
Abstract
Cellulose and associated polysaccharides, such as xylans, comprise the major portion of the plant cell wall as structural polymers. As the plants evolved and distributed first in the seas and then on land, following their demise, the accumulated cellulosic materials had to be assimilated and returned to nature. Thus the cellulose-degrading bacteria have evolved to complement lignin-degrading microbial systems for the purpose of restoring the tremendous quantities of organic components of the plant cell wall to the environment for continued life cycles of carbon and energy on the global scale. This chapter is a sequel to a previous chapter of the same title from the second edition of this treatise (Coughlan MP, Mayer F (1992) The cellulose-decomposing bacteria and their enzyme systems. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes, vol I, 2nd edn. Springer, New York, pp 459–516.) and represents an update of our own subsequent chapter (Bayer EA, Shoham Y, Lamed R (2006) Cellulose-decomposing prokaryotes and their enzyme systems. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes, vol 2, 3rd edn. Springer, New York, pp 578–617.) which appeared in the third edition. Although the basic elements of the previous chapters are still essentially up to date, the field of the cellulose-decomposing bacteria has since advanced greatly, owing to two major factors: (1) the advent, progression, and increasing facility of genome- and metagenome-sequencing efforts and (2) the current initiatives to utilize plant-derived biomass for the production of biofuels as an alternative to fossil fuels for an energy source.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Glycoside Hydrolase
- Enzyme Commission
- Glycoside Hydrolase Family
- Clostridium Thermocellum
- Catalytic Module
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
From an anthropocentric point of view, for millennia, human culture has been intricately involved with cellulose, the major component of the plant cell wall. The development of wood, paper, and textile industries has served to weave cellulosic materials into the fabric of our society. Within the past century, however, cellulosic wastes, derived mainly from the same industries, have also become a major source of environmental pollution. This chapter will concentrate mainly on cellulose and the cellulolytic bacteria, in view of their importance to mankind and world ecology. Nevertheless, the true substrate of these bacteria—i.e., the complement of plant cell wall polysaccharides in general—is much more complex than cellulose alone. Likewise, the complement of enzymes—both the cellulolytic and the non-cellulolytic glycoside hydrolases (GHs)—are produced concurrently in these bacteria for the purpose of efficient synergistic degradation of the complete substrate composite as it appears in nature. Consequently, when we discuss the cellulose-decomposing bacteria and their enzyme systems, we cannot ignore the related non-cellulolytic enzymes, and these will also be treated, albeit secondarily, in this chapter.
The plant cell wall consists of an intricate mixture of polysaccharides (Carpita and Gibeaut 1993); cellulose, hemicellulose, and lignin are its major constituents. These polymers are of a very robust nature. They both equip the plant with a stable structural framework and protect the plant cell from the perils of its environment. Despite its recalcitrant nature, in the guise of dead or dying plant matter, the polysaccharides of the plant cell wall provide an exceptional source of carbon and energy, and a multitude of different microorganisms has evolved which are capable of degrading plant cell wall polysaccharides.
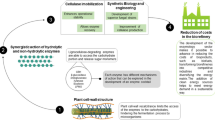
In any given ecosystem, the polysaccharide-degrading microbes are not alone, but rely on the complementary contribution of other bacterial and/or fungal species (Bayer and Lamed 1992; Bayer et al. 1994; Ljungdahl and Eriksson 1985). The polymer-degrading strains play a primary and crucial role in the ecosystem by converting the plant cell wall polysaccharides to the respective simple sugars and other degradation products (Fig. 6.1 ). They are assisted by satellite microbes, which cleanse the microenvironment from the breakdown products, producing, in the final analysis, methane and carbon dioxide.


Simplified schematic description of a typical ecosystem comprising degrading plant matter. Cellulolytic, xylanolytic, and other hemicellulosic microbes combine to decompose the major polysaccharide components to soluble sugars. “Satellite” microorganisms assimilate the excess sugars and other cellular end products, which are ultimately converted to methane and carbon dioxide
In a given polysaccharide-degrading microorganism, the enzymes that catalyze the degradation may occur in several possible states, according to recognized paradigms (Himmel et al. 2010). These include (1) enzymes in the free state and (2) multifunctional polypeptides in discrete multi-enzyme complexes and/or enzymes attached directly to the bacterial cell surface. All of these paradigms exhibit similar types of enzyme components, which usually comprise modular proteins that contain a multiplicity of functional modules. The “free” enzymes comprise a single polypeptide chain, which contains a catalytic module usually connected to a cellulose-binding module (CBM). The multifunctional enzyme paradigm includes more than one catalytic module per polypeptide chain. Cellulosomes are exocellular macromolecular machines, designed for efficient degradation of cellulose and associated plant cell wall polysaccharides (Bayer et al. 1998a, 2004, 2008; Shoham et al. 1999; Doi and Kosugi 2004). In contrast to the free and multifunctional enzymes, the cellulosome complex is composed of a collection of subunits, each of which comprises a set of interacting functional modules. Thus, one type of cellulosomal module, the CBM, is selective for binding to the substrate. Another family of modules, the catalytic modules, is specialized for the hydrolysis of the cellulose chains. Yet another complementary pair of modules—the cohesins and dockerins—serves to integrate the enzymatic subunits into the complex and the complex, in turn, into the cell surface. Multiple copies of the cohesins form a unique type of nonenzymatic integrating subunit called scaffoldin to which the dockerin-containing enzymes are attached. This “Lego™”-like arrangement of the modular subunits generates an intricate multicomponent complex, the enzymes of which are bound en bloc to the insoluble substrate and act synergistically toward its complete digestion. Finally, single enzymes and cellulosomes can both be attached to the bacterial cell surface using one of several mechanisms. In the case of a single enzyme, the catalytic module is attached to the cell surface via a specialized binding module. In the cellulosome, a similar type of binding can occur, but this is done so via an anchoring scaffoldin which then binds to the primary (enzyme-integrating) scaffoldin. Alternatively, in some cases, the enzyme or anchoring scaffoldin is bound covalently to the cell surface enzymatically. Finally, the attachment of some types of cellulosome to the bacterial surface has not yet been elucidated. The above paradigms will be discussed in more detail later in this chapter.
A list of cellulose-degrading bacteria is presented in Table 6.1 , together with their dominant enzyme paradigm (some species show numerous types of enzymes, e.g., free and cellulosomal) and their distinctive types of modules (i.e., CBMs, cohesins, dockerins).
Inherent to the study of cellulases and related enzymes is their potential industrial application—particularly toward conversion of cellulosic biomass to biofuels. For reviews on the potential uses of these enzymes, the reader is referred to appropriate reviews on the subject (Bhat 2000; Himmel et al. 1999, 2007; Lynd et al. 1991, 2008; Perlack et al. 2005; Ragauskas et al. 2006; Schubert 2006; Galbe and Zacchi 2007; DOE 2008; Himmel 2008; Wall et al. 2008; Nordon et al. 2009; Sheehan 2009; Wilson 2009; Xu et al. 2009; Klein-Marcuschamer et al. 2011).
Plant Cell Wall Polysaccharides
Plant cells produce a composite matrix of hardy and durable polysaccharides on the outer surface of the plasma membranes, called the cell wall (Carpita and Gibeaut 1993). The cell wall confers a protective coating to the plant cell, providing structure, turgidity, and durability, which render the cell resistant to the outer elements, including mechanical, chemical, and microbial assault. Different types of plant cell tissues exhibit different ratios of the three major types of cell wall component; on the average, the cell wall contains roughly 40 % cellulose, 30 % hemicellulose, and 20 % lignin, but the exact composition of an individual type of plant varies greatly. The first two polymers are indeed polysaccharides. On the other hand, lignin is a heterogeneous, high-molecular-weight hydrophobic polymer, which consists of non-repeating aromatic monomers connected via phenoxy linkages (Higuchi 1990; Lewis and Yamamoto 1990). Unlike cellulose and hemicellulose, which are degraded aerobically or anerobically, lignin degradation requires oxygen and is limited to filamentous prokaryotes (e.g., the Actinomycetes Streptomyces viridans) and fungi (e.g., Phanerochaete chrysosporium, Bejerkendera adusta, and Pleurotus ostreatus), which produce a complicated set of enzymes that hydrolyze the polymer. In fact, the recalcitrant lignin interferes severely with the access of enzymes to the cellulose component, and is rate-limiting for anaerobic degradation of cellulose. In any case, the lignin component must be degraded or removed, before efficient degradation of cellulose can take place. Nevertheless, since lignin is not a polysaccharide, it will not be discussed further in this chapter.
Cellulose
Cellulose is the major constituent of plant matter and thus represents the most abundant organic polymer on Earth. Cellulose is a remarkably stable homopolymer, consisting of a linear (unbranched) polymer of β-1,4-linked glucose units. Chemically, the repeating unit is simply glucose, but, structurally, the repeating unit is the disaccharide cellobiose, i.e., 4-O-(β-d-glucopyranosyl)-d-glucopyranose, since each glucose residue is rotated 180° relative to its neighbor (Fig. 6.2 ). The individual cellulose chains contain from about 100 to more than 10,000 glucose units, packed tightly in parallel fashion into microfibrils by extensive inter and intrachain hydrogen bonding interactions, which account for the rigid structural stability of cellulose. The microfibrils exhibit variable amounts of crystalline and amorphous components, again depending on the degree of polymerization, the extent of hydrogen bonding and, ultimately, on the source of the cellulose. Cellulose of the plant cell wall is composed of two different forms: cellulose Iα and cellulose Iβ. Cellulose Iα is in a triclinic state with a single chain per unit cell and is of higher energy than cellulose Iβ, which is in a monoclinic state and much more stable (Atalla and VanderHart 1984; Sugiyama et al. 1991; Atalla 1999; Ding and Himmel 2006). Enzymatic hydrolysis of the Iα form occurs more readily, but the cellulose of the plant cell wall comprises mainly the more stable Iβ form. The hydroxyl groups of glucose are in the equatorial position, as opposed to the axial positions which are all nonpolar protons that do not participate in hydrogen bonding interactions. Thus, owing to the packing of the glucose chains in the microfibrils, the “sides” are polar and hydrogen bonding whereas the “tops and bottoms” are hydrophobic in character (Matthews et al. 2006). The microfibrils themselves are further assembled into plant cell walls, the tunic of some sea animals, pellicles from bacterial origin, etc. Highly crystalline forms of cellulose include cotton, bacterial cellulose (from Acetobacter xylinum) and the cellulose from the algae Valonia ventricosa, which exhibit crystallinity levels of about 45 %, 75 %, and 95 %, respectively. The following reviews are available for more information on the structure of cellulose: A talla (1999); Atalla and VanderHart (1984); Chanzy (1990); O’Sullivan (1997); Ding and Himmel (2006); Moon et al. (2011).


Structure of cellulose. Three parallel chains are shown, and a glucose moiety and repeating cellobiose unit are indicated. The model was built by Dr. José Tormo, based on early crystallographic data
Hemicellulose
Hemicelluloses are relatively low-molecular-weight, branched heteropolysaccharides that are associated with both cellulose and lignin and together build the plant cell wall material (Puls and Schuseil 1993; Timell 1967). The main backbone of hemicellulose is usually made of one or two sugars, which determines their classification. For example, the main backbone of xylan is composed of 1,4-linked-β-d-xylopyranose units. Similarly, the backbone of galactoglucomannan is made of linear 1,4-linked β-d-glucopyranose and β-d-mannopyranose units with α-1,6-linked galactose residues. Other common hemicelluloses include arabinogalactan, lichenins (mixed 1,3-1,4-linked β-d-glucans), and glucomannan. Most hemicellulases are based on a 1,4-β-linkage and the main backbone is branched, whereas the individual sugars may be acetylated or methylated. For example, the linear xylan backbone is highly substituted with a variety of saccharide and nonsaccharide components (Fig. 6.3 ). In the plant cell wall, xylan is closely associated with other wall components. The 4-O-methyl-α-d-glucuronic acid residues can be ester-linked to the hydroxyl groups of lignin, providing cross-links between the cell walls and lignin (Das et al. 1984). Similarly, feruloyl substituents serve as cross-linking sites to either lignin or other xylan molecules. Thus, the chemical complexity of xylan is in direct contrast to the chemical simplicity of cellulose. Likewise, the structural diversity of the xylans is in contrast to the structural integrity of the cellulose microfibril. Consequently, unlike the crystalline-like character of cellulose, the hemicellulose component adopts a gel-like consistency, providing an amorphous matrix in which the rigid crystalline cellulose microfibrils are embedded.


Composition of a typical xylan component of hemicellulose. The xylobiose unit (β-Xylp–β-Xylp) is indicated by the blue-sided box, as are major substituents: Me-α-GlcA, methylglucuronic acid; αAraf, arabinofuranosyl group; OAc, acetyl group. A presumed lignin attachment site to a feruloyl substituent of xylan is also illustrated. Sites of cleavage by selected hemicellulases and carbohydrate esterases are also shown: Xyn, xylanase; Abn, arabinofuranosidase; Glr, glucuronidase; Axe, acetyl xylan esterase; Fae, ferulic acid esterase
Pectin
Pectin is a structural polysaccharide which is another major component of the primary cell wall of terrestrial plants. Pectin derivatives serve to mediate plant defense responses and regulate plant development (Ridley et al. 2001). The pectins are heteropolysaccharides composed of α-(1-4)-linked galacturonic acid, substituted with numerous constituent groups, e.g., xylose, rhamnose, and galactose. In addition, a large percentage of the galacturonic carboxyl groups are methylated. During the normal physiological processes of the plant (including plant growth, maturation, fruit ripening, and aging), the distribution, quantity, chemical composition, and structure of pectin is altered.
Cellulose-Degrading Bacteria
The cellulolytic microbes occupy a broad range of habitats. Some are free living and rid the environment of plant polysaccharides by converting them to the simple sugars which they assimilate. Others are linked closely with cellulolytic animals, residing in the digestive tracts of ruminants and other grazers or in the guts of wood-degrading termites and worms (Haigler and Weimer 1991). Cellulose-based ecosystems include soils, swamps, marshes, rivers, lakes, and seawater sediments; rotting grasses, leaves, and wood; cotton bales; sewage sludge; silage; compost heaps; muds; and decaying vegetable matter in hot and volcanic springs, acid springs, and alkaline springs (Ljungdahl and Eriksson 1985; Stutzenberger 1990).
The cellulolytic microorganisms include protozoa, fungi, and bacteria and are ubiquitous in nature. The cellulose-decomposing bacteria include aerobic, anaerobic, mesophilic, and thermophilic strains, inhabiting a great variety of environments, including the most extreme, vis-à-vis temperature, pressure, and pH. Cellulolytic bacteria have also been found in the gut of wood-eating worms, termites, and vertebrate herbivores, all of which exploit anaerobic symbionts for the digestion of wood and fodder.
In nature, many cellulolytic species exist in symbiotic relationships with secondary microorganisms (Ljungdahl and Eriksson 1985). The primary microorganisms degrade cellulose directly to cellobiose and glucose. Only part of the breakdown products is assimilated by the polymer degrading strain(s), and the rest is utilized by the satellite microorganisms. Removal of the excess of sugars promotes further cellulose degradation by the primary species, since cellobiose-induced inhibition of cellulase action and repression of cellulase synthesis are precluded.
Modern interest in cellulolytic microorganisms was spawned by the decay of cotton fabric in army tents and military clothing in the South Pacific jungles during World War II. The basic research program that resulted from this military problem led to the establishment of the US Army Natick Laboratories (Reese 1976). The resultant research led to the discovery that the causative agent for the costly problem was a cellulolytic fungi, Trichoderma viride (subsequently renamed Trichoderma reesei). Subsequent research, originally from the Natick Laboratories and later spreading to other research institutes and universities, led to the identification and classification of thousands of different strains of cellulolytic fungi and bacteria. Many of the major types of cellulolytic bacteria have been listed in the chapter published in the second edition of The Prokaryotes (Coughlan and Mayer 1992). During the interim period until publication of the chapter in the third edition (Bayer et al. 2006), the major emphasis in the area did not concentrate on the discovery or description of new cellulolytic strains but centered on characterizing the enzymes and enzyme systems from selected bacteria that degrade cellulose and plant cell wall polysaccharides in general. More recently, however, the emerging simplicity of genome sequencing efforts and metagenomic prospecting (Li et al. 2009) has supplanted the more tedious biochemical approaches.
Enzymes That Degrade Plant Cell Wall Polysaccharides
The chemical and structural intricacy of plant cell wall polysaccharides is matched by the diversity and complexity of the enzymes that degrade them. The cellulases and hemicellulases are family members of the broad superfamily of glycoside hydrolases (see Table 6.2 ), which catalyze the hydrolysis of oligosaccharides and polysaccharides (Gilbert and Hazlewood 1993; Kuhad et al. 1997; Ohmiya et al. 1997; Schülein 1997; Tomme et al. 1995a; Viikari and Teeri 1997; Warren 1996; Wilson and Irwin 1999). In the past decade, numerous bacterial genomes were sequenced (see Table 6.1 ), and databases for the rapidly spiraling accumulation sequences and structures of cellulolytic and hemicellulolytic enzymes are readily available online (see discussion below).
Historically, the type of substrate and manner in which a given enzyme interacts with its substrate were decisive in the classification of the glycosidases, as established first by the Enzyme Commission (EC) and later by the Nomenclature Committee of the International Union of Biochemistry. Enzymes were usually named and grouped according to the reactions they catalyzed. Thus, cellulases, xylanases, mannanases, and chitinases were grouped a priori in different categories. Moreover, enzymes which cleave polysaccharide substrates in the middle of the chain (“endo”-acting enzymes) versus those which clip at the chain ends (“exo”-acting enzymes) were also placed in different groups. For example, in the case of cellulases, the endoglucanases were grouped in EC 3.2.1.4, whereas the exoglucanases (i.e., cellobiohydrolases) were classified as EC 3.2.1.91.
It is interesting that the distinction between endo- and exo-acting enzymes is also reflected by the architecture of the respective class of active site, even within the same family of enzymes (Fig. 6.4 ). The endoglucanases, e.g., are commonly characterized by a groove or cleft, into which any part of a linear cellulose chain can fit. On the other hand, the exoglucanases bear tunnel-like active sites, which can only accept a substrate chain via its terminus. The exo-acting enzyme apparently threads the cellulose chain through the tunnel, wherein successive units (e.g., cellobiose) would be cleaved in a sequential manner. The sequential hydrolysis of a cellulose chain is a relatively new notion of growing importance, which has earned the term “processivity” (Davies and Henrissat 1995), and processive enzymes are considered to be key components which contribute to the overall efficiency of a given cellulase system.


Structures of a typical endoglucanase and exoglucanase. In each case, the structure is viewed from a perspective, which demonstrates the comparative architecture of the respective active site. Despite the sequence similarity of both enzymes and their classification as Family-6 glycoside hydrolases, their respective active-site architecture is different. The Family-6 endoglucanase (endoglucanase Cel6A from the bacterium Thermomonospora fusca, PDB code 1TML) is characterized by a deep cleft to accommodate the cellulose chain at any point along its length, whereas the active site of the Family-6 exoglucanase (cellobiohydrolase CBHI from the cellulolytic fungus, Trichoderma reesei, PDB code 1CEL) bears an extended loop that forms a tunnel, through which one of the termini of a cellulose chain can be threaded
Though instructive, there is growing dissatisfaction with the endo/exo terminology. As our understanding of the nature of catalysis by these enzymes progresses, it has become clear that some enzymes are capable of both endo- and exo-action (Johnson et al. 1996; Morag et al. 1991; Reverbel-Leroy et al. 1997; Sakon et al. 1997). Moreover, some glycoside hydrolase families include both endo- and exo-enzymes, again indicating that the mode of cleavage can be independent of sequence homology and structural fold. In this context, relatively minor changes in the lengths of relevant loops in the general proximity of the active site, may dictate the endo- or exo-mode of action without significant differences in the overall fold.
Due to subtle but diverse chemical and structural aspects of the substrates involved, plant cell wall–degrading enzymes do not follow the same rules as common enzyme standards, such as simple proteases, DNAse, RNAse, and lysozyme. In fact, the cellulases and hemicellulases are usually very large enzymes, whose molecular masses often exceed those of proteases by factors of 2 to 5 and more. Their polypeptide chains partition into a series of functional modules and linker segments (frequently glycosylated), which together determine their overall activity characteristics and interaction with their substrates and/or with other components of the cellulolytic and hemicellulolytic system.
However, the historical division of enzymes is inappropriate for the classification of the cellulases and other glycoside hydrolases. Like other enzymes (e.g., proteases), previous classification systems of the glycoside hydrolases centered on the types of substrates and the bonds cleaved by a given enzyme. The problem with the glycoside hydrolases is that the polysaccharide substrates and particularly the bonds they cleave are all quite similar, and classification of the different types of enzymes according to conventional criteria often misses the mark. Consequently, alternative approaches were pursued. Over the past decade or so, the definitive trend has evolved to classify the different glycoside hydrolases into groups based on common sequence, structural fold and mechanistic themes (Davies and Henrissat 1995; Henrissat 1991; Henrissat and Bairoch 1996; Henrissat and Davies 1997; Henrissat et al. 1998).
A comprehensive, authoritative website that provides a complete and growing catalog of the different glycoside hydrolase families is available (Coutinho and Henrissat 1999a, b, c; Coutinho et al. 2003a, b, c; Henrissat and Coutinho 2001; Henrissat et al. 2003; Cantarel et al. 2009): The Carbohydrate-Active Enzymes server (http://www.cazy.org/). The website also provides similar sequence information for additional types of enzymes that participate in the degradation of plant cell wall polysaccharides, namely, carbohydrate esterases (e.g., that cleave acetyl, feruloyl and cinnamoyl groups from xylans) and polysaccharide lyases (that act on pectin). Additional associated modular components of these enzymes, particularly the carbohydrate-binding modules (CBMs), are also classified into families and documented exhaustively. An extensive list of sequenced genomes is included, which contains the carbohydrate-active enzymes encoded by the genome (“CAZome”) of the given bacterium and facilitates insight into the nature and extent of the metabolism of complex carbohydrates of the species and comparison between both related and unrelated species. The site contains excellent introductory explanatory material, and the interested reader is encouraged to use this site extensively. Moreover, a companion website, CAZypedia (http://www.cazypedia.org/index.php/Main_Page), provides an encyclopedic resource for detailed understanding of the different glycoside hydrolase families.
Cellulases
The cellulases include the large number of endo- and exo-glucanases which hydrolyze β-1,4-glucosidic bonds within the chains that comprise the cellulose polymer (Béguin and Aubert 1994; Haigler and Weimer 1991; Tomme et al. 1995a). Thus, in principle, the degradation of cellulose requires the cleavage of a single type of bond. Nevertheless, in practice, we find that cellulolytic microorganisms produce a variety of complementary cellulases of different specificities from many different families. The major glycoside hydrolase families of cellulases include GH5, GH6, GH7 (found in fungi), GH8, GH9, GH12, GH44, GH45, GH48, GH74, and GH124.
It may seem somewhat surprising that the combined effect of so many different enzymes is required to degrade such a chemically simplistic substrate. This complexity reflects the difficulties an enzyme system encounters upon degrading such a highly crystalline substrate as cellulose. As described in the previous section, cellulases that degrade the cellulose chain can be either “endo-acting” or “exo-acting.” Moreover, the degradation of crystalline cellulose should be viewed three-dimensionally and in situ, where the cellulose chains are packed within the microcrystal, thus generating the remarkably stable physical properties of the crystalline substrate. The enzymes have to bind to the cellulose surface, localize and isolate suitable chains, destined for degradation. It would seem logical that amorphous regions or defects in the crystalline portions of the substrate would be favorable sites for initiation of the process. The structural as opposed to chemical heterogeneity of the substrate dictates the synergistic action of a complex set of complementary enzymes toward its complete digestion.
Various models have been suggested to account for the observed synergy between and among two or more different types of cellulases. For example, an endo-acting enzyme can produce new chain ends in the internal portion of a polysaccharide backbone, and the two newly exposed chains would then be available for action of exo-acting enzymes. In addition, two different types of exo-glucanases may exhibit different specificities by acting on a cellulose chain from opposite ends (i.e., the reducing versus the nonreducing end of the polymer). Likewise, an endoglucanase may be selective for only one of the two sterically distinct glucosidic bonds on the cellulosic surface. In addition, some cellulases may display high levels of activity at the beginning of the degradative process, i.e., on the highly crystalline material, whereas others would be selective for newly exposed, partially degraded chains, otherwise embedded within the crystal. Still others would show very high levels of activity after the degradative process has advanced, and cellulose chains which have been freed of the crystalline setting would then be hydrolyzed quite rapidly. A collection of various enzymes, which exhibit complementary specificities and modes of action, would account for the observed synergistic action of the complete cellulase “system” in digesting the cellulosic substrate.
In addition to endo- and exo-glucanases, included in the overall group of cellulases are the β-glucosidases (EC 3.2.1.21), which hydrolyzes terminal, nonreducing β-d-glucose residues from cellooligodextrins. These enzymes are members of the following glycoside hydrolase families: GH1, GH3, and GH116. In particular, this type of enzyme cleaves cellobiose—the major end product of cellulase digestion—to generate two molecules of glucose. Some β-glucosidases are specific for cellobiose whereas others show broad specificity for other β-d-glycosides, e.g., xylobiose. Often, the β-glucosidases are associated with the microbial cell surface and hydrolyze cellobiose to glucose before, during or after the transport process.
Among the novel glycoside hydrolase families, a new and important oxidative family, previously classified as a glycoside hydrolase family (GH61), was found in fungi which break internal glucan bonds (Beeson et al. 2012). A similar oxidative family of enzymes has also been proposed to exist in bacteria – the Family-33 CBMs, which were originally considered to be CBMs (Forsberg et al. 2011). Both the GH61 and the CBM33s will have to be reclassified separately as oxidative enzymes associated to the other CAZymes.
Hemicellulases
Strictly speaking, hemicellulases are not the precise subject of this chapter, since they do not directly sever the β-1,4-glucosidic bond of cellulose. Nevertheless, in nature, they are essential to the bacterial degradation of insoluble cellulose, since the natural bacterial substrate—the plant cell wall—comprises an architecturally cogent composite of cellulose and hemicelluloses. In natural systems, the two types of polysaccharides cannot be easily separated, and microbial systems have to deal simultaneously with both. The xylan component is particularly of interest for several reasons: (a) xylan is a major hemicellulosic component of the plant cell wall; (b) the xylanases are well-defined enzymes, closely associated with the cellulase; and (c) the repeating units (both xylose and xylobiose) bear striking structural resemblance to their cellulosic counterparts (i.e., glucose and cellobiose).
In contrast to cellulose degradation, the degradation of the hemicelluloses imposes a somewhat different challenge, since this group of polysaccharides includes widely different types of sugars or nonsugar constituents with different types of bonds. Thus, the complete degradation of hemicellulose requires the action of different types of enzymes. These enzymes, the hemicellulases, can differ in the chemical bond they cleave, or, as in the case of the cellulases, they may cleave a similar type of bond but with different substrate or product specificity (Biely 1985; Coughlan and Hazlewood 1993; Eriksson et al. 1990; Gilbert and Hazlewood 1993).
Hemicellulases can be divided into two main types, those that cleave the main chain backbone, i.e., xylanases or mannanases, and those that degrade side chain substituents or short end-products, such as arabinofuranosidase, glucuronidase, acetyl esterases, and xylosidase. Like the cellulases, hemicellulases can be of the endo or exo types. A schematic view of the types of bonds that would be hydrolyzed by different types of hemicellulases is presented in Fig. 6.3 .
Xylan-Degrading Enzymes
The xylanases are by far the most characterized and studied of the hemicellulases and involve the cleavage of a major main chain backbone. Endoxylanases (1,4-β-d-xylan xylanhydrolase, EC 3.2.1.8) hydrolyze the 1,4-β-d-xylopyranosyl linkage of xylans, such as d-glucurono-d-xylans and l-arabino-d-xylan. These single-subunit enzymes from both fungi and bacteria exhibit a broad range of physiochemical properties, whereby two main classes have been described: alkaline proteins of low Mr (<30,000) and acidic proteins of high Mr. This general classification scheme correlates with their assignment into glycoside hydrolase Families 10 and 11 (http://www.cazy.org/), whereby the former represents the high Mr xylanases and the latter coincides with the low Mr enzymes. The two families also differ in their catalytic properties, such that the Family 10 enzymes seem to display a greater versatility toward the substrate than that observed for those of Family 11, and are thus typically able to hydrolyze highly substituted xylan more efficiently. The Family 10 xylanases exhibit a (β/α)8 topology whereas those from Family 11 form a β-jelly roll fold. Both families show a retaining catalytic mechanism of hydrolysis. In addition to the GH10 and GH11 families which comprise xylanases exclusively, a small portion of the GH5 and GH8 enzymes exhibit xylanase activity.
Mannan- and Galactan-Degrading Enzymes
Glucomannans and galactoglucomannans are branched heteropolysaccharides found in hardwood and softwood. The degradation of these polymers again involve many hydrolytic enzymes, including endo-1,4-β−mannanase (EC 3.2.1.78), β−mannosidase (EC 3.2.1.25), β−glucosidase (EC 3.2.1.21), and α−galactosidase (EC 3.2.1.22). 1,4-β−d-Mannanases hydrolyze main chain linkages of d-mannans and d-galacto-d-mannans. These enzymes, both of the endo or exo types, are produced in various microorganisms, including Bacillus subtilis, Aspergillus niger, and intestinal and rumen bacteria and commonly occur in Families GH5, GH26, and GH113 (http://www.cazy.org/). Likewise, 1,4-β-galactanases hydrolyze 1,4-linked β−d-galactosyl groups from the nonreducing end and are members of Family GH53.
Lichenin-Degrading Enzymes
Lichenase (1,3-1,4-β−d-glucan 4-glucanohydrolase, EC 3.2.1.73) is a mixed linkage β−glucanase, which cleaves the β−1,4 linkages adjacent to the β−1,3 bonds of the lichenin substrate. According to modern structure-based classification, lichenases can be members of Families 8, 16, or 17 (http://www.cazy.org/).
Other Polysaccharide-Degrading Activities
Other types of enzyme activities, in which polysaccharides characterized by other types of linkages (i.e., α- or β-, 1,2, 1,3, 1,6, etc.) are cleaved and are classified in various additional glycoside hydrolase families.
β−D-Xylosidases
The 1,4-β−d-xylosidases (1,4-β−d-xylan xylohydrolase, EC 3.2.1.37) hydrolyze xylooligosacharides (i.e., xylan breakdown products and mainly xylobiose) to xylose. These enzymes are either intracellular or extracellular components and are closely associated with hemicellulolytic activities. Monomeric, dimeric, and tetrameric xylosidases have been found with Mrs of 26,000–360,000. Many of the xylosidases act on a variety of substrates. For example Aspergillus niger produces an enzyme, classified as a β−xylosidase, that can hydrolyze β−galactosides, β−glucosides, and α−arabinosides, in addition to β−xylosides. The β−xylosidases are members of numerous glycoside hydrolase families, including GH3, GH30, GH39, GH43, GH52, GH54, GH116, and GH120 (http://www.cazy.org/).
Other Side Chain–Degrading Enzymes
α-d-Glucuronidases (EC 3.2.1.39) catalyze the cleavage of the α-1,2 glucosidic bond of 4-O-methyl-α-d-glucuronic acid side chain. This bond has a stabilizing effect on the neighboring xylosidic bonds of the main chain. Several α-glucuronidase genes have recently been cloned and sequenced and usually occupy Families GH67 and GH115.
α-l-Arabinofuranosidases (α-l-arabinofuranoside arabinofuranohydrolase, EC 3.2.1.55) is another important enzyme that cleaves nonreducing terminal α-l-arabinfuranosidic linkages in arabinoxylan, l-arabinan, and other l-arabinose-containing polysaccharides. These enzymes are found either in the cell-associated or extracellular form and can be members of Families GH43, GH51, GH54, GH62, or GH127. 1, 4-β-Mannosidases hydrolyze 1,4-linked β−d-mannosyl groups from the nonreducing end. These enzymes (similar to β−xylosidases) hydrolyze mainly the end products of the mannanases, i.e., mannobiose and mannotriose and are members of Families GH1, GH2, and GH5.
Pectin-Degrading Enzymes
Polysaccharide Lyases (PLs) cleave uronic acid-containing polysaccharide chains via a β-elimination mechanism to generate an unsaturated hexanuronic acid residue creating a new reducing end. Among the 22 PL families, only a few are relevant to the degradation of plant cell wall polysaccharides, specifically pectins and rhamnogalacturonan (Table 6.3 ). Like the other CAZymes, the PLs are modular proteins, which serve to complement the activities of the cellulases and other enzymes to better degrade the plant cell wall polysaccharide components.
Carbohydrate Esterases
The side chain substituents of xylan are composed not only of sugars but also of acidic residues, such as acetic, ferulic (4-hydroxy-3-methoxycinnamic), or p-coumaric (4-hydroxycinnamic) acids. Carbohydrate esterases that cleave these residues (see Fig. 6.3 ) are found in enzyme preparations from both hemicellulolytic and cellulolytic cultures (Borneman et al. 1993). Such enzymes sometimes represent separate modules, separated by linker segments from other cellulolytic or hemicellulolytic catalytic modules in the same polypeptide chain. Like the glycoside hydrolases, the carbohydrate esterases are classified into families according to sequence homology and common structural fold (http://www.cazy.org/) and they frequently appear together with other modular components, notably xylanases from glycoside hydrolase families 10 and 11, on the same polypeptide chain. A list of the important CE families is given in Table 6.4 . Most of the families contain enzymes that exhibit acetyl xylan esterase activity. Family CE1 also has members that cleave cinnamoyl and feruloyl bonds. These enzymes are very important ones, since this would allow a bacterium to sever the xylan components that are attached covalently to lignin. As lignin and its degradation products are frequently deleterious to enzymes that degrade plant-derived polysaccharides and to the bacterium itself, the action of the ferulic and coumaric acid esterases would promote more effective degradation of the xylan upon its separation from the lignin.
Cellulases and Hemicellulases Are Modular Enzymes
The initial contribution of biochemical methods for determining the characteristics of a given cellulase was extended immeasurably by the contribution of molecular biology and bioinformatics. By comparing the sequences of the cellulases and related enzymes, an entirely new view of these enzymes emerged.
Cellulases and hemicellulases are composed of a series of separate modular components. This fact explains the very large size of some of these enzymes and gives us some insight into their complex mode of action. Each module comprises a consecutive portion of the polypeptide chain and forms an independently folding, structurally and functionally distinct unit (Coutinho and Henrissat 1999a, b, c; Gilkes et al. 1991; Teeri et al. 1992). Each enzyme contains at least one catalytic module, which catalyzes the actual hydrolysis of the glycosidic bond and provides the basis for classification of the simple enzymes (i.e., those containing a single catalytic module). Other accessory or “helper” modules assist or modify the primary hydrolytic action of the enzyme, thus modulating the overall properties of the enzyme. Some of the different themes illustrating the modular compositions of the cellulases and related enzymes are presented in Fig. 6.5 . In many cases, certain patterns can be observed between the catalytic module(s) and the types of ancillary modules, notably the CBMs, which consistently occur in natural enzyme systems of the cellulolytic bacteria. Knowledge of the different modular components that comprise a given enzyme and thus modulate its activity can thus suggest the functional characteristics of the enzyme.


