

Abstract
Nitrification, the oxidation of ammonia via nitrite to nitrate, has always been considered to be a two-step process catalysed by chemolithoautotrophic microorganisms oxidizing either ammonia or nitrite. No known nitrifier carries out both steps, although complete nitrification should be energetically advantageous. This functional separation has puzzled microbiologists for a century. Here we report on the discovery and cultivation of a completely nitrifying bacterium from the genus Nitrospira, a globally distributed group of nitrite oxidizers. The genome of this chemolithoautotrophic organism encodes the pathways both for ammonia and nitrite oxidation, which are concomitantly activated during growth by ammonia oxidation to nitrate. Genes affiliated with the phylogenetically distinct ammonia monooxygenase and hydroxylamine dehydrogenase genes of Nitrospira are present in many environments and were retrieved on Nitrospira-contigs in new metagenomes from engineered systems. These findings fundamentally change our picture of nitrification and point to completely nitrifying Nitrospira as key components of nitrogen-cycling microbial communities.
Similar content being viewed by others


Main
Nitrification is catalysed by ammonia-oxidizing bacteria (AOB)1 or archaea (AOA)2 and nitrite-oxidizing bacteria (NOB)1. Since the pioneering studies by Winogradsky more than a century ago3, nitrifying microorganisms are generally perceived as specialized chemolithoautotrophs that obtain energy for growth by oxidizing either ammonia or nitrite. The known ammonia-oxidizing microbes (AOM) and NOB are phylogenetically not closely related, and none of these organisms can oxidize both substrates. This separation of the two nitrification steps in different organisms leads to a tight cross-feeding interaction and the frequently observed co-aggregation of AOM with NOB in nitrifying consortia4. However, the functional separation is a puzzling phenomenon since complete nitrification would yield more energy (∆G°′ = −349 kJ mol−1 NH3) than either single step (∆G°′ = −275 kJ mol−1 NH3 for ammonia oxidation to nitrite and ∆G°′ = −74 kJ mol−1 NO2− for nitrite oxidation to nitrate). Thus, an organism catalysing complete nitrification should have growth advantages over the ‘incomplete’ AOM and NOB. Based on kinetic theory of optimal pathway length5,6, Costa et al.7 argued that a hypothetical complete nitrifier would likely be outcompeted by incomplete, cross-feeding AOM and NOB in many environments. However, the same authors7 also pointed out that a complete nitrifier might be competitive under conditions that favour the maximization of growth yield rather than growth rate and coined the term “comammox” (complete ammonia oxidizer) to describe such a hypothetical microbe. Conditions selecting for comammox may be characterized by slow, substrate influx-limited growth with a spatial clustering of biomass in microbial aggregates and biofilms7. A prerequisite for the existence of comammox would also be that any biochemical incompatibilities of ammonia and nitrite oxidation can be overcome by adaptations of enzymes or cellular compartmentalization7. Aside from these theoretical considerations, the old question of whether comammox exists in nature has not been resolved.
The globally distributed genus Nitrospira represents the most diverse known group of NOB. Nitrospira members have been found in terrestrial8 and limnic habitats9,10, marine waters11, deep sea sediments, sponge tissue12, geothermal springs13, drinking water distribution systems14, corroded iron pipes15, and wastewater treatment plants (WWTPs)10,16. At least six phylogenetic sublineages of Nitrospira exist, of which lineage II seems to be most widely distributed in both natural and engineered ecosystems10. The ecological success of Nitrospira has been linked to an economical pathway for nitrite oxidation17 and a substantial metabolic versatility, which includes the utilization of various organic compounds in addition to nitrite and CO210,11,17,18,19, cyanate or urea degradation and nitrification by reciprocal feeding with AOM19,20, and chemolithoautotrophic aerobic hydrogen oxidation21.
Enrichment of conspicuous Nitrospira
A microbial biofilm developing on the walls of a pipe under the flow of hot water (56 °C, pH 7.5) raised from a 1,200 m deep oil exploration well (Aushiger, North Caucasus, Russia) was sampled and incubated at 46 °C in ammonium-containing mineral medium to enrich moderately thermophilic AOM. After a series of subcultivation steps, we obtained enrichment culture ‘ENR4’ that oxidized ammonia to nitrate and contained a dense population of cells morphologically resembling described Nitrospira species13,15 (Extended Data Fig. 1a, b). A second abundant population consisted of rod-shaped cells, but no organism in ENR4 displayed the typical morphologies of known AOM. Inspection by fluorescence in situ hybridization (FISH) with nitrifier-specific ribosomal RNA-targeted probes10,22 confirmed that ENR4 contained Nitrospira (Extended Data Fig. 1c) but no other detectable nitrifiers. Moreover, known bacterial or archaeal genes of ammonia monooxygenase (AMO) subunit alpha (amoA) and 16S rRNA genes of AOA were not detected by PCR in ENR4. Considering the intriguing possibility that the Nitrospira population might be responsible for both ammonia and nitrite oxidation, we sequenced the metagenome of the enrichment (Supplementary Tables 1–7) to identify the ammonia oxidizer. Sequence assembly and differential coverage binning23 showed that the ENR4 metagenome was dominated by two organisms (one Nitrospira strain and a betaproteobacterium affiliated with the family Hydrogenophilaceae) and revealed two additional rare populations (an alphaproteobacterium related to Tepidamorphus gemmatus, family Rhodobiaceae, and an actinobacterium affiliated with Thermoleophilum, family Thermoleophilaceae). Archaea were not detected (Extended Data Fig. 2a). Based on the relative genome sequence coverage in three sequenced samples of the culture, Nitrospira was the most abundant population in ENR4 (68 to 80% of the community), followed by the betaproteobacterium (18 to 29%) and the other two organisms (≤2%). Subsequent FISH identified the relatively abundant rod-shaped cells as the betaproteobacterium (Extended Data Fig. 1c), whereas the two rare populations were encountered only sporadically by microscopy. Further subcultivation led to enrichment ‘ENR6’ that also oxidized ammonia to nitrate and, according to metagenome analysis, contained only Nitrospira (60% according to relative sequence coverage) and the betaproteobacterium (40%) (Extended Data Fig. 2b). The time of enrichment, from sampling of the source biofilm to ENR6, was four years. The high sequence coverage (Extended Data Fig. 2) allowed us to reconstruct complete and closed Nitrospira genomes and almost complete genomes of the other bacteria from the metagenomes of cultures ENR4 and ENR6, respectively. The Nitrospira genomes retrieved from the two enrichments were identical. We provisionally classify this highly enriched Nitrospira strain as ‘Candidatus Nitrospira inopinata’ (in.o.pi.na'ta. L. fem. adj. inopinata unexpected, surprising).
Discovery of comammox
The obtained bacterial genomes were screened for the key functional genes of autotrophic nitrification. As expected, Ca. N. inopinata possesses the key enzyme for nitrite oxidation, nitrite oxidoreductase (NXR). Its genome contains the nxrA and nxrB genes coding for the subunits alpha and beta, respectively, of the periplasmic Nitrospira NXR17 and genes of four candidate Nitrospira NxrC gamma subunits17 (Extended Data Fig. 3). Unlike other cultured Nitrospira, which possess two to five paralogous copies of the nxrAB genes8,17, Ca. N. inopinata has only one copy of these genes. Much more surprisingly, Ca. N. inopinata also possesses homologues to the hallmark enzymes of ammonia oxidation, AMO and hydroxylamine dehydrogenase (also referred to as hydroxylamine oxidoreductase, HAO)24 (Extended Data Fig. 1d). Its amoA gene is dissimilar to those of canonical AOM and was thus not picked up in the initial amoA PCR screening of ENR4. The three AMO subunits alpha (AmoA), beta (AmoB) and gamma (AmoC) are encoded by a single amoCAB gene cluster and by two additional amoC genes at other genomic loci (the AmoC copies share amino acid sequence identities of 99.63 to 100%) (Fig. 1, Extended Data Fig. 3). The amoCAB gene order is conserved in all AOB24. Ca. N. inopinata also has homologues of the putative membrane-associated proteins AmoD and AmoE of AOB, which may interact with the ammonia-oxidizing machinery or the electron transport chain25, and a homologue of the putative membrane protein OrfM found in all AOB24 (Fig. 1, Extended Data Fig. 3). Similar to betaproteobacterial AOB24, genes of the copper resistance proteins CopC/D are located close to the amo locus (Fig. 1). The single hao gene of Ca. N. inopinata encodes a predicted octahaem cytochrome c protein resembling the HAO of AOB26. Like in AOB, hao shares a genomic locus with gene haoB of a putative membrane protein found in all AOB27 and with two genes of tetrahaem c-type cytochromes, which resemble cytochrome c554 (CycA) and cytochrome cm552 (CycB) of AOB24 (Fig. 1). HAO, CycA, and CycB form the hydroxylamine ubiquinone reduction module (HURM) in AOB, which transfers electrons from hydroxylamine to the quinone pool28. The full genetic complement for both ammonia and nitrite oxidation strongly suggested that Ca. N. inopinata is a comammox organism (Extended Data Fig. 1d). No canonical nitrification genes were found in the genomes of the other three bacteria detected in ENR4, suggesting that these co-enriched organisms were heterotrophs that used organic substrates produced by the autotrophic Nitrospira29. The betaproteobacterial genome, which was identical in enrichments ENR4 and ENR6, encodes a membrane-associated nitrate reductase that is highly similar to the known nitrate reductases of E. coli and other Proteobacteria.

Gene alignments of the amoCAB, hao, and nxrAB loci with flanking genes are shown. Only two or three of up to nine syntenic cytochrome c biogenesis genes upstream of the hao loci are displayed. Colours identify homologous genes. Genes without homologues in the analysed data set are white if their function is known, otherwise grey. Transposases are magenta irrespectively of homology. Numbers below genes represent amino acid sequence identities (in per cent) of the predicted gene products compared to Ca. N. inopinata. Asterisks mark comammox clade B amoA genes. Wiggly lines indicate ends of metagenomic contigs. Underlined gene products of Ca. N. inopinata have homologues in AOB genomes (amino acid identities in per cent to AOB are indicated in parentheses), but gene arrangements can differ from AOB24. Genes and noncoding regions are drawn to scale. Metagenomic bins are numbered as in Supplementary Table 8. MBR, membrane bioreactor; WWTP, wastewater treatment plant; GWW, groundwater well.
