
Abstract
This chapter covers one of the microbiological steps of the nitrogen cycle, nitrification, which is the biological oxidation of reduced forms of inorganic nitrogen to nitrite and nitrate. Nitrifying bacteria use the oxidation of inorganic nitrogen compounds as their major energy source. Reactions are catalyzed by two physiological groups of bacteria: ammonia-oxidizing bacteria, which gain energy from oxidation of ammonia to nitrite, and nitrite-oxidizing bacteria, which thrive by oxidizing nitrite to nitrate. Because of the toxic nature of nitrite, its rapid conversion to nitrate, assimilated by plants and microorganisms, is essential. Ammonia oxidizers are lithoautotrophic organisms using carbon dioxide as the main carbon source; ammonia monooxygenase oxidizes ammonia to hydroxylamine, which is converted to nitrite by the hydroxylamine oxidoreductase. When grown lithotrophically with nitrite, nitrite is oxidized to nitrate by the nitrite oxidoreductase and the oxygen atom in the nitrate molecule is derived from water. The enzyme also reduces nitrite to nitrate when Nitrobacter strains are grown heterotrophically in the presence of nitrate. Detailed schemes for electron flow and energy transduction as well as energy generation schemes are outlined and the role of nitrifying bacteria in the environment highlighted. The two groups of nitrifying bacteria are phylogenetically unrelated, as they are found in different classes of Proteobacteria and members of the nitrite oxidizers are even found in different phyla. This chapter also covers the physiology and phylogeny of recently detected anaerobic ammonium-oxidizing deep-branching members of the phylum Planctomycetes and of Nitrosomonas eutropha.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Life depends on the element nitrogen. In nature, nitrogen exists mainly in the oxidation states -III (NH3), O (N2), +I (N2O), +II (NO), +III (NO −2 ), +IV (NO2), and +V (NO −3 ). Owing to nitrogen transformations by the activity of living organisms and to chemical instability, any form of oxidation state has only a transient existence. Dinitrogen (N2) is the most inert and frequent constituent of the atmosphere.
Taking into account also abiotic transformations, three cycles of nitrogen can be distinguished:
-
1.
The cycle of the atmosphere
-
2.
The interaction between the atmosphere and the biosphere
-
3.
The cycle of the biosphere
The nitrogen cycle mediated by the biosphere (Fig. 3.1 ) can also be characterized by mobilization and immobilization of nitrogen compounds. Most of the reactions are catalyzed exclusively by prokaryotes. By microbial nitrogen fixation, dinitrogen is reduced to ammonia and subsequently transferred to amino acids and assimilated into cell material. On the other hand, ammonia is released from organic nitrogen compounds by microbial activity called “ammonification” or “mineralization.” Ammonia (NH3)/ammonium (NH +4 ) is the most frequently found form of nitrogen in the biosphere and is transferred efficiently over long distances via volatilization. In contrast, nitrite is usually found in trace amounts in aerobic habitats and only accumulates at low oxygen partial pressure, for example, in soil with high water potential. Because of the toxicity of nitrite for living organisms, the maintenance of low nitrite concentration in aerobic habitats is essential. Under oxic conditions, ammonia and nitrite are not stable and are converted to nitrate by nitrifying bacteria. Nitrification, the biological oxidation of reduced forms of inorganic nitrogen to nitrite and nitrate, is catalyzed by two physiological groups of bacteria. Ammonia-oxidizing bacteria, which use ammonia and not ammonium as substrate (Suzuki et al. 1974), gain energy from oxidation of ammonia to nitrite, and nitrite-oxidizing bacteria thrive by oxidizing nitrite to nitrate. In seawater and freshwater as well as in soil, nitrite produced by the ammonia oxidizers is immediately consumed by nitrite oxidizers, and thus, the nitrite concentration is extremely low in these environments (El-Demerdash and Ottow 1983; Schmidt 1982). Nitrate can be assimilated by plants and microorganisms. Under anoxic or oxygen-limited conditions, nitrate is used as electron acceptor for anaerobic respiration (if organic matter is available) and thereby converted to ammonia (respiratory ammonification) or dinitrogen (denitrification).


Nitrogen cycle mediated by the biosphere
This chapter focuses on nitrifying bacteria, which use the oxidation of inorganic nitrogen compounds as their major energy source. Lithotrophic nitrifiers are Gram-negative bacteria and conventionally have been placed in the family Nitrobacter iaceae (Buchanan 1917; Watson 1971; Watson et al. 1989). However, phylogenetically the lithoautotrophic ammonia oxidizers, characterized by the prefix Nitroso-, and nitrite oxidizers, characterized by the prefix Nitro-, are not closely related (Teske et al. 1996; Purkhold et al. 2000). Comparative 16S rRNA sequence analysis demonstrated that all recognized ammonia oxidizers are either members of the β- or γ-subclass of Proteobacteria (Fig. 3.2 ). The genera Nitrosomonas (including Nitrosococcus mobilis), Nitrosospira, Nitrosolobus, and Nitrosovibrio form a closely related monophyletic assemblage within the β-subclass of Proteobacteria (Head et al. 1993; Woese et al. 1984; Teske et al. 1994; Utåker et al. 1995; Pommerening-Röser et al. 1996; Purkhold et al. 2000), whereas the genus Nitrosococcus constitutes a separate branch within the γ-subclass of Proteobacteria (Woese et al. 1985; Purkhold et al. 2000). Among the nitrite oxidizers, the genera Nitrobacter, Nitrococcus, and Nitrospina were assigned to the α-, γ-, and γ-subclass of Proteobacteria, respectively (Orso et al. 1994; Teske et al. 1994). Nitrite oxidizers of the genus Nitrospira are affiliated with the recently described Nitrospira phylum, which represents an independent line of descent within the domain Bacteria (Ehrich et al. 1995).


16S rRNA-based tree reflecting the phylogenetic relationship of ammonia- and nitrite-oxidizing bacteria. The consensus tree is based on the results of a maximum likelihood analysis of the 16S rRNA primary structure data from the nitrifying bacteria shown in the tree and a selection of reference sequences. Only homologous positions that share identical residues in at least 50 % of all available almost complete bacterial 16S rRNA sequences were included for tree reconstruction. In the tree, ammonia oxidizers are labeled green, and nitrite oxidizers are depicted in red. It should be noted that the assignment of the genus Nitrospina to the δ-Proteobacteria is tentative and might change if additional reference sequences become available. Multifurcations connect lineages for which no unambiguous branching order could be retrieved using different treeing methods. Bar represents 10 % estimated sequence divergence
The most important character of lithotrophic nitrifying bacteria is energy generation via ammonia oxidation to nitrite (section “Biochemistry of Ammonia-Oxidizing Bacteria”) and nitrite oxidation to nitrate (section “Biochemistry of Nitrite-Oxidizing Bacteria”), respectively, according to the following equations:
Equations 3.1 and 3.2 describe the two half-reactions of ammonia oxidation to the intermediate hydroxylamine (NH2OH). The total reaction is given in Eq. 3.3. For hydroxylamine oxidation, no oxygen is consumed (Eq. 3.4). Subsequently two electrons are transferred back to reaction 3.2, and the remaining two electrons pass to the respiratory chain (Eq. 3.5). The second step of ammonia oxidation, the hydroxylamine oxidation, is depicted in Eq. 3.6. The overall reaction (Eq. 3.7) shows that biogenic ammonia oxidation causes nitric acid production. The δG0′ value of reaction 3.7 is significantly higher than that of nitrite oxidation (Eq. 3.10). Nitrite oxidation starts with Eq. 3.8. Electrons are released and penetrate the respiratory chain at the cytochrome c level (Eq. 3.9). There is no acid production when nitrite is oxidized to nitrate (Eq. 3.10).
Ammonia oxidation is initiated by the enzyme ammonia monooxygenase (AMO; sections “Enzymes Involved in Ammonia Oxidation”; “Ammonia Monooxygenase”), which oxidizes ammonia to hydroxylamine. Substrates for AMO are ammonia (Wood 1986), dioxygen, and two electrons. One atom of molecular oxygen is reduced to water, while the second oxygen atom is incorporated to form hydroxylamine. The intermediate hydroxylamine is further oxidized to nitrite by hydroxylamine oxidoreductase (HAO; sections “Enzymes Involved in Ammonia Oxidation”; “Hydroxylamine Oxidoreductase”). Two of the four electrons derived are required for AMO activity, and the other two are used for energy generation (section “Electron Flow and Energy Transduction”). The AmoA protein is assumed to contain the active site of AMO (Hyman and Arp 1992). A second AMO subunit named “AmoB” has been identified (Bergmann and Hooper 1994a). The gene cluster encoding AMO contains a third open reading frame termed “amoC,” which is located upstream of the genes amoA and amoB (Klotz et al. 1997; section “Genetics of Ammonia Oxidizers”). Neither AmoA nor AmoB has been purified in the active state as yet. The enzyme HAO is a trimer of 63-kDa subunits, including seven c-type hemes and a novel heme (P-460) per monomer (Arciero and Hooper 1993; Hoppert et al. 1995; Igarashi et al. 1997). The enzyme is located in the periplasmic space but anchored in the cytoplasmic membrane.
Nitrite oxidation is initiated by the enzyme nitrite oxidoreductase (NO2-OR; section “Enzymes Involved in Nitrite Oxidation”) which occurs as characteristic membrane-associated two-dimensional crystals in all nitrite oxidizers. These regularly arranged particles are located on the surface of the cytoplasmic—and if present—intracytoplasmic membranes of nitrite-oxidizing bacteria. In all Nitrobacter species and in Nitrococcus, particles are arranged in rows, whereas in Nitrospina and both Nitrospira species, hexagonal patterns were observed. The NO2-OR consists of two subunits (Meincke et al. 1992). For Nitrobacter hamburgensis, the molecular weight of one particle was found to be 186 kDa representing an αβ-heterodimer (Spieck et al. 1996). The full sequence of the β-subunit as well as a partial sequence of the α-subunit of the NO2-OR of Nitrobacter hamburgensis shows similarities to nitrate reductases of several chemoorganotrophic bacteria (section “Genetics of Nitrite Oxidizers”). During oxidation of nitrite to nitrate, the additional oxygen atom of nitrate is derived from water (Aleem 1965) and two electrons are released for energy generation.
In addition to lithotrophic nitrifiers, various heterotrophic bacteria, fungi, and algae (Focht and Verstraete 1977; Killham 1986; Papen et al. 1989) are capable of oxidizing ammonia to nitrate. However, in contrast to lithotrophic nitrification, heterotrophic nitrification is not coupled to energy generation (section “Heterotrophic Nitrification”). Consequently, heterotrophic nitrifiers are dependent on the oxidation of organic substrates (Focht and Verstraete 1977; Kuenen and Robertson 1987). During heterotrophic nitrification, ammonia or reduced nitrogen from organic compounds (e.g., the amino group of amino acids) is co-oxidized to hydroxylamine, gaseous nitrogen oxides, nitrite, or nitrate. For example, methane-oxidizing bacteria were shown to co-oxidize ammonia to nitrite by a biochemically well-characterized particulate (membrane-bound) methane monooxygenase and a unique hydroxylaminoxidoreductase (Anthony 1982; Yoshinari 1985; O’Neil and Wilkinson 1977; Zahn et al. 1994; Bergmann et al. 2000). The methane monooxygenase is assumed to be biochemically related to the AMO of ammonia-oxidizing bacteria, and methane-oxidizing bacteria are potential contributors to nitrification in the rhizosphere of rice plants (Bodelier and Frenzel 1999). Conversely, ammonia oxidizers are able to oxidize methane to methanol (Hyman and Wood 1983; Ward 1987; Jones and Morita 1983; Steudler et al. 1996), but up to now, there is no evidence that ammonia oxidizers significantly contribute to the oxidation of atmospheric methane (CH4) in natural systems (Jlang and Bakken 1999; Bodelier and Frenzel 1999). In general, heterotrophic nitrification is considered to contribute only marginally to the global nitrogen cycle (Brady 1984; Brown 1988) but nevertheless might be of local importance especially in heath and conifer forest soils (e.g., see Van de Dijk and Troelstra 1980; Schimel et al. 1984).
Lithotrophic nitrifiers are autotrophic bacteria that fix carbon dioxide (CO2) via the Calvin-Benson cycle (Harms et al. 1981) and, to a lesser extent, via phosphoenolpyruvate carboxylase (Takahashi et al. 1993). In the past, they were thus described as obligate lithoautotrophs and were thought to find organic compounds toxic. However, this assumption is not correct for several nitrifier species. Clark and Schmidt (1967) demonstrated that ammonia oxidizers of the genus Nitrosomonas and nitrite oxidizers of the genus Nitrobacter are capable of growing mixotrophically with ammonia or nitrite as electron donors and with a combination of carbon dioxide and organic compounds as carbon source. Compared to purely autotrophic growth, the addition of organic compounds stimulated cell growth and increased cell yield (Steinmüller and Bock 1976; Matin 1978; Krümmel and Harms 1982; Watson et al. 1986). Furthermore, the nitrite oxidizers Nitrobacter winogradskyi, N. hamburgensis, and N. vulgaris can grow chemoorganotrophically with acetate or pyruvate as electron donor and dioxygen or nitrate (in absence of dioxygen) as electron acceptor (Bock 1976; Freitag et al. 1987). However, for these organisms, heterotrophic growth was always slower than lithotrophic growth. Recently, Daims and coworkers (Daims et al. 2000, 2001) showed that in nitrifying activated sludge, not yet cultured Nitrospira-related nitrite oxidizers fix CO2 and simultaneously take up pyruvate but not acetate, butyrate, and propionate. In addition, some strains of Nitrosomonas can utilize organic substances like urea or glutamine as source of their substrate ammonia for lithotrophic growth (Koops et al. 1991).
The transformation of ammonia to nitrate via nitrite by the nitrifying bacteria has various direct and indirect implications for natural and man-made systems. For example, nitrifying bacteria contribute directly or indirectly to loss of nitrogen compounds from various environments due to:
-
1.
Leaching of mobile nitrogen compounds produced by nitrifiers. Leaching is the mobilization and transfer of nitrate to rivers, lakes, seawater, and groundwater. Nitrification is not desirable in agricultural soil because it induces loss of soil nitrogen. Fertilizer ammonium, which is required for plant growth, adsorbs well to clay particles of soil owing to its positive charge (Fig. 3.1 ). When converted to nitrate, the inorganic soil nitrogen becomes mobile and thus susceptible to denitrification and leaching. In some countries, nitrification inhibitors like nitrapyrin (N-Serve) are used in agriculture to minimize nitrogen loss (Huber et al. 1977; Keeny 1986; Slangen and Kerkhoff 1984; Lipschultz et al. 1981; Poth and Focht 1985).
-
2.
Denitrification. Denitrification is the microbial reduction of nitrate via nitrite, nitric oxide (NO) and nitrous oxide (N2O) to dinitrogen (N2). This type of anaerobic respiration can be performed by a variety of phylogenetically different heterotrophic microorganisms. By aerobic oxidation of ammonia to nitrate, nitrifying bacteria produce the electron acceptor for subsequent denitrification in many natural and engineered systems. During the last years, nitrifiers also have been shown to be able to denitrify (section “Denitrification Catalyzed by Ammonia Oxidizers”).
-
3.
Chemodenitrification of nitrite (produced by the ammonia oxidizers) in acidic environments. Chemodenitrification is defined as nonenzymatically catalyzed loss of nitrogen due to dismutation of nitric acid at pH values <4.5 leading to the formation of nitrate and gaseous nitric oxide. In the atmosphere, nitric oxide is unstable and reacts with oxygen or ozone to form gaseous nitrogen dioxide, which chemically dismutates to nitrous and nitric acid in the presence of water.
In addition to the well-recognized metabolic activity, nitrifiers react to and produce gaseous nitrogen oxides like N2O, NO, and nitrogen dioxide (NO2). For example, the presence of NO is required for ammonia oxidation of Nitrosomonas eutropha (Zart et al. 2000). Furthermore, cell growth and the ammonia-oxidizing activity of this species are enhanced by NO2 (Zart and Bock 1998). On the other hand, NO is always produced during ammonia oxidation (Stüven and Bock 2001; Schmidt et al. 2001b). In the absence of oxygen, Nitrosomonas eutropha and, to a certain extent, Nitrobacter sp. are able to denitrify concomitant with the production of nitric oxide, nitrous oxide, and dinitrogen (in the case of N. eutropha; Poth and Focht 1985; Freitag et al. 1987; Zart and Bock 1998; Bock et al. 1995). Furthermore, NO2 has recently been reported to be produced by Nitrosomonas europaea if grown in coculture with Paracoccus denitrificans (Stüven and Bock 2001). Owing to the above-mentioned activities, ammonia-oxidizing bacteria are considered to contribute to the increasing nitrous oxide level in the atmosphere (Bouwman et al. 1993). While flux mechanisms for nitric oxide exchange are frequently studied (e.g., Conrad 1996), the processes of nitrogen dioxide production and release from soil have rarely been investigated (Williams et al. 1992; Baumgärtner 1991). Nitric oxide, nitrogen dioxide, and nitrous oxide circles between soil and atmosphere are of great importance because N2O is a greenhouse gas and NO and NO2 act as effectors for metabolic activity of microorganisms. The latter effect is of particular importance considering the increasing amounts of the nitrogen oxides NO and NO2 in the anthropogenically polluted atmosphere originating from methane, oil, and coal combustion. Compared to these sources, the contribution of microbiologically produced NO and NO2, however, is marginal.
The production of nitric and nitrous acid by nitrifiers also contributes to biodeterioration and can cause harmful effects for plants. With an outdoor exposure experiment lasting for 7 years, Mansch and Bock (1998) could demonstrate that the ammonia concentration of the atmosphere in the city of Duisburg (Germany) was high enough to support cell growth of lithotrophic ammonia oxidizers in natural sandstone. The formation of nitrous and nitric acid by such endolithic nitrifiers causes biodeterioration of carbonaceous masonry (Bock and Sand 1993). Furthermore, in unbuffered environments (e.g., forest soils), the oxidation of ammonia to nitric and nitrous acid leads to acidification followed by the formation of aluminum ions (Al3+) from insoluble aluminates, which are toxic to the roots of trees (Mulder et al. 1989; Stams et al. 1991). However, it should be kept in mind that plants can also benefit from nitrifying activity. Especially, many tree species prefer nitrate instead of ammonia as nitrogen source.
Nitrification is also important in biotechnology for efficient removal of ammonium from sewage (Painter 1988; Eighmy and Bishop 1989). Nitrifiers oxidize ammonium, which together with urea is the most frequently found nitrogen compound in sewage, to nitrate which can subsequently be removed from the sewage by denitrifying bacteria via anaerobic respiration. This treatment, which is an integral part of modern nutrient removal at wastewater treatment plants, prevents environments from increasing amounts of ammonia (causing eutrophication and oxygen depletion) and reduces the toxic effects of ammonium to aquatic life. However, the slow growth rate of nitrifiers and their susceptibility to changes in pH and temperature as well as to toxic sewage compounds causes frequent failure of nitrification in municipal and industrial wastewater treatment plants. In the future, wastewater treatment plants also might exploit the unique physiology of recently identified but not yet cultured novel planctomycetes that can catalyze anaerobic oxidation of ammonium to dinitrogen with nitrite as electron acceptor (Strous et al. 1999; Schmid et al. 2000; see section “Anaerobic Ammonium Oxidation Catalyzed by Deep Branching Planctomycetes” in this chapter).
Nitrifying bacteria are slow-growing organisms because their cell growth is inefficient. For example, nitrite oxidizers oxidize 85–115 mol of nitrate to generate the energy required for assimilation of 1 mol of carbon dioxide (Bömeke 1954). Thus, it is not surprising that the shortest generation times measured in laboratory experiments did not exceed 7 h for Nitrosomonas and 10 h for Nitrobacter (Bock et al. 1990). For cell division in natural environments, most nitrifier species even need several days to weeks depending on substrate, oxygen availability, the temperature, and pH values. The slow growth rates of nitrifiers have severely hampered cultivation-dependent approaches to investigate the number, community composition, and dynamics of nitrifiers in different environments. The number of nitrifiers in complex systems has been traditionally determined by the most probable number (MPN) technique (Matulewich et al. 1975). However, this method is time-consuming, and the nitrifier cell counts determined usually do not correlate well with nitrifying potential estimated for the same environmental sample under optimized laboratory conditions (Belser and Mays 1982; Belser 1979; Groffmann 1987; Mansch and Bock 1998). These discrepancies illustrate that not all nitrifiers can be cultivated using standard methods (Stephen et al. 1998; Juretschko et al. 1998; Purkhold et al. 2000). Furthermore, in many environments, nitrifiers form dense microcolonies of ten to several thousand cells embedded in extracellular polymeric substances (EPS; Fig. 3.3 ). Since these microcolonies are resistant to the dispersal techniques implemented in standard cultivation protocols, the use of these protocols dramatically underestimates the number of nitrifiers occurring in microcolonies (Watson et al. 1989; Stehr et al. 1995; Wagner et al. 1995).


Transmission electron micrographs of ultrathin sections of an ammonia oxidizer microcolony in activated sludge (a) Arrows indicate intracytoplasmic membranes (b) Modified from Wagner et al. (1995)
For direct microscopic enumeration of nitrifiers in complex samples, the fluorescent antibody (FA) technique can be applied (Belser 1979; Fliermanns et al. 1974), but for antibody production, the target cells have to be isolated first as pure culture and the produced antibodies often recognize only a few strains of a species (Belser and Schmidt 1978). Recently monoclonal antibodies targeting the nitrite oxidoreductase were developed that allow group-specific detection of nitrite-oxidizing bacteria (Bartosch et al. 1999). In addition, polyclonal antibodies specifically recognizing the AmoB protein of β-subclass ammonia oxidizers are available (Pinck et al. 2001). Alternatively, nitrifiers can be detected in environmental samples independent from their culturability by using a variety of different polymerase chain reaction (PCR) techniques for specific amplification of 16S rRNA gene fragments (e.g., Degrange and Bardin 1995; Hiorns et al. 1995; Voytek and Ward 1995; McCaig et al. 1994; Kowalchuk et al. 1997; Utåker and Nes 1998) or a fragment of the amoA gene (e.g., Rotthauwe et al. 1997; Purkhold et al. 2000). Quantitative population structure analysis of nitrifying bacteria within their natural habitat can most precisely be obtained by applying the recently developed set of rRNA-targeted oligonucleotide probes for fluorescence in situ hybridization (FISH; Wagner et al. 1995, 1996; Mobarry et al. 1996; Juretschko et al. 1998; Daims et al. 2000; Fig. 3.4 ).