Scheme illustrating the diversity of the modular architecture of cellulases and other glycoside hydrolases. The different modules are grouped into families according to conserved sequences as shown by the pictograms in the Figure. (a) One of the most common types of cellulases consists of a catalytic module, flanked by a CBM at its N- or C-terminus. The particular enzyme shown in (a) comprises a catalytic module from Family-48 and a Family-2 CBM. (b) Cellulosomal enzymes are characterized by a “dockerin module” attached to a catalytic module. In this case, the same type of enzyme as in (a), carrying a Family-48 catalytic module, harbors a dockerin module instead of a CBM. (c) Many cellulases contain “X domains,” i.e., domains of unknown (as yet undefined) function. Often such domains prove to be a CBM when the appropriate binding specificity is determined experimentally. (d) Some enzymes have more than one CBM. Often, one CBM, such as the Family-3 CBM shown in the Figure, serves to bind the cellulase strongly to the flat surface of the insoluble substrate, whereas the other one (the Family-3c CBM) acts in concert with the catalytic module by binding transiently to a single cellulose or to a hemicellulose chain. (e) Some cellulosomal cellulases have a CBM together with a dockerin in the same polypeptide chain. (f) Some cellulases have more than one type of catalytic module, such as the Family-5 and Family-44 modules shown in the Figure, and the two probably work in concerted fashion to degrade the substrate efficiently
The Catalytic Modules: Families of Enzymes
The definitive component of a given enzyme is the catalytic module. Former EC-based classification schemes according to substrate specificity are now considered somewhat obsolete, since they fail to take into account the structural features of the enzymes themselves and for the compound reasons listed in the previous sections. The catalytic modules of glycoside hydrolases are now categorized into families according to amino acid sequence homology (Cantarel et al. 2009; Coutinho and Henrissat 1999a, b, c; Henrissat 1991; Henrissat and Bairoch 1996; Henrissat and Davies 1997; Henrissat et al. 1998). For more information, see the website server for the Carbohydrate-Active Enzymes (CAZy), designed and maintained by Bernard Henrissat and Pedro Coutinho (http://www.cazy.org/).
The enzymes of a given glycoside hydrolase family are similar in sequence, they display the same structural topology, and the positions of the catalytic residues are conserved with respect to the common fold. X-ray crystallography has provided a general overview of the structural themes of the glycoside hydrolases and their interaction with their intriguing set of substrates (Bayer et al. 1998; Davies and Henrissat 1995; Henrissat and Davies 1997).
The mechanism of cellulose and hemicellulose hydrolysis occurs via general acid catalysis and is accompanied by either an overall retention or an inversion of the configuration of the anomeric carbon (Davies and Henrissat 1995; McCarter and Withers 1994; White and Rose 1997; Withers 2001). In both cases, cleavage is catalyzed primarily by two active-site carboxyl groups. One of these acts as a proton donor and the other as a nucleophile or base. Retaining enzymes function via a double-displacement mechanism, by which a transient covalent enzyme-substrate intermediate is formed (Fig. 6.6a ). In contrast, inverting enzymes employ a single-step mechanism as shown schematically in Fig. 6.6b . The distance between the acid catalyst and the base represents the major structural difference between the two mechanisms. In retaining enzymes, the distance between the two catalytic residues is about 5.5 Å, whereas in inverting enzymes, the distance is about 10 Å. In the inverting enzymes, additional space is provided for a water molecule, involved directly in the hydrolysis, and the resultant product exhibits a stereochemistry opposite to that of the substrate. In all cases, the mechanism of hydrolysis is conserved within a given glycoside hydrolase family (Coutinho and Henrissat 1999a, b, c; Davies and Henrissat 1995; Henrissat and Davies 1997).


The two major catalytic mechanisms of glycosidic bond hydrolysis. (a) The retaining mechanism involves initial protonation of the glycosidic oxygen via the acid/base catalyst with concomitant formation of a glycosyl-enzyme intermediate through the nucleophile. Hydrolysis of the intermediate is then accomplished via attack by a water molecule, resulting in a product which exhibits the same stereochemistry as that of the substrate. (b) The inverting mechanism involves the single-step protonation of the glycosidic oxygen via the acid/base catalyst and concomitant attack of a water molecule, activated by the nucleophile. The resultant product exhibits a stereochemistry opposite to that of the substrate. The type of mechanism is conserved within a given glycoside hydrolase family and dictated by the active-site architecture and atomic distance between the acid/base and nucleophilic residues (aspartic and/or glutamic acids)
Carbohydrate-Binding Modules (CBMs)
In addition to the catalytic module, free cellulases and hemicellulases usually contain at least one carbohydrate-binding module (CBM) as an integral part of the polypeptide chain (Linder and Teeri 1997; Tomme et al. 1995b). The CBM serves predominantly as a targeting agent to direct and attach the catalytic module to the insoluble crystalline substrate. Like the catalytic modules, the CBMs are categorized into a series of families according to sequence homology and consequent structural fold.
For historical reference, until the year 2000 or so, the original term used in the literature for such substrate-binding modules was CBD, as an indication of cellulose-binding domain. However, CBD is deceptive, since not all of them bind to cellulose, and some families have members that bind to cellulose as well as other types of polysaccharides. It became clear that a more general term was required, and the term CBM (carbohydrate instead of cellulose, module instead of domain) was chosen and is clearly more appropriate on both counts.
Some CBM families (or subfamilies or family members) bind either preferentially or additionally to other insoluble polysaccharides, e.g., xylan or chitin. For example, the Family-5 CBM and some of the members of the Family-3 CBMs bind to chitin as well as cellulose (Brun et al. 1997; Morag et al. 1995). Moreover, the Family-2 CBMs can be divided into two subfamilies, one of which indeed binds preferentially to insoluble cellulose, but the other binds to xylan (Boraston et al. 1999). The molecular basis for this was proposed to reflect the fact that in the first subfamily, three surface-exposed tryptophans contribute to cellulose binding (Simpson et al. 1999; Williamson et al. 1999). However, in the case of the xylan-binding members, one of these tryptophans is missing, whereas the other two assume a different conformation, thereby allowing them to stack against the hydrophobic surfaces of two xylose rings of a xylan substrate.
Other types of CBM prefer less crystalline substrates (e.g., acid-swollen cellulose), single cellulose chains, and/or soluble oligosaccharides, e.g., laminarin (1,3-β-glucan) and barley 1,3/1,4-β-glucan (Tomme et al. 1996a, b; Zverlov et al. 2001). Still others exhibit alternative accessory function(s), a topic to be described below in more detail. Moreover, the CBMs responsible for the primary binding event may further disrupt hydrogen-bonding interactions between adjacent cellulose chains of the microfibril (Din et al. 1994), thereby increasing their accessibility to subsequent attack by the hydrolytic module.
Consequently, the concept of CBM has been broadened and redefined as CBM—i.e., carbohydrate-binding module (Boraston et al. 1999, 2004; Coutinho and Henrissat 1999a, b, c). In the previous edition of The Prokaryotes, more than a decade ago, 26 different CBM families were described. To date (March 2012), the number of CBM families have increased to 64 (http://www.cazy.org/). A CBM can be identified as a member of the family on the basis of sequence and position of binding residues before the binding function itself is established; nevertheless, it is imperative to confirm experimentally the specificity of binding of an individual CBM.
The structures of CBMs from a number of families and subfamilies have been determined, and an understanding of their structures has provided interesting information regarding the mode of binding to cellulose. Those that bind to crystalline substrates, appear to do so via a similar type of mechanism. One of the surfaces of such CBMs is characteristically flat and appears to complement the flat surface of crystalline cellulose. A series of aromatic amino acid residues on this flat surface form a planar strip (Mattinen et al. 1997; Simpson and Barras 1999; Tormo et al. 1996; Koyama et al. 1997; Lehtio et al. 2003; Ding et al. 2006) that stack opposite the glucose rings of a single cellulose chain. In addition, to the planar aromatic strip, several polar amino acid residues on the same surface appear to anchor the CBM to two adjacent cellulose chains. The binding of the CBM to crystalline cellulose would thus involve precisely oriented, contrasting hydrophobic and hydrophilic interactions between the reciprocally flat surfaces of the protein and the carbohydrate substrate. Together they provide a selective biological interaction, which contributes to the specificity that a CBM exhibits toward its structure. In some cases, the putative binding surface turns out to be irregular instead of flat, which may obstruct binding (Petkun et al. 2010).
In contrast to the interaction with the crystalline cellulose surface, other CBMs seem to interact with single cellulose chains. The Family-3c and Family-4 CBMs preferentially bind to non-crystalline forms of cellulose and clearly have a different function in nature (Johnson et al. 1996; Sakon et al. 1997; Tomme et al. 1996a, b). For example, the role of Family-4 CBM may be to recognize, bind to, and deliver an appropriate catalytic module to a cellulose chain, which has been loosened or liberated from a more ordered arrangement within the cellulose microfibril. The binding of the Family-3c CBM to single cellulose chains and its remarkable role in cellulose hydrolysis will be discussed later (Fig. 6.9 : section on “Helper Modules”). The role of the CBMs has been expanded recently from the conventional substrate targeting function to cell-surface attachment (Ezer et al. 2008; Montanier et al. 2009) and vital important biomass sensing functions leading to transcriptional regulation (Kahel-Raifer et al. 2010; Nataf et al. 2010; Bahari et al. 2011).
The Family-9 Cellulases: An Example
This section pertains to enzyme diversity and how a single type of catalytic module can be modified by the class of helper module(s) that flank its C- or N-terminus. We are only at the beginning in our understanding of how the modular arrangement affects the overall activity and function of a given enzyme.
In its simplest form, an enzyme would presumably consist of a single catalytic module, usually with a standard CBM, which would target the enzyme to the crystalline substrate. Indeed, this is the norm for many individual glycoside hydrolase families. However, in others, e.g., the Family-9 cellulases, the catalytic modules commonly occur in tandem with a number of accessory modules. Although the story remains rather incomplete, we can discuss the currently available information regarding Family 9 and draw several interesting conclusions from the few publications on this currently developing subject.
Family-9 Theme and Variations
The crystal structure of the Family-9 catalytic module displays an (α/α)6-barrel fold and inverting catalytic machinery. There are numerous Family-9 cellulases of plant origin (Coutinho et al. 2003a, b, c) the great majority of which are lone catalytic modules that lack accessory modules. Another type of eukaryotic Family-9 cellulase that lacks helper modules is produced by the termite (Ni et al. 2005). Only a few of the prokaryotic Family-9 enzymes consist of a solitary catalytic module (Fig. 6.7a ). The prokaryotic Family-9 enzymes, however, are almost invariably decorated with a variety of subsidiary modules that modulate the activity of the catalytic module.


Theme and variations: schematic view of some of the modular arrangements of the Family-9 glycoside hydrolases. (a) The solitary catalytic module; (b) the catalytic module and fused Family-3c CBM; (c) immunoglobulin-like (Ig) domain, fused to the catalytic module; (d) successive Family-4 CBM, Ig, and catalytic modules. The representations of the different modules are based on their known structures and are presented sequentially, left-to-right, from the N- to C-terminus. Structures in (a) and (b) are derived from cellulase E4 from Thermomonospora fusca (PDB code, 1TF4), those in (c) and (d) are from the CelD endoglucanase of C. thermocellum (PDB code, 1CLC). The Figure used for the Family-4 CBM in (d) is derived from the nmr structure of the N-terminal CBM of Cellulomonas fimi β-1,4-glucanase CenC (PDB code, 1ULO). The structures in (b) and (c) are authentic views of the respective crystallized bi-modular protein components. The CBM in (d) has been placed manually to indicate its N-terminal position in the protein sequence, but its spatial position in the quaternary structure and the structure of the linker segment remains unknown
Microbial Family-9 cellulases commonly conform to one of the themes shown in Fig. 6.7 , which were recognized in the previous edition. In one of these, the catalytic module is followed immediately downstream by a fused Family-3c CBM (Fig. 6.7b ). This particular type of CBM imparts special characteristics to the enzyme (see below). A second theme consists of an immunoglobulin-like (Ig) domain (of unknown function) immediately upstream to the catalytic module (Fig. 6.7c ). A variation of the latter theme includes a Family-4 CBM at the N-terminus of the enzyme, followed by an Ig domain and Family-9 catalytic module (Fig. 6.7d ). In addition to the above-described modular arrangement, each of the free prokaryotic enzyme systems includes a standard CBM that binds strongly to crystalline cellulose. In the last decade, several additional themes have been described, notably GH9-CBM3c′-CBM3b′ (i.e., a GH9 catalytic module followed by two successive subtypes of CBM3) with a C-terminal dockerin. This theme is present in the genomes of Clostridium thermocellum, Acidothermus cellulolytics, and Clostridium clariflavum.
The Family-9 glycoside hydrolase of the cellulosomal scaffoldin from the cellulolytic anaerobic bacterium Acetivibrio cellulolyticus contains no helper module (Ding et al. 1999). The A. cellulolyticus enzyme forms part of a multi-modular scaffoldin, but the catalytic module appears to be a functionally distinct entity that lacks adjoining helper modules. The other modules are conventional scaffoldin-associated modules, e.g., cohesins and a true cellulose-binding CBM.
This thematic arrangement of the Family-9 cellulases is mirrored in the respective sequences of the catalytic modules. The divergent sequences are reflected by the phylogenetic relationship of the parent cellulases (Fig. 6.8 ). Thus, the simplest cellulases (the Group A eukaryotic cellulases from plants) that lack adjacent helper modules are all phylogenetically related (Theme A). Interestingly, the catalytic module of ScaA from A. cellulolyticus is distinct from the other groups designated in Fig. 6.8 , but closest to the plant enzymes, as might be anticipated from its lack of a helper module. In a similar manner, catalytic modules from cellulases that are fused to a Family-3c CBM (Group B), all map within the same branch (Theme B). On the other hand, the catalytic modules that bear an adjacent Ig-like domain all fall into a cluster on the opposite side of the tree. Cellulases which have the Ig-like domain only (Theme C) occupy a small separate branch and those that also include a Family-4 CBM (Theme D) that develops distally to form a separate subcluster.


Phylogenetic analysis of the N-terminal Family-9 catalytic module of ScaA and its relationship with other Family-9 members. The various theme groupings roughly follow the groups shown in Fig. 6.7 . Theme A (Group A) enzymes lack associated helper modules. Theme B (Group B) enzymes carry a fused Family-3c CBM downstream to the catalytic module. Theme C (Group C) and Theme D (Group D) enzymes carry an Ig domain upstream to the catalytic module, the Theme D enzymes having an additional N-terminal Family-4 CBM. Theme A enzymes: ScaA Acece, ScaA scaffoldin from the cellulolytic bacterium A. cellulolyticus (AF155197); and plant (eukaryotic) cellulases from Prunus persica (X96853), Populus alba (D32166), Citrus sinensis (AF000135), Persea americana (M17634), Pinus radiata (X96853), Arabidopsis thaliana (X98543), Phaseolus vulgaris (M57400), Capsium annuum (X97189), Lycopersicon esculentum (U20590). Theme B enzymes: CelF Clotm, endoglucanase F from Clostridium thermocellum (X60545); CelZ Closr, exoglucanse Z from Clostridium stercorarium (X55299); CelA Calsa, cellulase A from Caldocellum saccharolyticum (L32742); CelG Cloce, endoglucanase G from Clostridium cellulolyticum (M87018); CelI Clotm, endoglucanase I from Clostridium thermocellum (L04735); CelB Celfi, endoglucanase B from Cellulomonas fimi (M64644); E4 Thefu, endo/exoglucanase E4 from Thermomonospora fusca (M73322). Theme C enzymes: CelJ Clotm, cellulase J from Clostridium thermocellum (D83704); CelD Clotm, endoglucanase D from Clostridium thermocellum (X04584); CelC Butfi, endoglucanase C from Butyrivibrio fibrisolvens (X55732). Theme D enzymes: CbhA Clotm, cellobiohydrolase A from Clostridium thermocellum (X80993); CelA Psefl, endoglucanase A from Pseudomonas fluorescens (X12570); CelC Celfi, endoglucanase C from Cellulomonas fimi (X57858); CelI Strre, endoglucanase I from Streptomyces reticuli (X65616); E1 Thefu, endoglucanase E1 from Thermomonospora fusca (L20094). The analysis of the designated catalytic modules was performed using GenBee, based on the respective GenBank sequences (accession codes in parentheses)


Structural aspects of Family-9 Theme-B cellulase E4 from T. fusca. (a) “Side view” of the E4 molecule. Shown are the Family-9 catalytic module (turquoise, on the left), the Family-3c CBM (in yellow, on the right) and the intermodular linker (dark blue strip). The presumed path of a single cellulose chain, from the CBM to the catalytic module, is shown at the bottom of the structure (arrows). The enzyme also possesses a fibronectin-like domain (FN3) and a cellulose-binding Family 2 CBM (not shown). Note that the linker appears to serve a defined structural role by which the Family-3c CBM is clamped tightly to the catalytic module. Selected surface residues on the catalytic module along the interface of both the linker and the CBM3c also serve to fasten both features tightly to the catalytic module. (b) “Bottom view” of the E4 molecule (∼90° rotation of a). From this perspective, the proposed catalytic residues (red), positioned in the active site cleft, are clearly visible. The path of the cellulose chain (arrows) passes through a succession of polar residues (green) on the bottom surface of the CBM which would conceivably bind to the incoming cellulose chain and serve to direct it toward the active-site acidic residues of the catalytic module
Family-9 Crystal Structures
Crystal structures of Family-9 cellulases have been elucidated, representing two subtypes of this particular family of glycoside hydrolase. These are cellulase E4 (or Cel9A) from Thermobifida fusca (Sakon et al. 1997) and Cel9D from Clostridium thermocellum (Juy et al. 1992). These two examples are architecturally distinct—the T. fusca Cel9A cellulase being an example of a Theme B Family-9 enzyme (see Figs. 6.7b and 6.8 ) and the C. thermocellum Cel9D cellulase being a Theme C enzyme. Fortunately, in both cases, one of the neighboring modules co-crystallized with the catalytic module, thus providing primary insight into their combined structures. In the case of T. fusca Cel9A, the catalytic module and neighboring Family-3c CBM were found to be interconnected by a long, rigid linker sequence, which envelops about half of the catalytic module until it connects to the adjacent CBM (Fig. 6.9a ). In contrast, in the C. thermocellum Cel9D, the catalytic module is adjoined at its N-terminus by a 7-stranded immunoglobulin-like (Ig) domain of unknown function. The comparison between the E4 and CelD cellulases indicates that a given type of catalytic module can be structurally and functionally modulated by different types of accessory modules.
Helper Modules
The Family-3c CBM is special. To date, this particular type of CBM has been found in nature associated exclusively with the Family-9 catalytic module. Structurally, the CBM is homologous to the other Family-3 CBMs, but contains substitutions in many important surface residues. The three-dimensional crystal structure of the T. fusca Cel9A cellulase revealed the close interrelationship between the Family-9 catalytic module and the Family-3c CBM, thus suggesting a functional role as a helper module. This CBM seems not to bind directly to crystalline cellulose but appears to act in concert with the catalytic module by binding transiently to the incoming cellulose chain, which is then fed into the active-site cleft pending hydrolysis (Fig. 6.9b ) (Gal et al. 1997a; Irwin et al. 1998; Sakon et al. 1997).
The information derived from the Family-9 enzymes suggests that the activity of catalytic modules can be modulated by accessory modules. The accessory modules can either supplement or otherwise alter the overall properties of an enzyme (Bayer et al. 1998b, c). The recurrent appearance in nature of a given type of module adjacent to a specific type of neighboring catalytic module may indicate a functionally significant theme. These observations raise the possibility of a more selective role for certain types of CBM and other modules, whereby their association with certain types of catalytic modules could signify a “helper” role. The helper module would provide hydrolytic efficiency and alter the catalytic character of the enzyme. Interestingly, in recent work on a Theme B enzyme, Cel9I from C. theromcellum (Burstein et al. 2009), recombinant forms of the individual GH9 catalytic module and CBM3c (together with the intermodular linker) were expressed individually, and the two modules underwent self-assembly to form a complex. Before complexation, the GH9 essentially lacked activity. Physical association of the two modules was shown to recover 60–70 % of the intact Cel9I endoglucanase activity.
Cellulase Analysis
The biochemical characterization of cellulases is in many cases a difficult task owing to the large variety of enzyme types and modes of action. At first glance, it is an intriguing phenomenon that for such a simple reaction (i.e., the hydrolysis of the β-1,4-glucose linkage in a linear glucose chain), nature has evolved so many types of cellulases. The vast varieties of enzymes are found not only among the different species of cellulolytic bacteria but also within the same organism. The reason for this extensive diversity comes from the insoluble nature of cellulose and the fact that although the chemical composition of the homopolymer is rather trivial, the physical and three-dimensional arrangement of the chains within the crystalline and amorphous regions of the microfibril can differ significantly.
Regarding the enzymes that degrade the substrate, the modular nature of the cellulases contributes additional degrees of complexity in our quest to characterize a given enzyme. Thus, the number, types, and arrangement of the accessory modules vis-à-vis the catalytic module are important structural features that modulate the overall activity of the enzyme in question. This descriptive information should always be defined for a recombinant enzyme. Whenever possible, it is desirable to determine the relative contribution of the individual accessory modules to the activity of the enzyme. In this regard, the affiliation of a given module, e.g., CBM, into a defined family does not necessarily define its contribution to enzyme activity, as different specificities and functions have been attributed to different members of the same family of module. Moreover, sequences for the different “X” modules (i.e., modules for which the function remains undefined) are widespread, most of which probably play a carbohydrate binding or processing role in assisting the catalytic module(s) in its capacity to hydrolyze the substrate.
Two decades ago, the range of cellulases and hemicellulases within a given species was assessed mainly by biochemical techniques. In some cases, individual enzymes were isolated and their properties assessed using desired insoluble or soluble substrates. Another approach involved electrophoretic separation of cell-derived or cell-free extracts, and analysis of desired activities using zymograms. There are advantages and disadvantages with each of these strategies, and the employment of combined complementary approaches is always advisable.
Molecular biology techniques are also used to reveal cellulase and hemicellulase genes, which can often be characterized on the basis of sequence homology with related, known genes (Béguin 1990; Hazlewood and Gilbert 1993) or according to their GH family membership (Table 6.2 ). If further information is required on the structure or action of a given enzyme, the gene can then be expressed in an appropriate host organism, and the properties of the product can be characterized.
It is always instructive to compare the properties of an expressed gene product with those of the same protein isolated from the original bacterial culture. The results may be surprising; there are hazards inherent to both approaches. Expression of a gene may yield preparations with reduced or altered enzymatic properties. In this context, the expressed gene product may not have been folded properly. It is of course assumed that the investigator has taken the time and trouble to sequence the cloned gene to ensure no mutations have occurred.
Unlike a gene expressed in a host cell environment, the native counterpart may have undergone posttranslational modifications (e.g., glycosylation, proteolytic truncation, etc.) that improve its physicochemical properties. Moreover, since the cellulase system in the native environment includes numerous enzyme types, often exhibiting similar molecular masses and other physical characteristics, the reputed purification of a given extracellular cellulase may still include contaminating enzymes that alter (usually increasing greatly due to synergistic action of two or more enzymes) the true enzymatic properties of the desired enzyme. The onus belongs to the conscience of the investigating scientist when publishing the properties of a given enzyme. Too often, erroneous data that enter the scientific literature are taken as fact. One should particularly be wary of comparing enzymatic activities of the same or similar types of enzymes (e.g., members of the same family) that have been published at different times and by different laboratories.
During the past decade, the phenomenal decrease in the costs associated with genomic and metagenomic sequencing efforts has completely altered accepted methodology for enzyme discovery. Today, sequencing of a cellulolytic microbe with concomitant bioinformatic annotation yields dozens and sometimes hundreds of new enzymes which can generally be included into the known families of glycoside hydrolases. The gargantuan efforts in establishing the CAZy database (http://www.cazy.org/) (Cantarel et al. 2009) have today provided the informed researcher with tools to determine the general features of a given enzyme. Nevertheless, researchers who seriously seek to understand more deeply the action of a newly discovered enzyme must perform the biochemical, structural, and enzymological studies in a meticulous manner.
The establishment of novel families – i.e., glycoside hydrolases as well as other carbohydrate-active enzyme superfamilies, requires much more intensive and elegant studies of this nature. This is particularly evident for many types of cellulases, where no simple colorimetric assays exist. In some cases, chromogenic substrates or assays are available and the detection of cellulolytic assays are, in this case, more straightforward; in others, the activity is much more subtle. This is reflected in the fact that most of the known glycoside hydrolase families which include genuine cellulase were identified early on, since their members were identified colorimetrically.
Since that time, new families of cellulases were difficult to establish, mainly due to the lack of a simple comprehensive assay or sets of assays that would definitively identify new types of cellulolytic activity. In the early 1990s, an important family was discovered that includes exoglucanases (glycoside hydrolase Family 48). The founding member of this family was a predominant component of the C. thermocellum cellulosome (Morag et al. 1991; Wang et al. 1993, 1994). Subsequent research has established that a member of this family is consistently a major component of each newly discovered cellulosome. In addition, members of this family have been discovered in both free and multifunctional cellulases.
Nearly two decades then passed until a new type of cellulase was discovered (Brás et al. 2011), which allowed formation of a new family (glycoside hydrolase Family 124). In this case, the actual cellulose-degrading function was somewhat cryptic and its detection required a combined approach until the enzyme could be verified as a cellulase.
Clearly, with continuing genomic and metagenomic sequencing, there are myriads of unknown and novel types of cellulases and other associated plant-derived polysaccharide-degrading enzymes that await future discovery. Novel, preferably medium- or high-throughput approaches will be required to promote this endeavor.
The assessment of cellulase activity is indeed a complicated undertaking, and there is no clear or standard methodology for doing so. This predicament apparently reflects a combination of factors, including the complex nature of the substrate, the multiplicity of enzymes and their synergistic action, and the variety of products formed. The fact that cellulose is an insoluble substrate converted to lower-order cellooligosaccharide products is a further complication. It must be noted that as the cellooligomers increase in length, they become less soluble, such that cellooctaose of eight glucose units is no longer soluble in aqueous solutions. Moreover, the accumulation of one (particularly cellobiose) or more of the cellulose degradation products may be inhibitory toward enzymatic activity.
Today, the study of cellulase action usually includes, in addition to conventional biochemical assays, the analysis of the primary structure and the assignment of the various modules into known families. The catalytic modules can usually be assigned into one of the known glycoside hydrolase families (Henrissat and Bairoch 1996; Henrissat and Davies 1997). Whenever the sequence of a known polysaccharide-degrading enzyme failed to match a known family, a new family of glycoside hydrolase was established. This approach was extensively developed in the last decade, due to the increasing number of available DNA sequences and bioinformatics analysis tools. At the same time, an increasing number of crystal or solution structures of various catalytic and accessory modules were published that allow us to examine a new protein sequence in light of its structure. Sometimes, the publication of the structure of an accessory module precedes determination of its function.
We can divide the analysis of a newly described prospective cellulase into several stages, such that a variety of complementary approaches are currently in use in order to classify the enzyme. Some of the questions one may ask are:
-
1.
What is the primary structure (the amino acid sequence) of the enzyme? What are the binding residues and/or binding module(s) associated with the enzyme? What are its other accessory modules and their respective role(s) in catalysis or stability?
-
2.
Is the enzyme a “true” cellulase, i.e., is its preferred substrate a cellulose or a cellulose-degradation product, and can the enzyme act alone on insoluble cellulose. This is to be distinguished from simple endoglucanases and exoglucanases and their activities on model substrates.
-
3.
What is the mode of action? Does the enzyme act as an endoglucanase, an exoglucanase, or a processive enzyme?
-
4.
What is the stereochemistry of the reaction? Does the enzyme exhibit an inverting or retaining mechanism?
-
5.
What are the catalytic residues: the acid/base residue and the nucleophile that characterize a glycoside hydrolase?
In the early years of cellulase research, several extensive reviews and book chapters dealt with different assays of cellulose degradation (Ghose 1987; Wood and Kellogg 1988). In this treatise, we will briefly summarize the various approaches currently in use and direct the reader to the relevant literature.
While characterizing the activity of a new enzyme preparation, one has to bear in mind several secondary or indirect issues, such as the purity of the protein preparation, the sensitivity of the assay used, and the cross-reactivity of the expected enzymatic activities. In some cases, only detailed kinetic analysis can provide appropriate characterization of the enzyme. As for many other types of glycoside hydrolases, cellulases can exhibit cross-reactivity with substrates of similar structure. This is particularly true when using, e.g., p-nitrophenyl derivatized substrates that provide highly sensitive assays. However, in many cases, such a soluble synthetic chromogenic substrate can fit the active-site pocket of a related but atypical enzyme, which catalyzes its hydrolysis. For example, Family-10 glycoside hydrolases are typically xylanases but individual members of this family can readily hydrolyze p-nitrophenyl cellobioside which is a typical cellulase substrate. Without a detailed comparative kinetic analysis (kcat/km) using different substrates, the true specificity of the enzyme might be overlooked.
Today, given the amino acid sequence of the protein, its assignment to a given glycoside hydrolase family can in many cases provide a reasonable general indication of its activity. The description of the modular structure provides additional knowledge that can imply how the catalytic function might be modulated, but this knowledge can also be misleading. In the final analysis, there is no substitute for extensive biochemical and biophysical characterization of the given protein (recombinant or native) and its catalytic properties. In case of a native enzyme, it is imperative to ensure that contaminating enzymatic activities have been removed. This is not a trivial undertaking. In the case of a recombinant form of an enzyme, it is imperative to ensure that the enzyme is correctly folded and the activity(ies) is (are) indicative of the parent protein. In the case of multi-modular enzymes, wherein the ancillary modules may alter the character of the catalytic module, again, these efforts are nontrivial.
General procedures for assaying for cellulase and hemicellulase activities are very well documented in the Methods in Enzymology Volume 160 (Wood and Kellogg 1988) and a new Volume of this series is forthcoming (Gilbert, 2012). Conventional procedures for cellulase assay have been defined precisely by IUPAC (Ghose 1987). However, due to the complexity of the substrate and enzyme systems, these procedures can only provide a starting point for understanding the true nature of the enzyme in question.
Since the publication of Part A of this treatise (Coughlan and Mayer 1992), many of the previously reported assays of cellulase activity are still in common use. These include the use of soluble, derivatized forms of cellulose, e.g., carboxymethyl cellulose and hydroxymethyl cellulose, as conventional substrates for determining endoglucanase activity. In addition, a derivatized, colored form of insoluble cellulose, i.e., azure cellulose, is frequently used as an indication of cellulase activity. Zymograms with such colored embedded substrates are useful in detecting endoglucanase or xylanase activities (Béguin 1983). Individual soluble cellooligomers (cellotetraose, cellopentaose, cellohexaose, etc.) are still used as substrates for analyzing enzyme action, but the reliance on these substrates as determinants for assessing cellulase activity is no longer a definitive approach. Substrate analogues and reagents were developed that include the use of thioglycoside substrates (Driguez 1997), fluoride-derivatized sugars (Williams and Withers 2000), and chromophoric and fluorescent cellooligosaccharides (Claeyssens and Henrissat 1992; O’Neill et al. 1989; van Tilbeurgh et al. 1985). An ultraviolet-spectrophotometric method and an enzyme-based biosensor have also been described (Bach and Schollmeyer 1992; Hilden et al. 2001). In addition, a novel and intriguing bifunctionalized fluorogenic tetrasaccharide has been developed as an effective reagent for measuring the kinetic constants of cellulases by resonance energy transfer (Armand et al. 1997).
The thiooligosaccharides serve as competitive inhibitors that mimic natural substrates but are enzyme resistant (Driguez 1997). In this type of oligosaccaride, the oxygen of a bond to be cleaved is replaced by sulfur. The thiooligodextrins are sometimes more soluble than the native cellodextrins and longer chains can be synthesized. The modified sugars can be used in biochemical studies or crystallographic studies to gain some information about the geometry of the active site or determine the mechanism of action of an enzyme.
Determination of “True” Cellulase Activity: Solubilization of Crystalline Cellulose Substrates
True cellulase activity is usually defined as the ability to solubilize to an appreciable degree insoluble, “crystalline” forms of cellulose. The extent of hydrolysis can be evaluated by turbidity assays, weight loss of insoluble material, generation of reducing power, and accumulation of soluble sugars. It is important to realize that crystalline cellulose is not of uniform composition and therefore the rate of catalysis is in most cases not linear with time or enzyme concentration. Notably, the different preparations of crystalline cellulose contain varying levels of loosely associated loops and chains. The latter are readily accessible to hydrolysis by a given enzyme and lead to relatively high initial rates of activity, which do not reflect the actual degree of true cellulase activity. For example, such loose chains can be degraded by a relatively ineffectual enzyme, whereas the crystalline portions of the substrate will be immune to further hydrolysis by the same enzyme. To overcome these difficulties, IUPAC suggests determining the amount of enzyme required to achieve digestion of 5.2 % of the insoluble substrate (e.g., filter paper) in 16 h (Ghose 1987; Irwin et al. 1993).
Cellulose substrates commonly in use include Avicel, filter paper, cotton, Solka Floc, as well as bacterial cellulose from Acetobacter aceti and algal cellulose prepared from Valonia. Consequently, these assays should be treated as a relative and not quantitative assessment. The nature of the original substrate selected—especially its extent of crystallinity—should always be taken into account. Proper controls and reference substrates should always be used. One should be wary about comparison among results reported by different laboratories and even by different researchers in the same laboratory. Nevertheless, such assays give an excellent indication of whether a given enzyme preparation exhibits substantial activity toward crystalline cellulose substrates.
Endoglucanase Versus Exoglucanase Activity
As discussed earlier in this chapter, the cellulases have traditionally been divided into either endoglucanases or exoglucanases (Fig. 6.4 ). The biochemical or enzymatic assays that discriminate between these two modes of action usually involve soluble forms of cellulose, i.e., carboxymethyl or hydroxymethyl derivatives of cellulose. The action of a given enzyme on these substrates is followed by determining the amount of reducing ends generated by the enzyme and the degree of polymerization (DP). The reducing power is usually determined either by using reagents such as 3,5-dinitrosalicylic acid (DNS) (Miller et al. 1960), ferricyanide (Kidby and Davidson 1973), or copper-arseno molybdate (Green et al. 1989; Marais et al. 1966).
Despite their traditional popularity, these two methods are intrinsically disadvantageous, owing to interference by metal ions and certain buffers. Moreover, such assays are sensitive to the chain length of the reducing end. A more recent approach involves the use of disodium 2,2′-bicinchoninate (BCA) for determination of reducing sugar. This procedure is more sensitive than the conventional methods and gives comparable values of reducing sugars for cellodextrins of different lengths (Doner and Irwin 1992; Garcia et al. 1993; Vlasenko et al. 1998; Waffenschmidt and Jaenicke 1987).
Viscosity-based measurements represent the most common approach for assessing the degree of polymerization. This approach is highly sensitive for internal bond cleavage, which leads to significant reduction of the average molecular weight of the substrate. The comparison between the amount of reducing sugars generated and the average molecular weight (i.e., viscosity or fluidity of the soluble cellulose substrate) gives a very good indication whether an enzyme is essentially exo- or endo-acting.
The average degree of polymerization can also be evaluated by size-exclusion chromatography either alone (Srisodsuk et al. 1998; Teeri 1997) or combined with multi-angle laser light scattering (Vlasenko et al. 1998). Mass spectrometric procedures can also be applied to determine the identity and distribution of degradation products following hydrolysis of cellulosic substrates by an enzyme (Hurlbert and Preston III 2001; Rydlund and Dahlman 1997). The mode of enzymatic action can also be appraised by determining the increase in reducing power associated with the insoluble versus the soluble fraction of the substrate. Increase in the proportion of reducing sugars associated with the soluble fraction indicates an exo type of activity whereas a relatively large increase in the insoluble fraction would suggest an endo type of activity (Barr et al. 1996).
Exocellulases can exhibit different specificities depending on their preference for the reducing or nonreducing end of the cellulose chain (Barr et al. 1996; Teeri 1997). This feature of an exocellulase can be determined either by using oligosaccharide substrates labeled by tritium or 18O at the reducing end. Other procedures involve NMR, HPLC, and/or mass spectrometric analysis of products released from native (unlabeled) cellooligosaccharides. In previous studies, the 3D structures of enzyme-substrate complexes have been obtained, and the specificities of the enzyme can be interpreted directly from the data (Davies and Henrissat 1995; Davies et al. 1998; Divne et al. 1998; Juy et al. 1992; Notenboom et al. 1998; Parsiegla et al. 1998; Rouvinen et al. 1990; Sakon et al. 1997; Zou et al. 1999). These efforts have since continued as novel families of the glycoside hydrolases were established; selected members of these families were subjected to crystallization studies in order to characterize the overall structural features and mode of action of the entire family.
Processivity
One of the major recent conceptual advances in assessing the mode of enzymatic action of a cellulase is the concept of processivity. Processive enzyme action can be defined as the sequential cleavage of a cellulose chain by an enzyme. In effect, exoglucanases are by nature and structure processive enzymes. Their tunnel-like active site thus allows processive action on the cellulose chain. Endoglucanases, however, were thought to be intrinsically non-processive. However, the traditional distinction between exo- and endo-cellulases has been modified.
Experiments combining two or more purified cellulases have shown that synergism can even be detected upon mixing two different types of exo-acting enzymes. Such experiments led to the recognition that the exo enzymes can operate on both ends (i.e., the reducing and nonreducing ends) of the cellulose chain. Some enzymes, however, exhibit both endo and exo activities, although in such cases the endo-cellulase activity is usually very low. In attempts to explain these phenomena, the concept of processivity was proposed, by which the activity of the enzyme is characterized by the sequential hydrolysis of the cellulose chain. Implicit in this concept is the notion that the catalytic site of the enzyme remains in continual and intimate contact with a given chain of the cellulose substrate.
A more complete mechanistic picture of the processive nature of such cellulases was revealed with the advent of high-resolution 3D structures. It was thus demonstrated that the cellulose chain makes contact with the protein at multiple sites, either via a tunnel-shaped structural element (such as that observed in the Family-48 enzymes) or by a special type of CBM (such as the Family-9 Theme B cellulases). These arrangements allow the threading of the cellulose chain into the active site, and, following initial cleavage at the end of the chain, the enzyme can move along the chain and position itself for the next cleavage. In addition to this processive nature of the active site, these enzymes can also make classic endo cleavages thus generating new ends.
Biochemically processive enzymes exhibit characteristics between endo and exo enzymes. They have low but detectable endo activity toward soluble derivatives of cellulose (i.e., CMC), and may or may not possess exo activity on such substrates. With insoluble substrates, they will generate reducing power with a ratio between the soluble to the insoluble fractions of about 7. Endocellulases usually give a ratio of less than 2, whereas exocellulases produce a ratio of 12–23 (Irwin et al. 1998).
Once the processive nature of an enzyme has been indicated experimentally, molecular insight into the mechanisms responsible for this feature can be gained by determining the 3D crystal structure of the active site together with model cellodextrins. In the case of the cellulases, the crystal structure of the catalytic module together with the fused CBM, combined with accumulating enzymatic activity data, allowed further postulation as to the accessory role of the fused module. The fused CBM presumably interacts with a single cellulose chain and feeds it into the active site. Interestingly, this module does not bind crystalline cellulose, but is inferred to act in dynamic binding of the single cellulose chain prior to its hydrolysis, thereby imparting the quality of processivity to the enzyme. Once such a property is associated with a given type of enzyme, the primary structure of the protein can now be used as an indication for all such enzymes. In the case of the Family-9 Theme B enzymes, it is now possible to identify the catalytic module (e.g., glycoside hydrolase Family 9) and the additional accessory modules (in this case, Family-3c CBM). Thus, the primary structure may by itself give a strong indication of the nature of the enzyme itself. Of course, the ultimate identification as to the mechanism of enzyme activity will come from detailed 3-D structure of the enzyme-substrate complex.
An intriguing recent development in the analysis of the cellulolytic action of a given cellulase or a mixture of cellulase is the direct transmission electron microscopic (TEM) observation of the enzymatic action on bacterial cellulose ribbons. The approach provides information as to the endo or exo preference of the enzyme, the extent of processivity, as well as the directionality of hydrolysis (i.e., from the reducing to the nonreducing ends or vice versa). This strategy has been used to study the hydrolysis of bacterial cellulose ribbons by individual purified enzymes, mixtures of purified enzymes, and intact cellulosomes.
Mechanism of Catalysis
The mechanism of catalysis of cellulases address issues such as stereochemistry, binding- and active-site residues, and transition state intermediates. Excellent reviews have been published covering many of these issues (Ly and Withers 1999; McCarter and Withers 1994; Rye and Withers 2000; Sinnott 1990; White and Rose 1997; Withers 2001; Withers and Aebersold 1995; Zechel and Withers 2000). The fact that the stereochemistry and catalytic residues are conserved between members of the same family allows the putative identification of these elements if one member of the given (glycoside hydrolase) has been characterized biochemically (Henrissat and Bairoch 1996; Henrissat et al. 1995; Henrissat and Davies 1997).
The stereochemistry of the reaction can in most cases be determined by proton NMR spectroscopy or by using chromatography systems that allow the resolution of anomeric species. In the case of NMR, the reaction between the test enzyme and its substrate is carried out in D2O and the appearance of the anomeric proton can be easily detected. Thus, for the degradation of cellulose, a retaining enzyme would produce a product in the β configuration whereas an inverting enzyme would yield the α-sugar.
The catalytic residues can be identified by performing site-directed mutagenesis on conserved acidic residues and studying the catalytic properties of the mutants with substrates bearing different leaving groups. Commonly used phenol substituents include the following, listed in order of leaving group ability (pKa values shown parenthetically): 2,4-dinitro (3.96) > 2,5-dinitro (5.15) > 3,4-dinitro (5.36) > 2-chloro4-nitro (5.45) > 4-nitro (7.18) > 2-nitro (7.22) > 3,5-dichloro (8.19) > 3-nitro (8.39) > 4-cyano (8.49) > 4-bromo (9.34) (Tull and Withers 1994). In retaining enzymes, the nucleophilic residue can be identified directly by trapping the intermediate with an appropriate inhibitor. Such inhibitors include model saccharides containing a fluorine substituent in the 2- or 5-position and a good leaving group, such as fluoride or dinitrophenolate (Williams and Withers 2000). The substituted substrate forms a relatively stable covalent substrate-enzyme complex, involving the nucleophile residues. The complex is then subjected to proteolytic cleavage and sequencing of the glycosylated peptide. The use of protocols involving combined liquid chromatography and mass spectrometry has facilitated the identification of the modified residues.
The acid-base residue in a retaining enzyme can be identified by a combination of kinetics-based methodologies. Mutation of this residue (usually to alanine) should affect the rate of both chemical steps, i.e., glycosylation and deglycosylation, though the effect on each step should be different. The effect on the glycosylation step will depend strongly on the leaving group ability of the aglycon. Thus, rates of hydrolysis for substrates with a poor leaving group should be affected much more strongly than those for substrates with a good leaving group. The deglycosylation step, however, will be affected equally for all substrates carrying different leaving groups, because the same glycosyl enzyme intermediate is hydrolyzed during this step. Thus, detailed kinetic analysis (i.e., determination of k cat and Km) with substrates bearing different leaving groups can reveal whether the corresponding mutation is the acid-base residue. It should be noted that this approach requires synthetic substrates that are not necessarily recognized by all families of enzymes and are not necessarily commercially available. For example, the Family-11 xylanases fail to hydrolyze p-nitrophenyl xylobioside, which is an excellent substrate for the Family-10 xylanases. The assignment of the acid-base catalyst can also be examined by use of external nucleophilic anions, such as azide. In this approach, termed “azide rescue,” the small azide anion enters the vacant space created by alanine replacement of the acidic amino acid residue. The azide reacts with the anomeric carbon instead of a water molecule to form the corresponding β-glycosyl azide product. In the absence of an acid-base catalyst, which normally provides general base catalysis during the second step, the deglycosylation step is severely affected. Thus, the acceleration of the reaction by the mutant enzymes in the presence of these external anions (provided that the second step is rate limiting) is a good indication that a mutant residue is the acid-base catalyst. Finally, the assignment of the acid-base catalyst can be tested by comparing the pH-dependence profiles for the wild-type and mutant enzymes. The profile for the native enzyme would approximate a perfect bell-shape curve, reflecting the ionization of the two active site carboxylic acids, whereas the no reduction of activity at high pH values would be observed for the mutant. This pH dependency approach is also applicable for identifying the nucleophile residues and the catalytic residues in inverting enzymes.
Prokaryotic Cellulase Systems
The cellulolytic bacteria produce a variety of different cellulases and related enzymes, which together convert the plant cell wall polysaccharides to simple soluble sugars that can subsequently be assimilated. The complement of cellulases and hemicellulases that are synthesized by a given bacterium for this purpose is referred to as its cellulase system. Different bacteria exploit different strategies for the ultimate degradation of their substrates. The given strategy is reflected by the complement and type(s) of enzymes produced by a given bacterium. The bacterial cellulase system may be characterized by free enzymes, cell-bound enzymes, multifunctional enzymes, cellulosomes, or any combination of the latter. Collectively, these four types of enzymes represent the major paradigms of plant cell wall polysaccharide-degrading enzymes (Himmel et al. 2010).
Cellulase enzyme systems are comprised of several different types of components, each type may exist in a multiplicity of forms. To add to the complexity, the same component may exist as free individual entities in the culture fluid, as individual entities bound to cellulose, or associated with the cell surface. Alternatively, an individual component may be organized as part of a multicomponent cellulosome complex attached to the cell surface, to the cellulose, to both, or as free complexes in the culture fluid. Furthermore, the situation existing during growth under one set of conditions (e.g., pH, temperature, distribution of carbon source) may not exist under another, or may change considerably during the course of cultivation. The bacterium reacts to these changes and its production of cellulases and/or cellulosomes may reflect the dynamics of the growth conditions.
Free Enzymes
As mentioned earlier in this chapter, the free enzymes in their simplest form comprise a catalytic module alone with no accessory modules. Such enzymes often specialize on degrading soluble oligosaccharide breakdown products. Alternatively, such single-modular enzymes may rely on an intrinsic association with insoluble polysaccharide substrate such as cellulose, perhaps related to the active site of the enzyme.
A higher-order level of organization and activity are free enzymes composed of a polypeptide chain that includes both a catalytic module together with a CBM. This basic bi-modular arrangement can be further extended by the inclusion of additional types of modules or repeating units of the same module, all of which serve to modulate the activity of the catalytic module on the substrate. The intact free enzyme, however, remains unattached to other enzymes and can work in an independent manner on a given substrate. Free enzymes containing larger numbers of ancillary modules are also prevalent in components of bacterial cellulase systems.
Examples of bacteria that possess free carbohydrate-degrading enzymes include the well-established actinomyces, Thermobifida fusca and Cellulomonas fimi. More details of their enzyme systems will be presented in a forthcoming section.
The more recent discovery of Saccharophagus degradans 2-40 has provided a particularly intriguing and elaborate cellulolytic bacterium that can grow alone on cellulose without the assistance of other microorganisms. S. degradans 2-40 is the first free-living marine bacterium demonstrated to be capable of degrading cellulosic algae and higher plant material, and its genome codes 15 extraordinarily long polypeptides, ranging from 274 to 1,600 kDa (Weiner et al. 2008). This bacterium has a remarkable range of catabolic capabilities, and many of the enzymes exhibit unusual modular architectures including novel combinations of catalytic and substrate-binding modules. S. degradans 2-40 can degrade different complex polysaccharides (at least 10), including agar, chitin, alginic acid, cellulose, β-glucan, laminarine, pectin, pullulan, starch, and xylan and utilize them as sole carbon and energy sources (Ensor et al. 1999).
The genome of S. degradans encodes abundant glycoside hydrolases families mainly GH5 (20 in number) followed by GH43 (13), GH13 (10), GH16 (9), GH2 (7), and GH3 (6) (Taylor et al. 2006). The CAZymes of this bacterium are generally extracellular free enzymes, many of which are decorated with at least one CBM. One of the GH5 enzymes is believed to exhibit processive endoglucanase activity and contains two such catalytic modules together with three copies of family-6 CBMs in the same polypeptide chain (Watson et al. 2009). Interestingly, the chitinases, agarases, and alginases produced by this bacterium are not exported into the extracellular matrix but are localized in surface protuberances, resembling those of the cellulosome-producing bacteria.
The genome of S. degradans encodes the largest set of identifiable CBMs so far reported (Weiner et al. 2008). Carbohydrate binding modules of Family 6 (CBM6) are the most numerous (43 copies) followed by CBM 32 (26) and CBM2 (19). Among the long polypeptides encoded by S. degradans 2-40 genome, five of them contain at least 52 bacterial cadherin (CA) and cadherin-like (CADG) domains. Both domain types exhibit Ca2+-dependent binding to different complex polysaccharides which serve as growth substrates (Fraiberg et al. 2010, 2011).
Recent evidence suggests that the regulatory mechanisms that control the expression of the various enzymes of the cellulolytic system are very complex and contain an intricate chemotaxis signal transduction network for detecting both extracellular and intracellular signals and numerous chemotactic response regulators (Zhang and Hutcheson 2011).
Multifunctional Enzymes
Some cellulases exhibit a more complex architecture in that more than one catalytic module and/or CBM may be included in the same protein. Examples of such enzymes are the very similar cellulases from Anaerocellum thermophilum (Zverlov et al. 1998) and Caldocellum saccharolyticum (Te’o et al. 1995), both of which contain a Family-9 and a Family-48 catalytic module. Additional examples of the latter type of multifunctional enzyme have been found in A. cellulolyticus and C. clariflavum. Other paired catalytic modules include those from Family 44 and either Family 5 or 9. Such an arrangement might indicate a close cooperation between two particular catalytic modules, which may lead to synergistic action on the cellulosic substrate, thus portending on a smaller scale the advent of cellulosomes.
Like the cellulases, xylanases also tend to exhibit a modular structure, being composed of multiple modules joined by linker sequences. Family-10 and Family-11 xylanases may be linked in the same polypeptide chain either to each other, to catalytic modules from Families 5, 16, and 43, or to carbohydrate esterases (Flint et al. 1993; Laurie et al. 1997). One particularly interesting combination of multifunctional catalytic modules that appear in the same polypeptide chain is a typical xylanase together with a feruloyl esterase. Such a combination would allow the rapid cleavage of hemicellulose from the lignin in natural systems, i.e., the plant cell wall (see Fig. 6.3 ). In this manner, the xylan chain would be severed by the xylanase component (Xyn in Fig. 6.3 ) and the lignin-xylan association would be disconnected simultaneously by the feruloyl acid esterase (Fae in Fig. 6.3 ).
Indeed, some xylanases are extremely complex in their modular architecture (Fig. 6.10 ). In addition to multiple catalytic modules, these enzymes often contain several different types of CBMs. Why would such a xylanase contain several types of CBM? And why would a xylanase contain a cellulose-specific CBM? Unlike the case of various cellulases, for which the CBM is usually essential for degrading insoluble crystalline cellulose, the CBMs of a hemicellulase do not necessarily bind the hemicellulose component (xylan). In some cases, its CBM is in fact an authentic CBM that situates the hemicellulase on the insoluble plant cell wall material by utilizing the most abundant and most stable cell-wall component—cellulose. Indeed, the three Family-3 CBMs (CBM3) shown in Fig. 6.10 apparently bind to crystalline cellulose. Why would this xylanase require three tandem copies of the same type of CBM is yet another mystery that should eventually be addressed experimentally. At any rate, once bound via the cellulose component of the plant cell wall composite substrate, the immobilized enzyme then acts on the accessible and appropriate hemicellulose components. Once thus situated on the plant cell wall, another type of CBM on the same molecule would then assist in the binding to the xylan (or mannan, etc.) component in order to direct the appropriate catalytic module to its true substrate. Hence, the modular proximity of the xylanase shown in Fig. 6.10 would presumably indicate that the two CBM22 would modulate the action of the Family-10 catalytic module, and the C-terminal CBM6 would facilitate the catalysis by the Family-43 module. Together, the two catalytic modules would act synergistically to degrade susceptible plant cell wall components. In this context, the complex architecture of a xylanase would reflect the complex chemistry of its substrate and the neighboring polymers of its immediate environment in the plant cell wall.


A very large, cell-surface enzyme from Thermoanaerobacter thermosulfurogenes. The 1861-residue enzyme contains an SLH module, which is believed to mediate the attachment of the enzyme to the cell surface in Gram-positive bacteria. The enzyme contains a multiplicity of modules, which apparently serve to regulate the hydrolytic action of its single Family-13 catalytic module with the complex substrate. Several X domains of unknown function may either represent as yet undescribed catalytic functions, carbohydrate-binding activities, or structural entities
Cellulosomes
Cellulosomes are multienzyme complexes, which bind to and catalyze the efficient degradation of cellulosic substrates. The first cellulosome was discovered while studying the anaerobic thermophilic bacterium, Clostridium thermocellum (Bayer et al. 1983; Lamed et al. 1983a, b). Since its initial description in the literature, the cellulosome concept has been subject to numerous reviews (Bayer et al. 1996; Béguin and Lemaire 1996; Belaich et al. 1997; Doi et al. 1994; Doi and Tamura 2001; Felix and Ljungdahl 1993; Karita et al. 1997; Lamed and Bayer 1988a, b, 1991, 1993; Lamed et al. 1983; Shoham et al. 1999).
Cellulosomes in C. thermocellum exist in both cell-associated and extracellular forms, the cell-associated form being associated with polycellulosomal protuberance-like organelles on the cell surface. Later, cellulosomes were detected in other cellulolytic organisms (Lamed et al. 1987a, b; Mayer et al. 1987), including Acetivibrio cellulolyticus, Bacteroides cellulosolvens, Clostridium cellulovorans, and Ruminococcus albus, all of which contained protuberance-like organelles on their surfaces (Bayer et al. 1994; Lamed and Bayer 1988) (Fig. 6.11 ). The role of surface functions was further shown to be important in increasing the efficiency of cellulose fermentation (Lu et al. 2006).


A very large, multi-modular xylanase from Caldicellulosiruptor. The 1795-residue enzyme contains 8 separate modules, including 2 catalytic modules from Family-10 (invariably a xylanase) and Family-43 (frequently an arabinofuranosidase). These are modulated by numerous CBMs, which include three from Family-3 (likely for binding to crystalline cellulose), two from Family-22 (shown to function in xylan binding) and one from Family-6
The cellulosomes contain numerous components, many of which were shown to display enzymatic activity. They also contain a characteristic nonenzymatic high-molecular-weight component. This component proved to be highly antigenic and glycosylated (Bayer et al. 1985). The cellulosomal enzymatic subunits from this organism showed a broad range of different cellulolytic and xylanolytic activities (Morag et al. 1990). Ultrastructural evidence indicated the multi-subunit nature of the cellulosome (Fig. 6.12 ).


Scanning electron microscopy (SEM) of Acetivibrio cellulolyticus showing the presence of large characteristic protuberance-like structures on the cell surface. Cells are shown in the free state (a) or bound to cellulose (b). Cell preparations were treated with cationized ferritin before processing. Cationized ferritin has been shown to stabilize such surface structures, thus allowing their ultrastructural visualization (Lamed et al. 1987a, b). Without pretreatment with cationized ferritin, these structures are invisible. In (b), the cellulose-bound cells appear to be connected to the substrate via structural extensions of the cell-surface protuberances. Such a mechanism was originally observed for other cellulolytic prokaryotes, notably C. thermocellum (Bayer and Lamed 1986)
Eventually, genetic engineering techniques led to the sequencing of cellulosomal genes in C. thermocellum and several other bacteria, thus confirming the existence of cellulosomes as a major paradigm of prokaryotic degradation of cellulose and related plant cell wall polysaccharides. These efforts were further extended with the genome sequences of various Clostridia and Ruminococci species.
Cell-Bound Enzymes
Some enzymes are connected directly to the cell wall. In Gram-positive bacteria, this is frequently accomplished via a specialized type of module, the SLH (S-layer homology) module, previously shown to be associated with the cell surface of Gram-positive bacteria (Lupas et al. 1994). This arrangement may have evolved to provide a more economic degradation of insoluble substrates and to reduce competition with other bacteria for the soluble products, subject to diffusion in the media. As opposed to free enzymes, diffusion of an attached enzyme would itself be prevented.
Examples of enzymes, which are bound to the cell surface via an SLH module include, a Family-5 cellulase and a Family-13 amylase-pullulanase from Bacillus, a Family-10 xylanase from Caldicellulosiruptor (Saul et al. 1990), a Family-5 endoglucanase from Clostridium josui, a Family-16 lichenase and a Family-10 xylanase from Clostridium thermocellum (Jung et al. 1998), and a variety of enzymes (Family-10 xylanases, a Family-5 mannanase, and a Family-13 amylase-pullulanase) from different species of Thermoanaerobacter (Matuschek et al. 1996). The modular architecture of these enzymes may be particularly complicated, containing several different modules in a single polypeptide chain, thus forming extremely large enzymes sometimes comprising over 2,000 amino acids (Fig. 6.13 ). Other surface functions, such as adhesive properties, may also be associated with the same protein (Fraiberg et al. 2011; Ozdemir et al. 2012).


Comparison between negative staining (bottom) and cryo images (top) of the purified cellulosome from C. thermocellum, adsorbed on cellulose microcrystals from the algae Valonia ventricosa. The images illustrate the diversity of shapes of the cellulosomes, which adopt either compact or loosely organized ultrastructure. In the cryo images, the subunits of the cellulosomes (i.e., the individual enzymatic components) are clearly visible (Micrographs courtesy of Claire Boisset and Henri Chanzy (CNRS — CERMAV, Grenoble, France))
In a different bacterium, Ruminococcus flavefaciens, from the rumen of herbivores, the cellulosome is attached covalently to the bacterial cell surface by scaffoldin E (ScaE) (Rincon et al. 2005). ScaE is an anchoring scaffoldin that includes a C-terminal cell-anchoring signal motif for covalent attachment to the cell wall via the enzymatic action of an appropriate cell-associated sortase. Two key scaffoldins, scaffoldin B (ScaB) and cellulose-binding protein A (CttA), are attached to the cell-surface ScaE scaffoldin via a C-terminal X-dockerin (XDoc) modular dyad. ScaB is essentially an adaptor scaffoldin to which scaffoldin A (ScaA) and/or selected dockerin-bearing enzymes, including cellulases, are incorporated into the R. flavefaciens cellulosome. CttA is believed to mediate the attachment of the bacterium to cellulosic substrates (Rincon et al. 2007). Genome sequencing revealed several other structural proteins that include sortase signal motifs (Berg Miller et al. 2009; Rincon et al. 2010). In one case, a GH10 xylanase was sequenced that also bears a sortase signal motif at its C terminus.
An additional mechanism of bacterial surface attachment has recently been reported (Devillard et al. 2004; Xu et al. 2004; Ezer et al. 2008). Several CBMs were discovered following genomic sequencing of the rumen bacterium, R. albus. These CBMs were classified as Family-37 and found exclusively in R. albus. Half of the parent proteins are carbohydrate-acting enzymes (glycoside hydrolases, pectate lyases, and carbohydrate esterases). The involvement of CBMs in anchoring plant cell wall–degrading enzymes onto the bacterial cell surface extends the types of functions that this superfamily of protein modules performs in nature.
Clostridium thermocellum Cellulosomal Subunits and Their Modules
A simplified schematic view of the cellulosome from C. thermocellum and its interaction with its substrate is shown in Fig. 6.14 . The cellulosomal enzyme subunits are united into a complex by means of the primary scaffoldin subunit (Bayer et al. 1994; Shoseyov et al. 1992; Fujino et al. 1993; Gerngross et al. 1993). The scaffoldins usually contain a Family-3 CBM that provides the cellulose-binding function (Poole et al. 1992). The scaffoldins also contain multiple copies of a definitive type of cohesin module. The cellulosomal enzyme subunits, on the other hand, contain a complementary type of dockerin module. The interaction between the cohesin and dockerin modules provides the definitive molecular mechanism that integrates the enzyme subunits into the cellulosome complex (Salamitou et al. 1994b; Tokatlidis et al. 1991, 1993). Cohesin and dockerins are considered to be cellulosome “signature sequences”—i.e., their presence is a good indication of a cellulosome in a given bacterium (Bayer et al. 1998a). This has indeed been confirmed in many cases. However, non-cellulosomal cohesins and dockerins have been identified in many bacteria, as well as archaea and a few isolated cases of primitive eukarya, without a link to polysaccharide degradation (Bayer et al. 1999; Adams et al. 2008; Chitayat et al. 2008a, b; Peer et al. 2009; Voronov-Goldman et al. 2009).


Simplified schematic view of the molecular disposition of the cellulosome and one of the associated anchoring scaffoldins on the cell surface of C. thermocellum. The key defines the symbols used for the modules, from which the different cellulosomal proteins are fabricated. The progression of cell to anchoring scaffoldin to cellulosome to cellulose substrate is illustrated. The SLH module links the parent anchoring scaffoldin to the cell. The cellulosomal scaffoldin subunit performs three separate functions, each mediated by its resident functional modules: (1) its multiple type-I cohesins integrate the cellulosomal enzymes into the complex via their resident type-I dockerins, (2) its Family-3a CBM binds to the cellulose surface, and (3) its type-II dockerin interacts with the type-II cohesin of the exocellular anchoring scaffoldin
The major difference between free enzymes and cellulosomal enzymes is that the free enzymes usually contain a CBM for guiding the catalytic module to the substrate, whereas the cellulosomal enzymes carry a dockerin module that incorporates the enzyme into the cellulosome complex. Otherwise, both the free and cellulosomal enzymes contain very similar types of catalytic modules. The cellulosomal enzymes rely on the Family-3a CBM of the scaffoldin subunit for collective binding to crystalline cellulose.
The incorporation of the multiplicity of enzyme subunits into the cellulosome complex is a function of the repeated copies of the cohesin module borne by the scaffoldin subunit. For most species of scaffoldin, the cohesins have been classified as type-I on the basis of sequence homology. The cohesin module is composed of about 150 amino acid residues. The basic structure of the cohesin is known and comprises a nine-stranded beta sandwich with a jelly-roll topology (Shimon et al. 1997; Spinelli et al. 2000; Tavares et al. 1997).
The dockerin module contains about 70 amino acids and is distinguished by a 22-residue duplicated sequence (Chauvaux et al. 1990), which bears similarity to the well-characterized EF-hand motif of various calcium-binding proteins (e.g., calmodulin and troponin C). Within this repeated sequence is a 12-residue calcium-binding loop, indicating that calcium-binding is an important characteristic of the dockerin module. This assumption was eventually confirmed experimentally (Yaron et al. 1995). The specificity characteristics of the cohesin-dockerin interaction have also been investigated. The results showed that four suspected residues may serve as recognition codes for interaction with the cohesin module (Mechaly et al. 2000, 2001; Pagès et al. 1997). The three-dimensional solution structure of the 69-residue dockerin module of a Clostridium thermocellum cellulosomal cellulase subunit was determined (Lytle et al. 2001). As predicted earlier (Bayer et al. 1998; Lytle et al. 2000; Pagès et al. 1997), the structure consists of two Ca2+-binding loop-helix motifs connected by a linker; the E helices entering each loop of the classical EF-hand motif are absent from the dockerin module.
The scaffoldin of C. thermocellum also contains a special type of dockerin module. This dockerin failed to bind to the cohesins from the same scaffoldin subunit, but instead interacted with a different type of cohesin—termed “type-II” cohesins—identified on the basis of sequence homology (Salamitou et al. 1994a). These cohesins are somewhat different from those of type I, having an additional segment and diversity in the latter half of the sequence. Three-dimensional structures for several examples of type-II cohesins have been reported (Noach et al. 2003, 2005, 2008, 2009; 2010; Carvalho et al. 2005). The type-II cohesins were discovered as component parts of a group of noncatalytic cell-surface “anchoring” proteins on C. thermocellum (Leibovitz and Béguin 1996; Leibovitz et al. 1997; Lemaire et al. 1995; Salamitou et al. 1994a). The three known anchoring scaffoldins in C. thermocellum contain different copy-numbers of the type-II cohesins as illustrated in Fig. 6.15 . Each of these anchoring scaffoldins also contains an SLH (S-layer homology) module, analogous to those of the cell-bound enzymes mentioned above (see section on “Cell-Bound Enzymes”). The intervening sequences, however, between the cohesins and SLH modules are different. In any case, the type-II cohesins selectively bind the type-II dockerins, and the cellulosome (i.e., the scaffoldin subunit together with all of its enzyme subunits) is thereby incorporated into the cell surface of C. thermocellum.


Schematic view of the modular similarity and diversity of scaffoldins from different cellulosome species. Four major scaffoldins of the current C. thermocellum paradigm are shown. The type-I cohesin-dockerin pairs are shown in yellow, the type-II pairs are shown in pink, and the anchoring component (the SLH module) is in green. Anchoring scaffoldins are designated by the adjacent symbol of an anchor. Other mesophilic clostridial species are characterized by a single scaffoldin. The four scaffoldins of the A. cellulolyticus system are more cross-interactive than that of the C. thermocellum paradigm. The reversed types of cohesin-dockerin pairings are evident in the B. cellulosolvens system, as are its two exceptionally large scaffoldins. The type-III cohesin-bearing scaffoldins of the R. flavefaciens system are especially elaborate. The single-cohesin ScaC “adaptor” scaffoldin provides the means with which to modify the repertoire of cellulosomal components, and the monovalent ScaE cohesin attaches the ScaB adaptor scaffoldin to the cell surface. Each of the seven ScaB cohesins binds to a cohesin of the trivalent ScaA primary scaffoldin which incorporates dockerin-bearing enzymes into the complex. Micrographs of the different bacteria are included in the figure
In recent years, structures for cohesin-dockerin complexes have been reported, which represent a significant breakthrough in our understanding of how the scaffoldins are organized and cellulosome architecture in general. In this context, cohesin-dockerin complexes for both type I and type II have been elucidated. Moreover, the structures provide insight on the molecular level regarding the specificity of this high-affinity interaction.
The crystal structure of C. thermocellum scaffoldin-borne cohesin two module together with the dockerin module from xylanase 10B was the first cohesin-dockerin complex reported (Carvalho et al. 2003). Interestingly, very little conformational change was observed in the cohesin module relative to the known structure of the same cohesin alone. The dockerin bound to the 8-3-6-5 face of the cohesin via an extensive hydrogen-bonding network and supporting hydrophobic interactions. Surprisingly, the twofold symmetry observed for the type-I dockerin sequences of this bacterium reflected a 180° rotation on cohesin surface, resulting in a dual mode of binding, in which the parent enzyme can attain one of two very different conformations in space, with respect to the interacting modular couterparts (Carvalho et al. 2007; Pinheiro et al. 2008).
The subsequent crystal structure of the type-II complex between the C. thermocellum SdbA cohesin module and the CipA scaffoldin XDoc modular dyad provided additional surprises (Adams et al. 2005). The resultant complex structure exhibited striking differences from that of the type-I complex. Notably, the lack of sequential symmetry of the dockerin module appeared to preclude a dual mode of binding. Indeed, as opposed to the type-I cohesin-dockerin interaction, the type-II dockerin contacts the cohesin counterpart across the entire length of both helices, which appears to result in a higher affinity and a single mode of interaction.
Similarity and Diversity of Scaffoldins from Different Species
The modular architecture of the known scaffoldins and their comparison to that of Clostridium thermocellum is presented in Fig. 6.15 . Two scaffoldins for Acetivibrio cellulolyticus and Bacteroides cellulosolvens, like C. thermocellum, carry dockerin modules at their C terminus (Ding et al. 1999, 2000). The A. cellulolyticus genome also includes a gene (immediately downstream of the scaffoldin gene) coding for an anchoring scaffoldin, that contains type-II cohesins. It thus seems that the arrangement of the cellulosome on the cell surface of these latter strains may be analogous to that of C. thermocellum. It is interesting to note that the cohesins of the Bacteroides cellulosolvens scaffoldin are clearly type-II cohesins and not of type I. This infers that there is not a clear linkage between the type-II cohesins and anchoring scaffoldins.
The scaffoldins from the other clostridial species thus far described all lack “type-II” dockerin modules, the inference being that cells of C. cellulovorans, e.g., would apparently not bear anchoring scaffoldins which contain type-II cohesins. Since their cellulosomes appear to be surface bound, their anchoring thereto is likely accomplished via an alternative molecular mechanism. In subsequent publications (Doi and Tamura 2001; Tamaru and Doi 1999; Tamaru et al. 1999), a cell-surface binding function was proposed for a module of unknown function [designated X2 (Coutinho and Henrissat 1999a, b, c)] of the scaffoldin from C. cellulovorans. On the basis of sequence alignment of a few conserved identical amino acids with S-layer proteins from Mycoplasma hyorhinis and Plasmodium reichenowi, the authors consider that this module may be recognized as an SLH module. The four X2 modules of the C. cellulovorans scaffoldin are very similar in sequence to the X-modules from the scaffoldins of Clostridium cellulolyticum and C. josui, which contain only two and one copy of this module, respectively. If this module functions in attaching the scaffoldin with its complement of enzymes to the cell surface, it is unclear why there would be different copy numbers of the module in the different scaffoldins. Likewise, one of the C. cellulovorans cellulosomal enzyme components (EngE) also contains a triplicated segment of unknown function [designated X48 (Coutinho and Henrissat 1999a, b, c)] that the authors consider to be involved in cell-surface attachment (Tamaru and Doi 1999). In any case, final proof of the function of the X2 and X48 modules awaits biochemical examination, as has been clearly achieved for the SLH module of the C. thermocellum anchoring scaffoldins (Chauvaux et al. 1999; Lemaire et al. 1998).
Finally, two novel scaffoldins were sequenced from the rumen bacterium, Ruminococcus flavefaciens strain 17 (Ding et al. 2001; Rincon et al. 2003, 2004, 2007). Although the proteins contain multiple cohesins, their sequences indicate that they are neither of type-I or type-II, but occupy their own phylogenetic branch. Interestingly, the ruminococcal scaffoldins lack a known type of CBM. The lack of a scaffoldin CBM and the question as to how the ruminococcal cellulosome(s) and/or the bacterium bind to the substrate were eventually resolved at least partially by the discovery of an additional CBM-bearing scaffoldin coded by a gene in the scaffoldin gene cluster of this bacterium (Rincon et al. 2007). Furthermore, a draft genome sequence of a similar strain of the same species was recently reported (Berg Miller et al. 2009), which revealed an exceptionally elaborate cellulosome system with a multitude of dockerin-bearing components (Rincon et al. 2010), roughly threefold of that observed in the C. thermocellum genome.
Schematic Comparison of Prokaryotic Cellulase Systems
In this section, we will describe schematically the similarity of and diversity in representative enzyme systems, demonstrating different strategies, from different plant cell wall–degrading bacteria. It is emphasized that the accumulating information is based on what is known currently from biochemical data combined with gene sequencing and bioinformatics. The information is still rather sketchy but quite revealing when compared with different bacteria. As time progresses and the entire genomes of cellulolytic microorganisms become known, the data concerning the complement of enzymes produced by a given bacterium will be complete, and we will be able to speculate with heightened certainty how the various cellulase systems might have evolved. Indeed, during the past decade, the genomes of many cellulolytic species have been sequenced (see Table 6.1 ), thereby supplementing our knowledge of the cellulase and cellulosome components. Representative schematic lists of the latter components will be provided below in forthcoming figures. More extensive descriptions of the total content of carbohydrate-active enymes, i.e., the CAZome, of the different cellulolytic bacteria, are now readily obtainable via the CAZy database (http://www.cazy.org/). A survey of genes, however, does not inform us how a given bacterial system is regulated and what role(s) the bacterium and its enzyme system may play in nature. The explosive development of molecular biology techniques, however revealing, cannot supplant the fundamental contribution of biochemical and ecological approaches to the study of microbial degradation of cellulose and other plant cell wall polysaccharides.
Free Enzyme Systems
Many cellulolytic microorganisms show a very similar pattern in the types of enzymes that comprise the complement of their cellulase system. For the purposes of this discussion, the concept of “cellulase system” will include the complement of all plant cell wall hydolyzing enzymes and other glycoside hydrolases, including the different cellulases, per se; the hemicellulases (e.g., xylanases, mannanases); pectin-degrading enzymes; etc.
The cellulase system of the mesophilic cellulolytic aerobe, Cellulomonas fimi, is one of the first studied, and for many years has been one of the most studied bacterial cellulase systems (O’Neill et al. 1986; Shen et al. 1995; Whittle et al. 1982). The enzymes of this bacterium are essentially free enzymes, which allowed their early isolation and characterization. Moreover, the genes of the cellulases from this bacterium were of the earliest to have been sequenced. The modular composition and family associations of representative glycoside hydrolases from this bacterium are shown symbolically in Fig. 6.16 . As an example of a free enzyme system, most of the enzymes bear a substrate-targeting CBM, which, in Cellulomonas fimi, are mainly from Family-2. Several of the enzymes have multiple copies of the FN3 domain (fibronectin 3 domain), the function of which is still unknown.


Cellulomonas fimi cellulase system – an example of a cell-free enzyme system: Pictographic view of the enzyme components and their modular architecture. The modular content of the enzymes in this and subsequent figures is shown from (left to right) the N-terminus to the C-terminus of the polypeptide chain. The family numbers of the given modules are enumerated; the catalytic modules are in red. Key to symbols: GH glycoside hydrolase (e.g., cellulase, xylanase, mannanase), CE carbohydrate esterase (e.g., acetyl xylan esterase and ferulic acid esterase), CBM carbohydrate-binding module), SLH S-layer homology (module), FN3 fibronectin-3 (domain), Ig immunoglobulin-like domain, X domain of unknown function
The Cellulomonas system includes four Family-6 enzymes. Two of these are shown in the figure—an endoglucanase and an exoglucanase (cellobiohydrolase) of the types described in Fig. 6.4 . The modularity of the endoglucanase is very simple, having the Family-6 catalytic module together with a Family-2 CBM. The cellobiohydrolase is a bit more complex with 3 additional FN3 domains that separate the same two types of modules. The two additional Cel6 enzymes appear to lack CBMs and are not included in the figure. Another cellobiohydrolase (that exhibits processive cleavage of the substrate) is from Family-48. Its general modular architecture is similar to that of the Family-6 cellobiohydrolase with the substitution of the catalytic module from a different family. The cellulase system from this organism also includes two Family-9 cellulases with modular themes B and D, familiar to us from the earlier description (Fig. 6.7 ). Two additional Family-9 cellulases are included in Cellulomonas fimi; one contains a simple GH9 catalytic module with a single CBM2 and the other has no additional ancillary modules (neither are described in the figure). In addition, a simple Family-5 cellulase and an interesting cell-borne Family-26 mannanase are components of the system. An additional Family-5 enzyme bears a CBM13 and two other Family-26 enzymes are present (not shown). The fact that an enzyme, i.e., the Family-26 enzyme, bears an SLH module and is presumably cell-associated would underscore its importance to the cell. Finally, 3 xylanases are part of the enzymatic apparatus of Cellulomonas fimi. One of these xylanases is a simple enzyme consisting of a Family-10 catalytic module connected to a Family-2 CBM. The other two are more complicated, each containing two catalytic modules—either a Family-10 or a Family-11 module and a carbohydrate esterase (in both cases, probably an acetyl xylan esterase (Fig. 6.3 )—plus several CBMs. The genome for this bacterium has recently been sequenced, and its enzymatic system is much more extensive than that shown in the figure. For example, seven members of GH43 have been detected in its genome. For more information regarding the CAZome of Cellulomonas fimi, the reader is referred to the CAZy database (http://www.cazy.org/).
A second example of a free enzyme system, from the aerobic thermophilic bacterium Thermobifida fusca (formerly classified as Thermomonospora fusca), has also been studied extensively (Wilson 1992, 2004, 2008, 2009; Wilson and Irwin 1999). A brief comparison of its known enzyme components (Fig. 6.17 ) shows a striking resemblance to those of Cellulomonas (compare Figs. 6.16 and 6.17 ). Both species produce similar types of cellulases from families 5, 6, 9, and 48 plus xylanases from families10 and 11. Nevertheless, the modular repertoire of the corresponding enzyme in T. fusca is generally somewhat simpler. For example, two of the T. fusca cellulases include single FN3 domains, whereas several Cellulomonas cellulases harbor multiple copies of the same module. Some T. fusca enzymes lack accessory modules other than a cellulose-binding CBM, whereas the corresponding Cellulomonas enzyme is elaborated by multiple copies of accessory modules. In some cases though, the respective CBMs appear on opposite termini of the polypeptide chain (i.e., the Family-48 and Family-5 cellulases). The T. fusca genome has now been sequenced, and more extensive information is available regarding its CAZome (http://www.cazy.org/). In contrast to the numerous members of the GH43 enzymes in Cellulomonas fimi, there is only one GH43 enzyme in T. fusca.


Thermobifida fusca cellulase system. A cell-free enzyme system. Compare with the Cellulomonas system (Fig. 6.16 ). Key to symbols: GH glycoside hydrolase (e.g., cellulase, xylanase, mannanase), CBM carbohydrate-binding module, FN3 fibronectin-3 (domain), Ig immunoglobulin-like domain, X domain of unknown function
The complement of enzymes and their modular content of the free enzyme systems from Cellulomonas and T. fusca are not necessarily similar in other free enzyme systems. Many free enzyme systems, such as those of Butyrivibrio fibrisolvens, Pseudomonas fluorescens, Fibrobacter succinogenes, Saccharophagus degradans, and various species of Streptomyces, Erwinia, and Thermatoga, appear to have several cellulases, xylanases, and mannanases from the common families, together with other glycoside hydrolases, e.g., arabinosidases, lichenases, amylases, pullulanases, galactanases, polygalacturonase, glucuronidases, and pectate lyases. In many of these bacterial enzymes, the Family-2 CBM appears to predominate as a common cellulose-binding module, but in others (e.g., Erwinia), relevant enzymes usually bear a cellulose-binding CBM from Family-3. Nevertheless, in many of the free systems, many enzymes are characterized by CBMs from other families as well as other noncatalytic modules of unknown function (X modules). Once again, until the genome sequences of cellulolytic prokaryotes are widely available, we are still limited in our capacity to compare among the enzyme systems, due to our incomplete knowledge of their enzyme sequences.
Multifunctional Enzyme Systems
In an hyperthermophilic bacterium, classified as Caldicellulosiruptor, the enzymes currently characterized in this system also appear to be free enzymes, but their modular organization is of a higher order (Daniel et al. 1996; Gibbs et al. 2000; Reeves et al. 2000). Many of the enzymes of this system are “bifunctional” in that they contain two separate catalytic modules in the same polypeptide chain (Fig. 6.18 ). As mentioned earlier (see section “Multifunctional Enzymes”), the appearance of two catalytic modules in the same enzyme would infer a distinctive synergistic action between the two. Thus, in Caldicellulosiruptor CelA, the Family-9 and Family-48 catalytic modules would be expected to work in concerted fashion on crystalline cellulose. In another type of enzyme, the Family-10 xylanase and Family-5 cellulase would likely be most effective on regions of the plant cell wall that are characterized by cellulose-xylan junctions. The diversity in the modular architecture of the Family-10 xylanases is particularly striking, and the various combinations of this type of catalytic module are apparently important to the sustenance of the bacterium in its environment. One of these xylanases appears to be attached to the cell surface via an SLH module (Ozdemir et al. 2012). In contrast to the Cellulomonas and T. fusca enzymes that often harbor a Family-2 CBM, the module responsible for binding to cellulosic substrates in Caldicellulosiruptor enzymes is usually one or more copies of a Family-3 CBM. The presence of more than one copy of a CBM in this case may reflect the extreme temperatures of the ecosystem.


Caldicellulosiruptor enzyme system: An example of a cell-free enzyme system that includes several multifunctional enzymes. Key to symbols: GH glycoside hydrolase (e.g., cellulase, xylanase, mannanase), CBM carbohydrate-binding module, SLH S-layer homology (module). See also Table 6.5
Other bacterial strains that include at least one free bifunctional enzyme in their enzyme systems are Anaerocellum thermophilum (now considered a species of Caldicellulosiruptor), Bacillus stearothermophilus, Fibrobacter succinogenes, Prevotella ruminicola, Ruminococcus albus, Ruminococcus flavefaciens, Streptomyces chattanoogensis, and thermophilic anaerobe NA10. The genomes of several species of Caldicellulosiruptor have now been sequenced (Kataeva et al. 2009; Blumer-Schuette et al. 2011). Each is characterized by different sets of bifunctional enzymes (Table 6.5 ), and some of these genomes either lack gene coding for such enzymes altogether or contain only one or two. Others carry up to seven bifunctional enzymes in their respective genomes (Dam et al. 2011). The different bifunctional enzymes include the various combinations (Himmel et al. 2010), notably cellulase-cellulase, cellulase-hemicellulase, hemicellulase-hemicellulase, hemicellulase-carbohydrate esterase, and even polysaccharide lyase-hemicellulase forms. The multiplicity of these genomes indicates the diverse nature of this genus of hyperthermophilic bacteria and reflects different patterns of substrate utilization.
Cellulosomal Systems
The inclusion of enzymes into a cellulosome via the noncatalytic scaffoldin subunit represents a higher level of organization. The association of complementary enzymes into a complex is considered to contribute sterically to their synergistic action on cellulose and other plant cell wall polysaccharides. As mentioned earlier (see earlier section “Similarity and Diversity of Scaffoldins from Different Species”), in the case of Clostridium thermocellum, Acetivibrio cellulolyticus, Bacteroides cellulosolvens, and Ruminococcus flavefaciens, the cellulosomes appear to be attached to the cell surface. The cellulosomes of C. cellulolyticum, C. cellulovorans, and C. josui may also be cell-associated, but, if so, the lack of a scaffoldin-borne dockerin and reciprocal anchoring scaffoldin would suggest an alternative mechanism.
The cellulosomes of some mesophilic clostridia, such as C. cellulolyticum, C. cellulovorans, C. josui, and C. papyroslvens are very similar. The genes encoding for many or most of the enzymes in the latter cellulosomal systems are arranged in a large cluster on the chromosome (Fig. 6.19 ). Additional cellulosomal genes, however, are located outside of the cluster in other regions of the chromosome. The majority of the cellulosome gene clusters from C. cellulolyticum and C. cellulovorans have been sequenced (Bagnara-Tardif et al. 1992; Belaich et al. 1999; Tamaru et al. 2000). In contrast, the cellulosomal genes from C. thermocellum are generally scattered over a large portion of the chromosome (Guglielmi and Béguin 1998). A few small clusters of cellulosomal genes are apparent in the genome, including a scaffoldin-containing cluster (Fig. 6.19 ) that also contains several cell-surface anchoring proteins (Fujino et al. 1993). The following descriptive analysis serves to compare the cellulosomal system of these three microorgansims. The genomes of all three bacteria have been sequenced, and the genomes of other cellulosome-producing bacteria are forthcoming in the near future.


Cellulosome-related gene clusters. Enzyme-linked gene clusters of the mesophilic Clostridia include an initial primary scaffoldin gene followed downstream by a series of genes encoding for various dockerin-bearing enzymes. Note the extensive similarity and subtle differences in the succession of enzyme-encoding genes. Multiple-scaffoldin gene clusters of the indicated bacteria comprise two or more genes in tandem that encode for scaffoldins. Scaffoldin genes and genes for enzymes are shown as light blue and pink arrows, respectively, whose length gives the approximate proportional size of the given gene relative to the others
Cellulosomal Components from Clostridium cellulolyticum
All of the sequenced enzymes from this organism are relatively common cellulases (Belaich et al. 1999). None of the known cellulosomal enzymes for this species contains more than one catalytic module (Fig. 6.20 ). The largest one, Cel9E (estimated at 94 kDa), is a Theme-D Family-9 cellulase (Gaudin et al. 2000). The critical Family-48 cellulase (Cel48F) is also a major cellulosome component (Reverbel-Leroy et al. 1997). The gene cluster of C. cellulolyticum contains several copies of other Family-9 cellulases, including Cel9G, Cel9H, and Cel9J, all of which contain the Theme-B fused Family-3c CBM (Belaich et al. 1998) (Fig. 6.8 ). The cellulosome system in this bacterium also contains numerous Family-5 cellulases (including Cel5A and Cel5D), a Family-5 mannanase (Man5K, which bears an N-terminal rather than C-terminal dockerin) and a Family-8 cellulase (Cel8C).


Clostridium cellulolyticum enzyme system. An example of a cellulosomal system. Key to symbols: GH glycoside hydrolase (e.g., cellulase, xylanase, mannanase), CBM carbohydrate-binding module, Doc dockerin module
Biochemical characterization of the C. cellulolyticum cellulosome demonstrated by SDS-PAGE a 160-kDa scaffoldin band and up to 16 smaller bands, representing putative enzyme subunits (Gal et al. 1997b). Many of these were clearly identified as known gene products. Early biochemical evidence suggested that xylanases from C. cellulolyticum are also organized in a cellulosome-like complex (Mohand-Oussaid et al. 1999). The genome sequence of this bacterium and subsequent proteomics studies revealed 62 dockerin-containing proteins, most of which are enzymes, including cellulases, xylanases, and other glycoside hydrolases, as well as carbohydrate esterases and polysaccharide lyases (Desvaux 2005; Blouzard et al. 2010).
Cellulosomal Components from Clostridium cellulovorans
Like C. cellulolyticum, the cellulases from this organism are relatively simple (see pictographical description of representative enzymes in Fig. 6.21 ). In addition to the cellulosomal enzymes thus described, several non-cellulosomal endoglucanases have also been partially or totally sequenced (Doi et al. 1998; Tamaru et al. 1999), notably those from Family-9 (Kosugi et al. 2002; Han et al. 2004, 2005).


Clostridium cellulovorans: A second cellulosomal system. Key to symbols: GH glycoside hydrolase (e.g., cellulase, xylanase, mannanase), CBM carbohydrate-binding module, Doc dockerin module, SLH S-layer homology (module), Ig immunoglobulin-like domain, X domain of unknown function
Several of the cellulosomal enzymes are architecturally synonymous to those of the C. cellulolyticum system (compare Figs. 6.20 and 6.21 ). This includes the critical Family-48 cellulase (Exg48S) (Liu and Doi 1998), two copies of the Theme-B Family-9 cellulase (Eng9H and Eng9Y), a Family-5 endoglucanase, and a Family-5 mannanase that bears an N-terminal dockerin (Tamaru and Doi 2000). Rather than a single Theme-D Family-9 cellulase as in C. cellulolyticum, the C. cellulovorans system contains two such enzymes (Eng9K and Eng9M). The C. cellulovorans cellulosome also appears to contain an unusual Theme-A Family-9 cellulase (Eng9L) that lacks helper modules. A dockerin-bearing pectate lyase (LyaA) infers that the bacterium would degrade pectin (Tamaru and Doi 2001). Indeed, early evidence (Sleat et al. 1984) indicated that, in addition to cellulose, C. cellulovorans is capable of assimilating a wide variety of other plant cell wall polysaccharides, including, xylans, pectins, and mannans.
The genome of C. cellulovorans was sequenced recently (Tamaru et al. 2010). Interestingly, 57 cellulosomal genes were identified in the genome, which, in addition to carbohydrate-active enzymes, also coded for lipases, peptidases, and proteinase inhibitors.
Cellulosomal Components from Clostridium thermocellum
Compared to the cellulosomal systems of C. cellulovorans and C. cellulolyticum, the enzymes from C. thermocellum are relatively large proteins, ranging in molecular size from about 40 to 180 kDa (Bayer et al. 1998c, 2000; Béguin and Lemaire 1996; Felix and Ljungdahl 1993; Lamed and Bayer 1988; Shoham et al. 1999). Examination of Fig. 6.22 reveals why these enzymes are so big—many of the larger ones contain multiple types of catalytic modules as well as other functional modules as an integral part of a single polypeptide chain [see Table I in (Bayer et al. 1998c) for a list of relevant references]. In addition to the cellulosomal enzymes, several noncellulosomal enzymes have also been described from this organism (Morag et al. 1990). These include two free enzymes (one of which lacks a CBM) and two cell-associated (SLH-containing) enzymes. Consequently, the potent cellulose- and plant cell wall–degrading activities of C. thermocellum are clearly reflected in its cellulase system, which displays an exceptional wealth, diversity, and intricacy of enzymatic components, thus representing the premier cellulose-degrading organism currently known.


Clostridium thermocellum: A complex cellulosomal system. Key to symbols: GH glycoside hydrolase (e.g., cellulase, xylanase, mannanase), CE carbohydrate esterase (e.g., acetyl xylan esterase and ferulic acid esterase), CBM carbohydrate-binding module, Doc dockerin module, SLH S-layer homology (module), Ig immunoglobulin-like domain, X domain of unknown function
Many of the C. thermocellum cellulosomal enzymes are cellulases, which include both endo- and exo-acting β-glucanases. Some of the important exoglucanases and processive cellulases include Cel48S, and various Family-9 cellulases. The Cel48S subunit is a member of the Family-48 glycoside hydrolases, and this particular family is recognized as a critical component of bacterial cellulosomes (Morag et al. 1991, 1993; Wang et al. 1993, 1994; Wu et al. 1988). Several other processive cellulases are members of the Family-9 glycoside hydrolases. Cel9F and Cel9N are Theme-B Family-9 enzymes (Fig. 6.7 ; Navarro et al. 1991). The other two are remarkably similar Theme-D enzymes, which exhibit nearly 95 % similarity along their common regions (Kataeva et al. 1999a, b; Zverlov et al. 1998c, 1999). The main difference between Cbh9A and Cel9K is the presence in the former of three extra modules (a Family-3 CBM and two modules of unknown function) (Kataeva et al. 2002, 2003, 2004, 2005). The functional significance of these supplementary modules to the activity of CbhA has not been elucidated.
The fact that the cellulosome from this organism contains many different types of cellulases is, of course, to be expected if we consider that growth of C. thermocellum is restricted to cellulose and its breakdown products, particularly cellobiose. Consequently, it is surprising to discover, in addition to the cellulases, numerous classic xylanases, i.e., those belonging to glycoside hydrolase families 10 and 11. In addition, two of the larger enzymes, Cel26H and Cel9/44J, contain hemicellulase components, i.e., Family-26 and Family-44 catalytic modules (a mannanase and a xylanase, respectively), together with a standard Family-5 and Family-9 (respectively) cellulase module in the same polypeptide chain (Ahsan et al. 1996; Yagüe et al. 1990). It is also interesting to note the presence of carbohydrate esterases together with xylanase modules in some of the enzyme subunits (i.e., XynU/A, XynY, XynZ and Cel5E), thus conferring the capacity to hydrolyze acetyl or feruloyl groups from hemicellulose substrates (Blum et al. 2000; Fernandes et al. 1999). Finally, the C. thermocellum cellulosome includes a typical Family-16 lichenase, a Family-26 mannanase, and a Family-18 chitinase.
The non-cellulosomal enzymes include another Theme-B Family-9 cellulase (Cel9I), and cell-bound forms of a xylanase (Xyn10X) and a lichenase (Lic16A), both of which contain multiple CBMs adjacent to the catalytic module. An additional non-cellulosomal Family-48 cellulase, Cel48Y, has also been described (Berger et al. 2007; Vazana et al. 2010). In the midst of all this complexity, the C. thermocellum non-cellulosomal cellulase system includes a simple Family-5 cellulase, Cel5C, which is completely devoid of additional accessory modules (Zverlov et al. 2005a; Feinberg et al. 2011). Why does this bacterium—which subsists exclusively on cellulosic substrates—need all these hemicellulases? The inclusion of such an impressive array of non-cellulolytic enzymes in a strict cellulose-utilizing species would suggest that their major purpose would be to collectively purge the unwanted polysaccharides from the milieu and to expose the preferred substrate—cellulose. The ferulic acid esterases, in concert with the xylanase components of the parent enzymes, could grant the bacterium a relatively simple mechanism by which it could detach the lignin component from the cellulose-hemicellulose composite. The lichenase (Lic16B) and chitinase (Chi18A) are also intriguing components of the cellulosome (Zverlov et al. 1991, 1998, 2002, 2005). The former would provide the bacterium with added action on cell-wall β-glucan components from certain types of plant matter. It is not clear whether the presence of the latter cellulosomal enzyme would reflect chitin-derived substrates from the exoskeletons of insects and/or from fungal cell walls. Whatever the source, the chitin breakdown products, like those of the hemicelluloses, would presumably not be utilized by the bacterium itself, but would be passed on to appropriate satellite bacteria for subsequent assimilation.
Subsequent genome sequencing of various strains of C. thermocellum has enhanced our understanding of the full complement of cellulosomal and non-cellulosomal enzymes produced by this bacterium (Zverlov et al. 2005a; Feinberg et al. 2011). Dockerin-containing components that are not directly involved in degradation of plant cell wall polysaccharides have also been identified (Kang et al. 2006; Schwarz and Zverlov 2006; Meguro et al. 2011). It is clear that the components and functions of the cellulosome system are much more complex than originally considered.
Gene Regulation of Cellulosomal Components
Over the past 10 years, the genomic revolution has provided the complete sequence of numerous bacterial genomes. Recent analysis of ∼1,500 of these genomes indicated that ∼40 % of the genomes encode for at least one cellulase gene (Medie et al. 2012). Within the cellulosome-producing bacteria, there are dozens of different cellulosome-related genes, and their expression appears to be highly regulated. Our ability to elucidate the regulatory mechanisms have changed dramatically in recent years due to the availability of new genomic sequences, the development of genetic tools for some of the classical cellulosome-producing strains and the establishment of workable proteomic procedures which allow the identification and quantification of numerous gene products in a single experiment. Much of the incentive for elucidating the regulatory mechanisms of cellulosome-producing bacteria is connected to their industrial potential for solubilizing lignocellulose for bioenergy production. In the context of this chapter, we will concentrate on new findings in C. thermocellum.
Regulation of Cellulase and Cellulosomal Genes in C. thermocellum
The various cellulosomal genes in C. thermocellum are, for the most part, mono-cistronic and scattered throughout the chromosome (Brown et al. 2007; Raman et al. 2009). Since the number of known dockerin-bearing enzymes is almost ten times the number of cohesins in the scaffoldin subunit, a unique interaction between cohesin-dockerin pairs is unlikely. This has indeed been substantiated for C. thermocellum in which all of the scaffoldin-borne cohesins recognize nearly all of the dockerin-containing enzymes. Thus, the composition of the cellulosome is governed by the relative amounts of the available dockerin-containing polypeptides that can be incorporated randomly into the complex (Bassen et al. 1995; Mitchell 1998). Regulation studies in C. thermocellum have indicated that the level and composition of the cellulosomal proteins vary with the composition of the growth media and cellobiose availability (Johnson et al. 1982; Nochur et al. 1990; Mishara et al. 1991; Bhat and Wood 1992; Nochur et al. 1992a, b; 1993; Raman et al. 2009).
Recent studies (Dror et al. 2003a, b; 2005; Stevenson and Weimer 2005; Zhang and Lynd 2005; Brown et al. 2007; Raman et al. 2011; Riederer et al. 2011) have demonstrated that expression of many cellulose-related genes is influenced by growth rate and the presence of extracellular polysaccharides. The molecular regulatory mechanisms in C. thermocellum were, until recently, very much obscure, and the bacterium does not appear to encode many of the well-characterized global regulatory elements found in Gram-positive bacteria, including the pleiotropic regulator CodY (Sonenshein 2007). In this regard, one of the LacI homologues, GlyR3, was shown to be a negative regulator of celC, a non-cellulosomal cellulase gene, and laminaribiose (a β-1-3 linked glucose dimer) appears to be its molecular inducer (Newcomb and Wu 2004; Demain et al. 2005; Newcomb et al. 2011). Remarkably, this was the first cellulose-related transcriptional factor identified in C. thermocellum.
Regulating by Sensing and the Involvement of Alternative Sigma Factors
As outlined above, it was postulated that C. thermocellum must possess a regulatory system that allows it to sense and react to the presence of high-molecular-weight polysaccharides in the extracellular environment presumably without importing their low-molecular-weight degradation products. While searching the C. thermocellum genome for the presence of carbohydrate binding modules, several Family-3 CBMs (CBM3s) were observed that were part of undefined polypeptides annotated as hypothetical proteins or membrane-associated proteins. Bioinformatic examination of these hypothetical peptides indicated possible homology to membrane-associated anti-σ factors. Following this initial observation, searching the public nucleotide and protein databases revealed that three strains of C. thermocellum contain a unique set of multiple ORFs resembling both Bacillus subtilis sigI and rsgI genes known to encode an alternative σI factor and its negative membrane-associated regulator RsgI, respectively (Asai et al. 2007).
Bioinformatic analysis of over 1,200 bacterial genomes revealed that the C. thermocellum RsgI-like proteins are unique to this species and are not present in several other cellulolytic clostridial species (e.g., Clostridium cellulolyticum and Clostridium papyrosolvens) (Kahel-Raifer et al. 2010). However, several new genome sequences of other cellulosome-producing bacteria, e.g., Acetivibrio cellulolyticus and Clostridium clariflavum, have revealed similar types of multiple biomass-sensing systems. Indeed, the possible involvement of alternative σ-factors in C. thermocellum was already suggested over 20 years ago by Mishara et al., following transcript analysis of three cellulosomal genes (celA, celD, celF) (Mishara et al. 1991). The C. thermocellum putative alternative σ factors are homologous to the recently characterized σI gene in B. subtilis (Zuber et al. 2001; Asai et al. 2007; Schirner and Errington 2009). Each of the genes encoding σI-like factors is positioned adjacent to a downstream gene encoding a multi-modular protein that contains only one strongly predicted trans-membrane helix (TMD) (Fig. 6.23 ). The ∼165-residue N-terminus of these trans-membrane proteins is homologous to the N-terminal segment of the B. subtilis anti-σI factor, RsgI. The C-terminal modules of these RsgI-like proteins, purportedly located outside the cell membrane, contain predicted polysaccharide-related functions including carbohydrate-binding modules (CBM3, CBM42), sugar-binding elements (PA14), and glycoside hydrolase modules of families 10 and 5.


Alternative σ-factor operons in C. thermocellum. The operons are made of two genes, the σ-factor gene (sig) and a transmembrane protein with an intracellular anti-σ factor at the N-terminus, followed by a transmembrane domain (TMD) and an extracellular sensor module at the C-terminus. Many of these proteins contain carbohydrate-related modules: i.e., a CBM, a glycoside hydrolase (GH), or sugar-binding proteins (PA)
The functional properties of the various elements of this system were verified experimentally (Kahel-Raifer et al. 2010; Nataf et al. 2010; Bahari et al. 2011). The binding properties of the extracellular sensing modules have been established with various polysaccharides including pectin, cellulose, arabinoxylan, and xylan (Kahel-Raifer et al. 2010; Bahari et al. 2011). Using isothermal titration calorimetry (ITC), it was possible to determine binding specificity and the dissociation constants (in the range of 0.02–1 μM) between the putative anti-σI factors to their corresponding σ factors (Nataf et al. 2010). The expression of the relevant alternative σ factor genes increased 3- to 30-fold in the presence of cellulose and xylan in the growth media, thus connecting their expression to direct detection of their extracellular polysaccharide substrates. Finally, the ability of σI1 to direct transcription from the σI1 promoter and from the promoter of celS (that encodes the Family 48 cellulase, Cel48S) was demonstrated in vitro by runoff transcription assays (Nataf et al. 2010).
Since many alternative sigma factors auto-regulate their own expression, it is possible to identify the signature of their promoter sequence by determining the transcriptional start sites of the target genes. Using this approach, over 60 cellulosomal genes were assigned to their corresponding alternative sigma factor.
In view of the above observations, a plausible model was proposed whereby the extracellular CBMs of putative anti-σI-like proteins can serve as biosensors that help assess the status of the biomass in the extracellular medium (Fig. 6.24 ). When the target substrate is unavailable, the σI-like factor is attached to the N-terminal cytoplasmic domain of the RsgI-like protein. Upon interaction with the target polysaccharide, the corresponding RsgI-borne CBM may undergo a conformational change, leading to the release of the σI factor, which then associates with RNA polymerase. Target gene(s) are then transcribed including those that code for various carbohydrate-active enzymes (CAZymes) and cellulosomal scaffoldins, as well as the σI/RsgI-like operon itself. These various CBMs recognize and bind different plant cell wall polysaccharides, which then induce different sets of CAZyme genes, thus activating the synthesis of the relevant glycoside hydrolases, carbohydrate esterases, and/or polysaccharide lyases.


Proposed mechanism for the activation of σ factors by extracellular polysaccharides. The carbohydrate-binding sensing module – the CBM – is positioned on the outer surface of the bacterium and linked via a short transmembrane domain to a short anti-σ peptide that binds and inactivates its cognate σ-factor (off state). In the presence of various target polysaccharides, the CBM binds the polymers which induces a conformational change that results in the release of the σ-factor, which now can initiate transcription of cellulose-utilization related promoters
New Genetic Tools for C. thermocellum
One of the major obstacles in studying gene regulation in C. thermocellum was the lack of reliable transformation procedures and genetic tools. Recently, the laboratory of Prof. Lee Lynd from Dartmouth College developed two procedures for obtaining knockout mutants in C. thermocellum (Olson et al. 2010; Tripathi et al. 2010; Argyros et al. 2011). Both procedures allow selection and counter selection for an integration event. The first system uses the elegant approach devised initially for yeast, taking advantage of the fact that mutants lacking orotidine-5′-phosphate decarboxylase (Pyr) not only require uracil for their growth (uracil auxotrophs) but are also resistant to the pyrimidine analog 5-fluoro-orotic acid (5-FOA) (Boeke et al. 1984). Thus, when working with a background-strain lacking orotidine-5′-phosphate decarboxylase (the pyrF gene in C. thermocellum), the presence of the pyrF gene can be selected for or against simply by the inclusion of uracil or 5-FOA, respectively, in the growth medium (Kondo et al. 1991; Schneider et al. 2005). The second approach utilizes the activity of hypoxanthine phosphoribosyl transferase (Hpt), which is required for purine metabolism and makes purine antimetabolites, such as 8-azahypoxanthine (AZH), toxic. Another component of the system is the gene thymidine kinase (Tdk) (which conveniently C. thermocellum lacks). Tdk converts fluoro deoxyuracil (FUDR) to fluoro-dUMP which is a suicide inhibitor of thymidylate synthetase, and this can be used for counter selection in the presence of FUDR. This second approach allows obtaining multiple deletion mutants without the presence of selection markers.
Genomics and Metagenomics
In the past decade, major strides for enzyme discovery have been achieved by genomic and metagenomic approaches, combined with bioinformatic analyses. An early work on a bacterial genome involved a plant cell wall polysaccharide-degrading species (Nelson et al. 1999). This initial work was eventually followed by genome sequencing studies of additional cellulolytic bacteria (Lykidis et al. 2007; Xie et al. 2007; Berg Miller et al. 2009; Kataeva et al. 2009; Hemme et al. 2010; Morrison et al. 2010; Tamaru et al. 2010; Feinberg et al. 2011). Combined bioinformatics, proteomics, and transcriptomics characterization can serve to reveal the components of the relevant enzyme system(s) in a given bacterium (Marcotte et al. 1999; Zverlov et al. 2005; Flint et al. 2008; Li et al. 2009; Raman et al. 2009; 2011; Rincon et al. 2010; Brulc et al. 2011; Dam et al. 2011).
The metagenomic approach utilizes genetic material directly from complex natural ecosystems, rather than using cultivated cells (Schloss and Handelsman 2003; Handelsman 2004). The advantage of this approach is that bias against unculturable bacteria is avoided and enzyme discovery is more representative. However, metagenomic libraries can introduce new types of biases due to nonuniform recovery of inserts and large numbers of clones required to cover the metagenome. Metagenomic analyses of different cellulose-containing ecosystems can serve to provide insight into novel types of enzymes that can be used for degradation of plant cell wall polysaccharides (Cottrell et al. 2005; Ferrer et al. 2005; Warnecke et al. 2007; Brulc et al. 2009; Li et al. 2009; Berg Miller et al. 2012).
Table 6.1 provides a list of cellulose-degrading bacteria whose genomes have been sequenced. It is clear that new genome sequences of cellulase- and cellulosome-producing bacteria will continue to accumulate, at least until the current involvement with cellulosic biomass-to-biofuel efforts remains in vogue.
Phylogenetics of Cellulase and Cellulosomal Systems
Early in the history of the development and establishment of the cellulosome concept, it was noted that the apparent occurrence of cellulosomes in different microorganisms tended to cross ecological, physiological, and evolutionary boundaries (Lamed et al. 1987). Initial biochemical and immunochemical evidence to this effect has been supported by the accumulated molecular biological studies.
Various lines of evidence indicate that the modular enzymes that degrade plant cell wall polysaccharides have evolved from a restricted number of common ancestral sequences. Much of the information in this direction remains a legacy, inherently encoded in the sequences of the functional modules that comprise the different enzymes. By comparing sequences of the various cellulosomal and noncellulosomal enzymes within and among the different strains, we can gain insight into the evolutionary rationale of the multigene families that comprise the glycoside hydrolases.
Horizontal Gene Transfer
It is clear that very similar enzymes which comprise a given glycoside hydrolase family are prevalent among a variety of different bacteria and fungi, thus indicating that they were not inherited through conventional evolutionary processes. The widespread occurrence of such conserved enzymes among phylogenetically different species argues that horizontal transfer of genes has been a major process by which a given microorganism can acquire a desirable enzyme. Once such a transfer event has taken place, the newly acquired gene would then be subjected to environmental pressures of its new surroundings, i.e., the genetic and physiological constitution of the cell itself. Following such selective pressure, the sequence of the gene would be adjusted to fit the host cell.
Gene Duplication
Sequence comparisons have also revealed the presence of very similar genes within a genome that may have very similar or even identical functions. One striking example is the tandem appearance of cbhA and celK genes in the chromosome of Clostridium thermocellum. Other examples are xynA and xynB also of C. thermocellum and xynA of the anaerobic fungus Neocallimastix patriciarum, which includes two very similar copies of Family-11 catalytic modules within the same polypeptide chain. These examples imply a mechanism of gene duplication (Chen et al. 1998; Gilbert et al. 1992), whereby the duplicated gene can serve as a template for secondary modifications that could result in two very similar enzymes with different properties, such as substrate and product specificities. A similar process could also account for the multiplicity of other types of modules (i.e., CBMs, cohesins or helper modules) within a polypeptide chain. Comparison of the modular architectures of similar genes from different species would suggest that individual modules can undergo a duplication process. This is exemplified by the multiple copies of FN3 in CelB from Cellulomonas fimi versus the single copy of the same module in cellulase E4 from Thermobifida fusca. But innumerable other examples are evident from the databases, whenever multiple copies of the same modular type exist in the same protein.
Domain Shuffling
Another observation from the genetic composition of the glycoside hydrolases argues for an alternative type of process, which would propagate new or modified types of enzymes. It is clear that many microbial enzyme systems contain individual hydrolases that carry very similar catalytic modules but include different types of accessory modules (Gilkes et al. 1991). An example that demonstrates this phenomenon is the observed species preference of otherwise very similar glycoside hydrolases for a given family of crystalline cellulose-binding CBM, which is entirely independent of the type of catalytic module borne by the complete enzyme. In this context, as we have seen above, the free enzymes of some bacteria, such as Cellulomonas fimi, Pseudomonas fluorescens, and Thermomonospora fusca, invariably include a Family-2 CBM, irrespective of the type of catalytic module. In contrast, those of other bacteria, e.g., Bacillus subtilis, Caldocellum saccharolyticum, Erwinia carotovora, and various clostridia, appear to prefer Family-3 CBMs. Moreover, the position of the CBM in the gene may be different for different genes. For example, the CBM may occur upstream or downstream from the catalytic module; it may be positioned either internally (sandwiched between two other modules) or at one of the termini of the polypeptide chain. The same pattern is characteristic of several other kinds of modules associated with the plant cell wall hydrolases. This is particularly evident in Family-9 cellulases and Family-10 xylanases, where the number and types of accessory modules may vary greatly within a given species. It seems that individual modules can be transferred en bloc and incorporated independently into appropriate enzymes. Once again, the modular architectures and sequence similarities between Clostridium thermocellum cellulosomal enzyme pairs (CbhA and CelK; XynA and XynB) are particularly revealing: in both cases, following an apparent gene duplication event, one or more additional modules appear to have been incorporated into the duplicated enzyme. Taken together, the information suggests that domain shuffling is an important process by which the properties of such enzymes can be modified and extended.
Proposed Mechanisms for Acquiring Cellulase and Cellulosomal Genes
Like the free enzyme systems, the phylogeny of cellulosomal components seems to have been driven by processes that include horizontal gene transfer, gene duplication, and domain shuffling. In cellulolytic/hemicellulolytic ecosystems, the resident microorganisms are usually in close contact, often under difficult conditions and in competition or cooperation with one another toward a common goal: the rapid degradation of recalcitrant polysaccharides and assimilation of their breakdown products.
A possible scenario for the molecular evolution of a cellulase/hemicellulase system in a prospective bacterium could involve the initial transfer of genetic material from one microbe to another in the same ecosystem. The size and type of transferred material could vary, such as a gene or part of gene (e.g., selected functional modules) or even all or part of a gene cluster. The process could then be sustained by gene duplication which would propagate the insertion of repeated modules, e.g., the multiple cohesin modules in the scaffoldins, or even smaller units, such as the linker sequences or the duplicated calcium-binding loop of the dockerin module. Domain shuffling can account for the observed permutations in the arrangement of modules in scaffoldin subunits from different species (Fig. 6.15 ). Finally, conventional mutagenesis would then render such products more suitable for the cellular environment or for interaction with other components of the cellulase system.
The available data suggest that there are no set of rules, which would, at this stage, enable us to anticipate the nature of a given cellulase system from a given microorganism. It seems that phylogenetically dissimilar organisms can possess similar types of cellulosomal or noncellulosomal enzyme systems, whereas phylogenetically related organisms that inhabit similar niches may be characterized by different types of enzyme systems. It is clear that in order to shed further light on this apparent enigma, we require more information about more types of enzyme systems. In addition to more sequences and structures, we will need more information—biochemical, physiological, and ecological—in order to sharpen existing notions regarding the enzymatic degradation of plant cell wall polysaccharides or to formulate new ones.
References
Adams JJ, Webb BA, Spencer HL, Smith SP (2005) Structural characterization of type II dockerin module from the cellulosome of Clostridium thermocellum: calcium-induced effects on conformation and target recognition. Biochemistry 44:2173–2182
Adams JJ, Gregg K, Bayer EA, Boraston AB, Smith SP (2008) Structural basis for a novel mode of Clostridium perfringens toxin complex formation. Proc Natl Acad Sci USA 105:12194–12199
Ahsan MM, Kimura T, Karita S, Sakka K, Ohmiya K (1996) Cloning, DNA sequencing, and expression of the gene encoding Clostridium thermocellum cellulase CelJ, the largest catalytic component of the cellulosome. J Bacteriol 178:5732–5740
Argyros DA, Tripathi SA, Barrett TF, Rogers SR, Feinberg LF, Olson DG, Foden JM, Miller BB, Lynd LR, Hogsett DA, Caiazza NC (2011) High ethanol titers from cellulose using metabolically engineered thermophilic, anaerobic microbes. Appl Environ Microbiol 77:8288–8294
Armand S, Drouillard S, Schulein M, Henrissat B, Driguez H (1997) A bifunctionalized fluorogenic tetrasaccharide as a substrate to study cellulases. J Biol Chem 272:2709–2713
Asai K, Ootsuji T, Obata K, Matsumoto T, Fujita Y, Sadaie Y (2007) Regulatory role of RsgI in sigI expression in Bacillus subtilis. Microbiology 153:92–101
Atalla RH (1999) Celluloses. In: Pinto BM (ed) Comprehensive natural products chemistry, vol 3. Elsevier, Cambridge, pp 529–598
Atalla RH, VanderHart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285
Bach E, Schollmeyer E (1992) An ultraviolet-spectrophotometric method with 2-cyanoacetamide for the determination of the enzymatic degradation of reducing polysaccharides. Anal Biochem 203(2):335–339
Bagnara-Tardif C, Gaudin C, Belaich A, Hoest P, Citard T, Belaich J-P (1992) Sequence analysis of a gene cluster encoding cellulases from Clostridium cellulolyticum. Gene 119:17–28
Bahari L, Gilad Y, Borovok I, Dassa B, Kahel-Raifer H, Jindou S, Nataf Y, Shoham Y, Lamed R, Bayer EA (2011) Glycoside hydrolases as components of putative carbohydrate biosensor proteins in Clostridium thermocellum. J Ind Microbiol Biotechnol 38:825–832
Barr BK, Hsieh Y-L, Ganem B, Wilson DB (1996) Identification of two functionally different classes of exocellulases. Biochemistry 35:586–592
Bassen RS, Romaniec MPM, Hazlewood GP, Freedman RB (1995) Characterization of the subunits in an apparently homogeneous subpopulation of Clostridium thermocellum cellulosomes. Enzyme Microb Technol 17:705–711
Bayer EA, Lamed R (1986) Ultrastructure of the cell surface cellulosome of Clostridium thermocellum and its interaction with cellulose. J Bacteriol 167:828–836
Bayer EA, Lamed R (1992) The cellulose paradox: pollutant par excellence and/or a reclaimable natural resource? Biodegradation 3:171–188
Bayer EA, Kenig R, Lamed R (1983) Adherence of Clostridium thermocellum to cellulose. J Bacteriol 156:818–827
Bayer EA, Setter E, Lamed R (1985) Organization and distribution of the cellulosome in Clostridium thermocellum. J Bacteriol 163:552–559
Bayer EA, Morag E, Lamed R (1994) The cellulosome—A treasure-trove for biotechnology. Trends Biotechnol 12:378–386
Bayer EA, Morag E, Shoham Y, Tormo J, Lamed R (1996) The cellulosome: a cell-surface organelle for the adhesion to and degradation of cellulose. In: Fletcher M (ed) Bacterial adhesion: molecular and ecological diversity. Wiley-Liss., New York, pp 155–182
Bayer EA, Chanzy H, Lamed R, Shoham Y (1998a) Cellulose, cellulases and cellulosomes. Curr Opin Struct Biol 8:548–557
Bayer EA, Morag E, Lamed R, Yaron S, Shoham Y (1998b) Cellulosome structure: four-pronged attack using biochemistry, molecular biology, crystallography and bioinformatics. In: Claeyssens M, Nerinckx W, Piens K (eds) Carbohydrases from Trichoderma reesei and other microorganisms. The Royal Society of Chemistry, London, pp 39–67
Bayer EA, Shimon LJW, Lamed R, Shoham Y (1998c) Cellulosomes: structure and ultrastructure. J Struct Biol 124:221–234
Bayer EA, Coutinho PM, Henrissat B (1999) Cellulosome-like sequences in Archaeoglobus fulgidus: an enigmatic vestige of cohesin and dockerin domains. FEBS Lett 463:277–280
Bayer EA, Shoham Y, Lamed R (2000) The cellulosome—an exocellular organelle for degrading plant cell wall polysaccharides. In: Doyle RJ (ed) Glycomicrobiology. Kluwer Academic/Plenum, New York, pp 387–439
Bayer EA, Belaich J-P, Shoham Y, Lamed R (2004) The cellulosomes: multi-enzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol 58:521–554
Bayer EA, Shoham Y, Lamed R (2006) Cellulose-decomposing prokaryotes and their enzyme systems. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E (eds) The prokaryotes, vol 2, 3rd edn. Springer, New York, pp 578–617
Bayer EA, Lamed R, White BA, Flint HJ (2008) From cellulosomes to cellulosomics. Chem Rec 8:364–377
Beeson WT, Phillips CM, Cate JH, Marletta MA (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J Am Chem Soc 134:890–892
Béguin P (1983) Detection of cellulase activity in polyacrylamide gels using Congo red-stained agar replicas. Anal Biochem 131:333–336
Béguin P (1990) Molecular biology of cellulose degradation. Annu Rev Microbiol 44:219–248
Béguin P, Aubert J-P (1994) The biological degradation of cellulose. FEMS Microbiol Lett 13:25–58
Béguin P, Lemaire M (1996) The cellulosome: an exocellular, multiprotein complex specialized in cellulose degradation. Crit Rev Biochem Molec Biol 31:201–236
Belaich J-P, Tardif C, Belaich A, Gaudin C (1997) The cellulolytic system of Clostridium cellulolyticum. J Biotechnol 57:3–14
Belaich A, Belaich J-P, Fierobe H-P, Gaudin C, Pagès S, Reverbel-Leroy C, Tardif C (1998) Cellulosome analysis and cellulases CelF and CelG from Clostridium cellulolyticum. In: Claeyssens M, Nerinckx W, Piens K (eds) Carbohydrases from Trichoderma reesei and other microorganisms. The Royal Society of Chemistry, London, pp 73–86
Belaich J-P, Belaich A, Fierobe H-P, Gal L, Gaudin C, Pagès S, Reverbel-Leroy C, Tardif C (1999) The cellulolytic system of Clostridium cellulolyticum. In: Ohmiya K, Hayashi K, Sakka K, Kobayashi Y, Karita S, Kimura T (eds) Genetics, biochemistry and ecology of cellulose degradation. Uni Publishers, Tokyo, pp 479–487
Berg Miller ME, Antonopoulos DA, Rincon MT, Band M, Bari A, Akaiko1 T, Hernandez A, Kim R, Liu L, Thimmapuram J, Henrissat B, Coutinho PM, Borovok I, Jindou S, Lamed R, Flint HJ, Bayer EA, White BA (2009) Diversity and strain specificity of plant cell wall degrading enzymes revealed by the draft genome of Ruminococcus flavefaciens FD-1. PLoS ONE 4:e6650
Berg Miller ME, Yeoman CJ, Tringe SG, Edwards RA, Flint HJ, Lamed R, Bayer EA, White BA (2012) Phage-bacteria relationships and CRISPR elements revealed by a metagenomic survey of the rumen microbiome. Environ Microbiol 14:207–227
Berger E, Zhang D, Zverlov VV, Schwarz WH (2007) Two noncellulosomal cellulases of Clostridium thermocellum, Cel9I and Cel48Y, hydrolyse crystalline cellulose synergistically. FEMS Microbiol Lett 268:194–201
Bhat MK (2000) Cellulases and related enzymes in biotechnology. Biotechnol Adv 18:355–383
Bhat KM, Wood TM (1992) The cellulase of the anaerobic bacterium Clostridium thermocellum: isolation, dissociation, and reassociation of the cellulosome. Carbohydr Res 227:293–300
Biely P (1985) Microbial xylanolytic systems. Trends Biotechnol 3:285–290
Blouzard JC, Coutinho PM, Fierobe HP, Henrissat B, Lignon S, Tardif C, Pagès S, de Philip P (2010) Modulation of cellulosome composition in Clostridium cellulolyticum: adaptation to the polysaccharide environment revealed by proteomic and carbohydrate-active enzyme analyses. Proteomics 10:541–554
Blum DL, Kataeva IA, Li XL, Ljungdahl LG (2000) Feruloyl esterase activity of the Clostridium thermocellum cellulosome can be attributed to previously unknown domains of XynY and XynZ. J Bacteriol 182(5):1346–1351
Blumer-Schuette SE, Ozdemir I, Mistry D, Lucas S, Lapidus A, Cheng JF, Goodwin LA, Pitluck S, Land ML, Hauser LJ, Woyke T, Mikhailova N, Pati A, Kyrpides NC, Ivanova N, Detter JC, Walston-Davenport K, Han S, Adams MW, Kelly RM (2011) Complete genome sequences for the anaerobic, extremely thermophilic plant biomass-degrading bacteria Caldicellulosiruptor hydrothermalis, Caldicellulosiruptor kristjanssonii, Caldicellulosiruptor kronotskyensis, Caldicellulosiruptor owensensis, and Caldicellulosiruptor lactoaceticus. J Bacteriol 193:1483–1484
Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197:345–346
Boraston AB, McLean BW, Kormos JM, Alam M, Gilkes NR, Haynes CA, Tomme P, Kilburn DG, Warren RA (1999) Carbohydrate-binding modules: diversity of structure and function. In: Gilbert HJ, Davies GJ, Henrissat B, Svensson B (eds) Recent advances in carbohydrate bioengineering. The Royal Society of Chemistry, Cambridge, pp 202–211
Boraston AB, Bolam DN, Gilbert HJ, Davies GJ (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J 382:769–781
Borneman WS, Ljungdahl LG, Hartley RD, Akin DE (1993) Feruloyl and p-coumaroyl esterases from the anaerobic fungus Neocallimastix strain MC-2: properties and functions in plant cell wall degradation. In: Coughlan MP, Hazlewood GP (eds) Hemicellulose and hemicellulases. Portland Press, London, pp 85–102
Brás JL, Cartmell A, Carvalho AL, Verzé G, Bayer EA, Vazana Y, Correia MA, Prates JA, Ratnaparkhe S, Boraston AB, Romão MJ, Fontes CM, Gilbert HJ (2011) Structural insights into a unique cellulase fold and mechanism of cellulose hydrolysis. Proc Natl Acad Sci USA 108:5237–5242
Brown SD, Raman B, McKeown CK, Kale SP, He ZL, Mielenz JR (2007) Construction and evaluation of a Clostridium thermocellum ATCC 27405 whole-genome oligonucleotide microarray. Appl Biochem Biotechnol 137:663–674
Brulc JM, Antonopoulos DA, Berg Miller ME, Wilson MK, Yannarell AC, Dinsdale EA, Edwards RE, Frank ED, Emerson JB, Walkin P, Coutinho PM, Henrissat B, Nelson KE, White BA (2009) Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc Natl Acad Sci USA 106:1948–1953
Brulc JM, Wilson MK, Yeoman CJ, Berg Miller ME, Jeraldo P, Jindou S, Goldenfeld N, Flint HJ, Lamed R, Borovok I, Vodovnik M, Nelson KE, Bayer EA, White BA (2011) Cellulosomics, a gene-centric approach to investigating the intraspecific diversity and adaptation of Ruminococcus flavefaciens within the rumen. PLoS One 6:e25329
Brun E, Moriaud F, Gans P, Blackledge M, Barras F, Marion D (1997) Solution structure of the cellulose-binding domain of the endoglucanase Z secreted by Erwinia chrysanthemi. Biochemistry 36:16074–16086
Burstein T, Shulman M, Jindou S, Petkun S, Frolow F, Shoham Y, Bayer EA, Lamed R (2009) Physical association of the catalytic and helper modules of a processive family-9 glycoside hydrolase is essential for activity. FEBS Lett 583:879–884
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238
Carpita NC, Gibeaut DM (1993) Structural models of primary cell walls in flowering plants: consistency of molecular structure with the physical properties of the walls during growth. Plant J 3:1–30
Carvalho AL, Dias FM, Prates JA, Nagy T, Gilbert HJ, Davies GJ, Ferreira LM, Romao MJ, Fontes CM (2003) Cellulosome assembly revealed by the crystal structure of the cohesin-dockerin complex. Proc Natl Acad Sci USA 100:13809–13814
Carvalho AL, Pires VM, Gloster TM, Turkenburg JP, Prates JA, Ferreira LM, Romao MJ, Davies GJ, Fontes CM, Gilbert HJ (2005) Insights into the structural determinants of cohesin-dockerin specificity revealed by the crystal structure of the type II cohesin from Clostridium thermocellum SdbA. J Mol Biol 349:909–915
Carvalho AL, Dias FMV, Nagy T, Prates JAM, Proctor MR, Smith N, Bayer EA, Davies GJ, Ferreira LMA, Ramão MJ, Fontes CMGA, Gilbert HJ (2007) Evidence for a dual binding mode of dockerin modules to cohesins. Proc Natl Acad Sci USA 104:3089–3094
Chanzy H (1990) Aspects of cellulose structure. In: Kennedy JF, Philips GO, Williams PA (eds) Cellulose sources and exploitation. Industrial utilization, biotechnology and physico-chemical properties. Ellis Horwood, New York, pp 3–12
Chauvaux S, Béguin P, Aubert J-P, Bhat KM, Gow LA, Wood TM, Bairoch A (1990) Calcium-binding affinity and calcium-enhanced activity of Clostridium thermocellum endoglucanase D. Biochem J 265:261–265
Chauvaux S, Matuschek M, Béguin P (1999) Distinct affinity of binding sites for S-layer homologous domains in Clostridium thermocellum and Bacillus anthracis cell envelopes. J Bacteriol 181:2455–2458
Chen H, Li XL, Blum DL, Ljungdahl LG (1998) Two genes of the anaerobic fungus Orpinomyces sp. strain PC-2 encoding cellulases with endoglucanase activities may have arisen by gene duplication. FEMS Microbiol Lett 159:63–68
Chitayat S, Adams JJ, Furness HS, Bayer EA, Smith SP (2008a) The solution structure of the C-terminal modular pair from Clostridium perfringens reveals a non-cellulosomal dockerin module. J Mol Biol 381:1202–1212
Chitayat S, Gregg K, Adams JJ, Ficko-Blean E, Bayer EA, Boraston AB, Smith SP (2008b) Three-dimensional structure of a putative non-cellulosomal cohesin module from a Clostridium perfringens family 84 glycoside hydrolase. J Mol Biol 375:20–28
Claeyssens M, Henrissat B (1992) Specificity mapping of cellulolytic enzymes: classification into families of structurally related proteins confirmed by biochemical analysis. Protein Sci 1(10):1293–1297
Cottrell MT, Yu L, Kirchman DL (2005) Sequence and expression analyses of Cytophaga-like hydrolases in a Western Arctic metagenomic library and the Sargasso Sea. Appl Environ Microbiol 71:8506–8513
Coughlan MP, Hazlewood GP (1993) β-1,4-D-Xylan-degrading enzyme systems: biochemistry, molecular biology and applications. Biotechnol Appl Biochem 17:259–289
Coughlan MP, Mayer F (1992) The cellulose-decomposing bacteria and their enzyme systems. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes, vol I, 2nd edn. Springer, New York, pp 459–516
Coutinho PM, Henrissat B (1999a) Carbohydrate-active enZYmes and associated MODular Organization server (CAZyModO Website). [Online]. http://afmb.cnrs-mrs.fr/∼pedro/DB/db.html. Accessed 17 Apr 2001
Coutinho PM, Henrissat B (1999b) Carbohydrate-active enzymes: an integrated database approach. In: Gilbert HJ, Davies GJ, Henrissat B, Svensson B (eds) Recent advances in carbohydrate bioengineering. The Royal Society of Chemistry, Cambridge, pp 3–12
Coutinho PM, Henrissat B (1999c) The modular structure of cellulases and other carbohydrate-active enzymes: an integrated database approach. In: Ohmiya K, Hayashi K, Sakka K, Kobayashi Y, Karita S, Kimura T (eds) Genetics, biochemistry and ecology of cellulose degradation. Uni Publishers, Tokyo, pp 15–23
Coutinho PM, Deleury E, Davies GJ, Henrissat B (2003a) An evolving hierarchical family classification for glycosyltransferases. J Mol Biol 328:307–317
Coutinho PM, Deleury E, Henrissat B (2003b) The families of carbohydrate-active enzymes in the genomic era. J Appl Glycosci 50:241–244
Coutinho PM, Stam M, Blanc E, Henrissat B (2003c) Why are there so many carbohydrate-active enzyme-related genes in plants? Trends Plant Sci 8:563–565
Dam P, Kataeva I, Yang SJ, Zhou F, Yin Y, Chou W, Poole FL, Westpheling J, Hettich R, Giannone R, Lewis DL, Kelly R, Gilbert HJ, Henrissat B, Xu Y, Adams MW (2011) Insights into plant biomass conversion from the genome of the anaerobic thermophilic bacterium Caldicellulosiruptor bescii DSM 6725. Nucleic Acids Res 39:3240–3254
Daniel RM, Toogood HS, Bergquist PL (1996) Thermostable proteases. Biotechnol Genet Eng Rev 13:51–100
Das NN, Das SC, Mukerjee AK (1984) On the ester linkage between lignin and 4-O-methyl-D-glucurono-D-xylan in jute fiber (Corchorus capsularis). Carbohydr Res 127:345–348
Davies G, Henrissat B (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3:853–859
Davies GJ, Mackenzie L, Varrot A, Dauter M, Brzozowski M, Schülein M, Withers SG (1998) Snapshots along an enzymatic reaction coordinate: analysis of a retaining β-glycoside hydrolase. Biochemistry 37:11707–11713
Demain AL, Newcomb M, Wu JH (2005) Cellulase, clostridia, and ethanol. Microbiol Mol Biol Rev 69:124–154
Desvaux M (2005) Clostridium cellulolyticum: model organism of mesophilic cellulolytic clostridia. FEMS Microbiol Rev 29:741–764
Devillard E, Goodheart DE, Karnati SK, Bayer EA, Lamed R, Miron J, Nelson KE, Morrison M (2004) Ruminococcus albus 8 mutants defective in cellulose degradation are deficient in two processive endocellulases, Cel48A and Cel9B, both of which possess a novel modular architecture. J Bacteriol 186:136–145
Din, N, Damude HG, Gilkes NR, Miller RCJ, Warren RAJ, and Kilburn DG (1994) C1-Cx revisited: intramolecular synergism in a cellulase. Proc. Natl. Acad. Sci. USA 91: 11383–11387
Ding SY, Himmel ME (2006) The maize primary cell wall microfibril: a new model derived from direct visualization. J Agric Food Chem 54:597–606
Ding S-Y, Bayer EA, Steiner D, Shoham Y, Lamed R (1999) A novel cellulosomal scaffoldin from acetivibrio cellulolyticus that contains a family-9 glycosyl hydrolase. J Bacteriol 181:6720–6729
Ding S-Y, Bayer EA, Steiner D, Shoham Y, Lamed R (2000) A scaffoldin of the Bacteroides cellulosolvens cellulosome that contains 11 type II cohesins. J Bacteriol 182:4915–4925
Ding S-Y, Rincon MT, Lamed R, Martin JC, McCrae SI, Aurilia V, Shoham Y, Bayer EA, Flint HJ (2001) Cellulosomal scaffoldin-like proteins from Ruminococcus flavefaciens. J Bacteriol 183:1945–1953
Ding S-Y, Xu Q, Mursheda KA, Baker JO, Bayer EA, Barak Y, Lamed R, Sugiyama J, Rumbles G, Himmel ME (2006) Versatile derivatives of carbohydrate-binding modules for imaging of complex carbohydrates approaching the molecular level of resolution. Biotechniques 41:435–440
Divne C, Stahlberg J, Teeri T, Jones T (1998) High-resolution crystal structures reveal how a cellulose chain is bound in the 50 Å long tunnel of cellobiohydrolase I from Trichoderma reesei. J Mol Biol 275:309–325
DOE, U. S. (2008) U.S. Department of Energy’s bioenergy research centers – an overview of the science. DOE Office of Science, Washington, DC
Doi RH, Kosugi A (2004) Cellulosomes: plant-cell-wall-degrading enzyme complexes. Nat Rev Microbiol 2:541–551
Doi RH, Tamura Y (2001) The Clostridium cellulovorans cellulosome: An enzyme complex with plant cell wall degrading activity. Chem Rec 1:24–32
Doi RH, Goldstein M, Hashida S, Park JS, Takagi M (1994) The Clostridium cellulovorans cellulosome. Crit Rev Microbiol 20:87–93
Doi RH, Park JS, Liu CC, Malburg LM, Tamaru Y, Ichiishi A, Ibrahim A (1998) Cellulosome and noncellulosomal cellulases of Clostridium cellulovorans. Extremophiles 2:53–60
Doner LW, Irwin PL (1992) Assay of reducing end-groups in oligosaccaride homolgues with 2,2'-bicinchoninate. Anal Biochem 202:50–53
Driguez H (1997) Thiooligosaccharides in glycobiology. Topics Curr Chem 187:85–116
Dror TW, Morag E, Rolider A, Bayer EA, Lamed R, Shoham Y (2003a) Regulation of the cellulosomal CelS (cel48A) gene of Clostridium thermocellum is growth rate dependent. J Bacteriol 185:3042–3048
Dror TW, Rolider A, Bayer EA, Lamed R, Shoham Y (2003b) Regulation of expression of scaffoldin-related genes in Clostridium thermocellum. J Bacteriol 185:5109–5116
Dror TW, Rolider A, Bayer EA, Lamed R, Shoham Y (2005) Regulation of major cellulosomal endoglucanases of Clostridium thermocellum differs from that of a prominent cellulosomal xylanase. J Bacteriol 187:2261–2266
Ensor L, Stosz S, Weiner R (1999) Expression of multiple insoluble complex polysaccharide degrading enzyme systems by a marine bacterium. J Ind Microbiol Biotechnol 23:123–126
Eriksson K-EL, Blanchette RA, Ander P (1990) Biodegradation of hemicelluloses, microbial and enzymatic degradation of wood and wood components. Springer, Heidelberg, pp 181–397
Ezer A, Matalon E, Jindou S, Borovok I, Atamna N, Yu Z, Morrison M, Bayer EA, Lamed R (2008) Cell-surface enzyme attachment is mediated by a family-37 carbohydrate-binding module, unique to Ruminococcus albus. J Bacteriol 190:8220–8222
Feinberg L, Foden J, Barrett T, Davenport KW, Bruce D, Detter C, Tapia R, Han C, Lapidus A, Lucas S, Cheng JF, Pitluck S, Woyke T, Ivanova N, Mikhailova N, Land M, Hauser L, Argyros DA, Goodwin L, Hogsett D, Caiazza N (2011) Complete genome sequence of the cellulolytic thermophile Clostridium thermocellum DSM1313. J Bacteriol 193:2906–2907
Felix CR, Ljungdahl LG (1993) The cellulosome – the exocellular organelle of Clostridium. Annu Rev Microbiol 47:791–819
Fernandes AC, Fontes CM, Gilbert HJ, Hazlewood GP, Fernandes TH, Ferreira LMA (1999) Homologous xylanases from Clostridium thermocellum: evidence for bi-functional activity, synergism between xylanase catalytic modules and the presence of xylan-binding domains in enzyme complexes. Biochem J 342:105–110
Ferrer M, Golyshina OV, Chernikova TN, Khachane AN, Reyes-Duarte D, Santos VA, Strompl C, Elborough K, Jarvis G, Neef A, Yakimov MM, Timmis KN, Golyshin PN (2005) Novel hydrolase diversity retrieved from a metagenome library of bovine rumen microflora. Environ Microbiol 7:1996–2010
Flint HJ, Martin J, McPherson CA, Daniel AS, Zhang JX (1993) A bifunctional enzyme, with separate xylanase and β(1,3-1,4)-glucanase domains, encoded by the xynD gene of Ruminococcus flavefaciens. J Bacteriol 175:2943–2951
Flint HJ, Bayer EA, Lamed R, White BA (2008) Polysaccharide utilization by gut bacteria: potential for new insights from genomic analysis. Nat Rev Microbiol 6:121–131
Forsberg Z, Vaaje-Kolstad G, Westereng B, Bunæs AC, Stenstrøm Y, MacKenzie A, Sørlie M, Horn SJ, Eijsink VG (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci 20:1479–1483
Fraiberg M, Borovok I, Weiner RM, Lamed R (2010) Discovery and characterization of cadherin domains in Saccharophagus degradans 2-40. J Bacteriol 192:1066–1074
Fraiberg M, Borovok I, Bayer EA, Weiner RM, Lamed R (2011) Cadherin domains in the polysaccharide-degrading, marine bacterium Saccharophagus degradans 2-40 are carbohydrate-binding modules. J Bacteriol 193:283–285
Fujino T, Béguin P, Aubert J-P (1993) Organization of a Clostridium thermocellum gene cluster encoding the cellulosomal scaffolding protein CipA and a protein possibly involved in attachment of the cellulosome to the cell surface. J Bacteriol 175:1891–1899
Gal L, Gaudin C, Belaich A, Pagès S, Tardif C, Belaich J-P (1997a) CelG from Clostridium cellulolyticum: a multidomain endoglucanase acting efficiently on crystalline cellulose. J Bacteriol 179:6595–6601
Gal L, Pagès S, Gaudin C, Belaich A, Reverbel-Leroy C, Tardif C, Belaich J-P (1997b) Characterization of the cellulolytic complex (cellulosome) produced by Clostridium cellulolyticum. Appl Environ Microbiol 63:903–909
Galbe M, Zacchi G (2007) Pretreatment of lignocellulosic materials for efficient bioethanol production. Adv Biochem Eng Biotechnol 108:41–65
Garcia E, Johnston D, Whitaker JR, Shoemaker SP (1993) Assessment of endo-1,4-β-D-glucanase activity by a rapid colorimetric assay using disodium 2,2'-bicinchoninate. J Food Biochem 17:135–145
Gaudin C, Belaich A, Champ S, Belaich JP (2000) CelE, a multidomain cellulase from Clostridium cellulolyticum: a key enzyme in the cellulosome? J Bacteriol 182:1910–1915
Gerngross UT, Romaniec MPM, Kobayashi T, Huskisson NS, Demain AL (1993) Sequencing of a Clostridium thermocellum gene (cipA) encoding the cellulosomal SL-protein reveals an unusual degree of internal homology. Mol Microbiol 8:325–334
Ghose TK (1987) Measurments of cellulase activity. Pure Appl Chem 59:257–268
Gibbs MD, Reeves RA, Farrington GK, Anderson P, Williams DP, Bergquist PL (2000) Multidomain and multifunctional glycosyl hydrolases from the extreme thermophile Caldicellulosiruptor isolate Tok7B.1. Curr Microbiol 40(5):333–340
Gilbert, HJ, Ed. (2012) Cellulases. San Diego, Elsevier.
Gilbert HJ, Hazlewood GP (1993) Bacterial cellulases and xylanases. J Gen Microbiol 139:187–194
Gilbert HJ, Hazlewood GP, Laurie JI, Orpin CG, Xue GP (1992) Homologous catalytic domains in a rumen fungal xylanase: evidence for gene duplication and prokaryotic origin. Mol Microbiol 6:2065–2072
Gilkes NR, Henrissat B, Kilburn DG, Miller RCJ, Warren RAJ (1991) Domains in microbial β-1,4-glycanases: sequence conservation, function, and enzyme families. Microbiol Rev 55(2):303–315
Green F 3rd, Clausen CA, Highley TL (1989) Adaptation of the Nelson-Somogyi reducing-sugar assay to a microassay using microtiter plates. Anal Biochem 182(2):197–199
Guglielmi G, Béguin P (1998) Cellulase and hemicellulase genes of Clostridium thermocellum from five independent collections contain few overlaps and are widely scattered across the chromosome. FEMS Microbiol Lett 161:209–215
Haigler CH, Weimer PJ (eds) (1991) Biosynthesis and biodegradation of cellulose. Marcel Dekker, New York
Han SO, Cho HY, Yukawa H, Inui M, Doi RH (2004) Regulation of expression of cellulosomes and noncellulosomal (hemi)cellulolytic enzymes in Clostridium cellulovorans during growth on different carbon sources. J Bacteriol 186:4218–4227
Han SO, Yukawa H, Inui M, Doi RH (2005) Molecular cloning and transcriptional and expression analysis of engO, encoding a new noncellulosomal family 9 enzyme, from Clostridium cellulovorans. J Bacteriol 187:4884–4889
Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685
Hazlewood GP, Gilbert HJ (1993) Molecular biology of hemicellulases. In: Coughlan MP, Hazlewood GP (eds) Hemicellulose and hemicellulases. Portland Press, London, pp 103–126
Hemme CL, Mouttaki H, Lee YJ, Zhang G, Goodwin L, Lucas S, Copeland A, Lapidus A, Glavina del Rio T, Tice H, Saunders E, Brettin T, Detter JC, Han CS, Pitluck S, Land ML, Hauser LJ, Kyrpides N, Mikhailova N, He Z, Wu L, Van Nostrand JD, Henrissat B, He Q, Lawson PA, Tanner RS, Lynd LR, Wiegel J, Fields MW, Arkin AP, Schadt CW, Stevenson BS, McInerney MJ, Yang Y, Dong H, Xing D, Ren N, Wang A, Huhnke RL, Mielenz JR, Ding SY, Himmel ME, Taghavi S, van der Lelie D, Rubin EM, Zhou J (2010) Sequencing of multiple clostridial genomes related to biomass conversion and biofuel production. J Bacteriol 192:6494–6496
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Henrissat B, Bairoch A (1996) Updating the sequence-based classification of glycosyl hydrolases. Biochem J 316:695–696
Henrissat B, Coutinho PM (2001) Classification of glycoside hydrolases and glycosyltransferases from hyperthermophiles. Methods Enzymol 330:183–201
Henrissat B, Davies G (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 7:637–644
Henrissat B, Callebaut I, Fabrega S, Lehn P, Mornon J-P, Davies G (1995) Conserved catalytic machinery and the prediction of a common fold for several families of glycosyl hydrolases. Proc Natl Acad Sci USA 92:7090–7094
Henrissat B, Teeri TT, Warren RAJ (1998) A scheme for designating enzymes that hydrolyse the polysaccharides in the cell walls of plants. FEBS Lett 425:352–354
Henrissat B, Coutinho PM, Deleury E, Davies GJ (2003) Sequence families and modular organisation of carbohydrate-active enzymes. In: Svendsen A (ed) Enzyme functionality: design, engineering and screening. Marcel Dekker, New York, pp 15–34
Higuchi T (1990) Lignin biochemistry, biosynthesis and biodegradation. Wood Sci Technol 24:23–63
Hilden L, Eng L, Johansson G, Lindqvist SE, Pettersson G (2001) An amperometric cellobiose dehydrogenase-based biosensor can be used for measurement of cellulase activity. Anal Biochem 290(2):245–250
Himmel ME (ed) (2008) Biomass recalcitrance: deconstructing the plant cell wall for bioenergy. Wiley, Inc
Himmel ME, Ruth MF, Wyman CE (1999) Cellulase for commodity products from cellulosic biomass. Curr Opin Biotechnol 10(4):358–364
Himmel ME, Ding S-Y, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315:804–807; Erratum: 316, 982
Himmel ME, Xu Q, Luo Y, Ding S-Y, Lamed R, Bayer EA (2010) Microbial enzyme systems for biomass conversion: emerging paradigms. Biofuels 1:323–341
Hurlbert JC, Preston III JF (2001) Functional characterization of a novel xylanase from corn strain of Erwinia chrysanthemi. J Bacteriol 183:2093–2100
Irwin D, Walker L, Spezio M, Wilson D (1993) Activity studies of eight purified cellulases: specificity, synergism, and binding domain effects. Biotechnol Bioeng 42:1002–1013
Irwin D, Shin D-H, Zhang S, Barr BK, Sakon J, Karplus PA, Wilson DB (1998) Roles of the catalytic domain and two cellulose binding domains of Thermomonospora fusca E4 in cellulose hydrolysis. J Bacteriol 180:1709–1714
Johnson EA, Sakojoh M, Halliwell G, Madia A, Demain AL (1982) Saccharification of complex cellulosic substrates by the cellulase system from Clostridium thermocellum. Appl Environ Microbiol 43:1125–1132
Johnson P, Joshi M, Tomme P, Kilburn D, McIntosh L (1996) Structure of the N-terminal cellulose-binding domain of Cellulomonas fimi Cen C determined by nuclear magnetic resonance spectroscopy. Biochemistry 35:14381–14394
Jung KH, Lee KM, Kim H, Yoon KH, Park SH, Pack MY (1998) Cloning and expression of a Clostridium thermocellum xylanase gene in Escherichia coli. Biochem Mol Biol Int 44(2):283–292
Juy M, Amit AG, Alzari PM, Poljak RJ, Claeyssens M, Béguin P, Aubert J-P (1992) Crystal structure of a thermostable bacterial cellulose-degrading enzyme. Nature 357:39–41
Kahel-Raifer H, Jindou S, Bahari L, Nataf Y, Shoham Y, Bayer EA, Borovok I, Lamed R (2010) The unique set of putative membrane-associated anti-σ factors in Clostridium thermocellum suggests a novel extracellular carbohydrate-sensing mechanism involved in gene regulation. FEMS Microbiol Lett 308:84–93
Kang S, Barak Y, Lamed R, Bayer EA, Morrison M (2006) The functional repertoire of prokaryote cellulosomes includes the serpin superfamily of serine proteinase inhibitors. Mol Microbiol 60:1344–1354
Karita S, Sakka K, Ohmiya K (1997) Cellulosomes, cellulase complexes, of anaerobic microbes: their structure models and functions. In: Onodera R, Itabashi H, Ushida K, Yano H, Sasaki Y (eds) Rumen microbes and digestive physiology in ruminants, vol 14. Japan Sci. Soc. Press, Tokyo/S.Karger, Basel, pp 47–57
Kataeva I, Li X-L, Chen H, Choi SK, Ljungdahl LG (1999a) Cloning and sequence analysis of a new cellulase gene encoding CelK, a major cellulosome component of clostridium thermocellum: evidence for gene duplication and recombination. J Bacteriol 181:5288–5295
Kataeva I, Li X-L, Chen H, Ljungdahl LG (1999b) CelK—a new cellobiohydrolase from clostridium thermocellum cellulosome: role of N-terminal cellulose-binding domain. In: Ohmiya K, Hayashi K, Sakka K, Kobayashi Y, Karita S, Kimura T (eds) Genetics, biochemistry and ecology of cellulose degradation. Uni Publishers, Tokyo, pp 454–460
Kataeva IA, Seidel RD III, Shah A, West LT, Li X-L, Ljungdahl LG (2002) The fibronectin type 3-like repeat from the Clostridium thermocellum cellobiohydrolase CbhA promotes hydrolysis of cellulose by modifying its surface. Appl Environ Microbiol 68:4292–4300
Kataeva IA, Uversky VN, Ljungdahl LG (2003) Calcium and domain interactions contribute to the thermostability of domains of the multimodular cellobiohydrolase, CbhA, a subunit of the Clostridium thermocellum cellulosome. Biochem J 372:151–161
Kataeva IA, Uversky VN, Brewer JM, Schubot F, Rose JP, Wang BC, Ljungdahl LG (2004) Interactions between immunoglobulin-like and catalytic modules in Clostridium thermocellum cellulosomal cellobiohydrolase CbhA. Protein Eng Des Sel 17:759–769
Kataeva IA, Brewer JM, Uversky VN, Ljungdahl LG (2005) Domain coupling in a multimodular cellobiohydrolase CbhA from Clostridium thermocellum. FEBS Lett 579:4367–4373
Kataeva IA, Yang SJ, Dam P, Poole FL, Yin Y, Zhou F, Chou WC, Xu Y, Goodwin L, Sims DR, Detter JC, Hauser LJ, Westpheling J, Adams MW (2009) Genome sequence of the anaerobic, thermophilic, and cellulolytic bacterium “Anaerocellum thermophilum” DSM 6725. J Bacteriol 191:3760–3761
Kidby DK, Davidson DJ (1973) A convenient ferricyanide estimation of reducing sugars in the nanomole range. Anal Biochem 55(1):321–325
Klein-Marcuschamer D, Oleskowicz-Popiel P, Simmons BA, Blanch HW (2011) The challenge of enzyme cost in the production of lignocellulosic biofuels. Biotechnol Bioeng 109:1083–1087
Kondo S, Yamagishi A, Oshima T (1991) Positive selection for uracil auxotrophs of the sulfur-dependent thermophilic archaebacterium Sulfolobus acidocaldarius by use of 5-fluoroorotic acid. J Bacteriol 173:7698–7700
Kosugi A, Murashima K, Doi RH (2002) Characterization of two noncellulosomal subunits, ArfA and BgaA, from Clostridium cellulovorans that cooperate with the cellulosome in plant cell wall degradation. J Bacteriol 184:6859–6865
Koyama M, Helbert W, Imai T, Sugiyama J, Henrissat B (1997) Parallel-up structure evidences the molecular directionality during biosynthesis of bacterial cellulose. Proc Natl Acad Sci USA 94:9091–9095
Kuhad RC, Singh A, Eriksson K-E (1997) Microorganisms and enzymes involved in the degradation of plant fiber cell walls. Adv Biochem Eng Biotechnol 57:45–125
Lamed R, Bayer EA (1988a) The cellulosome concept: exocellular/extracellular enzyme reactor centers for efficient binding and cellulolysis. In: Aubert J-P, Beguin P, Millet J (eds) Biochemistry and genetics of cellulose degradation. Academic, London, pp 101–116
Lamed R, Bayer EA (1988b) The cellulosome of Clostridium thermocellum. Adv Appl Microbiol 33:1–46
Lamed R, Bayer EA (1991) Cellulose degradation by thermophilic anaerobic bacteria. In: Haigler CH, Weimer PJ (eds) Biosynthesis and biodegradation of cellulose and cellulose materials. Marcel Dekker, New York, pp 377–410
Lamed R, Bayer EA (1993) The cellulosome concept—a decade later! In: Shimada K, Hoshino S, Ohmiya K, Sakka K, Kobayashi Y, Karita S (eds) Genetics, biochemistry and ecology of lignocellulose degradation. Uni Publishers, Tokyo, Japan, pp 1–12
Lamed R, Setter E, Bayer EA (1983a) Characterization of a cellulose-binding, cellulase-containing complex in Clostridium thermocellum. J Bacteriol 156:828–836
Lamed R, Setter E, Kenig R, Bayer EA (1983b) The cellulosome—a discrete cell surface organelle of Clostridium thermocellum which exhibits separate antigenic, cellulose-binding and various cellulolytic activities. Biotechnol Bioeng Symp 13:163–181
Lamed E, Naimark J, Morgenstern E, Bayer EA (1987a) Scanning electron microscopic delineation of bacterial surface topology using cationized ferritin. J Microbiol Methods 7:233–240
Lamed R, Naimark J, Morgenstern E, Bayer EA (1987b) Specialized cell surface structures in cellulolytic bacteria. J Bacteriol 169:3792–3800
Laurie JI, Clarke JH, Ciruela A, Faulds CB, Williamson G, Gilbert HJ, Rixon JE, Millward-Sadler J, Hazlewood GP (1997) The NodB domain of a multidomain xylanase from Cellulomonas fimi deacylates acetylxylan. FEMS Microbiol Lett 148:261–264
Lehtio J, Sugiyama J, Gustavsson M, Fransson L, Linder M, Teeri TT (2003) The binding specificity and affinity determinants of family 1 and family 3 cellulose binding modules. Proc Natl Acad Sci USA 100:484–489
Leibovitz E, Béguin P (1996) A new type of cohesin domain that specifically binds the dockerin domain of the Clostridium thermocellum cellulosome-integrating protein CipA. J Bacteriol 178:3077–3084
Leibovitz E, Ohayon H, Gounon P, Béguin P (1997) Characterization and subcellular localization of the Clostridium thermocellum scaffoldin dockerin binding protein SdbA. J Bacteriol 179:2519–2523
Lemaire M, Ohayon H, Gounon P, Fujino T, Béguin P (1995) OlpB, a new outer layer protein of Clostridium thermocellum, and binding of its S-layer-like domains to components of the cell envelope. J Bacteriol 177:2451–2459
Lemaire M, Miras I, Gounon P, Béguin P (1998) Identification of a region responsible for binding to the cell wall within the S-layer protein of Clostridium thermocellum. Microbiology 144:211–217
Lewis NG, Yamamoto E (1990) Lignin: occurrence, biogenesis and biodegradation. Annu Rev Plant Physiol Plant Mol Biol 41:455–496
Li LL, McCorkle SR, Monchy S, Taghavi S, van der Lelie D (2009) Bioprospecting metagenomes: glycosyl hydrolases for converting biomass. Biotechnol Biofuels 18:2–10
Lin C, Urbance JW, Stahl DA (1994) Acetivibrio cellulolyticus and Bacteroides cellulosolvens are members of the greater clostridial assemblage. FEMS Microbiol Lett 124:151–155
Linder M, Teeri TT (1997) The roles and function of cellulose-binding domains. J Biotechnol 57:15–28
Liu CC, Doi RH (1998) Properties of exgS, a gene for a major subunit of the Clostridium cellulovorans cellulosome. Gene 211:39–47
Ljungdahl LG, Eriksson K-E (1985) Ecology of microbial cellulose degradation. Adv Microb Ecol 8:237–299
Lu Y, Zhang YH, Lynd LR (2006) Enzyme-microbe synergy during cellulose hydrolysis by Clostridium thermocellum. Proc Natl Acad Sci USA 103:16165–16169
Lupas A, Engelhardt H, Peters J, Santarius U, Volker S, Baumeister W (1994) Domain structure of the Acetogenium kivui surface layer revealed by electron crystallography and sequence analysis. J Bacteriol 176:1224–1233
Ly HD, Withers SG (1999) Mutagenesis of glycosidases. Annu Rev Biochem 68:487–522
Lykidis A, Mavromatis K, Ivanova N, Anderson I, Land M, DiBartolo G, Martinez M, Lapidus A, Lucas S, Copeland A, Richardson P, Wilson DB, Kyrpides N (2007) Genome sequence and analysis of the soil cellulolytic actinomycete Thermobifida fusca YX. J Bacteriol 189:2477–2486
Lynd LR, Cushman JH, Nichols RJ, Wyman CE (1991) Fuel ethanol from cellulosic biomass. Science 251:1318–1323
Lynd LR, Laser MS, Bransby D, Dale BE, Davison B, Hamilton R, Himmel M, Keller M, McMillan JD, Sheehan J, Wyman CE (2008) How biotech can transform biofuels. Nat Biotechnol 26:169–172
Lytle B, Volkman BF, Westler WM, Wu JHD (2000) Secondary structure and calcium-induced folding of the Clostridium thermocellum dockerin domain determined by NMR spectroscopy. Arch Biochem Biophys 379:237–244
Lytle BL, Volkman BF, Westler WM, Heckman MP, Wu JH (2001) Solution structure of a type I dockerin domain, a novel prokaryotic, extracellular calcium-binding domain. J Mol Biol 307(3):745–753
Marais JP, De Wit JL, Quicke GV (1966) A critical examination of the Nelson-Somogyi method for the determination of reducing sugars. Anal Biochem 15(3):373–381
Marcotte EM, Pellegrini M, Ng H-L, Rice DW, Yeates TO, Eisenberg D (1999) Detecting protein function and protein-protein interactions from genome sequences. Science 285:751–753
Matthews JF, Skopec CE, Mason PE, Zuccato P, Torget RW, Sugiyama J, Himmel ME, Brady JW (2006) Computer simulation studies of microcrystalline cellulose Ibeta. Carbohydr Res 341:138–152
Mattinen M-L, Kontteli M, Kerovuo J, Linder M, Annila A, Lindeberg G, Reinikainen T, Drakenberg T (1997) Three-dimensional structures of three engineered cellulose-binding domains of cellobiohydrolase I from Trichoderma reesei. Protein Sci 6:294–303
Matuschek M, Sahm K, Zibat A, Bahl H (1996) Characterization of genes from Thermoanaerobacterium thermosulfurigenes EM1 that encode two glycosyl hydrolases with conserved S-layer-like domains. Mol Gen Genet 252(4):493–496
Mayer F, Coughlan MP, Mori Y, Ljungdahl LG (1987) Macromolecular organization of the cellulolytic enzyme complex of Clostridium thermocellum as revealed by electron microscopy. Appl Environ Microbiol 53:2785–2792
McCarter JD, Withers SG (1994) Mechanisms of enzymatic glycoside hydrolysis. Curr Opin Struct Biol 4:885–892
Mechaly A, Yaron S, Lamed R, Fierobe H-P, Belaich A, Belaich J-P, Shoham Y, Bayer EA (2000) Cohesin-dockerin recognition in cellulosome assembly: experiment versus hypothesis. Proteins 39:170–177
Mechaly A, Fierobe H-P, Belaich A, Belaich J-P, Lamed R, Shoham Y, Bayer EA (2001) Cohesin-dockerin interaction in cellulosome assembly: a single hydroxyl group of a dockerin domain distinguishes between non-recognition and high-affinity recognition. J Biol Chem 276:9883–9888
Medie FM, Davies GJ, Drancourt M, Henrissat B (2012) Genome analyses highlight the different biological roles of cellulases. Nat Rev Microbiol 10:227–234
Meguro H, Morisaka H, Kuroda K, Miyake H, Tamaru Y, Ueda M (2011) Putative role of cellulosomal protease inhibitors in Clostridium cellulovorans based on gene expression and measurement of activities. J Bacteriol 193:5527–5530
Miller GLR, Blum WE, Burton AL (1960) Measurements of carboxymethylcellulase activity. Anal Biochem 2:127–132
Mishara S, Beguin P, Aubert J-P (1991) Transcription of Clostridium thermocellum endoglucanase genes celF and celD. J Bacteriol 173:80–85
Mitchell WF (1998) Physiology of carbohydrate to solvent conversion by Clostridia. Adv Microbiol Physiol 39:31–130
Mohand-Oussaid O, Payot S, Guedon E, Gelhaye E, Youyou A, Petitdemange H (1999) The extracellular xylan degradative system in clostridium cellulolyticum cultivated on xylan: evidence for cell-free cellulosome production. J Bacteriol 181:4035–4040
Montanier C, van Bueren AL, Dumon C, Flint JE, Correia MA, Prates JA, Firbank SJ, Lewis RJ, Grondin GG, Ghinet MG, Gloster TM, Herve C, Knox JP, Talbot BG, Turkenburg JP, Kerovuo J, Brzezinski R, Fontes CM, Davies GJ, Boraston AB, Gilbert HJ (2009) Evidence that family 35 carbohydrate binding modules display conserved specificity but divergent function. Proc Natl Acad Sci USA 106:3065–3070
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Morag E, Bayer EA, Lamed R (1990) Relationship of cellulosomal and noncellulosomal xylanases of Clostridium thermocellum to cellulose-degrading enzymes. J Bacteriol 172:6098–6105
Morag E, Halevy I, Bayer EA, Lamed R (1991) Isolation and properties of a major cellobiohydrolase from the cellulosome of Clostridium thermocellum. J Bacteriol 173:4155–4162
Morag E, Bayer EA, Hazlewood GP, Gilbert HJ, Lamed R (1993) Cellulase SS (CelS) is synonymous with the major cellobiohydrolase (subunit S8) from the cellulosome of Clostridium thermocellum. Appl Biochem Biotechnol 43:147–151
Morag E, Lapidot A, Govorko D, Lamed R, Wilchek M, Bayer EA, Shoham Y (1995) Expression, purification and characterization of the cellulose-binding domain of the scaffoldin subunit from the cellulosome of Clostridium thermocellum. Appl Environ Microbiol 61:1980–1986
Morrison M, Daugherty SC, Nelson WC, Davidsen T, Nelson KE (2010) The FibRumBa database: a resource for biologists with interests in gastrointestinal microbial ecology, plant biomass degradation, and anaerobic microbiology. Microb Ecol 59:212–213
Nataf Y, Bahari L, Kahel-Raifer H, Borovok I, Lamed R, Bayer EA, Sonenshein AL, Shoham Y (2010b) Clostridium thermocellum cellulosomal genes are regulated by extracytoplasmic polysaccharides via alternative sigma factors. Proc Natl Acad Sci USA 107:18646–18651
Navarro A, Chebrou M-C, Béguin P, Aubert J-P (1991) Nucleotide sequence of the cellulase gene celF of Clostridium thermocellum. Res Microbiol 142:927–936
Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg SL, Smith HO, Venter JC, Fraser CM (1999) Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399:323–329
Newcomb M, Wu JHD (2004) Transcriptional regulators of the Clostridium thermocellum cellulase system. In: Ohmiya K, Sakka K, Karita S, Kimura T, Onishi Y (eds) Biotechnology of lignocellulose degradation and biomass utilization. Uni Publisher, Tokyo, pp 148–154
Newcomb M, Millen J, Chen CY, Wu JH (2011) Co-transcription of the celC gene cluster in Clostridium thermocellum. Appl Microbiol Biotechnol 90:625–634
Ni J, Takehara M, Watanabe H (2005) Heterologous overexpression of a mutant termite cellulase gene in Escherichia coli by DNA shuffling of four orthologous parental cDNAs. Biosci Biotechnol Biochem 69:1711–1720
Noach I, Lamed R, Xu Q, Rosenheck S, Shimon LJW, Kenig R, Bayer EA, Frolow F (2003) Preliminary X-ray characterization and phasing of a type II cohesin domain from the cellulosome of Acetivibrio cellulolyticus. Acta Crystallogr Sect D Biol Struct D59:1670–1673
Noach I, Frolow F, Jakoby H, Rosenheck S, Shimon LJW, Lamed R, Bayer EA (2005) Crystal structure of a type-II cohesin module from the Bacteroides cellulosolvens cellulosome reveals novel and distinctive secondary structural elements. J Mol Biol 348:1–12
Noach IO, Alber EA, Bayer R, Lamed M, Levy-Assaraf LJ, Shimon W, and Frolow F, (2008) Crystallization and preliminary X-ray analysis of Acetivibrio cellulolyticus cellulosomal type II cohesin module: Two versions having different linker lengths. Acta Cryst. F64: 58–61
Noach I, Frolow F, Alber O, Lamed R, Shimon LJW, Bayer EA (2009) Inter-modular linker flexibility revealed from crystal structures of adjacent cellulosomal cohesins of Acetivibrio cellulolyticus. J Mol Biol 391:86–97
Noach I, Levy-Assaraf M, Lamed R, Shimon L, Frolow F, Bayer EA (2010) Modular arrangement of a cellulosomal scaffoldin subunit revealed from the crystal structure of a cohesin dyad. J Mol Biol 399:294–305
Nochur SV, Demain AL, Roberts MF (1990) Mutation of Clostridium thermocellum in the presence of certain carbon sources. FEMS Microbiol Lett 71:199–204
Nochur SV, Demain AL, Roberts MF (1992a) Carbohydrate utilization by Clostridium thermocellum: importance of internal pH in regulating growth. Enzyme Microb Technol 14:338–349
Nochur SV, Jacobson GR, Roberts MF, Demain AL (1992b) Mode of sugar phosphorylation in Clostridium thermocellum. Appl Biochem Biotechnol 33:33–41
Nochur SV, Roberts MF, Demain AL (1993) True cellulase production by Clostridium thermocellum grown on different carbon sources. Biotechnol Lett 15:641–646
Nordon RE, Craig SJ, Foong FC (2009) Molecular engineering of the cellulosome complex for affinity and bioenergy applications. Biotechnol Lett 31:465–476
Notenboom V, Birsan C, Warren R, Withers S, Rose D (1998) Exploring the cellulose/xylan specificity of the β-1,4-glycanase Cex from Cellulomonas fimi through crystallography and mutation. Biochemistry 37:4751–4758
Ohmiya K, Sakka K, Karita S, Kimura T (1997) Structure of cellulases and their applications. Biotechnol Genet Eng Rev 14:365–414
Olson DG, Tripathi SA, Giannone RJ, Lo J, Caiazza NC, Hogsett DA, Hettich RL, Guss AM, Dubrovsky G, Lynd LR (2010) Deletion of the Cel48S cellulase from Clostridium thermocellum. Proc Natl Acad Sci USA 107:17727–17732
O'Neill G, Goh SH, Warren RA, Kilburn DG, Miller RC (1986) Structure of the gene encoding the exoglucanase of Cellulomonas fimi. Gene 44(2–3):325–330
O'Neill RA, Darvill A, Albersheim P (1989) A fluorescence assay for enzymes that cleave glycosidic linkages to produce reducing sugars. Anal Biochem 177(1):11–15
O'Sullivan AC (1997) Cellulose: the structure slowly unravels. Cellulose 4:173–207
Ozdemir I, Blumer-Schuette SE, Kelly RM (2012) S-layer homology domain proteins Csac_0678 and Csac_2722 are implicated in plant polysaccharide deconstruction by the extremely thermophilic bacterium Caldicellulosiruptor saccharolyticus. Appl Environ Microbiol 78:768–777
Pagès S, Belaich A, Belaich J-P, Morag E, Lamed R, Shoham Y, Bayer EA (1997) Species-specificity of the cohesin-dockerin interaction between Clostridium thermocellum and Clostridium cellulolyticum: prediction of specificity determinants of the dockerin domain. Proteins 29:517–527
Parsiegla G, Juy M, Reverbel-Leroy C, Tardif C, Belaich JP, Driguez H, Haser R (1998) The crystal structure of the processive endocellulase CelF of Clostridium cellulolyticum in complex with a thiooligosaccharide inhibitor at 2.0 Å resolution. EMBO J 17:5551–5562
Peer A, Smith SP, Bayer EA, Lamed R, Borovok I (2009) Non-cellulosomal cohesin and dockerin-like modules in the three domains of life. FEMS Microbiol Lett 291:1–16
Perlack RD, Wright LL, Turhollow AF, Graham RL, Stokes BJ, Erbach DC (2005) Biomass as feedstock for a bioenergy and bioproduct industry: the technical feasibility of a billion-ton annual supply. A joint study sponsored by the U.S. Department of Energy and the U.S. Department of Agriculture. Oak Ridge National Laboratory, Oak Ridge
Petkun S, Jindou S, Shimon LJW, Rosenheck S, Bayer EA, Lamed R, Frolow F (2010) Structure of a family 3b' carbohydrate-binding module from the Cel9V glycoside hydrolase from Clostridium thermocellum. Structural diversity and implications for carbohydrate binding. Acta Cryst D66:33–43
Pinheiro BA, Proctor MR, Martinez-Fleites CC, Prates JAM, Money VA, Davies GJ, Bayer EA, Fontes CMGA, Fierobe H-P, Gilbert HJ (2008) The Clostridium cellulolyticum dockerin displays a dual binding mode for its cohesin partner. J Biol Chem 283:18422–18430
Poole DM, Morag E, Lamed R, Bayer EA, Hazlewood GP, Gilbert HJ (1992) Identification of the cellulose binding domain of the cellulosome subunit S1 from Clostridium thermocellum. FEMS Microbiol Lett 99:181–186
Puls J, Schuseil J (1993) Chemistry of hemicellulases: relationship between hemicellulose structure and enzymes required for hydrolysis. In: Coughlan MP, Hazlewood GP (eds) Hemicellulose and hemicellulases. Portland Press, London, pp 1–27
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ Jr, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T (2006) The path forward for biofuels and biomaterials. Science 311:484–489
Raman B, Pan C, Hurst GB, Rodriguez M Jr, McKeown CK, Lankford PK, Samatova NF, Mielenz JR (2009) Impact of pretreated Switchgrass and biomass carbohydrates on Clostridium thermocellum ATCC 27405 cellulosome composition: a quantitative proteomic analysis. PLoS One 4:e5271
Raman B, McKeown CK, Rodriguez M, Brown SD, Mielenz JR (2011) Transcriptomic analysis of Clostridium thermocellum ATCC 27405 cellulose fermentation. BMC Microbiol 11:134
Reese RT (1976) History of the cellulase program at the U.S. Army Natick Development Center. Biotechnol Bioeng Symp 6:9–20
Reeves RA, Gibbs MD, Morris DD, Griffiths KR, Saul DJ, Bergquist PL (2000) Sequencing and expression of additional xylanase genes from the hyperthermophile Thermotoga maritima FjSS3B.1. Appl Environ Microbiol 66(4):1532–1537
Reverbel-Leroy C, Pagés S, Belaich A, Belaich J-P, Tardif C (1997) The processive endocellulase CelF, a major component of the clostridium cellulolyticum cellulosome: purification and characterization of the recombinant form. J Bacteriol 179:46–52
Ridley BL, O'Neill MA, Mohnen D (2001) Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 57:929–967
Riederer A, Takasuka TE, Makino S, Stevenson DM, Bukhman YV, Elsen NL, Fox BG (2011) Global gene expression patterns in Clostridium thermocellum as determined by microarray analysis of chemostat cultures on cellulose or cellobiose. Appl Environ Microbiol 77:1243–1253
Rincon MT, Ding S-Y, McCrae SI, Martin JC, Aurilia V, Lamed R, Shoham Y, Bayer EA, Flint HJ (2003) Novel organization and divergent dockerin specificities in the cellulosome system of Ruminococcus flavefaciens. J Bacteriol 185:703–713
Rincon MT, Martin JC, Aurilia V, McCrae SI, Rucklidge G, Reid M, Bayer EA, Lamed R, Flint HJ (2004) ScaC, an adaptor protein carrying a novel cohesin that expands the dockerin-binding repertoire of the Ruminococcus flavefaciens 17 cellulosome. J Bacteriol 186:2576–2585
Rincon MT, Cepeljnik T, Martin JC, Lamed R, Barak Y, Bayer EA, Flint HJ (2005) Unconventional mode of attachment of the Ruminococcus flavefaciens cellulosome to the cell surface. J Bacteriol 187:7569–7578
Rincon MT, Cepeljnik T, Martin JC, Barak Y, Lamed R, Bayer EA, Flint HJ (2007) A novel cell surface-anchored cellulose-binding protein encoded by the sca gene cluster of Ruminococcus flavefaciens. J Bacteriol 189:4774–7283
Rincon MT, Dassa B, Flint HJ, Travis AR, Jindou S, Borovok I, Lamed R, Bayer EA, Henrissat B, Coutinho PM, Antonopoulos DA, Berg ME, White BA (2010) Abundance and diversity of dockerin-containing proteins in the fiber-degrading rumen bacterium, Ruminococcus flavefaciens FD1. PLoS One 5:e12476
Rouvinen J, Bergfors T, Teeri T, Knowles JKC, Jones TA (1990) Three-dimensional structure of cellobiohydrolase II from Trichoderma reesei. Science 279:380–386
Rydlund A, Dahlman O (1997) Oligosaccharides obtained by enzymatic hydrolysis of birch kraft pulp xylan: analysis by capillary zone electrophoresis and mass spectrometry. Carbohydr Res 300:95–102
Rye CS, Withers SG (2000) Glycosidase mechanisms. Curr Opin Chem Biol 4:573–580
Sakon J, Irwin D, Wilson DB, Karplus PA (1997) Structure and mechanism of endo/exocellulase E4 from Thermomonospora fusca. Nat Struct Biol 4:810–818
Salamitou S, Lemaire M, Fujino T, Ohayon H, Gounon P, Béguin P, Aubert J-P (1994a) Subcellular localization of Clostridium thermocellum ORF3p, a protein carrying a receptor for the docking sequence borne by the catalytic components of the cellulosome. J Bacteriol 176:2828–2834
Salamitou S, Raynaud O, Lemaire M, Coughlan M, Béguin P, Aubert J-P (1994b) Recognition specificity of the duplicated segments present in Clostridium thermocellum endoglucanase CelD and in the cellulosome-integrating protein CipA. J Bacteriol 176:2822–2827
Saul DJ, Williams LC, Grayling RA, Chamley LW, Love DR, Bergquist PL (1990) celB, a gene coding for a bifunctional cellulase from the extreme thermophile “Caldocellum saccharolyticum”. Appl Environ Microbiol 56(10):3117–3124
Schirner K, Errington J (2009) The cell wall regulator {sigma}I specifically suppresses the lethal phenotype of mbl mutants in Bacillus subtilis. J Bacteriol 191:1404–1413
Schloss PD, Handelsman J (2003) Biotechnological prospects from metagenomics. Curr Opin Biotechnol 14:303–310
Schneider JC, Jenings AF, Mun DM, McGovern PM, Chew LC (2005) Auxotrophic markers pyrF and proC can replace antibiotic markers on protein production plasmids in high-cell-density Pseudomonas fluorescens fermentation. Biotechnol Prog 21:343–348
Schubert C (2006) Can biofuels finally take center stage? Nat Biotechnol 24:777–784
Schülein M (1997) Enzymatic properties of cellulases from Humicola insolens. J Biotechnol 57:71–81
Schwarz WH, Zverlov VV (2006) Protease inhibitors in bacteria: an emerging concept for the regulation of bacterial protein complexes? Mol Microbiol 60:1323–1326
Sheehan JJ (2009) Biofuels and the conundrum of sustainability. Curr Opin Biotechnol 20:318–324
Shen H, Meinke A, Tomme P, Damude HG, Kwan E, Kilburn DG, Miller RC Jr, Warren RAJ, Gilkes NR (1995) Cellulomonas fimi cellobiohydrolases. In: Saddler JN, Penner MH (eds) Enzymatic degradation of insoluble polysaccharides. American Chemical Society, Washington, DC, pp 174–196
Shimon LJW, Bayer EA, Morag E, Lamed R, Yaron S, Shoham Y, Frolow F (1997) The crystal structure at 2.15 Å resolution of a cohesin domain of the cellulosome from Clostridium thermocellum. Structure 5:381–390
Shoham Y, Lamed R, Bayer EA (1999) The cellulosome concept as an efficient microbial strategy for the degradation of insoluble polysaccharides. Trends Microbiol 7:275–281
Shoseyov O, Takagi M, Goldstein MA, Doi RH (1992) Primary sequence analysis of Clostridium cellulovorans cellulose binding protein A. Proc Natl Acad Sci USA 89:3483–3487
Simpson HD, Barras F (1999) Functional analysis of the carbohydrate-binding domains of Erwinia chrysanthemi Cel5 (Endoglucanase Z) and an Escherichia coli putative chitinase. J Bacteriol 181(15):4611–4616
Simpson PJ, Bolam DN, Cooper A, Ciruela A, Hazlewood GP, Gilbert HJ, Williamson MP (1999) A family IIb xylan-binding domain has a similar secondary structure to a homologous family IIa cellulose-binding domain but different ligand specificity. Structure Fold Des 7:853–864
Sinnott ML (1990) Catalytic mechanisms of enzymic glycosyl transfer. Chem Rev 90:1171–1202
Sleat R, Mah RA, Robinson R (1984) Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium cellulovorans, sp. nov. Appl Environ Microbiol 48:88–93
Sonenshein AL (2007) Control of key metabolic intersections in Bacillus subtilis. Nat Rev Microbiol 5:917–927
Spinelli S, Fierobe HP, Belaich A, Belaich JP, Henrissat B, Cambillau C (2000) Crystal structure of a cohesin module from Clostridium cellulolyticum: implications for dockerin recognition. J Mol Biol 304(2):189–200
Srisodsuk M, Kleman-Leyer K, Keranen S, Kirk TK, Teeri TT (1998) Modes of action on cotton and bacterial cellulose of a homologous endoglucanase-exoglucanase pair from Trichoderma reesei. Eur J Biochem 251(3):885–892
Stevenson DM, Weimer PJ (2005) Expression of 17 genes in Clostridium thermocellum ATCC 27405 during fermentation of cellulose or cellobiose in continuous culture. Appl Environ Microbiol 71:4672–4678
Stutzenberger F (1990) Bacterial cellulases. In: Fogarty WM, Kelly CT (eds) Microbial enzymes and biotechnology. Elsevier Applied Science, London/New York, pp 37–70
Sugiyama J, Vuong R, Chanzy H (1991) Electron diffraction study of the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 24:4168–4175
Tamaru Y, Doi RH (1999) Three surface layer homology domains at the N terminus of the Clostridium cellulovorans major cellulosomal subunit EngE. J Bacteriol 181:3270–3276
Tamaru Y, Doi RH (2000) The engL gene cluster of Clostridium cellulovorans contains a gene for cellulosomal ManA. J Bacteriol 182:244–247
Tamaru Y, RH Doi (2001) Pectate lyase A, an enzymatic subunit of the Clostridium cellulovorans cellulosome. Proc Natl Acad Sci USA 20: 20
Tamaru Y, Liu C-C, Ichi-ishi A, Malburg L, Doi RH (1999) The Clostridium cellulovorans cellulosome and non-cellulosomal cellulases. In: Ohmiya K, Hayashi K, Sakka K, Kobayashi Y, Karita S, Kimura T (eds) Genetics, biochemistry and ecology of cellulose degradation. Uni Publishers, Tokyo, pp 488–494
Tamaru Y, Karita S, Ibrahim A, Chan H, Doi RH (2000) A large gene cluster for the Clostridium cellulovorans cellulosome. J Bacteriol 182(20):5906–5910
Tamaru Y, H Miyake, K Kuroda, A Nakanishi, Y Kawade, K Yamamoto, M Uemura, Y Fujita, RH Doi, M Ueda (2010) Genome sequence of the cellulosome-producing mesophilic organism Clostridium cellulovorans 743B. J Bacteriol 192: 901–902
Tavares GA, Béguin P, Alzari PM (1997) The crystal structure of a type I cohesin domain at 1.7 Å resolution. J Mol Biol 273:701–713
Taylor LE 2nd, Henrissat B, Coutinho PM, Ekborg NA, Hutcheson SW, Weiner RM (2006) Complete cellulase system in the marine bacterium Saccharophagus degradans strain 2-40T. J Bacteriol 188:3849–3861
Teeri TT (1997) Crystalline cellulose degradation: new insight into the function of cellobiohydrolases. Trends Biotechnol 15:160–167
Teeri TT, Reinikainen T, Ruohonen L, Jones TA, Knowles JKC (1992) Domain function in Trichoderma reesei cellulases. J Biotechnol 24:169–176
Te'o VS, Saul DJ, Bergquist PL (1995) CelA, another gene coding for a multidomain cellulase from the extreme thermophile Caldocellum saccharolyticum. Appl Microbiol Biotechnol 43:291–296
Timell TE (1967) Recent progress in the chemistry of wood hemicelluloses. Wood Sci Technol 1:45–70
Tokatlidis K, Salamitou S, Béguin P, Dhurjati P, Aubert J-P (1991) Interaction of the duplicated segment carried by Clostridium thermocellum cellulases with cellulosome components. FEBS Lett 291:185–188
Tokatlidis K, Dhurjati P, Béguin P (1993) Properties conferred on Clostridium thermocellum endoglucanase CelC by grafting the duplicated segment of endoglucanase CelD. Protein Eng 6(8):947–952
Tomme P, Warren RAJ, Gilkes NR (1995a) Cellulose hydrolysis by bacteria and fungi. Adv Microb Physiol 37:1–81
Tomme P, Warren RAJ, Miller RC, Kilburn DG, Gilkes NR (1995b) Cellulose-binding domains—Classification and properties. In: Saddler JM, Penner MH (eds) Enzymatic degradation of insoluble polysaccharides. American Chemical Society, Washington, D.C., pp 142–161
Tomme P, Creagh AL, Kilburn DG, Haynes CA (1996) Interaction of polysaccharides with the N-terminal cellulose-binding domain of Cellulomonas fimi CenC. Biochemistry 35:13885–13894
Tormo J, Lamed R, Chirino AJ, Morag E, Bayer EA, Shoham Y, Steitz TA (1996) Crystal structure of a bacterial family-III cellulose-binding domain: a general mechanism for attachment to cellulose. EMBO J 15:5739–5751
Tripathi SA, Olson DG, Argyros DA, Miller BB, Barrett TF, Murphy DM, McCool JD, Warner AK, Rajgarhia VB, Lynd LR, Hogsett DA, Caiazza NC (2010) Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl Environ Microbiol 76:6591–6599
Tull D, Withers SG (1994) Mechanisms of cellulases and xylanases: a detailed kinetic study of the exo-β-1,4-glycanase from Cellulomonas fimi. Biochemistry 33(20):6363–6370
van Tilbeurgh H, G Pettersson, R Bhikabhai, H De Boeck, M Claeyssens (1985) Studies of the cellulolytic system of Trichoderma reesei QM 9414. Reaction specificity and thermodynamics of interactions of small substrates and ligands with the 1,4-β-glucan cellobiohydrolase II. Eur J Biochem 148(2):329–334
Vazana Y, Moraïs S, Barak Y, Lamed R, Bayer EA (2010) Interplay between Clostridium thermocellum family-48 and family-9 cellulases in the cellulosomal versus non-cellulosomal states. Appl Environ Microbiol 76:3236–3243
Viikari L, and Teeri T, Eds. (1997) Biochemistry and genetics of cellulases and hemicellulases and their application. J. Biotechnol. 57: 1–228
Vlasenko EY, Ryan AI, Shoemaker CF, Shoemaker SP (1998) The use of capillary viscometry, reducing end-group analysis, and size exclusion chromatography combined with multi-angle laser light scattering to characterize endo-1,4-β-D-glucanases on carboxymethylcellulose: a comparative evaluation of three methods. Enzyme Microb Technol 23:350–359
Voronov-Goldman M, Noach I, Lamed R, Shimon LJW, Borovok I, Bayer EA, Frolow F (2009) Crystallization and preliminary X-ray analysis of a cohesin-like module from ORF 2375 of the archaeon Archaeoglobus fulgidus. Acta Cryst F65:275–278
Waffenschmidt S, Jaenicke L (1987) Assay of reducing sugars in the nanomole range with 2,2′-bicinchoninate. Anal Biochem 165:337–340
Wall JD, Harwood CS, Demain A (eds) (2008) Bioenergy. ASM Press, Washington, DC
Wang WK, Kruus K, Wu JHD (1993) Cloning and DNA sequence of the gene coding for Clostridium thermocellum cellulase SS (CelS), a major cellulosome component. J Bacteriol 175:1293–1302
Wang WK, Kruus K, Wu JHD (1994) Cloning and expression of the Clostridium thermocellum cellulase celS gene in Escherichia coli. Appl Microbiol Biotechnol 42:346–352
Warnecke F, Luginbuhl P, Ivanova N, Ghassemian M, Richardson TH, Stege JT, Cayouette M, McHardy AC, Djordjevic G, Aboushadi N, Sorek R, Tringe SG, Podar M, Martin HG, Kunin V, Dalevi D, Madejska J, Kirton E, Platt D, Szeto E, Salamov A, Barry K, Mikhailova N, Kyrpides NC, Matson EG, Ottesen EA, Zhang X, Hernandez M, Murillo C, Acosta LG, Rigoutsos I, Tamayo G, Green BD, Chang C, Rubin EM, Mathur EJ, Robertson DE, Hugenholtz P, Leadbetter JR (2007) Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 450:560–565
Warren RAJ (1996) Microbial hydrolysis of polysaccharides. Annu Rev Microbiol 50:183–212
Watson BJ, Zhang H, Longmire AG, Moon YH, Hutchenson SW (2009) Processive endoglucanases mediate degradation of cellulose by Saccharophagus degradans. J Bacteriol 191:5697–5705
Weiner RM, Taylor LE, Henrissat B, Hauser L, Land M, Coutinho PM, Rancurel C, Saunders EH, Longmire AG, Zhang H, Bayer EA, Gilbert HJ, Larimer F, Zhulin IB, Lamed R, Richardson PM, Borovok I, Hutcheson S (2008) Complete genome sequence of the complex carbohydrate-degrading marine bacterium, Saccharophagus degradans strain 2-40. PLoS Genet 4:e1000087
White A, Rose DR (1997) Mechanism of catalysis by retaining β-glycosyl hydrolases. Curr Opin Struct Biol 7:645–651
Whittle DJ, Kilburn DG, Warren RA, Miller RC (1982) Molecular cloning of a Cellulomonas fimi cellulose gene in Escherichia coli. Gene 17(2):139–145
Williams SJ, Withers SG (2000) Glycosyl fluorides in enzymatic reactions. Carbohydr Res 327:27–46
Williamson MP, Simpson PJ, Bolam DN, Hazlewood GP, Ciruela A, Cooper A, Gilbert HJ (1999) How the N-terminal xylan-binding domain from C. fimi xylanase D recognises xylan. In: Gilbert HJ, Davies GJ, Henrissat B, Svensson B (eds) Recent advances in carbohydrate bioengineering. The Royal Society of Chemistry, Cambridge, pp 212–220
Wilson DB (1992) Biochemistry and genetics of actinomycete cellulases. Crit Rev Biotechnol 12:45–63
Wilson DB (2004) Studies of Thermobifida fusca plant cell wall degrading enzymes. Chem Rec 4:72–82
Wilson DB (2008) Three microbial strategies for plant cell wall degradation. Ann N Y Acad Sci 1125:289–297
Wilson DB (2009) Cellulases and biofuels. Curr Opin Biotechnol 20:295–299
Wilson DB, Irwin DC (1999) Genetics and properties of cellulases. Adv Biochem Eng 65:1–21
Withers SG (2001) Mechanisms of glycosyl transferases and hydrolases. Carbohydr Res 44:325–337
Withers SG, Aebersold R (1995) Approaches to labeling and identification of active site residues in glycosidases. Protein Sci 4(3):361–372
Wood WA, Kellogg ST (eds) (1988) Biomass (Part A: cellulose and hemicellulose), vol 160. Academic, San Diego
Wu JHD, Orme-Johnson WH, Demain AL (1988) Two components of an extracellular protein aggregate of Clostridium thermocellum together degrade crystalline cellulose. Biochemistry 27:1703–1709
Xie G, Bruce DC, Challacombe JF, Chertkov O, Detter JC, Gilna P, Han CS, Lucas S, Misra M, Myers GL, Richardson P, Tapia R, Thayer N, Thompson LS, Brettin TS, Henrissat B, Wilson DB, McBride MJ (2007) Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl Environ Microbiol 73:3536–3546
Xu Q, Morrison M, Bayer EA, Atamna N, Lamed R (2004) A novel family of carbohydrate-binding modules identified with Ruminococcus albus proteins. FEBS Lett 566:11–16
Xu Q, Singh A, Himmel ME (2009) Perspectives and new directions for the production of bioethanol using consolidated bioprocessing of lignocellulose. Curr Opin Biotechnol 20:364–371
Yagüe E, Béguin P, Aubert J-P (1990) Nucleotide sequence and deletion analysis of the cellulase-encoding gene celH of Clostridium thermocellum. Gene 89:61–67
Yaron S, Morag E, Bayer EA, Lamed R, Shoham Y (1995) Expression, purification and subunit-binding properties of cohesins 2 and 3 of the Clostridium thermocellum cellulosome. FEBS Lett 360:121–124
Zechel DL, Withers SG (2000) Glycosidase mechanisms: anatomy of a finely tuned catalyst. Acc Chem Res 33:11–18
Zhang H, Hutcheson SW (2011) Complex expression of the cellulolytic transcriptome of Saccharophagus degradans. Appl Environ Microbiol 77:5591–5596
Zhang YH, Lynd LR (2005) Regulation of cellulase synthesis in batch and continuous cultures of Clostridium thermocellum. J Bacteriol 187:99–106
Zou J, Kleywegt GJ, Stahlberg J, Driguez H, Nerinckx W, Claeyssens M, Koivula A, Teeri TT, Jones TA (1999) Crystallographic evidence for substrate ring distortion and protein conformational changes during catalysis in cellobiohydrolase Ce16A from Trichoderma reesei. Structure Fold Des 7:1035–1045
Zuber U, Drzewiecki K, Hecker M (2001) Putative sigma factor SigI (YkoZ) of Bacillus subtilis is induced by heat shock. J Bacteriol 183:1472–1475
Zverlov VV, Laptev DA, Tishkov VI, Velikodvorskaja GA (1991) Nucleotide sequence of the Clostridium thermocellum laminarinase gene. Biochem Biophys Res Commun 181:507–512
Zverlov VV, Liebl W, Bachleitner M, Schwarz WH (1998a) Nucleotide sequence of arfB of Clostridium stercorarium, and prediction of catalytic residues of α-L-arabinofuranosidases based on local similarity with several families of glycosyl hydrolases. FEMS Microbiol Lett 164:337–343
Zverlov VV, Mahr S, Riedel K, Bronnenmeier K (1998b) Properties and gene structure of a bifunctional cellulolytic enzyme (CelA) from the extreme thermophile Anaerocellum thermophilum with separate glycosyl hydrolase family 9 and 48 catalytic domains. Microbiology 144:457–465
Zverlov VV, Velikodvorskaya GV, Schwarz WH, Bronnenmeier K, Kellermann J, Staudenbauer WL (1998c) Multidomain structure and cellulosomal localization of the Clostridium thermocellum cellobiohydrolase CbhA. J Bacteriol 180:3091–3099
Zverlov VV, Velikodvorskaya GV, Schwarz WH, Kellermann J, Staudenbauer WL (1999) Duplicated Clostridium thermocellum cellobiohydrolase gene encoding cellulosomal subunits S3 and S5. Appl Microbiol Biotechnol 51:852–859
Zverlov VV, Volkov IY, Velikodvorskaya GA, Schwarz WH (2001) The binding pattern of two carbohydrate-binding modules of laminarinase Lam16A from Thermotoga neapolitana: differences in β-glucan binding within family CBM4. Microbiology 147(Pt 3):621–629
Zverlov VV, Fuchs K-P, Schwarz WH (2002) Chi18A, the endochitinase in the cellulosome of the thermophilic, cellulolytic bacterium Clostridium thermocellum. Appl Environ Microbiol 68:3176–3179
Zverlov VV, Kellermann J, Schwarz WH (2005a) Functional subgenomics of Clostridium thermocellum cellulosomal genes: identification of the major catalytic components in the extracellular complex and detection of three new enzymes. Proteomics 5:3646–3653
Zverlov VV, Schantz N, Schmitt-Kopplin P, Schwarz WH (2005b) Two new major subunits in the cellulosome of Clostridium thermocellum: xyloglucanase Xgh74A and endoxylanase Xyn10D. Microbiology 151:3395–3401
Acknowledgments
Grants from the Israel Science Foundation (administered by the Israel Academy of Sciences and Humanities, Jerusalem), the US-Israel Binational Foundation (BSF), the Israel Ministry of Science (IMOS), and by the Weizmann Institute of Science Alternative Energy Research Initiative (AERI) are greatly appreciated. The authors are also pleased to acknowledge the establishment of an Israeli Center of Research Excellence (I-CORE) managed by the Israel Science Foundation (grant No 152/11) and additional support by the Technion-Niedersachsen Research Cooperation Program. Y.S. holds the Erwin and Rosl Pollak Chair in Biotechnology at the Technion, E.A.B. is the incumbent of The Maynard I., and Elaine Wishner Chair of Bio-organic Chemistry.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Bayer, E.A., Shoham, Y., Lamed, R. (2013). Lignocellulose-Decomposing Bacteria and Their Enzyme Systems. In: Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30141-4_67
Download citation
DOI: https://doi.org/10.1007/978-3-642-30141-4_67
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30140-7
Online ISBN: 978-3-642-30141-4
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences