
Abstract
Transforming growth factor-β-induced gene product-h3 (TGFBI/BIGH3) is an extracellular matrix protein expressed in a wide variety of tissues. TGFBI binds to type I, II, and IV collagens, as well as to biglycan and decorin and plays important roles in cell-to-cell, cell-to-collagen, and cell-to-matrix interactions. Furthermore, TGFBI is involved in cell growth and migration, tumorigenesis, wound healing, and apoptosis. To investigate whether TGFBI is involved in the maintenance of skeletal tissues, Tgfbi knockout mice were generated by crossing male and female Tgfbi heterozygous mice. Skeletal preparation showed that the skeletal size in Tgfbi knockout mice was smaller than in wild-type and heterozygous mice. However, chondrocytic cell alignment in the growth plates, bone mineral density, and bone forming rates were similar in Tgfbi knockout, wild-type, and heterozygous mice. Alterations in skeletal tissue arrangements in Tgfbi knockout mice were estimated from safranin O staining, trichrome staining, and immunohistochemistry for type II and X collagen, and matrix metalloproteinase 13 (MMP13). Cartilage matrix degradation was observed in the articular cartilage of Tgfbi knockout mice. Although the detection of type II collagen in the articular cartilage was lower in Tgfbi knockout mice than wild-type mice, the detection of MMP13 was markedly higher, indicating that Tgfbi deficiency is associated with the degradation of cartilage matrix. These results suggest that TGFBI plays an important role in maintaining skeletal tissues and the cartilage matrix in mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Transforming growth factor-β-induced gene product-h3 (TGFBI/BIGH3) was first identified in human lung adenocarcinoma cells. The gene encodes a 683-amino acid protein that contains a secretory signal sequence, an N-terminal cysteine-rich EMI domain, four fasciclin 1 domains, and an RGD (Arg–Gly–Asp) motif [1, 2]. TGFBI is an extracellular matrix (ECM) protein that modulates cell growth [3], tumorigenesis [4], wound healing [5], and apoptosis [6]. Moreover, it controls cell adhesion and migration [7–9]. TGFBI is expressed in various tissues, including the bone, cartilage, heart, liver, and skin [1, 7, 10, 11].
TGF-β is an important regulator of chondrocyte and osteoblast differentiation during endochondral ossification. The protein is localized to hypertrophic chondrocytes, osteoblasts, osteoclasts, and vascular structures [12]. TGF-β inhibits the terminal differentiation and hypertrophy of chondrocytes, which results in the synthesis of cartilage matrix and the maintenance of articular cartilage [13, 14]. TGF-β also regulates bone formation by activating osteoblast recruitment, differentiation, and function and inhibiting terminal osteoblast differentiation, bone matrix synthesis, and osteocyte apoptosis [15]. TGF-β induces TGFBI, which is strongly expressed in the perichondrium, periosteum, and prehypertrophic chondrocytes in articular cartilage and in growth plate cartilage during endochondral ossification [11]. TGFBI has a high affinity for other ECM proteins, including collagen, laminin, and fibronectin [16]. These observations suggest that TGFBI expressed in chondrocytes and osteoblasts forms important interactions with ECM proteins and maintains ECM homeostasis in skeletal tissues. In the present study, we examined alterations in the skeletal tissue of Tgfbi knockout mice.
Materials and Methods
Generation of Tgfbi Knockout Mice
Tgfbi knockout mice were generated by crossing male and female Tgfbi heterozygous mice. Genotypes were determined by polymerase chain reaction (PCR) using genomic DNA from the tail. The primer sets used and the PCR genotyping reactions have been described previously [17]. All animal experiments were conducted after obtaining approval from the animal ethics committee of Kyungpook National University.
Skeletal Preparation
Skeletons from postnatal mice were prepared and stained with alcian blue for cartilage and alizarin red S for bone [18, 19]. Briefly, the skin and viscera were removed. Samples were fixed overnight in 95 % ethanol and then stained overnight in 150 mg/L alcian blue solution (Sigma-Aldrich, St. Louis, MO, USA) containing 20 % acetic acid and 80 % ethanol. After being rinsed in 95 % ethanol for 3 h, the samples were treated with 2 % KOH for 24 h to clarify the soft tissues. Finally, the bones were stained with 50 mg/L alizarin red S solution containing 1 % KOH.
Histological and Histomorphometric Observations
Postnatal mice were fixed in 4 % paraformaldehyde at 4 °C overnight. Undecalcified vertebrae were embedded in destabilized methyl methacrylate as previously described [20], sectioned at 5-μm, and stained with von Kossa stain. To determine the dynamic histomorphometric indices, mice were injected with calcein at 30 mg/kg body weight at 6 and 2 days before sacrifice. Static and dynamic histomorphometric analysis was conducted using the Bioquant image analysis system (Bio-Quant, Inc., San Diego, CA, USA) [21]. Long bones decalcified for 1 month with 0.5 M ethylenediaminetetraacetic acid solution were embedded in paraffin, sectioned at 5-μm, and stained with hematoxylin and eosin (H&E), alcian blue, and safranin O. Masson trichrome staining (Sigma-Aldrich) [22] was performed to measure the collagen content. Using this system, organized collagen fibers were stained blue, and the cytoplasm, muscle, and erythrocytes were stained red [23].
Immunohistochemical Analysis
To examine the level and localization of type II collagen (Col2), type X collagen (Col10), and matrix metalloproteinase 13 (MMP13), deparaffinized 5-μm sections were blocked with 3 % bovine serum albumin at room temperature for 1 h and then incubated overnight at 4 °C with anti-Col2 antibody (1:100; LSL-LB-1297; Cosmo Bio Co. Tokyo, Japan), anti-Col10 antibody (1:100; LSL-LB-0092; Cosmo Bio Co.), anti-MMP13 antibody (1:50; ab75606; Abcam, Cambridge, MA, USA) or anti-aggrecan antibody (1:50; AB1031; EMD Millipore, Billerica, MA, USA) diluted in 0.03 % phosphate-buffered saline (PBS). After incubation with the primary antibody, the slides were washed in PBS and incubated with a biotinylated anti-rabbit IgG secondary antibody (VectaStain ABC Kit, Vector Laboratories, Burlingame, CA, USA) at room temperature for 1 h. Signals for antibody binding were visualized using diaminobenzidine substrate (Invitrogen, Carlsbad, CA, USA). Samples were counterstained with 0.25 % methyl green.
Results
Reduced Bone Growth in Tgfbi Knockout Mice
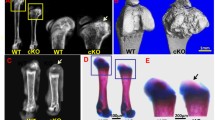
The Tgfbi heterozygous mouse model was generated by homologous recombination in C57BL/6 embryonic stem cells [17]. Although some heterozygous mice are slightly smaller and more nervous than wild-type mice, they have no specific phenotype. By crossing male and female Tgfbi heterozygous mice, wild-type, heterozygous, and knockout littermates were generated. In RT-PCR analysis, Tgfbi expression was not detected in the long bones, kidneys, or other tissues of Tgfbi knockout mice (Fig. S1 and data not shown). Whereas newborn Tgfbi knockout mice did not differ from their littermates (data not shown), mice at postnatal day 15 (P15) started to exhibit a reduction in body size (Fig. 1a). At P21, the body size of Tgfbi knockout mice was remarkably reduced compared with that of wild-type and heterozygous mice (Fig. 1a). To confirm the reduction in body size, bone lengths were observed using skeletons stained with alcian blue for cartilage and alizarin red for bone (Fig. 1b, c). Although the skeletal morphology was not altered, the long bones, including the humerus, radius, ulna, femur, and tibia were significantly shorter in Tgfbi knockout mice than wild-type mice at P15, P21, and P30 (Fig. 1b, c).

Gross appearance and skeletal preparation of Tgfbi knockout mice. a Gross appearance of Tgfbi knockout mice. Tgfbi knockout mice were smaller, overall, than wild-type mice on postnatal days 15 and 21 (P15 and P21). b For skeletal preparations of Tgfbi knockout mice at P15, P21, and P30, the skeleton was stained with alcian blue for cartilage and then with alizarin red for bone. The femur or tibia, representative of long bones, was significantly shorter in Tgfbi knockout mice. c Reduced lengths of the femur and tibia at P15 and P30. Both sides of the hindlimb from 3 to 5 mice per genotype were analyzed. Values are the mean and SD. *p < 0.05 versus wild-type mice. WT wild-type, HET heterozygotes, KO knockout (Color figure online)
No Altered Osteoblast Function for Bone Formation in Tgfbi Knockout Mice
To determine the cause of the shorter bone lengths in Tgfbi knockout mice, we investigated chondrocyte arrangements and osteoblast function. Growth plates are responsible for linear growth in vertebrates. H&E and alcian blue staining showed that chondrocytic cell arrangements in the growth plates were identical in the vertebrae of Tgfbi knockout mice and wild-type mice (Fig. 2a, b), indicating that knockout of Tgfbi had no evident effect on vertebral linear growth. Similar findings were obtained when the growth plates of long bones of the same age were assessed (data not shown).

Histological analysis and microarchitecture parameters of vertebrae from Tgfbi knockout mice at P30. a, b Chondrocyte arrangements stained with H&E/alcian blue in the vertebral growth plates were identical in Tgfbi knockout mice and wild-type mice. c, d The lumbar vertebrae of mice were analyzed with the von Kossa stain, which stains mineralized bones black. No differences in mineralized cancellous bone volume, trabecular thickness, or trabecular number were observed. e, f Distances between calcein double labeling were similar in Tgfbi knockout mice and wild-type mice. g Dynamic indices of bone formation based on fluorescent calcein labeling were quantified using the Bioquant program. MS/BS mineralized surface per bone surface MAR mineral apposition rate, BFR/BS bone formation rate per bone surfaces, WT wild-type, KO knockout. Scale bar 50-μm (Color figure online)
Mineralized bone patterns and calcein double labeling were analyzed to determine whether Tgfbi affected osteoblast function. Undecalcified lumbar vertebrae at P15 and P30 were stained with von Kossa stain to detect bone mineralization. The appearance of the mineralized trabecular bone was unchanged in Tgfbi knockout mice at P30 (Fig. 2c, d). Histomorphometric analysis of the fourth lumbar vertebra showed no significant reduction in cancellous bone volume, trabecular thickness, trabecular number, or trabecular separation (Fig. S2A). At P15, the findings in wild-type and knockout mice were identical (Fig. S2B). Mice were treated with calcein for double labeling to measure the dynamic and static parameters of bone formation. Identical patterns of calcein double labeling were clearly observed on the surfaces of the lumbar bones of Tgfbi knockout and wild-type mice (Fig. 2e, f). Furthermore, the bone forming rate (BFR), an indicator of osteoblast activity, and the mineral apposition rate (MAR), an indicator of the rate of new bone deposition in the radial direction, were similar for wild-type and Tgfbi knockout mice (Fig. 2g). These results indicated that the absence of the Tgfbi gene did not affect the functions of chondrocytes or osteoblasts.
Bone Matrix Degradation in Tgfbi Knockout Mice
The ECM of cartilage and bone, secreted by chondroblasts and osteoblasts, respectively, comprises a complex mixture of proteins and proteoglycans. Proteoglycans are glycoproteins with large clusters of carbohydrate chains, which are assessed with safranin O staining. The intensity and area of staining is directly proportional to the proteoglycan content in cartilage [24]. Masson trichrome staining was used to distinguish cells from the surrounding connective tissues, including collagen fibers [23, 25]. TGFBI is an ECM protein that is expressed and secreted by chondroblasts for incorporation into cartilage and osteoblasts for incorporation into bone [11]. To determine whether TGFBI is involved in the maintenance of cartilage and bone matrix, we assessed the proteoglycan content, collagen arrangement, and protein localization in the cartilage matrix of Tgfbi knockout and wild-type mice at P60. The proteoglycan content in the articular cartilage of Tgfbi knockout mice, as determined with safranin O staining, was markedly lower than that in wild-type mice (Fig. 3a). The proteoglycan content of cartilage samples was evaluated according to the modified Mankin method [26, 27]. The proteoglycan content was reduced in the articular cartilage of Tgfbi knockout mice, indicating increased cartilage degradation in Tgfbi knockout mice compared with that in wild-type mice (Fig. 3b). Furthermore, as determined with Masson trichrome staining, the assessment of deposited collagen was looser in Tgfbi knockout mice than in wild-type mice (Fig. 3c). In the articular cartilage during matrix degradation in cartilage or osteoarthritis progression, type X collagen and MMP13 are highly expressed and type II collagen expression is reduced [28, 29]. To assess bone matrix degradation in Tgfbi knockout mice, the levels and localizations of type II and X collagen and MMP13 were examined with immunostaining. The type II collagen level in the articular cartilage was notably lower in Tgfbi knockout mice than in wild-type mice (Fig. 4a, b). On the other hand, in chondrocytes of the superficial and middle zones of articular cartilage, MMP13 was present in Tgfbi knockout mice, but rarely detected in wild-type mice (Figs. 4e, f, S3). The level of aggrecan protein was increased in the articular cartilage of Tgfbi knockout mice, indicating that cartilage homeostasis was disrupted by Tgfbi deficiency (Fig. S3). However, the levels of type X collagen, a specific marker of hypertrophic chondrocytes, were similar in Tgfbi knockout and wild-type mice (Fig. 4c, d). These results suggest that cartilage matrix is degraded in the absence of Tgfbi.

Bone matrix degradation and loose collagen matrix in Tgfbi knockout mice at P60. a Safranin O staining (red) showed well-organized proteoglycans in skeletal tissues. However, safranin O staining was reduced in Tgfbi knockout mice compared with that in the wild-type mice, indicating degradation of the bone matrix in Tgfbi knockout mice. Scale bar 200-μm. b The proteoglycan content of cartilage samples was evaluated histologically with safranin‐O staining, as shown in a, according to the modified Mankin method, resulting in reduced proteoglycan content in Tgfbi knockout mice compared with that in wild-type mice. Both sides of the hindlimb from 3 to 5 mice per genotype were analyzed. Values are the mean and SD. *p < 0.05 versus wild-type mice. c Trichrome staining (blue) shows the organization of collagen matrix complexes. In Tgfbi knockout mice, a loosely arranged collagen matrix was observed (light blue), whereas a dense blue was observed in wild-type mice. Scale bar 200-μm. C1–C2 and C3–C4 are high magnification images of WT and KO, respectively. Scale bar 50-μm. WT wild-type, KO knockout (Color figure online)

Altered levels and localizations of bone matrix proteins in Tgfbi knockout mice at P60. Type II (Col2) (a, b) and type X (Col10) (c, d) collagens, and matrix metalloproteinase 13 (MMP13) (e, f) were detected in wild-type and Tgfbi knockout mice with immunostaining. The level of type II collagen was lower in Tgfbi knockout mice than in wild-type mice, whereas the level of MMP13 was markedly higher. In addition, the localization of MMP13 was detected in chondrocytes of the superficial and middle zones of the articular cartilage of Tgfbi knockout mice. However, the level of type X collagen was similar in both mice. Scale bar 50-μm
Discussion
The ECM is an important determinant of cellular behavior, because it regulates cellular adhesion, migration, proliferation, and differentiation. It is essential for maintaining connective tissue [30]. The actions of the ECM derive from its ability to sequester and modulate the activities of specific growth factors, including TGF-β [31]. TGF-β plays a critical role in ECM synthesis and degradation and in the cellular response to ECM, which is mediated by integrins, CD44, and annexin 5 [32]. Furthermore, several studies have shown that ECM proteins are associated with TGF-β signaling. The ECM and TGF-β signaling act in an integrated manner to maintain tissue homeostasis [33]. In addition, in cultured mesenchymal cells, TGF-β promotes matrix deposition by increasing the expression of ECM genes and suppressing the activities of genes that degrade the ECM, such as MMPs [34]. Interestingly, an inactive (latent) form of TGF-β, which is anchored to the ECM, has been reported to interact with latent TGF-β binding protein [20, 35]. Furthermore, ECM stiffness and TGF-β induce the expression of sox9, col2a1, and aggrecan and subsequent chondrocyte differentiation [36].
TGFBI is an ECM protein induced by TGF-β and expressed in a wide variety of tissues. TGFBI is involved in cell growth and migration, tumorigenesis, wound healing, and apoptosis, indicating that it plays roles in a wide range of physiological and pathological conditions, including diabetes, corneal dystrophy, and tumorigenesis [37]. Some TGFBI mutations have been associated with several forms of human autosomal dominant corneal dystrophies [38]. Moreover, in a study using Tgfbi-overexpressing transgenic mice, TGFBI was shown to be involved in anterior segment morphogenesis and eye development [39]. However, no abnormalities have been observed in cornea of Tgfbi knockout mice. TGFBI interacts strongly with other ECM proteins, including collagen, laminin, and fibronectin [16]. In many cell types, TGFBI functions as a linker protein that connects various matrix molecules and facilitates cell-collagen interactions [40–42]. Furthermore, TGFBI binds to type I, II, and IV collagens and to proteoglycans, such as, biglycan and decorin [42]. These findings indicate that interactions between TGFBI and other ECM proteins are important for the maintenance of ECM homeostasis. However, the role of TGFBI in ECM homeostasis in vivo has not been well characterized.
Tgfbi-deficient mice have been generated by two groups using a general strategy for targeted mutations: Zhang et al. [43] and Bae et al. [17]. The strategies for targeting Tgfbi allele were different. Bae et al. [17] targeted exon 3 with a neomycin cassette that was removed through Flp-mediated recombination; Zhang et al. [43] targeted exons 4–6 by replacement with a neomycin cassette. The two resulting mouse models exhibited identical phenotypes: smaller body size and shorter bone length than in wild-type mice. However, no other abnormalities were observed in the other tissues of Tgfbi-deficient mice. Yu and colleagues have reported that bone mass, size, and strength were reduced in their Tgfbi-deficient mice [43, 44], indicating that TGFBI plays a positive role in bone development. They also reported that Tgfbi-deficient mice exhibited a significant decrease in periosteal BFR, suggesting that the decreased bone size is due to reduced periosteal bone formation. However, Yu and colleagues did not perform a detailed study of alterations in bone matrix. In the present study, we studied the in vivo changes in bone matrix and skeletal size using the Tgfbi-deficient mouse model generated by our group [17]. Reduced bone growth is a consistent finding in both Tgfbi-deficient mouse models. As shown in this study, Tgfbi knockout mice also exhibited bone matrix degradation and reduced bone growth with lower proteoglycan and collagen contents and loose collagen deposition in the articular cartilage. Although the type II collagen level was reduced, the MMP13 level was higher in the articular cartilage of Tgfbi knockout mice than in that of wild-type mice, and it was localized in chondrocytes of the superficial and middle zones of the articular cartilage of Tgfbi knockout mice. MMPs play a role in the degradation of cartilage matrix components, including all types of collagen, proteoglycans, and other components of the ECM [45], indicating that MMPs regulate the balance between structural proteins in the articular cartilage matrix. MMP13 degrades ECM constituents, including type II collagen [46]. Whereas MMP13 deficiency protects against cartilage erosion [47], overexpression of constitutively active Mmp13 in murine cartilage induces cartilage degradation and promotes osteoarthritic pathology [48], indicating that MMP13 plays a role in maintaining cartilage matrix. In this study, the reduction in type II collagen and increase in MMP13 in Tgfbi knockout mice suggests that the cartilage matrix is degraded and impaired in the absence of Tgfbi. TGFBI might serve as a bridge to secure the connection with collagen and prevent degradation by MMPs. Taken together, our observations suggest that TGFBI plays a role in the maintenance of skeletal tissues, including the articular cartilage, in mice.
During endochondral ossification, cartilage grows via interstitial and appositional growth. Interstitial growth results from the continuous cell division of chondrocytes, which is accompanied by secretion of ECM within existing cartilage. On the other hand, appositional growth results in thickening of cartilage when ECM is added to the surface of the existing peripheral cartilage. Thus, the assembly and degradation of ECM molecules are important for the maintenance of the bone matrix. In the present study, we demonstrated that TGFBI positively regulates the maintenance of skeletal size and the bone matrix in mice.
References
Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF (1992) cDNA cloning and sequence analysis of betaig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol 11:511–522
Kawamoto T, Noshiro M, Shen M, Nakamasu K, Hashimoto K, Kawashima-Ohya Y, Gotoh O, Kato Y (1998) Structural and phylogenetic analyses of RGD-CAP/beta ig-h3, a fasciclin-like adhesion protein expressed in chick chondrocytes. Biochim Biophys Acta 1395:288–292
Skonier J, Benenett K, Rothwell V, Kosowski S, Plowman G, Wallace P, Edelhoff S, Disteche C, Neubauer M, Marquardt H, Rodgers J, Purchio AF (1994) beta ig-h3: a transforming growth factor-beta-responsive gene encoding a secreted protein that inhibits cell attachment in vitro and suppresses the growth of CHO cells in nude mice. DNA Cell Biol 13:571–584
Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ, Wang XF (2008) Extracellular matrix protein beta ig-h3/TGFBI promotes metastasis of colon cancer by enhancing cell extravasation. Genes Dev 22:308–321
Rawe IM, Zhan Q, Burrows R, Bennett K, Cintron C (1997) Beta-ig. Molecular cloning and in situ hybridization in corneal tissues. Invest Ophthalmol Vis Sci 38:893–900
Kim JE, Kim SJ, Jeong HW, Lee BH, Choi JY, Park RW, Park JY, Kim IS (2003) RGD peptides released from beta ig-h3, a TGF-beta-induced cell-adhesive molecule, mediate apoptosis. Oncogene 22:2045–2053
LeBaron RG, Bezverkov KI, Zimber MP, Pavele R, Skonier J, Purchio AF (1995) Beta IGH3, a novel secretory protein inducible by transforming growth factor-beta, is present in normal skin and promotes the adhesion and spreading of dermal fibroblasts in vitro. J Invest Dermatol 104:844–849
Ohno S, Noshiro M, Makihira S, Kawamoto T, Shen M, Yan W, Kawashima-Ohya Y, Fujimoto K, Tanne K, Kato Y (1999) RGD-CAP ((beta)ig-h3) enhances the spreading of chondrocytes and fibroblasts via integrin alpha(1)beta(1). Biochim Biophys Acta 1451:196–205
Kim JE, Kim SJ, Lee BH, Park RW, Kim KS, Kim IS (2000) Identification of motifs for cell adhesion within the repeated domains of transforming growth factor-β-induced gene, βig-h3. J Biol Chem 275:30907–30915
Schorderet DF, Menasche M, Morand S, Bonnel S, Buchillier V, Marchant D, Auderset K, Bonny C, Abitbol M, Munier FL (2000) Genomic characterization and embryonic expression of the mouse Bigh3 (Tgfbi) gene. Biochem Biophys Res Commun 274:267–274
Han MS, Kim JE, Shin HI, Kim IS (2008) Expression patterns of βig-h3 in chondrocyte differentiation during endochondral ossification. Exp Mol Med 40:453–460
Thorp BH, Anderson I, Jakowlew SB (1992) Transforming growth factor-beta 1, -beta 2 and -beta 3 in cartilage and bone cells during endochondral ossification in the chick. Development 114:907–911
Blaney Davidson EN, van der Kraan PM, van den Berg WB (2007) TGF-beta and osteoarthritis. Osteoarthr Cartil 15:597–604
Yang X, Chen L, Xu X, Li C, Huang C, Deng CX (2001) TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J Cell Biol 153:35–46
Tang SY, Alliston T (2013) Regulation of postnatal bone homeostasis by TGF-β. Bonekey Rep 255:1–5
Kim JE, Park RW, Choi JY, Bae YC, Kim KS, Joo CK, Kim IS (2002) Molecular properties of wild-type and mutant βIG-H3 proteins. Invest Ophthalmol Vis Sci 43:656–661
Bae JS, Lee W, Nam JO, Kim JE, Kim SW, Kim IS (2014) Transforming growth factor-β-induced protein promotes severe vascular inflammatory responses. Am J Resp Crit Care 189:779–786
Baek WY, Lee MA, Jung JW, Kim SY, Akiyama H, de Crombrugghe B, Kim JE (2009) Positive regulation of adult bone formation by osteoblast-specific transcription factor Osterix. J Bone Miner Res 24:1055–1065
Baek WY, de Crombrugghe B, Kim JE (2010) Postnatally induced inactivation of Osterix in osteoblasts results in the reduction of bone formation and maintenance. Bone 46:920–928
Erben RG (1997) Embedding of bone samples in methylmethacrylate: an improved method suitable for bone histomorphometry, histochemistry, and immunohistochemistry. J Histochem Cytochem 45:307–313
Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR (1987) Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2:595–610
Bancroft JD, Layton C (2012) Connective and mesenchymal tissue with their stains. In: Suvarna SK, Layton C, Bancroft JD (eds) Bancroft’s theory and practice of histological techniques. Churchill Livingstone Elsevier, Oxford, pp 187–214
Yu H, Yang X, Cheng J, Wang X, Shen SG (2011) Distraction osteogenesis combined with tissue-engineered cartilage in the reconstruction of condylar osteochondral defect. J Oral Maxillofac Surg 69:e558–e564
Shimizu S, Asou Y, Itoh S, Chung UI, Kawaguchi H, Shinomiya K, Muneta T (2007) Prevention of cartilage destruction with intraarticular osteoclastogenesis inhibitory factor/osteoprotegerin in a murine model of osteoarthritis. Arthritis Rheum 56:3358–3365
Estrada LE, Dodge GR, Richardson DW, Farole A, Jimenez SA (2001) Characterization of a biomaterial with cartilage-like properties expressing type X collagen generated in vitro using neonatal porcine articular and growth plate chondrocytes. Osteoarthr Cartil 9:169–177
Mankin HJ, Dorfman H, Lippiello L, Zarins A (1971) Biochemical and metabolic abnormalities in articular cartilage from osteoarthritic human hips. II. Correlation of morphology with biochemical and metabolic data. J Bone Joint Surg Am 53:523–537
Aigner T, Cook JL, Gerwin N, Glasson SS, Laverty S, Little CB, McIlwraith W, Kraus VB (2010) Histopathology atlas of animal model systems—overview of guiding principles. Osteoarthr Cartil 18(Suppl 3):S2–S6
Kamekura S, Hoshi K, Shimoaka T, Chung U, Chikuda H, Yamada T, Uchida M, Ogata N, Seichi A, Nakamura K, Kawaguchi H (2005) Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr Cartil 13:632–641
Chen B, Qin J, Wang H, Magdalou J, Chen L (2010) Effects of adenovirus-mediated bFGF, IL-1Ra and IGF-1 gene transfer on human osteoarthritic chondrocytes and osteoarthritis in rabbits. Exp Mol Med 42:684–695
Eyre DR, Weis MA, Wu JJ (2006) Articular cartilage collagen: an irreplaceable framework? Eur Cell Mater 12:57–63
Roberts AB, Mccune BK, Sporn MB (1992) TGF-β: regulation of extracellular matrix. Kidney Int 41:557–559
Wong M, Carter DR (2003) Articular cartilage functional histomorphology and mechanobiology: a research perspective. Bone 33:1–13
Munger JS, Sheppard D (2011) Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol 3:a005017
Varga J, Jimenez SA (1986) Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta. Biochem Biophys Res Commun 138:974–980
Ruiz-Ortega M, Rodríguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J (2007) TGF-β signaling in vascular fibrosis. Cardiovasc Res 74:196–206
Allen JL, Cooke ME, Alliston T (2012) ECM stiffness primes the TGFβ pathway to promote chondrocyte differentiation. Mol Biol Cell 23:3731–3742
Thapa N, Lee BH, Kim IS (2007) TGFBIp/betaig-h3 protein: a versatile matrix molecule induced by TGF-beta. Int J Biochem Cell Biol 39:2183–2194
Munier FL, Korvatska E, Djemai A, Le Paslier D, Zografos L, Pescia G, Schorderet DF (1997) Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 15:247–251
Kim JE, Han MS, Bae YC, Kim HK, Kim TI, Kim EK, Kim IS (2007) Anterior segment dysgenesis after overexpression of transforming growth factor-beta-induced gene, beta igh3, in the mouse eye. Mol Vis 13:1942–1952
Billings PC, Whitbeck JC, Adams CS, Abrams WR, Cohen AJ, Engelsberg BN, Howard PS, Rosenbloom J (2002) The transforming growth factor-beta-inducible matrix protein (beta)ig-h3 interacts with fibronectin. J Biol Chem 277:28003–28009
Hanssen E, Reinboth B, Gibson MA (2003) Covalent and non-covalent interactions of betaig-h3 with collagen VI. Beta ig-h3 is covalently attached to the amino-terminal region of collagen VI in tissue microfibrils. J Biol Chem 278:24334–24341
Reinboth B, Thomas J, Hanssen E, Gibson MA (2006) Beta ig-h3 interacts directly with biglycan and decorin, promotes collagen VI aggregation, and participates in ternary complexing with these macromolecules. J Biol Chem 281:7816–7824
Zhang Y, Wen G, Shao G, Wang C, Lin C, Fang H, Balajee AS, Bhagat G, Hei TK, Zhao Y (2009) TGFBI deficiency predisposes mice to spontaneous tumor development. Cancer Res 69:37–44
Yu H, Wergedal JE, Zhao Y, Mohan S (2012) Targeted disruption of TGFBI in mice reveals its role in regulating bone mass and bone size through periosteal bone formation. Calcif Tissue Int 91:81–87
McDonnell S, Morgan M, Lynch C (1999) Role of matrix metalloproteinases in normal and disease processes. Biochem Soc Trans 27:734–740
Vincenti MP, Coon CI, Mengshol JA, Yocum S, Mitchell P, Brinckerhoff CE (1998) Cloning of the gene for interstitial collagenase-3 (matrix metalloproteinase-13) from rabbit synovial fibroblasts: differential expression with collagenase-1 (matrix metalloproteinase-1). Biochem J 331:341–346
Little C, Barai A, Burkhardt D, Smith S, Fosang A, Werb Z, Shah M, Thompson E (2009) Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 60:3723–3733
Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, Turner J, Wu W, Billinghurst C, Meijers T, Poole AR, Babij P, DeGennaro LJ (2001) Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest 107:35–44
Acknowledgments
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI13C1874).
Conflict of Interest
Jung-Mi Lee, Eun-Hye Lee, In-San Kim and Jung-Eun Kim state that they have no conflict of interest.
Human and Animal Rights and Informed Consent
All procedures of animal experiments were approved by the Institutional Animal Care and Use Committee of Kyungpook National University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lee, JM., Lee, EH., Kim, IS. et al. Tgfbi Deficiency Leads to a Reduction in Skeletal Size and Degradation of the Bone Matrix. Calcif Tissue Int 96, 56–64 (2015). https://doi.org/10.1007/s00223-014-9938-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-014-9938-4