In situ detection (with fluorescently labeled 16S rRNA-targeted oligonucleotide probes) of ammonia-oxidizing and nitrite-oxidizing bacteria in a nitrifying biofilm from a municipal wastewater treatment plant. Ammonia oxidizers are stained red, whereas nitrite-oxidizing bacteria of the genus Nitrospira appear green. Bar = 10 μm
Nitrifying bacteria are present in oxic and even anoxic environments. They are widely distributed in freshwater, seawater, soils, on/in rocks, in masonry, and in wastewater treatment systems. Nitrifiers also could be enriched or isolated from extreme habitats like heating systems with temperatures of up to 47 °C (Ehrich et al. 1995; E. Lebedeva, personal communication) and permafrost soils up to a depth of 60 m at a temperature of down to −12 °C. Although the pH optimum for cell growth is 7.6–7.8, nitrifiers were frequently detected in environments with suboptimal pH (e.g., acid tea soils and forest soils at pH values below 4) but also in highly alkaliphilic soda lakes at a pH of 9.7–10.5 (Sorokin et al. 2001). Growth under suboptimal acidic conditions might be possible by ureolytic activity, by aggregate formation (De Boer et al. 1991), or as biofilms (e.g., on clay particles; Allison and Prosser 1993). In many environments, nitrifier sensitivity to sunlight is of ecological importance. The light sensitivity of ammonia and nitrite oxidizers increases from blue light to long wave UV (Hooper and Terry 1974; Hyman and Wood 1984a; Shears and Wood 1985). Based on spectroscopic similarities, Shears and Wood (1985) postulated a model of the ammonia monooxygenase light inhibition similar to the three-stage catalytic cycle of the tyrosinase reaction. In Nitrobacter, which is more sensitive to visible light than Nitrosomonas (Bock 1965), the photooxidation of c-type cytochromes is assumed to cause light-induced cell death (Bock 1970).
Although Nitrosomonas europaea and Nitrobacter sp. are the most commonly investigated ammonia and nitrite oxidizers in laboratory studies, molecular analysis revealed that other nitrifiers are of higher importance in many natural and engineered systems. For example, stone material of historical buildings and many soil systems seem to be dominated by members of the genera Nitrosovibrio and Nitrosospira, respectively (Spieck et al. 1992; Hiorns et al. 1995; Stephen et al. 1996; Meincke et al. 1989), whereas different Nitrosomonas species and Nitrosococcus mobilis are the most abundant ammonia oxidizers in wastewater treatment plants (Juretschko et al. 1998; Purkhold et al. 2000). Interestingly, not yet cultured members of the genus Nitrospira and not Nitrobacter are the most abundant nitrite oxidizers in sewage treatment plants and aquaria filters (Burrell et al. 1998; Juretschko et al. 1998; Wagner et al. 1996; Daims et al. 2000).
Phylogeny of Lithotrophic Nitrifying Bacteria
Traditionally, nitrifying bacteria have been lumped together into one coherent group, the family Nitrobacter iaceae (Watson 1971; Watson et al. 1989). Based on their ability to lithotrophically oxidize either ammonia to nitrite or nitrite to nitrate, nitrifying bacteria were separated into two groups, the ammonia and the nitrite oxidizers. The assignment of ammonia- and nitrite-oxidizing bacteria into genera was dependent primarily upon their morphological features like cell size, shape, and the arrangement of the intracytoplasmic membranes (Watson et al. 1989). The physiological and morphological grouping of the nitrifying bacteria is in contradiction to data obtained from molecular phylogenetic studies which show at least subdivision level diversity within and between the ammonia and nitrite oxidizers (Head et al. 1993; Orso et al. 1994; Teske et al. 1994; Purkhold et al. 2000; Ehrich et al. 1995). Significant differences between ammonia- and nitrite-oxidizing bacteria are also indicated by the fact that both physiological groups possess very different key enzyme systems for the energy-gaining oxidation of ammonia and nitrite, respectively (sections “Enzymes Involved in Ammonia Oxidation”; “Enzymes Involved in Nitrite Oxidation”). With the exception of the nitrite oxidizers of the genera Nitrospina and Nitrospira, all known nitrifiers are closely related to phototrophs and thus presumably originated in several independent events by conversion of photosynthetic ancestors to chemolithotrophs (Teske et al. 1994). Consistent with this conversion hypothesis, all nitrifying bacteria related to phototrophs retain the general structural features of the putative ancestor’s photosynthetic membrane complex, while nitrite oxidizers of the genera Nitrospina and Nitrospira lack intracytoplasmic membranes (ICMs). However, it should be noted that the ammonia oxidizers of the genera Nitrosospira and Nitrosovibrio lack an extensive intracytoplasmic membrane system (Koops and Möller 1992).
Phylogeny of Ammonia Oxidizers
Chemolithotrophic ammonia oxidizers were isolated for the first time at the end of the nineteenth century (Winogradsky 1892). Since then, 16 species of ammonia oxidizers have been described (Jones et al. 1988; Koops et al. 1976, 1990; 1991, Watson 1965), and according to DNA-DNA hybridization experiments, at least 15 additional genospecies are “hidden” in existing culture collections (Koops et al. 1991; Koops and Harms 1985; Stehr et al. 1995). Our current perception of evolutionary relationships of ammonia-oxidizing bacteria is mainly based on comparative sequence analysis of their genes encoding the 16S rRNA and the active site polypeptide of the ammonia monooxygenase (AmoA). During the last decade, the genes for both biopolymers were sequenced for all recognized ammonia oxidizer species (Alzerreca et al. 1999; Head et al. 1993; Pommerening-Röser et al. 1996; Teske et al. 1994; Purkhold et al. 2000; Rotthauwe et al. 1995, 1997; McTavish et al. 1993; Horz et al. 2000), and the deduced phylogeny now provides an encompassing and relatively robust framework for assignment of 16S rDNA and amoA sequences of (1) ammonia oxidizer isolates (Stehr et al. 1995; Suwa et al. 1997; Utåker et al. 1995; Juretschko et al. 1998) and (2) cloned sequence fragments directly retrieved from the environment (e.g., Stephen et al. 1996; Rotthauwe et al. 1995; Purkhold et al. 2000).
According to comparative 16S rRNA sequence analysis, all recognized ammonia oxidizers are members of two monophyletic lineages within the β- and γ-subclass of Proteobacteria (Fig. 3.5 ). The marine species Nitrosococcus halophilus and Nitrosococcus oceani, which are distantly related to methane-oxidizing bacteria, cluster together in the γ-subclass of Proteobacteria. All other ammonia oxidizers form a monophyletic assemblage within the β-subclass of Proteobacteria, most closely related to the iron oxidizer Gallionella ferruginea. This lineage encompasses the genera Nitrosomonas (including Nitrosococcus mobilis, which is actually a member of the genus Nitrosomonas), Nitrosovibrio, Nitrosolobus, and Nitrosospira. It has been suggested (Head et al. 1993) and subsequently questioned (Teske et al. 1994) that the latter three genera should be reclassified into the single genus Nitrosospira. The nitrosomonads can be further subdivided into the N. europaea/Nc. mobilis cluster, the N. marina cluster, the N. oligotropha cluster, and the N. communis cluster (Purkhold et al. 2000). Nitrosomonas cryotolerans forms a separate lineage within the β-Proteobacteria. The genera Nitrosospira, Nitrosolobus, and Nitrosovibrio are closely related and form a cluster to the exclusion of the nitrosomonads. Similar but not identical evolutionary relationships were obtained if comparative analysis of AmoA sequences were performed (Purkhold et al. 2000). In the AmoA tree, the N. europaea/Nc. mobilis cluster, the N. marina cluster, and the Nitrosospira cluster are retained, whereas the members of the N. oligotropha cluster and the N. communis cluster form no monophyletic assemblages.


Phylogenetic neighbor-joining 16S rRNA tree reflecting the relationships of ammonia-oxidizing bacteria and several reference organisms. The multifurcation connects branches for which a relative order could not be unambiguously determined by applying different treeing methods. Parsimony bootstrap values for branches are reported. Missing bootstrap values indicate that the branch in question was not recovered in the majority of bootstrap replicates by the parsimony method. AOB ammonia-oxidizing bacteria, MOB methane-oxidizing bacteria. The bar indicates 10 % estimated sequence divergence (Modified from Purkhold et al. 2000)
Phylogeny of Nitrite Oxidizers
Four different genera, Nitrobacter, Nitrococcus, Nitrospina, and Nitrospira, of lithotrophic nitrite-oxidizing bacteria have been described. From 16S rRNA sequence analysis, the first three genera were assigned to different subclasses of the Proteobacteria, whereas Nitrospira is the name-giving genus of an independent bacterial phylum (Fig. 3.5 ). The genus Nitrobacter contains the four closely related species (N. hamburgensis, N. vulgaris, N. winogradskii, and N. alkalicus) within the α-subclass of Proteobacteria. Nitrite oxidizers of the genus Nitrobacter are phylogenetically related to Bradyrhizobium japonicum, Blastobacter denitrificans, Afipia felis, Afipia clevelandensis, and the phototroph Rhodobacter palustris (Seewaldt et al. 1982; Orso et al. 1994; Teske et al. 1994) with which Nitrobacter shares a nearly identical arrangement of ICMs.
The genus Nitrococcus represented by the single marine species Nitrococcus mobilis is, like the marine ammonia oxidizers of the genus Nitrosococcus, a member of the ectothiorhodospira branch of the γ-subclass of Proteobacteria, consistent with an assumed photosynthetic ancestry of these nitrifiers. Nitrococcus and Nitrosococcus are the only nitrite and ammonia oxidizers that are relatively closely related, but the closest relatives of Nitrococcus mobilis are the phototrophic bacteria Arhodomonas aquaeoli, Ectthiorhodospira halochloris, and Ectthiorhodospira halophila (Teske et al. 1994).
The genus Nitrospina with the marine Nitrospina gracilis as the only species (represented by two isolates, one from the Atlantic and the other from the Pacific) has been provisionally assigned to the δ-subclass of Proteobacteria and is the only member of a deep branch within this subclass (Teske et al. 1994). Nitrospina gracilis shows no ICMs.
The genus Nitrospira encompasses the marine species Nitrospira marina and Nitrospira moscoviensis, isolated from a municipal water heating system. The genus Nitrospira forms a monophyletic grouping with the genera Thermodesulfovibrio, Leptospirillum, and with “Magnetobacterium bavaricum.” This phylogenetic assemblage has recently been identified as a novel phylum within the domain Bacteria and was named “Nitrospira phylum” (Ehrich et al. 1995). There is accumulating molecular evidence that Nitrospira-related nitrite oxidizers are of major importance for nitrite oxidation in wastewater treatment plants and aquarium filters (Burrell et al. 1998; Juretschko et al. 1998; Hovanec et al. 1998; Daims et al. 2000) and also occur in many natural environments including the rhizosphere (Fig. 3.6 ). Like Nitrospina gracilis, members of the genus Nitrospira do not possess ICMs and are apparently not closely related to phototrophic bacteria.


Phylogenetic tree of the genus Nitrospira based on comparative analysis of 16S rRNA sequences. The basic tree topology was determined by maximum likelihood analysis of all sequences longer than 1,300 nucleotides. Shorter sequences were successively added without changing the overall tree topology. Branches leading to sequences shorter than 1,315 nucleotides are dotted to point out that the exact affiliation of these sequences cannot be determined. Black spots on tree nodes symbolize high parsimony bootstrap support above 90 % based on 100 iterations. The scale bar indicates 0.1 estimated changes per nucleotide. The four sublineages of the genus Nitrospira are delimited by horizontal dashed lines and marked by the numbers I to IV. Two of the four sublineages entirely consist of 16S rDNA sequences amplified from environmental samples (Modified from H. Daims et al. 2001)
Biochemistry of Ammonia-Oxidizing Bacteria
Ammonia oxidizers are lithoautotrophic organisms using carbon dioxide as the main carbon source (Bock et al. 1991). Their only way to gain energy is the two-step oxidation of ammonia to nitrite (Hooper 1969). Investigations of the Km values and pH optima indicate that ammonia (NH3) rather than ammonium (NH +4 ) is the substrate of ammonia oxidizers (Suzuki et al. 1974; Drozd 1976). This is in accordance with results showing that the ammonia-oxidizing enzyme might be located in the cytoplasmic membrane (Suzuki and Kwok 1981; Tsang and Suzuki 1982), since membranes are highly permeable to ammonia but not to ammonium (Kleiner 1985). First, ammonia is oxidized to hydroxylamine (Kluyver and Donker 1926) by the ammonia monooxygenase (AMO; Hollocher et al. 1981). This enzyme does not possess high substrate specificity and also oxidizes several apolar compounds such as methane, carbon monoxide, or some aliphatic and aromatic hydrocarbons (Hooper et al. 1997). These compounds can act as competitive inhibitors of ammonia oxidation (Hyman et al. 1988; Keener and Arp 1993). The second step is performed by the hydroxylamine oxidoreductase (HAO). This enzyme oxidizes hydroxylamine to nitrite (Wood 1986). Two of the four electrons released (Andersson and Hooper 1983) are required for the AMO reaction (Tsang and Suzuki 1982), whereas the remaining ones are used for the generation of proton motive force (Hollocher et al. 1982) to regenerate ATP and NADH (Wheelis 1984; Wood 1986). Most of the investigations on energy metabolism of ammonia-oxidizing bacteria have been carried out with Nitrosomonas europaea. Keeping in mind that the ammonia oxidizers encompass five different genera affiliated to two proteobacterial subclasses (section “Phylogeny of Ammonia Oxidizers”), additional species should be investigated to obtain a more encompassing picture of the biochemistry of the ammonia-oxidizing system (Giannakis et al. 1985).
Ammonia and Hydroxylamine as Substrates
The overall process of ammonia oxidation to nitrite may be characterized as a two-stage process:
However, this two-stage scenario is a simplification. For lithotrophic ammonia oxidizers, ammonia is essential as the primary substrate. The intermediate hydroxylamine (NH2OH) is the real energy source. The coupling between ammonia and hydroxylamine oxidation, a complex mechanism not yet established in detail, is suggested by several observations. The addition of hydroxylamine to ammonia-oxidizing cells shortened the lag phase of ammonia oxidation (Hooper 1969), probably by providing reductants to the monooxygenase. It is generally assumed that partial reduction of c-type cytochromes is necessary to start ammonia oxidation. Cytochrome reduction was attained by addition of hydroxylamine to cell-free preparations of Nitrosomonas europaea (Suzuki et al. 1981). If both ammonia and hydroxylamine are used, the molar growth yield of hydroxylamine was found to be twice the amount of ammonia (Böttcher and Koops 1994; De Bruijn et al. 1995). On the other hand, increasing amounts of hydroxylamine are inhibitory to ammonia oxidation (Hyman and Wood 1984b; Poth and Focht 1985; Abeliovich and Vonshak 1993), probably due to imbalancing the redox state of AMO and HAO (Wood 1986). Another result is more difficult to understand. All attempts to grow ammonia oxidizers on hydroxylamine as the only substrate have failed, although hydroxylamine is oxidized to nitrite (Lees 1952; Hoffman and Lees 1953; Engel and Alexander 1958; Nicholas and Jones 1960). This failure is most likely not caused by the toxicity of hydroxylamine, because addition of hydroxylamine in the presence of ammonia promotes substrate oxidation and cell growth. As demonstrated recently, Nitrosomonas eutropha cells are capable of growing on hydroxylamine as the only substrate when AMO is simultaneously inhibited by acetylene (S. Oesterreicher, personal communication; Fig. 3.7 ). Without addition of acetylene, N. eutropha cells 3lyse within 3 days when hydroxylamine is oxidized to nitrite, although within the first day NADH and ATP are still formed (C. Look, personal communication). It is important to note that during these experiments, ammonia was present as nitrogen source because hydroxylamine could not be assimilated. The observation that reduction of a functionally active AMO in the absence of ammonia leads to cell death could be explained by the formation of toxic oxygen radicals by this enzyme under these conditions. This suicidal activity of ammonia oxidizers also might cause nitrification breakdown in wastewater treatment plants, if (1) plenty of organic substrate is available as additional alternative electron donor and (2) ammonia is present in very low concentrations.


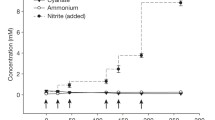
Growth of Nitrosomonas eutropha in the presence of ammonia, 2,315 parts per million (ppm) acetylene, and hydroxylamine (4 mmol) as substrate (48–96 h). The AMO was inhibited by acetylene, while cell growth was detectable after a lag phase of 2 days. Most of the hydroxylamine undergoes deterioration in contact with atmospheric oxygen. As calculated from additional nitrite formation, 400 μmol of hydroxylamine was oxidized to nitrite, resulting in an increase of cell number
Enzymes Involved in Ammonia Oxidation
Ammonia Monooxygenase
The first intermediate of ammonia oxidation is assumed to be hydroxylamine (section “Genes Encoding AMO, HAO, and Related Enzymes”). In the presence of hydrazine (an irreversible inhibitor of hydroxylamine oxidation; Nicholas and Jones 1960; Hynes and Knowles 1978), the production of small quantities of hydroxylamine from ammonia was observed (Hoffman and Lees 1953; Yoshida and Alexander 1964). Using 18O2, it could be demonstrated that more than 92 % of the oxygen in hydroxylamine originates from dioxygen (Dua et al. 1979). The enzyme AMO, catalyzing the conversion of ammonia to hydroxylamine, has not yet been purified as active protein, but Hyman and Wood (1985) were able to identify a membrane-associated 14C-labeled protein, putatively representing a component of AMO, when whole cells of Nitrosomonas europaea were incubated with [14C]acetylene. The N-terminal amino acid sequence of the [14C]acetylene-labeled protein (AmoA) was determined. Based on this sequence, an oligonucleotide was derived and was used to identify and clone the gene amoA. The AmoA protein is a 31.8 kDa (McTavish et al. 1993), probably containing the active site of AMO (Hyman and Arp 1992), and consists of five transmembrane sequences and one periplasmic loop. In the same operon, a second gene amoB is located adjacent to amoA. From the deduced amino acid sequence, the protein has a molecular weight of 43 kDa (Bergmann and Hooper 1994a) and is characterized by two transmembrane domains and two periplasmic loops (Vanelli et al. 1996). Upstream of the genes amoA and amoB, a third open reading frame amoC is located which might encode a chaperone helping the AmoA and AmoB protein subunits to integrate into the membrane properly (Klotz et al. 1997).
Indirect evidence indicates that AMO is a copper-containing monooxygenase (Rees and Nason 1966; Tomlinson et al. 1966; Loveless and Painter 1968; Dua et al. 1979; Hollocher et al. 1981; Wood 1988a; Hooper and Terry 1973). Quantitative immunoblot analysis using polyclonal antibodies revealed that total cell protein of Nitrosomonas eutropha consisted of approximately 6 % AmoA and AmoB, when cells were grown using standard conditions (Pinck et al. 2001). The specific cellular amount of AMO in cells of Nitrosomonas eutropha was regulated by ammonium concentration. At high ammonium concentrations, less AMO was found than under ammonium-limiting conditions. Furthermore, AMO seems to be strongly protected from degradation. Cells starving 1 year for ammonia still contained high amounts of AMO, although they showed far less ammonia oxidation activity than growing cells. Hence, the amount of AMO does not directly correlate with the activity of ammonia oxidation.
Most information about the reactions catalyzed by AMO originates from studies with intact cells. In addition to oxidizing ammonia, AMO can hydroxylate non-growth-supporting substrates such as hydrocarbons and alcohols (Hooper and Terry 1973; Suzuki et al. 1976; Tsang and Suzuki 1982; Hyman and Wood 1983, 1984a, 1984b; Hyman et al. 1985; Voysey and Wood 1987). This is not only of theoretical interest but also could be of importance for microbial ecology (Hall 1986). For example, pure cultures of ammonia oxidizers are able to oxidize methane but could not grow on this alternative electron donor (O’Neil and Wilkinson 1977; Hyman and Wood 1983; Jones and Morita 1983). Recent data, however, suggest that at least in the rice rhizosphere, ammonia oxidizers do not significantly contribute to the methane oxidation (Bodelier and Frenzel 1999; section “Co-oxidation and Inhibition of AMO”). This capability reflects structural and functional homologies between the ammonia and the methane monooxygenase of ammonia oxidizers and methanotrophs, respectively (Bedard and Knowles 1989). Since substrates or competitive inhibitors of AMO are apolar, it seems reasonable to assume that its active site is hydrophobic. As suggested by Hooper et al. (1997), the reaction is started by the activation of oxygen rather than the substrate. Oxygen might be activated by reduction with a reduced metal-containing center of the enzyme followed by the release of water to form a reactive oxygen species. This compound may extract an electron from the substrate (hydroxylation of the substrate) or interact with nitric oxide to form the real oxidant nitrogen dioxide/dinitrogen tetroxide (see also Fig. 3.17 ).
Hydroxylamine Oxidoreductase
The key enzyme of hydroxylamine oxidation, HAO, is a multiheme enzyme, located in the periplasmic space (Olson and Hooper 1983; Hooper et al. 1984; Hooper and DiSpirito 1985; section “Genes Encoding AMO, HAO, and Related Enzymes”). The enzyme complex has a relative molecular weight of 180,315–190,315 and consists of an α3 oligomer closely associated with three heme centers including seven c-type hemes and a novel heme, P-460, per monomer (Arciero and Hooper 1993; Hoppert et al. 1995; Igarashi et al. 1997; Bergmann and Hooper 1994b). The P-460 was found to be a CO-binding heme (Hooper et al. 1978; Lipscomb et al. 1982). According to spectroscopic and chemical investigations, the P-460 iron resides in a heme-like macrocycle, but the presumed porphyrin must have some unusual features (Andersson et al. 1984). In total, HAO constitutes about 40 % of the c-type heme of Nitrosomonas europaea (Hooper et al. 1978). The c-type hemes of HAO can be placed into two classes with different oxidation-reduction midpoint potentials and protein environments, respectively (Lipscomb and Hooper 1982; Prince et al. 1983; Hooper 1984a; Collins et al. 1993; Arciero et al. 1991). A detailed discussion of possible interactions of the described redox centers of the HAO can be found in Hooper (1989).
Hydroxylamine is supposed to bind at the HAO near the P-460 center. Electrons are released and transferred to c-hemes (Hooper and Terry 1977; Hooper and Balny 1982; Olson and Hooper 1983). Initially, Hooper and Balny (1982) postulated that HAO catalyzes a two-electron dehydrogenation of hydroxylamine and a subsequent net addition of one oxygen atom from dioxygen. Later, they favored a mechanism in which water was the source of the second oxygen atom of the metabolic final product nitrite (Andersson and Hooper 1983; Hooper 1984).
The oxidation of hydroxylamine to nitrite was postulated to be a two-step reaction with enzyme-bound nitroxyl (HNO) as an intermediate (Andersson and Hooper 1983):
However, in cell-free extracts of Nitrosomonas europaea, nitric oxide was suggested as another possible intermediate of hydroxylamine oxidation (Hooper and Terry 1979). Experiments with 15N-label showed that nitric oxide was produced by hydroxylamine oxidation and not by nitrite reduction. The authors discussed a mixed-function hydroxylation of nitric oxide to be involved in the oxidation from (HNO) to nitrite, with all intermediates being enzyme bound. Miller and Wood (1983) analyzed CO-binding cytochromes of the b type in Nitrosomonas europaea and discussed their possible function in binding nitric oxide resulting from hydroxylamine oxidation.
Electron Flow and Energy Transduction
Electron Flow
The first step of ammonia oxidation to nitrite, the conversion to hydroxylamine, is endergonic. Thus, hydroxylamine is the real energy-generating substrate. If all subsequent steps of the hydroxylamine oxidation to nitrite are coupled to electron transport chains, a maximum yield of four electrons can result. The number of electrons passing to the terminal oxidase(s), however, is uncertain because four systems (ammonia monooxygenase, nitrite reductase, cytochrome oxidase, and NADH production) are fed with electrons from the oxidation of hydroxylamine to nitrite (Wood 1986). Electrons from HAO reduce cytochrome c 554, a 25-kDa tetraheme protein (Andersson et al. 1986). Because both ammonia- and hydroxylamine oxidation seem to be balanced at a steady state, cytochrome c 554 is thought to be the first electron transfer branch point. Two of the four electrons released from the hydroxylamine oxidation must pass to the monooxygenase reaction, the latter two flow to a second branch point, for example, cytochrome c 552 and then to one of the terminal oxidases cytochrome aa 3 (DiSpirito et al. 1986) or nitrite reductase. Once per 5.7 cycles, two electrons are assumed to enter a reverse electron transfer pathway for NADH production (Wood 1986). Cytochrome c 554 is a probable candidate for the proposed central role because it is a two-electron carrier (Arciero et al. 1991; Bergmann et al. 1994). The electron carriers downstream have not been investigated in detail. However, the production of nitric and nitrous oxide by Nitrosomonas europaea and N. eutropha suggested that nitric oxide reductase as well as nitrous oxide reductase might be present (Hooper et al. 1997). Yamanaka and Shinra (1974) postulated the path of electrons from HAO to the terminal oxidase to be:
HAO → cytochrome c 554 → cytochrome c 552 → terminal oxidase. However, several other membrane-bound redox carriers have been identified in Nitrosomonas europaea. The function of a tetraheme c-type cytochrome (Cyt c B) is unknown (Bergmann et al. 1994). The periplasmic diheme cytochrome c peroxidase (Arciero and Hooper 1994) of Nitrosomonas europaea might protect enzymes like HAO, which are easily inactivated by hydrogen peroxide (H2O2; Hooper and Terry 1977). Wood (1986) suggested a more conventional construction of the electron transport chain, involving ubiquinone and membrane-bound b- and c-type cytochromes. Wood (1986) discussed a possible proton motive Q cycle as described by Mitchell (1975). Ubiquinone (Q8 species) and membrane-bound cytochromes of types b and c were identified in Nitrosomonas europaea (Hooper et al. 1972; Tronson et al. 1973; Miller and Wood 1983). In Fig. 3.8 , a model of the electron flow in ammonia oxidizers is depicted.


Model of the electron flow in ammonia-oxidizing bacteria. The part of the figure dealing with nitric oxide, nitrogen dioxide, and dinitrogen tetroxide is hypothetical (section “Novel Aspects”). CM cytoplasmic membrane, i inside the cell/cytoplasmic space, o outside of the cell/periplasmic space, and TCA the tricarboxylic acid cycle (Figure was kindly provided by I. Schmidt)
Energy Transduction
ATP synthesis driven by proton motive force occurs in Nitrosomonas (Drozd 1976, 1980; Hollocher et al. 1982; Kumar and Nicholas 1982). This energy transduction is assumed to proceed at the level of hydroxylamine oxidation, but the process is not as yet well understood.
A simplified scheme would be a two-proton release per electron pair translocated from hydroxylamine to the electron transport chain via the periplasmic located HAO outside the membrane. In addition, the consumption of two protons in the cytochrome oxidase reaction is probably located on the cytoplasmic site of the membrane. However, the exact amount of ATP gained by oxidation of hydroxylamine is not known, since the total number of electrons fed into the energy-generating respiration chain per mol hydroxylamine oxidized varies, depending upon growth stage and environmental conditions. This variation reflects the fact that the production of NADH by reverse electron flow is not constant and coupled to hydroxylamine oxidation by an unknown mechanism.
According to Hollocher et al. (1982), the H+/O ratio depends on the substrate concentration. From their measurements, they extrapolated the maximum values to be 3.4 and 4.4 for ammonia and hydroxylamine oxidation, respectively. In addition, Drozd (1976, 1980) stated the maximum P/O ratio from hydroxylamine oxidation to be only one. Measurements of respiration-driven proton translocation indicated the association of only one proton translocation loop, with the transport of two electrons channeled from hydroxylamine to the terminal oxidase.
NADH Production
Aleem (1966) showed that cell-free extracts of Nitrosomonas europaea catalyzed an ATP-dependent NAD(P)+ reduction with hydroxylamine as substrate. The reaction was interpreted as ATP-driven reverse electron flow. This hypothesis is in accordance with the postulate that the transmembrane oxidation-reduction loops of respiration chains are reversible, with the exception of the cytochrome c oxidase loop. However, in vivo, the proton motive force resulting from the hydroxylamine oxidation might perhaps drive the reverse direction of the electron flow directly, without support of ATP as previously demonstrated for the nitrite oxidizers Nitrobacter winogradskyi and Nitrobacter vulgaris (Freitag and Bock 1990).
Co-oxidation and Inhibition of AMO
The ammonia monooxygenase (AMO) is a nonspecific enzyme. Ammonia oxidizers are capable of co-oxidizing a range of hydrocarbons (including methane and even xenobiotics), which raised interest in exploiting these microorganisms for bioremediation (Vanelli et al. 1990). The broad substrate range of AMO also is responsible for inhibition of ammonia oxidizers by a variety of substances (Table 3.1 ). During oxidation of acetylene via AMO, reactive intermediates that bind irreversibly to AMO are formed in the presence of oxygen. The same mechanism causes the inhibition of AMO by trichlorethylene. The acetylene inhibition can be ameliorated by high ammonia concentrations via an unknown mechanism (Hyman and Wood 1985). Competitive inhibitors of AMO are methyl fluorides, dimethyl ether (Voysey and Wood 1987; Miller et al. 1993; Hyman et al. 1994), alkanes, alkenes (Hyman et al. 1988), and aromatic compounds (e.g., aniline; Keener and Arp 1994; Voysey and Wood 1987; Hyman and Wood 1983; Jones and Morita 1983). Carbon monoxide (CO) not only binds irreversibly to cytochromes but also competitively inhibits AMO, the enzyme that oxidizes it to carbon dioxide (Tsang and Suzuki 1982; Erickson et al. 1972). Since copper is a cofactor of AMO (Loveless and Painter 1968; Hooper and Terry 1973), metal chelators such as allylthiourea and diethyldithiocarbamate are noncompetitive, reversible inhibitors (Lees 1952).
In addition to some of the above-mentioned inhibitors, Table 3.1 lists other inhibitors of ammonia oxidation that do not directly interact with AMO. Ammonia oxidation is much more strongly inhibited by all listed physical parameters and chemical compounds than is hydroxylamine oxidation.
Denitrification Catalyzed by Ammonia Oxidizers
Ammonia-oxidizing bacteria not only catalyze aerobic ammonia oxidation but also show denitrifying activity with nitrite as electron acceptor. For example, small amounts of nitric oxide and nitrous oxide are produced during denitrification with ammonia as electron donor at reduced oxygen concentrations (Hooper 1968; Goreau et al. 1980; Remde and Conrad 1990; Stüven et al. 1992). When using 14NH +4 and 15NO −2 , Poth and Focht (1985) demonstrated that nitrous oxide was produced at low oxygen tension by nitrite reduction and not by hydroxylamine oxidation. The reaction is thought to be catalyzed by a periplasmic soluble cytochrome oxidase/nitrite reductase induced at low oxygen partial pressure (Miller and Wood 1982; Miller and Nicholas 1985; DiSpirito et al. 1985). Additionally, the formation of dinitrogen was observed (Poth 1986; Bock et al. 1995), indicating that at least some strains of Nitrosomonas possess a nitrous oxide reductase. However, this enzyme has not been isolated as yet from denitrifying ammonia oxidizers.
Ammonia oxidizers show relatively high denitrification activities when they are cultivated under oxygen-limited conditions in the presence of organic matter (mixotrophic growth conditions; Bock et al. 1995). However, under these conditions, ammonia oxidation rates are low (Zart et al. 1996). For this reason, the denitrifying potentials of ammonia oxidizers cannot be efficiently exploited for one-step nitrogen removal in wastewater treatment plants.
In the absence of dissolved oxygen, Nitrosomonas eutropha and Nitrosomonas europaea are capable of anoxic denitrification using molecular hydrogen, or simple organic compounds such as acetate, pyruvate, or formate as electron donors and nitrite as electron acceptor (Bock et al. 1995; Abeliovich and Vonshak 1992; Stüven et al. 1992).
Genetics of Ammonia Oxidizers
Relatively little information regarding the genetic makeup of ammonia oxidizers is available. Most studies focused on Nitrosomonas europaea (genome of ca. 2.2 Mb) whose genomic sequence is currently being determined [{spider.jgi-psf.org}]. For the other ammonia oxidizers of the β- and λ-subclasses of Proteobacteria (section “Phylogeny of Ammonia Oxidizers”), sequence information is restricted to the genes coding for the 16S rRNA (for a review, see Purkhold et al. 2000), the 16S-23S rDNA intergenic spacer region (Aakra et al. 1999), as well as the ammonia monooxygenase operon (Rotthauwe et al. 1995; Purkhold et al. 2000; Alzerreca et al. 1999). Recently, a gene for a copper-containing dissimilatory nitrite reductase (nirK) that has been detected by PCR and was sequenced for several β-subclass ammonia-oxidizing bacteria is under way (Casciotti and Ward 2001). Ammonia oxidizers can also harbor plasmids, as demonstrated by the isolation and characterization of two cryptic plasmids in a Nitrosomonas strain retrieved from activated sludge (Yamagata et al. 1999).
Genes Encoding AMO, HAO, and Related Enzymes
Genes coding for enzymes involved in the oxidation of ammonia, particularly the ammonia monooxygenase (AMO), the hydroxylamine oxidoreductase (HAO), and the accompanying cytochromes, have been most intensively studied in N. europaea, which has multiple copies of these primary nitrification genes (section “Enzymes Involved in Ammonia Oxidation”). Nitrosomonas europaea has a duplicated amo operon containing a continuous arrangement of the genes amoC, amoA, and amoB, which are cotranscribed as a 3.5-kb mRNA and encode the three subunits of AMO, AmoC, AmoB, and AmoA (McTavish et al. 1993; Klotz et al. 1997; Sayavedra-Soto et al. 1998). A third copy of amoC, which is not associated with the genes for the other subunits of this enzyme, has recently been identified (Sayavedra-Soto et al. 1998). Multiple amo operons also have been found in several other ammonia oxidizers (Table 3.2 ). Furthermore, N. europaea has at least three copies of each of the genes coding for the hydroxylamine oxidoreductase (hao) and cytochrome c 554 (cycA or hcy; McTavish et al. 1993; Hommes et al. 1994). Each copy of the hao gene is located 950 bp upstream of a copy of the hcy gene, but both genes are always found to be within different operons (Bergmann et al. 1994; Sayavedra-Soto et al. 1994). Downstream of two of the hcy genes, an ORF (cycB) predicted to encode another tetraheme cytochrome c was detected (Bergmann et al. 1994). The nucleic acid sequences of the multiple copies of all above-mentioned genes (except for the unlinked amoC genes) are either identical or highly similar within a single ammonia oxidizer species, whereas much lower similarities occur between the respective genes of different species. Thus, it is likely that the multiple gene copies originated from relatively recent gene duplication events and were not caused by lateral gene transfer (Klotz and Norton 1998). It has been speculated that the presence of multiple genes might (1) allow more-rapid generation of the respective mRNA during ammonia flushes within the local environment of the ammonia oxidizers (Hommes et al. 1998) or (2) be responsible for maintaining a certain ratio of the gene products (Bergmann et al. 1994).
In addition to those genes with products directly involved in ammonia oxidation, genes of N. europaea encoding the enolase (eno) and CTP synthase (pyrG) were sequenced (Mahony and Miller 1998). The enolase catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate, and its gene was found to be linked on the chromosome with the pyrG gene, albeit both genes are not cotranscribed. A similar arrangement of both genes is present in the Escherichia coli genome (where they are cotranscribed), though these genes are not linked in other investigated bacterial genomes.
Unfortunately, no sequence information regarding the genes involved in CO2 fixation/carboxysome formation of the autotrophic ammonia oxidizers is currently available.
Regulation of AMO and HAO
One unusual feature of N. europaea is that it possesses multiple copies of those genes directly involved in ammonia oxidation. This is remarkable, since, with the exception of rRNA and tRNA genes, only relatively few cases of gene duplications have been described for bacteria (e.g., Hass et al. 1992; Sela et al. 1989; Tubulekas and Hughes 1993; Kusian et al. 1995). The significance of the N. europaea ammonia oxidation genes being present in multiple copies has been investigated using techniques for transformation and insertional mutagenesis (Hommes et al. 1996, 1998). Disruption of each of the two amoA copies showed that each copy was functional in N. europaea and that neither copy is essential in the cell. However, knockout of one of the amoA copies, but not of the other, has a significant influence on the growth rates of the cells (Hommes et al. 1998), suggesting different regulation of each copy. Surprisingly, however, the putative σ70-type promotors of both amoA genes were found to be identical (Hommes et al. 2001), indicating that the differential transcription of both genes (Hommes et al. 1998) involves regions upstream of the promotor where the DNA sequences of both copies diverge (Hommes et al. 2001). Similar results were obtained with cells carrying single mutations in each of the amoB genes (Stein et al. 2000). Insertional mutagenesis of each of the three hao gene copies, all of which possess σ70-type promotors (Hommes et al. 2001), showed that none of them was essential and that their inactivation could be compensated fully by the two remaining hao genes (Hommes et al. 1996). However, owing to the presence of three hao gene copies, differences in their regulation might only become apparent after simultaneous inactivation of two of the copies.
Ammonia-oxidizing bacteria thrive in environments where ammonia is often present in very low concentrations. In these habitats, the capability to efficiently make use of temporal flushes of ammonia might represent an important selective advantage for an ammonia oxidizer. Therefore, the genetic and physiological responses of ammonia oxidizers under conditions of ammonium limitation (ammonium present in amounts that can be metabolized to completion), of starvation (absence of ammonium), and in the presence of excess ammonium were intensively investigated. Nitrosomonas cryotolerans and N. eutropha survive ammonia starvation for at least 25 weeks (Jones and Morita 1985) and 1 year, respectively (Pinck et al. 2001). In contrast to energy-starved heterotrophic bacteria, N. cryotolerans cells after 10 weeks of starvation (1) do not miniaturize, (2) maintain stable levels of intracellular ATP, and (3) show no changes in the total protein, DNA, or RNA levels (Johnstone and Jones 1988). Furthermore, quantitative FISH demonstrated that ammonia oxidizers in activated sludge maintain relatively stable cellular rRNA concentrations during starvation for 1 month or inhibition with allylthiourea for several days (Wagner et al. 1995; Morgenroth et al. 2000). During prolonged starvation for several months or years, ammonia oxidizers lose ammonia-oxidizing activity but still contain significant amounts of AMO inasmuch as this enzyme is degraded more slowly in comparison to the mean cellular protein (Pinck et al. 2001). Under conditions of ammonia starvation, the mRNA of the amo gene disappears within 8 h, though the ammonia and hydroxylamine oxidation activities do not change over a period of 24 h (Stein and Arp 1998a). Limiting ammonium concentrations results in a large loss of ammonia-oxidizing activity (85 %) after 24 h, but it neither affects the steady-state levels of amoA mRNA nor the result in degradation of the AmoA subunit (Stein and Arp 1998a). Interestingly, short-chain alkanes and other substrates having a high binding affinity for AMO ameliorate the inactivating effects of ammonia limitation by protecting the energy-generating activity of N. europaea from potentially toxic by-products of its metabolism (Stein and Arp 1998a, b; section “Ammonia and Hydroxylamine as Substrates”). Interestingly, N. europaea cells grown in biofilms recover much faster after ammonium starvation than their planktonic counterparts. Preliminary data suggest that this phenomenon might be caused by cell-to-cell communication via N-(3-oxohexanoyl)-L-homoserine lactone (Batchelor et al. 1997). As expected, ammonium/ammonia induces the transcription of the ammonia monooxygenase and hydroxylamine oxidoreductase genes as well as the transcription of several additional genes that were not further characterized in Nitrosomonas europaea (Sayavedra-Soto et al. 1996). Furthermore, the activity of AMO is regulated by the presence of ammonia at translational (Hyman and Arp 1995; Stein et al. 1997) and posttranslational (Stein et al. 1997) levels.
Biochemistry of Nitrite-Oxidizing Bacteria
The second step of nitrification, the oxidation of nitrite to nitrate, is performed by nitrite-oxidizing bacteria. Although at least four different genera of nitrite oxidizers exist in nature (section “Phylogeny of Nitrite Oxidizers”), most of our knowledge on the physiology and biochemistry of these organisms stems from research on Nitrobacter species and thus cannot be generalized for all nitrite oxidizers.
The key enzyme of nitrite-oxidizing bacteria is the membrane-bound nitrite oxidoreductase (Tanaka et al. 1983), which oxidizes nitrite with water as the source of oxygen to form nitrate (Aleem et al. 1965). The electrons released from this reaction are transferred via a- and c-type cytochromes to a cytochrome oxidase of the aa 3 type. However, the mechanism of energy conservation in nitrite oxidizers is still unclear. Neither Hollocher et al. (1982) nor Sone et al. (1983) were able to find an electron transport chain linked to proton translocation in nitrite-oxidizing cells of Nitrobacter winogradskyi. The first product of energy conservation was shown to be NADH and not ATP (Sundermeyer and Bock 1981).
Except for Nitrobacter, all other isolated nitrite oxidizers are obligate lithotrophs with nitrite serving as the only energy source. Although many strains of Nitrobacter are able to grow heterotrophically, growth is very inefficient and slow (Smith and Hoare 1968; Bock 1976). Additionally, inorganic substrates other than nitrite, namely, nitric oxide, can be used for lithotrophic growth, indicating metabolic diversity among Nitrobacter species (Freitag et al. 1987).
In anoxic environments, Nitrobacter cells are able to grow by denitrification (Freitag et al. 1987; Bock et al. 1988). Nitrate can be used as acceptor for electrons derived from organic compounds to promote anaerobic growth. Since the oxidation of nitrite is a reversible process, the nitrite oxidoreductase can reduce nitrate to nitrite in the absence of oxygen (Sundermeyer-Klinger et al. 1984). Furthermore, the nitrite oxidoreductase copurifies with a nitrite reductase that reduces nitrite to nitric oxide (Ahlers et al. 1990).
Nitrite as a Substrate
The utilization of nitrite as an energy source has been the subject of several reviews (Wood 1986; Hooper 1989; Bock et al. 1991; Yamanaka et al. 1981; Tanaka et al. 1983; Sundermeyer-Klinger et al. 1984; Fukuoka et al. 1987). Nitrite is oxidized to nitrate, and the oxygen atom in the nitrate molecule is derived from water (Aleem 1965; Kumar et al. 1983; Hollocher 1984) according to Eq. 3.15.
The two electrons released are transported to oxygen, as described in Eq. 3.16.
The produced nitrate is inhibitory for Nitrobacter species at concentrations between 30 and 65 mM, probably owing to feedback inhibition.
The electron flux from nitrite to oxygen could pass the following electron carriers (Bock and Koops 1992):
Enzymes Involved in Nitrite Oxidation
Nitrite Oxidoreductase
Nitrite oxidation is a reversible process. The enzyme nitrite oxidoreductase (NO2-OR) catalyzes the oxidation of nitrite to nitrate and the reduction of nitrate to nitrite (section “Genetics of Nitrite Oxidizers”). The NO2-OR is an inducible membrane protein present in Nitrobacter cells, which are either grown lithotrophically with nitrite or heterotrophically in the presence of nitrate. Depending upon the enzyme isolation technique, the molecular features of NO2-OR vary considerably. Cytochromes of the a and c type were present when the enzyme of Nitrobacter winogradskyi was solubilized with Triton X-100 and purified by ion exchange and size exclusion chromatography (Tanaka et al. 1983). The purified protein was composed of three subunits of 55, 29, and 19 kDa. Cytochromes a 1 and c were also found when n-octylglycoside was chosen as detergent. However, using sodium deoxycholate and subsequent isolation by sucrose gradient centrifugation, only cytochrome c could be detected (Sundermeyer-Klinger et al. 1984). In this preparation, the holoenzyme of Nitrobacter hamburgensis consisted of three subunits with relative weights of 116–130, 65, and 32 kDa. No cytochromes were found when the NO2-OR was isolated from membranes by heat treatment. In this case, only two subunits of 115–130 and 65 kDa were present for Nitrobacter winogradskyi, Nitrobacter vulgaris, and for Nitrobacter hamburgensis (Bock et al. 1990).
All preparations of the NO2-OR contain molybdenum (Mo) and iron-sulfur clusters. In membranes of Nitrobacter winogradskyi, signals attributed to molybdenum were detected by electron proton resonance spectroscopy (Ingledew and Halling 1976). In isolated NO2-OR, molybdenum occurred in the form of molybdopterin (Krüger et al. 1987). The molybdenum content varied between 0.13 (Sundermeyer-Klinger et al. 1984) and 1.4 γ-atoms per molecule (Fukuoka et al. 1987). This difference can probably be explained by the fact that molybdenum often is lost during the enzyme isolation procedure. Molybdenum is essential for nitrite oxidation, and when it is replaced by tungsten, lithoautotrophically growing cells of Nitrobacter hamburgensis are inhibited, whereas heterotrophically growing cells are not. Flavoproteins are absent in NO2-OR preparations. When isolated with Triton X-100, manganese was found to be associated with the NO2-OR (Tanaka et al. 1983). The pH optimum of the NO2-OR for nitrite oxidation differs from that for nitrate reduction. Optimal pH for nitrite oxidation with ferricytochrome c 550, ferricyanide, or chlorate as oxidants is about 8.0. With reduced methyl or benzyl viologen as reductants, the optimal pH for nitrate reduction ranges from 6.0 to 7.0. The apparent Km value for nitrite oxidized by the NO2-OR with the aid of different electron acceptors varied with the test conditions between 0.5 and 2.6 mM (Tanaka et al. 1983) or 0.5 and 3.6 mM (Sundermeyer-Klinger et al. 1984), whereas the Km value for nitrate amounted to about 0.9 mM.
It is important to note that the specific activities of NO2-OR are influenced by the purification steps of the isolation procedure. As shown in Table 3.3 , the nitrite oxidation activity and the nitrate reduction activity are highest in the membrane fraction. Both activities decrease to about 80 % when NO2-OR is isolated from membranes without detergent. If Triton X-100 or sodium deoxycholate is used for isolation, this effect is even more pronounced (Yamanaka and Fukumori 1988; Sundermeyer-Klinger et al. 1984).
Cytochrome c Oxidase
In Nitrobacter species, absorption peaks at 605 nm in difference spectra indicate a cytochrome c oxidase of the aa 3 type. This membrane-bound enzyme was purified to an electrophoretically homogeneous state (Yamanaka et al. 1981; Sewell et al. 1972), and the function of cytochrome aa 3 was determined as a terminal oxidase by photoactivation of CO-inhibited oxygen consumption. In contrast to mitochondrial terminal oxidases, cytochrome aa 3 of Nitrobacter winogradskyi is composed of two subunits with 40 and 27 kDa in a molar ratio of l:l (Yamanaka et al. 1979). One molecule of the enzyme contains two molecules of heme a, two atoms of copper, one atom of magnesium, but no zinc (Yamanaka and Fukumori 1988). The Km values were estimated to be 110 and 24 μM for horse heart cytochrome c and ferricytochrome c (both of which can serve as electron donors) of Nitrobacter winogradskyi, respectively. Phospholipids isolated from Nitrobacter winogradskyi did not stimulate the oxidation rate of native ferrocytochrome c or horse heart cytochrome c (Yamanaka and Fukumori 1988). If cytochrome aa 3 was incorporated in phospholipid vesicles or membrane vesicles, respiratory control was observed, but proton-pumping activity was not (Sone et al. 1983; Sone 1986).
Nitrite Reductase
In Nitrobacter vulgaris, a membrane-bound nitrate reductase (NiR) was copurified with the nitrite oxidoreductase (Ahlers et al. 1990). The NiR reduces nitrite to nitric oxide, which is released under reduced oxygen partial pressure from the cells to the environment. Therefore, this enzyme seems to be a dissimilatory nitrite reductase of the denitrification type.
In the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of NO2-OR and NiR, three bands are visible. In addition to the two proteins with Mr 115,000 and 65,000, which are constituents of the NO2-OR, a third protein with Mr 63,000, possibly representing the NiR, is detectable. The pH optimum of the NiR was shown to be 6.1, and the Km value for nitrite was 0.263 mM. The isoelectric point (IEP) was calculated to be at pH 5.5–6.0. Reduced horse heart cytochrome c can serve as an electron donor for nitrite reduction in Nitrobacter winogradskyi and Nitrobacter vulgaris. The biological function of NiR is difficult to understand, since ATP generation has not been detected during nitrite reduction (Freitag and Bock 1990).
Electron Flow and Energy Transduction
As shown in Fig. 3.9 , the first step, the electron transfer from nitrite to cytochrome a1 is catalyzed by the enzyme nitrite oxidoreductase. Cytochrome a 1 was shown to be necessary to channel electrons from nitrite to cytochrome c (Yamanaka and Fukumori 1988). Cytochrome a 1 of Nitrobacter winogradskyi is not autoxidizable (Tanaka et al. 1983) and shows a typical absorption maximum at 589 nm. It is always found in nitrite-oxidizing and nitrate-reducing cells of all Nitrobacter species. On the other hand, Nitrospira marina does not possess any cytochrome of the a type (Watson et al. 1986).


Model of the electron flow in Nitrobacter. Depicted are the pathways of denitrification and heterotrophic growth (upper part) and the nitrification pathway (lower part). CM cytoplasmic membrane, i inside the cell/cytoplasmic space, o outside of the cell/periplasmic space, NO 2 -OR nitrite oxidoreductase, and TCA the tricarboxylic acid cycle (Figure was kindly provided by I. Schmidt)
The electrons enter the underlying respiratory chain at the level of cytochrome c (Aleem 1968; Cobley 1976b; Aleem and Sewell 1981; Sundermeyer and Bock 1981; Tanaka et al. 1983). The reduction of cytochrome c is a thermodynamically unfavorable step, which is slow in cell-free extracts. Electrons derived from the nitrite/nitrate couple have a redox potential of Em,7 = +420 mV, whereas those of ferrocytochrome c/ferricytochrome c have a potential of Em,7 = +260 mV. A relatively high nitrite concentration would cause a lowering of the redox potential, but in natural habitats, high nitrite concentrations are rarely found (Schmidt 1982). Actually, a highly active cytochrome aa 3 pushes nitrite oxidation by the removal of electrons from cytochrome c. In addition, the concentration of cytochrome aa 3 also varies dependent upon the oxygen concentration. SDS-PAGE experiments demonstrated that cells of Nitrobacter vulgaris grown under high oxygen partial pressure possess high nitrite-oxidizing activity and a high cytochrome aa 3 content, whereas those cells grown under low oxygen tension have a low activity and a low cytochrome aa 3 content (E. Bock, unpublished observation).
The nitrite-oxidizing system of Nitrobacter vulgaris can be remodeled by reassociation of n-octylglycoside-isolated NO2-OR with cytochrome aa 3. The activity of the nitrite-oxidizing system increased with increasing amounts of cytochrome c oxidase (Fig. 3.10 ). Present alone, NO2-OR or cytochrome aa 3 was unable to oxidize nitrite to nitrate. The in vitro modeling of the nitrite-oxidizing system of Nitrobacter vulgaris shows clearly that both enzymes are essential for the oxidation of nitrite to nitrate, with oxygen as the terminal electron acceptor. At a fixed NO2-OR content, the enzyme activity is regulated by the concentration of cytochrome aa 3.


Increase of the specific nitrite-oxidizing activity in cell-free enzyme preparations of Nitrobacter vulgaris. Isolated nitrite oxidoreductase was complemented with increasing amounts of cytochrome oxidase (aa 3) for 20 h at 28 °C. The specific activity was measured as nitrite oxidized to nitrate with oxygen as the electron acceptor
In addition to oxygen, CO2 can serve as electron sink in Nitrobacter. Cytochrome c oxidation generates energy that is necessary for autotrophic carbon dioxide fixation. Since lithoautotrophic growth is inefficient, 85–115 mol of nitrite has to be oxidized to assimilate 1 mol of carbon dioxide (Bömeke 1954).
Electron Flow of the Conventional Respiratory Chain
The electrons from nitrite meet the underlying respiratory chain at the level of cytochrome c (not shown in Fig. 3.9 ). This chain functions in lithotrophically, mixotrophically, and heterotrophically growing cells as well as in endogenous respiring cells of Nitrobacter in the absence or presence of oxygen (Fig. 3.11 ). Electrons from NADH + H+ (NADH) pass via flavin mononucleotide (FMN) and ubiquinone to a cytochrome bc 1 complex and finally to the terminal oxidase.


Components of the conventional respiratory chain of Nitrobacter. The blocks symbolize proteins and their subunits in Mr
The responsible NADH oxidase has not yet been isolated. However, FMN was found to be present in heterotrophically grown Nitrobacter cells (Kirstein et al. 1986). Ubiquinone Q10 was the isoprenoid in the respiratory chain (Aleem and Sewell 1984).
The cytochrome bc 1 is supposed to consist of a cytochrome b 560 (55 kDa) and cytochrome c1-550 (32 kDa; M. Rudert, unpublished observations). The terminal oxidase (presented in Fig. 3.11 ) is a protein complex isolated from Nitrobacter vulgaris cells (M. Rudert, unpublished observations) consisting of a 25-kDa cytochrome c 552 and a 24.5-kDa cytochrome o. This oxidase is assumed to be active at high oxygen partial pressure and is also present in lithotrophically grown cells of Nitrobacter winogradskyi (Aleem and Sewell 1984) as well as in heterotrophically grown cells of Nitrobacter hamburgensis (Kirstein et al. 1986). At low oxygen tension, cytochrome aa 3 might perform this function (Fig. 3.9 ).
Additional electron carriers have been described by different authors. Kurokawa et al. (1987) isolated a flavin adenine dinucleotide-containing flavoenzyme from lithotrophically grown cells of Nitrobacter winogradskyi with NAD(P)H cytochrome c reductase and transhydrogenase activities. Two cytochromes of the b type, b 560 and b 564, were found in Nitrobacter hamburgensis. Cytochrome b 560 is typical for heterotrophically grown cells and might belong to the bc 1 complex (Kirstein et al. 1986). The function of cytochrome b 564 is unknown. Several additional membrane-bound and soluble cytochromes of the c type have been described (Chaudhry et al. 1981; Miller and Wood 1982). As reported by Tanaka et al. (1982) and Yamanaka et al. (1982), the amino acid composition of a soluble cytochrome c of Nitrobacter winogradskyi is similar to the mitochondrial cytochrome c.
ATP Production
A generally accepted concept for the mechanism of energy generation derived from the described electron flow system is not available. Cobley (1976a) reported proton release into the cytoplasm, whereas Wetzstein and Ferguson (1985) detected proton extrusion into the periplasmic space coupled to oxidation of nitrite with artificial electron donors. However, proton-pumping activity able to drive a membrane-bound ATPase could neither be measured for nitrite-oxidizing cells nor for nitrite-oxidizing vesicles (Hollocher et al. 1982).
Apart from the oxidation of exogenous organic substrates, Nitrobacter cells can oxidize endogenous material, for example, poly-β-hydroxybutyrate; this metabolic activity is called “endogenous respiration.” It has been shown that the oxidation of both exogenic and endogenic matter causes electron flow via a “normal” respiratory chain. Thus, nitrite-oxidizing Nitrobacter can be considered as a regulatory specialist because nitrite oxidation interferes with normal respiration, for example, nitrite oxidation might inhibit endogenous respiration (Eigener and Bock 1975). Changing from endogenous respiration to nitrite oxidation, active cells increased their ATP pool to a maximum of 1 mol of ATP by the oxidation of 1 mol of nitrite (Aleem 1968). All attempts to reproduce this result have failed, but in whole cells and in membrane vesicles, ATP was formed at the expense of NADH oxidation with nitrate (Kiesow 1964; Freitag and Bock 1990) and/or oxygen as electron acceptor (Sewell and Aleem 1979).
In Nitrobacter, phosphorylation of ADP is carried out by a membrane-bound ATP synthase. Isolated Nitrobacter ATPase is similar to the F1-ATPase from a thermophilic bacterium (Yamanaka and Fukumori 1988). With respect to ATP production, Nitrobacter might be best described as a “normal” respiring organism, but this does not explain why heterotrophic growth is so slow.
NADH Production and Cell Growth
Lithotrophically grown cells of Nitrobacter winogradskyi and Nitrobacter vulgaris possess an average poly-β-hydroxybutyrate (PHB) content of 10–30 % of the cell dry weight (E. Bock, unpublished observations). This relatively high content indirectly indicates overproduction of NADH. Kiesow (1964) demonstrated in vivo NADH synthesis in nitrite-oxidizing cells by measuring the increase in extinction at 340 nm. Repeating these experiments, we also found NADH formation but only at low oxygen partial pressure. The reaction was sensitive to the uncoupler 2,4-dinitrophenol and insensitive to the ATPase inhibitor N,N′-dicyclohexylcarbodiimide (DCCD; Freitag and Bock 1990).
In Fig. 3.12 , the classical scheme of reverse electron flow for generation of NADH is shown. The functional models proposed by Wood (1986) and Hooper (1989) leave many questions unanswered. For example, the authors cannot explain why nitrite-oxidizing cells or spheroplasts of Nitrobacter winogradskyi do not produce a proton gradient, which is necessary to understand reverse electron flow (Hollocher et al. 1982). As stated above, nitrite-oxidizing bacteria are able to grow with nitrite, although the electron transfer from nitrite to cytochrome c is more electronegative than the nitrite/nitrate couple (Aleem 1968; Ferguson 1982). In spite of the existing unfavorable redox potential of cytochrome c, the electrons released from nitrite are promptly removed by cytochrome aa 3, so that nitrite oxidation can proceed without energy consumption (O’Kelley et al. 1970). As shown by Cobley (Cobley 1976a, b), the membrane potential has a stimulatory effect on the rate of nitrite oxidation. In experiments with whole cells and membrane vesicles, the nitrite-oxidizing activity decreased in the presence of uncouplers, which collapsed the membrane potential. Thus, even the loss of activity of the isolated NO2-OR might be caused by the loss of the transmembrane electric field, which mediates a conformation transition between an “inactive” and an “active” form of the enzyme (Tsong and Astumian 1987).


Classical scheme of ATP-dependent NADH synthesis in nitrite-oxidizing cells of Nitrobacter
Sundermeyer and Bock (1981) were the first to present evidence that NADH synthesis is the primary energy-conserving process in nitrite-oxidizing cells. In addition to nitrite, nitric oxide was shown to be a suitable electron donor for NADH synthesis (Freitag and Bock 1990). Figures 3.13 and 3.14 show the formation of NADH (increase in absorption at 340 nm) in whole cells of Nitrobacter winogradskyi. When nitrite was added to aerobic cell suspensions, the dissolved oxygen tension dropped within 5 min to less than 4 % of saturation. As shown in Fig. 3.13 , the NADH pool of the cells first decreased for 5 min and then increased at a constant rate. When nitrite was added to anaerobic cell suspensions, the NADH content increased without any lag phase (Fig. 3.14 ). As shown in the figures, the rates of NADH formation with nitric oxide as substrate were faster than those with nitrite. Compared to Em,7 = +420 mV for the nitrite/nitrate couple, the redox potential of the nitric oxide/nitrite couple is Em,7 = +374 mV (Wood 1978), if water is the reactant. Thermodynamically, there is no great difference between the two reactions; nevertheless, nitric oxide was the better substrate than nitrite when Nitrobacter winogradskyi was grown lithoautotrophically.


NADH formation in whole cells of Nitrobacter winogradskyi in the presence of nitrite (a) and nitric oxide (b) under oxic conditions. NADH production was measured as the increase in absorption at 340 nm


NADH formation in whole cells of Nitrobacter winogradskyi in the presence of nitrite (a) and nitric oxide (b) under anoxic conditions. NADH production was measured as the increase in absorption at 340 nm
It is generally accepted that NADH generation in Nitrobacter cells is an ATP-independent reaction as shown for Thiobacillus ferrooxidans (Lu and Kelly 1988). But it is still unclear how NADH is synthesized.
Genetics of Nitrite Oxidizers
With exception of the 16S rRNA genes (Sorokin et al. 1998; Orso et al. 1994; Teske et al. 1994; Ehrich et al. 1995), no genetic data are available for nitrite-oxidizing bacteria other than Nitrobacter (section “Phylogeny of Nitrite Oxidizers”). For Nitrobacter species, sequence of the 16S-23S rRNA intergenic spacer and partial sequences of the 23S rRNA genes have been determined (Grundmann et al. 2000). Furthermore, the genes encoding the two catalytic core subunits of cytochrome c oxidase of Nitrobacter winogradskyi occur in the same operon. Similar to many ammonia-oxidizing bacteria, N. winogradskyi possesses at least two copies of these genes in its genome (Berben 1996). In addition, the sequences of the Calvin cycle genes were determined for Nitrobacter vulgaris (Strecker et al. 1994) and Nitrobacter winogradskyi (GenBank accession numbers [{http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Search=Nucleotide=AF109915=DocSum}{AF109915}] and [{http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Search=Nucleotide=AF109914=DocSum}{AF109914}]). In Nitrobacter vulgaris, the genes for the large and small subunits of ribulose-1,5-bisphosphate carboxylase/oxygenase, the glyceraldehyde-3-phosphate dehydrogenase, and a regulatory protein of the LysR family are located together within one cluster. Another cluster contains the genes for fructose-1,6- and sedoheptulose-1,7-biphosphatase, phosphoribulokinase, and fructose-1,6- and sedoheptulose-1,7-bisphosphatealdolase. Furthermore, the genes for both subunits (norA and norB) of the membrane-bound nitrite oxidoreductase (NO2-OR) from Nitrobacter hamburgensis were sequenced. These genes cluster together with an additional ORF (norX) in the order norA, norX, and norB. The deduced amino acid sequence of protein NorB contains four cysteine clusters with striking homology to those of iron-sulfur centers of bacterial ferredoxins. The β-subunit and the sequenced part of the α-subunit of NO2-OR exhibit significant sequence similarities with the β- and α-subunits of the two dissimilatory nitrate reductases of several chemoorganotrophic bacteria including E. coli (Kirstein and Bock 1993). This is consistent with biochemical data that suggest a close functional similarity between both enzyme complexes (Sundermeyer-Klinger et al. 1984; Hochstein and Tomlinson 1988).
Heterotrophic Nitrification
The oxidation of ammonia (van Niel et al. 1993), hydroxylamine (Ralt et al. 1981), or organic nitrogen compounds, for example, oximes (Castignetti and Hollocher 1984), to nitrite and nitrate by various chemoorganotrophic microorganisms is called “heterotrophic nitrification.” Heterotrophic nitrification is a cometabolism that is not coupled to energy conservation (Wood 1988b). Thus, growth of all heterotrophic nitrifiers is completely dependent on the oxidation of organic substrates (Focht and Verstraete 1977; Kuenen and Robertson 1987). The final product of heterotrophic nitrification often is nitrite (Castignetti and Gunner 1980), so that heterotrophic nitrification may supply the substrate for lithotrophic nitrite oxidizers and heterotrophic denitrifiers. This additional nitrite production (together with the ability of nitrite oxidizers to grow chemoorganotrophically) might explain why in many environments the number of lithoautotrophic nitrite oxidizers is much higher than that of lithoautotrophic ammonia oxidizers (Kuenen and Robertson 1987).
Recently, attention has been driven to heterotrophic nitrifiers because many of them are capable of aerobic denitrification in the presence of organic matter, leading to the complete elimination of dissolved nitrogen compounds with the formation of gaseous nitrogen oxides and/or dinitrogen gas (Castignetti and Hollocher 1984; Robertson et al. 1989; Andersson and Levine 1986; van Niel et al. 1987). Owing to the simultaneous nitrifying and denitrifying activity, nitrification rates of heterotrophic nitrifiers are often underestimated (Castignetti and Hollocher 1984; Kuenen and Robertson 1987). For example, Paracoccus denitrificans (formerly called Thiosphaera pantotropha) produces nitrite from urea, ammonia, and hydroxylamine and is also able to reduce nitrite even under aerobic conditions (Robertson and Kuenen 1983, 1984; Robertson and Kuenen 1988). Therefore, in cultures of this organism, nitrite only accumulates when the nitrite reductase activity is repressed.
Biochemically, the ammonia-oxidizing enzyme of Paracoccus denitrificans shows some similarities to the AMO of lithotrophic ammonia oxidizers, for example, the ability to oxidize alkanes, the apparent requirement for copper, and inhibition by light, diethyldithiocarbamate and allylthiourea (Moir et al. 1996a; Crossmann et al. 1997). The purified ammonia-oxidizing enzyme of P. denitrificans contains two polypeptides of 38 and 46 kDa, respectively (Moir et al. 1996a). However, the genes encoding for these polypeptides are not closely related to the amo genes of lithotrophic ammonia oxidizers (Crossmann et al. 1997). The hydroxylamine oxidoreductase from P. denitrificans is a monomeric protein of approximately 18.5 kDa containing nonheme iron (Wehrfritz et al. 1993; Moir et al. 1996b).
The environmental importance of heterotrophic nitrifiers is controversial in the literature. Generally, it is assumed that in most environments, the biological conversion of reduced forms of nitrogen to nitrite and nitrate is catalyzed mainly by the lithoautotrophic ammonia- and nitrite-oxidizing bacteria and not by heterotrophic nitrifiers. This reflects that the nitrification rates of heterotrophic nitrifiers are small compared to those of autotrophic nitrifiers (Robertson and Kuenen 1988). Therefore, heterotrophic nitrification was thought to occur preferentially under conditions unfavorable for autotrophic nitrification, for example, in acidic environments (Killham 1986). In such environments, heterotrophic bacteria, fungi, and even some algae might contribute considerably to nitrification (Schimel et al. 1984; Killham 1986, 1987; Robertson and Kuenen 1990; Spiller et al. 1976). But according to recent reports, even in acidic soils, heterotrophic nitrification contributes to overall nitrate production only to a minor extent (Stams et al. 1990; Barraclough and Puri 1995).
Novel Aspects
Nitrogenous Oxides Are Essential for Aerobic Ammonia Oxidation
Nitrifying as well as denitrifying bacteria contribute to the net production of nitrogenous oxides from soil (Kester et al. 1996, 1997a, b) and from aquatic environments (Xu et al. 1995). This is noteworthy since the gaseous compounds nitric oxide (NO), nitrogen dioxide (NO2), and nitrous oxide (N2O) are of significance for the chemistry of the atmosphere (Johnston 1972; Crutzen 1979; Galbally and Roy 1983). Additionally, nitrous oxide acts as a greenhouse gas (Wang et al. 1976; Andersson and Levine 1986). Furthermore, nitric oxide and to a more moderate extent nitrogen dioxide (Mancinelli and McKay 1983) have strong inhibitory effects on bacteria (Mancinelli and McKay 1983; Shank et al. 1962). Toxicity of nitric oxide is based on its capability to form metal nitrosyl complexes (mainly with heme proteins, iron-sulfur proteins, and copper-containing proteins; Henry et al. 1991), resulting, for example, in the inhibition of cytochrome oxidases (Carr and Ferguson 1990). Furthermore, nitric oxide was shown to form S-nitrosothiols from sulfhydryl groups (Stammler et al. 1992; Hausladen et al. 1996) and to cause C → T transitions in the DNA (Wink et al. 1991).
To protect themselves from toxic effects of nitric oxide, many bacteria possess detoxifying enzymes. Denitrifying organisms produce nitric oxide by the activity of the nitrite reductase but are able to keep the internal nitric oxide concentration low by reducing it to nitrous oxide via the NO reductase. Thus, these organisms are strongly dependent on a close functional coupling between both enzymes (Zumft 1993). Consistent with this finding, loss of NO reductase is lethal for denitrifying cells of Pseudomonas stutzeri (Braun and Zumft 1991). Other organisms, for example, Pseudomonas strain PS88, which do not possess the ability to denitrify, are able to detoxify nitric oxide by means of oxidative processes that convert it to nitrate (Baumgärtner et al. 1996; Hausladen et al. 1996; Koschorreck et al. 1996).
For ammonia-oxidizing bacteria under oxic conditions, NO is also toxic but only in the absence of ammonia (E. Bock, unpublished observation). If ammonia is present, NO and/or NO2 is even essential for aerobic ammonia oxidation. Therefore, the removal of nitric oxide (NO) from cultures of Nitrosomonas eutropha by intensive aeration leads to inhibition of ammonia oxidation. This phenomenon has implications on batch cultivation of ammonia oxidizers in the laboratory (Zart and Bock 1998). Usually, it is necessary to avoid intensive aeration or stirring during the first days of incubation; otherwise, the cells will grow very slowly or even not at all. It was possible to achieve recovery of ammonia oxidation by adding nitric oxide to the air supply. The grade of recovery was dependent on the concentration of nitric oxide supplied (Zart et al. 2000). Inhibition of ammonia oxidation was also observed, when nitric oxide was removed from nitrifying cells of N. eutropha by means of DMPS (2,3-dimercapto-1-propanesulfonic acid) in the presence of Fe3+ ions. The addition of nitric oxide lowered inhibition by DMPS. In another assay, ammonia oxidation of Nitrosomonas eutropha was inhibited by activity of the NO-detoxifying bacterium Pseudomonas PS88 (Baumgärtner et al. 1996; Koschorreck et al. 1996) and could be recovered by addition of NO or lowering the activity of the pseudomonad (Zart et al. 2000). The enhancing effect of nitric oxide and nitrogen dioxide on aerobic nitrification and cell growth of Nitrosomonas eutropha could also be demonstrated using fermenter cultures (Zart and Bock 1998). As shown in Fig. 3.15 , the specific activity of ammonia oxidation increased drastically in the presence of nitric oxide and even more if nitrogen dioxide was added instead of nitric oxide. Nitrosomonas eutropha was able to tolerate long exposure (up to 30 days) to nitrogen dioxide or nitric oxide at a concentration as high as 50 ppm when ammonia was oxidized. This is unusual since already 1 ppm nitric oxide is inhibitory for various chemoorganotrophic bacteria (Mancinelli and McKay 1983). This experiment also shows that nitrite is not a potent inhibitor for N. eutropha because nitrite accumulated in this experiment to 100 mM. Thus, ammonia oxidation in N. eutropha was obviously not inhibited by nitrite accumulation as described for N. europaea (Anthonisen et al. 1976; Wullenweber et al. 1978; Drozd 1980).


Increase in specific activity of ammonia oxidation in fermenter cultures of Nitrosomonas eutropha upon addition of nitric oxide or nitrogen dioxide to the air supply
The increase of nitrification rates upon nitrogen dioxide or nitric oxide addition was partly due to a significant increase of cell density. In the presence of 50 ppm of nitrogen dioxide, it was possible to obtain up to 2 × 1010 cells ml−1 of N. eutropha (Zart and Bock 1998), a cell density of ammonia oxidizers, which to our knowledge has never been reported before. Generally, in batch cultures of different Nitrosomonas strains, a cell density of about 2 × 108 cells ml−1 is rarely exceeded (Engel and Alexander 1958; Prosser 1989). In continuous cultures of N. europaea with complete biomass retention, Tappe et al. (1996) obtained about 5 × 109 cells ml–1. But it is not clear, so far, how nitric oxide and nitrogen dioxide affect the maximum cell density of these organisms.
Beyond this, nitric oxide and nitrogen dioxide had an enhancing effect on the increase of cell number of Nitrosomonas eutropha (Zart and Bock 1998). The specific growth rate (measured as increase in protein) slightly increased upon addition of nitrogen dioxide, but the fission rate increased in a much stronger way. Thus, it is obvious that cell growth was uncoupled from protein increase. Consequently, the cells were depleted of protein when growing in the presence of nitrogen dioxide. This is shown in Fig. 3.16 where the alteration of cell morphology of Nitrosomonas eutropha grown in presence of 50 ppm of nitrogen dioxide is depicted for a period of 4 weeks. Most striking are the reduction of cell material and the increase of electron-dense inclusion bodies. These inclusion bodies were storage material and resembled glycogen-like particles of Nitrospina gracilis (Watson and Waterbury 1971) and Nitrosolobus multiformis (Watson et al. 1971). Surprisingly, reduction of the protein content per cell was accompanied by increased specific activity of ammonia oxidation.


Electron micrograph of ultrathin sections of cells of Nitrosomonas eutropha showing the morphological alteration of cells grown in a fermenter aerated with 50 ppm NO2. The cells were harvested from the reactor after inoculation (a) and for 1 (b), 2 (c), 3 (d), and 4 weeks (e) of incubation. ICM intracytoplasmic membranes, C carboxysome, and P unknown particles/electron-dense inclusion bodies
Although nitrification and cell growth of ammonia oxidizers were enhanced by adding nitric oxide or nitrogen dioxide to the air supply of the cultures, the cell yield (cell protein produced per mol of ammonia oxidized) and the energy efficiency slightly decreased. This finding might be due to the aerobic production of N2 induced by the addition of NO2 (Zart and Bock 1998). More than 50 % of the ammonia was oxidized to dinitrogen (N2) and traces of nitrous oxide (N2O) by Nitrosomonas eutropha. Increasing amounts of supplementary nitrogen dioxide resulted in increasing nitrogen losses (Zart and Bock 1998). Previously, significant nitrogen losses were only obtained with extremely oxygen-limited cultures of ammonia-oxidizing bacteria (Bock et al. 1995; Zart et al. 1996).
Recently, it could be demonstrated that the addition of NO2 to cells of Nitrosomonas eutropha grown first under anoxic conditions with hydrogen as electron donor and nitrite as electron acceptor (Schmidt and Bock 1997) and then shifted to oxic conditions significantly reduced the lag for the initiation of ammonia oxidation (Schmidt et al. 2001b).
Formation of nitric oxide was up to now interpreted as formation of a by-product of ammonia oxidation without significance for the metabolism of ammonia oxidizers. Inhibition of ammonia oxidation of Nitrosomonas eutropha upon withdrawal of nitric oxide from the culture medium indicates that the production of nitric oxide by ammonia oxidizers seems to be not the formation of a “waste compound” but rather the provision of an important agent for the oxidation of ammonia (Zart et al. 1999). However, nitrogen dioxide rather than nitric oxide could represent the decisive agent, where the latter is acting as precursor for nitrogen dioxide. This hypothesis is based on reports of Schmidt and Bock (Schmidt and Bock 1997, 1998) who described anaerobic oxidation of ammonia by Nitrosomonas eutropha using nitrogen dioxide as an oxygen donor in the AMO reaction. It seems not unlikely that nitrogen dioxide might be involved in the conversion of ammonia to hydroxylamine under oxic conditions as well. In such a case, nitrogen dioxide might act as cosubstrate for the AMO reaction. But since atmospheric nitrogen dioxide concentration hardly exceeds 800 ppb (Galbally and Roy 1983; Baumgärtner 1991), the cells would not be able to cover their requirements by consuming it just from the atmosphere. They might rather produce nitric oxide, which can be oxidized chemically to nitrogen dioxide with dioxygen (Bodenstein 1918). Although the latter reaction proceeds predominantly in the gas phase (Ford et al. 1993; Wink et al. 1993), nitrogen dioxide-consuming reactions (Lewis and Deen 1994) and low concentrations of nitric oxide in the liquid (Pires et al. 1994) can alter the development of the aqueous reaction, so that nitrogen dioxide might be produced in biological systems by oxidation of nitric oxide as it is in the gas phase (Huie 1994).
Considering the finding of Dua et al. (1979) that up to 97% of the oxygen of hydroxylamine originates from molecular oxygen, it is important to note that oxidation of ammonia with nitrogen dioxide leads to the formation of hydroxylamine and nitric oxide (Schmidt and Bock 1997). Under oxic conditions, the latter might be reoxidized with dioxygen to form nitrogen dioxide, which would again be available for the oxidation of ammonia by the AMO. Thus, molecular oxygen would not react directly with ammonia but is hypothetically mediated by nitric oxide/nitrogen dioxide. Consequently, it is not necessary to provide nitric oxide or nitrogen dioxide and ammonia in the same ratio since the nitrogenous oxides are permanently recycled. In Fig. 3.17 , a hypothetical model of this NOx cycle is depicted which refers to the three-stage catalytic cycle of the tyrosinase reaction (Shears and Wood 1985). The AMO can have three oxidation states (Shears and Wood 1985; Bedard and Knowles 1989; Keener and Arp 1993). As already mentioned, copper is a constituent of the enzyme, and NO/N2O4 is involved in ammonia oxidation. Therefore, it can be speculated that NO is a cofactor of the AMO. In accordance with Zart and Bock (1998), enzyme-bound N2O4 is the final cosubstrate for oxidizing ammonia to hydroxylamine. Dinitrogen tetroxide (N2O4) is formed from NO by oxidation with molecular oxygen. The AMO consists of three stages: (1) the deoxy form (reduced), (2) the oxy form (oxidized), and (3) the metoxy form (oxidized). The first stage is oxygen sensitive, and the second and the third one are oxygen stable. The oxygen sensitivity of the deoxy form is caused by an excess of electrons that might lead to the formation of oxygen radicals, which directly or indirectly react with NO, forming peroxy nitrite (OONO). This compound is toxic and destroys the AMO. This working model is in accordance with the observation that purified AMO can oxidize ammonia to hydroxylamine with NADH (H+) as electron donor under anoxic conditions with N2O4 as oxidant. Oxygen was shown to be inhibitory (E. Bock and C. Pinck, unpublished data). Copper in the active center might be responsible for noncovalently binding the nitrogen oxides in form of nitrosyl complexes. As described, oxygen is reacting with NO of the deoxy form (Fig. 3.17 /3), transforming NO to N2O4 (oxy form). The copper ions are oxidized from Cu+ to Cu2+. The N2O4 molecule of the oxy form is the final oxygen donor for ammonia oxidation to hydroxylamine. By this reaction, the enzyme is transferred to the metoxy form (Fig. 3.17 /1), which is subsequently converted by reduction to the deoxy form, thereby completing the cycle (Fig. 3.17 /2). This model also provides an explanation for the inhibition of ammonia oxidation after withdrawal of nitric oxide. As demonstrated recently, acetylene inhibits the aerobic and not the anaerobic ammonia oxidation. Therefore, acetylene is assumed to bind to the deoxy form of the AMO (Schmidt et al. 2001a). Additionally, the oxidation of ammonia with nitrogen dioxide/dinitrogen tetroxide (DG0′ = −140 kJ · mol−1) is thermodynamically more favorable than the “classical” oxidation of ammonia with dioxygen (DG0′ = −120 kJ·mol−1).


Hypothetical model of an NOx-mediated three-stage cycle driving the oxidation of ammonia to hydroxylamine in ammonia-oxidizing bacteria. This cycle does not contribute to the release of NO during aerobic ammonia oxidation (Stüven and Bock 2001), which is probably caused by chemodenitrification of nitrite at the hydroxylamine oxidoreductase (HAO)
Anaerobic Ammonia Oxidation Catalyzed by Nitrosomonas eutropha
Anaerobic Ammonia Oxidation with Nitrogen Dioxide
In the absence of dissolved oxygen, ammonia oxidation and cell growth have been observed recently in cultures of Nitrosomonas eutropha (Schmidt and Bock 1997). Those cells were able to replace molecular oxygen by nitrogen dioxide or dinitrogen tetroxide, respectively. Hydroxylamine and nitric oxide were formed in this reaction. While nitric oxide was not further metabolized, hydroxylamine was oxidized to nitrite. However, anoxic ammonia oxidation with nitrogen dioxide was more than tenfold slower than ammonia oxidation with oxygen as electron acceptor.
The activity of ammonia oxidizers decreases when the oxygen concentration in the medium decreases too. This can be put down to the fact that the oxidant for ammonia oxidation was limited. Therefore, the organisms need another oxidant for anaerobic ammonia oxidation. For a long time, nitrite was discussed as a suitable oxidant (Broda 1977), but as shown in numerous experiments, it could not serve as electron acceptor for anaerobic ammonia oxidation and cell growth in Nitrosomonas (Bock et al. 1995; Zart et al. 1996).
Anaerobic Ammonia Oxidation in Cell-Free Extracts
To reveal the stoichiometry of anaerobic ammonia oxidation, consumption and production of ammonia, nitrogen dioxide, nitric oxide, nitrite, nitrous oxide, and dinitrogen were analyzed in cell-free extracts of N. eutropha. Many attempts were performed to prepare active cell-free extracts of Nitrosomonas europaea under oxic conditions (Suzuki et al. 1970, 1981). One of the most serious problems associated with the characterization of the AMO in extracts has been the instability of the enzyme activity (Suzuki et al. 1974, 1981; Ensign et al. 1993) caused by the sensitivity of reduced AMO to oxygen (C. Pinck and E. Bock, unpublished observation). In contrast, the ammonia-oxidizing enzyme system is stable and active in cell-free extracts under anoxic conditions and thus allowed to characterize the anaerobic ammonia oxidation (Schmidt and Bock 1998). In a helium atmosphere supplied with 25 ppm of nitrogen dioxide, ammonia and nitrogen dioxide were consumed in a ratio of approximately 1:2 by cell-free extracts of Nitrosomonas eutropha. The production of nitric oxide was closely related to the consumption of nitrogen dioxide. Nitric oxide was released in amounts nearly equimolar to the consumption of nitrogen dioxide. The production rate of nitrite was significantly lower than the oxidation rate of ammonia. It is assumed that nitrogen dioxide and nitrite served as acceptors for electrons derived from ammonia oxidation. Approximately 22 % of the nitrite was converted into gaseous nitrogen compounds (nitrogen loss). The main products of denitrification were dinitrogen and traces of nitrous oxide. During the anaerobic ammonia oxidation, hydroxylamine concentrations between 30 and 40 μM were measured. In control experiments with cell-free extracts of Ralstonia eutropha (formerly Alcaligenes eutrophus), Enterobacter aerogenes, and Pseudonocardia nitrificans, neither ammonia oxidation nor nitrogen dioxide consumption could be detected. In sterile control experiments, there was again no ammonia consumption. In addition, no formation of nitric oxide, dinitrogen, or nitrous oxide was measurable.
At 25 °C and atmospheric pressure, the ratio of nitrogen dioxide (NO2) to its dimer dinitrogen tetroxide (N2O4) is 30:70. To decide whether nitrogen dioxide or dinitrogen tetroxide is the electron acceptor for anaerobic ammonia oxidation, experiments were performed at a temperature of 4 °C, conditions under which the dinitrogen tetroxide concentration is almost 100 %. Experiments were performed with crude cell-free extracts in the presence of hydrazine as specific inhibitor for HAO. Although the specific activity of the cell-free extracts decreased significantly at 4 °C, anaerobic ammonia oxidation could be detected with a stoichiometry of the converted nitrogen compounds comparable to those observed at 25 °C. These results indicate that dinitrogen tetroxide can be used as electron acceptor for anaerobic ammonia oxidation.
Based on the observed correlations between ammonia and nitrogen dioxide/dinitrogen tetroxide consumption and nitric oxide and hydroxylamine production, the following equations are proposed. Equations 3.18 and 3.19 describe the two half-reactions of the anaerobic ammonia oxidation. The total AMO reaction is presented in Eq. 3.20:
For comparison, the aerobic ammonia oxidation to hydroxylamine is given in Eqs. 3.21–3.23.
The δG0′ values of the reactions (3.19 and 3.22) were calculated under the assumption that the reducing equivalents for the AMO are energetically near the ubiquinone level (+110 mV). Equations 3.18–3.23 indicate that there are only a few differences between the anaerobic and the aerobic ammonia oxidation of Nitrosomonas eutropha. Instead of molecular oxygen in the course of aerobic ammonia oxidation, dinitrogen tetroxide was used as the electron acceptor, and nitric oxide, an additional product, was released.
It appears likely that the same enzyme is responsible for both the aerobic and the anaerobic ammonia oxidation, since (1) hydroxylamine is an intermediate of both reactions, (2) acetylene inhibits the aerobic as well as the anaerobic ammonia oxidation, and (3) anaerobic ammonia oxidation starts immediately after transferring cell-free extracts to anoxic conditions.
For the oxidation of hydroxylamine under oxic conditions, no oxygen is needed. Therefore, it can be assumed that under anoxic conditions, hydroxylamine was oxidized according to reaction 3.24 as well.
Increasing pool sizes of intracellular ATP and NADH indicated energy conservation in the absence of oxygen but presence of nitrogen dioxide (Schmidt and Bock 1998). Under these conditions, reducing equivalents were also used for the reduction of carbon dioxide, resulting in cell growth and excretion of extracellular organic compounds like glycerol into the medium.
The relevance of the anaerobic ammonia oxidation with NO2/N2O4 as oxidant for microbial ecology cannot be assessed at present. The nitrogen dioxide concentration of maximal 400–800 ppb in the atmosphere (Crutzen 1979) should be too low to support anaerobic ammonia oxidation. However, this metabolism might occur in oxygen-limited zones, where locally restricted higher nitrogen dioxide concentrations might exist because of the reaction of molecular oxygen and nitric oxide (Nielsen 1992).
Anaerobic Ammonium Oxidation Catalyzed by Deep Branching Planctomycetes
Recently, a novel organism was discovered which is capable of catalyzing the anaerobic oxidation of ammonium, with nitrite as electron acceptor (Strous et al. 1999). This organism, which up to now cannot be obtained in pure culture, was enriched from a denitrifying plant reactor where the anaerobic ammonium oxidation process was observed for the first time (Mulder et al. 1995; van de Graaf et al. 1995). By comparative 16S rDNA sequence analysis, the organism was identified as a novel, deep-branching member of the order Planctomycetales, and the name “Candidatus Brocadia annamoxidans” was proposed (Jetten et al. 2001). The 16S rDNA-based molecular diversity surveys and subsequent fluorescence in situ hybridization analyses of several reactors and wastewater treatment plants with anaerobic ammonia-oxidizing activity demonstrated that at least two different genera that form a monophyletic lineage within the Planctomycetales (Fig. 3.18 ) can catalyze this process (Schmid et al. 2000, 2001). In most plants analyzed so far, “Candidatus Kuenenia stuttgartiensis” and not “Candidatus Brocadia annamoxidans” is the most abundant anaerobic ammonium oxidizer (Schmid et al. 2000; Jetten et al. 2001).


16S rDNA tree of the Planctomycetales. Anaerobic ammonium oxidizers are labeled green. The scale bar represents 10 % estimated sequence divergence (Modified from Schmid et al. 2001)
“Candidatus Brocadia annamoxidans” is a chemolithoautotrophic organism with a very low growth rate (0.003 h−1) and a conspicuous ultrastructure. It contains inside the cytoplasm so-called anammoxosomes, membrane-bounded compartments that make up 30–60% of the cell volume and harbor an unusual hydroxylamine oxidoreductase (Jetten et al. 2001). The metabolic pathway of the so-called ANAMMOX process proposed for “Candidatus Brocadia annamoxidans” differs significantly from the known pathway of aerobic, lithotrophic ammonia oxidizers, since ammonium is supposed to be oxidized with hydroxylamine to form hydrazine (N2H4), which is subsequently oxidized to dinitrogen, the main end product of the ANAMMOX process. The four reducing equivalents generated in this oxidation step are used for the initial reduction of nitrite to hydroxylamine (van de Graaf et al. 1997). Preliminary data suggest that the unusual hydroxylamine oxidoreductase of “Candidatus Brocadia annamoxidans” (Jetten et al. 2001), which has a smaller molecular mass than the respective enzyme of Nitrosomonas and contains several c-type cytochromes, catalyzes the oxidation of hydrazine to dinitrogen. Anaerobic ammonia oxidation occurs only if the cell density of “Candidatus Brocadia annamoxidans” is higher than 1010–1011 cells ml−1. The reason for this cell density-dependent activity is not clear so far. The following Eqs. 3.25–3.28 summarize the proposed pathway. The overall reaction resembles a process already proposed in 1977 based on theoretical considerations (Broda 1977):
From Eq. 3.28, it is obvious that the overall reaction is balanced. No reducing power is gained which is essential for the fixation of carbon dioxide by autotrophic organisms. To gain these reducing equivalents, the cells are required to oxidize nitrite to nitrate. Therefore, the amount of nitrite consumed is about 20 % higher than one could expect from Eq. 3.28, and nitrate is additionally formed (van de Graaf et al. 1996):
The metabolic activity of “Candidatus Brocadia annamoxidans” is strongly inhibited by oxygen, phosphate, acetylene, or shock loading. In addition, organic electron donors or high concentrations of nitrite are inhibitory (van de Graaf et al. 1996).
“Candidatus Brocadia annamoxidans” oxidizes ammonium anaerobically about 50-fold faster than Nitrosomonas eutropha with 25 ppm of nitrogen dioxide in the absence of oxygen (Anaerobic Ammonia Oxidation Catalyzed by Nitrosomonas eutropha). Since ANAMMOX was shown to exhibit rather efficient ammonium elimination rates of up to 3 kg of NH +4 m−3 day−1 (van de Graaf et al. 1996), it is suitable for the treatment of wastewater containing much ammonium and little organic chemical oxygen demand (COD). For such wastewater, it has been calculated that the replacement of the conventional nitrogen elimination steps by ANAMMOX would result in a reduction of the operational costs of up to 90 % (Jetten et al. 2001). However, the presence of anaerobic ammonium oxidizers in addition to ammonium nitrite is required in the wastewater. Therefore, partial conventional nitrification, converting approximately half of the ammonium to nitrite, and ANAMMOX have been combined for efficient nitrogen removal from high-strength organic wastewater (summarized in Jetten et al. 2001). It should, however, be noted that very long lag phases are required for obtaining ANAMMOX activity in such plants (Mulder et al. 1995; Jetten et al. 1997), which might be a disadvantage for applications.
References
Aakra A, Utaker JB, Nes IF (1999) RFLP of rRNA genes and sequencing of the 16S-23S rDNA intergenic spacer region of ammonia-oxidizing bacteria: a phylogenetic approach. Int J Syst Bacteriol 49:123–130
Abeliovich A, Vonshak A (1992) Anaerobic metabolism of Nitrosomonas europaea. Arch Microbiol 158:267–270
Abeliovich A, Vonshak A (1993) Factors inhibiting nitrification of ammonia in deep wastewater reservoirs. Water Res 27:1585–1590
Ahlers B, König W, Bock E (1990) Nitrite reductase activity in Nitrobacter vulgaris. FEMS Microbiol Lett 67:121–126
Aleem MIH (1965) Path of carbon and assimilatory power in chemosynthetic bacteria. I. Nitrobacter agilis. Biochim Biophys Acta 107:14–28
Aleem MIH, Hoch GE, Varner JE (1965) Water is the source of oxidant and reductant in bacterial chemosynthesis. Proc Natl Acad Sci USA 54:869–873
Aleem MIH (1966) Generation of reducing power in chemosynthesis II: energy-linked reduction of pyridine nucleotides in the chemoautotroph Nitrosomonas europaea. Biochim Biophys Acta 113:216–224
Aleem MIH (1968) Mechanism of oxidative phosphorylation in the chemoautotroph Nitrobacter agilis. Biochim Biophys Acta 162:338–347
Aleem MIH, Sewell DL (1981) Mechanism of nitrite oxidation and oxidoreductase-systems in Nitrobacter agilis. Curr Microbiol 5:267–272
Aleem MIH, Sewell DL (1984) Oxidoreductase systems in Nitrobacter agilis. In: Strohl WR, Tuovinen OH (eds) Microbial chemoautotrophy. Ohio State University Press, Columbus, pp 185–210
Allison SM, Prosser JI (1993) Survival of ammonia oxidizing bacteria in air-dried soil. FEMS Microbiol Lett 79:65–68
Alzerreca JJ, Norton JM, Klotz MG (1999) The amo operon in marine ammonia-oxidizing gamma-proteobacteria. FEMS Microbiol Lett 180:21–29
Andersson KK, Hooper AB (1983) O2 and H2O are each the source of one O in NO2-produced from NH3 by Nitrosomonas; 15N-NMR evidence. FEBS Lett 164:236–240
Andersson KK, Kent TA, Lipscomb JD, Hooper AB, Münck E (1984) Mössbauer, EPR and optical studies of the P-460 center of hydroxylamine oxidoreductase from Nitrosomonas. J Biol Chem 259:6833–6840
Andersson IC, Levine JS (1986) Relative rates of NO and N2O production by nitrifiers, denitrifiers and nitrate respirers. Appl Environ Microbiol 51:938–945
Andersson KK, Lipscomb DJ, Valentine M, Munck E, Hooper AB (1986) Tetraheme cytochrome c-554 from Nitrosomonas europaea: heme-heme interactions and ligand bindings. J Biol Chem 261:1126–1138
Anthonisen AC, Loehr RC, Prakasam TBS, Srinath EG (1976) Inhibition of nitrification by ammonia and nitrous acid. J Wat Poll Control Fed 48:835–852
Anthony C (1982) The biochemistry of methanotrophs. Academic, London
Arciero DM, Balny C, Hooper AB (1991) Spectroscopic and rapid kinetic studies of reduction of cytochrome c554 by hydroxylamine oxidoreductase from Nitrosomonas europaea. Biochemistry 30:11466–11472
Arciero DM, Hooper AB (1993) Hydroxylamine oxidoreductase from Nitrosomonas europaea is a multimer of an octa-heme subunit. J Biol Chem 268:14645–14654
Arciero DM, Hooper AB (1994) A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced state. J Biol Chem 269:11878–11886
Barraclough D, Puri G (1995) The use of 15N pool dilution and enrichment to separate the heterotrophic and autotrophic pathways of nitrification. Soil Biol Biochem 27:17–22
Bartosch S, Wolgast I, Spieck E, Bock E (1999) Identification of nitrite-oxidizing bacteria with monoclonal antibodies recognizing the nitrite oxidoreductase. Appl Environ Microbiol 65:4126–4233
Batchelor SE, Cooper M, Chhabra SR, Glover LA, Stewart GS, Williams P, Prosser JI (1997) Cell density-regulated recovery of starved biofilm populations of ammonia-oxidizing bacteria. Appl Environ Microbiol 63:2281–2286
Baumgärtner M (1991) Umsetzung von Stickoxiden (NOx) in Böden, auf Gebäudeoberflächen und in Mikroorganismen. Konstanzer Dissertationen Nr. 327. Hartung-Gorre Konstanz Germany. Konstanzer Dissertationen Nr. 327
Baumgärtner M, Koschorreck M, Conrad R (1996) Oxidative consumption of nitric oxide by heterotrophic bacteria in soil. FEMS Microbiol Ecol 19:165–170
Bedard C, Knowles R (1989) Physiology, biochemistry, and specific inhibitors of CH4, NH +4 , and CO oxidation by methanotrophs and nitrifiers. Microbiol Rev 53:68–84
Belser LW, Schmidt EL (1978) Serological diversity within a terrestrial ammonia-oxidizing population. Appl Environ Microbiol 36:589–593
Belser LW (1979) Population ecology of nitrifying bacteria. Ann Rev Microbiol 33:309–333
Belser LW, Mays EL (1982) Use of nitrifier activity measurements to estimate the efficiency of viable nitrifier counts in soils and sediments. Appl Environ Microbiol 43:945–948
Berben G (1996) Nitrobacter winogradskyi cytochrome c oxidase genes are organized in a repeated gene cluster. Ant v Leeuwenhoek 69:305–315
Bergmann DJ, Hooper AB (1994a) Primary structure of cytochrome P-460 of Nitrosomonas. FEBS Lett 353:324–326
Bergmann DJ, Hooper AB (1994b) Sequence of the gene amoB for the 43 kDa polypeptide of ammonia monooxygenase of Nitrosomonas europaea. Biochim Biophys Res Comm 204:759–762
Bergmann DJ, Arciero D, Hooper AB (1994) Organization of the HAO gene cluster of Nitrosomonas europaea: genes for two tetraheme cytochromes. J Bacteriol 176:3148–3153
Bergmann DJ, Zahn JA, DiSpirito AA (2000) Primary structure of cytochrome c′ of Methylococcus capsulatus bath: evidence of a phylogenetic link between P460 and c′-type cytochromes. Arch Microbiol 173:29–34
Blasco F, Lobbi C, Ratouchniak J, Bonnefoy V, Chippaux M (1990) Nitrate reductases of Escherichia coli: sequence of the second nitrate reductase and comparison with that encoded by the narGHJI operon. Molec Gen Genet 222:104–111
Bock E (1965) Vergleichende Untersuchungen über die Wirkung sichtbaren Lichtes auf Nitrosomonas europaea und Nitrobacter winogradskyi. Arch Mikrobiol 51:18–41
Bock E (1970) Untersuchungen über die Wechselwirkung zwischen Licht und Chemosynthese am Beispiel von Nitrobacter winogradskyi. Arch Mikrobiol 70:217–239
Bock E (1976) Growth of Nitrobacter in the presence of organic matter. II. Chemoorganotrophic growth of Nitrobacter agilis. Arch Microbiol 108:305–312
Bock E, Wilderer PA, Freitag A (1988) Growth of Nitrobacter in the absence of dissolved oxygen. Water Res 22:245–250
Bock E, Koops H-P, Möller UC, Rudert M (1990) A new facultatively nitrite oxidizing bacterium Nitrobacter vulgaris sp. nov. Arch Microbiol 153:105–110
Bock E, Koops H-P, Harms H, Ahlers B (1991) The biochemistry of nitrifying organisms. In: Shively JM (ed) Variations of autotrophic life. Academic, London, pp 171–200
Bock E, Koops H-P (1992) The genus Nitrobacter and related genera. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer KH (eds) The prokaryotes, 2nd edn. Springer, New York, pp 2302–2309, http://www.prokaryotes.com
Bock E, Sand W (1993) The microbiology of masonry biodeterioration. J Appl Bacteriol 74:503–514
Bock E, Stüven R, Schmidt I, Zart D (1995) Nitrogen loss caused by denitrifying Nitrosomonas cells using ammonium, or hydroxylamine as electron donors and nitrite as electron acceptor. Arch Microbiol 163:16–20
Bodelier PL, Frenzel P (1999) Contribution of methanotrophic and nitrifying bacteria to CH4 and NH +4 oxidation in the rhizosphere of rice plants as determined by new methods of discrimination. Appl Environ Microbiol 65:1826–1833
Bodenstein M (1918) Die Geschwindigkeit der Reaktion zwischen Stickoxid und Sauerstoff Z. f. Elektroch. 24:183–201
Bömeke H (1954) Über das Verhältnis des oxidierten Stickstoffs zum reduzierten Kohlenstoff beim Nitratbildner. Arch Mikrobiol 20:176–182
Böttcher B, Koops H-P (1994) Growth of lithotrophic ammonia-oxidizing bacteria on hydroxylamine. FEMS Microbiol Lett 122:263–266
Bouwman AF, Fung I, Matthews E, John J (1993) Global analysis of the potential for N2O production in natural soils. Global Biogeochem Cycles 7:557–597
Brady NC (1984) The nature and properties of soils. Macmillan, New York, pp 283–302
Braun C, Zumft WG (1991) Marker exchange of the structural genes for nitric oxide reductase blocks the denitrification pathway of Pseudomonas stutzeri at nitric oxide. J Biol Chem 266:22785–22788
Broda E (1977) Two kinds of lithotrophs missing in nature. Z Allg Mikrobiol 17:491–493
Brown CM (1988) Nitrate metabolism in aquatic bacteria. In: Austin B (ed) Methods in aquatic bacteriology. Wiley, New York, pp 367–388
Buchanan RE (1917) Studies on the nomenclature and classification of bacteria. J Bacteriol 2:347–350
Burrell PC, Keller J, Blackall LL (1998) Microbiology of a nitrite-oxidizing bioreactor. Appl Environ Microbiol 64:1878–1883
Carr GJ, Ferguson SJ (1990) Nitric oxide formed by nitrite reductase of Paracoccus denitrificans is sufficiently stable to inhibit cytochrome oxidase activity and is reduced by its reductase under aerobic conditions. Biochim Biophys Acta 1017:57–62
Casciotti KL, Ward BB (2001) Dissimilatory nitrite reductase genes from autotrophic ammonia-oxidizing bacteria. Appl Environ Microbiol 67:2213–2221
Castignetti D, Gunner HB (1980) Sequential nitrification by an Alcaligenes sp. and Nitrobacter agilis. Can J Microbiol 26:1114–1119
Castignetti D, Hollocher TC (1984) Heterotrophic nitrification among denitrifiers. Appl Environ Microbiol 47:620–623
Chaudhry GR, Suzuki I, Duckworth HW, Lees H (1981) Isolation and properties of cytochrome c553, cytochrome c550, and cytochrome c549, 554 from Nitrobacter agilis. Biochim Biophys Acta 637:18–27
Clark C, Schmidt EL (1967) Growth response of Nitrosomonas europaea to amino acids. J Bacteriol 93:1302–1309
Cobley JB (1976a) Energy-conserving reactions in phosphorylating electron-transport particles from Nitrobacter winogradskyi. Activation of nitrite oxidation by the electrical component of the proton motive force. Biochem J 156:481–491
Cobley JB (1976b) Reduction of cytochromes by nitrite in electron-transport particles from Nitrobacter winogradskyi. Biochem J 156:493–498
Collins MJ, Arciero DM, Hooper AB (1993) Optical spectropotentiometric resolution of the hemes of hydroxylamine oxidoreductase Heme quantitation and pH dependence of EM. J Biol Chem 268:14655–14662
Conrad R (1996) Metabolism of nitric oxide in soil and soil microorganisms and regulation of flux into the atmosphere. In: Murrell JC, Kelly DP (eds) Microbiology of atmospheric trace gases: sources, sinks and global change processes. NATO ASI Series, Springer, Berlin, pp 167–203
Crossmann LC, Moir JWB, Enticknap JJ, Richardson DJ, Spiro S (1997) Heterologous expression of heterotrophic nitrification genes. Microbiology 143:3775–3783
Crutzen PJ (1979) The role of NO and NO2 in the chemistry of the troposphere and stratosphere. Ann Rev Earth Planet Sci 74:443–472
Daims H, Nielsen PH, Nielsen JL, Juretschko S, Wagner M (2000) Novel Nitrospira-like bacteria as dominant nitrite-oxidizers in biofilms from wastewater treatment plants: diversity and in situ physiology. Wat Sci Tech 41:85–90
Daims H, Nielsen JL, Nielsen PH, Schleifer K-H, Wagner M (2001) In situ characterization of Nitrospira-like nitrite-oxidizing bacteria active in wastewater treatment plants. Appl Environ Microbiol 67(11):5273–5284
De Boer W, Klein Gunnewiek PJA, Veenhuis M, Bock E, Laanbroek HJ (1991) Nitrification at low pH by aggregated chemolithotrophic bacteria. Appl Environ Microbiol 57:3600–3604
De Bruijn P, van de Graaf AA, Jetten MSM, Robertson LA, Kuenen JG (1995) Growth of Nitrosomonas europaea on hydroxylamine. FEMS Microbiol Lett 125:179–184
Degrange V, Bardin R (1995) Detection and counting of Nitrobacter populations in soil by PCR. Appl Environ Microbiol 61:2093–2098
DiSpirito AA, Taaffe LR, Hooper AB (1985) Localization and concentration of hydroxylamine oxidoreductase and cytochromes c552, c554, cm553, cm552, and a in Nitrosomonas europaea. Biochim Biophys Acta 806:320–330
DiSpirito AA, Lipscomp JD, Hooper AB (1986) Cytochrome aa3 from Nitrosomonas europaea. J Bacteriol 261:17048–17056
Drozd JW (1976) Energy coupling and respiration in Nitrosomonas europaea. Arch Microbiol 101:257–262
Drozd JW (1980) Respiration in ammonia-oxidizing chemoautotrophic bacteria. In: Knowles R (ed) Diversity of bacterial respiratory systems, vol 2. CRC Press, Boca Raton, pp 87–111
Dua RD, Bhandari B, Nicholas DJD (1979) Stable isotope studies on the oxidation of ammonia to hydroxylamine by Nitrosomonas europaea. FEBS Lett 106:401–404
Ehrich S, Behrens D, Lebedeva E, Ludwig W, Bock E (1995) A new obligately chemolithoautotrophic, nitrite-oxidizing bacterium. Nitrospira moscoviensis sp. nov. and its phylogenetic relationship. Arch Microbiol 164:16–23
Eigener U, Bock E (1975) Study on the regulation of oxidation and CO2 assimilation in intact Nitrobacter winogradskyi cells. Arch Microbiol 102:241–246
Eighmy TT, Bishop PL (1989) Distribution and role of bacterial nitrifying populations in nitrogen removal in aquatic treatment systems. Water Res 23:947–955
El-Demerdash ME, Ottow JCG (1983) Einfluss einer hohen Nitratdüngung auf Kinetik und Gaszusammensetzung der Denitrifikation in unterschiedlichen Böden. Z Pflanzenernährung und Bodenkunde 146:138–150
Engel MS, Alexander M (1958) Growth and autotrophic metabolism of Nitrosomonas europaea. J Bacteriol 76:217–222
Ensign SA, Hyman MR, Arp DJ (1993) In vitro activation of ammonia monooxygenase from Nitrosomonas by copper. J Bacteriol 175:1971–1998
Erickson RH, Hooper AB, Terry KR (1972) Solubilization and purification of cytochrome a, from Nitrosomonas. Biochim Biophys Acta 283:155–166
Ferguson S (1982) Is a proton-pumping cytochrome oxidase essential for energy conservation in Nitrobacter? FEBS Lett 146:239–243
Fliermanns CB, Bohlool BB, Schmidt EL (1974) Autecological study of the chemoautotroph Nitrobacter by immunofluorescence. Appl Environ Microbiol 27:124–129
Focht DD, Verstraete W (1977) Biochemical ecology of nitrification and denitrification. Adv Microbial Ecol 1:135–214
Ford PC, Wink DA, Stanbury DM (1993) Autoxidation kinetics of aqueous nitric oxide. FEBS Lett 326:1–3
Freitag A, Rudert M, Bock E (1987) Growth of Nitrobacter by dissimilatoric nitrate reduction. FEMS Microbiol Lett 48:105–109
Freitag A, Bock E (1990) Energy conservation in Nitrobacter. FEMS Microbiol Lett 66:157–162
Fukuoka M, Fukumori Y, Yamanaka T (1987) Nitrobacter winogradskyi cytochrome a1c1 is an iron-sulfur molybdo-enzyme having hemes a and c. J Biochem 102:525–530
Galbally IE, Roy CR (1983) The fate of nitrogen compounds in the atmosphere. Devel Plant Soil Sci 9:263–284
Giannakis C, Miller DJ, Nicholas DJD (1985) Comparative studies on redox proteins from ammonia oxidizing bacteria. FEMS Microbiol Lett 30:81–85
Goreau TJ, Kaplan WA, Wofsy SC, McElroy MB, Valois FW, Watson SW (1980) Production of NO2 and N2O by nitrifying bacteria at reduced concentrations of oxygen. Appl Environ Microbiol 40:526–532
Groffmann PM (1987) Nitrification and denitrification in soil: a comparison of enzyme assay, incubation and enumeration methods. Plant Soil 97:445–450
Grundmann GL, Neyra M, Normand P (2000) High-resolution phylogenetic genetic analysis of NO2-oxidizing Nitrobacter species using the rrs-rrl IGS sequence and rrl genes. Int J Syst Microbiol 50:1893–1898
Hall GH (1986) Nitrification in lakes. In: Prosser JI (ed) Nitrification. IRL Press, Oxford, UK, pp 127–156
Harms H, Koops H-P, Martiny H, Wullenweber W (1981) D-Ribulose 1,5-biphosphate carboxylase and polhedral inclusions in Nitrosomonas spec. Arch Microbiol 128:280–281
Hass R, Veit S, Meyer TF (1992) Silent pilin genes of Neisseria gonorrhoeae MS11 and the occurrence of related hypervariant sequences among other gonococcal isolates. Molec Microbiol 6:197–208
Hausladen A, Privalle CT, Keng T, DeAngelo J, Stamler JS (1996) Nitrosative stress: activation of the transcription factor OxyR. Cell 86:719–729
Head IM, Hiorns WD, Embley TM, McCarthy AJ, Saunders JR (1993) The phylogeny of autotrophic ammonia-oxidizing bacteria as determined by analysis of 16S ribosomal RNA gene sequences. J Gen Microbiol 139:1147–1153
Henry Y, Ducrocq C, Drapier J-C, Servent D, Pellat C, Guissani A (1991) Nitric oxide, a biological effector—electron paramagnetic resonance detection of nitrosyl-iron-protein complexes in whole cells. Eur Biophys J 20:1–15
Hiorns WD, Hastings RC, Head IM, Hall GR, McCarthy AJ, Saunders JR, Pickup RW (1995) Amplification of 16S ribosomal RNA genes of autotrophic ammonia-oxidizing bacteria demonstrates the ubiquity of Nitrosospiras in the environment. Microbiol 141:2793–2800
Hochstein LI, Tomlinson GA (1988) The enzymes associated with denitrification. Ann Rev Microbiol 42:231–261
Hoffman T, Lees H (1953) The biochemistry of the nitrifying bacteria. Biochem J 54:579–583
Hollocher TC, Tate ME, Nicholas DJD (1981) Oxidation of ammonia by Nitrosomonas europaea. Definitive 18O-tracer evidence that hydroxylamine formation involves a monooxygenase. J Biol Chem 256:10834–10836
Hollocher TC, Kumar S, Nicholas DJD (1982) Respiration dependent proton translocation in Nitrosomonas europaea and its apparent absence in Nitrobacter agilis during inorganic oxidation. J Bacteriol 149:1013–1020
Hollocher TC (1984) Source of the oxygen atoms of nitrate in the oxidation of nitrite by Nitrobacter agilis and evidence against a P-O-N anhydride mechanism in oxidative phosphorylation. Arch Biochem Biophys 233:721–727
Hommes NG, Sayavedra-Soto LA, Arp DJ (1994) Sequence of hcy, a gene encoding cytochrome c-554 from Nitrosomonas europaea. Gene 146:87–89
Hommes NG, Sayavedra-Soto LA, Arp DJ (1996) Mutagenesis of hydroxylamine oxidoreductase in Nitrosomonas europaea by transformation and recombination. J Bacteriol 178:3710–3714
Hommes NG, Sayavedra-Soto LA, Arp DJ (1998) Mutagenesis and expression of amo, which codes for ammonia monooxygenase in Nitrosomonas europaea. J Bacteriol 180:3353–3359
Hommes NG, Sayavedra-Soto LA, Arp DJ (2001) Transcript analysis of multiple copies of amo (encoding ammonia monooxygenase) and hao (encoding hydroxylamine oxidoreductase) in Nitrosomonas europaea. J Bacteriol 183:1096–1100
Hooper AB (1968) A nitrite-reducing enzyme from Nitrosomonas europaea. Preliminary characterization with hydroxylamine as electron donor. Biochim Biophys Acta 162:49–65
Hooper AB (1969) Lag phase of ammonia oxidation of resting cells of Nitrosomonas europaea. J Bacteriol 97:968–969
Hooper AB, Erickson RH, Terry RH (1972) Electron transport systems in Nitrosomonas: isolation of a membrane-envelope fraction. J Bacteriol 110:430–438
Hooper AB, Terry KR (1973) Specific inhibitors of ammonia oxidation in Nitrosomonas. J Bacteriol 115:480–485
Hooper AB, Terry KR (1974) Photoinactivation of ammonia oxidation in Nitrosomonas. J Bacteriol 119:899–906
Hooper AB, Terry KR (1977) Hydroxylamine oxidoreductase from Nitrosomonas: inactivation by hydrogen-peroxide. Biochemistry 16:455–459
Hooper AB, Maxwell PC, Terry KR (1978) Hydroxylamine oxidoreductase from Nitrosomonas europaea: absorption spectra and content of heme and metal. Biochemistry 17:2984–2989
Hooper AB, Terry KR (1979) Hydroxylamine oxidoreductase of Nitrosomonas: production of nitric oxide from hydroxylamine. Biochim Biophys Acta 571:12–20
Hooper AB, Balny C (1982) Reaction of oxygen with hydroxylamine oxidoreductase of Nitrosomonas. FEBS Lett 144:299–303
Hooper AB (1984) Ammonia oxidation and energy transduction in the nitrifying bacteria. In: Strohl WR, Tuovinen OH (eds) Microbial chemoautotrophy. Ohio State University Press, Columbus, pp 133–167
Hooper AB, DiSpirito AA, Olson TC, Andersson KA, Cunningham W, Taaffe LR (1984) Generation of the proton gradient by a periplasmic dehydrogenase. In: Crawford RL, Hanson RS (eds) Microbial growth on C1 compounds. American Society for Microbiology, Washington, DC, pp 53–58
Hooper AB, DiSpirito AA (1985) In bacteria which grow on simple reductants generation of a proton gradient involves extracytoplasmic oxidation of substrate. Microbiol Rev 49:140–157
Hooper AB (1989) Biochemistry of the nitrifying lithoautotrophic bacteria. In: Schlegel HG, Bowien B (eds) Autotrophic bacteria. Science Tech, Madison, pp 239–265
Hooper AB, Vannelli T, Bergmann DJ, Arciero DM (1997) Enzymology of the oxidation of ammonia to nitrite by bacteria. Ant v Leeuwenhoek 71:59–67
Hoppert M, Mahony TJ, Mayer F, Miller DJ (1995) Quaternary structure of the hydroxylamine oxidoreductase from Nitrosomonas europaea. Arch Microbiol 163:300–306
Horz HP, Rotthauwe JH, Lukow T, Liesack W (2000) Identification of major subgroups of ammonia-oxidizing bacteria in environmental samples by T-RFLP analysis of amoA PCR products. J Microbiol Meth 39:197–204
Hovanec TA, Taylor LT, Blakis A, Delong EF (1998) Nitrospira-like bacteria associated with nitrite oxidation in freshwater aquaria. Appl Environ Microbiol 64:258–264
Huber DM, Warren HL, Nelson DW, Tsai CY (1977) Nitrification inhibitors – new tools for food production. BioScience 27:523–529
Huie RE (1994) The reaction kinetics of NO2. Toxicology 89:193–216
Hyman MR, Wood PM (1983) Methane oxidation by Nitrosomonas europaea. Biochem J 212:31–37
Hyman MR, Wood PM (1984a) Bromocarbon oxidation by Nitrosomonas europaea. In: Crawford RL, Hanson RS (eds) Microbial growth on C1 compounds. American Society for Microbiology, Washington, DC, pp 49–52
Hyman MR, Wood PM (1984b) Ethylene oxidation by Nitrosomonas europaea. Arch Microbiol 137:155–158
Hyman MR, Wood PM (1985) Suicidal inactivation and labeling of ammonia mono-oxygenase by acetylene. Biochem J 227:719–725
Hyman MR, Sansome-Smith AW, Shears JH, Wood RM (1985) A kinetic study of benzene oxidation to phenol by whole cells of Nitrosomonas europaea and evidence for further oxidation to hydroquinone. Arch Microbiol 43:302–306
Hyman MR, Murton IB, Arp DJ (1988) Interaction of ammonia monooxygenase from Nitrosomonas europaea with alkanes, alkenes, and alkynes. Appl Environ Microbiol 54:3187–3190
Hyman MR, Arp DJ (1992) 14C2H2-and 14CO2-labelling studies of the de novo synthesis of polypeptides by Nitrosomonas europaea during recovery from acetylene and light inactivation of ammonia monooxygenase. J Biol Chem 267:1534–1545
Hyman MR, Page CL, Arp DJ (1994) Oxidation of methyl fluoride and dimethyl ether by ammonia monooxygenase in Nitrosomonas eutropha. Appl Environ Microbiol 60:3033–3035
Hyman MR, Arp DJ (1995) Effects of ammonia on the de novo synthesis of polypeptides in cells of Nitrosomonas europaea denied ammonia as an energy source. J Bacteriol 177:4974–4979
Hynes RK, Knowles R (1978) Inhibition by acetylene of ammonia oxidation in Nitrosomonas europaea. FEMS Microbiol Lett 4:319–321
Igarashi N, Moriyama H, Fujiwara T, Fukumori Y, Tanaka N (1997) The 2.8 A structure of hydroxylamine oxidoreductase from a nitrifying chemoautotrophic bacterium, Nitrosomonas europaea. Nat Struct Biol 4:276–284
Ingledew WJ, Halling PJ (1976) Paramagnetic centers of the nitrite oxidizing bacterium Nitrobacter. FEBS Lett 67:90–93
Jetten MSM, Logemann S, Muyzer G, Robertson LA, de Vries S, van Loosdrecht MCM, Kuenen JG (1997) Novel principles in the microbial conversion of nitrogen compounds. Ant v Leeuwenhoek 71:75–93
Jetten M, Wagner M, Fuerst J, van Loosdrecht M, Kuenen G, Strous M (2001) Microbiology and application of the anaerobic ammonium oxidation (“anammox”) process. Curr Opin Biotechnol 12:283–288
Jlang QQ, Bakken LR (1999) Nitrous oxide production and methane oxidation by different ammonia-oxidizing bacteria. Appl Environ Microbiol 65:2679–2684
Johnston H (1972) Newly recognized vital nitrogen cycle. Proc Natl Acad Sci USA 69:2369–2372
Johnstone BH, Jones RD (1988) Physiological effects of long energy-source deprivation on the survival of a marine chemolithotrophic ammonium-oxidizing bacterium. Marine Ecol Prog Ser 49:295–303
Jones RD, Morita RY (1983) Methane oxidation by Nitrosococcus oceanus and Nitrosomonas europaea. Appl Environ Microbiol 45:401–410
Jones RD, Morita RY (1985) Survival of an marine ammonium oxidizer under energy source deprivation. Marine Ecol Prog Ser 26:175–179
Jones RD, Morita RY, Koops H-P, Watson SW (1988) A new marine ammonium-oxidizing bacterium, Nitrosomonas cryotolerans sp. nov. Can J Microbiol 34:1122–1128
Juretschko S, Timmermann G, Schmid M, Schleifer K-H, Pommerening-Röser A, Koops H-P, Wagner M (1998) Combined molecular and conventional analysis of nitrifying bacterium diversity in activated sludge: Nitrosococcus mobilis and Nitrospira-like bacteria as dominant populations. Appl Environ Microbiol 64:3042–3051
Keener WK, Arp DJ (1993) Kinetic studies of ammonia monooxygenase inhibition in Nitrosomonas europaea by hydrocarbons and halogenated hydrocarbons in an optimized whole-cell assay. Appl Environ Microbiol 59:2501–2510
Keener WK, Arp DJ (1994) Transformation of aromatic compounds by Nitrosomonas europaea. Appl Environ Microbiol 60:1914–1920
Keeny DR (1986) Inhibition of nitrification in soil. In: Prosser JJ (ed) Nitrification. IRL Press, Oxford, UK, pp 99–115
Kester RA, de Boer W, Laanbroek HJ (1996) Short exposure to acetylene to distinguish between nitrifier and denitrifier nitrous oxide production in soil and sediment samples. FEMS Microbiol Ecol 20:111–120
Kester RA, Meijer ME, Libochant JA (1997a) Contribution of nitrification and denitrification to the NO and N2O emissions of an acid forest soil, a river sediment and a fertilized grassland soil. Soil Biol Biochem 29:1655–1664
Kester RA, de Boer W, Laanbroek HJ (1997b) Production of NO and N2O by pure cultures of nitrifying and denitrifying bacteria during changes in aeration. Appl Environ Microbiol 63:3872–3877
Kiesow L (1964) On the assimilation of energy from inorganic sources in autotrophic forms of life. Proc Natl Acad Sci USA 52:980–988
Killham K (1986) Heterotrophic nitrification. In: Prosser JI (ed) Nitrification. IRL Press, Oxford, UK, pp 117–126
Killham K (1987) A new perfusion system for measurement and characterization of potential rates of soil nitrification. Plant Soil 97:267–272
Kirstein KO, Bock E, Miller DJ, Nicholas DJD (1986) Membrane-bound b-type cytochromes in Nitrobacter. FEMS Microbiol Lett 36:63–67
Kirstein K, Bock E (1993) Close genetic relationship between Nitrobacter hamburgensis nitrite oxidoreductase and Escherichia coli nitrate reductases. Arch Microbiol 160:447–453
Kleiner D (1985) Bacterial ammonium transport. FEMS Microbiol Rev 32:87–100
Klotz MG, Alzerreca J, Norton ML (1997) A gene encoding a membrane protein exists upstream of the amo A/amo B genes in ammonia oxidizing bacteria: a third member of the amo operon? FEMS Microbiol Lett 150:65–73
Klotz MG, Norton JM (1998) Multiple copies of ammonia monooxygenase (amo) operons have evolved under biased AT/GC mutational pressure in ammonia-oxidizing autotrophic bacteria. FEMS Microbiol Lett 168:303–311
Kluyver AJ, Donker HJK (1926) Die Einheit der Biochemie. Chem Zelle und Gewebe 13:134–190
Koops H-P, Harms H, Wehrmann H (1976) Isolation of a moderate halophilic ammonia-oxidizing bacterium Nitrosococcus mobilis nov. sp. Arch Microbiol 10:277–282
Koops HP, Harms H (1985) Deoxyribonucleic acid homologies among 96 strains of ammonia-oxidizing bacteria. Arch Microbiol 141(3):214–218
Koops H-P, Böttcher B, Möller UC, Pommerening-Röser A, Stehr G (1990) Description of a new species of Nitrosococcus. Arch Microbiol 154:244–248
Koops H-P, Böttcher B, Möller UC, Pommerening-Röser A, Stehr G (1991) Classification of eight new species of ammonia-oxidizing bacteria: Nitrosomonas communis sp. nov., Nitrosomonas ureae sp. nov., Nitrosomonas aestuarii sp. nov., Nitrosomonas marina sp. nov., Nitrosomonas nitrosa sp. nov., Nitrosomonas eutropha sp. nov., Nitrosomonas oligotropha sp. nov., and Nitrosomonas halophila sp. nov. J Gen Microbiol 137:1689–1699
Koops H-P, Möller UC (1992) The lithotrophic ammonia-oxidizing bacteria. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (eds) The prokaryotes, 2nd edn. Springer, New York, pp 2626–2637
Koschorreck M, Moore E, Conrad R (1996) Oxidation of nitric oxide by a new heterotrophic Pseudomonas sp. Arch Microbiol 166:23–31
Kowalchuk GA, Stephen JR, de Boer W, Prosser JI, Embley TM, Woldendorp JW (1997) Analysis of ammonia-oxidizing bacteria of the β subdivision of the class Proteobacteria in coastal sand dunes by denaturing gradient gel electrophoresis and sequencing of PCR-amplified 16S ribosomal DNA fragments. Appl Environ Microbiol 63:1489–1497
Krüger B, Meyer O, Nagel M, Andreesen JR, Meincke M, Bock E, Blümle S, Zumft WG (1987) Evidence for the presence of bactopterin in the eubacterial molybdoenzymes nicotinic acid dehydrogenase, nitrite oxidoreductase, and respiratory nitrate reductase. FEMS Microbiol Lett 48:225–227
Krümmel A, Harms H (1982) Effect of organic matter on growth and cell yield of ammonia-oxidizing bacteria. Arch Microbiol 133:50–54
Kuenen JG, Robertson LA (1987) Ecology of nitrification and denitrification. In: Cole JA, Ferguson S (eds) The Nitrogen and sulfur cycles. Cambridge University Press, Cambridge, UK, pp 162–218
Kumar S, Nicholas DJD (1982) A proton motive force-dependent adenosine-5′ triphosphate synthesis in spheroplasts of Nitrosomonas europaea. FEMS Microbiol Lett 14:21–25
Kumar S, Nicholas DJD, Williams EH (1983) Definitive 15N NMR evidence that water serves as a source of “O” during nitrite oxidation by Nitrobacter agilis. FEMS Microbiol Lett 152:71–74
Kurokawa T, Fukumori Y, Yamanaka T (1987) Purification of a flavoprotein having NADPH-cytochrome c reductase and transhydrogenase activities from Nitrobacter winogradskyi and its molecular and enzymatic properties. Arch Microbiol 148:95–99
Kusian B, Bednarski R, Husemann M, Bowien B (1995) Characterization of the duplicate ribulose-1,5-bisphosphate carboxylase genes and cbb promoters of Alcaligenes eutrophus. J Bacteriol 177:4442–4450
Lees H (1952) The biochemistry of the nitrifying organisms. The ammonia-oxidizing systems of Nitrosomonas. Biochem J 52:134–139
Lewis RS, Deen WM (1994) Kinetics of the reaction of nitric oxide with oxygen in aqueous solutions. Chem Res Toxicol 7:568–574
Lipschultz R, Zafiriou OC, Wofsy SC, McElroy MB, Valois W, Watson SW (1981) Production of NO and N2O by soil nitrifying bacteria. Nature 294:641–643
Lipscomb JD, Hooper AB (1982) Resolution of multiple heme centers of hydroxylamine oxidoreductase from Nitrosomonas. 1: Electron paramagnetic resonance spectroscopy. Biochemistry 21:3965–3972
Lipscomb JD, Andersson KK, Münck E, Kent TA, Hooper AB (1982) Resolution of multiple heme centers of hydroxylamine oxidoreductase from Nitrosomonas. 2: Mössbauer spectroscopy. Biochemistry 21:3973–3976
Loveless JE, Painter HA (1968) The influence of metal ion concentrations and pH value on the growth of a Nitrosomonas strain isolated from activated sludge. J Gen Microbiol 52:1–14
Lu WP, Kelly DP (1988) Chemolithotrophic ATP synthesis and NAD(P) reduction in Thiobacillus tepidarius and Thiobacillus versutus. Arch Microbiol 130:250–254
Mahony TJ, Miller DJ (1998) Linkage of genes encoding enolase (eno) and CTP synthase (pyr G) in the beta-subdivision proteobacterium Nitrosomonas europaea. FEMS Micrbiol Lett 165:153–157
Mancinelli RL, McKay CP (1983) Effects of nitric oxide and nitrogen dioxide on bacterial growth. Appl Environ Microbiol 46:198–202
Mansch R, Bock E (1998) Biodeterioration of natural stone with special reference to nitrifying bacteria. Biodegradation 9:47–64
Matin A (1978) Organic nutrition of chemoorganotrophic bacteria. Ann Rev Microbiol 32:433–468
Matulewich VA, Strom PF, Finstein MS (1975) Length of incubation for enumerating nitrifying bacteria present in various environments. Appl Environ Microbiol 29:265–268
McCaig AE, Embley TM, Prosser JI (1994) Molecular analysis of enrichment cultures of marine ammonia oxidizers. FEMS Microbiol Lett 120:363–368
McTavish H, Fuchs J, Hooper AB (1993) Sequence of the gene for ammonia monooxygenase of Nitrosomonas europaea. J Bacteriol 175:2436–2444
Meincke M, Krieg E, Bock E (1989) Nitrosovibrio s the dominant ammonia oxidizing bacteria in building stones. Appl Environ Microbiol 56:2108–2110
Meincke M, Bock E, Kastrau D, Kroneck PMH (1992) Nitrite oxidoreductase from Nitrobacter hamburgensis: redox centers and their catalytic role. Arch Microbiol 158:127–131
Miller DJ, Wood PM (1982) Characterization of the c-type cytochromes of Nitrosomonas europaea with the aid of fluorescent gels. Biochem J 207:511–517
Miller DJ, Wood PM (1983) Two membrane-bound b-type cytochromes in Nitrosomonas europaea. FEMS Microbiol Lett 20:323–326
Miller DJ, Nicholas DJD (1985) Further characterization of the soluble cytochrome oxidase/nitrite reductase from Nitrosomonas europaea. J Gen Microbiol 131:2851–2854
Miller LG, Coutlakis MD, Oremland RS, Ward BB (1993) Selective inhibition of ammonium oxidation and nitrification-linked N2O. Appl Environ Microbiol 59:2457–2464
Mitchell PD (1975) Protonmotive redox mechanism of the cytochrome bc1 complex in the respiratory chain: Protonmotive ubiquinone cycle. FEBS Lett 56:1–6
Mobarry BK, Wagner M, Urbain V, Rittmann BE, Stahl DA (1996) Phylogenetic probes for analyzing abundance and spatial organization of nitrifying bacteria. Appl Environ Microbiol 62:2156–2162, Published erratum appears in Appl Environ Microbiol Feb 1997;63(2), 815
Moir JWB, Crossmann LC, Spiro S, Richardson DJ (1996a) The purification of ammonia monooxygenase from Paracoccus denitrificans. FEBS Lett 387:71–74
Moir JWB, Wehrfritz J-M, Spiro S, Richardson DJ (1996b) The biochemical characterisation of a novel non-haem-iron hydroxylamine oxidase from Paracoccus denitrificans GB17. Biochem J 319:823–827
Morgenroth E, Obermayer A, Arnold E, Brühl A, Wagner M, Wilderer PA (2000) Effect of long-term idle periods on the performance of sequencing batch reactors. Wat Sci Tech 41:105–113
Mulder J, van Breemen N, Rasmussen W, Driscoll CT (1989) Aluminium chemistry of acidic sandy soils affected by atmospheric depositions in the Netherlands and Denmark. In: Lewis TE (ed) Environmental chemistry and toxicology of aluminium. Lewis, Chelsea, pp 171–194
Mulder A, van de Graaf AA, Robertson LA, Kuenen JG (1995) Anaerobic ammonia oxidation discovered in a denitrifying fluidized bed reactor. FEMS Microbiol Lett 16:177–184
Nicholas DJD, Jones OTG (1960) Oxidation of hydroxylamine in cell-free extracts of Nitrosomonas europaea. Nature 185:512–514
Nielsen LP (1992) Denitrification in sediment determined from nitrogen isotope pairing. FEMS Microbiol Ecol 86:357–362
Norton JM, Low JM, Klotz MG (1996) The gene encoding ammonia monooxygenase subunit A exists in three nearly identical copies in Nitrosospira sp. NpAv. FEMS Microbiol Lett 139:181–188
O’Kelley JC, Becker GE, Nason A (1970) Characterization of the particulate nitrite oxidase and its component activities from the chemoautotroph Nitrobacter agilis. Biochim Biophys Acta 205:409–425
Olson TC, Hooper AB (1983) Energy coupling in the bacterial oxidation of small molecules: an extracytoplasmic dehydrogenase in Nitrosomonas. FEMS Microbiol Lett 19:47–50
O’Neil JG, Wilkinson JF (1977) Oxidation of ammonia by methane-oxidizing bacteria and the effect of ammonia on methane oxidation. J Gen Microbiol 100:407–412
Orso S, Gouy M, Navarro E, Normand P (1994) Molecular phylogenetic analysis of Nitrobacter spp. Int J Syst Bacteriol 44:83–86
Painter HA (1988) Nitrification in the treatment of sewage and waste-waters. In: Prosser JI (ed) Nitrification. IRL Press, Oxford, UK, pp 185–211
Papen H, von Berg R, Hinkel I, Thoene B, Rennenberg H (1989) Heterotrophic nitrification by Alcaligenes faecalis: NO2-, NO3-, N2O, and NO production in exponentially growing cultures. Appl Environ Microbiol 55:2068–2072
Pinck C, Coeur C, Potier P, Bock E (2001) Polyclonal antibodies recognizing the AmoB protein of ammonia oxidizers of the beta-subclass of the class Proteobacteria. Appl Environ Microbiol 67:118–124
Pires M, Rossi MJ, Ross DS (1994) Kinetic and mechanistic aspects of the NO oxidation by O2 in aqueous phase. Int J Chem Kin 26:1207–1227
Pommerening-Röser A, Rath G, Koops H-P (1996) Phylogenetic diversity within the genus Nitrosomonas. Syst Appl Microbiol 19:344–351
Poth M, Focht DD (1985) 14 N kinetic analysis of N2O production by Nitrosomonas europaea: an examination of nitrifier denitrification. Appl Environ Microbiol 49:1134–1141
Poth M (1986) Dinitrogen production from nitrite by a Nitrosomonas isolate. Appl Environ Microbiol 52:957–959
Prince RC, Larroque C, Hooper AB (1983) Resolution of the hemes of hydroxylamine oxidoreductase by redox potentiometry and optical spectroscopy. FEBS Lett 163:25–27
Prosser JI (1989) Autotrophic nitrification in bacteria. In: Rose AH, Tempest DW (eds) Advances in microbial physiology, vol 30. Academic, London, pp 125–181
Purkhold U, Pommerening-Röser A, Juretschko S, Schmid MC, Koops H-P, Wagner M (2000) Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl Environ Microbiol 66:5368–5382
Ralt D, Gomez RF, Tannerbaum SR (1981) Conversion of acetohydroxamate and hydroxylamine to nitrite by intestinal microorganisms. Eur J Appl Microbiol Biotechnol 12:226–230
Rees M, Nason A (1966) Incorporation of atmospheric oxygen into nitrite formed during ammonia oxidation by Nitrosomonas europaea. Biochim Biophys Acta 1(13):398–401
Remde A, Conrad R (1990) Production of nitric oxide by Nitrosomonas europaea by reduction of nitrite. Arch Microbiol 154:187–191
Robertson LA, Kuenen JG (1983) Thiosphaera pantotropha gen. nov. sp. nov., a new facultative anaerobic, facultative autotrophic sulfur bacterium. J Gen Microbiol 129:2847–2855
Robertson LA, Kuenen JG (1984) Aerobic denitrification L. A controversy revived. Arch Microbiol 139:351–354
Robertson LA, Kuenen JG (1988) Heterotrophic nitrification in Thiosphaera pantotropha—oxygen uptake and enzyme studies. J Gen Microbiol 134:857–863
Robertson LA, Cornelisse R, de Vos P, Hadioetomo R, Kuenen JG (1989) Aerobic denitrification in various heterotrophic nitrifiers. Ant v Leeuwenhoek 56:289–300
Robertson LA, Kuenen JG (1990) Combined heterotrophic nitrification and aerobic denitrification in Thiosphaera pantotropha and other bacteria. Ant v Leeuwenhoek 57:139–152
Rotthauwe JH, de Boer W, Liesack W (1995) Comparative analysis of gene sequences encoding ammonia monooxygenase of Nitrosospira sp. AHB1 and Nitrosolobus multiformis C-71. FEMS Microbiol Lett 133:131–135
Rotthauwe J-H, Witzel K-P, Liesack W (1997) The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl Environ Microbiol 63:4704–4712
Sayavedra-Soto LA, Hommes NG, Arp DJ (1994) Characterization of the gene encoding hydroxylamine oxidoreductase in Nitrosomonas europaea. J Bacteriol 176:504–510
Sayavedra-Soto LA, Hommes NG, Russell SA, Arp DJ (1996) Induction of ammonia monooxygenase and hydroxylamine oxidoreductase mRNAs by ammonium in Nitrosomonas europaea. Molec Microbiol 20:541–548
Schimel JPM, Firestone K, Killham KS (1984) Identification of heterotrophic nitrification in a Sierran forest soil. Appl Environ Microbiol 48:802–806
Schmid M, Twachtmann U, Klein M, Strous M, Juretschko S, Jetten M, Metzger JW, Schleifer K-H, Wagner M (2000) Molecular evidence for a genus-level diversity of bacteria capable of catalyzing anaerobic ammonium oxidation system. Appl Microbiol 23:93–106
Schmid M, Schmitz-Esser S, Jetten M, Wagner M (2001) 16S-23S rDNA intergenic spacer and 23S rDNA of anaerobic ammonium oxidizers: Implications for phylogeny and in situ detection. Environ Microbiol 3:450–459
Schmidt EL (1982) Nitrification in soil. In: Stevenson FJ (ed) Nitrogen in agricultural soils. ASA-CSSA-SSSA, Madison, pp 253–288
Schmidt I, Bock E (1997) Anaerobic ammonia oxidation with nitrogen dioxide by Nitrosomonas eutropha. Arch Microbiol 167:106–111
Schmidt I, Bock E (1998) Anaerobic ammonia oxidation by cell-free extracts of Nitrosomonas eutropha. Ant v Leeuwenhoek 73:271–278
Schmidt I, Bock E, Jetten MSM (2001a) Ammonia oxidation by Nitrosomonas eutropha with NO2 as oxidant is not inhibited by acetylene. Microbiology 147:2247–2253
Schmidt I, Zart D, Bock E (2001b) Effects of gaseous NO2 on cells of Nitrosomonas eutropha previously incapable of using ammonia as an energy source. Ant v Leeuwenhoek 79:39–7
Seewaldt E, Schleifer K-H, Bock E, Stackebrandt E (1982) The close phylogenetic relationship of Nitrobacter and Rhodopseudomonas palustris. Arch Microbiol 131:287–290
Sela S, Yogev D, Razin S, Bercovier H (1989) Duplication of the tuf gene: a new insight into the phylogeny of eubacteria. J Bacteriol 177:581–584
Sewell DL, Aleem MIH, Wilson DF (1972) The oxidation-reduction potentials and rates of oxidation of the cytochromes of Nitrobacter agilis. Arch Biochem Biophys 153:312–319
Sewell DL, Aleem MIH (1979) NADH-linked oxidative phosphorylation in Nitrobacter agilis. Curr Microbiol 2:35–37
Shank JL, Silliker JH, Harper JH (1962) The effect of nitric oxide on bacteria. Appl Microbiol 10:185–189
Shears JH, Wood PM (1985) Spectroscopic evidence for a photosensitive oxygenated state of ammonia monooxygenase. Biochem J 226:499–507
Slangen JHG, Kerkhoff P (1984) Nitrification inhibitors in agriculture and horticulture: a literature review. Fertilizer Res 5:1–76
Smith AJ, Hoare DS (1968) Acetate assimilation by Nitrobacter agilis in relation to its “obligate autotrophy”. J Bacteriol 95:844–855
Sone N, Yanagita Y, Hon-nami K, Fukumori Y, Yamanaka T (1983) Proton-pump activity of Nitrobacter agilis and Thermus thermophilus cytochrome c oxidase. FEBS Lett 155:150–155
Sone N (1986) Measurement of proton pump activity of the thermophilic bacterium PS 3 and Nitrobacter agilis at the cytochrome oxidase level using total membranes and heptyl-thioglycoside. J Biochem 100:1465–1476
Sorokin DY, Myzer G, Brinkhoff T, Kuenen JG, Jetten MSM (1998) Isolation and characterization of a novel facultatively alkaliphilic Nitrobacter species N. alkalicus sp. nov. Arch Microbiol 170:345–352
Sorokin DY, Tourova T, Schmid M, Wagner M, Koops HP, Kuenen JG, Jetten M (2001) Isolation and properties of obligately chemolithoautotrophic and extremely alkalitolerant ammonia oxidizing bacteria from Mongolian soda lakes. Arch Microbiol 176:170–177
Spieck E, Meincke M, Bock E (1992) Taxonomic diversity of Nitrosovibrio strains isolated from building sandstone. FEMS Microbiol Ecol 102:21–26
Spieck E, Aamand J, Bartosch S, Bock E (1996) Immunocytochemical detection and location of the membrane-bound nitrite oxidoreductase in cells of Nitrobacter and Nitrospira. FEMS Microbiol Lett 139:71–76
Spiller H, Dietsch E, Kessler E (1976) Intracellular appearance of nitrite and nitrate in nitrogen-starved cells of Ankistrodesmus braunii. Planta 129:175–181
Stammler JS, Simon DJ, Osborne V, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci USA 82:7738–7742
Stams AJM, Flameling EM, Marnette ECL (1990) The importance of autotrophic versus heterotrophic oxidation of atmospheric ammonium in forest ecosystems with acid soil. FEMS Microbiol Ecol 74:337–344
Stams AJM, Booltink HWG, Lutke-Schipholt IJ, Beemsterboer B, Woittiez JRW, Van Breemen N (1991) A field study on the fate of 15N-ammonium to demonstrate nitrification of atmospheric ammonium in an acid forest soil. Biogeochemistry 13:241–255
Stehr G, Böttcher B, Dittberner P, Rath G, Koops H-P (1995) The ammonia-oxidizing nitrifying population of the river Elbe estuary. FEMS Microbiol Ecol 17:177–186
Stein LY, Arp DJ, Hyman MR (1997) Regulation of the synthesis and activity of ammonia monooxygenase in Nitrosomonas europaea by altering pH to affect NH3 availability. Appl Environ Microbiol 63:4588–4592
Stein LY, Arp DJ (1998a) Ammonium limitation results in the loss of ammonia oxidizing activity in Nitrosomonas europaea. Appl Environ Microbiol 64:1514–1521
Stein LY, Arp DJ (1998b) Loss of ammonia monooxygenase activity in Nitrosomonas europaea upon exposure to nitrite. Appl Environ Microbiol 64:4098–4102
Stein LY, Sayavedra-Soto LA, Hommes NG, Arp DJ (2000) Differential regulation of amoA and amoB gene copies in Nitrosomonas europaea. FEMS Microbiol Lett 192:163–168
Steinmüller W, Bock E (1976) Growth of Nitrobacter in the presence of organic matter. I. Mixotrophic growth. Arch Microbiol 108:299–304
Stephen JR, McCaig AE, Smith Z, Prosser JI, Embley TM (1996) Molecular diversity of soil and marine 16S rRNA gene sequences related to β-subgroup ammonia-oxidizing bacteria. Appl Environ Microbiol 62:4147–4154
Stephen JR, Kowalchuk GA, Bruns MAV, McCaig AE, Phillips CJ, Embley TM, Prosser JI (1998) Analysis of beta-subgroup proteobacterial ammonia oxidizer populations in soil by denaturing gradient gel electrophoresis analysis and hierarchical phylogenetic probing. Appl Environ Microbiol 64:2958–2965
Steudler PA, Jones RD, Castro MS, Mellilo JM, Lewis DL (1996) Microbial controls of methane oxidation in temperate forest and agriculture soils. NATO ASI Ser. Ser. I 39:69–84
Strecker M, Sickinger E, English RS, Shively JM, Bock E (1994) Calvin cycle genes in Nitrobacter vulgaris T3. FEMS Microbiol Lett 120:45–50
Strous M, Fuerst JA, Kramer EHM, Logemann S, Muyzer V, van de Pas-Schoonen KT, Webb R, Kuenen JG, Jetten MSM (1999) Missing lithotroph identified as new planctomycete. Nature 400:446–449
Stüven R, Vollmer M, Bock E (1992) The impact of organic matter on NO formation by Nitrosomonas europaea. Arch Microbiol 158:439–443
Stüven R, Bock E (2001) Nitrification and denitrification as a source for NO and NO2 production in high-strength wastewater. Water Res 35(8):1905–1914
Sundermeyer H, Bock E (1981) Energy metabolism of autotrophically and heterotrophically grown cells of Nitrobacter winogradskyi. Arch Microbiol 130:250–254
Sundermeyer-Klinger H, Meyer V, Warninghoff B, Bock E (1984) Membrane-bound nitrite oxidoreductase of Nitrobacter: evidence for a nitrate reductase system. Arch Microbiol 140:153–158
Suwa Y, Sumino T, Noto K (1997) Phylogenetic relationships of activated sludge isolates of ammonia oxidizers with different sensitivities to ammonium sulfate. J Gen Appl Microbiol 43:373–379
Suzuki I, Dular U, Kwok S-C (1970) Cell-free ammonia oxidation by Nitrosomonas europaea extracts: effects of polyamines, Mg2+ and albumin. Biochem Biophys Res Commun 39:950–955
Suzuki I, Dular U, Kwok S-C (1974) Ammonia or ammonium ion as substrate for oxidation by Nitrosomonas cells and extracts. J Bacteriol 120:556–558
Suzuki I, Kwok S-C, Dular U (1976) Competitive inhibition of ammonia oxidation in Nitrosomonas europaea by methane, carbon monoxide or methanol. FEBS Lett 72:117–120
Suzuki I, Kwok SC (1981) A partial resolution and reconstitution of the ammonia-oxidizing system of Nitrosomonas europaea: role of cytochrome c554. Can J Biochem 59:484–488
Suzuki I, Kwok S-C, Dular U, Tsang DCY (1981) Cell-free ammonia oxidizing system of Nitrosomonas europaea: general conditions and properties. Can J Biochem 59:477–483
Takahashi R, Ohmori T, Watanabe K, Tokuyama T (1993) Phosphoenolpyruvate carboxylase of ammonia oxidizing chemoautotrophic bacterium Nitrosomonas europaea ATCC 25978. J Ferm Bioeng 76:232–234
Tanaka Y, Fukumori Y, Yamanaka Y (1982) The complete amino acid sequence of Nitrobacter agilis cytochrome c550. Biochim Biophys Acta 707:14–20
Tanaka Y, Fukumori Y, Yamanaka T (1983) Purification of cytochrome a1c1 from Nitrobacter agilis and characterization of nitrite oxidation system of the bacterium. Arch Microbiol 135:265–271
Tappe W, Tomaschewski C, Rittershaus S, Groeneweg J (1996) Cultivation of nitrifying bacteria in the retentostat, a simple fermenter with internal biomass retention. FEMS Microbiol Ecol 19:47–52
Teske A, Alm E, Regan JM, Toze S, Rittmann BE, Stahl DA (1994) Evolutionary relationship among ammonia-and nitrite oxidizing bacteria. J Bacteriol 176:6623–6630
Teske A, Wawer C, Muyzer G, Ramsing NB (1996) Distribution of sulfate-reducing bacteria in a stratified fjord (Mariager Fjord, Denmark) as evaluated by most-probable-number counts and denaturing gradient gel electrophoresis of PCR-amplified ribosomal DNA fragments. Appl Environ Microbiol 62:1405–1415
Tomlinson TG, Boon AG, Trotman CNA (1966) Inhibition of nitrification in the activated sludge process of sewage disposal. J Appl Bacteriol 29:266–291
Tronson DA, Ritchie GAF, Nicholas DJD (1973) Purification of c-type cytochromes from Nitrosomonas europaea. Biochim Biophys Acta 310:331–343
Tsang DCY, Suzuki I (1982) Cytochrome c554 as a possible electron donor in the hydroxylation of ammonia and carbon monoxide in Nitrosomonas europaea. Can J Biochem 60:1018–1024
Tsong TY, Astumian RD (1987) Electroconformational coupling. Progr Biophys Molec Biol 50:1–45
Tubulekas I, Hughes D (1993) Growth and translation elongation rate are sensitive to the concentration of EF-Tu. Molec Microbiol 8:761–770
Utåker JB, Bakken L, Jiang QQ, Nes IF (1995) Phylogenetic analysis of seven new isolates of ammonia-oxidizing bacteria based on 16S rRNA gene sequences. Syst Appl Microbiol 18:549–559
Utåker JB, Nes IF (1998) A qualitative evaluation of the published oligonucleotides specific for the 16S rRNA gene sequences of the ammonia-oxidizing bacteria. Syst Appl Microbiol 21:72–88
Van de Dijk SJ, Troelstra SR (1980) Heterotrophic nitrification in a heath soil demonstrated by an in-situ method. Plant Soil 57:11–21
Van de Graaf AA, Mulder A, De Bruijn P, Jetten MSM, Robertson LA, Kuenen JG (1995) Anaerobic oxidation of ammonium is a biologically mediated process. Appl Environ Microbiol 61:1246–1251
Van de Graaf AA, De Bruijn P, Robertson LA, Kuenen JG (1996) Autotrophic growth of anaerobic ammonium-oxidizing micro-organisms in a fluidized bed reactor. Microbiology 142:2187–2196
Van de Graaf AA, De Bruijn P, Robertson LA, Jetten MSM, Kuenen JG (1997) Metabolic pathway of anaerobic ammonium oxidation on the basis of 15N studies in a fluidized bed reactor. Microbiology 143:2415–2421
Vanelli T, Logan M, Arciero M, Hooper AB (1990) Degradation of halogenated aliphatic compounds by ammonia-oxidizing bacterium Nitrosomonas europaea. Appl Environ Microbiol 56:1169–1171
Vanelli T, Bergmann DJ, Arciero DM, Hooper AB (1996) Mechanism of N-oxidation and electron transfer in the ammonia-oxidizing autotrophs. In: Lidstrom ME, Tabita FR (eds) Proceedings of the 8th international symposium on microbial growth on C1 compounds. Kluwer Academic, Dordrecht, pp 80–87
Van Niel EWJ, Robertson LA, Kuenen JG (1987) Heterotrophic nitrification in denitrifying bacteria. In: Proceeding 4th European Congress on Biotechnology, vol 3, p 363
Van Niel EWJ, Arts PAM, Wesselink BJ, Robertson LA, Kuenen JG (1993) Competition between heterotrophic and autotrophic nitrifiers for ammonia in chemostat cultures. FEMS Microbiol Ecol 102:109–118
Voysey PA, Wood PM (1987) Methanol and formaldehyde oxidation by an autotrophic nitrifying bacterium. J Gen Microbiol 133:283–290
Voytek MA, Ward BB (1995) Detection of ammonium-oxidizing bacteria of the beta-subclass of the class Proteobacteria in aquatic samples with the PCR. Appl Environ Microbiol 61:1444–1450
Wagner M, Rath G, Amann R, Koops H-P, Schleifer K-H (1995) In situ identification of ammonia-oxidizing bacteria. Syst Appl Microbiol 18:251–264
Wagner M, Rath G, Koops H-P, Flood J, Amann R (1996) In situ analysis of nitrifying bacteria in sewage treatment plants. Water Sci Tech 34(1–2):237–244
Wang WC, Yung YL, Lacis AL, Mo TM, Hanson JE (1976) Greenhouse effects due to man-made perturbations of trace gases. Science 194:685–689
Ward BB (1987) Kinetic studies on ammonia and methane oxidation by Nitrosococcus oceanus. Arch Microbiol 147:126–133
Watson SW (1965) Characteristics of a marine nitrifying bacterium, Nitrosocystis oceanus sp. n. Limnol Oceanogr 10(Suppl):R274–R289
Watson SW (1971) Taxonomic considerations of the family Nitrobacteraceae Buchanan: requests for opinions. Int J Syst Bacteriol 21:254–270
Watson SW, Waterbury JB (1971) Characteristics of two marine nitrite oxidizing bacteria. Nitrospina gracilis nov. gen. nov. sp. and Nitrococcus mobilis nov. gen. nov. sp. Arch Mikrobiol 77:203–230
Watson SW, Graham LB, Remsen CC, Valois FW (1971) A lobular, ammonia-oxidizing bacterium. Nitrosolobus multiformis nov. gen. nov. sp. Arch Mikrobiol 76:183–303
Watson SW, Bock E, Valois EW, Waterbury JB, Schlosser U (1986) Nitrospira marina gen. nov. sp. nov.: a chemolithotrophic nitrite-oxidizing bacterium. Arch Microbiol 144:1–7
Watson SW, Bock E, Harms H, Koops H-P, Hooper AB (1989) Nitrifying bacteria. In: Murray RGE, Brenner DJ, Bryant MP, Holt JG, Krieg NR, Moulder JW, Pfennig N, Sneath PHA, Staley JT, Williams S (eds) Bergey’s manual of systematic bacteriology, vol 3. Williams and Wilkins, Baltimore, pp 1808–1834
Wehrfritz J-M, Reilly A, Spiro S, Richardson DJ (1993) Purification of hydroxylamine oxidoreductase from Thiosphera pantotropha. Identification of electron acceptors that couple heterotrophic nitrification to aerobic denitrification. FEBS Lett 335:246–250
Wetzstein HG, Ferguson RJ (1985) Respiration-dependent proton translocation and the mechanism of proton motive force generation in Nitrobacter winogradskyi. FEMS Microbiol Lett 30:87–92
Wheelis M (1984) Energy conservation and pyridine nucleotide reduction in chemoautotrophic bacteria: a thermodynamic analysis. Arch Microbiol 138:166–169
Williams EJ, Hutchinson GL, Fehsenfeld FC (1992) NOx and N2O emissions from soil. Global Biogeochem Cycles 6:351–388
Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS, Keefer LK (1991) DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 254:1001–1003
Wink DA, Darbyshire JF, Nims RW, Saavedra JE, Ford PC (1993) Reactions of the bioregulatory agent nitric oxide in oxygenated aqueous media: determination of kinetics for oxidation and nitrosation by intermediates generated in the NO/O2 reaction. Chem Res Toxicol 6:23–27
Winogradsky S (1892) Archives des sciences biologique. Contributions à la morphologie des organismes de la nitrification. St Petersbourg 1:88–137
Woese CR, Weisburg WG, Paster BJ, Hahn CM, Tanner RS, Krieg NR, Koops H-P, Harms H, Stackebrandt E (1984) The phylogeny of purple bacteria: the beta subdivision. Syst Appl Microbiol 5:327–336
Woese CR, Weisburg WG, Hahn CM, Paster BJ, Zablen LB, Lewis BJ, Macke TJ, Ludwig W, Stackebrandt E (1985) The phylogeny of purple bacteria: the gamma subdivision. Syst Appl Microbiol 6:25–33
Wood PM (1978) Periplasmic location of the terminal reductase in nitrite respiration. FEBS Lett 92:214–218
Wood PM (1986) Nitrification as a bacterial energy source. In: Prosser JI (ed) Nitrification. IRL Press, Oxford, UK, pp 63–78
Wood PM (1988a) Chemolithotrophy. In: Anthony C (ed) Bacterial energy transduction. Academic, London, pp 183–230
Wood PM (1988b) Monooxygenase and free radical mechanism for biological ammonia oxidation. In: Cole JA, Ferguson S (eds) The nitrogen and sulfur cycles: 42nd symposium of the society of general microbiology. Cambridge University Press, Cambridge UK, pp 219–243
Wullenweber M, Koops H-P, Martiny H (1978) Der Einfluß von Nitrit auf den Verlauf des Wachstums von Nitrosomonas Stamm Nm1. Mitt Inst Allg Bot Hamburg 16:159–164
Xu B, Fortkamp U, Enfors S-O (1995) Continuous measurement of NOaq during denitrification by immobilized Pseudomonas stutzeri. Biotechnol Biotech 9:659–664
Yamagata A, Kato J, Hirota R, Kuroda A, Ikeda T, Takiguchi N, Ohtake H (1999) Isolation and characterization of two cryptic plasmids in the ammonia-oxidizing bacterium Nitrosomonas sp. strain ENI-11. J Bacteriol 181:3375–3381
Yamanaka T, Shinra M (1974) Cytochrome c552 and cytochrome c554 derived from Nitrosomonas europaea: purification, properties, and their function in hydroxylamine oxidation. J Biochem 75:1265–1273
Yamanaka T, Fugii K, Kamita Y (1979) Subunits of cytochrome a-type terminal oxidase derived from Thiobacillus novellus and Nitrobacter agilis. J Biochem 86:821–824
Yamanaka T, Kamita Y, Fukumori Y (1981) Molecular and enzymatic properties of “cytochrome aa3 type” terminal oxidase derived from Nitrobacter agilis. J Biochem 89:265–273
Yamanaka T, Tanaka Y, Fukumori Y (1982) Nitrobacter agilis cytochrome c550: isolation, physicochemical and enzymatic properties and primary structure. Plant Cell Physiol 23:441–449
Yamanaka T, Fukumori Y (1988) The nitrite oxidizing system of Nitrobacter winogradskyi. FEMS Microbiol Rev 54:259–270
Yoshida T, Alexander M (1964) Hydroxylamine formation by Nitrosomonas europaea. Can J Microbiol 10:923–926
Yoshinari T (1985) Nitrite and nitrous oxide production by Methylosinus trichosporium. Can J Microbiol 31:139–144
Zahn JA, Duncan C, DiSpirito AA (1994) Oxidation of hydroxylamine by cytochrome P-460 of the obligate methylotroph Methylococcus capsulatus. Bath J Bacteriol 176:5879–5887
Zart D, Schmidt I, Bock E (1996) Neue Wege vom Ammonium zum Stickstoff. In: Lemmer H, Griebe T, Flemming H-K (eds) Ökologie der Abwasserorganismen. Springer, Berlin, pp 183–192
Zart D, Bock E (1998) High rate of aerobic nitrification and denitrification by Nitrosomonas eutropha grown in a fermentor with complete biomass retention in the presence of gaseous NO2 or NO. Arch Microbiol 169:282–286
Zart D, Stüven R, Bock E (1999) Nitrification and denitrification—microbial fundamentals and consequences for application. In: Rehm HJ, Reed G, Pühler A, Stadler PJW (eds) Biotechnology: a multi-volume comprehensive treatise, 2nd revised edn. 11a Wiley-VCH Weinheim Germany, New York, pp 55–64
Zart D, Schmidt I, Bock E (2000) Significance of gaseous NO for ammonia oxidation by Nitrosomonas eutropha. Ant v Leeuwenhoek 77:49–55
Zumft WG (1993) The biological role of nitric oxide in bacteria. Arch Microbiol 160:253–264
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Bock, E., Wagner, M. (2013). Oxidation of Inorganic Nitrogen Compounds as an Energy Source. In: Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F. (eds) The Prokaryotes. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30141-4_64
Download citation
DOI: https://doi.org/10.1007/978-3-642-30141-4_64
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30140-7
Online ISBN: 978-3-642-30141-4
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences