
Abstract
In skeletal muscle a rise in the cytosolic calcium concentration is the first trigger able to initiate the contraction of the sarcomere. Intracellular calcium levels are tightly controlled by channels and pumps, and it is not surprising that many inherited skeletal muscle disorders arise from mutations altering the players regulating calcium ions concentration (Betzenhauser and Marks 2010). In this chapter, we will focus on the pathologies linked to the sarcoplasmic reticulum calcium channel-RyR1 mutations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 The Muscle Excitation-Contraction Coupling: Calcium Release Complex and RyR1
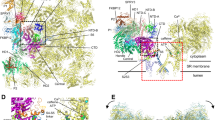
Skeletal muscle contraction is initiated by a burst of calcium out of its major store, the sarcoplasmic reticulum. This rise in intracellular calcium concentration is triggered by a post synaptic action potential which propagates along the muscle fiber and reaches plasma membrane invaginations called transverse tubules. Transverse tubules (T-tubules) form regular invaginations along the muscle axis and make close and periodic contacts with the sarcoplasmic reticulum (SR) that is wrapped around the myofibril apparatus. These peculiar membrane structures are called triads (Fig. 9.1). Triads are formed by the tight apposition of two terminal cisternae of SR on both sides of a T-tubule, and are specifically localized at the A-I bands transition in mammalian skeletal muscle. The transformation of an action potential into a massive calcium release, a process called the Excitation-Contraction (EC) coupling, is performed by a macromolecular complex, the Calcium Release Complex (CRC). This complex is anchored both in the T-tubules membrane and in the SR membrane, and has specific triad localization. The main players of CRC are the dihydropyridine receptor (DHPR), an L-type calcium channel of the T-tubules membrane, and the ryanodine receptor (RyR1), the sarcoplasmic reticulum calcium channel (Fig. 9.1). When an incoming action potential reaches the T-tubules, it activates the voltage gated DHPR, which functions both as a calcium channel and a voltage sensor. This first step triggers a conformational modification in the alpha 1 subunit of DHPR (Cav1.1) which is in direct contact with RyR1. The physical coupling allows a structural modification in the RyR1 channel upon DHPR activation that results in its opening and in a massive calcium efflux from the terminal cisternae of SR. The released calcium interacts with the myofilaments and induces the muscle contraction. Muscle relaxation occurs when RyR1 closes as a result of high cytosolic calcium concentration, and calcium is reuptake into the sarcoplasmic reticulum by the SERCA (Sarcoplasmic Endoplasmic Reticulum ATPase) calcium pump.

The calcium release complex in the triad of skeletal muscle
When analyzed by electron microscopy (EM), triad membranes appear as regular structures, with a 15 nm space between T-tubule and SR membrane. In this space, the cytosolic part of the 2.3 MDa RyR1 tetramer is so large that it forms an electron dense structure, the characteristic “feet” early observed between T-tubule and SR terminal cisternae (Franzini-Armstrong et al. 1983). The protein was identified thanks to its binding to ryanodine, an alkaloid extracted from the tropical plant Ryana speciosa, and shown to be the intracellular calcium channel of muscle cells (Inui et al. 1987). In mammals, three isoforms of ryanodine receptor have been cloned from three different genes arising from a common ancestor. RyR1 is the major skeletal muscle isoform, but is also expressed in lymphocytes and dendritic cells (Bracci et al. 2007; Uemura et al. 2007) or in specific areas of the central nervous system (Giannini et al. 1995). RyR2 is predominantly expressed in the heart, but it is also in the main brain isoform. RyR3 is weakly expressed in a large panel of tissues, among which skeletal muscle and brain. The repertoire of ryanodine receptors may vary in gene composition and expression among species. In teleost fishes RYR2 and RYR3 are conserved but the RYR1 gene has been duplicated in α and β isoforms, respectively expressed in fast- or slow-twitch muscles (Franck et al. 1998; Hirata et al. 2007), whereas in the genome of Caenorhabditis elegans and Drosophila melanogaster, only one RYR homologous gene is found (Hasan et al. 1992; Maryon et al. 1996).
The human RYR1 gene is located on chromosome 19q13.2. It spans 153 865 base pairs, and contains 106 exons. RYR1 is alternatively spliced for the exons 70 and 83, but the functional role of these splicing events is unclear. The 560 kDa protein RyR1 consists of 5038 amino acid that share 60–70 % identity with RyR2 and RyR3. The functional RyR1 channel is a homotetramer whose structure is so far only partially solved. Cryo-electron microcopy images have allowed 9.6 Å resolution reconstitutions of the 3D structure of the channel (Serysheva et al. 2008) in different activation states (Samsó et al. 2009). More recently, portions of the molecule have been crystalized and their structure identified at atomic resolution (Tung et al. 2010; Yuchi et al. 2012).
Nevertheless, in the absence of precise localization, most of the functional domains that have been identified for RyR1 are mapped on the linear sequence of the protein (Fig. 9.2): E–C domains allowing interaction with the DHPR or regulatory proteins of the CRC such as calmodulin and FKBP12, modulatory regions allowing the interaction with kinases (PKC and PKA), ATP, and calcium ions. The major part (4,000 first amino acids) of the RyR1 sequence is cytosolic, and forms the so-called “feet” of the molecule observed in EM. A number of modulators of RyR1 have their binding domain in this huge cytosolic region: Ca2+, ATP, Mg2+ as well as pharmacological agents (ryanodine, caffeine, halothane, dantrolene, or ruthenium red). It is also in the cytosolic part that RyR1 undergoes phosphorylation and nitrosylation, both modifications being involved in the regulation of the channel activity (Lanner et al. 2010). The last 1,000 amino acids, the C-terminal part of the protein, contain 6–8 transmembrane helices responsible for SR membrane anchoring and form the pore domain of the channel (Du et al. 2002). In its isolated form, the RyR1 channel is activated by Ca2+ or ryanodine at low concentration (μM), by ATP, caffeine, or halothane. On the other hand, elevated concentrations (>100 μM) of Ca2+, Mg2+, and ryanodine inhibit its activity (Meissner 1994). Hence the sensibility of RyR1 to different ranges of calcium concentration allows the channel to be in a closed state at resting calcium levels (50–150 nM) and to be inactivated when cytosolic calcium concentration rise above the mM range, preventing a deleterious prolonged activation (Fill and Copello 2002).

