
Abstract
The acid phosphatases in Piriformospora indica and Sebacina vermifera sensu were similar in their molecular mass. The raised antibody against acid phosphatase (ACPase) of P. indica showed maximum ELISA reading with S. vermifera sensu supporting strong relationship between these two fungi. The immunoblot analysis showed the strong reactivity of antiserum with S. vermifera sensu. The antiserum blotted the bands of 66 kDa in S. vermifera separated in denatured—PAGE at equidistance with P. indica. The enzyme assay zymogram of non-denatured-ACPase showed parallel bands at the same height in native PAGE. The antiserum also localized the enzyme in S. vermifera by immunofluorescence technique, showing strong relationship of this fungus with P. indica. The immunogold labeling of antiserum from P. indica precisely localized the enzyme in cytoplasm and vacuoles of S. vermifera supporting the strong immunological link between these two fungi. Two-dimensional map of crude protein of these two fungi showed some differences in minor proteins.
P. indica and S. vermifera sensu belonging to the same taxonomic group show similar morphology, isozyme characteristics, and various functions as well. However, they show distinct genetic variation based on the random amplified polymorphic DNA (RAPD) analysis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Extracellular Phosphatase
- Versus Constant Voltage
- Intraradical Hypha
- Immunological Relationship
- ACPase Activity
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Phosphatases are the enzymes of wide specificity which cleave phosphate ester bonds, and this plays an important role in the hydrolysis of polyphosphates and organic phosphates. Acid and alkaline phosphatases are the two forms of intracellular phosphatase active at acidic and alkaline conditions, respectively. ACPase was found to be mainly involved in uptake of P by the fungal mycelia, and alkaline phosphatase (ALPase) is linked with its assimilation (Fries et al. 1998).
Acid phosphatase in soil originates from both plants and fungi, while ALPase is believed to be of purely microbial origin (Gianinazzi-Pearson and Gianinazzi 1978; Tarafdar and Rao 1996). Indications are found that ACPase of fungal origin has a higher hydrolyzing efficiency than enzymes of plant origin (Tarafdar et al. 2001). Studying ACPase is difficult due to their multiform occurrence in organisms, their relative nonspecificity, their small quantity, and their instability in dilute solution. Their study is also complicated by wide variations in the activity and property of isozyme between species and between different stages in each plant’s development (Alves et al. 1994) (Fig. 3.1).

3D structure of purple acid phosphatase from animal (Lindqvist et al. 1999)
Alkaline phosphatase has been proposed as a marker for analyzing the symbiotic efficiency of colonization (Tisserant et al. 1993). The argument for this was that ALPase is an important enzyme in metabolic processes leading to P transfer to the host plant. The alkaline phosphatase activity is shown to be increased sharply prior to mycorrhizal stimulation of plant growth and then declined as the mycorrhizal colonization gets aged and P accumulated within the host. Arbuscule is speculated to be a site of nutrient exchange between the host plant and AM fungi (Cox et al. 1980). Phosphate efflux from the fungi to the host plant at arbuscules is supported by the recent discovery of novel plant Pi transporters that are localized around arbuscules and acquire Pi from the fungi (Rausch et al. 2001; Harrison et al. 2002; Paszkowski et al. 2002).
1.1 Sebacinales
Mycorrhizal taxa of Sebacinaceae, including mycobionts of ectomycorrhizas, orchid mycorrhizas (McCormick et al. 2004), ericoid mycorrhizas, and jungermannioid mycorrhizas, are distributed over two subgroups. One group contains species with microscopically visible basidiomes, whereas members of the other group probably lack basidiomes. Sebacina appears to be phylogenetic; current species concepts in Sebacinaceae are questionable. S. vermifera sensu consists of a broad complex of species possibly including mycobionts of jungermannioid and ericoid mycorrhizas. Extrapolating from the known rDNA sequences in Sebacinaceae, it is evident that there is a cosm of mycorrhizal biodiversity yet to be discovered in this group. Taxonomically, the Sebacinaceae recognized a new order, the Sebacinales (Weiß et al. 2004; Malla et al. 2005). The order primarily contains the genera Sebacina, Tremelloscypha, Efibulobasidium, Craterocolla, and Piriformospora. Proteomics and genomics data about P. indica fungus have recently been described (Peškan-Berghöfer et al. 2004; Shahollari et al. 2005; Kaldorf et al. 2005; Malla et al. 2007a).
Piriformospora indica and Sebacina vermifera from Sebacinales are documented to function as bio-fertilizer, bioregulator, bioprotector, and supplement to the health of the plant and the soil (Verma et al. 1998; Varma et al. 1999; Malla et al. 2002). More recently, they are documented to act as agent for biological hardening of tissue culture-raised plants. Despite their enormous potential, their biotechnological applications could not be exploited to the level they deserve. The axenic cultivability of these fungi provided ample opportunity to study the comparative immunological relationship and molecular marker like random amplified polymorphic DNA (RAPD) to establish variability in between these two fungi (Weiß et al. 2004; Malla and Varma 2004; Malla et al. 2004).
