
Abstract
Ryanodine receptors (RyR) are intracellular Ca2+-permeable channels that provide the sarcoplasmic reticulum Ca2+ release required for skeletal and cardiac muscle contractions. RyR1 underlies skeletal muscle contraction, and RyR2 fulfills this role in cardiac muscle. Over the past 20 years, numerous mutations in both RyR isoforms have been identified and linked to skeletal and cardiac diseases. Malignant hyperthermia, central core disease, and catecholaminergic polymorphic ventricular tachycardia have been genetically linked to mutations in either RyR1 or RyR2. Thus, RyR channelopathies are both of interest because they cause significant human diseases and provide model systems that can be studied to elucidate important structure–function relationships of these ion channels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Ryanodine receptors and excitation–contraction coupling
A regulated rise in intracellular Ca2+ is required for many physiological functions including muscle contraction, secretion, regulation of gene expression, and fertilization [10]. Intracellular Ca2+ can be elevated via the activation of plasma membrane Ca2+ permeable channels or via the release of Ca2+ from intracellular stores [22]. Unregulated or deficient Ca2+ signaling can lead to deleterious cellular outcomes, especially in excitable cells such as skeletal and cardiac myocytes [95]. The rise in Ca2+ required for myocyte contraction is provided by the activation of the sarcoplasmic reticulum (SR) Ca2+ release channels in the ryanodine receptors (RyR) [122]. Given the central role that RyR play in regulating Ca2+ release, it is not surprising that dysregulation of these channels can lead to severe muscle pathologies. In fact, mutations in RyR1 and RyR2 isoforms that are associated with human diseases have been identified [90, 96]. RyR channelopathies include malignant hyperthermia (MH) and central core disease (CCD) in skeletal muscle and catecholaminergic polymorphic ventricular tachycardia (CPVT) in cardiac muscle [17, 42, 96]. Furthermore, alterations in RyR post-translational modifications and remodeling of the RyR channel macromolecular complexes are associated with acquired muscle pathologies including skeletal muscle fatigue and heart failure (HF) [7, 112]. A more complete understanding of RyR channelopathies will allow a greater understanding of RyR function and may help in the development of therapeutic strategies designed to rescue normal RyR dysfunction.
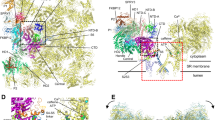
RyRs are a family of large, homotetrameric intracellular Ca2+ release channels that, upon activation, allow rapid release of Ca2+ from SR stores into the cytosol (Fig. 1) [34]. There are three mammalian isoforms (RyR1, RyR2, and RyR3) that exhibit subtype-specific tissue expression patterns. RyR1 is the predominant isoform expressed in mammalian skeletal muscle, and RyR2 is the exclusive subtype expressed in cardiac myocytes [34]. While RyR1 and RyR2 are required for myocyte contraction in their respective tissues, the role of RyR3 is less clear, although this isoform is present early in skeletal muscle development, and a role for RyR3 in learning and memory has been proposed based on murine knockout studies [5]. The three RyR subtypes exhibit a high degree of structural and functional homology. Each monomer is in excess of 560 Kd with the ∼90% of in the channel sequence comprising enormous cytosolic domains. Indeed, the N-terminal ∼4,100 amino acids comprise the cytosolic domain, and the transmembrane domain is contained in the C-terminal ∼800 residues [34]. The number of RyR transmembrane domains has been the subject of much debate, but it is now generally accepted that each monomer spans the SR membrane at least six times and possibly as many as eight times [28]. The ion conducting pore and the regions required for SR membrane localization are contained in this region, and the functional Ca2+ release channels are organized into homotetramers [34].

Disease-related mutations in human RyR1 and RyR2 are clustered in three mutational “hot spots.” a The tetrameric structure of RyR Ca2+ release channels with the membrane topology superimposed on one of the subunits. b The distribution of over 200 malignant hyperthermia and central core disease-causing mutations in human RyR1 and over 70 catecholaminergic polymorphic ventricular tachycardia-associated mutations in human RyR2
RyR1 and RyR2 release the SR Ca2+ required for muscle contraction, and as such, they are required for excitation–contraction (EC) coupling in skeletal and cardiac muscles, respectively [11, 77]. Even though RyR1 and RyR2 are structurally quite similar, they are activated by divergent mechanisms. Both isoforms are functionally coupled to changes in sarcolemmal membrane potential through voltage-gated Ca2+ channels (VGCC) of the L-type class, also known as dihydropyridine receptors (DHPR) [103]. DHPR physically interact with RyR1 in skeletal muscle, and conformational changes in the DHPR following membrane depolarization induce RyR1 to open [103]. Activation of L-type Ca2+ channels also induces RyR2 activation in cardiac myocytes, but there is no known physical interaction between the two channels. Instead, Ca2+ enters the myocyte through active LTCC and triggers RyR2 activation in a process known as Ca2+-induced Ca2+ release (CICR) [11]. All three isoforms exhibit a biphasic response to free Ca2+ and can participate in CICR in permeabilized systems. The three isoforms exhibit different sensitivities to Ca2+-dependent activation and inactivation, but Ca2+-dependent RyR activation generally occurs at ∼0.3–10 μM Ca2+, and the channels are typically inhibited by millimolar Ca2+ [34]. This Ca2+ sensitivity ensures that RyRs are normally closed at resting cytosolic Ca2+ (50–150 nM) and may also help prevent the sustained activation of RyR at high Ca2+ concentrations.
RyRs in myocytes are typically part of large macromolecular complexes and are regulated by endogenous and exogenous ligands [34, 122]. Endogenous regulatory molecules in addition to Ca2+ include Mg2+ ions, which are inhibitory, and adenine nucleotides and cyclic ADP-ribose, which both activate the channels [34, 77, 78]. Numerous proteins also interact with and regulate Ca2+ release from RyR including SR resident and cytosolic proteins. Notably, FKBP12 and FKBP12.6 (also known as calstabin1 and calstabin2) interact with and maintain the stability of RyR isoforms in muscle [12, 75]. Calstabin–RyR interactions also promote coupled gating of RyR from skeletal and cardiac muscles [73, 74]. Calmodulin binds to RyR1 and RyR2 and likely participates in Ca2+-dependent regulation of channel activity [6, 102]. Detailed studies of the effects of CaM on RyR1 have revealed that Ca2+-free CaM activates the receptor, while Ca2+-bound CaM inhibits channel function [82, 97]. Phosphorylation represents an additional mode of RyR regulation and kinases (PKA, CamK) and phosphatases (PP1, PP2A); the phosphodiesterase PDE4D3 and the muscle A-kinase anchoring protein are present in RyR macromolecular complexes [122]. RyR-mediated Ca2+ release may also be controlled by the cellular oxidative state as the channels can be modulated by oxidation and nitrosylation [8, 29, 32, 115]. RyRs are therefore a focal point for regulating Ca2+ release by multiple separate and interacting pathways.
Exogenous pharmacological agents have long been known to regulate RyR activity and are useful tools for examining RyR function [30, 34, 58, 78, 100]. The identification, purification, and early characterizations of RyR were greatly facilitated by use of the plant alkaloid ryanodine, which binds to RyR with high affinity [37, 62, 88]. Low (micromolar) concentrations of ryanodine lock RyR into a characteristic open one-half subconductance state [49], and higher concentrations of ryanodine inhibit RyR channels in a use-dependent manner. 3H-ryanodine binding, therefore, is a commonly used readout of channel activity [29]. Caffeine and 4-chloro-m-cresol (CmC) activate RyR channels and are thought to facilitate channel opening by increasing the sensitivity of RyR to Ca2+-dependent activation [100]. Ruthenium red is a commonly used RyR blocker [70], and local anesthetics such as procaine and tetracaine also block RyR channel activity in bilayers and in intact cells [58, 101, 116]. Additional pharmacological agents can exert toxic or beneficial effects on muscle function by acting on RyR. For example, volatile anesthetics such as halothane are thought to increase RyR activity and can precipitate MH in individuals carrying MH mutations in RyR1, and the muscle relaxant dantrolene is used to prevent MH crises because it inhibits SR Ca2+-leak via mutant RyR1 [60, 71]. While the exact mechanism(s) underlying the actions of these pharmacological agents is not completely understood, RyR activators and inhibitors are routinely used to study RyR function.
Skeletal muscle RyR channelopathies
MH, the first identified RyR channelopathy, is inherited in an autosomal dominant fashion and continues to be of major concern for anesthetic-induced deaths in otherwise healthy individuals [98]. Susceptibility estimates for MH range from 1 in 15,000 children to 1 in 50,000 adults undergoing anesthesia [14]. The exact prevalence of MH susceptibility is difficult to determine since the syndrome only becomes apparent after exposure to triggering agents such as halothane and succinylcholine, but as many as 1 in 2,000–3,000 individuals may be susceptible to anesthesia-induced hyperthermic episodes [81]. A related syndrome referred to as porcine stress syndrome (PSS) is found in certain lines of domestic swine where stressed pigs undergo stress-induced hyperthermia [71].
MH in humans and PSS in pigs are thought to develop following excessive skeletal muscle contraction that results from excess Ca2+ in the myoplasm following anesthesia in humans or during stress in pigs [71]. This excessive Ca2+ causes sustained contractions, which accounts for the rapid onset of muscle rigidity. Continued contraction and elevated Ca2+ exert a severe metabolic demand on myocytes, and ATP levels become depleted. Myocytes respond by increasing ATP production via oxidative phosphorylation and glycolysis, which leads to acidosis. This prolonged hypermetabolic state generates heat and is thought to underlie the elevated body temperatures observed in MH episodes [71].
MH episodes in both humans and pigs are typically rapid and severe. Individuals who experience an episode can reach core body temperatures of 43°C, which leads to organ failure and death if not quickly treated [80]. Anesthetic-induced death rates in excess of 80% were observed in MH episodes prior to the discovery of the preventative effects of the muscle relaxant dantrolene [46]. Rapid administration of dantrolene during MH episodes has reduced mortality to less than 10% [99]. Susceptibility can be determined in vitro by measuring the contractile response to caffeine or halothane in biopsied muscle fibers from humans and pigs [31]. Samples from MH cases exhibit an enhanced sensitivity to these agents. CICR from SR isolated from MH-susceptible pigs exhibited increased sensitivity to Ca2+, caffeine, and halothane, which implicated abnormal Ca2+ release as the source of MH. Alterations in 3H-ryanodine binding properties in porcine MH samples provided evidence linking RyR1 dysfunction to the disease [79]. 3H-ryanodine binding was increased at optimal [Ca2+] as well as in the presence of inhibiting [Ca2+] indicating an overall gain-of-function defect. Ryanodine-sensitive channels in MH samples are also dysfunctional, further implicating altered RyR1 function in the etiology of the disease [35, 36].
While hyperthermic responses to surgery where known since the early 1900s, the link between volatile anesthetics, family history, and MH was initially described in a letter to Lancet from Denborough and Lovell in 1960 [25]. Therein, they described a patient who had survived a severe hyperthermic response after receiving halothane in preparation for surgery to repair a broken leg. The patient's family history had numerous instances (ten of 38 individuals who underwent surgery) of deaths during or shortly following surgeries in which general inhaled anesthetics were administered [24]. MH has since been described as a pharmacogenetic disorder since the disease is only manifested in humans during or immediately following the application of a general anesthetic [98]. While it was long suspected that MH was caused by defective Ca2+ handling, the critical link between MH and RyR1 was provided by a molecular genetic study of the condition in swine. Comparison of the cDNA sequence of RyR1 in MH pigs to that of normal controls revealed a missense mutation (C to T) at nucleotide position 1,843 leading to a substitution of a cysteine residue for an arginine at position 615 (R615C) [39]. An analogous mutation in human ryr1 (R614C) was identified later [44].
Mutations in ryr1 are also associated with the rare congenital myopathy CCD and the related diseases multiminicore disease, nemaline myopathy, and centronuclear myopathy [54]. These rare diseases exhibit autosomal dominant and recessive modes of inheritance and have variable pathologies, but the defining characteristic is the presence of cores of metabolically inactive tissue in the center of muscle fibers [54]. These cores develop over time in CCD patients and are devoid of mitochondria and signs of oxidative metabolism. The pathological significance of these cores is unclear, but the most severe cases of CCD involve pronounced muscle weakness. CCD symptoms are present at a young age, but unlike MH, these symptoms are apparent in the absence of other factors. MH and CCD may result from similar molecular mechanisms since some CCD patients also exhibit MH symptoms [54].
The clinical overlap of MH and CCD and the severity of CCD-related symptoms may relate to the specific set of mutations that patients carry. Significant symptomatic heterogeneity exists in patient populations that carry ryr1 mutations. Dominant mutations in ryr1 that are associated with MH are typically localized to MH susceptibility regions 1 and 2 [123]. Conversely, a recent analysis of a group of patients with various core myopathies demonstrated that CCD mutations inherited in a dominant fashion were clustered in the C-terminus and were associated with patients who exhibited pronounced cores and a strong CCD phenotype [123]. In contrast, core-associated mutations inherited in a recessive fashion spanned the entire ryr1 gene, and patients with these mutations exhibited a wider range of clinical severity [123].
Molecular mechanisms of MH and CCD
To date, nearly 200 mutations in ryr1 have been linked to MH and/or CCD (Fig. 1) [96]. The bulk of the mutations are missense substitutions and are conserved in three “hot spots” located in the N-terminal (C35–R614), central (D2129–R2458), and C-terminal regions (I3916–G4942) in the amino acid sequence of RyR1 [96]. As shown in Fig. 1, however, many mutations are located outside these hot spots, and the apparent clustering in three regions may be the result of a sequencing bias [76]. Attempts to understand the molecular mechanisms behind MH and CCD have involved the examination of the effects of these mutations on RyR1 function. This initially involved the laborious methodology of obtaining muscle biopsies from affected patients or MH pigs and testing the sensitivity of contracture to activating agents such as caffeine or halothane [55, 84]. Sensitivity to halothane, caffeine, and CICR was measured in these samples, and 3H-ryanodine binding and single channel studies were also conducted [55, 79, 86]. These studies established that MH and CCD mutations typically increased the sensitivity of RyR1 to activation, indicating a gain-of-function. Recently, the use of recombinant techniques has rapidly increased the knowledge about the molecular mechanisms underlying MH and CCD.
The ability to clone and express mutant RyR1 cDNA in heterologous systems has allowed detailed analyses of RyR1 function and dysfunction. Recombinant RyR1 expressed in C2C12 [87], HEK-293 [106], and COS-7 [107] cell lines function as intracellular Ca2+ release channels and are activated by caffeine, halothane, and CmC. Ca2+ released from the ER via RyR1 can be monitored in individual cells by using Ca2+-sensing dyes combined with imaging or photometric detection. Detailed biophysical analyses can be performed on single recombinant RyR1 by isolating microsomes and fusing them to lipid bilayers. Single RyR1 can be measured under tightly controlled conditions to measure the sensitivity to Ca2+ activation or inhibition or the sensitivity to endogenous or pharmacological activators and inhibitors. Similar studies have been performed using recombinant RyR2.
Early experiments on heterologously expressed RyR1 established that the RyR1–R614C mutant channels were more sensitive to caffeine and halothane, similar to results from in vitro contracture tests [87]. Recombinant RyR1–R614C also exhibited an increase in the sensitivity to CICR similar to defects observed in muscle strips and microsomes isolated from MH patients and MH pigs. Later experiments showed that 15 different MH, CCD, or MH/CCD RyR1 mutations showed increased sensitivity to caffeine and halothane when expressed in HEK-293 [106]. Using the same methodology, a mutation in the C-terminal hot spot (I4898T) exhibited no response to caffeine and had severely reduced 3H-ryanodine binding [69]. Co-expression of the I4898T mutation with wild type RyR1 partially rescued these deficiencies. Responses to caffeine were reduced when compared to wild type RyR1 expressing cells, but enhanced when compared to I4898T expressing cells. These results led the authors to conclude that the I4898T mutation led to a severe gain-of-function effect [69]. This enhanced activity was thought to promote a constitutive “leak” of Ca2+ from the SR. The reduced 3H-ryanodine binding was proposed to result from the mutation disrupting the 3H-ryanodine binding site.
Experiments on RyR1 expressed in HEK-293 cells allow rapid and relatively straightforward analyses of receptor function, but HEK-293 cells fail to recapitulate the muscle environment. As such, this system does not allow RyR1 to be activated by DHPR activity or regulated by other muscle-specific factors. Expression of wild type and mutated RyR1 in dyspedic (RyR1 knockout) myotubes that can be activated by endogenous DHPR recapitulates EC coupling [4, 83]. Voltage clamp allows activation of DHPR and simultaneous intracellular Ca2+ measurements can be used to study MH and CCD mutations in a near-physiological setting. Numerous RyR1 mutations have been examined using this system, and in most cases, the results corroborated prior studies conducted in HEK-293 cells [26]. That is, most MH and CCD mutations led to a gain-of-function effect that promoted “leaky” RyR1 [3, 26, 27, 119]. In MH, this leak is uncovered when volatile anesthetics are administered, while in CCD cases, chronic leak is thought to lead to a reduction in SR store content and result in muscle weakness [68].
A careful examination of CCD mutations in dyspedic myotubes revealed a graded severity of mutations on voltage-activated Ca2+ release. N-terminal mutations typically reduced the amount of Ca2+ released in response to depolarization. This was coupled with an increase in the basal Ca2+ levels and a reduction in the SR content, consistent with leaky channels [3]. The effects of the C-terminal I4898T mutations were, however, quite different. Myotubes expressing this mutant showed severely deficient Ca2+ release in response to depolarization, but normal resting Ca2+ and SR store content [4]. These results are consistent with a mutation that reduces Ca2+ conductance. I4898 is located in the proposed pore region of RyR1, indicating that a reduction in permeation is behind the I4898T CCD mutation. The combined results of these studies led Avila and Dirksen to propose that CCD mutations could be grouped into two classes: leak-inducing mutations and mutations that uncouple RyR from DHPR activation (Fig. 2) [26]. In the case of CCD, either mechanism could explain the muscle weakness observed in patients with the disease.

Two different effects of central core disease-related mutations on skeletal muscle excitation–contraction coupling. a A cartoon depicting the effects of a leaky RyR1 mutation. This class of mutations exhibits an increased sensitivity to activation by membrane depolarization, Ca2+, caffeine, and halothane. b A cartoon depicting the effects of an uncoupled mutation. Mutations of this class lead to nonfunctional channels and increased sarcoplasmic reticulum Ca2+ store content
A crystal structure was recently solved for the N-terminus (residues 1–210) of rabbit RyR1 representing the first high resolution (∼2.5 A) structure for any region of RyR [1]. This structure contains part of the MH susceptibility region 1 of RyR1, and 11 known RyR1 mutations were mapped onto this structure. Further structural analysis demonstrated that three of the mutations (C36R, R164C, and R178C) did not measurably disrupt the folding of the N-terminal domain. These results suggest that the mutations disrupt function through alterations in intramolecular interactions instead of disrupting the structure immediately surrounding the mutations [1]. This is in line with the observations that mutations in divergent regions of RyR1 lead to similar channel dysfunction.
Cardiac RyR channelopathies
Defects in SR Ca2+ release in cardiac myocytes are also the source of human disease [92]. Inheritable arrhythmogenic disorders brought on by emotional or physical stress have been linked to mutations in RyR2 and its luminal binding partner calsequestrin2 [61, 92]. The hallmark of these diseases is PVT evident in electrocardiograms (ECGs) of patients in the absence of any known structural heart defects [17, 64]. Patients diagnosed with CPVT exhibit bidirectional VTs during exercise or stress and are at risk for syncope and sudden cardiac death [64]. Early diagnosis is critical since many CPVT mutation carriers die with the first episode at a young age and current therapeutic strategies involve β-blocker treatment to blunt the effects of catecholamines [17].
A genetic locus for CPVT was identified in chromosome 1q42–q43. Priori et al. identified four separate missense mutations in the gene for RyR2 in this region in DNA from CPVT patients [92]. More than 70 RyR2 mutations have since been identified and, similar to MH and CCD mutations, are spread throughout the entire amino acid sequence of human RyR2 (http://www.fsm.it/cardmoc) (Fig. 1). The finding that mutations in the gene for calsequestrin2 are also associated with CPVT further demonstrates that altered SR Ca2+ cycling is the root cause of the arrhythmias [61]. Cytosolic Ca2+ levels are tightly regulated to allow the rapid initiation and termination of muscle contraction during a heartbeat [11]. The cardiac action potential initiates VGCC opening to allow Ca2+ to enter the cell during systole. This Ca2+ rapidly activates RyR2 to flood the cytosol with Ca2+ and allow contraction. At the termination of the AP, Ca2+ is removed from the cytosol by plasmalemmal sodium calcium exchanger (NCX) and sarcolemmal calcium pump [11].
Under normal circumstances, β-adrenergic stimulation has positive ionotropic, chronotropic, and lusistropic effects [11]. This allows a rapid increase in contractility, heart rate, and sarcomere relaxation required during times of stress and is a major component of the sympathetic “fight-or-flight” response. Signaling events downstream of β-adrenergic receptors (β-AR) after norepinephrine binding facilitate this efficient upregulation. Activation of β-AR on cardiac myocytes stimulates the production of cAMP and subsequent activation of PKA. PKA phosphorylates numerous substrates involved in EC coupling including Cav1.2 [23], RyR2 [113], and phospholamban [59]. The combined effect of these modifications is to increase the magnitude and the rate of decline of the Ca2+ transient [15]. This allows the SR to be effectively re-filled during shorter diastolic periods brought on by increased AP frequency. This normally efficient upregulation in response to catecholamines is disrupted in CPVT. The basis of this disruption is thought to be the aberrant Ca2+ release from the SR during diastole [17]. Similar to digitalis intoxication, this elevation in cytosolic Ca2+ is thought to activate NCX to promote electrogenic influx of three Na+ and efflux of one Ca2+ that can result in delayed afterdepolarizations (DADs), which can trigger ventricular tachycardia and sudden cardiac death [38, 72].
The β-adrenergic cascade is downregulated and desensitized in HF [13]. When cardiac function begins to diminish, there is a compensatory increase in circulating catecholamines. This is initially protective, but becomes maladaptive over time. A major consequence of this prolonged adrenergic signaling is a remodeling of the RyR2 macromolecular complex. RyR2 becomes PKA hyperphosphorylated in HF [75]. The PKA hyperphosphorylation is exacerbated by the loss of PP1 and PP2A phosphatases and PDE4D3 phosphodiesterase from the RyR2 complex [75, 94]. Most importantly, the stabilizing subunit FKBP12.6 (calstabin2) is also depleted from the complex, which leads to leaky RyR2 channels [75]. Hyperphosphorylated, FKBP12.6-deficient RyR2 channels exhibit subconductance states and increased activity in the presence of diastolic levels of Ca2+ [75].
Divergent models to explain the molecular basis of CPVT
A major remodeling of the RyR2 macromolecular complex occurs during HF due to chronic PKA hyperphosphorylation causing dissociation of FKBP12.6 from the RyR2 complex [75]. It was subsequently found that mice with FKBP12.6 deletions (FKBP12.6−/− mice) exhibited ventricular arrhythmias and sudden cardiac death in response to exercise and catecholamine administration, therefore exhibiting similarities to human CPVT patents [110]. Catecholamine-treated FKBP12.6−/− cardiac myocytes also exhibited DADs during APs evoked at 12 Hz. Cardiac microsomes isolated from exercised FKBP12.6−/− contained RyR2 that exhibited increased sensitivity to 150 nM cytosolic Ca2+ and subconductance states (a hallmark of FKBP-depleted channels [12, 75]). Similar effects were observed in lipid bilayer studies with CPVT-mutated RyR2, as PKA treatment of three different CPVT-mutated RyR2 resulted in increased Po at diastolic Ca2+ levels and the appearance of subconductance states, indicating destabilized channels [110]. These results suggested that the common link between HF and CPVT was leaky RyR2 resulting from a loss of FKBP12.6. Consistent with this, all of the CPVT-mutated RyR2 tested exhibited reduced FKBP12.6 binding (Fig. 3a) [110]. Furthermore, repairing this interaction with a small molecule or overexpression of FKBP12.6 repaired the defective channels and exercise-induced arrhythmias [47, 110]. The potential role of RyR2 hyperphosphorylation and FKBP12.6 depletion in the progression of HF and CPVT has come under much scrutiny. Some studies support the general hypothesis [40, 121], but other studies have not recapitulated the main findings [41, 50, 114]. Much of this confusion likely derives from divergent experimental conditions that are employed by the different groups examining the problem.

Four different proposed mechanisms to explain how RyR2 mutations lead to catecholaminergic polymorphic ventricular tachycardia (CPVT). a CPVT mutations are more sensitive to PKA hyperphosphorylation, which leads to depletion of the channel stabilizing protein, calstabin2 (FKBP12.6), and sarcoplasmic reticulum (SR) Ca2+ leak. b CPVT mutations lead to an increase in sensitivity to store-overload-induced Ca2+ release that is exacerbated by the increased SR store content triggered by stressful situations. c CPVT mutations lead to increased RyR2 sensitivity to activating agents such as caffeine, 4-CmC, or cAMP-mobilizing agents. d CPVT mutations lead to an unzippering of intramolecular interactions, which leads to increased diastolic SR Ca2+ leak
Alternative mechanisms for the molecular basis for CPVT have also been proposed using recombinant RyR2 expressed in heterologous systems and more recently with the use of transgenic mice. Unlike the case with dyspedic myotubes, no muscle-specific null backgrounds exist for reconstitution experiments. Recombinant techniques have been employed that allow expression of mutated RyR2 in HEK-293 cells and atrial tumor HL-1 cells [41, 53]. Cellular based assays, single channel recordings, and 3H-ryanodine binding assays are typically used for functional studies. The majority of studies indicate that CPVT mutations induce gain-of-function changes in RyR2 function.
Chen and colleagues performed the first such studies using CPVT-mutated recombinant RyR2 [53]. They expressed murine RyR2 harboring the R4496C mutation (equivalent to human R4497C) in HEK-293 cells and found that the mutated RyR2 exhibited an increase in basal activity. Specifically, an increase in 3H-ryanodine binding and single channel Po was observed in R4496C samples. HEK-293 cells expressing the mutant receptor were also more likely to undergo spontaneous Ca2+ oscillations and were more sensitive to caffeine stimulation than cells expressing the wild type receptor. The effects of β-AR activation were not tested in these studies, but the authors proposed a mechanism for CPVT whereby an increase in SR store content induced by adrenergic activity would cause spontaneous release of Ca2+ via mutated RyR2 (Fig. 3b) [53]. Further studies by the same group refined the hypothesis and proposed that a reduced threshold for store-overload-induced Ca2+ release (SOICR) is the source of arrhythmias in CPVT [51].
Enhanced RyR2 activity in the face of elevated SR Ca2+ is a well-known phenomenon observed in response to numerous pathological stimuli including digitalis intoxication, elevated extracellular Ca2+, and ischemia/reperfusion [63]. An increase in myocyte sensitivity to SOICR is thought to occur as a result of altered luminal regulation of mutated RyR2 in CPVT [52]. The finding that mutations in calsequestrin can also lead to CPVT has supported the idea that altered SR luminal Ca2+ is an important determinant of CPVT. A loss of function in CSQ2 reduces SR Ca2+ buffering resulting in elevated SR Ca2+ [21]. Further increases in SR Ca2+ induced by adrenergic upregulation of SERCA would lead to luminal activation of RyR2 at resting diastolic cytosolic Ca2+and arrhythmias.
The sensitivity of mutated RyR2 to various activating agents was also tested after heterologous expression in HL-1 cells [41]. These conditions more closely mimicked the disease state in the sense that the mutated RyR2 was expressed in cells that contained endogenous, nonmutated human RyR2. These studies allowed the added advantage that RyR2 could be studied in cardiomyocyte-derived cells. George et al. found that the cells expressing the mutated RyR2 did not exhibit the increased activity at rest that had been observed in HEK-293 cells. Cells expressing the mutated receptors were, however, more responsive to caffeine, 4CmC, and the cAMP-mobilizing agents isoproterenol and forskolin (Fig. 3c). The authors also reported a disruption in the RyR2–FKBP12.6 interactions in response to isoproterenol or forskolin, but the wild type and mutant receptors were indistinguishable in this respect [41]. In subsequent experiments examining interdomain RyR2 interactions, the same group proposed that CPVT mutations increased the propensity of RyR2 to undergo Ca2+-induced conformational changes [43].
Matsusaki's group has explored the possibility of another mechanism involving altered interdomain interactions in CPVT and HF [85, 120]. They proposed that mutations causing CPVT induce an “unzipping” of interdomain interactions between the n-terminal (1–600) and central (2,000–2,500) domains that destabilizes the receptor at low cytosolic [Ca2+] (Fig. 3d) [85]. They based their hypothesis on the observation that regions in the N-terminal and central domains of RyR2 may interact and that a disruption of these domains occurs during channel activation. A similar scheme has been proposed for RyR1 interdomain interactions during activation [117]. Of note, many CPVT-associated RyR2 mutations are located in these two domains. A synthetic peptide corresponding to residues (G2460–P2495) that bound to the zipper domain in the N-terminus of RyR2 increased Ca2+ leak from cardiac microsomes [85]. Based on these results, Matsuzaki and colleagues have proposed that compounds that restore defective interdomain interactions may protect against pathological Ca2+ leak observed in HF [118].
Transgenic approaches to study RyR channelopathies
Major advances in our understanding of RyR channelopathies have been provided by the generation of MH/CCD and CPVT mouse models. Numerous transgenic mice harboring RyR mutations have been generated in recent years in an attempt to create animal models that recapitulate human RyR channelopathies. These mouse models have helped address some questions about molecular mechanisms, but discrepancies still remain.
The first transgenic mouse model harboring a disease-related mutation in RyR1 was created by Susan Hamilton and colleagues in 2006 where they examined mice engineered to express the Y522S MH mutation [20]. Mice homozygous for Y522S exhibited severe developmental abnormalities and died between E17 and P1, but humans with MH are typically heterozygous for RyR1 mutations. Heterozygous Y522S mice faithfully recapitulated many aspects of human MH. MH episodes, characterized by overcontraction and elevated core temperatures, were precipitated by isofluorane or stress with heat. In vitro contracture tests also demonstrated an increased sensitivity to caffeine and isofluorane. At the cellular level, heterozygous Y522S myotubes exhibited a hyperpolarizing shift in voltage-induced Ca2+ release experiments similar to prior experiments using recombinant expression systems. These experiments further demonstrated the “leaky” quality of RyR1 harboring Y522S. Unlike earlier experiments, however, Ca2+ stores were unaffected by the mutation in the mouse model [20]. A reduction in SR Ca2+ would be expected from a chronic leak associated with mutated RyR1. A likely explanation for this discrepancy is that some other compensatory process is evoked in the transgenic mice to maintain SR Ca2+ store content. These experiments highlight the importance of verifying transfection results with in vivo models. Subsequent studies using the RyR1-Y522S heterozygous mice have yielded novel insights into the susceptibility of these mice to heat stroke and also to the contribution to skeletal EC coupling of retrograde regulation of DHPR by Ca2+ released from RyR1 [2, 29].
As described above, RyR1 14895T mutations and other mutations in the C-terminus of RyR1 likely exert their effects via uncoupling EC coupling rather than by increasing SR Ca2+ leak. MacClennan and colleagues generated a transgenic mouse harboring the RyR1-14895T mutation to help determine whether this mutant is indeed uncoupled from DHPR in vivo [124]. Similar to the RyR1-Y522S mice, animals homozygous for the 14895T mutation exhibit severe developmental defects and die before birth further emphasizing the important role RyR1 plays in embryonic development. However, humans heterozygous for I4895T exhibit a severe form of CCD. A more recent study demonstrated that the heterozygous mice did in fact display a progressive myopathy that was more similar to the human condition [125]. As the mice aged, abnormalities in skeletal muscle structure and function became apparent. Similar to human CCD, muscles exhibited cores and some of the mice developed severe muscle deficiencies including complete hind limb paralysis [125].
Similar transgenic approaches have also been applied to CPVT-associated mutations in RyR2. Four different transgenic models have been generated in recent years to examine the in vivo effects of the R4496C, R176Q, P2328S, and R2474S RyR2 mutants [16, 45, 57, 65]. Comparisons among these three mutant mice allow the opportunity to examine mechanisms behind arrhythmogenic mutations in three divergent regions of RyR2. Priori's group generated the first RyR2 transgenic model in 2005 to test the effects of the R4496C mutation [16]. These mice largely recapitulated the main aspects of human CPVT. Mice heterozygous for R4496C exhibited a tendency towards bidirectional VT upon epinephrine or caffeine injections while resting ECGs were normal. Later experiments demonstrated that myocytes isolated from the mutant mice also exhibited DADs in response to isoproterenol [67]. Unlike the studies on whole animals, myocytes from R4496C heterozygotes exhibit some signs of dysfunction including DADs in the absence of adrenergic upregulation. These results highlight the notion that the channels exhibit abnormal activity at rest. Similar observations were made in myocytes from mice engineered to express the R176Q or R2474S RyR2 mutations [57, 65].
The R176Q mutation may be associated with the inherited cardiomyopathy arrhythmogenic right ventricular dysplasia (ARVD). This mutation is analogous to the R163C RyR1 mutation associated with MH. ARVD patients typically exhibit progressive ventricular replacement with fibrofatty deposits in addition to CPVT. ARVD patients with RyR2 mutations tend to have mild ARVD symptoms and are classified as ARVD2, although ARVD diagnosis of patients with RyR2 mutations is controversial given the lack of severe ARVD symptoms [91]. Nevertheless, these patients are at risk for bidirectional VTs and sudden cardiac death. Patients with R176Q mutations also carry a second mutation of T2504M [105]. Both of these mutations exhibit increased activity in vitro [104], but the generation of the R176Q mice allowed the determination of the contribution of this mutation to human disease. Hearts from mice heterozygous for R176Q were structurally normal. Catecholamines triggered ventricular tachycardias and isoproterenol elicited oscillatory Ca2+ signals in myocytes from these mice [57].
Additional studies with the R4496 mice have indicated that Ca2+ handling in the Purkinje fibers may be a source of cardiac arrhythmias [18]. Purkinje fibers and isolated Purkinje cells exhibit triggered and spontaneous Ca2+ release events. The physiological role of these signals is not fully understood, but they may be involved in triggering action potentials. The arrhythmogenic activity of the R4496C mutation was found to originate in the Purkinje fibers in detailed optical mapping studies with voltage-sensitive dyes on Langendorff-perfused hearts [18]. Epinephrine and caffeine-induced bidirectional VTs in the ECGs from of these mice were also changed to monophasic VTs upon Purkinje fiber chemical ablation. This study introduced the idea that the deadly arrhythmias triggered in CPVT patients may not originate in ventricles.
Transgenic mice expressing the P2328S mutation were generated to examine the effects of mutating a site in the central domain of RyR2. Mice homozygous for the mutation were viable, which allowed comparisons to be made with heterozygous animals [45]. Myocytes isolated from the homozygous mice exhibited the most severe alterations in Ca2+ handling. Hearts isolated from homozygous mice were also more prone to arrhythmias in Langendorff studies, indicating a gene dosage effect of the P2328S mutation.
Mice engineered to express the R2474S mutation exhibited ventricular tachycardia upon exercise and catecholamine treatment (Fig. 4) [65]. Cells isolated from these mice also exhibited aberrant Ca2+ waves, action potentials, and transient inward currents upon treatment with isoproteranol. Isoproterenol treatment also elicited an increase in the diastolic Ca2+ spark frequency. Ca2+ sparks represent the spontaneous activity small clusters of RyR2 under resting conditions. An increase in spark frequency is indicative of an increase in diastolic Ca2+ leak. Similar to human disease, these effects were apparent in mice heterozygous for the mutation. The majority of mice homozygous for R2474S mutation died before birth with only 3.5% of the animals being born, while the heterozygous mice were born at the Mendelian ratio.

Fixing sarcoplasmic reticulum (SR) Ca2+ leak by targeting RyR2–calstabin2 interactions. The traces in (a), (b), and (c) are representative telemetric electrocardiogram (ECG) recordings from wild type, R2474S heterozygous (R2474S+/−) mice, and R2474S+/− mice that were treated with S107. Mice were subjected to a stress protocol consisting of a treadmill exercise followed by epinephrine injections. Wild type mice did not exhibit irregular ECGs under these conditions, while a majority of R2474S+/− mice exhibited severe ventricular tachycardias and sudden cardiac death (single asterisk, bidirectional VT; double asterisk, polymorphic VT). Treatment with S107 largely prevented these effects as shown by the regular ECG. Below the traces are cartoons depicting the condition of the RyR–calstabin2 complex under each experimental condition. Data were from Lehnart et al. [65] and reprinted with permission from the American Society of Clinical Investigation
Some CPVT patients also exhibit neurological dysfunctions including epileptic seizures [64, 89]. Consistent with this observation, generalized tonic–clonic seizures were identified in the heterozygous R2474S mice [65]. Seizure activity was mapped to hippocampal regions in telemetry recordings, and brain slices isolated from R2474S heterozygotes exhibited increased spontaneous, ryanodine-sensitive Ca2+ signals. Of note, these defects were observed in the absence of catecholamine treatment. This is unlike the cardiac phenotype, which was only induced by exercise and catecholamines. One possible reason for this is that neurons are more prone to abnormal RyR2 dysfunction at rest than are cardiac myocytes. Another possibility is that there could be an increase in resting cAMP signaling in hippocampal regions that render the R2474S channels leaky.
Blocking RyR leak as a therapeutic approach
A common feature of RyR channelopathies is that most disease-causing mutations in RyR cause the channel to be active or leaky at low cytosolic Ca2+ levels. Dantrolene may be protective in MH episodes by inhibiting RyR function, although the exact mechanism of dantolene is unknown [60]. A protective effect of flecainide in CPVT has been proposed based on its ability to block leak from RyR2 in CSQ2-deficient mice [109]. Besides MH, CCD, and CPVT, RyR leak may also play a role in HF and some forms of muscular dystrophy and epilepsy [8, 65]. The design and development of novel therapeutics that fix RyR leak should be a major goal of future research to treat these diseases.
Results linking the loss of FKBP isoforms from RyR complexes with increased Ca2+ leak from RyR have led to the development of small molecules that restore FKBP/RyR interactions. JTV519 (a 1,4-benzothiazepine derivative) improved cardiac function in a canine HF model [120]. A possible mechanism for these protective effects was provided by evidence that JTV519 enhanced FKBP12.6 binding to RyR2, even when the receptor was hyperphosphorylated [111]. The functional consequence of this rescued binding was a reduced single channel open probability of RyR2 at low [Ca2+] in bilayers. Importantly, JTV519 also rescued FKBP12.6 binding to RyR2 harboring CPVT mutations [66]. In addition, JTV519 prevented pacing-induced arrhythmias in mice with a haploinsufficiency of calstabin2 (calstabin2+/−), but was without effect in mice homozygous for calstabin2 deletion (calstabin2−/−) [111].
Even though JTV519 has therapeutic potential for its ability to stabilize RyR2 function, it also blocks Na+, K+, and Ca2+ channels present in cardiac myocytes [56]. RyR2-specific molecules are required to rescue the RyR2 gain-of-function present in HF without altering other aspects of cardiac electrophysiology. The synthesis of a novel, orally bioavailable, benzothiazapine (S107) that meets this requirement has recently been described [9, 65]. S107 stabilizes RyR–calstabin interactions, but unlike JTV519, exhibits no activity when tested against hundreds of GPCRs, ion channels, or enzymes [9]. Importantly, S107 was protective in the R2474S mouse model of CPVT since mice pretreated with S107 failed to develop exercise and catecholamine-induced arrhythmias (Fig. 4) [65]. Furthermore, myocytes isolated from R2474S mice pretreated with S107 exhibited fewer of these aberrant Ca2+ release events further underscoring the protective effects of this compound.
These results support a model whereby JTV519 and S107 exert therapeutic effects through stabilizing RyR2–calstabin2 interactions that become disrupted by excessive PKA phosphorylation of S2808. It should be noted, however, that other groups have suggested alternative mechanisms of action for JTV519 [48, 120]. Specifically, it has been proposed that JTV519 can exert inhibitory effects on RyR2 function by limiting SOICR [48] or by stabilizing interdomain interactions [120]. The impact that S107 may or may not have on these parameters has not been tested, but the compound has been shown to promote RyR–calstabin interactions in the face of excessive PKA phosphorylation [9, 65] or nitrosylation [8, 33].
S107 was also effective in blocking Ca2+ leak and dysfunction in neuronal and skeletal muscles. For example, S107 was able to rescue the developmental and neurological effects observed in the R247S mice [65]. R2474S homozygous mice were born at a higher rate when the drug was administered to the dams prenatally. Furthermore, S107 was effective at blunting the seizure activity evident in these mice. RyR isoforms are present throughout the brain, but the physiological role of RyR function in learning, memory, and neurological dysfunction is largely unknown. Results from studies of R2474S mice suggest that fixing Ca2+ leak from RyR may represent a novel treatment strategy for some form of epilepsy. It remains to be determined if Ca2+ leak is evident in other neurodegenerative diseases, but an upregulation of RyR2 has been proposed as a pathological feature in mouse models of Alzheimer's disease [19]. Preventing Ca2+ leak may also hold promise as a treatment for skeletal muscle weakness found in HF and muscular dystrophy [8, 9]. Similar to RyR2, RyR1 in skeletal muscle is PKA hyperphosphorylated, calstabin-depleted, and leaky in HF [93, 108]. S107 successfully reduced muscle fatigue in mice after strenuous exercise indicating Ca2+ leak as a source of muscle weakness [9]. Similarly, muscle weakness was blunted by S107 in a mouse model of muscular dystrophy [8]. This was associated with a reduction in Ca2+ spark activity indicating that SR leak inhibition was the mechanism behind the therapeutic effects of S107.
An additional therapeutic approach was uncovered in studies examining calsequestrin2-mediated CPVT where increased Ca2+ leak was observed in myocytes isolated from mice expressing a calsequestrin2 mutation [109]. This leak is thought to result from increased Ca2+ stores caused by the loss of calsequestrin SR Ca2+ buffering resulting in an increase in RyR2 activity. Flecainide was found to block this Ca2+ leak at the cellular level and prevent CPVT symptoms in mice with the calsequestrin mutation. Importantly, flecainide was also effective at preventing CPVT episodes in human subjects, one of whom carried a calsequestrin mutation and the other carried a RyR2 mutation. Both were symptomatic even with β-blockade and Ca2+ channel blockers, demonstrating the utility in treating CPVT by targeting the underlying molecular cause of the disease [109].
Future outlook
Much work has been done in an attempt to elucidate the molecular mechanisms underlying the RyR channelopathies since the first disease-causing RyR mutations were discovered in 1991 [39, 44]. A wealth of experimental evidence has suggested that many facets of MH, CCD, and CPVT can be accounted for by an increase in SR Ca2+ leak from mutated RyR. Much work remains to be done in terms of defining the exact molecular basis for this leak. Continued efforts will address how alterations in channel activity lead to cellular and ultimately whole organ dysfunction. A complete understanding of the molecular underpinnings of RyR channelopathies will facilitate the generation of novel therapeutics designed to block leak from RyR. Additional structure–function studies and transgenic approaches are also needed to help solve some of the remaining mysteries concerning RyR regulation by cytosolic and luminal Ca2+, gating and permeation, and modulation by protein binding partners and kinases.
References
Amador FJ, Liu S, Ishiyama N, Plevin MJ, Wilson A, MacLennan DH, Ikura M (2009) Crystal structure of type I ryanodine receptor amino-terminal beta-trefoil domain reveals a disease-associated mutation “hot spot” loop. Proc Natl Acad Sci U S A 106:11040–11044
Andronache Z, Hamilton SL, Dirksen RT, Melzer W (2009) A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2+ release in muscle fibers of Y522S RyR1 knock-in mice. Proc Natl Acad Sci U S A 106:4531–4536
Avila G, Dirksen RT (2001) Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J Gen Physiol 118:277–290
Avila G, O'Brien JJ, Dirksen RT (2001) Excitation–contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc Natl Acad Sci U S A 98:4215–4220
Balschun D, Wolfer DP, Bertocchini F, Barone V, Conti A, Zuschratter W, Missiaen L, Lipp HP, Frey JU, Sorrentino V (1999) Deletion of the ryanodine receptor type 3 (RyR3) impairs forms of synaptic plasticity and spatial learning. EMBO J 18:5264–5273
Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G (2001) Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J Biol Chem 276:20144–20153
Bellinger AM, Mongillo M, Marks AR (2008) Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest 118:445–453
Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15:325–330
Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, LaCampagne A, Marks AR (2008) Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci U S A 105:2198–2202
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Bers DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205
Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77:513–523
Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB (1982) Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med 307:205–211
Britt BA, Kalow W (1970) Malignant hyperthermia: a statistical review. Can Anaesth Soc J 17:293–315
Callewaert G, Cleemann L, Morad M (1988) Epinephrine enhances Ca2+ current-regulated Ca2+ release and Ca2+ reuptake in rat ventricular myocytes. Proc Natl Acad Sci U S A 85:2009–2013
Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG (2005) Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res 96:e77–e82
Cerrone M, Napolitano C, Priori SG (2009) Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm 6:1652–1659
Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O'Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J (2007) Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res 101:1039–1048
Chakroborty S, Goussakov I, Miller MB, Stutzmann GE (2009) Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 29:9458–9470
Chelu MG, Goonasekera SA, Durham WJ, Tang W, Lueck JD, Riehl J, Pessah IN, Zhang P, Bhattacharjee MB, Dirksen RT, Hamilton SL (2006) Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J 20:329–330
Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC (2007) Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ Res 101:617–626
Clapham DE (2007) Calcium signaling. Cell 131:1047–1058
De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA (1996) Specific phosphorylation of a site in the full-length form of the alpha 1 subunit of the cardiac L-type calcium channel by adenosine 3′, 5′-cyclic monophosphate-dependent protein kinase. Biochemistry 35:10392–10402
Denborough MA (2008) Malignant hyperthermia. 1962. Anesthesiology 108:156–157
Denborough MA, Lovell RRH (1960) Anaesthetic deaths in a family. Lancet 276:45
Dirksen RT, Avila G (2002) Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca(2+) release channels? Trends Cardiovasc Med 12:189–197
Dirksen RT, Avila G (2004) Distinct effects on Ca2+ handling caused by malignant hyperthermia and central core disease mutations in RyR1. Biophys J 87:3193–3204
Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH (2002) Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc Natl Acad Sci U S A 99:16725–16730
Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N, Protasi F, Dirksen R, Hamilton SL (2008) RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knock in mice. Cell 133:53–65
Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I (1994) The pharmacology of intracellular Ca(2+)-release channels. Trends Pharmacol Sci 15:145–149
Ellis FR, Harriman DG (1973) A new screening test for susceptibility to malignant hyperpyrexia. Br J Anaesth 45:638
Eu JP, Sun J, Xu L, Stamler JS, Meissner G (2000) The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell 102:499–509
Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, Marks AR, Lacampagne A (2010) Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 107:1559–1564
Fill M, Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev 82:893–922
Fill M, Coronado R, Mickelson JR, Vilven J, Ma JJ, Jacobson BA, Louis CF (1990) Abnormal ryanodine receptor channels in malignant hyperthermia. Biophys J 57:471–475
Fill M, Stefani E, Nelson TE (1991) Abnormal human sarcoplasmic reticulum Ca2+ release channels in malignant hyperthermic skeletal muscle. Biophys J 59:1085–1090
Fleischer S, Ogunbunmi EM, Dixon MC, Fleer EA (1985) Localization of Ca2+ release channels with ryanodine in junctional terminal cisternae of sarcoplasmic reticulum of fast skeletal muscle. Proc Natl Acad Sci U S A 82:7256–7259
Fozzard HA (1992) Afterdepolarizations and triggered activity. Basic Res Cardiol 87(Suppl 2):105–113
Fujii J, Otsu K, Zorzato F, de Leon S, Khanna VK, Weiler JE, O'Brien PJ, MacLennan DH (1991) Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science 253:448–451
Gellen B, Fernandez-Velasco M, Briec F, Vinet L, LeQuang K, Rouet-Benzineb P, Benitah JP, Pezet M, Palais G, Pellegrin N, Zhang A, Perrier R, Escoubet B, Marniquet X, Richard S, Jaisser F, Gomez AM, Charpentier F, Mercadier JJ (2008) Conditional FKBP12.6 overexpression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation–contraction coupling. Circulation 117:1778–1786
George CH, Higgs GV, Lai FA (2003) Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res 93:531–540
George CH, Jundi H, Thomas NL, Fry DL, Lai FA (2007) Ryanodine receptors and ventricular arrhythmias: emerging trends in mutations, mechanisms and therapies. J Mol Cell Cardiol 42:34–50
George CH, Jundi H, Walters N, Thomas NL, West RR, Lai FA (2006) Arrhythmogenic mutation-linked defects in ryanodine receptor autoregulation reveal a novel mechanism of Ca2+ release channel dysfunction. Circ Res 98:88–97
Gillard EF, Otsu K, Fujii J, Khanna VK, de Leon S, Derdemezi J, Britt BA, Duff CL, Worton RG, MacLennan DH (1991) A substitution of cysteine for arginine 614 in the ryanodine receptor is potentially causative of human malignant hyperthermia. Genomics 11:751–755
Goddard CA, Ghais NS, Zhang Y, Williams AJ, Colledge WH, Grace AA, Huang CL (2008) Physiological consequences of the P2328S mutation in the ryanodine receptor (RyR2) gene in genetically modified murine hearts. Acta Physiol (Oxf) 194:123–140
Harrison GG (1975) Control of the malignant hyperpyrexic syndrome in MHS swine by dantrolene sodium. Br J Anaesth 47:62–65
Huang F, Shan J, Reiken S, Wehrens XH, Marks AR (2006) Analysis of calstabin2 (FKBP12.6)–ryanodine receptor interactions: rescue of heart failure by calstabin2 in mice. Proc Natl Acad Sci U S A 103:3456–3461
Hunt DJ, Jones PP, Wang R, Chen W, Bolstad J, Chen K, Shimoni Y, Chen SR (2007) K201 (JTV519) suppresses spontaneous Ca2+ release and [3H]ryanodine binding to RyR2 irrespective of FKBP12.6 association. Biochem J 404:431–438
Hymel L, Inui M, Fleischer S, Schindler H (1988) Purified ryanodine receptor of skeletal muscle sarcoplasmic reticulum forms Ca2 + -activated oligomeric Ca2+ channels in planar bilayers. Proc Natl Acad Sci U S A 85:441–445
Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH (2002) Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res 91:1015–1022
Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR (2005) Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res 97:1173–1181
Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR (2004) RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A 101:13062–13067
Jiang D, Xiao B, Zhang L, Chen SR (2002) Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res 91:218–225
Jungbluth H, Sewry CA, Muntoni F (2003) What's new in neuromuscular disorders? The congenital myopathies. Eur J Paediatr Neurol 7:23–30
Kalow W, Britt BA, Terreau ME, Haist C (1970) Metabolic error of muscle metabolism after recovery from malignant hyperthermia. Lancet 2:895–898
Kaneko N, Matsuda R, Hata Y, Shimamoto K (2009) Pharmacological characteristics and clinical applications of K201. Curr Clin Pharmacol 4:126–131
Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL (2006) Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A 103:12179–12184
Koulen P, Thrower EC (2001) Pharmacological modulation of intracellular Ca(2+) channels at the single-channel level. Mol Neurobiol 24:65–86
Kranias EG, Garvey JL, Srivastava RD, Solaro RJ (1985) Phosphorylation and functional modifications of sarcoplasmic reticulum and myofibrils in isolated rabbit hearts stimulated with isoprenaline. Biochem J 226:113–121
Krause T, Gerbershagen MU, Fiege M, Weisshorn R, Wappler F (2004) Dantrolene—a review of its pharmacology, therapeutic use and new developments. Anaesthesia 59:364–373
Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M (2001) A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 69:1378–1384
Lai FA, Erickson HP, Rousseau E, Liu QY, Meissner G (1988) Purification and reconstitution of the calcium release channel from skeletal muscle. Nature 331:315–319
Lakatta EG (1992) Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc Res 26:193–214
Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P (1995) Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 91:1512–1519
Lehnart SE, Mongillo M, Bellinger A, Lindegger N, Chen BX, Hsueh W, Reiken S, Wronska A, Drew LJ, Ward CW, Lederer WJ, Kass RS, Morley G, Marks AR (2008) Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J Clin Invest 118:2230–2245
Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR (2004) Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 109:3208–3214
Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG (2006) Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 99:292–298
Loke J, MacLennan DH (1998) Malignant hyperthermia and central core disease: disorders of Ca2+ release channels. Am J Med 104:470–486
Lynch PJ, Tong J, Lehane M, Mallet A, Giblin L, Heffron JJ, Vaughan P, Zafra G, MacLennan DH, McCarthy TV (1999) A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci U S A 96:4164–4169
Ma J (1993) Block by ruthenium red of the ryanodine-activated calcium release channel of skeletal muscle. J Gen Physiol 102:1031–1056
MacLennan DH, Phillips MS (1992) Malignant hyperthermia. Science 256:789–794
Marban E, Robinson SW, Wier WG (1986) Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest 78:1185–1192
Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR (2001) Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res 88:1151–1158
Marx SO, Ondrias K, Marks AR (1998) Coupled gating between individual skeletal muscle Ca2+ release channels (ryanodine receptors). Science 281:818–821
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR (2000) PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–376
McCarthy TV, Quane KA, Lynch PJ (2000) Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat 15:410–417
Meissner G (1994) Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol 56:485–508
Meissner G (2004) Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 35:621–628
Mickelson JR, Gallant EM, Litterer LA, Johnson KM, Rempel WE, Louis CF (1988) Abnormal sarcoplasmic reticulum ryanodine receptor in malignant hyperthermia. J Biol Chem 263:9310–9315
Mickelson JR, Louis CF (1996) Malignant hyperthermia: excitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol Rev 76:537–592
Monnier N, Krivosic-Horber R, Payen JF, Kozak-Ribbens G, Nivoche Y, Adnet P, Reyford H, Lunardi J (2002) Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology 97:1067–1074
Moore CP, Rodney G, Zhang JZ, Santacruz-Toloza L, Strasburg G, Hamilton SL (1999) Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry 38:8532–8537
Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD (1996) Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380:72–75
Nelson TE (1983) Abnormality in calcium release from skeletal sarcoplasmic reticulum of pigs susceptible to malignant hyperthermia. J Clin Invest 72:862–870
Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, Ohkusa T, Ikeda Y, Kobayashi S, Ikemoto N, Matsuzaki M (2005) Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation 111:3400–3410
Ohnishi ST, Taylor S, Gronert GA (1983) Calcium-induced Ca2+ release from sarcoplasmic reticulum of pigs susceptible to malignant hyperthermia. The effects of halothane and dantrolene. FEBS Lett 161:103–107
Otsu K, Nishida K, Kimura Y, Kuzuya T, Hori M, Kamada T, Tada M (1994) The point mutation Arg615→Cys in the Ca2+ release channel of skeletal sarcoplasmic reticulum is responsible for hypersensitivity to caffeine and halothane in malignant hyperthermia. J Biol Chem 269:9413–9415
Pessah IN, Francini AO, Scales DJ, Waterhouse AL, Casida JE (1986) Calcium-ryanodine receptor complex. Solubilization and partial characterization from skeletal muscle junctional sarcoplasmic reticulum vesicles. J Biol Chem 261:8643–8648
Postma AV, Denjoy I, Kamblock J, Alders M, Lupoglazoff JM, Vaksmann G, Dubosq-Bidot L, Sebillon P, Mannens MM, Guicheney P, Wilde AA (2005) Catecholaminergic polymorphic ventricular tachycardia: RYR2 mutations, bradycardia, and follow up of the patients. J Med Genet 42:863–870
Priori SG, Napolitano C (2005) Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J Clin Invest 115:2033–2038
Priori SG, Napolitano C (2005) Intracellular calcium handling dysfunction and arrhythmogenesis: a new challenge for the electrophysiologist. Circ Res 97:1077–1079
Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA (2001) Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103:196–200
Reiken S, Lacampagne A, Zhou H, Kherani A, Lehnart SE, Ward C, Huang F, Gaburjakova M, Gaburjakova J, Rosemblit N, Warren MS, He KL, Yi GH, Wang J, Burkhoff D, Vassort G, Marks AR (2003) PKA phosphorylation activates the calcium release channel (ryanodine receptor) in skeletal muscle: defective regulation in heart failure. J Cell Biol 160:919–928
Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, Burkhoff D, Marks AR (2003) Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 107:2459–2466
Rizzuto R, Pozzan T (2003) When calcium goes wrong: genetic alterations of a ubiquitous signaling route. Nat Genet 34:135–141
Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P (2006) Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 27:977–989
Rodney GG, Williams BY, Strasburg GM, Beckingham K, Hamilton SL (2000) Regulation of RYR1 activity by Ca(2+) and calmodulin. Biochemistry 39:7807–7812
Rosenberg H, Davis M, James D, Pollock N, Stowell K (2007) Malignant hyperthermia. Orphanet J Rare Dis 2:21
Rosero EB, Adesanya AO, Timaran CH, Joshi GP (2009) Trends and outcomes of malignant hyperthermia in the United States, 2000 to 2005. Anesthesiology 110:89–94
Rousseau E, Ladine J, Liu QY, Meissner G (1988) Activation of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Arch Biochem Biophys 267:75–86
Shannon TR, Ginsburg KS, Bers DM (2002) Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res 91:594–600
Smith JS, Rousseau E, Meissner G (1989) Calmodulin modulation of single sarcoplasmic reticulum Ca2 + -release channels from cardiac and skeletal muscle. Circ Res 64:352–359
Tanabe T, Beam KG, Adams BA, Niidome T, Numa S (1990) Regions of the skeletal muscle dihydropyridine receptor critical for excitation–contraction coupling. Nature 346:567–569
Thomas NL, George CH, Lai FA (2004) Functional heterogeneity of ryanodine receptor mutations associated with sudden cardiac death. Cardiovasc Res 64:52–60
Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A (2001) Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 10:189–194
Tong J, Oyamada H, Demaurex N, Grinstein S, McCarthy TV, MacLennan DH (1997) Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J Biol Chem 272:26332–26339
Treves S, Larini F, Menegazzi P, Steinberg TH, Koval M, Vilsen B, Andersen JP, Zorzato F (1994) Alteration of intracellular Ca2+ transients in COS-7 cells transfected with the cDNA encoding skeletal-muscle ryanodine receptor carrying a mutation associated with malignant hyperthermia. Biochem J 301(Pt 3):661–665
Ward CW, Reiken S, Marks AR, Marty I, Vassort G, Lacampagne A (2003) Defects in ryanodine receptor calcium release in skeletal muscle from post-myocardial infarct rats. FASEB J 17:1517–1519
Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC (2009) Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med 15:380–383
Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR (2003) FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113:829–840
Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR (2004) Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304:292–296
Wehrens XH, Marks AR (2003) Altered function and regulation of cardiac ryanodine receptors in cardiac disease. Trends Biochem Sci 28:671–678
Witcher DR, Kovacs RJ, Schulman H, Cefali DC, Jones LR (1991) Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J Biol Chem 266:11144–11152
Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H, Chen SR (2005) Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res 96:847–855
Xu L, Eu JP, Meissner G, Stamler JS (1998) Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279:234–237
Xu L, Jones R, Meissner G (1993) Effects of local anesthetics on single channel behavior of skeletal muscle calcium release channel. J Gen Physiol 101:207–233
Yamamoto T, El-Hayek R, Ikemoto N (2000) Postulated role of interdomain interaction within the ryanodine receptor in Ca(2+) channel regulation. J Biol Chem 275:11618–11625
Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, Oda T, Kobayashi S, Ikemoto N, Matsuzaki M (2008) Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation 117:762–772
Yang T, Ta TA, Pessah IN, Allen PD (2003) Functional defects in six ryanodine receptor isoform-1 (RyR1) mutations associated with malignant hyperthermia and their impact on skeletal excitation–contraction coupling. J Biol Chem 278:25722–25730
Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Matsuzaki M (2003) FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation 107:477–484
Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M (2000) Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation 102:2131–2136
Zalk R, Lehnart SE, Marks AR (2007) Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem 76:367–385
Zhou H, Jungbluth H, Sewry CA, Feng L, Bertini E, Bushby K, Straub V, Roper H, Rose MR, Brockington M, Kinali M, Manzur A, Robb S, Appleton R, Messina S, D'Amico A, Quinlivan R, Swash M, Muller CR, Brown S, Treves S, Muntoni F (2007) Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 130:2024–2036
Zvaritch E, Depreux F, Kraeva N, Loy RE, Goonasekera SA, Boncompagni S, Kraev A, Gramolini AO, Dirksen RT, Franzini-Armstrong C, Seidman CE, Seidman JG, Maclennan DH (2007) An Ryr1I4895T mutation abolishes Ca2+ release channel function and delays development in homozygous offspring of a mutant mouse line. Proc Natl Acad Sci U S A 104:18537–18542
Zvaritch E, Kraeva N, Bombardier E, McCloy RA, Depreux F, Holmyard D, Kraev A, Seidman CE, Seidman JG, Tupling AR, Maclennan DH (2009) Ca2+ dysregulation in Ryr1I4895T/wt mice causes congenital myopathy with progressive formation of minicores, cores, and nemaline rods. Proc Natl Acad Sci U S A 106:21813–21818
Acknowledgements
Dr. Marks is a consultant for ARMGO Pharma, Inc, a start-up company targeting RyR2 to treat HF and cardiac arrhythmias, and is the recipient of funding from the National Institutes of Health (HL 061503, HL 067849, HL 056180, and HL 083418) and from the Fondation Leducq.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Betzenhauser, M.J., Marks, A.R. Ryanodine receptor channelopathies. Pflugers Arch - Eur J Physiol 460, 467–480 (2010). https://doi.org/10.1007/s00424-010-0794-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-010-0794-4