Schematic representation of the structure of RyR1 showing the different functional domains on the linear structure
2 Implication of RYR1 in Muscle Pathologies
Two classes of pathologies that differ in their presentation have been so far associated to the RYR1 gene: The first one is triggered by external stimuli and the second one appears at birth (Fig. 9.3). The first pathology ever associated to RyR1 was Malignant Hyperthermia (MH) in 1991 (Gillard et al. 1991). Mutations in RyR1 were further identified in association with Central Core Disease (CCD), a congenital myopathy of moderate severity, characterized by the presence of “cores”, abnormalities in the muscle fiber revealed either by histology or electron microscopy studies (Magee et al. 1956). The description of severe forms of myopathies with cores and of other myopathies with histological abnormalities has then led to the notion that a whole class of myopathies, now called RyR1-Related Congenital Myopathies or RyR1-RCM, can be related to RyR1 dysfunction.

Pathologies associated to RyR1 dysfunction
2.1 Induced Myopathies
2.1.1 Malignant Hyperthermia
Malignant Hyperthermia (OMIM:145600) is a pharmacogenetic disease of the skeletal muscle with a dominant mode of inheritance. The MH crisis is triggered by exposition to inhalational anesthetics, like halothane or isoflurane, and results in a hypermetabolic state of skeletal muscles. It is actually the clinical expression of an otherwise infra-clinic metabolic myopathy (Rosenberg et al. 2007). The exposition of susceptible individuals to the triggering agent leads to an increase in the myoplasmic calcium concentration followed by a generalized muscle contracture and by hyperthermia. A MH crisis will rapidly result in acidosis of both respiratory and metabolic origin, in a progressive degradation of muscle with a major rhabdomyolysis, together with hyperkalemia responsible for cardiac arrhythmia, hypercapnia and central hyperthermia. Hence, a MH crisis can be lethal in absence of the prompt administration of sodium dantrolene, a calcium channel blocker.
The exact prevalence of MH susceptibility is difficult to evaluate because patients do not present with symptoms apart from exposition to the triggering agent. Empirically, it has been estimated that each year 1 MH crisis arise among a population of 2 million people in Europe or North-America. This estimation probably underestimates the genetic MH susceptibility in the general population. Indeed, it has been shown that some patients undergo a MH crisis only at the second or third exposition to the triggering agent. Based on the genetic studies, it was calculated that the frequency of RYR1 mutations responsible for MH could reach 1/2,000 in the general population (Monnier et al. 2002).
In vitro studies demonstrated that contraction of skeletal muscles fibers from MH susceptible patients in response to caffeine or halothane was enhanced compared to control. This observation was pivotal in the development of a biologic testing allowing the identification of individuals at risk for MH, the in vitro contracture test (IVCT). Two protocols are prevailing today, one used in North America and the other in Europe, and although they are based on the very same principle they differ in their experimental details and interpretation. In Europe, the contraction level of muscle fibers bathed in buffers containing increasing doses of caffeine or halothane allows to define phenotypes as normal (MHN), equivocal (MHE, when fibers contract in response to low doses of only one of the triggering agent), or susceptible (MHS, when muscle contracts in response to both agents). Noticeably, patients tested as MHE are regarded as clinically susceptible to MH. In the North American protocol, individuals are considered as susceptible to MH whenever the fibers contract with caffeine or halothane. Positive testing in IVCT is still the gold standard for MH diagnosis (EMHG 1984), but despite a 99 % sensitivity and a 94 % specificity, this test can still lead to false positive results. In Japan, a muscle biopsy is also required to test MH susceptibility, but the protocol uses the measure of calcium induced calcium release on skinned fibers (Ibarra et al. 2006). Independently of the protocol used to test MH susceptibility, genetic analysis is mandatory to identify the mutation thus allowing less invasive testing of relatives.
The RYR1 gene was linked to MH in 1990 (MacLennan et al. 1990; McCarthy et al. 1990), and the first mutation described 1 year later (Gillard et al. 1991). To date, more than 160 mutations have been associated to MH (Monnier et al. 2005; Robinson et al. 2006; Sambuughin et al. 2005). The mutations are clustered in three regions of the gene: MH1, corresponding to the N-terminal part of the protein between amino acids 35 and 614; MH2, a central zone between amino acids 2,163 and 2,458; and MH3, localized in the C-terminal transmembrane domain (Fig. 9.2). In the European population, exhaustive sequencing showed that 60–75 % of the MH susceptible families have a RYR1 mutation (Robinson et al. 2006), suggesting the involvement of other genes. MH genetic heterogeneity was confirmed when the gene encoding the α1-subunit of the DHPR, was also linked to the pathology, with different mutations being described in CACNA1S (Monnier et al. 1997; Pirone et al. 2011; Stewart et al. 2001; Toppin et al. 2010). However, genetic detection of a RYR1 mutation has been accepted as an established cause of MH only for 30 well-characterized RYR1 variants (http://www.emhg.org), and so far only the identification in a patient of one of these 30 mutations can replace a positive IVCT test as a formal MH diagnosis (Urwyler et al. 2001).
RYR1 mutations associated to MH are inherited on a dominant mode and are for most of them missense mutations or small DNA deletion leading to in-frame amino acids deletions. Some mutations are highly recurrent and patients with homozygous RyR1 MH mutation have been characterized. Their phenotype was similar to heterozygous patients, except for elevated plasma creatine kinase levels (Monnier et al. 2002). Although MH is considered as a non-symptomatic syndrome without exposition to the triggering agent, clinical manifestations such as myalgias, cramps, and rhabdomyolysis have been reported among few MH susceptible patients, most of them after physical exercise (Wappler et al. 2001).
2.1.2 Exertional Heat Stroke
Exertional Heat Stroke (EHS) is characterized by hyperthermia, central neurologic disorders, and variable levels of rhabdomyolysis during, or after, an intense or prolonged physical exercise. EHS is considered as exercise-induced hyperthermia.
EHS should be distinguished from Heat Stroke (HS) arising from exposure to overheated environment and Exercise-linked Rhabdomyolysis (ER), which usually happens after an intense exercise inducing skeletal muscles damages resulting to mechanical or metabolic stress. Notably, ER can precede EHS.