2 Materials and Methods
Actively growing colonies of P. indica and S. vermifera sensu were transferred in modified Aspergillus medium (Hill and Kaefer 2001; Pham et al. 2004b), incubated for 10 days at 28 ± 2 °C in the dark with constant shaking at 120 rpm. The morphological features of the fungi were studied with the aid of Leica microscope (Type 020–518.500, Germany).
2.1 Gel Filtration Chromatography
The crude protein extracted was fractionated by 80 % saturation of ammonium sulfate, dialyzed overnight against phosphate buffer (80 mM), and passed through column chromatography using Sephadex G-100 (Cutler 2001). The flow rate was 0.5 ml/min and fractions of 4 ml each were collected. The relative ACPase activity was measured in 25 μl aliquots of selected fractions at OD 420 nm using 2 mM p-NPP as substrate at standard condition of 37 °C. Fraction with higher activity was pooled and passed through ion exchange chromatography using DEAE Sephadex (Bollag et al. 1996). The column was neutralized with phosphate buffer (80 mM) and eluted with 0.1 M NaCl.
2.2 Molecular Mass
The active fractions pooled from ion exchanger were separated in 10 % non-denaturing PAGE (native PAGE). The gel after separation at 4 °C for 6 h was reacted with 2 mM p-NPP with sodium acetate buffer at room temperature. Yellow visible phosphatase bands thus formed were eluted from the native gel using elution buffer (ammonium carbonate, 100 mM) and separated in 12 % SDS-PAGE to determine molecular mass.
2.3 Developing Antiserum
New Zealand white rabbit were used for the production of antiserum. Polyclonal antibodies were raised against P. indica ACPase detected in native gel as yellow band. The band was manually cut by blade and prepared as immunogen, as described by Amero et al. 1996.
2.4 Preparation of Antiserum
The antiserum was prepared according to Hahn et al. (1998). The antiserum was precipitated up to 50 % by adding an equal amount of saturated ammonium sulfate solution. Further purification of antiserum was done by ion exchanger, DEAE—Sepharose CL-6B (Johnstone and Thorpe 1996).
2.5 Immunodetection
The quick method of detection of antiserum was done by single radial and double immunodiffusion (Vaerman 1981) and ELISA (Harlow and Lane 1999; Voller et al. 1980) method. The optimal concentrations of the conjugate as well as of primary antibody were determined by crisscross serial dilution analysis.
2.6 Immunoblotting
Western blotting of the antiserum was done according to Towbin et al. (1979) and Horst (2000). Nitrocellulose sheet (Schleicher and Schuell, Germany) 0.45 μm pore size was used for transblot of protein. The blots after transfer were detected with Ponceau S stain (Sigma), destained with deionized water. The purified antiserum using DEAE—Sephadex CL-6B served as primary antibody. Enzyme-conjugated secondary antibody (HRPO, anti-rabbit IgG, Sigma, A-9169) was used for binding of primary antiserum. 3, 3′-Diaminobenzidine tetrahydrochloride (DAB) in combination with urea peroxide (Sigma Co.) as substrate provided for visualization of immunological complex.
2.7 Immunofluorescence
The hyphae and spores were fixed for 15 min at 4 °C with 3.7 % paraformaldehyde, mixed with an equal amount of double-strength phosphate buffer, and passed through a 0.45 μm Millipore filter. Washed the treated sample with PBS (3 × 5 min) containing 100 mM glycine. Biomass was permeabilized with 0.1 % Triton X-100 in PBS for 4 m. Unspecific binding of the antibodies was blocked by 1 % BSA (Meyberg 1988). After several washings with PBST (containing 0.02 % Tween 20), the biomass was incubated with antiserum fractionated with ammonium sulfate diluted to 1:100. Fluorescein isothiocyanate (FITC) (Sigma Aldrich, F-0382) conjugated 2nd antibody diluted 1:100 was used for visualization of antigen–antibody complex and screened with confocal microscope (Olympus).
2.8 Immunogold
Four day old samples were fixed in 1 % glutaraldehyde and 2 % paraformaldehyde, filtered through a 0.45 μm pore-sized paper kept for 18 h at 4 °C, and postfixed for 2 h in 1 % osmium tetroxide. Samples were dehydrated with graded alcohol/acetone solutions, embedded in LR white resin (Botton and Chalot 1991). Ultrathin-sectioned (60–90 nm thick) labeled grid with primary antibody IgG (1:100 diluted) was kept overnight at 4 ° C and washed with 0.1 M PB four times. The grid was labeled with secondary antibody anti-rabbit-goat IgG conjugated with 15 nm gold particle (1:100) and stained in 0.5 % aqueous uranyl acetate for 10 min and lead citrate for 5 min. The stained sections were observed with Philips CM-10 electron microscope operated at 60–80 KV.
