisms underlying the beneficial effects of therapy on viral infections. It is possible that vitamin D signaling augments innate immune responses to viral infection directly. However, the effects may be indirect. For example, an intriguing animal study suggested that robust innate immune control of intestinal microbiota may influence the severity of pulmonary influenza infections. Mice treated with multiple antibiotics to eliminate commensal intestinal bacteria exhibited strongly reduced T-cell responses to sub-lethal doses of inhaled A/PR8 influenza virus relative to controls, along with increased viral titers [42]. Remarkably, local or distal administration of bacterial lipopolysaccharide (LPS), a ligand for Toll-like receptor 4 (TLR4), a pattern recognition receptor that drives innate immune responses to bacterial infection, attenuated the impairment in antibiotic-treated mice [42]. Given the increasingly well-established roles of vitamin D signaling in anti-bacterial innate immunity in humans (see below), this model study may provide insights into the mechanisms of action by which vitamin D boosts anti-viral immune responses.
4 Vitamin D signaling in T cells
We now know that the VDR and CYP27B1 are expressed in several cell types in the immune system, including in T lymphocytes, neutrophils and antigen presenting cells such as macrophages and dendritic cells, and that 1,25D signaling modulates both innate and adaptive immune responses [43–46]. 1,25D signaling regulates the function and phenotype of dendritic cells, the most potent of the antigen presenting cells. 1,25D enhances dendritic cell tolerogenicity [45], which promotes the production and function of T regulatory (Treg) cells, critical mediators of immune system tolerance. 1,25D also acts directly on T lymphocytes to inhibit their proliferation [43, 46]. 1,25D signaling represses the transcription of genes encoding key T helper 1 (Th1) proinflammatory cytokines, such as interferon-γ (IFN-γ) and interleukins 17 and 21 [46, 47]. T cells treated with 1,25D also acquire the characteristics of Treg cells [47]. Thus, 1,25D signaling directly and indirectly acts to suppress antigen presentation to, and activation and recruitment of Th1 cells, instead favoring a more regulatory T cell phenotype.
The effects of 1,25D on T cell phenotypes would be consistent with the increasing evidence that vitamin D sufficiency acts to suppress T cell-driven autoimmune diseases. Vitamin D or 1,25D analogues are used widely for the treatment of the disfiguring and inflammatory condition psoriasis [48], which is increasingly considered to have a T cell-driven autoimmune component [49, 50]. Several epidemiological studies have reported inverse correlations between circulating 25D levels and risk of other autoimmune conditions such as multiple sclerosis, rheumatoid arthritis, and type-1 diabetes [46, 51]. In particular, there are links between vitamin D deficiency in infancy and early childhood and increased risk of type-1 diabetes [46, 52]. While these findings are compelling, several authors have argued (e.g. [51]) that prospective studies are needed to more firmly establish links between vitamin D deficiency and autoimmunity.
Recent work has revealed that 1,25D signaling also regulates T cell antigen receptor function. Engagement of the T cell antigen receptor on naïve human T cells led to p38 MAP kinase-dependent stimulation of VDR expression, which in turn strongly induced expression of phospholipase C-γ1, a cofactor of the classical T cell antigen receptor signaling pathway, within 48 h of initial T cell receptor stimulation [53]. The authors speculated that the 48 h lag might be an evolutionarily conserved mechanism to prevent explosive T cell proliferation in the presence of antigen [53]. If innate immune responses quickly control infection during the lag period, the onset of T cell division would take place in a controlled manner in a relatively uninflammatory microenvironment characterized by limiting antigen concentrations. In the absence of a lag or under conditions of insufficient innate immune response and uncontrolled infection, elevated antigen concentration would produce a more proinflammatory microenvironment and more aggressive T cell proliferation. Given, as detailed below, that 1,25D signaling enhances innate immune responses to infection, the lag in T cell activation would represent a coordinated strategy to decrease the potential for T cell-driven immunopathology.
5 Vitamin D is an inducer of innate antimicrobial immune responses
The most rapidly evolving area of research into the immunomodulatory properties of vitamin D signaling has been in our understanding of the molecular mechanisms by which vitamin D metabolism and 1,25D signaling are implicated in innate immune responses. As indicated above we have known for over 20 years that 1,25D reduces M.tb. viability in infected macrophages [38]. We have also known since the early ‘90’s that 1,25D strongly induces expression of CD14, a co-receptor critical for recognition of LPS by TLR4 [54].
In 2003, we mapped the positions of promoter-proximal consensus VDREs in the human genome. This in silico screen identified VDREs adjacent to the transcription start-sites of two genes encoding antimicrobial peptides (AMPs) β-defensin 2 (DEFB2/DEFB4/HBD2) and cathelicidin antimicrobial peptide (CAMP/LL37) [55]. AMPs along with various cytokines and chemokines are the first-responders of the innate immune attack against invading pathogens [56–58]. CAMP and some β-defensins also have dual functions as chemoattractants for neutrophils, monocytes and other cellular components of immune responses [56–58]. We found that CAMP expression was strongly stimulated by 1,25D in all cell types examined [55], a finding subsequently borne out in several in vitro and in vivo studies [59–62]. One of the more interesting in vivo studies examined CAMP regulation in the biliary tract, which is normally microbe-free. D’Aldebert and colleagues founds that CAMP expression in biliary epithelial cells was regulated by physiological concentration of bile acids signaling through the VDR and a related nuclear receptor for bile acids, FXR [63]. The potential role of the VDR in this signaling is consistent with previous molecular studies that the VDR can act as a bile acid sensor [64]. Notably, neither the CAMP nor the HBD2 VDRE is conserved in mice, and Gombart and coworkers showed that the CAMP VDRE is embedded in a human/primate specific Alu repeat transposable element [59]. The insertion of the VDRE-containing Alu repeat in the CAMP gene originated in the primate lineage leading to humans, apes, and Old World and New World monkeys [65], which dates the event back 55–60 Ma.
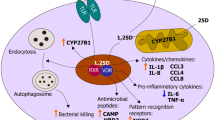
While 1,25D alone was sufficient for strong induction of CAMP expression in cell-based experiments, its effect in isolation on HBD2 expression was modest [54] or not detected [60]. However, 1,25D enhanced 2-fold the strong induction of HBD2 by IL-1β [54]. Subsequent work showed that signaling through TLR1/2 pattern recognition receptors in human monocytes induced IL-1β expression and signaling, and that the combination of IL-1β and 1,25D were required for strong induction of HBD2 expression [66]. IL-1β signaling was very likely mediated by induced binding of the NF-κB transcription factor to tandem binding sites in the HBD2 proximal promoter ([66]; Fig. 1).

Regulation of expression of the gene encoding antimicrobial peptide DEFB2/DEFB4/HBD2 by signaling through pattern recognition receptors and the VDR. As described in the text, the 1,25D-bound VDR can induce expression of the gene encoding pattern recognition receptor NOD2/CARD15 through binding to distal VDREs. NOD2 activated by binding of muramyl dipeptide (MDP) arising from breakdown of bacterial peptidoglycan can activate HBD2 expression through promoter-proximal NF-κB binding sites. Similar autocrine signaling by IL-1β, whose expression is induced by signaling through TLR2/1, also activates HBD2 expression via NF-κB. See text for details
The NF-kB binding sites in the HBD2 promoter represent an important hub of innate immune signaling. We recently found that the 1,25D-activated VDR directly induces expression of the gene encoding pattern recognition receptor NOD2/CARD15 (nucleotide oligomerization domain protein 2/caspase recruitment domain-containing protein 15) [67]. NOD2 is member of a class of pattern recognition receptors that is structurally distinct from the TLRs. It is an intracellular protein that is activated by muramyl dipeptide (MDP), a lysosomal breakdown product of bacterial peptidoglycan. 1,25D strongly induced NOD2 expression in human myeloid and epithelial cells via VDR binding to at least two distal high-affinity VDREs. Similar to IL-1β, NOD2 signaling induces NF-κB function and enhances HBD2 expression [68] (Fig. 1). We found that the combination of pretreatment with 1,25D to induce NOD2, followed by addition of MDP synergistically induced HBD2 expression [67].
The induction of the NOD2-HBD2 innate immune pathway by 1,25D is significant for several reasons. Importantly, attenuated or disrupted expression of NOD2 or HBD2 is associated with an enhanced risk of development of Crohn’s disease (CD), a chronic inflammatory condition [69, 70]. While often mislabeled as an autoimmune disease [71], CD likely arises from a defect in innate immune handling of intestinal bacterial load, which leads to intestinal inflammation in the absence of autoimmunity [72, 73]. The epithelial lining of the colon is a site of strong CYP27B1 expression [15], and vitamin D deficiency is associated with active disease, although this may arise through defective intestinal absorption. Rates of CD apparently increase with increasing latitude in Europe and North America [74, 75], suggestive of a contribution of vitamin D insufficiency/deficiency, although data concerning seasonal variations in disease relapse rates are conflicting [76–78]. There are also polymorphisms in the VDR gene that correlate with susceptibility to CD [79]. The direct and indirect regulation by 1,25D of the NOD2-HBD2 innate immune pathway strongly suggests that 1,25D deficiency or insufficiency may indeed contribute to the pathogenesis of CD, and provides further evidence that 1,25D signaling is important for optimal innate immune responses.
6 Intracrine 1,25D signaling driven by immune system regulation of CYP27B1 expression
Rather than acting predominantly as a renal endocrine hormone, it is likely that most of the physiological actions of 1,25D are mediated by locally produced hormone acting in an intracrine or paracrine manner. Nowhere is this truer than in cell types critical for innate immune responses. Several studies have documented the role of 1,25D in inducing monocytic differentiation [80]. Notably, both VDR expression and the capacity of monocyte-derived macrophages and dendritic cells to produce 1,25D from 25D are developmentally regulated [80–82]. The pathophysiological consequences of excessive macrophage 1,25D biosynthesis are manifested in sarcoidosis, a granulomatous inflammatory disease, in which elevated 1,25D can lead to hypercalcemia in extreme cases [83].
The last few years have seen a number of interesting reports detailing the immune signaling pathways leading to the induction of CYP27B1 expression, particularly in macrophages. Expression profiling studies of human macrophages revealed that activation of TLR1/2 toll-like receptor heterodimers with 19 kDa lipopeptide stimulated expression of genes encoding CYP27B1 and VDR ([60]; Fig. 2). Other work showed that LPS signaling through TLR4 receptors also induced CYP27B1 expression [84], our unpublished results), consistent with correlations between expression levels of TLR4 and CYP27B1 [85, 86]. In human macrophages cultured in human serum, downstream VDR-driven induction of CAMP expression observed upon TLR2/1 stimulation was strongly dependent on serum 25D concentrations [60]; responses were markedly attenuated in macrophages cultured in serum from vitamin D-deficient individuals. Importantly, VDR-regulated gene expression could be restored by supplementation with 25D. Notably, consistent with other studies [87, 88], serum levels of 25D in African Americans were approximately one half those of Caucasian Americans [60], a difference likely arising from reduced UVB-induced cutaneous synthesis of vitamin D3 in darker African-American skin. Subsequent work showed that IL-15 expression induced by TLR1/2 signaling was required for CYP27B1 induction [89] (Fig. 2). These findings also provided a mechanistic basis for the previously observed increased expression of CYP27B1 expression during macrophage development, as IL-15 is an inducer of monocyte-macrophage differentiation. This work is important because it reveals that naïve macrophages acquire the capacity to respond to circulating 25D upon detection of a microbial infection, and suggests that downstream VDR-driven gene expression is proportional to the levels of 25D throughout the physiological range.

Regulation of vitamin D metabolism and intracrine signaling by cytokine and pattern recognition receptors. Signaling through TLR2/1 induces expression of IL-15, which in turn induces expression of CYP27B1. CYP27B1 expression can also be induced by signaling through TLR4. Elevated intracrine production of 1,25D activates the VDR, which induces expression of genes encoding AMPs such as CAMP, pattern recognition receptor NOD2/CARD15, or TLR4 coreceptor CD14
More recent studies have provided evidence that T cell cytokines can influence TLR-regulated production of CYP27B1 in macrophages. IFN-γ, a cytokine secreted by proinflammatory Th1 cells, enhanced TLR2/1-dependent induction of 1,25D target genes CAMP and HDB2 expression in macrophages in vitamin D sufficient serum. In contrast, IL-17, secreted by the T helper subtype Th17, had no effect, whereas the Th2 cytokine IL-4 strongly suppressed the effect of TLR2/1 stimulation on CAMP and HBD2 expression [90]. Remarkably, IL-4 enhanced TLR2/1-stimulated expression of the CYP27B1 and VDR genes, as did IFN-γ. However, studies of bioconversion of 25D in cytokine-treated cells suggested that, while 1α-hydroxylation was enhanced in IFN-γ-treated cells, 25D was preferentially converted to 24,25D in the presence of IL-4. Gene ablation studies showed that the effect of IL-4 on 25D metabolism was abolished by siRNA-mediated knockdown of CYP24, although there was no apparent effect of IL-4 on CYP24 mRNA or protein expression. The authors speculated that the elevated levels of 24-hydroxylation induced by IL-4 may have arisen from enhanced delivery of 25D to the mitochondrial compartment containing CYP24 [90]. No matter what the mechanism by which IL-4 regulates CYP24 function, the paper clearly showed that T-cell cytokines can (differentially) control TLR-stimulated vitamin D metabolism and 1,25D-driven innate immune antimicrobial responses in macrophages.
7 Concluding remarks
The above has highlighted many of the exciting recent advances in our understanding of how the immune system regulates vitamin D metabolism and how, in turn, 1,25D modulates immune system function. In particular, vitamin D has emerged as a key inducer of human innate immune responses to microbial infection, and the next few years will certainly see more important findings in this area. It should be kept in mind, however, that research into the role of vitamin D signaling in controlling innate immunity may be complicated by the fact that many of the responses may be human- (or human/primate)-specific and that animal models such as mice may not recapitulate many of the vitamin D-dependent molecular-genetic mechanisms seen in humans. Unlike adaptive immunity, which is found in vertebrates only, innate immune responses are present in a vast array of plant and animal species [91, 92]. However, many details and mechanisms of regulation of innate immune function are species-specific. For example, TLR-driven induction of AMP expression in mouse macrophages is mediated by NO. However, this does not appear to be conserved in humans, where 1,25D signaling appears to play a key role as an intermediate [60]. Similarly, the promoter-proximal VDREs in the HBD2 and CAMP genes are not conserved in mice [55, 59, 65]. Indeed, the arrays of genes encoding AMPs vary considerably between species [93, 94], as does the conservation of certain cytokines and chemokines, and numbers of TLRs [95]. Therefore, mouse models, which have been powerful tools in furthering our understanding of the calcium homeostatic functions of vitamin D, may not be as appropriate for dissecting the roles of vitamin D signaling in immune system regulation.
References
Tavera-Mendoza L, White JH. Cell defenses and the sunshine vitamin. Sci Am. 2007;297:62–72.
Cheng JB, Levine MA, Bell NH, et al. Genetic evidence that the human CYP2R1 enzyme is a key vitamin D 25-hydroxylase. Proc Natl Acad Sci U S A. 2004;101:7711–5.
Holick MF. Vitamin D deficiency. New Engl J Med. 2007;357:266–81.
Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. 2004;29:664–73.
Shinkyo R, Sakaki T, Kamakura M, et al. Metabolism of vitamin D by human microsomal CYP2R1. Biochem Biophys Res Commun. 2004;324:451–7.
Zehnder D, Bland R, Williams MC, et al. Extrarenal expression of 25-hydroxyvitamin d(3)-1 alpha-hydroxylase. J Clin Endocrinol Metab. 2001;86:888–94.
Hewison M, Burke F, Evans KN, et al. Extra-renal 25-hydroxyvitamin D3-1alpha-hydroxylase in human health and disease. J Ster Biochem Mol Biol. 2007;103:316–21.
Malabanan A, Veronikis IE, Holick MF. Redefining vitamin D insufficiency. Lancet. 1998;351:805–6.
Bischoff-Ferrari HA, Giovannucci E, Willett WC, et al. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr. 2006;84:18–28.
Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81:353–73.
Chapuy MC, Preziosi P, Maamer M, et al. Prevalence of vitamin D insufficiency in an adult normal population. Osteoporos Int. 1997;7:439–43.
Thomas MK, Lloyd-Jones DM, Thadhani RI, et al. Hypovitaminosis D in medical inpatients. New Engl J Med. 1998;338:777–83.
Holick MF, Siris ES, Binkley N, et al. Prevalence of vitamin D inadequacy among postmenopausal North American women receiving osteoporosis therapy. J Clin Endocrinol Metab. 2005;90:3215–24.
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8.
Heaney RP, Holick MF. Why the IOM recommendations for vitamin D are deficient. J Bone Miner Res. 2011;26:455–7.
Slomski A. IOM endorses vitamin D, calcium only for bone health, dispels deficiency claims. JAMA. 2011;305:455–7.
Chawla A, Repa J, Evans RM, et al. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70.
Lin R, White JH. The pleiotropic actions of vitamin D. BioEssays. 2004;26:21–8.
Lin R, Nagai Y, Sladek R, et al. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 signaling on cell proliferation, differentiation and immune system regulation. Mol Endocrinol. 2002;16:1243–56.
Wang TT, Tavera-Mendoza L, Laperriere D, et al. Large-scale in silico and microarray-based genomic screening of 1,25-dihydroxyvitamin D3 target genes. Mol Endocrinol. 2005;19:2685–95.
Matthews D, LaPorta E, Zinser GM, et al. Genomic vitamin D signaling in breast cancer: insights from animal models and human cells. J Steroid Biochem Mol Biol. 2010;121:362–7.
Rochel N, Wurtz JM, Mitschler A, et al. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–9.
Drocourt L, Ourlin JC, Pascussi JM, et al. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the vitamin D receptor pathway in primary human hepatocytes. J Biol Chem. 2002;277:25125–32.
Thompson PD, Jurutka PW, Whitfield GK, et al. Liganded VDR induces CYP3A4 in small intestinal and colon cancer cells via DR3 and ER6 vitamin D responsive elements. Biochem Biophys Res Comm. 2002;299:730–8.
Thummel KE, Brimer C, Yasuda K, et al. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxyvitamin D3. Mol Pharmacol. 2001;60:1399–406.
Tavera-Mendoza L, Wang TT, Lallemant B, et al. Convergence of vitamin D and retinoic acid signaling at a common hormone response element. EMBO Rep. 2006;7:180–5.
Matilainen JM, Husso T, Toropainen S, et al. Primary effect of 1α,25(OH)2D3 on IL-10 expression in monocytes is short-term down-regulation. Biochim Biophys Acta. 2010;1803:1276–86.
Dilworth FJ, Chambon P. Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene. 2001;20:3047–54.
McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–74.
Kim S, Yamazaki M, Zella LA, et al. Activation of receptor activator of NF-kappaB ligand gene expression by 1,25-dihydroxyvitamin D3 is mediated through multiple long-range enhancers. Mol Cell Biol. 2006;26:6469–86.
Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. J Biol Chem. 2010;285:15599–610.
Takeuchi A, Reddy G, Kobayashi T, et al. Nuclear factor of activated T cells (NFAT) as a molecular target for 1,25-dihydroxyvitamin D3-mediated effects. J Immunol. 1998;160:209–18.
An B-S, Tavera-Mendoza LE, Dimitrov V, et al. Stimulation of SIRT1-regulated FoxO protein function by the ligand-bound vitamin D receptor. Mol Cell Biol. 2010;30:4890–900.
Masten AR. Sunlight in tuberculosis. Chest. 1935;1:8–23.
Guy RA. The history of cod liver oil as a remedy. Am J Dis Child. 1923;26:112–6.
Grad R. Cod and the consumptive: a brief history of cod-liver oil in the treatment of pulmonary tuberculosis. Pharm Hist. 2004;46:106–20.
Martineau AR, Honecker FU, Wilkinson RJ, Griffiths CJ. Vitamin D in the treatment of pulmonary tuberculosis. J Ster Biochem Mol Biol. 2007;103:793–8.
Rook GA, Steele J, Fraher L, Barker S, Karmali R, O'Riordan J, et al. Vitamin D3, interferon, and control of proliferation of mycobacterium tuberculosis by human monocytes. Immunol. 1986;57:159–63.
White JH. Vitamin D signaling, infectious diseases, and regulation of innate immunity. Infect Immun. 2008;76:3837–43.
Martineau AR, Timms PM, Bothamley GH, et al. High-dose vitamin D-3 during intensive-phase antimicrobial treatment of pulmonary tuberculosis: a double-blind randomised controlled trial. Lancet. 2011;377:242–50.
Urashima M, Segawa T, Okazaki M, et al. Randomized trial of vitamin D supplementation to prevent seasonal influenza A in schoolchildren. Am J Clin Nutr. 2010;91:1255–60.
Ichinohe T, Pang IK, Kumamoto Y, et al. Micobiota regulates immune defense against respiratory tract influenza A virus infection. Proc Nat Acad Sci U S A. 2011;108:5354–9.
Bhalla AK, Amento EP, Serog B, Glimcher LH. 1,25-Dihydroxyvitamin D3 inhibits antigen-induced T cell activation. J Immunol. 1984;133:1748–54.
Brennan A, Katz DR, Nunn JD, Barker S, et al. Dendritic cells from human tissues express receptors for the immunoregulatory vitamin D3 metabolite, dihydroxycholecalciferol. Immunol. 1987;61:457–61.
Adorini L, Penna G. Dendritic cell tolerogenicity: a key mechanism in immunomodulation by vitamin D receptor agonists. Human Immunol. 2009;70:345–52.
Baeke F, Takiishi T, Korf H, et al. Vitamin D: modulator of the immune system. Curr Opin Pharmacol. 2010;10:482–96.
Von Essen MR, Kongsbak M, Schjerling P, et al. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol. 2010;11:344–9.
Gorman S, Judge MA, Hart PH. Immune-modifying properties of topical vitamin D: focus on dendritic cell and T cells. J Ster Biochem Mol Biol. 2010;121:247–9.
Bowcock AM, Krueger JG. Getting under the skin: the immunogenetics of psoriasis. Nat Rev Immunol. 2005;5:699–711.
Prinz JC. From bench to bedside—translational research in psoriasis. J Eur Acad Dermatol Vernerol. 2010;24:1–4.
Solomon AJ, Whitham RH. Curr Neurol Neurosci Rep. 2010;10:389–96.
Hypponen E, Laara E, Reunanen E, et al. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet. 2001;358:1500–3.
Jeffery LE, Burke F, Mura M, et al. 1,25-dihydroxyvitamin D-3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol. 2009;183:5458–67.
Oberg F, Botlin J, Nilsson K. Functional antagonisms between vitamin-D3 and retinoic acid in the regulation of CD14 and CD23 expression during monocytic differentiation of U-937 cells. J Immunol. 1993;150:3487–95.
Wang TT, Nestel F, Bourdeau V, et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173:2909–12.
Hancock REW, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–7.
Doss M, White MR, Tecle T, Hartshorn KL. Human defensins and LL-37 in mucosal immunity. J Leuk Biol. 2010;87:79–92.
Liu PT, Krutzik SR, Modlin RL. Therapeutic implications of the TLR and VDR partnership. Trends Mol Med. 2007;13:117–24.
Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D-3. FASEB J. 2005;19:1067–77.
Liu PT, Stenger S, Li HY, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–3.
Weber G, Heilborn JD, Chamorro Jimenez CI, et al. Vitamin D induces the antimicrobial protein hCAP18 in human skin. J Invest Dermatol. 2005;124:1080–2.
White JH. Vitamin D as an inducer of cathelicidin antimicrobial peptide expression: past, present and future. J Ster Biochem Mol Biol. 2010;121:234–8.
D'Aldebert E, Biyeyeme Bi Mve MJ, Mergey M, et al. Bile salts control the antimicrobial peptide cathelicidin through nuclear receptors in the human biliary epithelium. Gastroenterol. 2009;136:1435–43.
Makishima M, Lu TT, Xie W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–6.
Gombart AF, Saito T, Koeffler HP. Exaptation of an ancient Alu short interspersed element provides a highly conserved vitamin D-mediated innate immune response in humans and primates. BMC Genomics. 2009;10:321.
Liu PT, Schenk M, Walker VP, et al. Convergence of IL-1 beta and VDR Activation Pathways in Human TLR2/1-Induced Antimicrobial Responses. PLoS ONE. 2009;4:e5810.
Wang TT, Dabbas B, Laperriere D, et al. Direct and indirect induction by 1,25-dihydroxyvitamin D3 of the NOD2/CARD15-beta defensin 2 innate immune pathway defective in Crohn’s disease. J Biol Chem. 2010;285:2227–31.
Voss E, Wehkamp J, Wehkamp K, et al. NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem. 2006;281:2005–11.
Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol. 2008;8:458–66.
Fellermann K, Stange DE, Schaeffeler E, et al. Am J Hum Genet. 2006;79:439–48.
Coulombe F, Behr MA. Crohn’s disease as an immune deficiency? Lancet. 2009;374:769–70.
Smith AM, Rahman FZ, Bu’Hussain H, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med. 2009;206:1883–97.
Marks DJB, Rahman FZ, Sewell GW, et al. Crohn’s disease: an immune deficiency state. Clin Rev Allergy Immunol. 2010;38:20–31.
Lim W-C, Hanauer SB, Li YC. Mechanisms of disease: vitamin D and inflammatory bowel disease. Nat Clin Pract Gastroenterol Hepatol. 2005;2:308–15.
Loftus Jr EV. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterol. 2004;126:1504–17.
Zeng L, Anderson FH. Seasonal change in the exacerbations of Crohn’s disease. Scand J Gastroenterol. 1996;31:79–82.
Lewis JD, Aberra FN, Lichtenstein GR, et al. Seasonal variation in flares of inflammatory bowel disease. Gastroenterol. 2004;126:665–73.
Aratari A, Papi C, Galletti B, et al. Seasonal variations in onset of symptoms in Crohn’s disease. Dig Liver Dis. 2006;38:319–23.
Simmons JD, Mullighan C, Welsh KL, Jelwell DP. Vitamin D receptor gene polymorphism: association with Crohn’s disease susceptibility. Gut. 2000;47:211–4.
Hewison M. Vitamin D and the immune system: new perspectives on an old theme. Endocrinol Metab Clin N Am. 2010;39:365–79.
Kreutz M, Andreesen R, Krause SW, et al. 1,25-dihydroxyvitamin-D3 production and vitamin-D3 receptor expression are developmentally regulated during differentiation of human monocytes into macrophages. Blood. 1990;76:2457–61.
Hewison M, Freeman L, Hughes SV, et al. Differential regulation of the vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol. 2003;170:5382–90.
Burke RR, Rybicki BA, Rao DS. Calcium and vitamin D in sarcoidosis: how to assess and manage. Sem Resp Crit Care. 2010;31:474–84.
Stoffels K, Overbergh L, Giulietti A, et al. Immune regulation of 25-hydroxyvitamin-D-3-1 alpha-hydroxylase in human monocytes. J Bone Min Res. 2006;21:37–47.
Evans KN, Taylor H, Zehnder D, et al. Increased expression of 25-hydroxyvitamin D-1 alpha-hydroxylase in dysgerminomas—a novel form of humoral hypercalcemia of malignancy. Am J Pathol. 2004;165:807–13.
Evans KN, Nguyen L, Chan J, et al. Effects of 25-hydroxyvitamin D-3 and 1,25-dihydroxyvitamin D-3 on cytokine production by human decidual cells. Biol Reprod. 2006;75:816–22.
Nesby-O'Dell S, Scanlon KS, Cogswell ME, et al. Hypovitaminosis D prevalence and determinants among African American and white women of reproductive age: Third National Health and Nutrition Examination Survey, 1988–1994. Am J Clin Nutr. 2002;76:187–92.
Stead WW, Senner JW, Reddick WT, Lofgren JP. Racial differences in sensitivity to infection by mycobacterium tuberculosis. N Engl J Med. 1990;322:422–7.
Krutzik SR, Hewison M, Liu PT, et al. IL-15 links TLR2/1-induced macrophage differentiation to the vitamin D-dependent antimicrobial pathway. J Immunol. 2008;181:7115–20.
Edfeldt K, Liu PT, Chun R, et al. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Nat Acad Sci U S A. 2010;107:22593–8.
Akira S. Innate immunity to pathogens: diversity in receptors for microbial recognition. Immunol Rev. 2009;227:5–8.
Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58.
Patil A, Hughes AL, Zhang G. Rapid evolution and diversification of mammalian α-defensins as revealed by comparative analysis of rodent and primate genomes. Physiol Genom. 2004;20:1–11.
Zanetti M. Cathelicidins: multifunctional peptides of the innate immunity. J Leuk Biol. 2004;75:39–48.
Kumar H, Kawai T, Akira S. Toll-like receptors and innate immunity. Biochem Biophys Res Comm. 2009;388:621–5.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
White, J.H. Vitamin D metabolism and signaling in the immune system. Rev Endocr Metab Disord 13, 21–29 (2012). https://doi.org/10.1007/s11154-011-9195-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-011-9195-z