MHS, EHS, and ER are characterized by abnormal metabolic states with enhanced oxidative stress and increased cytosolic calcium levels (Capacchione et al. 2009). A common genetic defect has been hypothesized to explain this hypermetabolic state. Accordingly, a few studies have shown that mutations in the RYR1 gene are detected in ER or EHS patients (Tobin et al. 2001; Wappler et al. 2001), without a clear evidence of clustering (Fig. 9.4). Our laboratory has recently shown in collaboration with the medical health service of the French Army that 15 % of the patients presenting with an EHS phenotype had a positive IVCT (unpublished data). Moreover, 25 % of these EHS/MHS patients had a RYR1 variation. Without further analysis of these RYR1 gene variations, it is however difficult to demonstrate their role in the pathophysiology of EHS. It can still be concluded form this study that individuals with RYR1 MH-related variations could be at risk of EHS during intensive physical exercise, which is in accordance with the usual recommendation made to MHS patients not to practice prolonged and intensive physical activities (Hopkins et al. 2007).

Occurrence of RyR1 mutations in association with different pathologies. Each mutation is represented by a vertical bar on the sequence of RyR1, the length of which correspond to the number of independent probands identified with the same mutation, and is classified by disease
2.2 Congenital Myopathies
2.2.1 A Heterogeneous Family of Pathologies
Congenital myopathies (CM) are a group of muscle diseases with heterogeneity in clinical, genetic, and histological presentations whose discovery has been tightly correlated with the development of histology and electron microscopy in the 1960s. The estimated incidence of CM is 6/100,000, which would rank CM among the most frequent myopathies. CM are often associated with a neonatal hypotonia of variable severity and a non-progressive muscle weakness. A primary diaphragm dysfunction can be the major sign for severe dominant phenotypes. Molecular genetic studies have revealed several genes responsible for the various forms of CM: ACTA1, NEB, RYR1, SEPN1, MTM1, DNM2 … A first attempt of CM classification has been performed based exclusively on the histological observations. However, the recent molecular studies have shown the existence of various clinical presentations with overlapping histological and genetic bases (Dubowitz and Swery 2007). The RYR1 gene was initially associated only with dominant forms of Central Core Disease (CCD), but is now also linked to Multi-minicore Disease (MmD), Core Myopathies with rods, CentroNuclear Myopathy (CNM), and Congenital Fiber Type disproportion (CFTD) (Wilmshurst et al. 2010; Clarke et al. 2010). Altogether, the pathologies involving the RYR1 gene, although variable in their clinical signs, should be classified as RyR1-Related Congenital Myopathies (RyR1-RCM).
2.2.2 Dominant Myopathies Linked to RYR1
2.2.2.1 Central Core Disease and Other Moderate Forms
In 1956, Shy and Magee described a new non-progressive myopathy, inherited on a dominant mode and affecting 5 patients from 2–65 years of age in three generations of the same family. The initial clinical description showed a congenital hypotonia, a delayed motor acquisition during childhood, and a slow evolution toward legs proximal muscles weakness (Shy and Magee 1956). Histological analysis revealed the presence in central portion of the fiber of numerous zones devoid of oxidative and phosphorylative activities (Fig. 9.5a), spanning along the entire longitudinal axis of the muscle. In 1958 the name “Central Core Disease” was proposed for such a clinical presentation (Greenfield et al. 1958). Cores morphology observed in electron microscopy is in agreement with histological data, showing absence of mitochondria and of sarcoplasmic reticulum in these regions associated with massive sarcomeric disorganization. Although the Central Core Disease initially described by Shy and Magee is the main dominant form, other histological presentations have been described (cores and rods, multiple cores, minicores) (Table 9.1 and Fig. 9.5) (Romero et al. 2005), associated with moderate hypotonia in early childhood, delay in motor milestone acquisition, and muscle weakness. The walking ability is usually preserved and the evolution is slowly progressive, although orthopedic complications often give rise to scoliosis. In less than 10 % of the cases, a severe neonatal hypotonia is associated to delay in motor function acquisition, however the evolution is usually favorable. The involvement of the 19q13.1 locus, which contains the RYR1 gene, has been proposed since 1990 by linkage analyses performed in families with dominant forms of CCD (Haan et al. 1990). To date, tenths of mutations in RYR1 have been associated with dominants forms of CCD, with more than 85 % of them being localized in the C-terminal pore forming region of the protein (Lynch et al. 1999; Monnier et al. 2000; Monnier et al. 2001; Scacheri et al. 2000). The identified CCD mutations are until now exclusively missense mutations or in-frame micro deletions of one or few amino acid. From our experience on a cohort of 116 patients with moderate forms of CCD, 40 % of the 71 mutations we have found are recurrent or highly recurrent (Fig. 9.4). Mutation p.Arg4861His was for instance identified in 13 % of the patients, and mutation p.Arg2508Cys in 6 % of the patients. De novo mutations are very frequent (25 %) in these dominant CCD forms.

Histological abnormalities observed in RyR1-RCM. a Central Cores (NADH-TR staining). b Minicores (NADH-TR staining). c Nuclear centralization (Hematoxyline-eosin staining). d Cores (star) and rods (arrow)
2.2.2.2 Severe Dominant Neonatal Cores Myopathy
Although the majority of dominant mutations of RYR1 are associated with moderate forms of the disease, in our experience 15 % of them were associated with a severe neonatal hypotonia that led to respiratory failure and could be lethal in the first weeks. Half of these patients required respiratory assistance, 2/3 did not acquire walking and 1/3 showed signs of multiple arthrogyposis from birth. These severe forms of CCD were associated with de novo missense mutations localized in the C-terminal domain (Fig. 9.2), half of them being clustered in the last luminal loop forming the pore of the RyR1 channel.
2.2.2.3 Inter- and Intra familial Phenotypic Heterogeneity
Recurrent mutations have allowed the study of interfamilial phenotypic heterogeneity for CCD. Among the 83 dominant mutations our laboratory has identified, 5 are responsible for both severe and moderate clinical signs. For instance the p.Arg4861Cys mutation detected in 30 % of de novo severe forms has also been identified in 3 % of the moderate forms of CCD. Along the same line, the p.Ile4898Thr mutation associated to moderate phenotypes has been associated to a lethal de novo myopathy in twins (Hernandez-Lain et al. 2011). Such variability in phenotypic expression can be observed among members of the same family and in 8 % of the families we have explored, clinical signs worsened from one generation to the next. Only once was the aggravation associated to another variation in the RYR1 gene (Monnier et al. 2008) showing that intrafamilial variability could arise from genetic background or variations in modifying genes.
2.2.3 Recessive Mutations Associated to RyR1
The description of recessive forms of congenital myopathies linked to RyR1 is more recent (Ferreiro et al. 2002a; Monnier et al. 2003; Romero et al. 2003). Congenital Multi-minicore Disease (MmD) of recessive mode of inheritance was defined on histological bases by the observation of multiple small regions devoid of oxidative activity, of only few sarcomers length, in both type I and II fibers (Ferreiro et al. 2000). The selenoprotein N gene (SEPN1) was first associated with 50 % of the severe so-called “classical” forms of MmD (Ferreiro et al. 2002b). However, RYR1 mutations were further identified in families with MmD forms associated with external ophtamloplegia, as an indication of the genetic heterogeneity of MmD (Monnier et al. 2003).
Further molecular studies showed that the number of MmD patients harboring RyR1 mutations was more important than initially suspected (Klein et al. 2012; Monnier et al. 2008; Zhou et al. 2006). Recessive RyR1 mutations have been identified in 30 % of the severe MmD cases we have analyzed. Clinical signs of MmD comprise neonatal hypotonia, severe respiratory failure, sucking and swallowing difficulties, eye muscle involvement (ophtalmoplegia, ophtalmoparesis, ptosis), arthrogryposis, and skeletal deformations in 1/3 of the cases (scoliosis, cyphoscoliosis, hyperlordosis). The generalized muscle weakness leads to lethality in 30 % of the severe cases. The clinical signs associated to moderate recessive forms of MmD linked to RyR1 mutations are similar to those of moderate dominant forms of CCD, except for eye muscle involvement, usually not associated to CCD.
Histologically, the analysis of MmD can be confusing because of the large spectrum of signs, which can even be associated to other forms of congenital myopathies. First the cores can vary in size, number or morphology, depending on the muscle explored or the age of the patient. Cores presentation can vary for a same patient on biopsies performed at different ages. Cores can also be associated with rods, or nuclei internalization, that are the main sign for the Centronuclear Myopathy (CNM). Nevertheless, opposite to CNM, internalizations of nuclei are usually associated in MmD with myofibrilar disorganization. At last, nonspecific histological presentations have also been reported, with a few centralized nuclei, local myofibrilar disorganization, predominance or hypotrophy of type I fibers. In rare cases a fiber type disproportion has also been described (Fig. 9.5).
Whereas mutations in the RYR1 gene leading to CCD are clustered in hot spots, mutations linked to MmD are spread all along the coding sequence (Fig. 9.4). Our laboratory has entirely sequenced the RYR1 gene in 106 families with recessive RyR1-RCM. Seventy five percent of the mutations we have detected are missense or small duplication/deletion mutations, among which 8 % are also known to be responsible for MH when expressed at the heterozygous state. The remaining 25 % of the mutations are nonsense mutation or are mutations affecting mRNA or protein stability, leading to a quantitative defect of RyR1 in the affected muscle (Monnier et al. 2008). A compound heterozygous status with 2 associated missense mutations was found for 58 % of the patient with a recessive form of RyR1-RCM. In 37 % of these cases a missense mutation was associated with a mutation affecting the quantity of RyR1 protein (nonsense or splicing mutation). Two mutations affecting RyR1 quantities were associated with only 5 % of the cases, (Klein et al. 2012; Monnier et al. 2003; Monnier et al. 2008; Zhou et al. 2006). However, a RyR1-RCM with complete loss of RyR1 protein has never been described so far, in good agreement with studies performed on mice showing that deletion of the RYR1 gene is lethal (Takeshima et al. 1994).
2.2.4 Complexity of RyR1-RCM Diagnosis
RyR1-RCM are among the most frequent congenital myopathies. Historically, RyR1-RCM were classified into clearly distinct entities: MH, which manifestation follows exposition to inhalational anesthetics, and CCD, characterized by muscle weakness and histologically diagnosed after analysis of muscle biopsies. The identification of core structures on muscle sections has remained a strong hint of a pathology linked to the RYR1 gene, especially with the increasing number of recessive mutations involved in MmD. The presence of cores as a sign of Core Myopathy should nonetheless be taken with care. A study realized in our laboratory on a panel of 242 Malignant Hyperthermia patients without any muscle weakness has shown the presence of cores in 20 % of the biopsies (Monnier et al. 2005). These patients harbored a RYR1 mutation, cores had been observed on muscle sections, but they did not show clinical signs of a myopathy. They should therefore be classified as “MHS patients with cores” rather than MH/CCD patients as it is sometimes stated in the literature. The “CCD/MH” phenotype should exclusively be attributed to patients that present with clear signs of both pathologies, on the histological and clinical point of view, which mean they have both a muscle weakness together with a positive susceptibility to MH.
With the increasing heterogeneity of clinical presentations that can be linked to RYR1 mutations, histopathology analysis has to be backed by other techniques to refine the diagnosis. Interestingly, the skeletal muscles MRI profile from RyR1-RCM patients has been described as typical and could be used as a strong indication for investigations of the RYR1 gene. The gluteus maximus from limb girdle, the vastus lateralis, adductor magnus, and sartorius from tight as well as the soleus from the leg are often altered, whereas the rectus femoris, biceps femoris, gracilus, and gastrocnemius are preserved (Jungbluth et al. 2004; Quijano-Roy et al. 2011). Genetic studies are nevertheless mandatory to show the involvement of RYR1. Although RyR1 mutations are frequent and spread all over the coding sequence, about 40 % of patients with well-characterized forms of CCD analyzed in our laboratory have no mutation in their entire RyR1 coding sequence, showing also a genetic heterogeneity of this congenital myopathy.
The ongoing introduction of massive parallel sequencing (or Next Generation Sequencing, NGS) in clinical diagnosis will most probably modify the strategies used to investigate heterogeneous muscle diseases. With such powerful sequencing capacity, it will be tempting to systematically sequence RYR1 gene for any clinical presentation with minimal signs of RyR1-RCM. This approach will probably be useful but must be balanced with the complexity of the pathophysiological mechanisms so far revealed. With an expected increase in the number of detected RyR1 variants, it will soon be mandatory to have clear information regarding the functional effect for each of these variations in order to unambiguously confirm their role in the pathology: disease causing mutation, modifying factor or polymorphisms. The detailed investigation of pathophysiological mechanisms for RyR1-RCM is thus a key step in the diagnosis process of these myopathies.
3 Pathophysiological Mechanisms Associated to RyR1 Mutations
Several mechanisms have been proposed to explain the impact of missense RyR1 mutations (Betzenhauser and Marks 2010; Lanner et al. 2010; MacLennan and Zvaritch 2011), as a result of functional studies performed using different tools. A number of studies used cells produced from the patients (primary muscle cultures, myotubes produced from patient’s fibroblasts transformed into muscle progenitors by MyoD expression, or immortalized lymphocytes). Other studies used expression of mutant RyR1 in non-muscle cell lines (HEK cells) or in mouse muscle cells devoid of endogenous RyR1 (primary culture from RyR1 KO mouse muscles). Some mutations have been studied with the different expression systems, and the proposed mechanisms are different, indicating that each expression system has its limitation and drawback. Therefore the actual pathophysiological mechanisms resulting from a precise mutation are probably more complex than the simplified and schematic mechanisms described thereafter.
3.1 RyR1 Gain of Function
Some of the studies led to the conclusion that mutant forms of RyR1 have a gain of function, i.e., the channel is releasing an increased amount of calcium in some conditions. This gain of function could have two expressions. First it can result in a hypersensitivity of the channel to pharmacologic agents (halothane, caffeine,…), which is characteristic of the so-called “MH mutation.” This is typically what is assayed when performing IVCT, and correspond to the shift in the dose response curve representing the amplitude of calcium released by increasing concentrations of caffeine or chlorocresol. It has been proposed that these MH mutations lower the threshold of RyR1 activation by luminal calcium in the presence of triggering agents, thus inducing massive and physiologically mis-regulated calcium efflux (Tong et al. 1997). This calcium overload would then be responsible for the generalized muscle contraction and the hypermetabolic state typical for the MH crisis. The second expression of a RyR1 gain of function is a calcium leak in physiological situations where the channel should be closed, at very low (<μM) or very high (>mM) calcium concentration. If this calcium leak is not compensated by an increased calcium flux toward the SR, it will result in a reduction in the amount of calcium stored in the SR. As a consequence the amount of calcium released upon stimulation will be reduced, and so will be the muscle contraction (Tong et al. 1999). Depending on the expression system used to assay such mutations (muscle cells equipped with all the calcium channels and pumps, or non-muscle cells expressing only few of them) and on the type of expression performed (transient expression or long term, chronic expression) a compensation of the calcium leak can be in place or not. This compensation can be performed for example by overexpression of SERCA pumps or of the membrane calcium channel involved in SOCE (Store Operated Calcium Entry), both mechanisms leading to an increase in the calcium flux toward the SR. But the compensatory overexpression of proteins will only occur upon chronic alteration. As a consequence, discordant results have been reported for the same mutation. For example, an increase in the basal cytosolic calcium concentration has been observed in some expression systems but not in other for the p.R2435L mutation (Dirksen et al. 2004; Ghassemi et al. 2009). The “physiological” calcium leak does not always lead to a detectable increase in the cytosolic calcium concentration at rest, but it is always translated in a reduction in the amount of calcium stored, explaining why mutations resulting in a gain of function in RyR1 at rest could be also associated with congenital myopathies (Ghassemi et al. 2009).
The molecular mechanisms leading to RyR1 gain of function and to calcium leak has been related to redox state of the muscle fiber. For many years, RyR1 has been known as sensitive to oxidizing agents (Eu et al. 2000), and has been proposed as a redox sensor in the skeletal muscle cell (Hidalgo et al. 2005). The amino acids involved in this sensibility have been identified (Aracena-Parks et al. 2006), and it has been shown that oxidative modification like S-nitrosylation of RyR1 results in a reduction in its association to FKBP12 (Aracena et al. 2005; Zissimopoulos et al. 2007). FKBP12 stabilizes RyR1 closed state (Brillantes et al. 1994), and dissociation of FKBP12 from RyR1 leads to a leaky calcium channel. This calcium leak increases the production of oxygen or nitrogen reactive species, which in turn increase the oxidative modifications of RyR1 and the calcium leak, in a deleterious circle. This mechanism has been observed in association with other pathologies such as Duchenne Muscular dystrophy (Bellinger et al. 2009) and aging (Anderson et al. 2011), where it amplified the muscle weakness but it has also been observed as a direct consequence of a RyR1 mutation, for example the p.Y522S mutation (Durham et al. 2008).
3.2 RyR1 Loss of Function
A second mechanism has been described to explain the effect of missense RyR1 mutation associated with congenital myopathies. An alteration of the E–C coupling, i.e., the coupling between the DHPR and RyR1, has been proposed to be at the origin of the pathology, and has been named “E–C uncoupling” (Dirksen et al. 2002). The observed effect of different mutations showed that membrane depolarization induced a reduction in calcium release although resting calcium levels were normal, excluding the presence of a leaky RyR1. The p.I4898T mutant form of RyR1 is for instance not able to mediate calcium efflux when stimulated by the DHPR (the coupling mechanism called “orthograde”). However the presence of the p.I4898T mutation does not alter the regulation of DHPR by RyR1, also called “retrograde coupling,” a mechanism that requires the sole presence of RyR1 channel in front of the DHPR, showing that the RyR1 tetramer is correctly targeted to the triad (Avila et al. 2001). More than an “E–C uncoupling,” this mechanism is in fact an alteration of the permeability of RyR to calcium, which has now been clearly demonstrated in a mouse model reproducing this p.I4898T mutation (Loy et al. 2011). The consequence of this kind of mutation, lethal at the homozygous state in mouse, is a muscle weakness, which most probably correlates to inefficient coupling between the excitation stimulus mediated by DHPR and RyR1 channel opening. A physical “E–C uncoupling,” namely the abolition of the interaction between DHPR and RyR1, has never been observed up to now, either because it too damaging for the cells, or because it relies on multiple contact points, involving different regions of RyR1, and could not be altered by a single point mutation.
3.3 Reduction in RyR1 Amount
The description of patients with mutations affecting RyR1 expression level has led to the most recent pathophysiological mechanism hypothesized (Monnier et al. 2008; Zhou et al. 2006). This mechanism is mostly associated to recessive forms, i.e., with two simultaneous RyR1 mutations. In this case, the presence of a “null” mutation (resulting in the complete absence of protein production from one allele) leads to the expression of the second RyR1 variation in a hemizygous context. The severity of the pathology in such a case depends on the nature of the expressed second mutation as well as on the residual expression level of the hypomorphic allele. In these patients, all the RyR1 expressed at the protein level could be mutated, as there is only one allele able to produce a protein. Therefore there is not only a reduction in the amount of channel expressed in the SR membrane, but also the expression of mutant RyR1 monomer in high proportion. This elevated percentage of mutant monomer can be very damaging, although its impact would have been milder if expressed together with a normal RyR1 allele. The overall reduction in RyR1 expression level could also have an impact on the stoichiometry of proteins of the CRC and its function. If the expression level of regulatory proteins (triadin, junctin, calmodulin…) is unchanged, reduced amounts of RyR1 could lead to an excess of these free regulatory proteins, which could have a negative effect on CRC function.
The tetrameric organization of RyR1 and its physical interaction with DHPR add complexity to the interpretation of the effect of mutations. The balance of combinations between normal or mutant monomers of RyR1 arising from dominant forms of myopathies, or between mutant monomers in recessive forms, is a factor that may influence the apparent discrepancy of clinical outcome and histological presentation for identical genotypes (MacLennan and Zvaritch 2011).
4 Conclusion
In the past decade, the class of pathologies involving RyR1 has expanded beyond its “classical” manifestation, MH, and CCD. While the complete analysis of RyR1 sequence became more accurate despite the complexity of the gene, it appeared that variations present on almost every exon of RYR1 could be associated to some form of myopathy. The heterogeneity of RyR1-related myopathies described so far may increase when routine massive parallel sequencing will be available in diagnosis, pointing to the necessity of understanding the intimate mechanisms leading to mis-regulation of calcium release in these pathologies. So far, the mouse models expressing some of the RyR1 mutations associated to human pathologies have confirmed important observations: morphologic alterations due to RyR1 mutation can have various forms, and structural abnormalities linked to different congenital myopathies (CCD, MmD, rods myopathies) could share the same etiology (MacLennan and Zvaritch 2011). These models, as well as future mice lines, will be pivotal in deciphering RyR1 pathophysiological mechanisms, together with cellular models reproducing the function of RyR1 in the calcium release complex. Although general mechanisms leading to RyR1 dysfunction seem to be understood, a detailed analysis of cellular and physiological models are still required to decipher the precise pathophysiology of RyR1-RCM. Several questions are in particular still unsolved such as: why is the characteristic feature of RyR1-RCM a depletion of mitochondria from focal zones of the muscle cells? Why are the myofibrillar apparatus of these cells also disorganized? How do the cellular default progress and why is the pathology usually not progressive?
Finally, it is tempting to speculate that if the RYR1 gene is associated to so many forms of muscle dysfunction due to calcium efflux dysregulation, it could also be associated to pathologies based on neuronal dysfunction. RyR1 is expressed in some neurons and recent data showed that a mutant form of RyR1 has indeed an impact on neuronal function (De Crescenzo et al. 2012). The future exploration of neuronal disorders with next generation sequencing technologies will probably provide new directions to explore in the pathophysiology of RyR1.
References
Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14:196–207
Aracena P, Tang W, Hamilton SL, Hidalgo C (2005) Effects of S-glutathionylation and S-nitrosylation on calmodulin binding to triads and FKBP12 binding to type 1 calcium release channels. Antioxid Redox Signal 7:870–881
Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL (2006) Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem 281:40354–40368
Avila G, O'Brien JJ, Dirksen RT (2001) Excitation-contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc Natl Acad Sci USA 98:4215–4220
Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15:325–330
Betzenhauser MJ, Marks AR (2010) Ryanodine receptor channelopathies. Pflugers Arch 460:467–480
Bracci L, Vukcevic M, Spagnoli G, Ducreux S, Zorzato F, Treves S (2007) Ca2+ signaling through ryanodine receptor 1 enhances maturation and activation of human dendritic cells. J Cell Sci 120:2232–2240
Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-bindingprotein. Cell 77:513–523
Capacchione JF, Muldoon SM (2009) The relationship between exertional heat illness, exertional rhabdomyolysis, and malignant hyperthermia. Anesth Analg 109:1065–1069
Clarke NF, Waddell LB, Cooper ST, Perry M, Smith RL, Kornberg AJ, Muntoni F, Lillis S, Straub V, Bushby K, Guglieri M, King MD, Farrell MA, Marty I, Lunardi J, Monnier N, North KN (2010) Recessive mutations in RYR1 are a common cause of congenital fiber type disproportion. Hum Mutat 31:E1544–E1550
De Crescenzo V, Fogarty KE, Lefkowitz JJ, Bellve KD, Zvaritch E, MacLennan DH, Walsh JV Jr (2012) Type 1 ryanodine receptor knock-in mutation causing central core disease of skeletal muscle also displays a neuronal phenotype. Proc Natl Acad Sci USA 109:610–615
Dirksen RT, Avila G (2002) Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca(2+) release channels? Trends Cardiovasc Med 12:189–197
Dirksen RT, Avila G (2004) Distinct effects on Ca2+ handling caused by malignant hyperthermia and central core disease mutations in RyR1. Biophys J 87:3193–3204
Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH (2002) Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc Natl Acad Sci USA 9926:16725–16730
Dubowitz V, Sewry C (2007) Mucle biopsy, a pratical approach. Saunders Elsevier, St.Louis, pp 407–443
Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N, Protasi F, Dirksen R, Hamilton SL (2008) RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell 133:53–65
Eu JP, Sun J, Xu L, Stamler JS, Meissner G (2000) The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell 102:499–509
European Malignant Hyperpyrexia Group (1984) Br J Anaesth 56:1267–1269
Ferreiro A, Estournet B, Chateau D, Romero NB, Laroche C, Odent S, Toutain A, Cabello A, Fontan D, dos Santos HG, Haenggeli CA, Bertini E, Urtizberea JA, Guicheney P, Fardeau M (2000) Multi-minicore disease–searching for boundaries: phenotype analysis of 38 cases. Ann Neurol 48:745–757
Ferreiro A, Monnier N, Romero NB, Leroy JP, Bönnemann C, Haenggeli CA, Straub V, Voss WD, Nivoche Y, Jungbluth H, Lemainque A, Voit T, Lunardi J, Fardeau M, Guicheney P (2002a) A recessive form of central core disease, transiently presenting as multi-minicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol 51:750–759
Ferreiro A, Quijano-Roy S, Pichereau C, Moghadaszadeh B, Goemans N, Bönnemann C, Jungbluth H, Straub V, Villanova M, Leroy JP, Romero NB, Martin JJ, Muntoni F, Voit T, Estournet B, Richard P, Fardeau M, Guicheney P (2002b) Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet 71:739–749
Fill M, Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev 82:893–922
Franck JP, Morrissette J, Keen JE, Londraville RL, Beamsley M, Block BA (1998) Cloning and characterization of fiber type-specific ryanodine receptor isoforms in skeletal muscles of fish. Am J Physiol 275:C401–C415
Franzini-Armstrong C, Nunzi G (1983) Junctional feet and particles in the triads of a fast-twitch muscle fibre. J Muscle Res Cell Motil 4:233–252
Ghassemi F, Vukcevic M, Xu L, Zhou H, Meissner G, Muntoni F, Jungbluth H, Zorzato F, Treves S (2009) A recessive ryanodine receptor 1 mutation in a CCD patient increases channel activity. Cell Calcium 45:192–197
Giannini G, Conti A, Mammarella S, Scrobogna M, Sorrentino V (1995) The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J Cell Biol 128:893–904
Gillard EF, Otsu K, Fujii J, Khanna VK, de Leon S, Derdemezi J, Britt BA, Duff CL, Worton RG, MacLennan DH (1991) A substitution of cysteine for arginine 614 in the ryanodine receptor is potentially causative of human malignant hyperthermia. Genomics 11:751–755
Greenfield JG, Cornmant T, Shy GM (1958) The prognostic value of the muscle biopsy in the floppy infant. Brain 81:461–484
Haan EA, Freemantle CJ, McCure JA, Friend KL, Mulley JC (1990) Assignment of the gene for central core disease to chromosome 19. Hum Genet 86:187–190
Hasan G, Rosbash M (1992) Drosophila homologs of two mammalian intracellular Ca(2+)-release channels: identification and expression patterns of the inositol 1,4,5-triphosphate and the ryanodine receptor genes. Development 116:967–975
Hernandez-Lain A, Husson I, Monnier N, Farnoux C, Brochier G, Lacène E, Beuvin M, Viou M, Manéré L, Claeys KG, Fardeau M, Lunardi J, Voit T, Romero NB (2011) De novo RYR1 heterozygous mutation (I4898T) causing lethal core-rod myopathy in twins. Eur J Med Genet 54:29–33
Hidalgo C, Donoso P, Carrasco MA (2005) The ryanodine receptors Ca2+ release channels: cellular redox sensors? IUBMB Life 57:315–322
Hirata H, Watanabe T, Hatakeyama J, Sprague SM, Saint-Amant L, Nagashima A, Cui WW, Zhou W, Kuwada JY (2007) Zebrafish relatively relaxed mutants have a ryanodine receptor defect, show slow swimming and provide a model of multi-minicore disease. Development 134:2771–2781
Hopkins PM (2007) Is there a link between malignant hyperthermia and exertional heat illness? Br J Sports Med 41:283–284, discussion 284
Ibarra MCA, Wu S, Murayama K, Minami N, Ichihara Y, Kikuchi H, Noguchi S, Hayashi YK, Ochiai R, Nishino I (2006) Malignant hyperthermia in Japan: mutation screening of the entire ryanodine receptor type 1 gene coding region by direct sequencing. Anesthesiology 104:1146–1154
Inui M, Saito A, Fleischer S (1987) Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem 262:1740–1747
Jungbluth H, Davis MR, Müller C, Counsell S, Allsop J, Chattopadhyay A, Messina S, Mercuri E, Laing NG, Sewry CA, Bydder G, Muntoni F (2004) Magnetic resonance imaging of muscle in congenital myopathies associated with RYR1 mutations. Neuromuscul Disord 14:785–790
Klein A, Lillis S, Munteanu I, Scoto M, Zhou H, Quinlivan R, Straub V, Manzur AY, Roper H, Jeannet PY, Rakowicz W, Jones DH, Jensen UB, Wraige E, Trump N, Schara U, Lochmuller H, Sarkozy A, Kingston H, Norwood F, Damian M, Kirschner J, Longman C, Roberts M, Auer-Grumbach M, Hughes I, Bushby K, Sewry C, Robb S, Abbs S, Jungbluth H, Muntoni F (2012) Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat 33:981–988
Lanner JT, Georgiou DK, Joshi AD, Hamilton SL (2010) Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol 2:a003996
Loy RE, Orynbayev M, Xu L, Andronache Z, Apostol S, Zvaritch E, MacLennan DH, Meissner G, Melzer W, Dirksen RT (2011) Muscle weakness in Ryr1I4895T/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ ion permeation and release from the sarcoplasmic reticulum. J Gen Physiol 137:43–57
Lynch PJ, Tong J, Lehane M, Mallet A, Giblin L, Heffron JJ, Vaughan P, Zafra G, MacLennan DH, McCarthy TV (1999) A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci USA 96:4164–4169
MacLennan DH, Duff C, Zorzato F, Fujii J, Phillips M, Korneluk RG, Frodis W, Britt BA, Worton RG (1990) Ryanodine receptor gene is a candidate for predisposition to malignant hyperthermia. Nature 343:559–561
MacLennan DH, Zvaritch E (2011) Mechanistic models for muscle diseases and disorders originating in the sarcoplasmic reticulum. Biochim Biophys Acta 1813:948–964
Magee KR, Shy GM (1956) A new congenital non-progressive myopathy. Brain 79:610-621
Maryon EB, Coronado R, Anderson P (1996) Unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contraction. J Cell Biol 134:885–893
McCarthy TV, Healy JM, Heffron JJ, Lehane M, Deufel T, Lehmann-Horn F, Farrall M, Johnson K (1990) Localization of the malignant hyperthermia susceptibility locus to human chromosome 19q12-13.2. Nature 343:562–564
Meissner G (1994) Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol 56:485–508
Monnier N, Procaccio V, Stieglitz P, Lunardi J (1997) Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet 60:1316–1325
Monnier N, Romero NB, Lerale J, Nivoche Y, Qi D, MacLennan DH, Fardeau M, Lunardi J (2000) An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet 9:2599–2608
Monnier N, Romero NB, Lerale J, Landrieu P, Nivoche Y, Fardeau M, Lunardi J (2001) Familial and sporadic forms of central core disease are associated with mutations in the C-terminal domain of the skeletal muscle ryanodine receptor. Hum Mol Genet 10:2581–2592
Monnier N, Krivosic-Horber R, Payen JF, Kozak-Ribbens G, Nivoche Y, Adnet P, Reyford H, Lunardi J (2002) Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology 97:1067–1074
Monnier N, Ferreiro A, Marty I, Labarre-Vila A, Mezin P, Lunardi J (2003) A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet 12:1171–1178
Monnier N, Kozak-Ribbens G, Krivosic-Horber R, Nivoche Y, Qi D, Kraev N, Loke J, Sharma P, Tegazzin V, Figarella-Branger D, Roméro N, Mezin P, Bendahan D, Payen JF, Depret T, Maclennan DH, Lunardi J (2005) Correlations between genotype and pharmacological, histological, functional, and clinical phenotypes in malignant hyperthermia susceptibility. Hum Mutat 26:413–425
Monnier N, Marty I, Faure J, Castiglioni C, Desnuelle C, Sacconi S, Estournet B, Ferreiro A, Romero N, Laquerriere A, Lazaro L, Martin JJ, Morava E, Rossi A, Van der Kooi A, de Visser M, Verschuuren C, Lunardi J (2008) Null mutations causing depletion of the type 1 ryanodine receptor (RYR1) are commonly associated with recessive structural congenital myopathies with cores. Hum Mutat 29:670–678
Pirone A, Schredelseker J, Tuluc P, Gravino E, Fortunato G, Flucher BE, Carsana A, Salvatore F, Grabner M (2010) Identification and functional characterization of malignant hyperthermia mutation T1354S in the outer pore of the Cavalpha1S-subunit. Am J Physiol Cell Physiol 299:C1345–C1354
Quijano-Roy S, Carlier RY, Fischer D (2011) Muscle imaging in congenital myopathies. Semin Pediatr Neurol 18:221–229
Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P (2006) Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 27:977–989
Romero NB, Monnier N, Viollet L, Cortey A, Chevallay M, Leroy JP, Lunardi J, Fardeau M (2003) Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain 126:2341–2349
Romero NB, Herasse M, Monnier N, Leroy JP, Fischer D, Ferreiro A, Viollet L, Eymard B, Laforêt P, Monges S, Lubieniecki F, Taratuto AL, Guicheney P, Lunardi J, Fardeau M (2005) Clinical and histopathological aspects of central core disease associated and non-associated with RYR1 locus. Acta Myol 24:70–73
Rosenberg H, Davis M, James D, Pollock N, Stowell K (2007) Malignant hyperthermia. Orphanet J Rare Dis 2:21
Sambuughin N, Holley H, Muldoon S, Brandom BW, de Bantel AM, Tobin JR, Nelson TE, Goldfarb LG (2005) Screening of the entire ryanodine receptor type 1 coding region for sequence variants associated with malignant hyperthermia susceptibility in the north american population. Anesthesiology 102:515–521
Samsó M, Feng W, Pessah IN, Allen PD (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol 7:e85
Scacheri PC, Hoffman EP, Fratkin JD, Semino-Mora C, Senchak A, Davis MR, Laing NG, Vedanarayanan V, Subramony SH (2000) A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology 55:1689–1696
Serysheva II, Ludtke SJ, Baker ML, Cong Y, Topf M, Eramian D, Sali A, Hamilton SL, Chiu W (2008) Subnanometer-resolution electron cryomicroscopy-based domain models for the cytoplasmic region of skeletal muscle RyR channel. Proc Natl Acad Sci U S A 105:9610–9615
Shy GM, Magee KR (1956) A new congenital non-progressive myopathy. Brain 79:610–621
Stewart SL, Hogan K, Rosenberg H, Fletcher JE (2001) Identification of the Arg1086His mutation in the alpha subunit of the voltage-dependent calcium channel (CACNA1S) in a North American family with malignant hyperthermia. Clin Genet 59:178–184
Takeshima H, Iino M, Takekura H, Nishi M, Kuno J, Minowa O, Takano H, Noda T (1994) Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 369:556–559
Tobin JR, Jason DR, Challa VR, Nelson TE, Sambuughin N (2001) Malignant hyperthermia and apparent heat stroke. JAMA 286:168–169
Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy TV, MacLennan DH (1997) Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem 272:26332–26339
Tong J, McCarthy TV, MacLennan DH (1999) Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J Biol Chem 274:693–702
Toppin PJ, Chandy TT, Ghanekar A, Kraeva N, Beattie WS, Riazi S (2010) A report of fulminant malignant hyperthermia in a patient with a novel mutation of the CACNA1S gene. Can J Anaesth 57:689-693
Tung CC, Lobo PA, Kimlicka L, Van Petegem F (2010) The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 468:585-588
Uemura Y, Liu TY, Narita Y, Suzuki M, Ohshima S, Mizukami S, Ichihara Y, Kikuchi H, Matsushita S (2007) Identification of functional type 1 ryanodine receptors in human dendritic cells. Biochem Biophys Res Commun 362:510-515
Urwyler A, Deufel T, McCarthy T, West S (2001) European Malignant Hyperthermia Group. Guidelines for molecular genetic detection of susceptibility to malignant hyperthermia. Br J Anaesth 86:283-287
Wappler F, Fiege M, Steinfath M, Agarwal K, Scholz J, Singh S, Matschke J, Schulte Am Esch J (2001) Evidence for susceptibility to malignant hyperthermia in patients with exercise-induced rhabdomyolysis. Anesthesiology 94:95-100
Wilmshurst JM, Lillis S, Zhou H, Pillay K, Henderson H, Kress W, Müller CR, Ndondo A, Cloke V, Cullup T, Bertini E, Boennemann C, Straub V, Quinlivan R, Dowling JJ, Al-Sarraj S, Treves S, Abbs S, Manzur AY, Sewry CA, Muntoni F, Jungbluth H (2010) RYR1 mutations are a common cause of congenital myopathies with central nuclei. Ann Neurol 68:717-726
Yuchi Z, Lau K, Van Petegem F (2012) Disease mutations in the ryanodine receptor central region: crystal structures of a phosphorylation hot spot domain. Structure 20:1201-1211
Zhou H, Yamaguchi N, Xu L, Wang Y, Sewry C, Jungbluth H, Zorzato F, Bertini E, Muntoni F, Meissner G, Treves S (2006) Characterization of recessive RYR1 mutations in core myopathies. Hum Mol Genet 15:2791–2803
Zissimopoulos S, Docrat N, Lai FA (2007) Redox sensitivity of theryanodine receptor interaction with FK506-binding protein. J Biol Chem 282:6976–6983
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Fauré, J., Lunardi, J., Monnier, N., Marty, I. (2014). Ryanodine Receptor 1 and Associated Pathologies. In: Weiss, N., Koschak, A. (eds) Pathologies of Calcium Channels. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-40282-1_9
Download citation
DOI: https://doi.org/10.1007/978-3-642-40282-1_9
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40281-4
Online ISBN: 978-3-642-40282-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)