2.9 Two-Dimensional PAGE
The first dimension was performed with the tube cell model 175 (Bio-Rad, Hercules, CA) and with glass capillary tube (1.0–1.4 mm internal diameter and 210 mm long) as described by Gravel and Golaz 1996. Ampholyte pH 5.0–7.0 and 3.0–10.0 were from Bio-Rad. The samples were loaded on the top of the capillary (cathodic side). 80 μg of the sample was loaded using Hamilton syringe. IEF was carried out at 200 V constant voltage for 2 h, followed by 500 V constant voltage for 2 h, and finally 800 V constant voltage for 16 h (overnight). Tube gels were equilibrated in equilibration buffer for ½ h at RT. The protean II chamber (Bio-Rad) is employed for second dimension. The gel (160 × 200 × 1.5 mm) was casted in the casting chamber (Bio-Rad). Tube gels were transferred on the top of SDS gels and separated at 90 V constant voltage in stacking gel and 120 V constant voltage in separating gel. Proteins in the 2D gel were stained with silver-staining method.
2.10 Staining of Acid Phosphatase with Fast Garnet GBC
The crude extract of biomass was separated in 10 % non-denaturing PAGE. The gel after separation was stained with Fast Garnet GBC in sodium acetate buffer containing α-naphthyl phosphate as substrate at room temperature (Ho and Zak 1979).
2.11 Detection of Phosphatase by ELF-97 Substrate
The fungal cultures grown for 48 h in broth were fixed in 3.7 % formaldehyde in phosphate buffer saline for 1 h in 4 °C. The samples were permeabilized in 0.02 % Tween 20 in PBS buffer for 10 min at room temperature. The samples were rinsed four times with PBS. The diluted ELF-97 substrate was filtered through 0.2 μm pore-sized spin filters E-6606 (Ingrid and van Aarle, 2001). Since the reaction occurs very fast, the reaction was performed on the microscope using excitation filter and emission filter from the fluorescein set, which provided the appropriate UV excitation and transmits wavelengths greater than 400 nm. 50 μl of substrate solution was added. Immediately, the sample was placed on the microscope and the development of the signal observed was monitored under the fluorescence microscope (Olympus, model, FV-300).
2.12 RAPD Technique
Isolation and purification of fungal DNA was done following modified CTAB protocol of Moller et al. (1992). DNA amplification was performed in a total volume of 25 μl containing (in μl) 2.5, buffer (10× without MgCl2); 2.5, MgCl2; 0.8, dNTPs (10 mM); 1.0, primer (30 ng/μl); 0.5 Taq polymerase (3U/μl); and DNA concentration ranging from 5 to 25. Random 10 bp oligonucleotide primers (Operon Technologies Alameda, California) were used to produce amplification. The DNA was amplified in PTC-200 Thermal Cycler (Techne, UK) electrophoresed on 1.5 % agarose gel in 1 % TAE at 3.5 V/cm for 2 h, stained with ethidium bromide.
Statistical analysis was performed using the NTSYS-pc program (Rohlf 1992). The degree of genetic relatedness or similarity was estimated using the Jaccard coefficient. Clustering of similarity matrices was done by UPGMA (unweighted pair group method with arithmetic mean) and projection by TREE program of NTSYS-pc.
3 Results
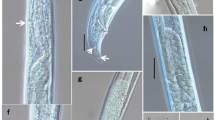
P. indica and S. vermifera sensu belonging to the same taxonomic group show similar morphology (Fig. 3.2). The molecular mass of ACPase in P. indica and S. vermifera sensu was identical. The enzyme was eluted from 8 % native gel and was separated in SDS gel. The identical molecular mass of acid phosphatase in both of the fungi suggests the close relationship of those fungi (Figs. 3.3 and 3.4).

Autofluorescence shown by P. indica (left) and S. vermifera (right) chlamydospores (arrow) seen under Leica microscope (model, 020–518.500) using I3 filter with excitations range 450–490 λ at ×400. The characteristic pear-shaped chlamydospores are more frequently common in P. indica and S. vermifera sensu

50 μg crude protein extracted from fresh biomass of fungus, loaded in native PAGE, was separated in 10 % gel for 6 h at 4 °C, was washed with 50 mM sodium acetate buffer to neutralize, and was shaken in water bath with solution of 2 mg/ml P-NPP. Yellow color is p-nitrophenol, product of enzyme reaction upon substrate. Lane 1 P. indica, Lane 2 S. vermifera. The banding pattern of the fungus shows precise similarity in between two fungi in their molecular mass and ionic strength

Molecular weight profile of ACPase eluted from native gel after several purification procedures. Lane 1 P. indica, Lane 2 S. vermifera sensu, Lane 3 molecular marker (Sigma wide range). The crude enzyme after selective precipitation of ammonium sulfate followed by gel filtration (Sephadex G-100) and ion exchange chromatography (DEAE-Sephadex) was separated in 8 % native PAGE and detected by assay using p-NPP. The eluted protein from native PAGE was separated by 12 % SDS-PAGE along with wide-range marker. The pure acid phosphatase showed 66kD molecular mass
The antibody raised against acid phosphatase of P. indica showed maximum ELISA reading with S. vermifera sensu. The immunoblot showed the strong reactivity of antiserum with own protein of cytoplasmic (CF) and wall membrane (W/MF) fraction and with protein of CF and W/MF of S. vermifera sensu. The P. indica antiserum blotted the bands of S. vermifera sensu at precise location of P. indica ACPase in 12 % SDS-PAGE (Fig. 3.5). Using fluorescein isothiocyanate (FITC)-labeled antibodies, localization of the enzyme was found on spores and hyphal walls. The antiserum localized the enzyme in S. vermifera sensu showing strong relationship of this fungus with P. indica. The precise localization was not distinct in hyphae. The antibody densely bound the spore wall in comparison to the inner spore may be due to poor penetration of antibody to wall. The antiserum also precisely localized the enzyme in cytoplasm and vacuoles of S. vermifera sensu supporting the immunological link between these two fungi (Fig. 3.6).

Western blot analysis of P. indica and Sebacina vermifera sensu separated by 10 % SDS-PAGE. Lane 1 CF. Lane 2 W/MF of P. indica reacted with homologous antiserum. Lane 3 CF. Lane 4 W/MF of S. vermifera sensu cross-reacted with P. indica antiserum. The result shows precisely defined bands in all samples. All blotted bands represent similarity in their molecular mass supporting immunologically highly related species. CF cytoplasmic fraction, W/MF wall membrane fraction

Immunolocalization of acid phosphatase in P. indica (top) and S. vermifera sensu (bottom). Electron micrograph of an ultrathin section of P. indica and S. vermifera sensu treated with secondary antibody (goat anti-rabbit) coupled to colloidal gold 15 nm size. Dark dots are gold particles indicating localization of the enzyme acid phosphatase. Localization is prominent in cytoplasm and vacuoles. The cells were fixed with 1 % glutaraldehyde and postfixed with 1 % osmium tetroxide. The primary antibody was raised against acid phosphatase of P. indica
Silver-stained 2-dimensional maps of mycelial protein of P. indica and S. vermifera sensu showed similarity in most of the major protein bands. The gel indicated differences in some of the minor proteins (Fig. 3.7).

Silver-stained two-dimensional maps of mycelial protein of P. indica and S. vermifera sensu loaded with 80 μg of protein onto IEF gels. Separation in the horizontal dimension was achieved by isoelectric focusing using carrier ampholyte in the pH range of 3–10 in the presence of 9.2 M urea and separation in the vertical dimension by 12 % SDS-PAGE
Using α-naphthyl phosphate as substrate, different isoforms of ACPase were obtained by Fast Garnet GBC staining. P. indica and S. vermifera sensu showed three distinct isoforms of ACPase each, one with higher molecular mass and two other with lower molecular mass. The pattern of isoforms was similar in both the fungi (Fig. 3.8). The higher molecular mass isoform is suspected to be dimeric form of the enzyme and lower is suspected to be monomeric form of ACPase on the basis of the position they formed in native gel.

Fast Garnet GBC staining of ACPase. The ACPase isoforms of P. indica and S. vermifera sensu show similar banding pattern. Lane 1 P. indica. Lane 2 S. vermifera sensu. The native PAGE separated for 6 h at 4 °C was neutralized with 50 mM sodium acetate buffer, stained with Fast Garnet GBC using α-naphthyl phosphate as substrate. The precisely localized band shows similar molecular mass and ionic strength of isoforms between P. indica and S. vermifera sensu
The 48 h old hyphae of P. indica grown in Aspergillus medium when reacted with enzyme-labeled fluorescence (ELF-97) substrate developed thick fluorescent precipitation in P. indica hyphae with dense production of ELF crystal. The reaction was comparable with control (Fig. 3.9). The activities were prominent but exact position or location of phosphatase was not confirmed. No any extracellular phosphatases and alkaline phosphatase activity were detected by this method.

Detection of ACPase activity by enzyme-labeled fluorescent substrate (ELF 97, molecular probe). (a) Control without substrate. (b) P. indica. (c). S. vermifera sensu. The result observed under Olympus standard microscope using excitation filter and dichroic mirror from the DAPI filter set and emission filter from the fluorescein set provides the appropriate UV excitation and transmits wavelengths greater than 400 nm. The microscope is equipped for epi-fluorescence with a high pressure Hg lamp. The results show uniform activity of phosphatase throughout the mycelium
The RAPD result showed about 60 % similarity between these two fungi according to the dendrogram (data not given). The RAPD data confirmed that even among these two species of Sebacinales belonging to the same morpho-zymographical groups, the level of variation was substantially high according to RAPD.
4 Discussion
The present experiment was undertaken to have an insight to the immunological relationship of P. indica and S. vermifera sensu grown in vitro. Soil fungi release the extracellular phosphatases in their environment (Nahas et al. 1982). These phosphatases are introduced into the soil by active exudation, leakage, or cell lysis (Dick et al. 1983; Tadano et al. 1993; Malla et al. 2007a). The phosphatase from P. indica and S. vermifera sensu separated in native PAGE was assayed with 2 mM of p-NPP at pH 5.3. The finding suggests that the mobility of the ACPase enzymes was identical in those two fungi exhibiting the same overall banding pattern. The bands separated were appearing to be homologous for both the isolates. Certain enzymes and isozymes are more suitable for taxonomic studies of the organisms (Malla 2008). These enzymes were especially important since they allowed closely related species to be readily separated. Usually, however, the species within each genus had very similar (probably homologous) banding patterns for various enzymes in terms of band number, relative mobility, and definition (Loxdale et al. 1983). The enzyme of ACPase had identical mobilities (which are based on molecular size and charge) between these two species. When p-NPP was used to visualize the bands in native gel, P. indica and S. vermifera sensu gave single precisely identical banding pattern of 66 kDa molecular mass.
Phosphatase activities, both ALPase and ACPase, have been visualized and shown to be associated with the hyphal wall and also in vacuoles of the extraradical mycelium using the enzyme-labeled fluorescent (ELF) substrate. In the intraradical mycelium, ACPase activity has been localized in arbuscules (Ezawa et al. 1995) and ACPase activity seemed to decrease with aging of the colonization. In the intraradical hyphae this activity is mainly located in the vacuoles (Saito 1995). The polyclonal antibodies raised against cytosolic ACPase of P. indica immuno-localized the enzyme on the vacuoles and cytoplasm of the mycelium. The antiserum precisely localized the enzyme in S. vermifera sensu, displaying strong relationship between these two fungi.
Separation of protein in two-gel system is a useful technique for distinguishing different fungi. The protein patterns of mycelium in two-dimensional PAGE of P. indica and S. vermifera sensu gave the massage that those two fungi are different in some minor proteins. So far one-dimensional PAGE showed no distinct bands of dissimilarity. Because of its resolution, the 2D PAGE has been applied to a great number of biological problems ranging from the analysis of proteins in different tissues under various hormonal states. The result obtained from 2D PAGE was comparable for the placement of those two fungi in the same genus not in species.
Staining of the native gel from both the fungi with Fast Garnet GBC using α-naphthyl phosphate as reaction substrate showed three isoforms of ACPases. It is noticeable that all those three bands of P. indica were identical to S. vermifera sensu. Stricto P. indica and S. vermifera sensu stricto, CF and W/MF showed three distinct isoforms of ACPase each, one with higher molecular mass and two with lower molecular mass. The mobility of the isozymes and patterns were similar in both the cases, supporting the closeness of these fungi to each other.
Several attempts were made to purify and characterize the enzyme from AM fungi; most of the result were based on activities of the enzyme related to extra- and intraradical hyphae, cytochemical localization, use of enzyme-labeled fluorescent substrate (ELF), and through expression of transporter genes responsible for ACPase (Aono et al. 2004). Due to unculturability of the arbuscular mycorrhizal fungus, very little is known about its purification. The enzyme-labeled fluorescent (ELF) substrate method could be used for visualizing phosphatase activity associated with colonized as well as noncolonized hyphae of fungus. Its high sensitivity provides the opportunity to study more in details the exudation or release of phosphatases by fungal mycelium in mono-axenic root organ cultures. Furthermore, the use of in situ hybridization may enable localization of the expression of phosphatase gene(s) at the cellular level. The ELF substrate method has proven to be a suitable and sensitive method in the determination of ACPase activity associated with axenic culture of P. indica. An advantage of ELF substrate compared to fast blue RR salt is its high sensitivity. When P. indica and S. vermifera sensu hyphae were incubated with ELF acid buffer solution, ELF precipitated all over the internal hyphal structure within 30 s except spores. The use of the ELF substrate in combination with laser scanning confocal microscopy could probably provide better insight into the location of phosphatase activity of fungi inside the roots and external hyphae. Comparing ELF with FB staining the ELF crystals was more abundant than those of FB. Almost similar observation was noticed in S. vermifera sensu showing closeness of these fungi to each other.
PCR-based techniques have already been applied to endo- and ectomycorrhizal fungi where morphological characters are in conflict, ambiguous, and missing (Podila and Lanfranco 2004). This approach has allowed the development of molecular tools for their identification (Hartmann et al. 2004; Malla et al. 2005; Malla and Varma 2007). The RAPD data confirmed that even among these two species of Sebacinales belonging to the same morpho-zymographical group, the level of variation was substantially high. The genetic variation of the isolates was grouped in the dendrogram (data not given). P. indica and S. vermifera sensu showed low similarity. The polymorphism showed by RAPD of these two fungi P. indica and S. vermifera sensu suggests placing these fungi in far-related group or separated genus although morphologically and immunologically they are closely related (Malla et al. 2006; Malla et al. 2007b).
5 Conclusion
The application of serological technique for localization and characterization of ACPase in these two fungi has provided new imminent into important aspects of this field. Result presented in this study provides, antibodies raised against protein in native gel band was an important technique. The benefits derived from this technology supply specific antibody which is difficult by ordinary purification procedure. Piriformospora indica and Sebacina vermifera sensu belonging to the same taxonomic group show similar morphology, functions, protein profiles, and isozymes characterization along with close immunological relationships. However, they show distinct genetic variation based on the random amplified polymorphic DNA analysis. Thus, it is suggested that such isolates should be considered into separate species. Molecular characterization offers an alternative approach for more reliable and reproductive identification at species level. We therefore suggest that along with morphology and immunological characterization, RAPD analysis could be used to distinguish indigenous species in natural ecosystem.
References
Alves JM, Siachakr CD, Allot M, Tizroutine S, Mussio I, Servaes A (1994) Isozyme modifications and plant regeneration through somatic embryogenesis in sweet potato. Plant Cell Rep 13:437–441
Amero SA, James TC, Elgin SCR (1996) Production of antibody using proteins in gel bands. In: Walker JM (ed) The protein protocols handbook. Humana, NJ, pp 717–720
Aono T, Maldonado-Mendoza IE, Dewbre GR, Harrison MJ, Saito M (2004) Expression of alkaline phosphatase genes in arbuscular mycorrhizas. New Phytol 162:525–534
Bollag DM, Rozycki MD, Edelstein SJ (1996) Protein methods, 2nd edn. Wiley-Liss, New York, pp 231–269
Botton B, Chalot M (1991) Techniques for the study of nitrogen metabolism in ectomycorrhiza. In: Norris JR, Read DJ, Varma AK (eds) Methods in microbiology 23. Academic, New York, pp 203–252
Cox G, Kg M, Sanders F, Nokolds C, Tinker PB (1980) Translocation and transfer of nutrients in vesicular arbuscular mycorrhizas.III. Polyphosphate granules and phosphorus translocation. New Phytol 84:649–659
Cutler P (2001) Chromatography on the basis of size. In: Simon R (Eds) Protein purification techniques. a practical approach, 2nd Edn. pp 187–211
Dick WA, Juma NG, Tabatabai MA (1983) Effects of soil and acid phosphatase and inorganic pyrophosphatase of corn roots. Soil Sci 136:19–25
Ezawa T, Saito M, Yoshida T (1995) Comparison of phosphatase localization in the intraradical hyphae of arbuscular mycorrhizal fungi Glomus spp and Gigaspora spp. Plant Soil 176:57–63
Fries LLM, Pacovsky RS, Safir GR, Kaminski J (1998) Phosphorus effect on phosphatase activity in endomycorrhizal maize. Physiol Plantarum 103:162–171
Gianinazzi-Pearson V, Gianinazzi S (1978) Enzymatic studies on the metabolism of vesicular-arbuscular mycorrhiza. II. Soluble alkaline phosphatase specific to mycorrhizal infection in onion roots. Physiol Plant Pathol 12:45–53
Gravel P, Golaz G (1996) Two-Dimensional PAGE using carrier ampholyte pH gradients in the first dimension. In: Walker JM (ed) The protein protocols handbook. Humana, NJ, pp 127–32
Hahn A, Goebel C, Hock B (1998) Polyclonal antibodies for the detection of arbuscular mycorrhizal fungi. In: Varma A (ed) Mycorrhiza manual. Springer, Berlin, pp 255–270
Harlow ED, Lane D (1999) Using antibodies. Cold Spring Harbour Laboratory Press, NY, pp 386–388
Harrison MJ, Dewbre GR, Liu JY (2002) A phosphate transporter from Medicago truncatula involved in the acquisition of phosphate released by arbuscular mycorrhizal fungi. Plant Cell 14:2413–2429
Hartmann A, Pukall R, Rothballer M, Gantner S, Metz S, Schloter MB (2004) Microbial community analysis in the rhizosphere by in situ and ex situ application of molecular probing, biomarker and cultivation technique. In: Varma A, Abbott L, Werner D, Hampp R (eds) Plant surface microbiology. Springer, Berlin, Heidelberg, Germany, pp 449–469
Hill TW, Kaefer E (2001) Improved protocols for aspergillus medium: trace elements and minimum medium salt stock solutions. Fungal Genet News Lett 48:20–21
Ho I, Zak B (1979) Acid phosphatase activity of six ectomycorrhizal fungi. Can J Bot 57:1203–1205
Horst MN (2000) Molecular approaches to glycobiology. pp 2–30
Ingrid M, van Aarle IM (2001) Microscopic detection of phosphatase activity of saprophytic and arbuscular mycorrhizal fungi using a fluorogenic substratr. Mycologia 93:17–24
Johnstone A, Thorpe R (1996) Immunochemistry in practice, 3rd edn. Blackwell Scientific, Oxford
Kaldorf M, Koch B, Rexer K-H, Kost G, Varma A (2005) Patterns of interaction between Populus Esch5 and Piriformospora indica: a transition from mutualism to antagonism. Plant Biol 7:210–218
Lindqvist Y, Johansson E, Kaija H, Vihko P, Schneider G (1999) Three-dimensional structure of a mammalian purple acid phosphatase at 2.2 Å resolution with a mu-(Hydr)oxo bridged di-iron center. J Mol Biol 291:135–147
Loxdale HD, Castaňera P, Brookes CP (1983) Electrophoretic study of enzyme from cereal aphid population. Bull Ent Res 73:645–657
Malla R (2008) Isozymes involved in phosphate metabolism in Piriformospora indica. J Inst Sci Technol 15:1–13
Malla R, Varma A (2004) Phosphatase (s) in microorganisms. Biotechnological applications of microbes. IK International/Kluwer academic Press, India, New York/Holland pp. 125–50
Malla R, Varma A (2007) Use of short oligonucleotide primers in random amplified polymorphic DNA techniques for species identification. In: Oelmüller R, Varma A (eds) Advanced techniques in soil microbiology, vol 11. Springer, Germany, pp 235–244
Malla R, Singh A, Md Z, Yadav V, Suniti, Verma A, Rai M, Varma A (2002) Piriformospora indica and plant growth promoting Rhizobacteria: An Appraisal. In: Rao GP, Bhat DJ, Lakhanpal TN, Manoharichari C (Eds) Frontiers of fungal diversity in India. 401–19
Malla R, Prasad R, Giang PH, Pokharel U, Oelmueller R, Varma A (2004) Characteristic features of symbiotic fungus Piriformospora indica. Endocytobiosis Cell Res 15:579–600
Malla R, Pokharel U, Varma A (2005) Random amplified polymorphic DNA of the two fungi from Sebacinales. J Institute Sci Technol 14:34–43
Malla R, Zeaullah MD, Pokharel U, Varma A (2006) Immunological characterization of acid phosphatase in an endophytic fungus. Nepal J Sci Technol 7:77–84
Malla R, Pokharel U, Prasad R, Varma A (2007a) Proteomics and genomics approach to study plant–microbe cross communication. In: Chauhan AK, Harsha K, Varma A (eds) Microbes for human life, vol 4. IK International, India, pp 609–636
Malla R, Pokharel U, Prasad R, Oelmüller R, Varma A (2007b) Immuno-technology for the localization of acid phosphatase using native gel bands in Piriformospora indica and other Soil Microorganism. In: Modern tools and techniques. Springer, Germany, pp 211–234
McCormick MK, Whigham DF, O’Neill J (2004) Mycorrhizal diversity in photosynthetic terrestrial orchid. New Phytol 163:425–438
Meyberg M (1988) Selective staining of fungal hyphae in parasitic and symbiotic plant fungus associations. Histochemistry 88:197–199
Moller EM, Bahnweg G, Sandermann H, Geiger HH (1992) A simple and efficient protocol for isolation of high molecular weight DNA from filament fungi, fruit bodies and infected plant tissue. Nucleic Acids Res 20:6115–6116
Nahas E, Terenzi HF, Rossi A (1982) Effect of carbon source and pH on the production and secretion of acid phosphatase and alkaline phosphatase in neurospora crassa. J Gen Microbiol 128:2017–2021
Paszkowski U, Kroken S, Roux C, Briggs SP (2002) Rice phosphate transporters includes an evolutionarily divergent gene specifically activated in arbuscular mycorrhizal symbiosis. Proc Natl Acad Sci 99:13324–13329
Peškan-Berghöfer T, Shahollari B, Pham HG, Hehl S, Markent C, Blank V, Kost G, Varma A, Oelmueller R (2004) Association of Piriformospora indica with Arabidopsis thaliana roots represent a novel system to study beneficial plant-microbe interactions and involve in early plant protein modifications in the endocytoplasmic reticulum and in the plasma membrane. Physiol Plant 122:465–71
Pham GH, Kumari R, Singh A, Malla R, Prasad R, Sachdev M, Kaldorf M, Buscot F, Oelmüller R, Hampp R, Saxena AK, Rexer K-H, Kost G, Varma A (2004b) Axenic cultures of Piriformospora indica. In: Plant Surface Microbiology. Springer, Germany. pp 593–613
Podila GK, Lanfranco L (2004) Functional genomic approaches for studies of mycorrhizal symbiosis. Plant surface microbiology. In: Varma A, Abbott L, Werner D, Hampp R (eds) Plant surface microbiology. Springer, Berlin, Heidelberg, Germany, pp 567–592
Rausch C, Daram P, Brunner S, Jansa J, Laloi M, Leggewie G, Amrhein N, Bucher M (2001) A phosphate transporter expressed in arbuscule-containing cells in potato. Nature 414:462–466
Rohlf F.J, NTSYS-pc. (1992) Numerical taxonomy and multivariate analysis system. Vers. 1.70. Exeter Software, Setauket, New York
Saito M (1995) Enzyme activities of the internal hyphae and germinated spores of an arbuscular mycorrhizal fungus. Gigaspora margarita Becker and Hall. New Phytologist 129:425–431
Shahollari B, Varma A, Oelmüller R (2005) Expression of a receptor kinase in Arabidopsis roots is stimulated by the basidiomycete Piriformospora indica and the protein accumulates in Triton X-100 insoluble plasma membrane microdomains. J Plant Physiol Holland 162:945–958
Tadano T, Ozawa K, Sakai H, Osaki M, Matsui H (1993) Secretion of acid phosphatase by the roots of crop plants under phosphorus-deficient conditions and some properties of the enzyme secreted by lupin roots. Plant Soil 156:95–98
Tarafdar JC, Rao AV (1996) Contribution of Aspergillus strains to acquisition of phosphorus by wheat (Triticum aestivum L.) and chick pea (Cicer arietinum L.) grown in a loamy soil. Appl Soil Ecol 3:190–214
Tarafdar JC, Yadav RS, Meena SC (2001) Comparative efficiency of acid phosphatase originated from plant and fungal sources. J Plant Nutr Soil Sci 164:279–282
Tisserant B, Gianinazzi-Pearson V, Gianinazzi S, Gollotte A (1993) In planta histochemical staining of fungal alkaline phosphatase activity for analysis of efficient arbuscular mycorrhizal infections. Mycol Res 97:245–250
Towbin H, Staehlin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci 76:4350–4354
Vaerman JP (1981) Single radial immunodiffusion. Meth Enzymol 73:291–305
Varma A, Verma S, Sudha, Sahay N, Britta B, Franken P (1999) Piriformospora indica—a cultivable plant growth promoting root endophyte with similarities to arbuscular mycorrhizal fungi. Appl Environ Microbiol 65:2741–2744
Verma S, Varma A, Rexer KH, Hassel A, Kost G, Sarabhoy A, Bisen P, Bütenhorn B, Franken P (1998) Piriformospora indica, gen. et sp. nov., a new root colonizing fungus. Mycologia 90:896–903
Voller A, Bidwell DE, Bartlett A (1980) Enzyme linked immunosorbent assay. In: Rose NR, Friedmans H (eds) Manual of clinical immunology. American Society for Microbiology, Washington, DC, pp 359–371
Weiß M, Selosse MA, Rexer KH, Urban A, Oberwinkler F (2004) Sebacinales: a hitherto overlooked cosm of heterobasidiomycetes with abroad mycorrhizal potential. Mycol Res 108:1–8
Acknowledgements
Rajani Malla is thankful to Dr. Ashok K. Chauhan, founder president, Amity—RBEF New Delhi—for his support. The author is also thankful to Dr. Upendra and Sweta from All India Institute of Medical Sciences and Dr. Ram Prasad from Amity University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Malla, R., Pokharel, U., Varma, A. (2013). Similarity Between Piriformospora indica and Sebacina vermifera Sensu Members of the Order Sebacinales Based on Immunological Techniques. In: Varma, A., Kost, G., Oelmüller, R. (eds) Piriformospora indica. Soil Biology, vol 33. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-33802-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-642-33802-1_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-33801-4
Online ISBN: 978-3-642-33802-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)