
Abstract
Hyaluronan (HA) is present throughout the body, including all the bones and cartilages of the skeleton, where it may fulfill both a structural and metabolic role depending on its molecular size. In mammals, HA is produced by three hyaluronan synthases (Has), of which Has2 is the predominant form in cartilage and bone. HA can be degraded by hyaluronidases (Hyal) and free radicals. Mammals possess five hyaluronidases, of which Hyal1 and Hyal2 are thought to be predominant in cartilage and bone. The structural role of HA in cartilage is dependent on its ability to form proteoglycan aggregates, whereas its metabolic role involves intracellular signaling induced by interaction with receptors such as CD44 and RHAMM. Such signaling differs between high-molecular-weight HA and its fragments. HA and its fragments play a major role in endochondral bone formation and possibly intramembranous bone formation, as they can regulate the differentiation and action of chondrocytes, osteoblasts, and osteoclasts. Cartilage-specific depletion of HA synthesis has been studied in floxed Has2 mice that have been crossed with mice expressing Cre under control of either the Prx1 or the Col2a1 promoter. Such deletion of Has2 gene expression results in a chondrodysplastic phenotype, in which all endochondral bones of the skeleton are severely truncated. The phenotype is characterized by severely impaired longitudinal growth of the bones due to abnormal organization and differentiation within the growth plates, particularly in the process of chondrocyte hypertrophy. The Col2a1-driven mice also exhibit defective modeling of the endochondral bone.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Many bones in the appendicular and axial skeleton are formed from a hyaline cartilage precursor, which subsequently develop and grow by endochondral ossification. The remaining bones of the skeleton, particularly those of the skull, do not involve a cartilage precursor and arise directly and grow by intramembranous bone formation. Hyaline cartilage persists in the endochondral bones at their growth plates during juvenile development and as articular cartilage at the bone surface in diarthrodial joints, where it allows smooth motion. In contrast, the more restricted motion of the spine is provided by the fibrocartilaginous intervertebral discs.
Cartilage and bone function depend on the unique structure of their extracellular matrix (ECM). In mature articular cartilage and intervertebral disc, the ECM provides the ability to resist compressive loading, and in the growth plate, it provides the scaffold that maintains the unique cellular organization responsible for long bone elongation. The cartilage ECM is characterized by the presence of proteoglycan aggregates that are formed via the interaction of hyaluronan (hyaluronic acid, HA) with numerous aggrecan molecules. While aggrecan is absent in bone, HA is present, though it is not thought to play a structural role as in cartilage but rather may participate in cell signaling. Irrespective of whether HA fulfills a structural or metabolic role in the skeleton, it is essential for the normal development and function of both cartilage and bone.
2 HA Structure and Metabolism
2.1 HA Structure and Distribution
HA was first identified in the vitreous humor of the eye in 1934 (Meyer and Palmer 1953), but its structure was not elucidated until 1954 (Weissmann et al. 1954). It is a linear polysaccharide composed of repeating disaccharides of d-glucuronic acid (GlcA) and N-acetyl d-glucosamine (GlcNAc), which are linked by β(1–3) and β(1–4) bonds, respectively (Fig. 9.1), and is a member of the glycosaminoglycan (GAG) family. HA is distinguished from other GAGs, such as chondroitin sulfate (CS), dermatan sulfate (DS), keratan sulfate (KS), heparan sulfate (HS), and heparin, by its long chain length, lack of sulfation, and mode of synthesis (Fraser et al. 1997). A typical HA molecule may possess in excess of 10,000 disaccharide units with a molecular weight of over 5 MDa, whereas the sulfated GAGs rarely exceed 50 kDa. HA is synthesized as a free polysaccharide chain at the plasma membrane of the cell, unlike the sulfated GAGs which are all synthesized on a protein primer within the Golgi. Its chain elongation is also reported to occur at the reducing terminus, rather than the nonreducing terminus as occurs in the sulfated GAGs (Prehm 2006).

The structure of HA. The figure illustrates the structure of glucuronic acid (GlcA) and N-acetyl-glucosamine (GlcNAc) that form the repeating disaccharide unit of HA and their interaction by β1 → 3 and β1 → 4 bonds. The figure also depicts the cleavage site of HA by mammalian hyaluronidases (Hyal1 and Hyal2)
HA is present in all vertebrates but has so far not been detected in invertebrates. It is however produced by some species of bacteria. HA is a ubiquitous molecule in vertebrates, being present in all tissues and body fluids (Fraser et al. 1997). In man, the abundance of HA is highest in the umbilical cord, a tissue often used for its preparation, and synovial fluid, where it is present at concentrations greater than 1 mg/ml. Among the body’s connective tissues, HA is most abundant in skin, vitreous, and cartilages of the skeleton. In the rat, about 27 % of the HA present in the body is found in the skeleton and its supporting structures (Reed et al. 1988). The tissue abundance of HA varies throughout life, but it is present from the embryo to the adult. HA can have many diverse functions throughout life, which depend on both its abundance and molecular weight. HA abundance can also vary with the site within a tissue. In the growth plate, its abundance increases from the proliferative to the hypertrophic zone (Matsui et al. 1991), where it is thought to contribute to cell hypertrophy in addition to its more conventional role in proteoglycan aggregate formation within the ECM (Pavasant et al. 1996).
2.2 HA Synthesis
HA is produced by hyaluronan synthase (Has) residing at the cell membrane (Weigel et al. 1997). Three distinct mammalian Has have been described, Has1, Has2, and Has3 (Shyjan et al. 1996; Watanabe and Yamaguchi 1996; Spicer et al. 1997), and while all produce HA of an identical composition, it can differ in its rate of synthesis, chain length, and the ease with which it can be released from the cell surface depending on the Has involved (Itano et al. 1999). Has1, Has2, and Has3 are integral membrane proteins, with a similar size and structure (Weigel and DeAngelis 2007). Each possesses seven transmembrane or membrane-associated domains and a large cytoplasmic domain containing the active site for HA synthesis (Fig. 9.2). Although the active conformation of Has at the cell membrane has not been defined precisely, there is evidence that at least Has2 is capable of forming homodimers and heterodimers with Has3 and that this and ubiquitination are important for maximal activity (Karousou et al. 2010). Synthesis takes place from cytoplasmic UDP-glucuronic acid and UDP-N-acetyl glucosamine. The monosaccharides are added to the reducing end of the nascent HA chain in the cytoplasm, and the growing chain is continually extruded into the extracellular space through a pore in the plasma membrane formed by the enzyme. The newly synthesized HA may be retained at the cell surface via interaction with Has or a HA receptor such as CD44, or it may be released and diffuse away from the cell. It is not clear what determines when release occurs. In cartilage, proteoglycan aggregate formation occurs via the interaction of aggrecan and link protein with the extracellular HA.

HA synthesis via Has. Has is depicted as an integral plasma membrane enzyme with multiple intracellular, extracellular, and transmembrane domains. The largest intracellular domain possesses the catalytic site for HA synthesis. HA is synthesized in the cytosol by the alternating addition of glucuronic acid (GlcA) and N-acetyl glucosamine (GlcNAc), with the resulting growing polymer being extruded directly from the cell
All Has genes are expressed by chondrocytes (Hiscock et al. 2000; Nishida et al. 1999; Recklies et al. 2001), with Has2 having the highest expression level and being principally responsible for HA production in all cartilages. It is less clear which Has is predominant in bone and whether osteoblasts, osteoclasts, and osteocytes are all involved in HA production. All three Has are expressed during embryonic development, albeit at different times and in different locations (Tien and Spicer 2005). Has2 appears to be by far the most important Has during embryonic development, as mice lacking Has2 gene expression die during mid-gestation (Camenisch et al. 2000), whereas those lacking Has1 or Has3 gene expression are viable and appear normal. Mice lacking both Has1 and Has3 also appear normal, though there is reported to be accelerated wound healing (Mack et al. 2012). Conditional inactivation of the Has2 gene in cartilage (Matsumoto et al. 2009; Moffatt et al. 2011) or over expression of Has2 in limb bud mesoderm (Li et al. 2007) results in severe chondrodystrophic phenotypes. This illustrates both the crucial role that Has2 plays in cartilage development and endochondral bone formation and the need for HA production to be regulated during limb development.
2.3 HA Degradation
In vivo, HA can be degraded by the action of hyaluronidases (Hyal) and free radicals (Stern et al. 2007). Six hyaluronidases have been described in mammals—Hyal1, Hyal2, Hyal3, Hyal4, HyalP1, and PH20, though PH20 is a testicular enzyme confined to the acrosome of sperm and in humans HyalP1 in an inactive pseudogene (Csoka et al. 2001). All the active hyaluronidases are endo-β-N-acetyl hexosaminidases, that utilize hydrolysis to cleave HA at the β(1–4) bonds (Fig. 9.1). The enzymes are also able to degrade CS via cleavage of the β(1–4) bond between N-acetyl galactosamine and glucuronic acid, albeit with lower efficiency than they cleave HA. The exception may be Hyal4, which may possess a greater chondroitinase activity.