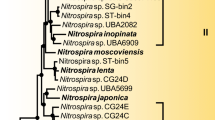
Phylogenetic inference based on 16S rRNA gene sequences showed that Ca. N. inopinata belongs to the widely distributed lineage II of the genus Nitrospira10 (Extended Data Fig. 4). The other cultured Nitrospira strains in lineage II are N. moscoviensis15, N. lenta30, and N. japonica31, which are NOB and do not possess the enzymatic repertoire to utilize ammonia as energy source. Consistently, the affiliation of Ca. N. inopinata with Nitrospira lineage II was supported by phylogenies based on nxrB gene and NxrA protein sequences (Extended Data Fig. 5). The nxrB gene is a suitable phylogenetic marker to differentiate the Nitrospira lineages8. NxrA phylogenies reliably distinguish Nitrospira NxrA from related enzymes17, but their resolution within the genus Nitrospira has not been evaluated and assignments of NxrA sequences to specific Nitrospira lineages must be treated with caution. Ca. N. inopinata represents a different species from the two comammox Nitrospira strains described by van Kessel et al.32, on the basis of the low pairwise average nucleotide identities (70.3 to 71.6%) between the genomes of Ca. N. inopinata and these organisms.
Full nitrification by Ca. N. inopinata
Complete nitrification by Ca. N. inopinata was demonstrated by incubation experiments in mineral media containing ammonium as the sole source of energy and reductant, and bicarbonate/CO2 as the sole carbon source. Consistent with the anticipated activity of comammox, Ca. N. inopinata nearly stoichiometrically oxidized 1 mM or 0.1 mM ammonium to nitrate (Fig. 2a, b). A transient accumulation of nitrite (up to 30% of the added ammonium) was observed in parallel to nitrate production, but nitrite was completely oxidized after all ammonium had been consumed. Much lower nitrite accumulation was detected in an experiment with 10 μM ammonium (Fig. 2c), suggesting that experimental parameters strongly influence this phenomenon and that nitrite accumulation might actually not occur under environmentally relevant conditions. Apparent nitrogen loss caused by formation of gaseous compounds was not observed in any experiment (Fig. 2), suggesting that NO formation from nitrite by NirK (Extended Data Fig. 1d) was not quantitatively important for Ca. N. inopinata. Growth of Ca. N. inopinata during oxidation of ammonium to nitrate was demonstrated by quantitative PCR targeting its single-copy amoA gene and continued after consumption of ammonium in the presence of nitrite until the end of the experiment (Fig. 2d). A pure culture of the betaproteobacterium, the only non-Nitrospira microbe in ENR6, was isolated in acetate-containing medium and showed no nitrifying activity after inoculation into ammonium- or nitrite-media at cell densities higher than the density of Ca. N. inopinata in the growth experiment (Fig. 2d, Extended Data Fig. 6). The function of Ca. N. inopinata as comammox was further confirmed by metaproteome analysis of ENR4, which showed that all key proteins of Ca. N. inopinata for ammonia and nitrite oxidation were expressed during incubation with ammonium and that NXR, HAO and AmoB were among the 50 most abundant proteins of this organism (Extended Data Fig. 7).

a–c, Near-stoichiometric oxidation of 1 mM, 0.1 mM, or 10 μm ammonium to nitrate with transient accumulation of nitrite. d, Growth of Ca. N. inopinata on ammonium (initial concentration 0.6 mM) as determined by qPCR of the amoA gene. Ammonia oxidation was slow because this experiment was started with a highly diluted culture. Significance of difference was calculated by a paired t-test (*P < 0.05; ***P < 0.01) between data points as indicated by horizontal lines. e, Near-stoichiometric oxidation of 0.5 mM nitrite to nitrate by ammonia-grown Ca. N. inopinata. The cells were washed to remove residual ammonium before nitrite addition. Data points show means, error bars show 1 s.d. of n = 4 (a, b, e) or n = 3 (c, d) biological replicates. If not visible, error bars are smaller than symbols.
When a culture grown on ammonium was transferred into mineral medium containing only 0.5 mM nitrite as energy source and electron donor, Ca. N. inopinata stoichiometrically oxidized nitrite to nitrate (Fig. 2e). However, subsequent additions of nitrite did not result in further nitrite oxidation. We hypothesize that nitrite was first oxidized by residual NXR activity, but metabolic activity and biosynthetic processes finally stalled in the absence of ammonium. This would be consistent with an incapability of Ca. N. inopinata to use nitrite as nitrogen source for assimilation due to the absence of genes for a nitrite transporter and assimilatory nitrite reductase. It is interesting to note that Ca. N. inopinata could theoretically utilize nitrite as nitrogen source by respiratory ammonification catalysed by a periplasmic cytochrome c nitrite reductase (NrfA) using electrons from quinol (Extended Data Fig. 1d). Genes for respiratory ammonification have not been detected in the genomes of the two comammox strains reported by van Kessel et al.32 and of other Nitrospira17,19. However, the Ca. N. inopinata genome lacks a second copy of respiratory complex III, which may be needed for the reverse transport of high-potential electrons from nitrite to quinone in other Nitrospira17. If nitrite is the only available electron donor, this gap in the reverse electron transport chain likely prevents nitrite reduction by NrfA and CO2 fixation by the reductive tricarboxylic acid cycle, which is the autotrophic pathway in Ca. N. inopinata (Extended Data Fig. 1d) and other Nitrospira17. Thus, Ca. N. inopinata may grow on nitrite only in the presence of a low-potential electron donor for quinone reduction such as H2 (ref. 21) or intracellular storage compounds (Extended Data Fig. 1d). In contrast, electrons derived from ammonia should be transferred to quinone as in AOB1 and thus enable autotrophic growth (Extended Data Fig. 1d). An ammonium transporter also enables Ca. N. inopinata to use ammonium as nitrogen source. The incapability to grow on nitrite distinguishes Ca. N. inopinata from the strictly nitrite-oxidizing Nitrospira that grow on nitrite and CO215,17.
Distinct AMO and HAO of Ca. N. inopinata
The amoA gene is a functional and phylogenetic marker for AOM33,34, which has been used in numerous studies as a cultivation-independent tool to detect and identify AOM in microbial communities. Intriguingly, phylogenetic analyses revealed that Ca. N. inopinata possesses a new type of AmoA that differs from the AmoA forms of known AOB and AOA. It belongs to a distinct clade (‘comammox AmoA clade A’) that contains numerous environmental sequences and shares a common ancestor with the AmoA lineage of the betaproteobacterial AOB (Fig. 3, Extended Data Fig. 8). Similar to the AmoA phylogeny, the amoB and amoC as well as the hao genes of Ca. N. inopinata fell into distinct lineages that are related to the respective homologues of AOB (Extended Data Fig. 9).

Bayesian inference tree showing the phylogenetic relationship of comammox AmoA to other members of the AmoA superfamily (202 taxa, 238 alignment positions). Comammox AmoA sequences formed clades A (posterior probability, PP = 0.99) and B (PP = 0.97) that grouped together (PP = 0.91) and with betaproteobacterial AmoA (PP = 0.70). Scale bar indicates estimated change per nucleotide.
A fascinating question is whether the unusual enzymes for ammonia oxidation are ancestral features of the genus Nitrospira or were acquired by Ca. N. inopinata by lateral gene transfer. The first scenario would imply that these genes have been lost by the strictly nitrite-oxidizing Nitrospira members. Indications for a possible lateral gene transfer event in Ca. N. inopinata are putative transposase genes directly upstream of the amoCAB genes (Fig. 1) and a tetranucleotide pattern of the amoCAB-containing region that significantly deviates from the genome-wide signature (Extended Data Fig. 10). The tetranucleotide pattern of the amoCAB region also clearly differs from the genome-wide signature of the betaproteobacterium found in ENR4 and ENR6, strongly suggesting that these genes did not originate from this strain. Putative transposases are also located downstream of the nxrAB operon, whose tetranucleotide pattern also deviates from the genome-wide signature (Fig. 1, Extended Data Fig. 10). Ca. N. inopinata belongs to Nitrospira lineage II (Extended Data Fig. 4), and its NXR is also affiliated with lineage II (Extended Data Fig. 5). Thus, if lateral gene transfer occurred, the nxr genes must have been received from another Nitrospira lineage II member. No indications were found for lateral gene transfer of the other two amoC copies or of hao and the other HURM genes of Ca. N. inopinata (Fig. 1, Extended Data Fig. 10).
Distribution of comammox Nitrospira
A screening of public databases retrieved sequences within comammox AmoA clade A, which originated from paddy and other agricultural soils, forest soils, paddy field floodwater, freshwater environments such as wetlands, river beds, aquifers and lake sediments, and from engineered systems (activated sludge and drinking water treatment plants) (Extended Data Fig. 8). For most of these sequences no quantitative information regarding their abundance is available. However, for three metagenomic data sets from Rifle soils35, relative abundances could be estimated from raw sequence data. In these soils, archaeal amoA sequences were found to be 3.8 to 10.5 fold more abundant than comammox amoA sequences. Interestingly, only very low numbers of betaproteobacterial amoA sequences were found and those were retrieved exclusively from the unassembled Rifle data sets. Additional database searches retrieved sequences from soil, freshwater and engineered environments that clustered in phylogenetic trees with the amoB, amoC and hao genes of Ca. N. inopinata (Extended Data Fig. 9). These results are consistent with a wide environmental distribution of comammox organisms with the possible exception of oceanic environments, as no comammox marker genes were identified in marine metagenomes.
To further elucidate the distribution of putative comammox Nitrospira in engineered systems, we sequenced total community metagenomes from a pilot-scale membrane bioreactor (MBR) at the municipal WWTP Aalborg West (Denmark) and of nitrifying activated sludge from the full-scale WWTP of the University of Veterinary Medicine, Vienna, Austria (“WWTP VetMed”). A great diversity of lineage I and II Nitrospira had previously been detected in WWTP VetMed18. Quantitative FISH in the VetMed sample used for metagenomics revealed dominance of Nitrospira (7.5 ± 3% of all detectable bacteria, 1 s.d.) over AOB (2.5 ± 1.2%). AOA did not occur in this WWTP at an abundance relevant for nitrification as no sequences affiliated with Thaumarchaeota were detected in the metagenomic data set. Additionally, we sequenced metagenomes of pasty and suspended iron sludge from a groundwater well (GWW) of a waterworks (Wolfenbüttel, Germany) (Supplementary Tables 1 and 2). Nitrospira population genome bins were retrieved by differential coverage binning23 from all metagenomes (Supplementary Table 8). According to 16S rRNA and NXR phylogenies, these Nitrospira belonged to lineages I and II (Extended Data Figs 4 and 5). Intriguingly, amo and hao genes similar to those of Ca. N. inopinata were found in one or more Nitrospira bins from every sample, suggesting that comammox Nitrospira frequently occur in engineered systems (Extended Data Figs 8 and 9, Fig. 1). However, not all Nitrospira bins contained genes for ammonia oxidation (Supplementary Table 8). Since nxr genes were found in all bins except GWW Nitrospira bin 7, we assume that comammox coexisted in these communities with Nitrospira that were strict nitrite oxidizers or used alternate metabolisms19,21. In several Nitrospira bins with sufficiently long contigs, the amo, hao and nxr loci and flanking genes were syntenic with Ca. N. inopinata (Fig. 1). In particular, a gene cluster for cytochrome c biogenesis is located directly upstream of the hao gene in Ca. N. inopinata and metagenomes (Fig. 1). This gene arrangement is not found in genome-sequenced AOB36,37,38,39,40, suggesting that it may be diagnostic for comammox Nitrospira. Transposases were found close to the amo and nxr genes, respectively, in two Nitrospira bins (Fig. 1). Moreover, a second type of AmoA was identified in some of the GWW Nitrospira bins. These sequences fell into a phylogenetic sister lineage of comammox AmoA clade A, which also contains other environmental sequences from soil and freshwater ecosystems (Extended Data Fig. 8, Fig. 3), and showed considerably lower identities to the AmoA of Ca. N. inopinata (Fig. 1). Consequently, we refer to this lineage as ‘comammox AmoA clade B’. The amoB and amoC sequences from those Nitrospira bins, which contained clade B-amoA, also fell separately in the respective phylogenetic trees (Extended Data Fig. 9a, b). Thus, two different and related new types of AMO occur in bacteria from the genus Nitrospira, and both share a common ancestor with the AMO of the betaproteobacterial AOB (Fig. 3).
We have noticed that sequences in comammox AmoA clades A and B were previously assigned as particulate methane monooxygenase (PmoA) to uncultured gammaproteobacterial (clade A, Crenothrix polyspora) and alphaproteobacterial (clade B) methanotrophs41,42. While these assignments were based on indirect evidence, our study provides direct physiological proof that an organism expressing an enzyme in clade A oxidizes ammonia and metagenomic evidence for a Nitrospira origin of genes in both clades. However, it remains possible that genes in these clades were exchanged by lateral gene transfer between nitrifiers and methanotrophs.
Discussion
The first cultured comammox organism Ca. N. inopinata is a moderately thermophilic Nitrospira member, and uncultured mesophilic comammox Nitrospira were identified by metagenomics in this study, too. The genus Nitrospira is one of the most diverse8,18 known nitrifier groups and colonizes virtually all oxic ecosystems10, including high-temperature environments13,15. It is tempting to speculate that the environmental distribution of comammox is largely congruent with that of Nitrospira, which are mostly uncultured and poorly characterized. Previous research was based on the dogma that all Nitrospira use nitrite, but not ammonia, as energy source. Due to this firm expectation, comammox Nitrospira were overlooked for decades and some repeatedly observed phenomena could not be well explained. For example, conspicuously high in situ abundances of uncultured Nitrospira, which exceeded the abundances of known AOM in the same samples, were detected in nitrifying biofilms, activated sludge, freshwater sediments, and drinking water distribution systems14,18,43,44,45. These puzzling observations are inconsistent with the classical concept of nitrification, which suggests a AOM:NOB ratio greater than one46. Aside from other energy-conserving metabolic activities of NOB in addition to nitrite oxidation10,21,46, the presence of comammox organisms in those Nitrospira communities would be a plausible explanation for the increased ratio of Nitrospira over known AOM. Indeed, we detected amo and hao genes in the Nitrospira metagenome from WWTP VetMed (Fig. 1, Extended Data Figs 8 and 9), a system in which Nitrospira outnumber AOB according to FISH and comammox represents 43 to 71% of the Nitrospira population as estimated from gene abundances in the metagenomic data sets. A high relative abundance of comammox (58 to 74% of all Nitrospira) was also estimated for the GWW based on metagenome analysis. More precise analyses of comammox abundance as well as its spatial interactions with other community members will require the development of assays to rapidly differentiate in situ between strictly nitrite-oxidizing and comammox Nitrospira.
Studies with strictly nitrite-oxidizing representatives of this genus characterized Nitrospira as slow-growing microbial K-strategists adapted to low substrate concentrations18,43,47,48. Many Nitrospira, including Ca. N. inopinata, also form microcolonies, flocs, and biofilms10,43. These properties, if generally shared by comammox Nitrospira, would be in agreement with the theoretically predicted7 ecological niche of comammox. The engineered systems surveyed in this study are characterized by biofilm or floc formation. Diffusion barriers and ammonium or nitrite concentration gradients47 in biofilms could create niches with limited substrate influx, where comammox might outcompete incomplete nitrifiers. Complex biofilm or floc architectures with numerous microenvironments may support diverse nitrifier communities like in WWTP VetMed, which consist of comammox as well as canonical AOB and NOB. Future comammox isolates from the Ca. N. inopinata culture and from other enrichments may offer chances to experimentally define the conditions that select for these organisms and to study the competition of comammox with other nitrifiers, including strictly nitrite-oxidizing Nitrospira and AOA adapted to low substrate concentrations48,49.
The discovery of comammox has revealed that the division of metabolic labour in nitrification is not obligate and will thus have far-reaching implications for future studies on the microbiology of nitrogen cycling. It opens a new field in nitrification research and some of the most pressing open questions range from the biochemistry, regulation, inhibition, and kinetics of complete nitrification to the diversity, population dynamics, metabolic versatility, and biological interactions of comammox organisms. In particular, the integration of comammox in studies on the niche specialization and niche partitioning of AOB and AOA50 or NOB43 will be crucial to obtain a picture of nitrification as it actually occurs in nature. Such insights may lead to refined strategies to manage nitrification in sewage treatment, drinking water supply, and agriculture. The presence of new AMO and HAO types, which share common ancestry with these enzymes of betaproteobacterial AOB, in the phylogenetically deep-branching genus Nitrospira15 impressively exemplifies the modular evolution of the nitrogen cycle28 and adds further complexity to the intricate evolutionary history of nitrification17,28.
Methods
Sampling sites
The inoculum for the Ca. N. inopinata enrichment culture was sampled from a microbial biofilm that grew on the metal surface of a pipe and was covered by hot water, which was raised from a 1,200 m deep oil exploration well. The water temperature was 56 °C and the pH 7.5. The well was located in Aushiger, North Caucasus, Russia (43°22′45.0′′ N, 43°43′26.1′′ E). The biofilm samples were taken in April 2011. Activated sludge, membrane biofilm, and foam (from a foaming event) samples were taken in August and October 2014 from a pilot-scale membrane bioreactor (MBR) performing nitrogen removal and enhanced biological phosphorus removal (EBPR) at the conventional full-scale WWTP Aalborg West, Aalborg, Denmark (57°02′59.9′′ N, 9°51′55.4′′ E). The influent wastewater for this MBR came from the primary settling tank of the full-scale plant, entering an anoxic/denitrification (2 m3) tank and going to an oxic/nitrification (2 m3) tank. An anaerobic tank (1.8 m3) used for return sludge sidestream hydrolysis provided easily degradable substrate for EBPR and denitrification. Activated sludge was also sampled from an aerated activated sludge basin (tank no. 2) of the full-scale WWTP of the University of Veterinary Medicine, Vienna, Austria (48°15′17.8′′ N, 16°25′45.6′′ E) in January 2015 (WWTP VetMed). The two continuously operated activated sludge tanks of this WWTP have a volume of 254 m3 each. The wastewater composition and nitrogen load vary with the amounts of animal faeces and other sewage. This WWTP was known to host a large diversity of Nitrospira18. Iron sludge samples were taken from groundwater well (GWW) no. 1 of the well field of the Wolfenbüttel waterworks (Wolfenbüttel, Germany) (52°08′55.9′′ N, 10°32′33.9′′ E). The well has a depth of 50 m below ground level (bgl) and a diameter of 600 mM. Groundwater is extracted through two well intake screens in 28 to 38 m bgl and 46 to 48 m bgl. The normal well capacity is 160 m3 h−1. Before sampling, the well had been out of operation for about three weeks. The well water is a mixture of aerobic and anaerobic groundwater from two different ground water storeys and is characterized by the following parameters (values from years 2012 to 2014): pH about 7.2, about 10 °C, 5 to 10 mg l−1 dissolved oxygen, 0.13 to 0.17 mg l−1 ammonium, <0.01 mg l−1 nitrite, 12 to 16 mg l−1 nitrate, 0.16 to 0.42 mg l−1 total iron, 0.03 to 0.08 mg l−1 manganese, 0.64 to 0.99 mg l−1 total organic carbon, 0.44 to 0.78 mg l−1 dissolved organic carbon, 71 to 81 mg l−1 dissolved inorganic carbon, 121 to 138 mg l−1 calcium. The drop pipe, through which the extracted water is pumped to ground level, was drawn out of the well on 27 April 2015 and had deposits of pasty iron sludge on the inner surface. A sample was taken from these deposits at several points corresponding to depths between 20 and 10 m bgl. A second sample consisted of suspended iron sludge deposits that had been flushed away from the upper well intake screen and retained on a fleece filter during pumping out of the turbid water on 28 April 2015.
Enrichment and cultivation of Ca. N. inopinata
The biofilm used as inoculum was suspended and incubated at 46 °C with 0.5 mM NH4Cl in a modified AOM medium51 containing (per litre): 50 mg KH2PO4; 75 mg KCl; 50 mg MgSO4 × 7H2O; 584 mg NaCl; 4 g CaCO3 (mostly undissolved, acting as a solid buffering system and growth surface); 1 ml of specific trace element solution (TES); and 1 ml of selenium-wolfram solution (SWS)52. The composition of TES and SWS is described below. Both solutions were added to the autoclaved medium by sterile filtration using 0.2 μm pore-size cellulose acetate filters (Thermo Scientific). The pH of the medium was around 8.2 after autoclaving and was kept around 7.8 by the CaCO3 buffering system during growth of the enrichment. TES contained (per litre): 34.4 mg MnSO4 × 1H2O; 50 mg H3BO3; 70 mg ZnCl2; 72.6 mg Na2MoO4 × 2H2O; 20 mg CuCl2 × 2H2O; 24 mg NiCl2 × 6H2O; 80 mg CoCl2 × 6H2O; 1 g FeSO4 × 7H2O. All salts except FeSO4 × 7H2O were dissolved in 997.5 ml Milli-Q water and 2.5 ml of 37% HCl was added before dissolving the FeSO4 × 7H2O salt. SWS contained (per litre): 0.5 g NaOH; 3 mg Na2SeO3 × 5H2O; 4 mg Na2WO4 × 2H2O. The primary ammonium-consuming enrichment was subsequently treated with antibiotics (one treatment with 50 mg l−1 vancomycin, two treatments with 50 mg l−1 bacitracin). The ammonium concentration was increased to 1 mM NH4Cl for these and all further cultivation steps. After these treatments and repeated serial dilutions in AOM medium without antibiotics, enrichment culture ENR4 was obtained that was characterized in this study. An aliquot of ENR4 was incubated at 50 °C for four weeks and then subjected to serial dilution at 46 °C. Propagation of the most diluted (10−8) ammonia-oxidizing culture was followed by serial dilution in AOM medium containing 1 mM urea instead of ammonium. The most diluted (10−7) urea-consuming (that is, nitrifying) culture was again cultivated in AOM medium with 1 mM NH4Cl and subjected to repeated serial dilutions, which resulted in culture ENR6 that was also characterized in this study. Enrichments ENR4 and ENR6 were further cultivated in 100 ml or 250 ml Schott bottles in AOM medium containing 1 mM NH4Cl. To obtain enough biomass for DNA extraction, enrichment ENR4 was up-scaled in 1 l and 2 l Schott bottles. The composition of enrichment cultures was analysed by phase contrast microscopy, electron microscopy, FISH with rRNA-targeted probes, amoA- and 16S rRNA-specific PCR, and metagenomics (see later for methodological details).
Physiological experiments with Ca. N. inopinata
To study nitrification by Ca. N. inopinata, an actively nitrifying ENR4 stock culture was harvested by centrifugation (9,300g, 30 min, 10 °C) and the biomass was suspended in AOM medium (see above) without ammonium. Aliquots (25 ml) of this suspension were distributed to 100 ml Schott bottles (all glassware was rinsed twice in 6 M HCl and three times in Milli-Q water, autoclaved, and dried at 60 °C before use). After addition of NH4Cl to final concentrations of 1 mM, 0.1 mM, or 10 μm, respectively, or of NaNO2 to a final concentration of 0.5 mM, the biomass was incubated at 46 °C for 9 h (10 μm NH4Cl) or 48 h (other experiments) without agitation in the dark. Samples (500 μl) for chemical analyses (see below) were taken directly after ammonium or nitrite addition and during the incubations. The samples were centrifuged (22,000g, 10 min, 4 °C) to remove cells and undissolved CaCO3 and 450 μl of the supernatant was transferred to plastic tubes and stored at −20 °C until analysis. Each incubation condition except 10 μm NH4Cl was performed in parallel with four biological replicates (biological triplicates for 10 μm NH4Cl), two dead biomass controls (cells were killed by autoclaving), and two abiotic controls that contained only medium and substrate, but no biomass. After the experiments, the remaining biomass was harvested by centrifugation (9,300g, 30 min, 10 °C), frozen immediately at −80 °C, and shipped on dry ice for proteome analysis. To quantify growth of Ca. N. inopinata by complete nitrification, culture ENR4 was incubated in mineral NOB medium, which has been used to cultivate nitrite-oxidizing Nitrospira21. In this experiment, the NOB medium was amended with ammonium instead of nitrite. The NOB medium was chosen because it contains less CaCO3, which can affect quantitative PCR (qPCR) efficiency and accuracy. Nitrifying activity of ENR4 in NOB medium was confirmed in preceding tests. Biomass from the supernatant (without undissolved CaCO3) from an ammonia-oxidizing culture was washed once in NOB medium, harvested by centrifugation (9,300g, 30 min, 10 °C), and prepared for incubation as described above. Following the addition of NH4Cl to a final concentration of 0.6 mM, samples (100 μl) for quantitative PCR were taken immediately and after 4, 5, 7, and 8 days of incubation. Samples for chemical measurements (see below) were taken immediately and after 1, 4, 5, 7, and 8 days of incubation. All samples were stored at −20 °C until analysis. These incubation experiments were performed in biological triplicates. Copy numbers of the Ca. N. inopinata amoA gene were determined by qPCR using the newly designed Ca. Nitrospira inopinata amoA gene-specific primers Nino_amoA_19F (5′-ATAATCAAAGCCGCCAAGTTGC-3′) and Nino_amoA_252R (5′-AACGGCTGACGATAATTGACC-3′). The qPCR reactions were run with three technical replicates in a Bio-Rad C1000 CFX96 Real-Time PCR system, using the Bio-Rad iQ SYBR Green Supermix kit (Bio-Rad). Each qPCR reaction was performed in 20 μl reaction mix containing 10 μl SYBR Green Supermix, 2 μl of the sampled ENR4 cell suspension, 0.1 μl of each primer (50 μM), and 7.9 μl of autoclaved double-distilled ultrapure water. Cells were lysed and DNA was released for 10 min at 95 °C, followed by 43 PCR cycles of 40 s at 94 °C, 40 s at 52 °C, and 45 s at 72 °C. Plasmids carrying the Ca. N. inopinata amoA gene were obtained by PCR-amplifying the gene from the ENR4 culture and cloning the product into the pCR4-TOPO TA vector (Invitrogen). The M13-PCR product from these plasmids containing the amoA gene was used as standard for qPCR (the amoA copy number in the standard was calculated from DNA concentration). Tenfold serial dilutions of the standard were subjected to qPCR in triplicates to generate an external standard curve. The amplification efficiency was 92.6%, and the correlation coefficient (r2) of the standard curve was 0.999.
Isolation of the betaproteobacterium from ENR4
A 1 ml aliquot of the ENR4 culture was transferred to 25 ml modified AOM medium (see above) containing 6 mM sodium acetate. After three weeks of incubation at 46 °C, a 1 ml aliquot of the betaproteobacterial primary enrichment was transferred into 25 ml of fresh modified AOM medium containing 6 mM sodium acetate. After three more weeks, a 5 ml aliquot of this culture was centrifuged (9,300g, 10 min, 10 °C) and the cells were resuspended in 25 ml NOB medium (see above) containing 1 ml of SWS and 4 mM sodium acetate. Thereafter, 1 ml of the betaproteobacterial enrichment was transferred into fresh NOB medium containing 4 mM sodium acetate every 2 weeks. The fourth transfer was checked for purity by FISH with the betaproteobacterium-specific probe Nmir1009, which showed 100% overlap with the EUB338 probe mix and DAPI signals. No Nitrospira cells were detected by FISH in the culture.
Physiological experiments with the betaproteobacterium
To test whether the betaproteobacterium had the capability to nitrify, 20 ml of a dense pure culture of this organism was centrifuged (9,300g, 10 min, 10 °C), washed once in modified AOM medium without solid CaCO3, and resuspended in modified AOM medium without ammonium and solid CaCO3. Aliquots of this suspension were distributed into 100 ml Schott bottles, which had been rinsed twice in 6 M HCl, washed 3 times in Milli-Q water, closed with aluminium caps, autoclaved, and dried at 60 °C before use. Subsequently, the following substrates were added: 1 mM NH4Cl; or 0.5 mM NaNO2 and 0.1 mM NH4Cl; or 4 mM sodium acetate and 0.1 mM NH4Cl (the 0.1 mM NH4Cl was added to the nitrite and acetate incubations to provide the organism with a nitrogen source for assimilation). The biomass was incubated at 46 °C in the dark without agitation. All experiments were performed in parallel with biological triplicates. Samples (700 μl) for qPCR and chemical analyses (see below) were taken immediately after experimental set-up and after 19, 24, 30, 42, and 48 h of incubation. The samples were stored at −20 °C until analysis. Cell densities of the betaproteobacterium were quantified by qPCR targeting the soxB gene, which encodes the SoxB component of the periplasmic thiosulfate-oxidizing Sox enzyme complex. SoxB is a single-copy gene in the genome of the betaproteobacterium. The primers used to quantify the soxB gene were soxB_F1 (5′-GGACCAGACCGCCATCACTTACCC-3′) and soxB_R1 (5′-GCACCATGTCCCCGCCTTGCT-3′). The qPCR protocol and conditions were the same as described above.
Chemical analyses
Ammonium levels were measured photometrically as described previously53,54 with adjusted volumes of sample and reagents. Standards were prepared in AOM or NOB medium and ranged from 7.25 to 1,000 μm NH4Cl. Nitrite concentrations were determined photometrically by the acidic Griess reaction55. Nitrate was reduced to nitrite by vanadium chloride and measured as NOx by the Griess assay. Nitrate concentrations were calculated from the NOx measurements as described elsewhere56. Standards were prepared in AOA or NOB medium and ranged from 7.8 to 1,000 μm for NOx and from 3.9 to 500 μm for nitrite.
Replication of physiological experiments
The number of replications are detailed in the subsections for each specific experiment, and were mostly determined by the amount of biomass available for the different cultures. In all experiments, a minimum of three biological replications were used. No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
FISH and microscopy
FISH with rRNA-targeted oligonucleotide probes was performed as described elsewhere57 using the EUB338 probe mix58,59 for the detection of Bacteria, probes Ntspa662 and Ntspa712 specific for Nitrospira10, and probes Nso1225, Nso190, and NEU specific for betaproteobacterial AOB22. The betaproteobacterium in ENR4 and ENR6 was detected by FISH with the specific probe Nmir1009 (5′-CACTCCCCCGTCTCCGGG-3′) with 35% of formamide in the hybridization buffer. If required, unlabelled competitor oligonucleotides were added in equimolar amounts as probes. Cells were counterstained by incubation for 5 min in a 0.1 μg ml−1 DAPI (4′,6-diamidino-2-phenylindole) solution. Fluorescence micrographs were recorded by using a Leica SP7 confocal laser scanning microscope equipped with a white light laser. To determine the relative abundances of Nitrospira and AOB in WWTP VetMed by quantitative FISH, 20 confocal images of FISH probe signals were taken at random positions in the sample and analysed as described elsewhere60 by using the digital image analysis software daime61. For whole-cell electron microscopy, cells were positively stained with 1% (w/v) uranyl acetate. Electron microscopy of thin sections was carried out as described elsewhere62.
PCR assays for marker genes of AOB and AOA
To check whether the Ca. N. inopinata enrichments contained known AOB or AOA, PCR tests were performed using primer sets amoA-1F/amoA-2R targeting betaproteobacterial amoA63, CamoA-19f/CamoA-616r targeting thaumarchaeal amoA33,64, and 771F/957R for thaumarchaeal 16S rRNA genes65 and the respective published reaction conditions. DNA was extracted for these PCR assays by using the PowerSoil DNA Isolation Kit (MoBio) according to the manufacturer’s instructions.
Metaproteomic analysis
Protein extraction from concentrated ENR4 biomass, proteolytic digestion, analysis of peptide lysates by mass spectrometry (MS), processing of MS raw files, and analysis of MS spectra were carried out as described elsewhere20. MS spectra were searched against a database of predicted gene products on the ENR4 metagenome scaffolds containing 12,234 sequence entries and a common Repository of Adventitious Proteins (cRAP) database using the Sequest HT algorithm. The PROPHANE pipeline (http://www.prophane.de/index.php) was used to classify the lowest common phylogenetic ancestor of each protein group and to calculate the normalized spectral abundance factor (NSAF).
DNA extraction for metagenomics
Biomass of enrichment ENR4 was collected from three culture bottles (samples ENR4_A, ENR4_E, ENR4_F) by centrifugation and frozen over night at −80 °C before total nucleic acids were extracted by bead beating in the presence of phosphate buffer, 10% (w/v) SDS and phenol as described elsewhere66 (see ref. 67 for full protocol). Bead beating was repeated twice to break remaining intact cells, the supernatants from each step were pooled, and nucleic acids purified by phenol/chloroform/isoamyl alcohol and chloroform/isoamyl alcohol extraction. Nucleic acids were precipitated using 20% (w/v) polyethylene glycol, washed in ice-cold 75% (v/v) ethanol, and resuspended in sterile 10 mM TRIS buffer. RNA was digested with RNase I (Promega) and the purity of DNA assessed by spectrophotometry. The same protocol was used to extract DNA from concentrated biomass of enrichment ENR6 (sample ENR6_N3), with the modification that bead beating was not repeated, and from an activated sludge sample of WWTP VetMed collecting only the supernatants of the second and third bead beating steps (DNA extract Vetmed_23). DNA was extracted from a second aliquot of the WWTP VetMed sample (DNA extract Vetmed_Pskit), and from pasty (sample GWW_HP_F1) or suspended (sample GWW_HP_D) iron sludge from the GWW, by using the PowerSoil DNA Isolation Kit (MoBio). DNA was extracted from all MBR samples by using the FastDNA SPIN Kit for Soil (MP Biomedicals) following the manufacturer’s instructions.
Metagenome sequencing
Sequencing libraries were prepared using the Nextera or TruSeq PCR free kits (Illumina Inc.) following the manufacturer’s recommendations. For the TruSeq PCR free kits, the 550 bp protocol was used with 1 μg of input DNA. The prepared libraries were sequenced using either an Illumina MiSeq with MiSeq Reagent Kit v3 (2x301 bp; Illumina Inc.) or an Illumina HiSeq2000 using the TruSeq PE Cluster Kit v3-cBot-HS and TruSeq SBS kit v.3-HS sequencing kit (Illumina Inc.). Nanopore sequencing was performed in addition to facilitate completion of the Ca. N. inopinata genome sequence. Library preparation was done using the Nanopore Sequencing kit (SQK-MAP005, Oxford Nanopore) following the manufacturer's recommendations (v. MN005_1124_revC_02Mar2015) with shearing in an Eppendorf MiniSpin plus centrifuge at 8,000 rpm and including the optional PreCR treatment step, as well as Ampure XP Bead purification after dA-tailing. The libraries were sequenced using nanopore flow cells (FLO-MAP003, Oxford Nanopore) using the MinION device (Oxford Nanopore) with the MinKNOW software (v. 0.50.1.15). Flow cells were primed twice with a mixture of 3 μl Fuel Mix, 75 μl 2 × Running Buffer, and 72 μl nuclease-free water for 10 min. Libraries were prepared for loading onto the flow cell by mixing 75 μl 2 × Running Buffer, 66 μl nuclease-free water, 3 μl Fuel Mix, and 6 μl Library (Pre-sequencing Mix). A sequencing run was started (MAP_48Hr_Sequencing_Run.py) after loading the library. Additional DNA library top-ups and restart of the run script was carried out to maximize yield by allowing a new selection of active pores. Base calling was carried out using Metrichor and the 2D Basecalling workflow (Rev 1.16). Details for each metagenome can be found in Supplementary Table 1.
Metagenome scaffold assembly and binning
Paired-end Illumina reads were imported into CLC Genomics Workbench v. 8.0 (CLCBio, Qiagen) and trimmed using a minimum phred score of 20 and a minimum length of 50 bp, with allowing no ambiguous nucleotides and trimming off Illumina sequencing adaptors if found. FASTQ files for the Oxford Nanopore 2D reads were obtained using the R package poRe v. 0.668 and error corrected using Illumina reads through Proovread v. 2.1369. For each environment, all trimmed Illumina reads were co-assembled using CLCs de novo assembly algorithm, using a kmer of 63 and a minimum scaffold length of 1 kbp. Trimmed reads were mapped to the assembled scaffolds using CLCs map reads to reference algorithm, with a minimum similarity of 95% over 70% of the read length. Open reading frames (ORFs) were predicted in the assembled scaffolds using Prodigal70. A set of 107 hidden Markov models (HMMs) of essential single-copy genes71 were searched against the ORFs using HMMER3 (http://hmmer.janelia.org/) with default settings, except option (-cut_tc) was used. Identified proteins were taxonomically classified using BLASTP against the RefSeq (v. 52) protein database with a maximum e-value cutoff of 10−5. MEGAN72 was used to extract class-level taxonomic assignments from the BLAST output. The script network.pl (http://madsalbertsen.github.io/mmgenome/) was used to extract paired-end read connections between scaffolds. PhyloPythiaS+73 was used to taxonomically classify all scaffolds of selected samples. In addition, selected metagenome assemblies were binned based on ESOM maps74. After training the ESOM using scaffolds >5 kbp and large scaffolds chopped into 5 kbp pieces, all scaffolds were projected back to the ESOM map to retrieve a single coordinate for all scaffolds. Individual genome bins were extracted using the multi-metagenome principles23 implemented in the mmgenome R package (http://madsalbertsen.github.io/mmgenome/). All genome bins are fully reproducible from the raw metagenome assemblies using Rmarkdown files available on http://madsalbertsen.github.io/mmgenome/. The script extract.fastq.reassembly.pl was used to extract paired-end reads from the binned scaffolds, which were used for re-assembly using SPAdes75. For selected samples, error-corrected Oxford Nanopore 2D reads were used for scaffolding using SSPACE-LongRead76. For all genomes, quality was assessed using coverage plots through the mmgenome R package and through the use of QUAST77 and CheckM78. Details for each metagenome assembly can be found in Supplementary Table 2, and further details for the reconstructed bacterial genomes (including CheckM results) in Supplementary Tables 3–7. Relative genome sequence coverage was calculated as the fraction of sequence coverage of a reconstructed genome compared to the summed coverage of all genomes in these low-complexity metagenomes. The reconstructed bacterial genomes were uploaded to the MicroScope platform79 for automatic annotation and for manual annotation refinement17 of key pathways of Ca. N. inopinata.
To test for the presence of additional organisms capable of nitrification, the raw reads for each enrichment ENR4 and ENR6 were mapped to the amoA, amoB, amoC, hao and nxrB sequences used to generate the trees in Extended Data Figs 5b,d, 8, and 9. Reads were required to align to any one member of a target data set over at least 70% of read length with BLASTN (word size = 7). Reads that mapped with >97% nucleotide identity were automatically classified. Reads with lower identity were placed with the Evolutionary Placement Algorithm (EPA) using RAxML80. Using this procedure, no indication was found for the presence of any nitrifier other than Ca. N. inopinata in these enrichments.
Sequence collection and phylogenetics
For phylogenetic analyses of AMO and HAO, full amino acid data sets were downloaded from the Pfam81 site for bacterial (pfam02461) and archaeal (pfam12942) amoA. Additional amino acid sequences were identified from the NCBI GenBank82 and the Integrated Microbial Genomes databases (IMG-ER and -MER)83 that were returned using the search words ‘ammonia, methane, amo, pmo or monooxygenase’ (GenBank) or had been annotated with one of the target pfams (IMG). A BLASTP84 search was performed using the Ca. Nitrospira inopinata amoA sequence as a query, word size = 2, BLOSUM 45, E = 10 and the top 1,000 returned sequences were downloaded. Comparable procedures were performed to generate a comprehensive set of amoB (pfam04744) and amoC (pfam04896) sequences. For construction of the hao (pfam13447) data set, query words were changed to ‘hydroxylamine’ and ‘Hao’. For each gene set, amino acid sequences were filtered using hmmsearch (http://hmmer.janelia.org/) with the respective pfam HMMs, requiring an expect value < 0.001. Amino acid sequences were clustered at 75% identity using USEARCH85 and aligned using Mafft86. Phylogenetic trees were calculated using PhyloBayes87, running 5 independent chains for 21,000 cycles each, using 11,000 cycles for burn-in and sampling every 20 cycles. Sequences that mapped to centroids that clustered within the comammox clade were used for additional phylogenetic calculations along with an outgroup of 27 betaproteobacterial amoA and 29 diverse pmoA sequences. Corresponding nucleotide sequences for this set were aligned according to their amino acid translations using MUSCLE88 and manually corrected for frameshifts. Nucleotide alignments were then used for constructing consensus trees in Phylobayes, running 5 independent chains for 21,000 cycles each, using 11,000 cycles for burn-in and sampling every 20 cycles.
To estimate relative abundances of amoA genes, comammox-type amoA sequences were identified from three publicly available Rifle soil metagenomic data sets (3300002121, 3300002122 and 3300002124) available within IMG. Functional profiles were generated within IMG using pfam12942 (archaeal amoA) and pfam02461 (bacterial amoA/pmoA) against the assembly and unassembled reads. All identified amoA/pmoA nucleotide sequences were downloaded as nucleic acid sequences and added to the existing amoA alignment used to generate Extended Data Fig. 8 with the -add option in Mafft. EPA in RAxML was used to assign downloaded sequences into the reference tree that is the basis for Extended Data Fig. 8. AmoA abundance for each amoA type (comammox, archaeal, betaproteobacterial AOB) was estimated by taking the sum of the estimated copy numbers of each assembled amoA gene of a given type as well as the number of unassembled reads assigned to a given amoA type.
Comammox, betaproteobacterial, and archaeal amoA sequences from the metagenomes of WWTP VetMed and the GWW were identified using the same procedure as above. Comammox amoA read abundances were then used to calculate an estimate of the fraction of Nitrospira that are comammox. AmoA was assumed to be a single copy gene in all comammox (as it is in Ca. N. inopinata). Total Nitrospira were enumerated by mapping raw reads from metagenomic samples using the first 700 nucleotides of the predicted ATP-citrate lyase subunit beta (aclB) gene from Ca. N. inopinata. Reads were required to align to Ca. N. inopinata aclB over at least 70% of read length and with >60% alignment identity with BLASTN (word size = 7). AclB was chosen on the basis that this gene has a restricted taxonomic distribution, encodes a key enzyme of the reductive tricarboxylic acid cycle employed by all known Nitrospira for CO2 fixation, and is present in single copy within known Nitrospira genomes. To test its utility, all 150 nt segments (pos 1:150, 2:151…1,051:1,200) of the Ca. N. inopinata aclB gene was used as a query against the nr database (BLAST, word size = 7, 70% read length and 60% alignment identity). Over the first 700 nucleotides of the aclB gene, test fragments mapped only to reference Nitrospira organisms. Downstream of this region, the aclB mapping was less specific, mapping to Nitrospira and Chlorobi with high (>90%) identity. Coverage of each gene was calculated by dividing the number of mapped reads by gene length of the query (843 nt for comammox amoA and 700 nt for Nitrospira aclB). Adjusted coverage was calculated by dividing gene coverage by total number of reads in the metagenomic data set. Ratios discussed in the main text are then the adjusted coverage of comammox (as calculated from comammox amoA) divided by the adjusted coverage for all Nitrospira (as calculated from aclB).
For phylogenetic analyses of NXR, the NxrA and nxrB sequences of Ca. N. inopinata were imported into existing NxrA17 and nxrB8 sequence databases using the software ARB89. NxrA sequences were aligned using Mafft, nxrB sequences were manually aligned according to their amino acid translations. Maximum likelihood trees were calculated using RAxML with the GAMMA model of rate heterogeneity using empirical base frequencies and the LG substitution model (NxrA) or with the GAMMA model of rate heterogeneity and the GTR substitution model (nxrB). Bayesian inference trees were calculated using PhyloBayes, running 3 independent chains for 32,200 cycles each, using 6,440 cycles for burn-in (NxrA) or 3 independent chains for 35,500 cycles each, using 7,000 cycles for burn-in (nxrB). Nitrospira 16S rRNA genes from this study were added to an existing Nitrospira 16S rRNA sequence database and aligned in ARB. Phylogenetic trees were calculated using RAxML with the GAMMA model of rate heterogeneity and the GTR substitution model, and using MrBayes90 v.3.2.1, running 4 independent chains for 5 million generations each, with 1.25 million cycles for burn-in and sampling every 100 generations.
Average nucleotide identity and tetranucleotide signature analyses
Pairwise average nucleotide identity (ANI) values were calculated for comammox Nitrospira genomes using BLAST (ANIb) in JSpecies91. Genome-wide tetranucleotide signatures were calculated for the forward and reverse strand for each genome with the oligonucleotideFrequency(width = 4) command using the Biostrings package in R92. Tetranucleotide patterns were also calculated across the length of the genome with a sliding window of 5 kb (step = 1 kb). The tetranucleotide pattern for each window was compared to the global tetranucleotide signature by calculating the Pearson correlation (r) of log(1+counts) of each window against the log(1+counts) of the global signature. P values, indicating a significantly low correlation for tetranucleotide signature of a window, were calculated by modelling 1 − r across all windows as a log-normal distribution. Multiple testing was accounted for by using the Benjamini–Hochberg procedure with a false discovery rate of 5%.
Accession codes
Primary accessions
European Nucleotide Archive
Data deposits
All raw sequence data is available in the European Nucleotide Archive (ENA) under the project accession number PRJEB10139. The genome sequence of Ca. N. inopinata has been deposited at ENA under the project PRJEB10818, sequence accession LN885086. The draft genome of the betaproteobacterium from ENR4 and ENR6 is available in the JGI Integrated Microbial Genomes Database (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) under the IMG Genome ID 2636415980.
References
Bock, E. & Wagner, M. in The Prokaryotes: A Handbook on the Biology of Bacteria (eds Dworkin, M. et al.) 457–495 (Springer, 2001)
Könneke, M. et al. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437, 543–546 (2005)
Winogradsky, S. Contributions a la morphologie des organismes de la nitrification. Arch. Sci. Biol. (St. Petersb.) 1, 87–137 (1892)
Arp, D. & Bottomley, P. J. Nitrifiers: more than 100 years from isolation to genome sequences. Microbe 1, 229–234 (2006)
Pfeiffer, T. & Bonhoeffer, S. Evolution of cross-feeding in microbial populations. Am. Nat. 163, E126–E135 (2004)
Heinrich, R. & Schuster, S. The regulation of cellular systems . (Chapman & Hall, 1996)
Costa, E., Pérez, J. & Kreft, J. U. Why is metabolic labour divided in nitrification? Trends Microbiol. 14, 213–219 (2006)
Pester, M. et al. NxrB encoding the beta subunit of nitrite oxidoreductase as functional and phylogenetic marker for nitrite-oxidizing Nitrospira . Environ. Microbiol. 16, 3055–3071 (2014)
Hovanec, T. A., Taylor, L. T., Blakis, A. & Delong, E. F. Nitrospira-like bacteria associated with nitrite oxidation in freshwater aquaria. Appl. Environ. Microbiol. 64, 258–264 (1998)
Daims, H., Nielsen, J. L., Nielsen, P. H., Schleifer, K. H. & Wagner, M. In situ characterization of Nitrospira-like nitrite-oxidizing bacteria active in wastewater treatment plants. Appl. Environ. Microbiol. 67, 5273–5284 (2001)
Watson, S. W., Bock, E., Valois, F. W., Waterbury, J. B. & Schlosser, U. Nitrospira marina gen. nov. sp. nov.: a chemolithotrophic nitrite-oxidizing bacterium. Arch. Microbiol. 144, 1–7 (1986)
Taylor, M. W., Radax, R., Steger, D. & Wagner, M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347 (2007)
Lebedeva, E. V. et al. Isolation and characterization of a moderately thermophilic nitrite-oxidizing bacterium from a geothermal spring. FEMS Microbiol. Ecol. 75, 195–204 (2011)
Martiny, A. C., Albrechtsen, H. J., Arvin, E. & Molin, S. Identification of bacteria in biofilm and bulk water samples from a nonchlorinated model drinking water distribution system: detection of a large nitrite-oxidizing population associated with Nitrospira spp. Appl. Environ. Microbiol. 71, 8611–8617 (2005)
Ehrich, S., Behrens, D., Lebedeva, E., Ludwig, W. & Bock, E. A new obligately chemolithoautotrophic, nitrite-oxidizing bacterium, Nitrospira moscoviensis sp. nov. and its phylogenetic relationship. Arch. Microbiol. 164, 16–23 (1995)
Schramm, A., De Beer, D., Wagner, M. & Amann, R. Identification and activities in situ of Nitrosospira and Nitrospira spp. as dominant populations in a nitrifying fluidized bed reactor. Appl. Environ. Microbiol. 64, 3480–3485 (1998)
Lücker, S. et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl Acad. Sci. USA 107, 13479–13484 (2010)
Gruber-Dorninger, C. et al. Functionally relevant diversity of closely related Nitrospira in activated sludge. ISME J. 9, 643–655 (2015)
Koch, H. et al. Expanded metabolic versatility of ubiquitous nitrite-oxidizing bacteria from the genus Nitrospira . Proc. Natl Acad. Sci. USA 112, 11371–11376 (2015)
Palatinszky, M. et al. Cyanate as an energy source for nitrifiers. Nature 524, 105–108 (2015)
Koch, H. et al. Growth of nitrite-oxidizing bacteria by aerobic hydrogen oxidation. Science 345, 1052–1054 (2014)
Mobarry, B. K., Wagner, M., Urbain, V., Rittmann, B. E. & Stahl, D. A. Phylogenetic probes for analyzing abundance and spatial organization of nitrifying bacteria. Appl. Environ. Microbiol. 62, 2156–2162 (1996)
Albertsen, M. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nature Biotechnol. 31, 533–538 (2013)
Arp, D. J., Chain, P. S. G. & Klotz, M. G. The impact of genome analyses on our understanding of ammonia-oxidizing bacteria. Annu. Rev. Microbiol. 61, 503–528 (2007)
Berube, P. M. & Stahl, D. A. The divergent AmoC3 subunit of ammonia monooxygenase functions as part of a stress response system in Nitrosomonas europaea . J. Bacteriol. 194, 3448–3456 (2012)
Klotz, M. G. et al. Evolution of an octahaem cytochrome c protein family that is key to aerobic and anaerobic ammonia oxidation by bacteria. Environ. Microbiol. 10, 3150–3163 (2008)
Bergmann, D. J., Hooper, A. B. & Klotz, M. G. Structure and sequence conservation of hao cluster genes of autotrophic ammonia-oxidizing bacteria: evidence for their evolutionary history. Appl. Environ. Microbiol. 71, 5371–5382 (2005)
Klotz, M. G. & Stein, L. Y. Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol. Lett. 278, 146–156 (2008)
Rittmann, B. E., Regan, J. M. & Stahl, D. A. Nitrification as a source of soluble organic substrate in biological treatment. Water Sci. Technol. 30, 1–8 (1994)
Nowka, B., Off, S., Daims, H. & Spieck, E. Improved isolation strategies allowed the phenotypic differentiation of two Nitrospira strains from widespread phylogenetic lineages. FEMS Microbiol. Ecol. 91, fiu031 (2015)
Ushiki, N., Fujitani, H., Aoi, Y. & Tsuneda, S. Isolation of Nitrospira belonging to sublineage II from a wastewater treatment plant. Microbes Environ. 28, 346–353 (2013)
van Kessel, M. A. H. J. et al. Complete nitrification by a single microorganism. Nature http://dx.doi.org/10.1038/nature16459 (2015)
Pester, M. et al. amoA-based consensus phylogeny of ammonia-oxidizing archaea and deep sequencing of amoA genes from soils of four different geographic regions. nviron. Microbiol . 14, 525–539 (2012)
Purkhold, U. et al. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl. Environ. Microbiol. 66, 5368–5382 (2000)
Wrighton, K. C. et al. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665 (2012)
Stein, L. Y. et al. Whole-genome analysis of the ammonia-oxidizing bacterium, Nitrosomonas eutropha C91: implications for niche adaptation. Environ. Microbiol. 9, 2993–3007 (2007)
Chain, P. et al. Complete genome sequence of the ammonia-oxidizing bacterium and obligate chemolithoautotroph Nitrosomonas europaea . J. Bacteriol. 185, 2759–2773 (2003)
Suwa, Y. et al. Genome sequence of Nitrosomonas sp. strain AL212, an ammonia-oxidizing bacterium sensitive to high levels of ammonia. J. Bacteriol. 193, 5047–5048 (2011)
Norton, J. M. et al. Complete genome sequence of Nitrosospira multiformis, an ammonia-oxidizing bacterium from the soil environment. Appl. Environ. Microbiol. 74, 3559–3572 (2008)
Klotz, M. G. et al. Complete genome sequence of the marine, chemolithoautotrophic, ammonia-oxidizing bacterium Nitrosococcus oceani ATCC 19707. Appl. Environ. Microbiol. 72, 6299–6315 (2006)
Radajewski, S. et al. Identification of active methylotroph populations in an acidic forest soil by stable-isotope probing. Microbiology 148, 2331–2342 (2002)
Stoecker, K. et al. Cohn’s Crenothrix is a filamentous methane oxidizer with an unusual methane monooxygenase. Proc. Natl Acad. Sci. USA 103, 2363–2367 (2006)
Schramm, A., de Beer, D., van den Heuvel, J. C., Ottengraf, S. & Amann, R. Microscale distribution of populations and activities of Nitrosospira and Nitrospira spp. along a macroscale gradient in a nitrifying bioreactor: quantification by in situ hybridization and the use of microsensors. Appl. Environ. Microbiol. 65, 3690–3696 (1999)
Altmann, D., Stief, P., Amann, R., De Beer, D. & Schramm, A. In situ distribution and activity of nitrifying bacteria in freshwater sediment. Environ. Microbiol. 5, 798–803 (2003)
Foesel, B. U. et al. Nitrosomonas Nm143-like ammonia oxidizers and Nitrospira marina-like nitrite oxidizers dominate the nitrifier community in a marine aquaculture biofilm. FEMS Microbiol. Ecol. 63, 192–204 (2008)
Winkler, M. K. H., Bassin, J. P., Kleerebezem, R., Sorokin, D. Y. & van Loosdrecht, M. C. M. Unravelling the reasons for disproportion in the ratio of AOB and NOB in aerobic granular sludge. Appl. Microbiol. Biotechnol. 94, 1657–1666 (2012)
Maixner, F. et al. Nitrite concentration influences the population structure of Nitrospira-like bacteria. Environ. Microbiol. 8, 1487–1495 (2006)
Nowka, B., Daims, H. & Spieck, E. Comparison of oxidation kinetics of nitrite-oxidizing bacteria: nitrite availability as a key factor in niche differentiation. Appl. Environ. Microbiol. 81, 745–753 (2015)
Martens-Habbena, W., Berube, P. M., Urakawa, H., de la Torre, J. R. & Stahl, D. A. Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria . Nature 461, 976–979 (2009)
Prosser, J. I. & Nicol, G. W. Archaeal and bacterial ammonia-oxidisers in soil: the quest for niche specialisation and differentiation. Trends Microbiol. 20, 523–531 (2012)
Lebedeva, E. V. et al. Enrichment and genome sequence of the group I.1a ammonia-oxidizing Archaeon “Ca. Nitrosotenuis uzonensis” representing a clade globally distributed in thermal habitats. PLoS One 8, e80835 (2013)
Widdel, F. Anaerober Abbau von Fettsäuren und Benzoesäure durch neu isolierte Arten Sulfat-reduzierender Bakterien. PhD thesis, Univ. Göttingen (1980)
Kandeler, E. & Gerber, H. Short-term assay of soil urease activity using colorimetric determination of ammonium. Biol. Fertil. Soils 6, 68–72 (1988)
Hood-Nowotny, R., Hinko-Najera Umana, N., Inselbacher, E., Oswald-Lachouani, P. & Wanek, W. Alternative methods for measuring inorganic, organic, and total dissolved nitrogen in soil. Soil Sci. Soc. Am. J . 74, 1018–1027 (2010)
Griess-Romijn van Eck, E. Physiological and chemical tests for drinking water. NEN 1056 IV-2 Nederlands Normalisatie Instituut, Rijswijk, The Netherlands (1966)
Miranda, K. M., Espey, M. G. & Wink, D. A. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 5, 62–71 (2001)
Daims, H., Stoecker, K. & Wagner, M. in Molecular Microbial Ecology (eds Osborn, A. M. & Smith, C. J. ) 213–239 (Bios-Garland, 2005)
Daims, H., Brühl, A., Amann, R., Schleifer, K.-H. & Wagner, M. The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444 (1999)
Amann, R. I. et al. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56, 1919–1925 (1990)
Daims, H. Use of fluorescence in situ hybridization and the daime image analysis program for the cultivation-independent quantification of microorganisms in environmental and medical samples. Cold Spring Harb. Protoc . 2009, t5253 (2009)
Daims, H., Lücker, S. & Wagner, M. daime, a novel image analysis program for microbial ecology and biofilm research. Environ. Microbiol. 8, 200–213 (2006)
Sorokin, D. Y. et al. Nitrification expanded: discovery, physiology and genomics of a nitrite-oxidizing bacterium from the phylum Chloroflexi . ISME J. 6, 2245–2256 (2012)
Rotthauwe, J.-H., Witzel, K.-P. & Liesack, W. The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microbiol. 63, 4704–4712 (1997)
Tourna, M., Freitag, T. E., Nicol, G. W. & Prosser, J. I. Growth, activity and temperature responses of ammonia-oxidizing archaea and bacteria in soil microcosms. Environ. Microbiol. 10, 1357–1364 (2008)
Ochsenreiter, T., Selezi, D., Quaiser, A., Bonch-Osmolovskaya, L. & Schleper, C. Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ. Microbiol. 5, 787–797 (2003)
Angel, R., Claus, P. & Conrad, R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 6, 847–862 (2012)
Angel, R. Total nucleic acid extraction from soil. Protoc. Exch . http://dx.doi.org/10.1038/protex.2012.046 (2012)
Watson, M. et al. poRe: an R package for the visualization and analysis of nanopore sequencing data. Bioinformatics 31, 114–115 (2015)
Hackl, T., Hedrich, R., Schultz, J. & Förster, F. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011 (2014)
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010)
Dupont, C. L. et al. Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J. 6, 1186–1199 (2012)
Huson, D. H., Mitra, S., Ruscheweyh, H. J., Weber, N. & Schuster, S. C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560 (2011)
Gregor, I., Dröge, J., Schirmer, M., Quince, C. & McHardy, A. C. PhyloPythiaS+: A self-training method for the rapid reconstruction of low-ranking taxonomic bins from metagenomes. Preprint at http://arxiv.org/abs/1406.7123 (2014)
Dick, G. J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009)
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012)
Boetzer, M. & Pirovano, W. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 211 (2014)
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013)
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015)
Vallenet, D. et al. MicroScope—an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 41, D636–D647 (2013)
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014)
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014)
Benson, D. A. et al. GenBank. Nucleic Acids Res. 43, D30–D35 (2015)
Markowitz, V. M. et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567 (2014)
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990)
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010)
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002)
Lartillot, N., Lepage, T. & Blanquart, S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25, 2286–2288 (2009)
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004)
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004)
Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012)
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl Acad. Sci. USA 106, 19126–19131 (2009)
The R Project. R: A language and environment for statistical computing. https://www.r-project.org/ (R Foundation for Statistical Computing, 2013)
Acknowledgements
We thank T. K. Lee and M. Steinberger for help with PCR analyses, N. V. Grigor’eva and M. Pogoda for assistance with culture maintenance, N. A. Kostrikina for assistance with electron microscopy, K. Kitzinger for support with FISH analyses, M. Mooshammer for help with chemical analyses, R. Hatzenpichler for designing probe Nmir1009, K. Eismann for help with proteome sample preparation, B. Scheer for help with mass spectrometer maintenance, Purena GmbH (Wolfenbüttel, Germany) for cooperation, N. Chernyh and J. Rosenthal for taking samples, and H. Koch and E. Bock for discussion. The authors are grateful for using the analytical facilities of the Centre for Chemical Microscopy (ProVIS) (Helmholtz Centre for Environmental Research), which is headed by H. Richnow (Department of Isotope Biochemistry) and supported by European Regional Development Funds (EFRE–Europe funds Saxony) and the Helmholtz Association. P.P. and H.D. were supported by the Austrian Science Fund (FWF) projects P27319-B21 and P25231-B21 (to H.D.). M.P., J.V., P.H., and M.W. were supported by the European Research Council Advanced Grant project NITRICARE 294343 (to M.W.). M.A., R.H.K., and P.H.N. were supported by the Danish Council for Independent Research, DFF – 4005-00369 and Innovation Fund Denmark (EcoDesign).
Author information
Authors and Affiliations
Contributions
H.D. did (meta)genomic analysis of Ca. N. inopinata and comammox Nitrospira, contributed to phylogenetic and proteomics data analyses, designed the study and wrote the paper; E.V.L. enriched Ca. N. inopinata; E.V.L., P.P., P.H., A.B. and M.P. performed physiological experiments, analysed data, and characterized enrichments; M.A., R.H.K. and P.H.N. carried out metagenome sequencing, assembly and binning; C.H. performed bioinformatic and phylogenetic analyses; N.J. and M.vB. performed proteomics measurements and data analysis; T.R. performed bioinformatic analyses; P.H., M.P. and J.V. maintained enrichment cultures and performed experiments; J.V. carried out database analyses; B.B. organized sampling and characterized environmental samples; M.W. designed the study, analysed data, and wrote the paper. All authors discussed the results and commented the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Photomicrographs and cell diagram of Ca. Nitrospira inopinata.
a, Transmission electron micrograph of a spiral-shaped cell with a flagellum. The size of Ca. N. inopinata cells is 0.18 to 0.3 μm in width and 0.7 to 1.6 μm in length. Scale bar represents 200 nm. b, Transmission electron micrograph of a thin section preparation. Microcolony showing the wide periplasmic space (PS), which is a characteristic feature of Nitrospira15. Scale bar represents 200 nm. c, Fluorescence image of cells from enrichment ENR4 after hybridization with oligonucleotide probes targeting Nitrospira (Ntspa662 and Ntspa712 both labelled with Cy3, red), the betaproteobacterium (Nmir1009 labelled with Cy5, blue), and Bacteria (EUB338 probe mix labelled with FLUOS, green). Ca. N. inopinata cells and microcolonies appear yellow and the betaproteobacterial cells appear cyan due to simultaneous hybridization to the respective specific probe and the EUB338 probe mix. Scale bar represents 2 μm. d, Cell metabolic cartoon constructed from the annotation of the Ca. N. inopinata genome. Enzyme complexes of the electron transport chain are labelled by Roman numerals.
Extended Data Figure 2 Sequence composition-independent binning of the metagenome scaffolds from the nitrifying enrichment cultures.
Circles represent scaffolds, scaled by the square root of their length. Only scaffolds ≥5 kbp are shown. Clusters of similarly coloured circles represent potential genome bins. These differential coverage plots were the starting points for further refinement and finishing of genome assemblies as described elsewhere23. a, binning of the scaffolds from enrichment culture ENR4 containing Ca. N. inopinata and three heterotrophic populations related to the Betaproteobacteria, Alphaproteobacteria, and Actinobacteria. b, binning of the scaffolds from enrichment culture ENR6 containing only Ca. N. inopinata and the betaproteobacterial accompanying heterotrophic organism. Enrichment ENR4, sample A, was used for comparison in differential coverage binning of culture ENR6.
Extended Data Figure 3 Circular representation of the Ca. N. inopinata chromosome.
Predicted coding sequences (CDS; rings 1+2), genes of enzymes involved in nitrification and other pathways of catabolic nitrogen metabolism (ring 3), RNA genes (ring 4), and local nucleotide composition measures (rings 5+6) are shown. Very short features were enlarged to enhance visibility. Clustered genes, such as several transfer RNA genes, may appear as one line owing to space limitations. The tick interval is 0.2 Mbp. Amo, ammonia monooxygenase; HAO, hydroxylamine dehydrogenase; CycA and CycB, tetraheme c-type cytochromes that form the hydroxylamine ubiquinone redox module together with HAO; NirK, Cu-dependent nitrite reductase; Nrf, cytochrome c nitrite reductase; Nxr, nitrite oxidoreductase; Orf, open reading frame.
Extended Data Figure 4 Phylogenetic affiliation of Ca. N. inopinata.
The maximum likelihood tree, which is based on 16S ribosomal RNA sequences of cultured and uncultured representative members of the genus Nitrospira, shows that the comammox organism Ca. N. inopinata (highlighted green) is a member of Nitrospira lineage II. Another 16S rRNA gene sequence was extracted from MBR metagenomic Nitrospira bin 1 (also highlighted green). This sequence bin also contained amo and hao genes (main text Fig. 1, Extended Data Figs 8 and 9). The cultured Nitrospira strains other than Ca. N. inopinata, which are not known to use ammonia as a source of energy and reductant, are highlighted blue. Nitrospira lineages are labelled red. Pie charts indicate statistical support of branches based on maximum likelihood (ML; 1,000 bootstrap iterations) and Bayesian inference (BI; posterior probability, 4 independent chains). In total, 95 taxa and 1,543 nucleotide sequence alignment positions were considered. Numbers in wedges indicate the numbers of taxa. The scale bar indicates 0.1 estimated substitutions per nucleotide.
Extended Data Figure 5 Phylogeny of NXR from Ca. N. inopinata and related proteins.
a, b, Maximum likelihood trees showing the alpha (a) and beta (b) subunits of selected enzymes from the DMSO reductase type II family. Names of validated enzymes are indicated (Clr, chlorate reductase; Ddh, dimethylsulfide dehydrogenase; NAR, nitrate reductase; NXR, nitrite oxidoreductase; Pcr, perchlorate reductase; Ser, selenate reductase). More distantly related molybdoenzymes were used as outgroup. Black dots on branches indicate high maximum likelihood bootstrap support (≥90%; 1,000 iterations). Known NXR forms are highlighted in red. The inset in a contains a subtree, which shows the phylogenetic affiliation of the NAR of the betaproteobacterium from enrichments ENR4 and ENR6 (highlighted in blue) with canonical nitrate reductases of Proteobacteria. In total, 1,279 (a) and 556 (b) amino acid sequence alignment positions, and 134 (a) and 99 (b) taxa (including outgroups), were considered. c, d, Maximum likelihood trees showing only Nitrospira NxrA (c) and nxrB (d) phylogenies. The tree in d was calculated using nucleotide sequences aligned according to their amino acid translations. Ca. N. inopinata is highlighted in red, sequences from metagenomic Nitrospira bins obtained in this study are highlighted in green. Asterisks mark metagenomic bins that also contain amo genes. Metagenomic bins are numbered as in Supplementary Table 8. Sublineages of the genus Nitrospira are indicated. As recognized earlier8, lineage II is paraphyletic with respect to lineage I in nxrB phylogenies, but differentiation of the lineages is stable. Pie charts indicate statistical support of branches based on maximum likelihood (ML; 1,000 bootstrap iterations) and Bayesian inference (BI; posterior probability, 3 independent chains). In total, 1,279 amino acid sequence alignment positions (c) and 1,290 nucleotide sequence alignment positions (d), and 30 (c) and 40 (d) taxa (including outgroups), were considered. All panels: numbers in or next to wedges indicate the numbers of taxa. The scale bars indicate 0.1 estimated substitutions per residue.
Extended Data Figure 6 Absence of nitrifying activity in the betaproteobacterium found in enrichments ENR4 and ENR6.
a, b, Incubation of a pure culture of the betaproteobacterium in mineral medium containing 1 mM ammonium (a) or 0.5 mM nitrite plus 0.1 mM ammonium as nitrogen source (b). No conversion of ammonium to nitrite or nitrate, or of nitrite to nitrate, was observed. Data points in a and b show means, error bars show 1 s.d. of n = 3 biological replicates. If not visible, error bars are smaller than symbols. The mean initial densities of the cultures, as determined by qPCR of the single-copy soxB gene, were 7.15 ± 0.01 (log(soxB copies) ml−1, 1 s.d., n = 3) for the 1 mM ammonium experiment (a) and 7.22 ± 0.02 (log(soxB copies) ml−1, 1 s.d., n = 3) for the 0.5 mM nitrite plus 0.1 mM ammonium experiment (b). After 48 h of incubation, the mean densities were 7.06 ± 0.10 and 7.15 ± 0.29, respectively. A slight decrease in the ammonium concentration was observed in these experiments and also in an abiotic control incubation containing only medium and 1 mM ammonium, but no cells (data points for this control show means of two technical replicates). It might be explained by adsorption of ammonium to the glass bottles or by outgassing of NH3. c, Photographs of incubation bottles after 53 h of incubation. The mean optical density at 600 nm (OD600) of the cultures at this time point was 0.006 ± 0.003 (1 s.d., n = 3) for the 1 mM ammonium experiment and 0.007 ± 0.008 (1 s.d., n = 3) for the 0.5 mM nitrite plus 0.1 mM ammonium experiment. Control incubations were carried out in medium containing 4 mM acetate and 0.1 mM ammonium as nitrogen source for assimilation (three biological replicates). The inoculum for these cultures was 2.5-fold diluted compared to the experiments with ammonium or nitrite. After incubation, the acetate-grown cultures were visibly turbid with a mean OD600 of 0.068 ± 0.011 (1 s.d., n = 3) and the mean density was 8.12 ± 0.03 (log(soxB copies) ml−1, 1 s.d., n = 3). Thus, the culture of the betaproteobacterium, which was used to inoculate all experiments, was physiologically active and grew on acetate. d, Fluorescence images showing the culture of the betaproteobacterium after FISH with the EUB338 probe mix (labelled with FLUOS, green), probe Nmir1009 that is specific for this organism (labelled with Cy3, red), and DAPI counterstaining (blue). The images show the same field of view after splitting the colour channels. According to FISH, all detected cells were the betaproteobacterium.
Extended Data Figure 7 Protein abundance levels of Ca. N. inopinata during growth on ammonia.
Displayed are the 450 most abundant proteins from Ca. N. inopinata in the metaproteome from culture ENR4 after incubation with 1 mM ammonium for 48 h. Red arrows and labels highlight key proteins for ammonia and nitrite oxidation. Columns show the mean normalized spectral abundance factor (NSAF), error bars show 1 s.d. of n = 4 biological replicates. In total 1,083 proteins in the metaproteome were unambiguously assigned to Ca. N. inopinata. Only one of the four putative NXR gamma subunits (NxrC) was among the top 450 expressed proteins. The other three NxrC candidates ranked at positions 561, 605 and 931. The AmoE1 protein was ranked at position 520, and HaoB at position 653.
Extended Data Figure 8 Phylogenetic affiliation of comammox amoA sequences to amoA sequences from different environments.
Bayesian inference tree showing the phylogenetic relationship of the amoA sequences from Ca. N. inopinata and metagenomic bins from this study (224 taxa, 939 nucleotide alignment positions). Ca. N. inopinata clusters confidently into comammox amoA clade A. Comammox amoA clade B (116 taxa) has been collapsed for clarity and the proportion of database sequences from soil (95 taxa), freshwater (13 taxa), and engineered environments (4 taxa) is represented as a proportion of the collapsed clade. AmoA from the metagenomic Nitrospira bins generated for this study (5 taxa in clade A, 4 taxa in clade B) are numbered as in Supplementary Table 8. Scale bar indicates estimated change per nucleotide. The outgroup consists of 27 betaproteobacterial amoA and 29 diverse pmoA sequences.
Extended Data Figure 9 Phylogenetic relationship of comammox amoB, amoC and hao sequences to corresponding gene family members.
Trees were calculated with PhyloBayes using nucleotide sequences aligned according to their amino acid translations. Support values indicate the consensus probability from 5 independent chains. Sequences outside the comammox clades are coloured as in main text Fig. 3. Metagenomic bins are numbered as in Supplementary Table 8. Scale bars indicate the estimated substitutions per nucleotide. a, Phylogenetic relationship of Ca. N. inopinata amoB to other amoB and pmoB genes (57 taxa, 1,518 alignment positions). b, Phylogenetic relationship of Ca. N. inopinata amoC to other amoC and pmoC genes (81 taxa, 993 alignment positions). c, Phylogenetic relationship of Ca. N. inopinata hydroxylamine dehydrogenase (hao) to other hao genes (37 taxa, 2,875 alignment positions).
Extended Data Figure 10 Genome-wide tetranucleotide analysis of Ca. N. inopinata and other Nitrospira.
Correlation of tetranucleotide patterns in a 5 kb sliding window (step size 1 kb) against genome-wide tetranucleotide signatures. The positions of key nitrification genes are indicated. Regions where the tetranucleotide patterns significantly deviate from the genome-wide signature, and nitrification genes located in such regions, are highlighted in green. Asterisks mark genes that are outside significantly deviating regions but may appear to be inside due to space limitations in the figure. a, Ca. N. inopinata (member of Nitrospira lineage II). The hao, cycA, and cycB genes are located in a region whose tetranucleotide pattern deviates slightly but not significantly from the genome-wide signature. The P value cutoff from the Benjamini–Hochberg procedure, indicating a significantly low correlation for a window’s tetranucleotide signature, was 0.00065 for this genome. b, N. moscoviensis (member of Nitrospira lineage II). The P value cutoff for this genome was 0.0013. c, N. defluvii (member of Nitrospira lineage I). The P value cutoff for this genome was 0.00072. In N. moscoviensis (b) and N. defluvii (c), all nxr genes are outside regions with significantly deviating tetranucleotide patterns.
Supplementary information
Supplementary Tables
This file contains Supplementary Tables 1-3 and Supplementary Table 8. (PDF 242 kb)
Supplementary Table 4
This file contains Supplementary Table 4, which lists marker genes and their copy numbers detected by CheckM in the closed Ca. N. inopinata genome. (XLSX 17 kb)
Supplementary Table 5
This file contains Supplementary Table 5, which lists marker genes and their copy numbers detected by CheckM in the genome of the betaproteobacterium found in enrichment cultures ENR4 and ENR6. (XLSX 26 kb)
Supplementary Table 6
This file contains Supplementary Table 6, which lists marker genes and their copy numbers detected by CheckM in the genome of the alphaproteobacterium found in enrichment culture ENR4. (XLSX 26 kb)
Supplementary Table 7
This file contains Supplementary Table 7, which lists marker genes and their copy numbers detected by CheckM in the genome of the actinobacterium found in enrichment culture ENR4. (XLSX 17 kb)
Rights and permissions
About this article

Cite this article
Daims, H., Lebedeva, E., Pjevac, P. et al. Complete nitrification by Nitrospira bacteria. Nature 528, 504–509 (2015). https://doi.org/10.1038/nature16461
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature16461
- Springer Nature Limited