
Abstract
Fuel cells are power generation devices converting chemical energy into electric energy by electrochemical reactions. Among various types of fuel cells, hydrogen-oxygen (H2-O2) based proton exchange membrane (PEM) fuel cells have attracted special attention due to their high efficiency, low temperature operation, and suitability for low to medium power generation. However, the requirement of high cost catalysts (platinum and its alloys) for both cathodic and anodic reactions makes them unsuitable for commercial applications. Development of efficient catalysts with reduced cost has drawn considerable scientific attention. This chapter reviews the PEM fuel cell cathode catalysis in terms of challenges and progress in the field with an emphasis on the application of carbon nanomaterials such as carbon nanotubes and graphene. Owing to their promising properties such as high electronic conductivity, corrosion resistance, and large surface area, carbon nanomaterials are suitable for catalyst support materials. Apart from this, doped carbon nanomaterials show potential toward the development of metal-free catalysts.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Fuel cells
- PEM fuel cell
- Electrochemical reactions
- Oxygen reduction reaction
- ORR catalyst
- CNT
- Nitrogen-doped CNT
- Nitrogen-doped graphene
- Platinum-free catalyst
1 Introduction
In view of the increasing world energy requirements and depleting fossil fuel resources, it is crucial to use the available energy resources efficiently and to explore the renewable energy sources. Presently, fossil fuels, i.e., petroleum oil, coal, and natural gas, contribute nearly 80 % of our energy requirements [1]. Heat engines are mostly used to generate power from these fuels. For example, the electric power is generated by steam and gas turbines, whereas internal combustion engines (ICEs) are used for mechanical work as well as electric power generation. In these heat engines, the combustion of fuel takes place, which converts the stored chemical energy into thermal energy. Finally, the thermal energy is used to perform mechanical work or to generate power. There are two major drawbacks of the combustion-based power generation, i.e., the low power conversion efficiency (η) and the emission of pollutants hazardous to the environment. The η of the thermal engines cannot exceed the Carnot limit given by η = (1 − T 2/T 1), where T 1 and T 2 are the absolute temperatures of source and sink, respectively [2]. For practical thermal engines, the efficiencies are well below the theoretical limit. Diesel and petrol engines, for example, have efficiencies ~40 % and ~30 %, respectively. Furthermore, the combustion of fossil fuels produces various pollutants and greenhouse gases, causing serious environmental concerns such as global warming, climate change, and health problems. In view of this, the development of efficient and environmentally friendly power generation technologies is of prime interest to the scientific community. Among others, hydrogen is being explored as a renewable energy source to reduce the greenhouse emissions and other pollutants. In a sustainable and emission-free energy cycle, hydrogen can be produced from electrochemical or photocatalytic hydrolysis of water. Although, the power generation from hydrogen is possible by using heat engines, but in the last decade, more efficient electrochemical energy conversion devices are being explored.
1.1 Fuel Cells
As an alternative of heat engines, power generation through the electrochemical conversion has attained significant interest. Galvanic cells are being used commercially for the small-scale power requirements since long back. Similar concept can be employed for the large-scale power generation. Fuel cells (FCs) are a class of such devices converting chemical energy to electric energy through electrochemical route. They differ from the dry cells in terms of fuel supply and by-product removal. Dissimilar to dry cells, where the cell becomes useless once all the reactants are consumed, FCs use a continuous feeding of reactants and removal of by-products. The basic advantage of FCs over the heat engines is the absence of the combustion step and hence, their efficiency is not limited by the Carnot limit (Fig. 15.1). This makes it possible to generate the power at very high efficiencies. Thermodynamically, the η for FCs is given by: η = (ΔG f /ΔH f ), where ΔG f and ΔH f represent the Gibbs free energy and the enthalpy of formation, respectively [2]. For a H2-O2 fuel cell (FC), the thermodynamical η can be as high as 91 %.

Principal difference between FCs and heat engines
1.2 Evolution of Fuel Cells
History of FCs goes back to 1800 when Nicholson and Carlisle, using a silver-zinc voltaic pile, have observed bubbles of gases formed on both the silver as well as the zinc side of the piles, essentially splitting water to its components by electricity [3]. Later studies have proved the gases to be hydrogen and oxygen produced by the electrolysis of water, according to the following electrochemical reactions.
The inverse process – formation of water by combining hydrogen and oxygen – has been demonstrated by Grove in 1839 [4, 5]. Using a setup shown in Fig. 15.2, with lower ends of each platinum (Pt) electrodes immersed in sulfuric acid (H2SO4) electrolyte and upper ends covered with glass tubes containing oxygen and hydrogen, Grove has observed a constant current flow between the electrodes with a consumption of both gases.

Schematic of gas voltaic battery
The electrochemical reactions occurring in an H2-O2 FC are:
In a further development in 1889, Mond and Langer have developed a FC based on hydrogen/coal gas and achieved a power conversion efficiency of ~50 % at a close circuit voltage of 0.73 [6]. Their system can be considered as the first real FC system since the design allowed supplying oxidant and fuel continuously with a provision to simultaneous removal of by-products. In another significant advancement in 1893, Ostwald has studied the thermodynamical aspects of the FCs. He has suggested the possibility of a combustion-free energy conversion device, which is not subjected to the limitations of second law of thermodynamics, leading to higher efficiencies compared to that of heat engines [7]. The first practical H2-O2 FC, having a power capacity of up to 5 kW, has been fabricated by Bacon [8]. Starting experiments on alkaline FCs (AFCs) in 1932, he has built a cell with nickel gauze electrodes in 1939. His successful work on nickel electrodes to replace costly Pt catalyst is considered another milestone in FC history as it highlights the need to develop Pt-free catalysts with other abundant materials [9]. Nevertheless the FCs research has received a real thrust when National Aeronautics and Space Administration (NASA) decided to use FCs as an auxiliary power source in their space missions. The Gemini earth-orbiting mission (1962–1965) has used FCs based on solid polymer electrolytes made of sulfonated polystyrene resin. This is an example of the first polymer electrolyte membrane (PEM) FC (PEMFC) used for space applications. Later, highly efficient AFCs (η ~70 %) based on Bacon’s technology have been used in Apollo mission [8, 9].
The obvious advantages of FCs have led intense research and development of various types of FCs covering a vast range of power outputs (few mW to MW). Based on the types of the fuels and/or the electrolytes, FCs have a range of operating temperatures, power outputs, and applications. Table 15.1 summarizes some of the important types of the FCs with their operating parameters. Selection of a suitable type of the FCs for a particular application depends on their power output, ease of operation, cost, and related safety issues. High temperature FCs such as solid oxide FCs (SOFCs), phosphoric acid FCs (PAFCs), and molten carbonate FCs (MCFCs) are suitable for stationary power generation plants with a few MW capacity. Hydrogen-based PEMFCs are more suitable for the medium power outputs such as transport vehicles, whereas direct methanol FCs (DMFCs), another type of PEM-based FCs, suit to portable devices and household applications (laptops, mobiles, etc.).
1.3 Polymer Electrolyte Membrane Fuel Cells
PEMFCs are low operating temperature FCs consisting of a polymer membrane as electrolyte [10]. As the polymer membranes conduct H+ ions, PEMFCs are also termed as “proton exchange membrane FCs.” They use hydrogen or low molecular weight hydrocarbons as the fuel at anode and oxygen/air as the oxidizer at cathode. Among others, hydrogen, being one of the potential futuristic energy sources, is the key fuel for PEMFCs. Grubb and Niedrach have developed the first PEMFC in the early 1960s. They have used sulfonated polystyrene membrane as electrolyte and Pt-deposited membrane as the catalyst for oxygen reduction and hydrogen oxidation reactions [11]. Apart from H2-based PEMFCs, methanol-based PEMFCs, namely, DMFCs, have been studied thoroughly due to their feasibility for small-scale portable applications. In principle, both of these fuel cells have similar structures. This report is focused on the H2-based PEMFCs due to their better performance in terms of futuristic power devices.
1.4 Working Principle and Components
In a PEMFC, hydrogen is oxidized at anode to produce H+ ions, which migrate through the PEM to the cathode. Reduction of oxygen takes place at cathode to form O2− ions, which combine with the H+ ions to form H2O (Eqs. 15.4 and 15.5). This develops a net cell potential (Eq. 15.6) that equals the difference between electrochemical potentials of hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR). When connected to an external load, a constant current can be drawn if the reactant gases are supplied and the reaction product (H2O) is removed continuously to maintain the system in steady state.
Figure 15.3a shows the schematic of H2-O2 PEMFC with various components such as bipolar plates (BPs) with flow channels, gas diffusion layer (GDL), and membrane electrode assembly (MEA). The MEA is the part of FC consisting of the cathode and anode catalyst layers (CLs), and the PEM itself. Each of these components uses different materials based on their particular functions [12]. Additionally, a practical FC consists of a few auxiliary components for continuous power generation. As shown in Fig. 15.3b, it has to perform a number of tasks such as (i) supply and transport of fuel and oxidant to the appropriate electrodes, (ii) removal of by-products, (iii) electron transfer to the external load, (iv) H+ ion transport through the membrane, (v) proper humidification of membrane, and (vi) maintaining the temperature at an optimum level. For high power generation, FC units are stacked in a series by using BPs. In a FC stack, other auxiliary components such as the humidifier, heat exchanger, compressor, heating system, etc. may be required occasionally [13].

(a) Schematic of a PEMFC and (b) transport of reactants and products in the PEMFC electrodes (part (b) has been reprinted with permission from [12])
1.4.1 Bipolar Plates
BPs, being an important component of PEMFC, contribute a significant weight (80 %) [14] and cost (25 %) of the FC stack [15]. As shown in Fig. 15.3a, in a single FC, two plates are placed on either side (cathode and anode) of the gas diffusion electrodes (GDE) containing gas flow channels. In a FC stack, each of such plate separates the GDE of two adjacent cells such that the one side works as anode plate, whereas the other side serves as cathode plate, signifying the name “bipolar plate” (BP). Gas flow channels formed on either side of the BP work as H2 flow channel for the anode side and O2/air flow channel for the cathode side. BPs perform a number of functions such as supply and uniform distribution of the reactants (fuel and oxidizer) to the GDL, facilitation of water and heat management, separation of individual cells, and transportation of electrons from cell to external load [16]. Also, BPs provide strength to the mechanically weak MEA [17]. For uniform distribution of gases, the flow channels of BPs have to be designed properly. Again, the materials should possess certain properties such as high electronic conductivity (>100 S/cm), high tensile and flexural strengths (>41 MPa and > 59 MPa, respectively), low corrosion rate (<1 μA/cm2), low hydrogen permeability (<2 × 10−6 cm3/(cm2s)), low contact resistance (<20 MΩ cm2), low density (<5 g/cm3), and high thermal conductivity (>10 W/mk) [15]. Based on these criteria, a number of materials have been investigated for BPs, which can be classified as metals, non-metals, and composites. Metallic BPs have good mechanical strength and electronic conductivity compared to the graphite and composite materials. In addition, they are easy to machine and are cost effective as well. However, the lower chemical resistance of metals makes them corrosion prone in the harsh FC environment [15, 18]. In addition to this, metallic BPs increase the weight of the stack, which is undesired for portable applications. Improving corrosion resistance of the metals has been another active area of FC research. The corrosion resistance of metallic BPs is improved by coating corrosion resistive materials on the metal surfaces. The frequently used coating materials include carbon-based materials such as graphite, diamond-like carbon; noble metals such as gold and Pt; metal carbides such as WC; metal nitrides (TiN, CrN, NbN, ZrN) [19, 20]; conductive polymers and organic self-assembled monopolymers [16]. However, the high specific density of metals remains an issue.
Graphite is considered a standard material for BPs due to its low resistivity and excellent corrosion resistance. However, due to the brittle nature and lack of mechanical strength, graphite BPs are not suitable for transport applications, where vibrations may lead to their failure. Again, it becomes difficult to fabricate gas flow channels through machining and the process is not cost effective as well [16]. To overcome these issues, graphite-reinforced polymer composites have been proposed as the alternatives to graphite BPs. The polymer matrix provides mechanical stability to the plates by reducing their brittle nature. Moreover, the polymers exhibit high corrosion resistance and a cost-effective fabrication is possible. The main drawback of these composite materials is the reduced electronic and thermal conductivities, which affects the cell performance adversely. Other reinforcing materials, such as carbon black (CB), carbon nanotubes (CNTs), and graphene, have been used to improve the mechanical strength and electronic as well as thermal conductivities of polymer composite BPs [21–23]. CNT-reinforced polymer composites exhibit large improvements in the properties [24].
1.4.2 Gas Diffusion Layer
GDL is a multifunctional component of a PEMFC. It is a thin layer (thickness ranging between 100 and 300 μm) of porous conducting material sandwiched between the BP and the CL [12]. GDL facilitates the diffusion of reactant gases to the CL and controls the humidity level of the cell. In addition, it provides a conducting path for the electrons from CL to BP and supports CL physically. Apart from this, GDL also plays a key role in transporting the heat generated at the electrodes to the gas flow channels [25, 26]. To perform these functions, a combination of suitable materials and proper designing the GDL is required. Material parameters affecting the GDL performance include hydrophobicity, porosity, pore-size distribution, pore connectivity, contact resistance, thermal and electronic conductivity, and corrosion resistance. Carbon-based materials, namely, carbon fiber (CF), carbon paper (CP), carbon cloth (CC), CNTs, CB, and graphite, are most widely used for GDL. These materials exhibit good corrosion resistance, pore connectivity, and electronic conductivity, which are appropriate for the GDL [27]. Proper functioning of GDL with improved performance can be obtained by modifying its porosity and pore-size distribution, hydrophobicity, morphology, and electronic conductivity [28]. The commonly used GDLs are having both single and double layered structures. Single layered GDL generally consists of a 150–400-μm thick macroporous layer of CF/CP [29]. Double layered GDL includes a woven or unwoven CF layer and a second microporous layer (MPL) comprising of CB and PTFE. Here, the first layer works as the GDL while the second microporous layer functions as water management layer [30]. Moreover, the MPL also provides a better contact with the CL [31]. Structure of GDL plays a crucial role in FC water management. Effective water management through GDL has found significant scientific interest in PEMFC research. Carefully designed GDL may eliminate the need of humidifier in PEMFC [32]. Efforts toward the improved water management include the use of novel GDL materials such as CNTs, graphene, etc., altering the contact angle by surface modifications such as polymer coatings, and changing the pore-structure and morphology. In single layered GDL, coating of polytetrafluoroethylene (PTFE) on CF is applied to increase the hydrophobicity. A balanced combination of hydrophobic and hydrophilic pores is required. Lin et al. have found that under flooding conditions, coating of PTFE enhances the gas and water transport [33]. Excessive loading of PTFE reduces the hydrophilic pores responsible for water transport and leads to more electrode flooding. Park et al. have found an optimum PTFE loading of 20 wt% to balance the water saturation in GDL and CL giving best cell performance. Porosity measurements have revealed that the PTFE loading decreases porosity and increases the hydrophobic pore-volume [34]. Moreover, the loading of PTFE increases the contact resistance of the GDL due to its insulating nature. The through-plane (along the thickness) conductivity of the layer decreases significantly. The in-plane (parallel to the GDL plane) electronic conductivity, however, is not affected significantly as the charge is conducted by the carbon fibers (CFs) [30].
1.4.3 Electrolyte Membrane
Electrolytes with good ionic conductivity are one of the essential parts of devices such as batteries and FCs. In H2-O2 PEMFCs, transport of H+ ions from anode to cathode is performed through a polymer-based membrane. Polymer electrolyte membranes (PEMs) are solid state electrolytes developed initially by GE for the Gemini space missions. The basic material requirements of an electrolyte for PEMFCs include high H+ ion conductivity and no electronic conductivity. In order to form a robust structure and to provide enough support to the CL, the membrane should have adequate mechanical strength. To sustain the corrosive FC environment, high corrosion resistance and chemical inertness are also needed. In addition, the membrane should be impermeable to the fuel/oxygen to prevent the crossover. The methodologies opted for the synthesis of electrolyte membranes seek their large-scale production with low costs [35].
Initially, polystyrene sulfonic acid membranes were used in PEMFCs, which suffered a drawback of being less durable. This issue was addressed well by perfluorinated sulfonic acid membranes (trade name Nafion®) developed by DuPont in 1960 [36]. Nafion® consists of a PTFE backbone with perfluorinated vinyl polyether side-chains, bonded by the oxygen atoms. The chemical structure of the Nafion® monomer unit is shown in Fig. 15.4. The sulfonic acid groups at the end of the side-chains provide free H+ ions for conduction in the hydrated conditions. The conductivity depends on the degree of hydration of the membrane. Membrane thickness is another important parameter affecting the cell performance. Smaller thickness is preferred due to the reduced membrane resistance, ease of hydration, and lower cost. However, the risk of fuel crossover increases with decreasing the thickness. The thickness has to be optimized to best performance without the fuel crossover.

Chemical structure of perfluorinated polymer (Nafion®) from DuPont
Cost reduction, improved durability and cell performance, and proper water management are the major issues related to the electrolyte membrane. Various PEM materials being studied for FC membrane are [35, 37]:
-
(i)
Perfluorinated ionomers: Sulfonic group attached perfluorinated polymers such as Nafion® are use most frequently for PEMs.
-
(ii)
Partially fluorinated polymers: These polymers have fluorocarbon backbones and hydrocarbon or aromatic side-chains. The ionic conductivity is lower than that of the perfluorinated ones.
-
(iii)
Non-fluorinated polymers: These polymers have hydrocarbon or aromatic backbones. The side-chains are modified with polar or sulfonic acid groups. These membranes show good mechanical properties but have poor conductivity.
-
(iv)
Acid–base blends: These membranes consist of an alkaline backbone modified by incorporation of an acid component. The acid component imparts the ionic conductivity comparable to that of Nafion® membrane.
1.4.4 Catalyst Layer
The anode and cathode CLs are coated on either sides of an electrolyte membrane. Properties such as high catalytic activity, high surface area, high corrosion resistance, good chemical inertness, excellent electronic conductivity, etc. are mandatory for the CLs. Conventionally, high surface area Pt particles coated on CB are being used as catalysts for both ORR and HOR in a PEMFC. CB, being electrically conductive and corrosion resistant, is used as support material for the nanosized Pt. However, Pt is not a suitable catalyst material for large-scale applications due to its high cost and scarce nature. For commercial applications, the exploration of alternate catalysts is indispensable as the high efficiency of FCs is of less importance without their cost effective fabrication [38]. Significant efforts have been made toward the synthesis of low Pt or Pt-free catalysts during the last few decades.
2 PEM Fuel Cell Catalysis
Electrochemical reactions on both the electrodes of a PEMFC have an activation energy barrier. Thermal energy at normal working temperatures of a PEMFC (80 °C) is not significant to cross this barrier. Catalysts, in general, alter the activation energy by changing the reaction path. In PEMFCs, heterogeneous catalysis is used to attain a practically significant reaction rate. Both the electrochemical half-cell reactions, namely, HOR at the anode and ORR at the cathode, require catalysts. Among the cathode and anode reactions, the sluggish nature of the former reaction makes its catalysis more challenging. Large Pt-loading as compared to anode is required at the cathode.
2.1 ORR Mechanism in PEMFCs
The ORR has been studied extensively, and a number of possible pathways are proposed. Its mechanism depends on the nature of the media as well as the catalyst sites. The two pathways – four-electron and two-electron pathways – are possible in both acidic and alkaline media [39]. While the four-electron pathway is a single step process, the two-electron process involves two steps with H2O2 as an intermediate. In PEMFCs (acidic media), the ORR pathways are represented as
Four-electron pathway:
Two-electron pathway:
-
(i)
$$ {\mathrm{O}}_2+2{\mathrm{H}}^{+}+2{\mathrm{e}}^{-}\to {\mathrm{H}}_2{\mathrm{O}}_2 $$(15.8)
-
(ii)
$$ {\mathrm{H}}_2{\mathrm{O}}_2+2{\mathrm{H}}^{+}+2{\mathrm{e}}^{-}\to 2{\mathrm{H}}_2\mathrm{O} $$(15.9)
The reactions need high activation energies, which necessitates the presence of a catalyst to attain practically significant reaction rates. Since the catalyst systems should have practically significant stability under the acidic media, Pt/Pt-alloy-based catalysts remain the most suitable ORR catalysts for PEMFCs. In the presence of the heterogeneous catalyst, molecular oxygen (O2) is adsorbed on the catalyst surface to form O2*, which either can be desorbed or reduced. The most accepted possible reduction pathways for Pt surface, proposed by Wroblowa et al., are shown in Fig. 15.5 [40, 41]. The ORR can proceed either by a single step four-electron process (Eq. 15.7) or by a couple of two-electron reactions (Eqs. 15.8 and 15.9). The former process, where the O2* is directly reduced to H2O, is shown in Fig. 15.5 with a rate constant of k1. The latter two-electron process proceeds via an intermediate step forming adsorbed hydrogen peroxide (H2O2*) as an intermediate with a rate constant of k2. The H2O2* either can be reduced to form H2O with a rate constant k3 or desorbed with a rate constant k5. In addition to this, the H2O2* can decompose chemically with a rate constant of k4. In PEMFC, the direct pathway (k1) is the most preferred pathway as the production of H2O2 lowers the system performance.

ORR pathways proposed by Wroblowa et al. [40]
The above reactions involve several sub-steps. Norskov et al. have proposed dissociative and associative pathways for ORR on Pt surface [42]. In the dissociative pathway, the O-O bond is broken to form adsorbed O (O*), which, on reacting with an H+, forms adsorbed HO (HO*). Finally, HO* forms H2O by combining with another H+ [42, 43]. The dissociative mechanism is given in the equation below, where the catalytic site is represented by (*) [44].
The dissociative process reduces the oxygen directly without the formation of H2O2 intermediate, and hence, it is a four-electron process with rate constant k1 (Fig. 15.5). In this process, the step involving the hydrogenation of OH is the rate-determining step due to its high activation energy. While the O-O bond breaks in the dissociative mechanism, the associative mechanism involves the adsorption of O2 without breaking the O-O bond. The reduction of adsorbed O2 (O2*) takes place according to the following chain of reactions:
Similar to the dissociative mechanism, this process does not produce H2O2. However, the presence of molecular O2 (O-O bond) may result in H2O2 formation with the following reaction:
The catalytic activity of any catalyst is a wise interplay between the adsorption energies of various intermediate species. A good catalyst adsorbs the reactants in such a way that the binding energy of most stable species (O and OH) remains minimum to obtain a lower overpotential (potential required to overcome the activation barrier).
2.2 Issues with PEMFC Catalysis
Owing to their high stability and catalytic activity, Pt-based systems are the mostly used catalysts for PEMFC cathode and anode CLs. Apart from high cost, the catalysts suffer from other issues such as low durability, carbon monoxide (CO) poisoning, and other contaminations. The CO poisoning occurs mostly at the anode side due to the presence of CO impurity in the steam-reformed H2. For cathode CL, requirement of high Pt-loading and low durability are the major concerns.
2.2.1 High Cost and Scarcity of Platinum
Among other issues with FC catalysts, the high cost and scarcity of Pt is of utmost concern. For large-scale application of FCs, such as public transport, it is estimated that the Pt amount as large as one fourth of the total estimated Pt deposits would be required to produce one billion cars [45]. The available amount of Pt is insufficient to meet its possible demand by the FC industry. In addition, the scarcity of Pt combined with its high demand makes it one of the most valuable metals having high cost (last 15 years average price: ~1,000 $/Oz). In case of the adoption of FC-based power sources, owing to increased demand, the Pt cost would further increase significantly. This would increase the fractional cost of CL in a FC stack, which is estimated to be ~35 % of the stack cost based on present market prices. Therefore, search for novel Pt-free or low-level Pt catalysts is inevitable.
2.2.2 Durability
During PEMFC operation, the performance of the CL decreases with time due to the reduction in the effective surface area of the catalyst. This causes a serious concern to PEMFC catalysis. The performance change is due to the irreversible changes happening in the CL. Numerous studies exploring the durability have revealed several mechanisms responsible for the catalyst degradation. Studies show that the important mechanisms responsible for catalytic activity decay in PEMFC are: (i) dissolution of Pt during the cyclic or steady-state operation, (ii) detachment of catalyst particles by corrosion of carbon support, (iii) reduction in the effective surface area due to the particle size growth, and (iv) poisoning/contamination of catalyst particles. The PEMFC catalyst degradation becomes more prominent under variable loads, which is a serious concern toward their application in vehicles.
The dissolution of Pt takes place primarily during the potential cycling at high potentials and is more prominent at the cathode. With increasing temperature, the dissolution rate also increases. Dissolved Pt-ionomers are either redeposited on the larger sized particles or diffused through the membrane. The previous case, where larger particles grow at the cost of smaller ones, is termed as Ostwald ripening [46]. Parthasarathy et al. have studied the coarsening of graphite-supported Pt-nanoparticles in a solution containing Pt ions (0.1 M PtCl4 in dimethyl sulfoxide) [47, 48]. TEM images of Pt samples reveal a significant coarsening effect (Fig. 15.6). The average particle size increases by a factor of ~5 by the deposition of Pt from the solution. In another stability test of Pt/C catalyst, Zhai et al. have observed a roughly 100 % increment in the Pt particle size after 300 h of testing [49]. The electrochemical surface area is reduced from 17.2 to 7.8 m2/gpt. XRD analysis of FC electrodes shows smaller FWHM for samples tested for 300 h. Moreover, the rate of change of the particle size decreases with increasing testing time as there is no significant change in FWHM for higher durations. The increased particle size effectively reduces the surface area, which in turn leads to the lower electrocatalytic activity [50]. On the other hand, when ionomers are diffused in the membrane, they either react with the crossed over hydrogen to form Pt deposits inaccessible for the catalysis or replace H+ ions of the membrane [51]. As a result, the H+ ion concentration in the membrane is reduced, which in turn reduces the ionic conductivity of the PEM. Secondly, at nanometer scale, small particles can form agglomerates by random cluster–cluster collisions to form larger clusters. In a third mechanism, nanoparticles may grow at atomic level to minimize their Gibb’s free energy.

TEM images of graphite-supported Pt (a) before and after the electrochemical test (Reprinted with permission from [48])
The corrosion of carbon support material is another important process responsible for the loss of the catalyst surface area [52]. In acidic media, carbon support corrodes slowly due to the electrochemical oxidation in the presence of Pt [53]. This leads to unsupported catalyst nanoparticles, which remain either unconnected electrically to the outer circuit or covered by the carbon particles to become inaccessible to the reactants. The oxidation behavior of highly oriented pyrolytic graphite (HOPG) under potential cycling in an acidic media has been studied by Choo et al. [54]. The study reveals that the HOPG surface corrodes severely for a potential cycling while remaining almost unaffected when kept at a constant potential. Scanning electron microscope (SEM) images of HOPG reveal that the samples remain unaffected when kept at a constant potential of 1.0 V, whereas severe corrosion is observed for a potential cycling between 0.8 and 1.0 V at a sweep rate of 90 mV/s (Fig. 15.7). Moreover, the corrosion becomes more prominent under high sweep rate of potential cycling. The atomic force microscope (AFM) images of the HOPG surfaces before and after a potential cycling between 0.8 and 1.0 V at a sweep rate of 10 mV/s are shown in Fig. 15.8a, b, respectively. Formation of pits by surface oxidation due to the potential cycling is clearly visible.

SEM image of HOPG after (a) holding at 1.0 V and (b) potential cycling for 22.2 h (scale bar: 1 μm) (Reprinted with permission from [54])

AFM images of HOPG surface (a) before and (b) after electrochemical corrosion (Reprinted with permission from [54])
2.3 CNT as Catalyst Support
The cost issue of the FC is mainly due to the expensive Pt-based catalyst. Hence, in order to reduce the cost, loading of catalyst must be minimized. One of the important challenges towards reducing the Pt-loading in FCs is the low durability of catalysts due to the catalyst support corrosion [55]. In case of catalyst support corrosion, high Pt-loading is required to obtain a reasonable durability. Owing to their high corrosion resistance, CNTs-based thin CLs can be used with less Pt-loading [56]. In addition, when compared to CC/CP-based CL, more uniform dispersion of Pt nanoparticles without agglomeration is possible to achieve. Hence, in the case of CNT-supported CL, the size distribution of Pt nanoparticles is found to be narrower [57]. This improves the Pt catalyst surface area and activity to reduce the total catalyst amount to be loaded. Use of CNTs shows promising improvement in terms of the size miniaturization, chemical and electrochemical stability, improved durability, and cost reduction of FCs.
CNTs, owing to their high specific surface area (~1,000–1,500 m2/g) [58–60] combined with conductive nature, are utilized in the CL of the PEMFCs. Effective surface area of the CL can be increased significantly by using CNTs as catalyst support. The increased effective area of the CL results in an improved FC performance [61]. The catalyst particles are coated on CNTs by various physical and chemical techniques such as deposition by chemical or electrochemical reduction of catalyst precursors, sputtering, thermal CVD, etc. [62]. Uniformity of coating, particle size distribution, CNTs–catalyst particle interaction, and electric connectivity of CNTs with GDL are the key parameters governing the catalyst performance. Sun et al. have synthesized a 3D network of electrically connected multiwalled CNTs (MWCNTs) on CP by a two-step CVD process (Fig. 15.9). A good electrical connectivity of such a network in PEMFC catalyst support provides effective electron transport [63]. Catalyst metal particles can be deposited on the CNTs coated CP/CC by various physical and chemical techniques [62]. In order to utilize the large, surface area of CNTs, a uniform loading of catalyst particles is mandatory. A comparatively high loading of Pt nanoparticles (2–4 nm) have been obtained by Saha et al. using glacial acetic acid to reduce Pt acetylacetonate [64]. Figure 15.10 shows the transmission electron microscope (TEM) images of the Pt-nanoparticle-loaded CNTs with uniform particle distribution without a sign of agglomeration. Moreover, the density of Pt-nanoparticles can be controlled by monitoring the Pt-precursor concentration during deposition. Table 15.2 summarizes a few research works using CNTs as catalyst support for reduced Pt-loading. For similar Pt-loading, the Pt/CNTs catalysts show up to 50 % enhanced (in terms of maximum power output) performance as compared to CB based catalysts.

TEM images of two stage MWCNTs network grown on the fibers of a CP at different magnifications (Reprinted with permission from [63])

TEM images showing CNTs coated with Pt nanoparticles for varying Pt-precursor concentration (a) 1 mM and (b) 4 mM (Reprinted with permission from [64])
CL may be applied either to the electrolyte membrane or to the GDL. Improvement in cell performance has been observed by using CNTs in both the cases. Lebert et al. have obtained improved cell performance by combining Pt/CNTs-based CL with Nafion membrane [67]. Similarly, Tang and coworkers have obtained improved polarization curve by using Pt/CNTs layer and Pt/CNTs-CB (Pt coated on CNT–CB blend), which works as both CL and GDL [68, 69]. Similar improvements have been observed in DMFCs using CNT-supported Pt/Ru as anode catalyst [70].
To resolve the durability issues, novel materials for catalyst support as well as catalyst are being explored. Carbon nanomaterials such as CNTs are one of the well-studied and promising catalyst support materials for improved durability. These have been investigated with higher thermal stability and durability for CLs [74, 75]. Table 15.3 summarizes a few of such studies. The long-term chemical stability of the CNT-coated CF-based electrodes has been found to improve significantly [81]. This is attributed to the improved thermal stability and higher oxidation resistance of support layer, and less agglomeration of catalyst particles. The migration of catalyst particles can be minimized by increasing their adhesion to the supporting CNT/CP layer. This can be obtained by using functionalized CNTs having active groups [65]. CNT-supported catalyst particles covered with silica layer have been found to be highly active with increased durability under potential cycling [46, 76].
2.4 Pt/Non-Precious Metals Alloys for ORR Catalysts
Transition metals such as Sc, Cr, Fe, Co, Ni, Cu, W, Sn. etc. are termed as non-precious metals (NPMs). Pt/NPM-alloys have been explored for ORR catalysts with low Pt-loading. The Pt/NPM-alloys exhibit improved activities in terms of the power density per unit Pt-loading. An enhancement by a factor of 3–5 has been observed. However, the enhancement factor varies widely, which depends on particle size, shape, composition, and crystallographic orientation. To compare the catalytic activity of Pt vs. Pt-alloy a simple normalization over mass or surface area is insufficient. Furthermore, the effect of surface electronic structure on the reactivity is still unknown.
The specific activity of Pt alloy catalysts exhibits a strong correlation with the Pt-Pt nearest neighbor (n-n) distance. As revealed by Min et al., smaller n-n distance helps the oxygen adsorption, leading to high specific activity. Among the studied Pt-alloy catalysts, with its smallest n-n distance, Pt3Ni exhibits the highest activity [82].
Li et al. have studied a tri-metallic alloy (Pt-Fe-Ni) ORR catalyst synthesized by electrodeposition. Power output of ~0.5 W/cm2 at 0.5Vwith an ultra-low Pt loading of 0.05 mg Pt cm−2 has been reported [83]. Similar to Pt, carbon nanomaterials have been used as catalysts supports for Pt/NPMs-alloy catalysts as well [84]. While the alloying can help reducing the required Pt amount, the leaching of base metals form the Pt-alloy catalyst is of major concern. In PEMFC operating potentials, the Pt-alloys are thermodynamically unstable. Incomplete alloying, presence of excess amounts of base metal, are the other factors leading to the leaching of base metals. The leached metal ions may replace the conducting H+ ions in the electrolyte membrane, effectively reducing its conductivity significantly. Furthermore, this also leads to the CL depletion and low durability [85].
2.5 Platinum-Free Catalysts
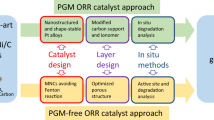
The replacement of Pt-based catalysts with Pt-free catalysts has been appreciated since the discovery of PEMFCs. The requirement of larger Pt-loading at cathode has fascinated special attention toward the development of Pt-free ORR catalysts. Intensive efforts toward the Pt-free ORR catalysis have offered a number of such material systems. The key problem toward the replacement of Pt is the acidic environment of PEMFCs, which discards the use of NPMs as ORR catalysts. While NPMs have low corrosion resistance, their compounds and complexes with the non-metals such as nitrogen (N), boron (B) and chalcogens exhibit the opposite nature, which makes them stable in the PEMFC environment. The development of Pt-free catalysts for ORR dates back to the study by Jasinski, showing the ORR catalytic activity of transition metal porphyrins and phthalocyanines [86]. Since then, transition metal-/N-containing carbon-based materials have been studied significantly as a substitute of Pt for ORR catalysis. The reported Pt-free catalysts are either the NPMs-based or the metal-free materials. Among others, the transition metal–based complexes exhibit promising results. A review on the developments in the NPM-based catalysts for PEMFC cathode by Othman et al. suggests that NPM-based ORR catalysts can be classified as NPM-based macrocycles, chalcogenides, electroconductive polymers, nitrides, and carbons [87]. The metal-free catalysts include carbon nanomaterials (porous carbon, CNTs, graphene, etc.) doped with various impurities such as N- and B-atoms. A classification of various Pt-free ORR catalysts is shown in Fig. 15.11.

Classification of Pt-free ORR catalysts
2.5.1 NPMs-Based Pt-Free Catalysts
In 1964, Jasinski has reported the use of metal porphyrins and phthalocyanines as NPM catalysts for ORR for the first time [86]. This has led to a new class of NPM catalysts for ORR named metal-N4-macrocycles (planer molecules), having a central metal ion surrounded by four N-atoms symmetrically. Figure 15.12 shows a few cobalt-N4-macrocycle molecules explored for ORR activity.

Structures of (a) cobalt phthalocyanine (CoPc), (b) cobalt tetrapyridinoporphyrazine (CoTAP), and (c) cobalt tetrapyrazinoporphyrazine (CoPTpz) (Reprinted with permission from [88])
Among other macrocycles, transition metal (Fe, Co, Ni and Cu)-N4-macrocyclic molecules have been explored more frequently as PEMFC cathode catalysts [89, 90]. Numerous studies during the last few decades have revealed that Fe– and Co–macrocyclic complexes possess high electrocatalytic activity. The metal-N4 macrocycles show both two-step and one-step pathways for reduction of oxygen. Xu et al. have studied the ORR activity of MWCNT supported cobalt phthalocyanine (CoPc), cobalt tetrapyridinoporphyrazine (CoTAP), and cobalt tetrapyrazinoporphyrazine (CoPTpz) and suggested a two-step pathway for CoPc while a one-step pathway for CoTAP- and CoPTpz-based catalysts [88]. The reaction mechanism and active site structures in metal-N4 macrocycles are, however, debatable due to their complex structures. The metal-N bonds participate in the process. Different types of active sites are proposed. More than one type of active sites may exist simultaneously in a catalyst material. The ORR activity depends strongly on the oxidation state of the metal ions and atomic arrangements [43].
Recently, transition metal complexes of heterocyclic electroconductive polymers such as polyaniline (PANI), polypyrrole (PPY), polythiophenes (PThs), poly(3-methylthiophene), etc. have been reported to have high ORR activity and durability. Bashyam et al. have used Co-PPY composite as an ORR catalyst for PEMFC [91]. The composite is prepared by entrapping cobalt precursor such as CoNO3.6H2O in the polymer matrix followed by the reduction with NaBH4. The catalytic activity is attributed to the interaction of Co atoms with polypyrrole, forming the CoNx active sites. Millan et al. have found that among various catalyst systems based on cobalt-modified electroconductive polymer supported on CB, PPY-based catalysts show superior ORR properties [92]. The Co-PPY composites having ORR activity and stability comparable to that of the commercial Pt/C catalysts (30 wt% Pt supported on Vulcan XC-72, a commercially available high surface area carbon) have been reported for direct borohydride fuel cells (DBFCs) [93]. The superior properties are attributed to the specific structure of the composites having N-Co-N bonds, as shown in Fig. 15.13.

Proposed structure of Co-PPY composites [92]
Pyrolysis of N-containing organic compounds in the presence of the transition metals such as Fe, Co, Ni, etc. produces metal/N-C composites having ORR activity. Studies by Wu et al. have revealed such ORR catalysts synthesized by pyrolysis of PANI in the presence of Fe/Co precursors [94–97]. The catalysts show high ORR activity with high selectivity for four-electron pathway.
2.5.2 Metal-Free ORR Catalysts
Metal-free ORR catalysts are principally the graphitic carbons with impurity (N, B, etc.) doping in the regular graphitic structure. N-containing carbon materials exhibit catalytic properties toward the ORR. As shown in Fig. 15.14, three different types of N-C sites, graphitic-N, pyridinic-N and pyrrolic-N, are possible. The graphitic-N is formed when the N-atom replaces a C-atom of the regular hexagonal network. Here, three C-atoms form bonds with the N-atom. On the other hand, the pyridinic-N site consists of C-N bonds located at the edge of the graphitic network. The N-atom, bonded by two C-atoms, becomes a part of the six-membered ring. Finally, the pyrrolic-N is formed by incorporation of the N-atom in a five-membered ring, as shown in Fig. 15.14.

Possible N-sites in N-doped graphitic carbon
Mutter et al., by studying various N- and metal-containing carbons, have determined that the pyridinic-N sites exhibit highest catalytic activity. Since the pyridinic-N sites are located at the edges of graphitic planes, the number of these sites is governed by the extent of edge plane exposure [98, 99].
Recently, N-doped CNTs (NCNTs) have been reported to have high catalytic activity toward ORR [100]. Vertically aligned NCNTs produced by pyrolysis of iron (II) phthalocyanine are employed as metal-free electrodes for ORR in AFCs. The NCNTs electrodes exhibit superior electrocatalytic activity with 4.1 mA/cm2 at −0.22 V compared to 1.1 mA/cm2 at −0.22 V for the standard Pt/C electrode in 0.1 M KOH saturated with air. Again, dissimilar to Pt/C, NCNTs catalysts exhibit immunity to CO poisoning. The electrocatalytic properties of NCNTs are attributed to the modified charge distribution in the presence of the electron-accepting N-atoms. The doping modifies the energy band structure, spin density, and charge density in the proximity of the dopant, forming high charge/spin density sites [101]. These sites are responsible for the enhanced ORR activity. For example, incorporation of N-atoms in a regular hexagonal carbon network of CNTs has been found to induce positive charges on the adjacent C-atoms. Density functional theory (DFT) calculations by Gong et al. have revealed the presence of the small positive charges on the C-atoms adjacent to the N-atom in NCNTs [100]. This positive charge facilitates the oxygen adsorption and weakens the O-O bond [102]. Apart from this, NCNT contain pyridinic-N, which acts as the ORR active site [103–105]. The number of these sites depends on the extent of N-doping. The catalytic activity exhibits increasing trend with increasing the N-doping [106, 107]. Rao et al. have studied the effect of N-content in vertically aligned NCNTs with a varying dopant concentration (0, 4.3, 5.6, 8.4, and 10.7 at.%) and obtained highest ORR activity for 8.4 at.% N-doping. Here, the catalytic activity is attributed to the presence of pyridinic-N sites, which adsorb O2 and weaken the O-O bond. However, the activity of NCNTs remains inferior to that of Pt/C [105]. The number of active sites, and hence the catalytic activity of NCNTs, is affected by the synthesis parameters such as precursor and temperature. Kundu et al. have reported the ORR activity of NCNT synthesized by pyrolysis of acetonitrile at 550 oC to be 10 % of that of a commercial Pt/C (20 wt% Pt). At higher processing temperature (750 oC), the activity decreases due to the reduced number of pyridinic-N sites [103].
Similar to NCNTs, N-doped graphene (NG) is another potential ORR catalyst material. Despite being comparatively emerging material, the ORR activity of NG has attained considerable scientific interest. NG is generally synthesized by pyrolyzing graphene oxide (GO) with an N-containing precursor [108]. Recently, the NG-based catalysts having ORR activity comparable or higher than that of Pt-based catalysts have been reported [109–111]. The ORR activity of NCNTs/NG can also be exploited by using it as a catalyst support material for both noble as well as non-noble metal-based catalysts. High ORR activity with nearly four-electron transfer mechanism has been observed for iron-incorporated NG [112]. Byon et al., for example, have used NG, prepared by reducing graphite oxide (GO), to form an NPM-based catalysts [113]. Fe-N-C moieties containing 40 % pyridinic-N and an average N/Fe coordination of three (FeN3) are used as ORR catalysts. For comparison, various support materials such as CB, oxidized CB, and NG are used. The ORR activity is measured by rotating disk electrode (RDE) in O2-saturated 0.05 M H2SO4 electrolyte. Compared to the carbon black (CB), the NG-supported catalyst (Fe-NG) exhibits ~13 times enhanced ORR activity (Fig. 15.15).

ORR mass activity of graphene-supported catalyst vs. carbon black–supported catalysts (Reprinted with permission from [113])
Apart from N-doping, co-doping of B, sulfur (S), halogens, etc. with nitrogen improves the ORR activity further [114, 115]. Wang et al. have reported an NG-based ORR catalyst having superior catalytic activity when compared with a commercial Pt/C (20 wt% Pt) by co-doping of graphene with B and N. The catalysts are prepared by heating GO in the presence of boric acid (H3BO3) and ammonia (NH3). Similar co-doping of B and N is obtained by Xue et al. using melamine diborate as a precursor for CVD synthesis of graphene foams (GFs) [115]. Separate samples of the N-/B-doped GFs are prepared by using ammonia and triethyl borate as the N-/B- precursors, respectively. Cyclic voltammetry (CV) measurements show much higher ORR activity for the doped GFs as compared to the undoped ones. Moreover, the co-doped GFs show highest catalytic activity when compared with the single N- or B-doped samples. Figure 15.16a, b show the SEM image and cyclic voltammograms (N2- and O2-saturated 0.1 M KOH) of BN-doped GFs. A large reduction peak in presence of oxygen exhibits the high ORR activity. The BN-doped GFs show high CO tolerance when compared to the Pt-based catalysts.

(a) SEM image and (b) cyclic voltammograms of BN-doped GFs (Reprinted with permission from [115])
Similar to pyridinic-N catalytic active sites, ORR active edge-halogenated graphene has been reported recently by Jeon et al. [116]. The halogenated graphenes having halogen atoms (Cl/Br/I) at the graphene edges have been synthesized by ball-milling the pristine graphene in the presence of the respective halogens (Cl2/Br2/I2). The catalysts show high stability and low CO poisoning along with promising ORR activity. Similar to NG, the halogens help in the O2 adsorption and weaken the O-O bonds.
Materials with a combination of CNTs and NG may possess unique properties. In a study, Li et al. have synthesized NG nanosheets attached with CNTs by a heat treatment in NH3 [117]. The outer layers of CNTs are partially unzipped to form NG nanosheets, which remain attached to the parent CNT. The exfoliated outer walls contain small amounts of catalysts (Fe) and N impurities acting as catalytic sites, whereas the intact inner CNT facilitates the charge transport. The other forms of carbon nanomaterials such as carbon nanocups, and carbon nano-onions, when doped with N also exhibit catalytic activities similar to that of NCNTs. Tang et al. have studied the catalytic activity of N-doped carbon nanocups (NCNCs) for H2O2 oxidation, a reverse process to the ORR [118]. With an activity comparable to that of Pt/CNT (11.5 wt% Pt), NCNCs show potential to replace Pt-based catalysts. The high activity of NCNCs over NCNTs originates from the inhomogeneous N distribution within the cups. N-atoms are concentrated on the rims of the nanocups, which work as ORR active sites [119].
3 Conclusions
Owing to their high efficiency, PEMFCs are promising prospective power generation devices. However, the dependence on noble metal-based catalysts hinders their large-scale applicability. For successful commercialization of PEMFC, replacement of Pt-based CL with other low-cost and abundant catalysts is inevitable. Considerable efforts are being made toward the development of low-cost and stable catalysts for PEMFCs. Numerous low Pt/Pt-free ORR catalysts are widely investigated. ORR catalysts based on the transition metals demonstrate promising properties. Recently, carbon nanomaterials have shown considerable potential for ORR catalysis. The CNT-supported CLs show improved ORR activity and high durability due to their large surface area, high electronic conductivity, and high corrosion resistance. In addition, the ORR activity and stability of NCNTs and NG is promising toward complete replacement of Pt-based catalysts. However, large-scale production of CNTs/graphene-based catalysts for commercial applications remains challenging. The knowledge regarding the nature of the active sites and their dependence on experimental parameters is not well established. More efforts toward the development and performance evolution of FC stacks based on these catalysts are required.
References
Key World Energy Statistics (2011) International Energy Agency, Paris, p 6
Larminie J, Dicks A, Knovel (2003) Fuel cell systems explained. Wiley, Chichester, p 406
Macinnis P (2009) 100 discoveries: the greatest breakthroughs in history. Pier 9, Millers Point, p 287
Grove WR (1843) On the gas voltaic battery. Experiments made with a view of ascertaining the rationale of Its action and its application to eudiometry. Philos Trans R Soc Lond 133:91
Grove WR, Esq MA (1839) XXIV. On voltaic series and the combination of gases by platinum. Philos Mag 14:127
Mond L, Langer C (1889) A new form of gas battery. Philos Trans R Soc Lond 46:296
Bagotsky VS (2009) Fuel cells: problems and solutions. Wiley, Hoboken, p 320
Warshay M, Prokopius PR (1989) The fuel cell in space: yesterday, today and tomorrow. In: Grove anniversary (1839–1989) fuel cell symposium, London
Bacon FT, Fry TM (1973) Review lecture: the development and practical application of fuel cells. Philos Trans R Soc Lond A 334:427
Barbir F (2005) Chapter 1 – introduction. In: PEM fuel cells. Academic, Burlington, pp 1–16
Andujar JM, Segura F (2009) Fuel cells: history and updating. A walk along two centuries. Renew Sustain Energy Rev 13:2309
Litster S, McLean G (2004) PEM fuel cell electrodes. J Power Sources 130:61
Li H, Tang Y, Wang Z, Shi Z, Wu S, Song D, Zhang J, Fatih K, Zhang J, Wang H (2008) A review of water flooding issues in the proton exchange membrane fuel cell. J Power Sources 178:103
Tsuchiya H, Kobayashi O (2004) Mass production cost of PEM fuel cell by learning curve. Int J Hydrogen Energy 29:985
Antunes RA, Oliveira MCL, Ett G, Ett V (2010) Corrosion of metal bipolar plates for PEM fuel cells: a review. Int J Hydrogen Energy 35:3632
Hermann A, Chaudhuri T, Spagnol P (2005) Bipolar plates for PEM fuel cells: a review. Int J Hydrogen Energy 30:1297
Li X, Sabir I (2005) Review of bipolar plates in PEM fuel cells: flow-field designs. Int J Hydrogen Energy 30:359
Tawfik H, Hung Y, Mahajan D (2007) Metal bipolar plates for PEM fuel cell – a review. J Power Sources 163:755
Cha B-C, You Y-Z, Hong S-T, Kim J-H, Kim D-W, Lee B-S, Kim S-K (2011) Nitride films as protective layers for metallic bipolar plates of polymer electrolyte membrane fuel cell stacks. Int J Hydrogen Energy 36:4565
Dur E, Cora M, Koç M (2011) Experimental investigations on the corrosion resistance characteristics of coated metallic bipolar plates for PEMFC. Int J Hydrogen Energy 36:7162
Ramasubramaniam R, Chen J, Liu H (2003) Homogeneous carbon nanotube/polymer composites for electrical applications. Appl Phys Lett 83:2928
Lee JH, Jang YK, Hong CE, Kim NH, Li P, Lee HK (2009) Effect of carbon fillers on properties of polymer composite bipolar plates of fuel cells. J Power Sources 193:523
Antunes RA, de Oliveira MCL, Ett G, Ett V (2011) Carbon materials in composite bipolar plates for polymer electrolyte membrane fuel cells: a review of the main challenges to improve electrical performance. J Power Sources 196:2945
Wu M, Shaw LL (2005) A novel concept of carbon-filled polymer blends for applications in PEM fuel cell bipolar plates. Int J Hydrogen Energy 30:373
Cindrella L, Kannan AM, Lin JF, Saminathan K, Ho Y, Lin CW, Wertz J (2009) Gas diffusion layer for proton exchange membrane fuel cells – a review. J Power Sources 194:146
Park S, Lee J-W, Popov BN (2012) A review of gas diffusion layer in PEM fuel cells: materials and designs. Int J Hydrogen Energy 37:5850
Lysenko V (2008) Current trends in the design of gas-diffusion layers for fuel cells. Fibre Chem 40:226
Jordan LR, Shukla AK, Behrsing T, Avery NR, Muddle BC, Forsyth M (2000) Diffusion layer parameters influencing optimal fuel cell performance. J Power Sources 86:250
Pozio A, Cemmi A, Carewska M, Paoletti C, Zaza F (2010) Characterization of gas diffusion electrodes for polymer electrolyte fuel cells. J Fuel Cell Sci Technol 7:041003
Ismail MS, Damjanovic T, Ingham DB, Pourkashanian M, Westwood A (2010) Effect of polytetrafluoroethylene-treatment and microporous layer-coating on the electrical conductivity of gas diffusion layers used in proton exchange membrane fuel cells. J Power Sources 195:2700
Zhan Z, Xiao J, Li D, Pan M, Yuan R (2006) Effects of porosity distribution variation on the liquid water flux through gas diffusion layers of PEM fuel cells. J Power Sources 160:1041
Chen J, Matsuura T, Hori M (2004) Novel gas diffusion layer with water management function for PEMFC. J Power Sources 131:155
Lin G, Van Nguyen T (2005) Effect of thickness and hydrophobic polymer content of the gas diffusion layer on electrode flooding level in a PEMFC. J Electrochem Soc 152:A1942
Park S, Lee J-W, Popov BN (2008) Effect of PTFE content in microporous layer on water management in PEM fuel cells. J Power Sources 177:457
Smitha B, Sridhar S, Khan AA (2005) Solid polymer electrolyte membranes for fuel cell applications - a review. J Membr Sci 259:10
Hoogers G (2003) Fuel cell technology handbook. CRC Press, Boca Raton, Florida
Neburchilov V, Martin J, Wang H, Zhang J (2007) A review of polymer electrolyte membranes for direct methanol fuel cells. J Power Sources 169:221
Mench MM (2008) Fuel cell engines. Wiley, Hoboken, New Jersey, pp 1–26
Song C, Zhang J (2008) Electrocatalytic oxygen reduction reaction. In: Zhang J PEM fuel cell electrocatalysts and catalyst layers. Springer, London, pp 89–134
Wroblowa HS, Yen Chi P, Razumney G (1976) Electroreduction of oxygen: a new mechanistic criterion. J Electroanal Chem Interfacial Electrochem 69:195
Marković NM, Schmidt TJ, Stamenković V, Ross PN (2001) Oxygen reduction reaction on Pt and Pt bimetallic surfaces: A selective review. Fuel Cells 1:105
Norskov JK, Rossmeisl J, Logadottir A, Lindqvist L, Kitchin JR, Bligaard T, Jonsson H (2004) Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J Phys Chem B 108:17886
Shi Z, Liu H, Lee K, Dy E, Chlistunoff J, Blair M, Zelenay P, Zhang J, Liu Z-S (2011) Theoretical study of possible active site structures in cobalt- polypyrrole catalysts for oxygen reduction reaction. J Phys Chem C 115:16672
Nilekar AU, Mavrikakis M (2008) Improved oxygen reduction reactivity of platinum monolayers on transition metal surfaces. Surf Sci 602:L89
Jaouen F, Proietti E, Lefevre M, Chenitz R, Dodelet J-P, Wu G, Chung HT, Johnston CM, Zelenay P (2011) Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells. Energy Environ Sci 4:114
Takenaka S, Matsumori H, Matsune H, Tanabe E, Kishida M (2008) High durability of carbon nanotube-supported Pt electrocatalysts covered with silica layers for the cathode in a PEMFC. J Electrochem Soc 155:B929
Parthasarathy P, Virkar AV (2013) A study of electrochemical Ostwald ripening in Pt and Ag catalysts supported on carbon. ECS Trans 50:1547
Parthasarathy P, Virkar AV (2013) Electrochemical Ostwald ripening of Pt and Ag catalysts supported on carbon. J Power Sources 234:82
Zhai Y, Zhang H, Xing D, Shao Z-G (2007) The stability of Pt/C catalyst in H3PO4/PBI PEMFC during high temperature life test. J Power Sources 164:126
Akita T, Taniguchi A, Maekawa J, Siroma Z, Tanaka K, Kohyama M, Yasuda K (2006) Analytical TEM study of Pt particle deposition in the proton-exchange membrane of a membrane-electrode-assembly. J Power Sources 159:461
Borup RL, Davey JR, Garzon FH, Wood DL, Inbody MA (2006) PEM fuel cell electrocatalyst durability measurements. J Power Sources 163:76
Wilson MS, Garzon FH, Sickafus KE, Gottesfeld S (1993) Surface area loss of supported platinum in polymer electrolyte fuel cells. J Electrochem Soc 140:2872
Stevens DA, Dahn JR (2005) Thermal degradation of the support in carbon-supported platinum electrocatalysts for PEM fuel cells. Carbon 43:179
Choo H-S, Kinumoto T, Nose M, Miyazaki K, Abe T, Ogumi Z (2008) Electrochemical oxidation of highly oriented pyrolytic graphite during potential cycling in sulfuric acid solution. J Power Sources 185:740
Borup R, Meyers J, Pivovar B, Kim YS, Mukundan R, Garland N, Myers D, Wilson M, Garzon F, Wood D, Zelenay P, More K, Stroh K, Zawodzinski T, Boncella J, McGrath JE, Inaba M, Miyatake K, Hori M, Ota K, Ogumi Z, Miyata S, Nishikata A, Siroma Z, Uchimoto Y, Yasuda K, Kimijima KI, Iwashita N (2007) Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem Rev 107:3904
Lin JF, Kamavaram V, Kannan AM (2010) Synthesis and characterization of carbon nanotubes supported platinum nanocatalyst for proton exchange membrane fuel cells. J Power Sources 195:466
Li L, Xing Y (2006) Electrochemical durability of carbon nanotubes in noncatalyzed and catalyzed oxidations. J Electrochem Soc 153:A1823
Yin YF, Mays T, McEnaney B (1999) Adsorption of nitrogen in carbon nanotube arrays. Langmuir 15:8714
Peigney A, Laurent C, Flahaut E, Bacsa RR, Rousset A (2001) Specific surface area of carbon nanotubes and bundles of carbon nanotubes. Carbon 39:507
Pinero R, Amoro´s C, Linares Solano A, Delpeux S, Frackowiak E, Szostak K, Beguin F (2002) High surface area carbon nanotubes prepared by chemical activation. Carbon 40:1614
Maheshwari PH, Mathur RB (2009) Improved performance of PEM fuel cell using carbon paper electrode prepared with CNT coated carbon fibers. Electrochim Acta 54:7476
Lee K, Zhang J, Wang H, Wilkinson DP (2006) Progress in the synthesis of carbon nanotube- and nanofiber-supported Pt electrocatalysts for PEM fuel cell catalysis. J Appl Electrochem 36:507
Sun X, Li R, Stansfield B, Dodelet JP, Désilets S (2004) 3D carbon nanotube network based on a hierarchical structure grown on carbon paper backing. Chem Phys Lett 394:266
Saha MS, Li R, Sun X (2008) High loading and monodispersed Pt nanoparticles on multiwalled carbon nanotubes for high performance proton exchange membrane fuel cells. J Power Sources 177:314
Saha MS, Li R, Sun X, Ye S (2009) 3-D composite electrodes for high performance PEM fuel cells composed of Pt supported on nitrogen-doped carbon nanotubes grown on carbon paper. Electrochem Commun 11:438
Wang CH, Du HY, Tsai YT, Chen CP, Huang CJ, Chen LC, Chen KH, Shih HC (2007) High performance of low electrocatalysts loading on CNT directly grown on carbon cloth for DMFC. J Power Sources 171:55
Lebert M, Kaempgen M, Soehn M, Wirth T, Roth S, Nicoloso N (2009) Fuel cell electrodes using carbon nanostructures. Catal Today 143:64
Tang Z, Poh CK, Lee KK, Tian Z, Chua DHC, Lin J (2010) Enhanced catalytic properties from platinum nanodots covered carbon nanotubes for proton-exchange membrane fuel cells. J Power Sources 195:155
Tang Z, Ng HY, Lin J, Wee ATS, Chua DHC (2010) Pt/CNT-based electrodes with high electrochemical activity and stability for proton exchange membrane fuel cells. J Electrochem Soc 157:B245
Jeng KT, Chien CC, Hsu NY, Yen SC, Chiou SD, Lin SH, Huang WM (2006) Performance of direct methanol fuel cell using carbon nanotube-supported Pt-Ru anode catalyst with controlled composition. J Power Sources 160:97
Tsai HY, Wu CC, Lee CY, Shih EP (2009) Microbial fuel cell performance of multiwall carbon nanotubes on carbon cloth as electrodes. J Power Sources 194:199
Jeng KT, Chien CC, Hsu NY, Huang WM, Chiou SD, Lin SH (2007) Fabrication and impedance studies of DMFC anode incorporated with CNT-supported high-metal-content electrocatalyst. J Power Sources 164:33
Girishkumar G, Rettker M, Underhile R, Binz D, Vinodgopal K, McGinn P, Kamat P (2005) Single-wall carbon nanotube-based proton exchange membrane assembly for hydrogen fuel cells. Langmuir 21:8487
Shao Y, Yin G, Gao Y, Shi P (2006) Durability study of Pt/C and Pt/CNTs catalysts under simulated PEM fuel cell conditions. J Electrochem Soc 153:A1093
Wang X, Li W, Chen Z, Waje M, Yan Y (2006) Durability investigation of carbon nanotube as catalyst support for proton exchange membrane fuel cell. J Power Sources 158:154
Matsumori H, Takenaka S, Matsune H, Kishida M (2010) Preparation of carbon nanotube-supported Pt catalysts covered with silica layers; application to cathode catalysts for PEFC. Appl Catal Gen 373:176
Zhu W, Zheng JP, Liang R, Wang B, Zhang C, Au G, Plichta EJ (2009) Durability study on SWNT/nanofiber buckypaper catalyst support for PEMFCs. J Electrochem Soc 156:B1099
Kaempgen M, Lebert M, Roth S, Soehn M, Nicoloso N (2008) Fuel cells based on multifunctional carbon nanotube networks. J Power Sources 180:755
Wang J, Yin G, Shao Y, Wang Z, Gao Y (2008) Investigation of further improvement of platinum catalyst durability with highly graphitized carbon nanotubes support. J Phys Chem C 112:5784
Li L, Xing Y (2008) Electrochemical durability of at 80 °C. J Power Sources 178:75
Chen J, Wang JZ, Minett AI, Liu Y, Lynam C, Liu H, Wallace GG (2009) Carbon nanotube network modified carbon fibre paper for Li-ion batteries. Energy Environ Sci 2:393
Min M-k, Cho J, Cho K, Kim H (2000) Particle size and alloying effects of Pt-based alloy catalysts for fuel cell applications. Electrochim Acta 45:4211
Li B, Chan SH (2013) PtFeNi tri-metallic alloy nanoparticles as electrocatalyst for oxygen reduction reaction in proton exchange membrane fuel cells with ultra-low Pt loading. Int J Hydrogen Energy 38:3338
Vinayan BP, Jafri RI, Nagar R, Rajalakshmi N, Sethupathi K, Ramaprabhu S (2012) Catalytic activity of platinum/cobalt alloy nanoparticles decorated functionalized multiwalled carbon nanotubes for oxygen reduction reaction in PEMFC. Int J Hydrogen Energy 37:412
Gasteiger HA, Kocha SS, Sompalli B, Wagner FT (2005) Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl Catal, B 56:9
Jasinski R (1964) A new fuel cell cathode catalyst. Nature 201:1212
Othman R, Dicks AL, Zhu Z (2012) Non precious metal catalysts for the PEM fuel cell cathode. Int J Hydrogen Energy 37:357
Xu Z, Li H, Cao G, Zhang Q, Li K, Zhao X (2011) Electrochemical performance of carbon nanotube-supported cobalt phthalocyanine and its nitrogen-rich derivatives for oxygen reduction. J Mol Catal A Chem 335:89
Wiesener K, Ohms D, Neumann V, Franke R (1989) N4 macrocycles as electrocatalysts for the cathodic reduction of oxygen. Mater Chem Phys 22:457
Bezerra CWB, Zhang L, Lee K, Liu H, Marques ALB, Marques EP, Wang H, Zhang J (2008) A review of Fe-N/C and Co-N/C catalysts for the oxygen reduction reaction. Electrochim Acta 53:4937
Bashyam R, Zelenay P (2006) A class of non-precious metal composite catalysts for fuel cells. Nature 443:63
Martinez Millan W, Toledano Thompson T, Arriaga LG, Smit MA (2009) Characterization of composite materials of electroconductive polymer and cobalt as electrocatalysts for the oxygen reduction reaction. Int J Hydrogen Energy 34:694
Qin HY, Liu ZX, Yin WX, Zhu JK, Li ZP (2008) A cobalt polypyrrole composite catalyzed cathode for the direct borohydride fuel cell. J Power Sources 185:909
Wu G, More KL, Johnston CM, Zelenay P (2011) High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 332:443
Wu G, Artyushkova K, Ferrandon M, Kropf AJ, Myers D, Zelenay P (2009) Performance durability of polyaniline-derived non-precious cathode catalysts. ECS Trans 25:1299
Wu G, Chen Z, Artyushkova K, Garzon FH, Zelenay P (2008) Polyaniline-derived non-precious catalyst for the polymer electrolyte fuel cell cathode. ECS Trans 16:159
Wu G, Johnston CM, Mack NH, Artyushkova K, Ferrandon M, Nelson M, Lezama-Pacheco JS, Conradson SD, More KL, Myers DJ, Zelenay P (2011) Synthesis-structure-performance correlation for polyaniline-Me-C non-precious metal cathode catalysts for oxygen reduction in fuel cells. J Mater Chem 21:11392
Matter PH, Wang E, Arias M, Biddinger EJ, Ozkan US (2007) Oxygen reduction reaction activity and surface properties of nanostructured nitrogen-containing carbon. J Mol Catal A Chem 264:73
Matter PH, Zhang L, Ozkan US (2006) The role of nanostructure in nitrogen-containing carbon catalysts for the oxygen reduction reaction. J Catal 239:83
Gong K, Du F, Xia Z, Durstock M, Dai L (2009) Nitrogen-doped carbon nanotube arrays with high electrocatalytic activity for oxygen reduction. Science 323:760
Wang S, Zhang L, Xia Z, Roy A, Chang DW, Baek J-B, Dai L (2012) BCN graphene as efficient metal-free electrocatalyst for the oxygen reduction reaction. Angew Chem Int Ed 51:4209
Zheng Y, Jiao Y, Jaroniec M, Jin Y, Qiao SZ (2012) Nanostructured metal-free electrochemical catalysts for highly efficient oxygen reduction. Small 8:3550
Kundu S, Nagaiah TC, Xia W, Wang Y, Dommele SV, Bitter JH, Santa M, Grundmeier G, Bron M, Schuhmann W, Muhler M (2009) Electrocatalytic activity and stability of nitrogen-containing carbon nanotubes in the oxygen reduction reaction. J Phys Chem C 113:14302
Subramanian NP, Li X, Nallathambi V, Kumaraguru SP, Colon-Mercado H, Wu G, Lee J-W, Popov BN (2009) Nitrogen-modified carbon-based catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. J Power Sources 188:38
Rao CV, Cabrera CR, Ishikawa Y (2010) In search of the active site in nitrogen-doped carbon nanotube electrodes for the oxygen reduction reaction. J Phys Chem Lett 1:2622
Chen Z, Higgins D, Tao H, Hsu RS, Chen Z (2009) Highly active nitrogen-doped carbon nanotubes for oxygen reduction reaction in fuel cell applications. J Phys Chem C 113:21008
Chen Z, Higgins D, Chen Z (2010) Nitrogen doped carbon nanotubes and their impact on the oxygen reduction reaction in fuel cells. Carbon 48:3057
Lin Z, Song M-k, Ding Y, Liu Y, Liu M, Wong C-p (2012) Facile preparation of nitrogen-doped graphene as a metal-free catalyst for oxygen reduction reaction. Phys Chem Chem Phys 14:3381
Ci S, Wu Y, Zou J, Tang L, Luo S, Li J, Wen Z (2012) Nitrogen-doped graphene nanosheets as high efficient catalysts for oxygen reduction reaction. Chin Sci Bull 57:3065
Lin Z, Waller GH, Liu Y, Liu M, Wong C-p (2012) Simple preparation of nanoporous few-layer nitrogen-doped graphene for use as an efficient electrocatalyst for oxygen reduction and oxygen evolution reactions. Carbon 53:130
Zheng B, Wang J, Wang F-B, Xia X-H (2013) Synthesis of nitrogen doped graphene with high electrocatalytic activity toward oxygen reduction reaction. Electrochem Commun 28:24
Parvez K, Yang S, Hernandez Y, Winter A, Turchanin A, Feng X, Müllen K (2012) Nitrogen-doped graphene and its iron-based composite as efficient electrocatalysts for oxygen reduction reaction. ACS Nano 6:9541
Byon HR, Suntivich J, Shao-Horn Y (2011) Graphene-based non-noble-metal catalysts for oxygen reduction reaction in acid. Chem Mater 23:3421
Su Y, Zhang Y, Zhuang X, Li S, Wu D, Zhang F, Feng X (2013) Low-temperature synthesis of nitrogen/sulfur co-doped three-dimensional graphene frameworks as efficient metal-free electrocatalyst for oxygen reduction reaction. Carbon 62:296
Xue Y, Yu D, Dai L, Wang R, Li D, Roy A, Lu F, Chen H, Liu Y, Qu J (2013) Three-dimensional B, N-doped graphene foam as a metal-free catalyst for oxygen reduction reaction. Phys Chem Chem Phys 15:12220
Jeon I-Y, Choi H-J, Choi M, Seo J-M, Jung S-M, Kim M-J, Zhang S, Zhang L, Xia Z, Dai L, Park N, Baek J-B (2013) Facile, scalable synthesis of edge-halogenated graphene nanoplatelets as efficient metal-free eletrocatalysts for oxygen reduction reaction. Sci Rep 3:1
Li Y, Zhou W, Wang H, Xie L, Liang Y, Wei F, Idrobo J-C, Pennycook SJ, Dai H (2012) An oxygen reduction electrocatalyst based on carbon nanotube-graphene complexes. Nat Nano 7:394
Tang Y, Allen BL, Kauffman DR, Star A (2009) Electrocatalytic activity of nitrogen-doped carbon nanotube cups. J Am Chem Soc 131:13200
Allen BL, Kichambare PD, Star A (2008) Synthesis, characterization, and manipulation of nitrogen-doped carbon nanotube cups. ACS Nano 2:1914
Acknowledgements
The authors acknowledge the financial support provided by Department of Science and Technology Nano Mission, India, for carrying out this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Sharma, R., Cherusseri, J., Kar, K.K. (2015). Polymer Electrolyte Membrane Fuel Cells: Role of Carbon Nanotubes/Graphene in Cathode Catalysis. In: Kar, K., Pandey, J., Rana, S. (eds) Handbook of Polymer Nanocomposites. Processing, Performance and Application. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-45229-1_81
Download citation
DOI: https://doi.org/10.1007/978-3-642-45229-1_81
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-45228-4
Online ISBN: 978-3-642-45229-1
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)