Hyal1, Hyal2, Hyal3, and Hyal4 have been shown to be expressed in both cartilage and bone (Bastow et al. 2008). However, Hyal1 and Hyal2 are ubiquitously expressed and are considered to be the major hyaluronidases in somatic tissues. The two enzymes are distinct in their cellular location and the size of the HA fragments that they produce. Hyal1 is a lysosomal enzyme, whereas Hyal2 is a glycosylphosphatidylinositol (GPI)-anchored enzyme on the outer surface of the plasma membrane. Hyal2 may be responsible for the initial degradation of extracellular HA, which is then internalized for subsequent degradation by Hyal1. Hyal2 has a limited capacity to degrade HA and produces fragments of about 20 kDa (50 disaccharide units). In contrast, Hyal1 results in more extensive degradation and fragments of smaller size. Hyal2 may also be released from the cell membrane and function in the more remote ECM, where it has been postulated to play a role in HA degradation during inflammation (Durigova et al. 2011). Genetic ablation of Hyal1 (Martin et al. 2008), Hyal2 (Jadin et al. 2008), or Hyal3 (Atmuri et al. 2008) in the mouse has very few phenotypic consequences on development, reproduction, and aging. In the skeleton, only subtle changes were observed in the Hyal1 and Hyal2 knockout mice: a slight loss of articular cartilage proteoglycan for Hyal1 and the appearance of an extraosseous structure in the frontonasal process and abnormally shaped cervical vertebrae for Hyal2.
HA is also susceptible to nonenzymic degradation by free radicals generated during cell metabolism (Stern et al. 2007). Superoxide is produced by many cells via NADPH oxidase and may undergo dismutation to form hydrogen peroxide. Neither superoxide nor hydrogen peroxide by themselves degrades HA, but when present together or when hydrogen peroxide is present with a transition metal, rapid degradation of HA may occur via the production of hydroxyl radicals. Superoxide also plays a role in the conversion of nitric oxide to peroxynitrite, which can also result in the degradation of HA. Because of the relatively short half-life of free radicals, HA near the cell surface will be most susceptible to radical-mediated degradation.
It is now clear that HA fragments can have distinct functions compared to the high-molecular-weight HA generated by Has (Stern et al. 2006; Noble 2002). For example, high-molecular-weight HA plays a structural role, is anti-inflammatory and antiangiogenic, and impedes cell differentiation, whereas HA fragments are pro-inflammatory and pro-angiogenic. Even the fragments may differ in function among themselves, depending on their molecular size, with the small HA tetrasaccharides being anti-apoptotic. HA fragment function may not be confined to the extracellular environment, as intracellular HA fragments may also influence cell metabolism (Hascall et al. 2004).
3 HA Functional Roles
3.1 HA in ECM Structure
HA forms the backbone of the proteoglycan aggregates that characterize all cartilages (Hascall 1988). A typical proteoglycan aggregate is composed of a central filament of HA with up to 100 aggrecan molecules radiating from it, with each interaction being stabilized by the presence of a link protein (LP) (Fig. 9.3) (Morgelin et al. 1988). The aggrecan core protein possesses three globular regions (Doege et al. 1987, 1991; Watanabe et al. 1995; Hering et al. 1997), termed G1, G2, and G3. The G1 region is responsible for the interaction with HA (Watanabe et al. 1997) and is separated from the G2 region by a relatively short interglobular domain (IGD) (Flannery et al. 1998) The G3 region is separated from the G2 region by a long GAG-attachment region substituted with CS and KS chains. The G3 region is essential for normal trafficking of the newly synthesized aggrecan through the cell (Zheng et al. 1998), while the GAG chains provide the molecule with the osmotic properties essential for tissue swelling (Hascall 1988). In addition to aggrecan, the proteoglycan aggregates may contain versican, which belongs to the same hyalectin family as aggrecan (Wight 2002). The versican core protein possesses terminal globular regions analogous to the G1 and G3 regions of aggrecan but contains far fewer CS chains in its central domain. In both cartilage and intervertebral disc, versican is present at a lower abundance than aggrecan (Sztrolovics et al. 2002), and its contribution to tissue function is not clear.

The structure of proteoglycan aggregates. The figure depicts the interaction of aggrecan with HA and the stabilization of the interaction by a link protein (LP). The disulfide-bonded globular regions (G1, G2, and G3) of the aggrecan core protein and the interglobular domain (IGD) are indicated. The sites of substitution by chondroitin sulfate (CS) and keratan sulfate (KS) chains are also depicted together with the location of the two subdomains for CS substitution (CS1 and CS2)
The large size of the proteoglycan aggregates results in their entrapment by the collagen framework of the tissue, and the high charge conferred by the GAGs results in tissue swelling and water retention. Proteoglycan aggregate function is impaired by glycolytic cleavage of the HA or proteolytic degradation of aggrecan, as occurs in the arthritides. Proteolytic processing by aggrecanases and matrix metalloproteinases (MMPs) (Sztrolovics et al. 1997; Lark et al. 1997; Hughes et al. 1995) results in the accumulation of aggregates enriched in G1 regions, which do not possess the osmotic properties associated with more intact molecules. The G1 regions remaining bound to HA may accumulate in the ECM for many years (Maroudas et al. 1998; Sivan et al. 2006). While the size of HA in the proteoglycan aggregates does decrease with age (Holmes et al. 1988), it is partially protected from both free radical and hyaluronidase cleavage by the presence of the link proteins (Rodriguez and Roughley 2006). Aggrecan fragments not possessing a G1 region are no longer bound to HA and may diffuse within the tissue. These GAG-rich fragments are rapidly lost from articular cartilage but may be retained for many years in the intervertebral disc (Roughley et al. 2006).
Proteoglycan aggregate function can also be perturbed by impaired synthesis, resulting in decreased size or abundance of the HA or decreased abundance or charge of the aggrecan. The consequence of such defects of aggrecan on skeletal development is illustrated by the chondrodystrophic phenotypes that result when gene mutations occur in aggrecan itself—spondyloepiphyseal dysplasia in humans (Gleghorn et al. 2005; Tompson et al. 2009), cartilage matrix deficiency (cmd) in mice (Watanabe et al. 1994; Krueger et al. 1999), and nanomelia in chickens (Primorac et al. 1994; Li et al. 1993)—or in the molecules responsible for its sulfation (Wallis 1995; Superti-Furga et al. 1996; Karniski 2001). Mutations in the versican gene also result in heritable disorders in man (Mukhopadhyay et al. 2006), but the phenotype extends beyond the skeleton, as expected for its expression in many extraskeletal tissues. To date, no human skeletal dysplasia has been attributed to HA deficiency due to a mutation in a Has gene, possibly because of the ubiquitous nature of HA and the lethal consequences that its deficiency could have on embryonic development.
3.2 HA Receptors and Signaling
HA should not be viewed as merely the scaffold upon which proteoglycan aggregate formation occurs, as it is a multifunctional molecule (Lee and Spicer 2000) that can interact with many other HA-binding proteins (Day and Prestwich 2002) and so influence cell metabolism (Fig. 9.4). HA has been shown to interact with several unrelated proteins generically called hyaladherins. Two of the most studied are CD44 and RHAMM (receptor for hyaluronan-mediated motility, also called CD168), which exist in many different isoforms through alternative splicing. CD44 is an integral cell surface membrane protein that contains a “link” module and can be substituted with a GAG chain on its extracellular domain, depending on whether the appropriate exon is present or not. The standard CD44 variant encodes a ubiquitously expressed protein, whereas all other isoforms, possessing variable extensions within the extracellular portion of the molecule, generally have a more restricted tissue and cell-type expression pattern. CD44 is thought to act as a cell adhesion molecule and to serve as a clearance receptor for internalization and degradation of HA (Knudson et al. 2002). RHAMM can be found both intracellularly and extracellularly, even though it does not possess a signal peptide sequence and is not targeted to the typical secretory pathway (Maxwell et al. 2008). It is generally accepted that RHAMM is present extracellularly at the cell membrane, where it can interact with CD44. While HA binding to CD44 occurs through its link domain, binding to RHAMM involves a basic region.

Schematic representation of signaling functions of HA through CD44 and RHAMM. CD44 can signal directly or indirectly by interaction with a diverse repertoire of other molecules. CD44 or its intracellular domain (ICD) can translocate to the nucleus and modulate gene expression (left). CD44 can also act as a co-receptor “presenting” growth factors or transducer proteins to their corresponding receptor tyrosine kinase (RTK) (middle). Ensuing is a cascade of phosphorylation events that culminate in transcriptional regulation of gene expression. The intracellular portion of CD44 also tethers with the ezrin/radixin/moesin complex (ERM) and the actin network to promote cell motility (right). Intracellular RHAMM can participate in cell proliferation/division events through interaction with kinases and the mitotic spindle
CD44 and RHAMM do not present inherent catalytic activity and thus signal indirectly through clustering and interaction with various membrane co-receptors, cytoskeletal components, and cytoplasmic molecules (Aruffo et al. 1990; Slevin et al. 2007; Turley et al. 2002). Some of the intracellular effects mediated by CD44 and RHAMM are through modulation of the phosphorylation status of a variety of kinases. For instance, CD44 is known to engage in protein–protein interaction with and modulate the activity of several membrane receptors possessing intrinsic kinase activity (MAPK, Src, ErbB2, EGFR, c-Met, PDGFR, VEGFR, TGFB-R). The effects of CD44 are extremely complex, being cell-type specific and dependent on the isoform produced and whether its interaction is with high molecular weight or fragmented HA. Accordingly, downstream effectors solicited by CD44 activation are numerous. Apart from its interaction with other membrane receptors, CD44 is known to associate with the ezrin/radixin/moesin (ERM) protein complex. ERM is localized just beneath the plasma membrane of cells and is believed to link the plasma membrane with the cytoskeleton through interaction with cell surface receptors and actin filaments. CD44 and other cell adhesion molecules (CD43, ICAM-1, syndecans, and l-selectin) bind to ERM triggering actin filament reorganization and subsequently affecting various processes, such as cell adhesion and motility, signaling, phagocytosis, and apoptosis.
CD44 has also been shown to signal directly after its nuclear translocation. Cleavage of the cell surface CD44 by metalloproteases was reported to generate a soluble fragment and a 25 kDa ectodomain, which undergoes further juxtamembrane processing to liberate an intracellular domain (ICD) (Okamoto et al. 2001; Lammich et al. 2002; Murakami et al. 2003). Upon stimulation of cell signaling through protein kinase C and Ca2+ activation, intramembrane gamma-secretase cleavage at the transmembrane domain of CD44 releases the 72-residue ICD, which is found to accumulate in the nucleus. Nuclear CD44 ICD has been reported to activate a TPA responsive element luciferase reporter construct. The CD44 gene itself has been shown to be a target of the ICD transcriptional activity, thus forming a feed-forward loop. Recently, the entire CD44 molecule was also shown to be present in the nucleus. The mechanism leading to the nuclear accumulation of full length CD44 has been shown to depend on its active endocytosis and transport from the cell membrane and on interaction with the nuclear membrane transportin molecule (Janiszewska et al. 2010). Although the mechanisms involved are still not fully understood, they seem to be independent of HA binding. In addition, the full length CD44 molecule has been shown to be internalized and to interact with cytosolic STAT3 and indirectly with the P300 acetyltransferase (Lee et al. 2009). The complex was found to migrate into the nucleus to regulate expression of cyclin D1 and promote cell proliferation. Other target genes also regulated by the CD44/STAT3/P300 complex include those for MMP2, VEGF, and BCL. The CD44 ICD has also been shown to be a docking site for SMAD1 (Peterson et al. 2004). Treatment of bovine articular chondrocytes with BMP7 induced the translocation of CD44-associated SMAD1 to the nucleus with transcriptional activation of a SMAD-binding element reporter plasmid. The appearance of nuclear SMAD1 was dependent on HA-CD44 interaction, as it was abolished by hyaluronidase treatment. It was proposed that SMAD1 binding to the ICD of CD44 would anchor it close to the plasma membrane for rapid presentation to the type I BMPR upon BMP7 stimulation (Andhare et al. 2009). However, the mechanism governing the CD44-SMAD1 interaction was not defined but was suggested to involve a phosphoserine in the CD44 ICD.
Incubation of articular chondrocytes with low molecular weight HA fragments has also been shown to stimulate gene expression through different signaling pathways (Fieber et al. 2004; Ohno et al. 2005, 2006; Schmitz et al. 2010). Phosphorylation and activation of Akt and NFkB stimulate distinct anabolic and catabolic responses, respectively, as reflected by the induction of Has2 and MMP gene expression. Although the upstream signaling cascades have not been clearly identified, the effects of the HA fragments appear to be partly related to HA displacement from CD44. It has been proposed that HA fragment binding results in “declustering” of CD44 and signaling through cytoskeletal (ERM) reorganization and possibly activation of kinases such as PKC and NFkB. It is not known whether degradative HA fragments are produced in the growth plate and whether they would be at a sufficient concentration to have any significant impact on either chondrocytes or osteoblasts.
RHAMM is supposedly expressed at very low levels, and its distribution appears mostly limited to injured tissues and pathologic cases involving inflammation and cancer (Fieber et al. 1999). Intracellular RHAMM can interact directly with ERK and is associated with mitotic spindle and microtubule assembly, where it interacts with BRCA1 (Tolg et al. 2010; Maxwell et al. 2011). Hence, RHAMM has been ascribed as a tumor-susceptibility gene in breast cancer, and its elevated expression and effects on the disorganization of the mitotic spindle have been postulated as one of the putative mechanisms involved in tumorigenicity. The HA-binding properties of RHAMM and the interaction with CD44 have also been linked to the neoplastic transforming capability of RHAMM.
Because of the relatively wide tissue distribution of CD44 and RHAMM and their roles in many seemingly crucial aspects of cell differentiation and activity, such as proliferation, migration, and invasion, it was anticipated that their gene deletion would be detrimental to life. Surprisingly, the knockout mouse models for CD44 and RHAMM did not display any overt defects under normal conditions (Protin et al. 1999; Schmits et al. 1997). However, several studies have documented subtle to more severe defects when the mice, or isolated knockout cells, are subjected to different insults, either in vivo or in vitro. For instance, the CD44 and RHAMM knockout mice presented striking problems in repair processes after bleomycin-induced lung damage (Teder et al. 2002), angiogenesis (Cao et al. 2006), and skin wound injury (Tolg et al. 2006). Excessive accumulation of HA was found in the lungs of CD44 KO mice, suggesting clearance problems. RHAMM deficient fibroblasts have impaired motility and blunted signaling through ERK1/2 phosphorylation. Moreover, arthritis was found to be exacerbated in CD44 knockout mice, when induced by collagen immunization (Nedvetzki et al. 2004) or TNF-α overexpression (Hayer et al. 2005). Given the close relationship in terms of function for CD44 and RHAMM, it is perhaps not surprising that RHAMM was shown to functionally substitute for the absence of CD44. ICAM-1, another molecule with adhesion properties, was also found to compensate for the lack of CD44 in vivo and act as a substitute co-receptor for c-Met in CD44 null mice (Olaku et al. 2011).
4 HA in Chondrogenesis and Osteogenesis
4.1 HA in Chondrogenesis and Endochondral Bone Formation
Chondrogenesis (Fig. 9.5) begins in mice with mesenchymal cell condensation at about embryonic day 9.5 (E9.5) in a process that is driven by bone morphogenetic proteins (BMPs) (Barna and Niswander 2007). By E10.5, the templates for most skeletal elements have been formed, and the mesenchymal cells within the condensations differentiate into chondrocytes to form the cartilaginous anlagen of the skeleton, under stimulation by Sox9, Sox5, and Sox6 (Bi et al. 1999; Lefebvre et al. 1998; Han and Lefebvre 2008). Chondrocyte differentiation is accompanied by a change from type I collagen (Col1) production to type II collagen (Col2) and aggrecan. In contrast, the cells at the periphery of the condensations differentiate to form the surrounding perichondrium. The embryonic cartilages grow by chondrocyte proliferation until about E13.5, when cells in the center of the future bones cease proliferation and undergo a transformation to first prehypertrophic and then hypertrophic chondrocytes, which are characterized by their expression of type X collagen (Col10) and the calcification of their surrounding ECM (Karsenty et al. 2009). At the same time as chondrocyte hypertrophy occurs, cells in the perichondrium differentiate into osteoblasts, under stimulation by Runx2 (Ducy et al. 1997), and enclose the hypertrophic cells with a bone collar. Vascular endothelial growth factor (VEGF) production by the hypertrophic chondrocytes stimulates blood vessel ingrowth (Gerber et al. 1999; Dai and Rabie 2007) and the arrival of osteoblastic and osteoclastic precursor cells. As the hypertrophic chondrocytes die, osteoblasts deposit bone on the calcified cartilage and a primary center of ossification is formed. This process of endochondral ossification is recapitulated later in the epiphyses of many long bones by the formation of secondary centers of ossification and continues throughout juvenile life within the growth plates. In all cases, chondrocyte proliferation and hypertrophy are controlled by a variety of regulatory factors, including fibroblast growth factor receptor 3 (FGFR3), Indian hedgehog (Ihh), and parathyroid hormone-related peptide (PTHrP) (Ornitz and Marie 2002; Lanske et al. 1996; Vortkamp et al. 1996; St Jacques et al. 1999).

Development of a fetal long bone. The figure depicts mesenchymal condensation (a); chondrocyte and perichondrial differentiation (b); the development of proliferative, maturation, and hypertrophic zones and the onset of vascularization into the perichondrium (c); the formation of the periosteal bone collar and vascular invasion of the calcified hypertrophic cartilage (d); and replacement of the calcified hypertrophic cartilage by bone (e)
HA is associated with many stages of chondrogenesis and endochondral ossification (Bastow et al. 2008). In the developing limb bud, Has2 secretes the HA required to facilitate mesenchymal cell migration (Toole et al. 1972; Kosher et al. 1981), and the formation of precartilaginous condensations is associated with the expression of CD44 by the mesenchymal cells and interaction with the HA (Rousche and Knudson 2002). Subsequent removal of the HA by hyaluronidase cleavage then occurs to allow chondrocyte differentiation (Li et al. 2007). During endochondral ossification, an increase in HA production is associated with chondrocyte hypertrophy and appears to be necessary to facilitate cell swelling (Pavasant et al. 1996). Hypertrophy is also associated with aggrecan loss from the surrounding ECM (Matsui et al. 1991), and this may be mediated by hyaluronidase cleavage of the proteoglycan aggregates (Buckwalter et al. 1987). HA fragments generated in the hypertrophic zone may serve to terminate the hypertrophic process by suppressing the expression of Runx2 (Tanne et al. 2008). The HA fragments may also facilitate vascular invasion, as such fragments are known to be angiogenic (West et al. 1985; Pardue et al. 2008).
The temporal and region-specific expression pattern of the various components of the Has2/HA/CD44/RHAMM cascade in the forming and developing skeleton has not been extensively studied. During early development in the chick limb, the expression of Has2 by in situ hybridization was highest in the region corresponding to the apical ectodermal ridge (AER) and was excluded from the condensing mesenchyme (Li et al. 2007). This result is consistent with HA being produced mostly by cells of the AER, where it is proposed to maintain the underlying layer of mesenchymal cells in a proliferative and non-differentiating state. When out of reach of this HA-rich environment, condensation of mesenchymal cells becomes favorable and acts as one of the events initiating the chondrogenic program. Therefore, a gradient of HA diffusion could help control limb bud outgrowth and patterning. The role of CD44 in the developing limb bud appears to be related to that of Has2 and HA. The standard CD44 isoform is expressed throughout the limb bud, but many different splice variants are restricted to the AER (Sherman et al. 1998). Neutralization of CD44 with an anti-CD44 antibody caused diminished proliferation of mesenchymal cells and reduced the growth of the limb bud in mice. This may be related to the capacity of FGF8 to bind to or be sequestered by the V3 variant of CD44 which possesses a HS chain. Thus, CD44 serves an important function for FGF8 presentation and indirectly regulates the proliferation of neighboring mesenchymal cells.
Little is also known about the role of Has2, HA, RHAMM, and CD44 during later limb development in the establishment of the growth plate and in longitudinal growth. In situ hybridization for Has2 gene expression in the mouse femur and tibia at birth showed that highest expression was localized to the prehypertrophic chondrocytes, with some signal in the articular surfaces (Dy et al. 2010). Very little if any signal was detected elsewhere in the long bones, including regions of the primary spongiosa where osteoclasts, osteoblasts, and osteocytes are present. Considering that HA can be detected throughout the growth plate, it is possible that levels of Has2 mRNA were below the detection limit inherent to in situ hybridization. These low levels may still produce sufficient Has2 protein to generate considerable amounts of HA. Unfortunately, no studies have reported on the localization of the Has2 protein in the growth plate. At present, it is unclear why a high level of Has2 expression is needed by the prehypertrophic cells, but it is possible that this relates to a unique function of HA in the pericellular environment of these cells and that this differs from its role in the rest of the growth plate.
In developing mouse long bones, CD44 expression appears to be restricted to the chondro-osseous junction (Jamal and Aubin 1996; Nakamura and Ozawa 1996; Noonan et al. 1996). At this interface, both osteoclast and osteoblast precursors express cell surface CD44. As growth and development proceed, osteocytes have also been identified as expressing high levels of CD44 (Hughes et al. 1994; Nakamura et al. 1995). Even though CD44 appears to be expressed in cultured articular chondrocytes, it was not detected in vivo in growth plate chondrocytes. If true, this would suggest that the effects of HA in the growth plate chondrocytes are likely not mediated through CD44 interaction. Consistent with studies conducted in the mouse and rat, CD44 in chick joints showed high levels in the articular fibrocartilage and weak expression in the epiphyseal chondrocytes (Dowthwaite et al. 1998). RHAMM just showed slightly lower expression in epiphyseal than articular chondrocytes. To our knowledge, the specific role of RHAMM in the skeleton in vivo has not been studied and could represent an alternative molecule mediating HA signaling. A recent study has shown that RHAMM overexpression in MC3T3 osteoblasts slightly enhanced ERK signaling and inhibited differentiation and subsequent mineralization (Hatano et al. 2011). While CD44 knockout mice do not possess histomorphometric defects associated with osteoblast and osteoclast parameters (de Vries et al. 2005), a μCT analyses revealed a subtle reduction in endocortical bone resorption associated with reduced RANKL production (Cao et al. 2005).
4.2 HA in Intramembranous Bone Formation
In contrast to endochondral bone formation, intramembranous ossification occurs without the formation of an initial cartilaginous template (Hall and Miyake 1992; Dunlop and Hall 1995), but in both cases, the eventual synthesis of mineralized bone occurs through the same process. The cranial vault, mandible, and parts of the clavicle typify bony elements formed purely through an intramembranous process, and periosteal bone development which forms the thickened cortical midshaft of long bones is also intramembranous in nature. Intramembranous bones arise directly through differentiation of mesenchymal cells, initially compacted in sheets or membranes, into osteoblasts. Although less well understood, the process of intramembranous bone formation seems to share many developmental cues and signals (BMPs, TGFβ, FGFs, Wnt, Runx2, Sox9) with endochondral bone formation. It has even been proposed that a chondrocyte-like cell, presenting a gene expression signature normally found in chondrocytes, is detected as an intermediate stage within the sequence of events leading to fully differentiated osteoblasts (Abzhanov et al. 2007).
The role of HA during intramembranous bone formation in vivo is unknown. However, osteoblasts do express Has2 message, and HA is also known to influence the behavior of both osteoclasts and osteoblasts in vitro. HA may participate in osteoclast binding to the ECM (Prince 2004), and HA fragments induce bone resorption (Ariyoshi et al. 2005). In contrast, HA has been reported to be an inhibitor of osteoblast differentiation (Falconi and Aubin 2007).
4.3 Has Expression in Chondrogenic and Osteogenic Cell Lines
The ATDC5 cell line has been used extensively to study chondrogenesis, as in culture it can recapitulate the differentiation from a mesenchymal cell to a chondrocyte. Has2 has been shown to be expressed at low levels in non-differentiated cells and to increase three- to fourfold with differentiation to chondrocytes. Has1 expression is 2–3 orders of magnitude lower than Has2, suggesting that Has1 is unlikely to compensate for a lack of Has2. shRNA-mediated knockdown of Has2 gene expression in ATDC5 cells using a lentiviral delivery system induces a sustained knockdown and results in up to 67 % reduction in HA production. ATDC5 cells with the highest degree of knockdown display an altered morphological appearance and a dramatic reduction in Alcian blue staining indicative of proteoglycan depletion in the ECM (Fig. 9.6). Thus, failure to produce normal HA levels impairs chondrogenesis and reduces cartilage ECM formation.

Knockdown of Has2 expression in ATDC5 cells. Stable pools of ATDC5 cells expressing the lentiviral shRNA were analyzed after 14 days for Has2 message levels (a) and HA secretion in the culture media during the last 24 h (b). Alcian blue staining of the day 14 cultures reveals considerably less proteoglycan production in the shRNA expressing cells (c)
The MC3T3-E1 cell line is commonly used to study osteogenesis, as in culture it can recapitulate the differentiation from an osteoblast precursor to a mature osteoblast producing mineralized bone. The expression levels of Has1 and Has2 in differentiating MC3T3-E1 osteoblasts were relatively constant, but the absolute levels of Has2 expression were 2–3 orders of magnitude higher than Has1. Thus, Has2 is likely the major contributor to HA production by osteoblasts in osteoid during bone formation and modeling.
5 Has2 Knockout Mice
5.1 Prx1-driven Has2 Knockout Mice
The early embryonic lethal nature of the global Has2 knockout mouse prevents the role of HA production by Has2 in skeletal development from being studied. To overcome this problem, two cartilage-specific knockout mice have been generated using the Cre-loxP system. The first of these utilized a mouse line with a floxed exon 2 in the Has2 gene, which was crossed with a line expressing Cre recombinase driven by a Prx1 enhancer (Matsumoto et al. 2009). The Prx1 enhancer results in Cre expression in the early limb bud mesenchyme and a subset of craniofacial mesenchyme (Logan et al. 2002). As such, it allows gene function to be studied in the developing limbs of the appendicular skeleton but does not result in gene excision in the axial skeleton. Exon 2 of the Has2 gene contains the start codon and two transmembrane domains, and its excision generates a null allele. The knockout mouse limbs show no evidence of Has2 message bearing exon 2, and HA content is barely detectable.
By E16.5, all bones of both the hindlimbs and forelimbs of the mutant Prx1-Cre-Has2 mice are extremely short compared to wild-type mice, and Alcian blue staining of the cartilaginous regions is very much reduced compared to wild-type mice. This suggests that the aggrecan content of the cartilage is severely diminished, as might be expected if the HA content of the cartilage is depleted and HA is essential for aggrecan retention via proteoglycan aggregate formation. In support of this conclusion, aggrecan expression is not itself altered in the mutant mice, confirming that the aggrecan depletion is due to loss from the ECM rather that lack of production by the chondrocytes.
The limb phenotype of the mutant mice indicates that longitudinal bone growth is perturbed in the absence of HA. It is therefore not surprising that the growth plates of the developing bones are abnormal, with the normal columnar cellular organization being perturbed and the disorganized cells being more densely packed. Both the cellular disorganization and the dense packing could be related to the loss of aggrecan from the ECM. Indeed, mice lacking aggrecan, or possessing unstable proteoglycan aggregates due to the absence of link protein, exhibit a similar growth plate disorganization (Watanabe and Yamada 2002).
The abnormality in the mutant growth plates is not restricted to cellular organization but also involves cell differentiation, as the normal progression from resting to hypertrophic chondrocytes is perturbed. While there is little difference in chondrocyte proliferation in the wild-type and mutant growth plates, subsequent chondrocyte hypertrophy within the mutant growth plates is impaired. This results in a reduction in the number of hypertrophic chondrocytes. At least in part, this appears to be due to a reduction in the expression of Indian hedgehog (Ihh), which is produced by the prehypertrophic chondrocytes and initiates hypertrophy. Thus, the phenotype of the mutant mice due to the absence of HA in the growth plates is not only due to impairment of its structural role in the ECM but also to impairment in its role in cell signaling.
In addition, the mutant mice exhibit a patterning defect in the proximal phalanges of some digits, though the link between such a specific patterning defect and HA depletion is unclear. The mutant mice also exhibit impaired joint cavitation, a process that is driven by HA (Dowthwaite et al. 2003). In the normal mouse, HA is elevated in the interzone between two developing bones, and its production results in a cell-free space that drives cavity formation in this region. It is therefore not surprising that in the absence of HA, the cells of the interzone remain closely packed. These changes are probably independent of a depletion in HA production by chondrocytes but reflect the early expression of Prx1 and perturbation of HA production by the mesenchymal stem cells prior to their patterning or differentiation into various skeletal elements.
5.2 Col2a1-Driven Has2 Knockout Mice
The second cartilage-specific Has2 knockout mouse was generated by crossing the floxed Has2 mice described above with mice possessing Cre under control of the Col2a1 promoter (Moffatt et al. 2011; Roughley et al. 2011). Excision of the floxed exon 2 in the Has2 gene will therefore occur later in skeletogenesis than with the Prx1-driven mice. Thus, while the Prx1-Cre-Has2 mouse undergoes excision in the mesenchymal stem cells of the limb buds, the Col2-Cre-Has2 mouse is expected to involve only committed chondrocytes. However, unlike the Prx1-Cre-Has2 mouse, which only generates a phenotype in the limbs, the Col2-Cre-Has2 mouse is expected to also involve the intervertebral discs and vertebrae of the spine. Heterozygous Col2-Cre-Has2 mice appeared normal at birth but showed a slightly diminished growth rate with minimal skeletal abnormality. In contrast, homozygous knockout animals developed in utero but died near birth and exhibited a severe chondrodystrophic phenotype, with abnormalities throughout the skeleton, including the limbs, spine, and rib cage (Fig. 9.7). The severely compressed rib cage, with its detrimental consequence on lung development, may account for the premature death of these mice compared to the Prx1-Cre-Has2 mice.

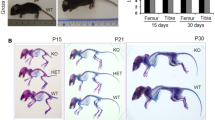
Appearance and skeleton of Has2 knockout mice. Wild-type mice (WT) and Has2 knockout mice (KO) at E18.5 are compared for their external appearance (a) and for the structure of their skeletons (b). Note the severe chondrodysplasia in the knockout mice, with shortened body length, limbs, snout, and rib cage. Cartilage is stained with Alcian blue and bone with Alizarin red
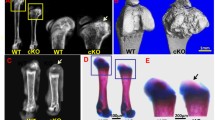
The long bones of the limbs of the mutant mice were short and wide, and while the cartilaginous anlagen for the bones appeared to develop normally, there was little evidence of further growth during embryonic development or for bone modeling within the diaphysis (Fig. 9.8). In the wild-type bones, a distinct primary center of ossification with adjacent growth plates had formed in the diaphysis by E15.5, whereas in the mutant, the primary center of ossification had yet to form, though hypertrophic cells were present. By E18.5, the diaphysis of the wild-type bones possessed a cortical rim with a well-formed trabecular center, whereas the mutant bones possessed an extensively mineralized tissue throughout the diaphysis, suggesting that the initial mineralized bone matrix had not been modeled. Perhaps surprisingly, these defects appear to be more severe than those reported for the long bone of the Prx1-Cre-Has2 mouse. The reason for the impaired modeling is not clear, as there was no apparent deficiency in vascular invasion or osteoclasts to account for such a defect. However, both osteoblast and osteoclast functions are influenced by the presence of HA in their surrounding ECM, and their attachment to a HA-deficient cartilage matrix may promote osteoid formation and impair bone resorption.

Histological analysis of the long bones and spine. Non-decalcified sections of femurs from wild-type mice (WT) and Has2 knockout mice (KO) were stained with Goldner stain (a). The femurs of the Has2 KO mice possess a thick cortical bone adjacent to the periosteum, with little evidence for modeling at the endosteal surface. Decalcified sections of femurs (b) and spines (c) from wild-type mice and Has2 knockout mice were stained with Safranin O. There was little evidence of an organized growth plate at the femoral epiphysis, and the formation of the vertebral primary center of ossification and the appearance of the IVDs were abnormal in the KO mice
The increased mineralization in the mutant diaphysis could also be due to excessive periosteal bone formation. This raises the intriguing possibility of decreased production of HA by the osteoblasts themselves in the Col2-Cre-Has2 mouse. In support of this possibility, Col2-driven lacZ expression has been transiently detected in the periosteal osteoblasts (Nakamura et al. 2006). Col2 has also been shown to be transiently expressed during intramembranous bone formation of the skull (Abzhanov et al. 2007), suggesting that cranial vault osteoblasts might also be affected in the Col2-Cre-Has2 knockout mice. Interestingly, the heads of the Has2 knockout mice appear shorter with a dome-shaped cranial vault (Fig. 9.7). Alternatively, lack of HA in the cartilage could create an environment where signaling events from chondrocytes to periosteal osteoblasts are disturbed. In this respect, it is interesting to note that the presence of CS can regulate the diffusion of Ihh between prehypertrophic chondrocytes and the periosteum (Cortes et al. 2009). CS localization in the growth plate cartilage is dependent on proteoglycan aggregate formation, which would be prevented in the absence of HA.
To fully assess the role of diminished HA production by osteoblasts on bone formation and turnover, osteoblast-specific knockout mice will be needed. They could be generated using an osterix(Osx)-Cre (Rodda and McMahon 2006) or a bone-specific Col1a1-Cre (Dacquin et al. 2002) mouse line to excise the floxed Has2 gene in the osteoblast lineage. The Osx-Cre line should excise Has2 early in committed osteoblast precursors but, at the same time, should not affect chondrogenesis. Such studies can show whether deletion of Has2 only in osteoblasts affects both intramembranous ossification and endochondral ossification. Indirectly, it could also indicate whether the presence of HA in osteoid has any influence on osteoclast remodeling activity.
At E18.5, there was also no evidence for normal growth plate formation and organized endochondral ossification in the mutant long bones, as expected in view of the lack of linear growth (Fig. 9.8). As with the Prx1-Cre-Has2 mice, there did not appear to be a deficiency in cell proliferation in the region where the growth plate should be, but the proliferating chondrocytes appeared unable to organize into linear columns. The cells did however begin to undergo hypertrophy, though the resulting cells were smaller than normal hypertrophic chondrocytes, more irregular in shape, and appeared to die prematurely. They also did not efficiently calcify their surrounding ECM. Thus, the absence of HA affects the formation, function, and fate of the hypertrophic chondrocytes and impairs the normal progression from endochondral cartilage to endochondral bone.
Both the vertebrae and intervertebral discs of the spine also appeared abnormal at E18.5 (Fig. 9.8). All vertebrae were reduced in height, and there was no evidence of an organized primary center of ossification with surrounding proliferative and hypertrophic chondrocytes. As with the mutant long bones, chondrocyte proliferation and hypertrophy surrounding the primary centers of ossification in the mutant vertebrae occurred in a haphazard and incomplete manner, with evidence for excessive mineralized tissue in its center. Development of the mutant intervertebral discs appeared to be delayed, with an increased abundance of large vacuolated cells (presumably notochordal cells) in their nucleus pulposus and remnants of the notochord persisting between adjacent discs.
In contrast to the Prx1-Cre-Has2 mouse, aggrecan was still abundant in the epiphyseal cartilage ECM despite the depletion in HA synthesis, though in both mice the amount of ECM was drastically reduced. In the absence of HA, aggrecan would be expected to diffuse from the tissue, and therefore it is likely that some HA persists in the Col2-Cre-Has2 mouse. Such HA might have been produced by mesenchymal cells in the cartilage anlagen of the bones prior to chondrocyte differentiation and then persist in the developing cartilage. However, such residual HA is insufficient to maintain normal function.
5.3 Inducible Has2 Knockout Mice
While HA production via Has2 gene expression is essential for normal cartilage and long bone development in the fetus (Moffatt et al. 2011; Matsumoto et al. 2009), it is not known whether a decrease in Has2 activity or HA production must be present from the onset of chondrogenesis for a pathologic phenotype to develop or whether depletion at later stages of development can also be problematic. Deficient HA synthesis by growth plate chondrocytes in the juvenile could impair long bone and vertebral growth, and deficient HA synthesis by articular chondrocytes and intervertebral disc cells in the adult could result in premature joint degeneration.
To address this issue, an inducible cartilage-specific Has2 knockout mouse, in which Has2 expression can be inactivated specifically in cartilage at various stages of postnatal development and growth, is needed. For this purpose, a mouse expressing the Cre transgene under control of both the Col2 promoter (for tissue specificity) and doxycycline administration (for temporal selectivity) has been generated (Grover and Roughley 2006). The inducible Col2-rtTA-Cre mouse can be crossed with the floxed Has2 line to generate the conditional knockout line. Although such mice develop a phenotype similar to that of the Col2-Cre-Has2 mice when fed doxycycline from the moment of conception, no major phenotype has been observed when such mice have been fed doxycycline postnatally.
One of the underlying premises upon which the predicted outcomes for postnatal HA depletion are based is that HA turnover is taking place in cartilage throughout life at a sufficient rate for diminished HA synthesis to deplete HA levels. If this is not the case, then no phenotype would be observed. It is certainly possible that HA turnover in the remote cartilage ECM could be slow and this could have contributed to the residual Safranin O staining present in the Col2-Cre-Has2 knockout mice. However, HA turnover is thought to be rapid at the cell surface (Morales and Hascall 1988), and depletion at this site might be expected to influence cell signaling and result in an abnormal phenotype irrespective of whether ECM changes occur or not.
It is also possible that postnatal administration of doxycycline is not able to promote complete excision of the floxed Has2 alleles in all chondrocytes, either because of poor delivery into larger avascular cartilages or because the activity of the Col2 promoter decreases with age. An alternative strategy to circumvent the latter problem would be the use of an alternative inducible Cre mouse line, such as the Agc1-Cre-Tet mouse, which uses an aggrecan gene enhancer to drive Cre expression (Han and Lefebvre 2008), or the aggrecan-Cre-ERT2 mouse, which possesses an inducible Cre construct within the 3′-UTR of the aggrecan gene (Henry et al. 2009). Both of these lines allow Cre expression in mature cartilage. The lack of a phenotype does, however, raise the question as to what level HA must be depleted in order for an abnormal phenotype to develop?
6 Disorders Due to Impaired HA Metabolism
Even though Has gene mutations have not been associated with skeletal disorders in humans, perturbation in Has expression and HA production has been associated with several nonskeletal disorders, particularly malignancies (Adamia et al. 2008; Ghosh et al. 2009; Yamane et al. 2010). Furthermore, in the Shar-Pei dog, a mutation upstream of the Has2 gene results in increased expression of Has2 and increased HA production, resulting in the characteristic thickened skin and a predisposition for periodic fever syndrome (Olsson et al. 2011). This latter feature may be a consequence of the pro-inflammatory nature of increased HA fragment generation during hyaluronidase-mediated turnover of the HA. An inability to degrade HA to small fragments, due to Hyal1 deficiency, results in mucopolysaccharidosis type IX (Triggs-Raine et al. 1999), which does show skeletal abnormalities leading to short stature.
It is also possible that deficient HA production could be associated postnatally with skeletal problems involving impaired osteogenesis, such as delayed bone formation during fracture healing and distraction osteogenesis, which both involve endochondral ossification. This could be due to abnormalities in HA production or fragmentation. In such cases, there is the intriguing possibility of using local HA administration to enhance bone repair. Local administration of HA has been used clinically in several situations for many years, including eye surgery and the arthritic joint (Laurent and Fraser 1992).
References
Abzhanov A, Rodda SJ, McMahon AP, Tabin CJ (2007) Regulation of skeletogenic differentiation in cranial dermal bone. Development 134:3133–3144
Adamia S, Reichert AA, Kuppusamy H, Kriangkum J, Ghosh A, Hodges JJ, Pilarski PM, Treon SP, Mant MJ, Reiman T, Belch AR, Pilarski LM (2008) Inherited and acquired variations in the hyaluronan synthase 1 (HAS1) gene may contribute to disease progression in multiple myeloma and Waldenstrom macroglobulinemia. Blood 112:5111–5121
Andhare RA, Takahashi N, Knudson W, Knudson CB (2009) Hyaluronan promotes the chondrocyte response to BMP-7. Osteoarthr Cartil 17:906–916
Ariyoshi W, Takahashi T, Kanno T, Ichimiya H, Takano H, Koseki T, Nishihara T (2005) Mechanisms involved in enhancement of osteoclast formation and function by low molecular weight hyaluronic acid. J Biol Chem 280:18967–18972
Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B (1990) CD44 is the principal cell surface receptor for hyaluronate. Cell 61:1303–1313
Atmuri V, Martin DC, Hemming R, Gutsol A, Byers S, Sahebjam S, Thliveris JA, Mort JS, Carmona E, Anderson JE, Dakshinamurti S, Triggs-Raine B (2008) Hyaluronidase 3 (HYAL3) knockout mice do not display evidence of hyaluronan accumulation. Matrix Biol 27:653–660
Barna M, Niswander L (2007) Visualization of cartilage formation: insight into cellular properties of skeletal progenitors and chondrodysplasia syndromes. Dev Cell 12:931–941
Bastow ER, Byers S, Golub SB, Clarkin CE, Pitsillides AA, Fosang AJ (2008) Hyaluronan synthesis and degradation in cartilage and bone. Cell Mol Life Sci 65:395–413
Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B (1999) Sox9 is required for cartilage formation. Nat Genet 22:85–89
Buckwalter JA, Rosenberg LC, Ungar R (1987) Changes in proteoglycan aggregates during cartilage mineralization. Calcif Tissue Int 41:228–236
Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A Jr, Kubalak S, Klewer SE, McDonald JA (2000) Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 106:349–360
Cao JJ, Singleton PA, Majumdar S, Boudignon B, Burghardt A, Kurimoto P, Wronski TJ, Bourguignon LY, Halloran BP (2005) Hyaluronan increases RANKL expression in bone marrow stromal cells through CD44. J Bone Miner Res 20:30–40
Cao G, Savani RC, Fehrenbach M, Lyons C, Zhang L, Coukos G, Delisser HM (2006) Involvement of endothelial CD44 during in vivo angiogenesis. Am J Pathol 169:325–336
Cortes M, Baria AT, Schwartz NB (2009) Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development 136:1697–1706
Csoka AB, Frost GI, Stern R (2001) The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol 20:499–508
Dacquin R, Starbuck M, Schinke T, Karsenty G (2002) Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn 224:245–251
Dai J, Rabie AB (2007) VEGF: an essential mediator of both angiogenesis and endochondral ossification. J Dent Res 86:937–950
Day AJ, Prestwich GD (2002) Hyaluronan-binding proteins: tying up the giant. J Biol Chem 277: 4585–4588
de Vries TJ, Schoenmaker T, Beertsen W, van der Neut R, Everts V (2005) Effect of CD44 deficiency on in vitro and in vivo osteoclast formation. J Cell Biochem 94:954–966
Doege K, Sasaki M, Horigan E, Hassell JR, Yamada Y (1987) Complete primary structure of the rat cartilage proteoglycan core protein deduced from cDNA clones. J Biol Chem 262: 17757–17767
Doege KJ, Sasaki M, Kimura T, Yamada Y (1991) Complete coding sequence and deduced primary structure of the human cartilage large aggregating proteoglycan, aggrecan. Human-specific repeats, and additional alternatively spliced forms. J Biol Chem 266:894–902
Dowthwaite GP, Edwards JC, Pitsillides AA (1998) An essential role for the interaction between hyaluronan and hyaluronan binding proteins during joint development. J Histochem Cytochem 46:641–651
Dowthwaite GP, Flannery CR, Flannelly J, Lewthwaite JC, Archer CW, Pitsillides AA (2003) A mechanism underlying the movement requirement for synovial joint cavitation. Matrix Biol 22:311–322
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Dunlop LL, Hall BK (1995) Relationships between cellular condensation, preosteoblast formation and epithelial-mesenchymal interactions in initiation of osteogenesis. Int J Dev Biol 39: 357–371
Durigova M, Troeberg L, Nagase H, Roughley PJ, Mort JS (2011) Involvement of ADAMTS5 and hyaluronidase in aggrecan degradation and release from OSM-stimulated cartilage. Eur Cell Mater 21:31–45
Dy P, Smits P, Silvester A, Penzo-Mendez A, Dumitriu B, Han Y, de la Motte CA, Kingsley DM, Lefebvre V (2010) Synovial joint morphogenesis requires the chondrogenic action of Sox5 and Sox6 in growth plate and articular cartilage. Dev Biol 341:346–359
Falconi D, Aubin JE (2007) LIF inhibits osteoblast differentiation at least in part by regulation of HAS2 and its product hyaluronan. J Bone Miner Res 22:1289–1300
Fieber C, Plug R, Sleeman J, Dall P, Ponta H, Hofmann M (1999) Characterisation of the murine gene encoding the intracellular hyaluronan receptor IHABP (RHAMM). Gene 226:41–50
Fieber C, Baumann P, Vallon R, Termeer C, Simon JC, Hofmann M, Angel P, Herrlich P, Sleeman JP (2004) Hyaluronan-oligosaccharide-induced transcription of metalloproteases. J Cell Sci 117:359–367
Flannery CR, Little CB, Caterson B (1998) Molecular cloning and sequence analysis of the aggrecan interglobular domain from porcine, equine, bovine and ovine cartilage: comparison of proteinase-susceptible regions and sites of keratan sulfate substitution. Matrix Biol 16: 507–511
Fraser JRE, Laurent TC, Laurent UBG (1997) Hyaluronan: its nature, distribution, functions and turnover. J Intern Med 242:27–33
Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N (1999) VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 5:623–628
Ghosh A, Kuppusamy H, Pilarski LM (2009) Aberrant splice variants of HAS1 (Hyaluronan Synthase 1) multimerize with and modulate normally spliced HAS1 protein: a potential mechanism promoting human cancer. J Biol Chem 284:18840–18850
Gleghorn L, Ramesar R, Beighton P, Wallis G (2005) A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet 77:484–490
Grover J, Roughley PJ (2006) Generation of a transgenic mouse in which Cre recombinase is expressed under control of the type II collagen promoter and doxycycline administration. Matrix Biol 25:158–165
Hall BK, Miyake T (1992) The membranous skeleton: the role of cell condensations in vertebrate skeletogenesis. Anat Embryol 186:107–124
Han Y, Lefebvre V (2008) L-Sox5 and Sox6 drive expression of the aggrecan gene in cartilage by securing binding of Sox9 to a far-upstream enhancer. Mol Cell Biol 28:4999–5013
Hascall VC (1988) Proteoglycans: the chondroitin sulfate/keratan sulfate proteoglycan of cartilage. ISI Atlas Sci Biochem 1:189–198
Hascall VC, Majors AK, De la Motte CA, Evanko SP, Wang AM, Drazba JA, Strong SA, Wight TN (2004) Intracellular hyaluronan: a new frontier for inflammation? Biochim Biophys Acta Gen Subj 1673:3–12
Hatano H, Shigeishi H, Kudo Y, Higashikawa K, Tobiume K, Takata T, Kamata N (2011) Overexpression of receptor for hyaluronan-mediated motility (RHAMM) in MC3T3-E1 cells induces proliferation and differentiation through phosphorylation of ERK1/2. J Bone Miner Metab 30(3):293–303
Hayer S, Steiner G, Gortz B, Reiter E, Tohidast-Akrad M, Amling M, Hoffmann O, Redlich K, Zwerina J, Skriner K, Hilberg F, Wagner EF, Smolen JS, Schett G (2005) CD44 is a determinant of inflammatory bone loss. J Exp Med 201:903–914
Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, Decrombrugghe B (2009) Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 47(12): 805–814
Hering TM, Kollar J, Huynh TD (1997) Complete coding sequence of bovine aggrecan: comparative structural analysis. Arch Biochem Biophys 345:259–270
Hiscock DRR, Caterson B, Flannery CR (2000) Expression of hyaluronan synthases in articular cartilage. Osteoarthr Cartil 8:120–126
Holmes MWA, Bayliss MT, Muir H (1988) Hyaluronic acid in human articular cartilage. Age-related changes in content and size. Biochem J 250:435–441
Hughes DE, Salter DM, Simpson R (1994) CD44 expression in human bone: a novel marker of osteocytic differentiation. J Bone Miner Res 9:39–44
Hughes CE, Caterson B, Fosang AJ, Roughley PJ, Mort JS (1995) Monoclonal antibodies that specifically recognize neoepitope sequences generated by `aggrecanase' and matrix metalloproteinase cleavage of aggrecan: application to catabolism in situ and in vitro. Biochem J 305:799–804
Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M, Shinomura T, Hamaguchi M, Yoshida Y, Ohnuki Y, Miyauchi S, Spicer AP, McDonald JA, Kimata K (1999) Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem 274: 25085–25092
Jadin L, Wu X, Ding H, Frost GI, Onclinx C, Triggs-Raine B, Flamion B (2008) Skeletal and hematological anomalies in HYAL2-deficient mice: a second type of mucopolysaccharidosis IX? FASEB J 22:4316–4326
Jamal HH, Aubin JE (1996) CD44 expression in fetal rat bone: in vivo and in vitro analysis. Exp Cell Res 223:467–477
Janiszewska M, De VC, Le Bitoux MA, Fusco C, Stamenkovic I (2010) Transportin regulates nuclear import of CD44. J Biol Chem 285:30548–30557
Karniski LP (2001) Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene: correlation between sulfate transport activity and chondrodysplasia phenotype. Hum Mol Genet 10:1485–1490
Karousou E, Kamiryo M, Skandalis SS, Ruusala A, Asteriou T, Passi A, Yamashita H, Hellman U, Heldin CH, Heldin P (2010) The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J Biol Chem 285:23647–23654
Karsenty G, Kronenberg HM, Settembre C (2009) Genetic control of bone formation. Annu Rev Cell Dev Biol 25:629–648
Knudson W, Chow G, Knudson CB (2002) CD44-mediated uptake and degradation of hyaluronan. Matrix Biol 21:15–23
Kosher RA, Savage MP, Walker KH (1981) A gradation of hyaluronate accumulation along the proximodistal axis of the embryonic chick limb bud. J Embryol Exp Morphol 63:85–98
Krueger RC Jr, Kurima K, Schwartz NB (1999) Completion of the mouse aggrecan gene structure and identification of the defect in the cmd-Bc mouse as a near complete deletion of the murine aggrecan gene. Mamm Genome 10:1119–1125
Lammich S, Okochi M, Takeda M, Kaether C, Capell A, Zimmer AK, Edbauer D, Walter J, Steiner H, Haass C (2002) Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Abeta-like peptide. J Biol Chem 277:44754–44759
Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, Karperien M, Defize LH, Ho C, Mulligan RC, Abou-Samra AB, Juppner H, Segre GV, Kronenberg HM (1996) PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 273: 663–666
Lark MW, Bayne EK, Flanagan J, Harper CF, Hoerrner LA, Hutchinson NI, Singer II, Donatelli SA, Weidner JR, Williams HR, Mumford RA, Lohmander LS (1997) Aggrecan degradation in human cartilage - Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J Clin Invest 100:93–106
Laurent TC, Fraser JRE (1992) Hyaluronan. FASEB J 6:2397–2404
Lee JY, Spicer AP (2000) Hyaluronan: a multifunctional, megaDalton, stealth molecule. Curr Opin Cell Biol 12:581–586
Lee JL, Wang MJ, Chen JY (2009) Acetylation and activation of STAT3 mediated by nuclear translocation of CD44. J Cell Biol 185:949–957
Lefebvre V, Li P, de Crombrugghe B (1998) A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J 17:5718–5733
Li H, Schwartz NB, Vertel BM (1993) cDNA cloning of chick cartilage chondroitin sulfate (aggrecan) core protein and identification of a stop codon in the aggrecan gene associated with the chondrodystrophy, nanomelia. J Biol Chem 268:23504–23511
Li Y, Toole BP, Dealy CN, Kosher RA (2007) Hyaluronan in limb morphogenesis. Dev Biol 305: 411–420
Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ (2002) Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33:77–80
Mack JA, Feldman RJ, Itano N, Kimata K, Lauer M, Hascall VC, Maytin EV (2012) Enhanced inflammation and accelerated wound closure following tetraphorbol ester application or full-thickness wounding in mice lacking hyaluronan synthases has1 and has3. J Invest Dermatol 132:198–207
Maroudas A, Bayliss MT, Uchitel-Kaushansky N, Schneiderman R, Gilav E (1998) Aggrecan turnover in human articular cartilage: use of aspartic acid racemization as a marker of molecular age. Arch Biochem Biophys 350:61–71
Martin DC, Atmuri V, Hemming RJ, Farley J, Mort JS, Byers S, Hombach-Klonisch S, Csoka AB, Stern R, Triggs-Raine BL (2008) A mouse model of human mucopolysaccharidosis IX exhibits osteoarthritis. Hum Mol Genet 17:1904–1915
Matsui Y, Alini M, Webber C, Poole AR (1991) Characterization of aggregating proteoglycans from the proliferative, maturing, hypertrophic, and calcifying zones of the cartilaginous physis. J Bone Joint Surg Am 73:1064–1074
Matsumoto K, Li Y, Jakuba C, Sugiyama Y, Sayo T, Okuno M, Dealy CN, Toole BP, Takeda J, Yamaguchi Y, Kosher RA (2009) Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development 136:2825–2835
Maxwell CA, McCarthy J, Turley E (2008) Cell-surface and mitotic-spindle RHAMM: moonlighting or dual oncogenic functions? J Cell Sci 121:925–932
Maxwell CA, Benitez J, Gomez-Baldo L, Osorio A, Bonifaci N, Fernandez-Ramires R et al (2011) Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol 9:e1001199
Meyer K, Palmer JW (1953) The polysaccharide of the vitreous humor. J Biol Chem 107:629–634
Moffatt P, Lee ER, St Jacques B, Matsumoto K, Yamaguchi Y, Roughley PJ (2011) Hyaluronan production by means of Has2 gene expression in chondrocytes is essential for long bone development. Dev Dyn 240:404–412
Morales TI, Hascall VC (1988) Correlated metabolism of proteoglycans and hyaluronic acid in bovine cartilage organ cultures. J Biol Chem 263:3632–3638
Morgelin M, Paulsson M, Hardingham TE, Heinegard D, Engel J (1988) Cartilage proteoglycans. Assembly with hyaluronate and link protein as studied by electron microscopy. Biochem J 253: 175–185
Mukhopadhyay A, Nikopoulos K, Maugeri A, de Brouwer AP, van Nouhuys CE, Boon CJ, Perveen R, Zegers HA, Wittebol-Post D, van den Biesen PR, van der Velde-Visser SD, Brunner HG, Black GC, Hoyng CB, Cremers FP (2006) Erosive vitreoretinopathy and wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest Ophthalmol Vis Sci 47:3565–3572
Murakami D, Okamoto I, Nagano O, Kawano Y, Tomita T, Iwatsubo T, De SB, Yumoto E, Saya H (2003) Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 22:1511–1516
Nakamura H, Ozawa H (1996) Immunolocalization of CD44 and the ERM family in bone cells of mouse tibiae. J Bone Miner Res 11:1715–1722
Nakamura H, Kenmotsu S, Sakai H, Ozawa H (1995) Localization of CD44, the hyaluronate receptor, on the plasma membrane of osteocytes and osteoclasts in rat tibiae. Cell Tissue Res 280:225–233
Nakamura E, Nguyen MT, Mackem S (2006) Kinetics of tamoxifen-regulated Cre activity in mice using a cartilage-specific CreER(T) to assay temporal activity windows along the proximodistal limb skeleton. Dev Dyn 235:2603–2612
Nedvetzki S, Gonen E, Assayag N, Reich R, Williams RO, Thurmond RL, Huang JF, Neudecker BA, Wang FS, Turley EA, Naor D (2004) RHAMM, a receptor for hyaluronan-mediated motility, compensates for CD44 in inflamed CD44-knockout mice: a different interpretation of redundancy. Proc Natl Acad Sci USA 101:18081–18086
Nishida Y, Knudson CB, Nietfeld JJ, Margulis A, Knudson W (1999) Antisense inhibition of hyaluronan synthase-2 in human articular chondrocytes inhibits proteoglycan retention and matrix assembly. J Biol Chem 274:21893–21899
Noble PW (2002) Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biol 21: 25–29
Noonan KJ, Stevens JW, Tammi R, Tammi M, Hernandez JA, Midura RJ (1996) Spatial distribution of CD44 and hyaluronan in the proximal tibia of the growing rat. J Orthop Res 14:573–581
Ohno S, Im HJ, Knudson CB, Knudson W (2005) Hyaluronan oligosaccharide-induced activation of transcription factors in bovine articular chondrocytes. Arthritis Rheum 52:800–809
Ohno S, Im HJ, Knudson CB, Knudson W (2006) Hyaluronan oligosaccharides induce matrix metalloproteinase 13 via transcriptional activation of NFkappaB and p38 MAP kinase in articular chondrocytes. J Biol Chem 281:17952–17960
Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, Wong AJ, Saya H (2001) Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol 155:755–762
Olaku V, Matzke A, Mitchell C, Hasenauer S, Sakkaravarthi A, Pace G, Ponta H, Orian-Rousseau V (2011) c-Met recruits ICAM-1 as a coreceptor to compensate for the loss of CD44 in Cd44 null mice. Mol Biol Cell 22:2777–2786
Olsson M, Meadows JR, Truve K, Rosengren PG, Puppo F, Mauceli E, Quilez J, Tonomura N, Zanna G, Docampo MJ, Bassols A, Avery AC, Karlsson EK, Thomas A, Kastner DL, Bongcam-Rudloff E, Webster MT, Sanchez A, Hedhammar A, Remmers EF, Andersson L, Ferrer L, Tintle L, Lindblad-Toh K (2011) A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet 7:e1001332
Ornitz DM, Marie PJ (2002) FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 16:1446–1465
Pardue EL, Ibrahim S, Ramamurthi A (2008) Role of hyaluronan in angiogenesis and its utility to angiogenic tissue engineering. Organogenesis 4:203–214
Pavasant P, Shizari T, Underhill CB (1996) Hyaluronan contributes to the enlargement of hypertrophic lacunae in the growth plate. J Cell Sci 109:327–334
Peterson RS, Andhare RA, Rousche KT, Knudson W, Wang W, Grossfield JB, Thomas RO, Hollingsworth RE, Knudson CB (2004) CD44 modulates Smad1 activation in the BMP-7 signaling pathway. J Cell Biol 166:1081–1091
Prehm P (2006) Biosynthesis of hyaluronan: direction of chain elongation. Biochem J 398: 469–473
Primorac D, Stover ML, Clark SH, Rowe DW (1994) Molecular basis of nanomelia, a heritable chondrodystrophy of chicken. Matrix 14:297–305
Prince CW (2004) Roles of hyaluronan in bone resorption. BMC Musculoskelet Disord 5:12
Protin U, Schweighoffer T, Jochum W, Hilberg F (1999) CD44-deficient mice develop normally with changes in subpopulations and recirculation of lymphocyte subsets. J Immunol 163: 4917–4923
Recklies AD, White C, Melching L, Roughley PJ (2001) Differential regulation and expression of hyaluronan synthases in human articular chondrocytes, synovial cells and osteosarcoma cells. Biochem J 354:17–24
Reed RK, Lilja K, Laurent TC (1988) Hyaluronan in the rat with special reference to the skin. Acta Physiol Scand 134:405–411
Rodda SJ, McMahon AP (2006) Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133: 3231–3244
Rodriguez E, Roughley P (2006) Link protein can retard the degradation of hyaluronan in proteoglycan aggregates. Osteoarthr Cartil 14:823–829
Roughley PJ, Melching LI, Heathfield TF, Pearce RH, Mort JS (2006) The structure and degradation of aggrecan in human intervertebral disc. Eur Spine J 15(Suppl 3):326–332
Roughley PJ, Lamplugh L, Lee ER, Matsumoto K, Yamaguchi Y (2011) The role of hyaluronan produced by Has2 gene expression in development of the spine. Spine 36:E914–E920
Rousche KT, Knudson CB (2002) Temporal expression of CD44 during embryonic chick limb development and modulation of its expression with retinoic acid. Matrix Biol 21:53–62
Schmits R, Filmus J, Gerwin N, Senaldi G, Kiefer F, Kundig T, Wakeham A, Shahinian A, Catzavelos C, Rak J, Furlonger C, Zakarian A, Simard JJ, Ohashi PS, Paige CJ, Gutierrez-Ramos JC, Mak TW (1997) CD44 regulates hematopoietic progenitor distribution, granuloma formation, and tumorigenicity. Blood 90:2217–2233
Schmitz I, Ariyoshi W, Takahashi N, Knudson CB, Knudson W (2010) Hyaluronan oligosaccharide treatment of chondrocytes stimulates expression of both HAS-2 and MMP-3, but by different signaling pathways. Osteoarthr Cartil 18:447–454
Sherman L, Wainwright D, Ponta H, Herrlich P (1998) A splice variant of CD44 expressed in the apical ectodermal ridge presents fibroblast growth factors to limb mesenchyme and is required for limb outgrowth. Genes Dev 12:1058–1071
Shyjan AM, Heldin P, Butcher EC, Yoshino T, Briskin MJ (1996) Functional cloning of the cDNA for a human hyaluronan synthase. J Biol Chem 271:23395–23399
Sivan SS, Tsitron E, Wachtel E, Roughley PJ, Sakkee N, Van der Ham F, DeGroot J, Roberts S, Maroudas A (2006) Aggrecan turnover in human intervertebral disc as determined by the racemization of aspartic acid. J Biol Chem 281:13009–13014
Slevin M, Krupinski J, Gaffney J, Matou S, West D, Delisser H, Savani RC, Kumar S (2007) Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol 26:58–68
Spicer AP, Olson JS, McDonald JA (1997) Molecular cloning and characterization of a cDNA encoding the third putative mammalian hyaluronan synthase. J Biol Chem 272:8957–8961
St-Jacques B, Hammerschmidt M, McMahon AP (1999) Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 13:2072–2086
Stern R, Asari AA, Sugahara KN (2006) Hyaluronan fragments: an information-rich system. Eur J Cell Biol 85:699–715
Stern R, Kogan G, Jedrzejas MJ, Soltes L (2007) The many ways to cleave hyaluronan. Biotechnol Adv 25:537–557
Superti-Furga A, Rossi A, Steinmann B, Gitzelmann R (1996) A chondrodysplasia family produced by mutations in the diastrophic dysplasia sulfate transporter gene: Genotype/phenotype correlations. Am J Med Genet 63:144–147
Sztrolovics R, Alini M, Roughley PJ, Mort JS (1997) Aggrecan degradation in human intervertebral disc and articular cartilage. Biochem J 326:235–241
Sztrolovics R, Grover J, CS-Szabo G, Zhang YP, Mort JS, Roughley PJ (2002) The characterization of versican and its message in human articular cartilage and intervertebral disc. J Orthop Res 20:257–266
Tanne Y, Tanimoto K, Tanaka N, Ueki M, Lin YY, Ohkuma S, Kamiya T, Tanaka E, Tanne K (2008) Expression and activity of Runx2 mediated by hyaluronan during chondrocyte differentiation. Arch Oral Biol 53:478–487
Teder P, Vandivier RW, Jiang D, Liang J, Cohn L, Pure E, Henson PM, Noble PW (2002) Resolution of lung inflammation by CD44. Science 296:155–158
Tien JY, Spicer AP (2005) Three vertebrate hyaluronan synthases are expressed during mouse development in distinct spatial and temporal patterns. Dev Dyn 233:130–141
Tolg C, Hamilton SR, Nakrieko KA, Kooshesh F, Walton P, McCarthy JB, Bissell MJ, Turley EA (2006) Rhamm−/− fibroblasts are defective in CD44-mediated ERK1,2 motogenic signaling, leading to defective skin wound repair. J Cell Biol 175:1017–1028
Tolg C, Hamilton SR, Morningstar L, Zhang J, Zhang S, Esguerra KV, Telmer PG, Luyt LG, Harrison R, McCarthy JB, Turley EA (2010) RHAMM promotes interphase microtubule instability and mitotic spindle integrity through MEK1/ERK1/2 activity. J Biol Chem 285: 26461–26474
Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D (2009) A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet 84:72–79
Toole BP, Jackson G, Gross J (1972) Hyaluronate in morphogenesis: inhibition of chondrogenesis in vitro. Proc Natl Acad Sci USA 69:1384–1386
Triggs-Raine B, Salo TJ, Zhang H, Wicklow BA, Natowicz MR (1999) Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc Natl Acad Sci USA 96:6296–6300
Turley EA, Noble PW, Bourguignon LYW (2002) Signaling properties of hyaluronan receptors. J Biol Chem 277:4589–4592
Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ (1996) Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 273:613–622
Wallis GA (1995) Cartilage disorders: the importance of being sulphated. Curr Biol 5:225–227
Watanabe H, Yamada Y (2002) Chondrodysplasia of gene knockout mice for aggrecan and link protein. Glycoconj J 19:269–273
Watanabe K, Yamaguchi Y (1996) Molecular identification of a putative human hyaluronan synthase. J Biol Chem 271:22945–22948
Watanabe H, Kimata K, Line S, Strong D, Gao L, Kozak CA, Yamada Y (1994) Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat Genet 7:154–157
Watanabe H, Gao L, Sugiyama S, Doege K, Kimata K, Yamada Y (1995) Mouse aggrecan, a large cartilage proteoglycan: protein sequence, gene structure and promoter sequence. Biochem J 308:433–440
Watanabe H, Cheung SC, Itano N, Kimata K, Yamada Y (1997) Identification of hyaluronan-binding domains of aggrecan. J Biol Chem 272:28057–28065
Weigel PH, DeAngelis PL (2007) Hyaluronan synthases: a decade-plus of novel glycosyltransferases. J Biol Chem 282:36777–36781
Weigel PH, Hascall VC, Tammi M (1997) Hyaluronan synthases. J Biol Chem 272:13997–14000
Weissmann B, Meyer K, Sampson P, Linker A (1954) Isolation of oligosaccharides enzymatically produced from hyaluronic acid. J Biol Chem 208:417–429
West DC, Hampson IN, Arnold F, Kumar S (1985) Angiogenesis induced by degradation products of hyaluronic acid. Science 228:1324–1326
Wight TN (2002) Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol 14:617–623
Yamane T, Kobayashi-Hattori K, Oishi Y, Takita T (2010) High-fat diet reduces levels of type I tropocollagen and hyaluronan in rat skin. Mol Nutr Food Res 54(Suppl 1):S53–S61
Zheng J, Luo W, Tanzer ML (1998) Aggrecan synthesis and secretion - a paradigm for molecular and cellular coordination of multiglobular protein folding and intracellular trafficking. J Biol Chem 273:12999–13006
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Roughley, P.J., Moffatt, P. (2013). The Role of HA and Has2 in the Development and Function of the Skeleton. In: DeSimone, D., Mecham, R. (eds) Extracellular Matrix in Development. Biology of Extracellular Matrix. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-35935-4_9
Download citation
DOI: https://doi.org/10.1007/978-3-642-35935-4_9
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-35934-7
Online ISBN: 978-3-642-35935-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)