
Abstract
Cellular damage to renal tubular epithelial cells (TECs) is commonly seen in the pathogenesis of renal pathologies, including various forms of acute kidney injury (AKI) and kidney diseases; however, the mechanisms of TEC injury are not fully understood. This chapter summarizes recent advances in understanding the impact of oxidative stress on these renal pathologies with a major focus on renal TECs in the cortex, the site in the kidney of high oxidative metabolic activity, and we also discuss the outcomes of antioxidant therapy in renal pathologies. Recent literature indicates that oxidative stress, induced by the excess levels of reactive oxygen species (ROS) from mitochondrial injury and/or nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, mediates cellular oxidative damage in AKI, nephrolithiasis, and diabetic nephropathy, and that antioxidant therapy has varying degrees of success depending on the bioavailability and activity specificity of the antioxidant agent. The hypothesis is that the oxidative stress is a common feature for these renal pathologies, and ROS-activated molecular pathways in TECs are potential targets for antioxidant therapeutic approaches that can slow the progression of AKI and kidney diseases.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
The nephron is a functional unit of the kidney and is composed of a renal corpuscle and a tubule; therefore, the homeostasis of renal tubular epithelial cells (TECs) is essential for kidney function. Indeed, tubular atrophy is a fundamental part of kidney transplant rejection (Ganji and Harririan 2012; Pascual et al. 2012), chronic kidney diseases (Farris and Colvin 2012) including diabetic nephropathy (Najafian et al. 2011), and nephrolithiasis (Evan et al. 2005), but the pathways for TEC death in these pathophysiological conditions are not fully understood. It has been well documented that under oxidative stress, reactive oxygen species (ROS) cause cellular damage by reacting with and denaturing cellular macromolecules including lipid, protein, and nucleic acids and/or even mediating or activating intracellular death signaling pathways (Rhee 1999). Our hypothesis is that oxidative stress is a common pathway mediating cell death of TECs that have a high oxidative metabolic activity in the kidney. Hence, identification or dissection of this pathway may be critical for understanding the susceptibility of the kidney to nephrotoxic drugs/chemicals, infection/immune response, trauma, mineral crystal aggregation (kidney stone), and hyperglycemia and for developing therapeutic solutions to these renal disorders.
Cellular ROS Generation in TECs
During the course of normal metabolism in cells, superoxide (O2 •−), the precursor of ROS, can be produced as inevitable by-products from energy production within the mitochondria. Such ROS production can be normally neutralized by natural antioxidant enzymes but causes oxidative stress under the condition of mitochondrial injury (Rocha et al. 2010). In addition to mitochondria, there are many cytosolic enzyme systems (e.g., cytochrome p450s, xanthine oxidase, 5-lipoxygenase) that produce O2 •−, but the major sources of this molecule production in response to stimuli including inflammation (e.g., hypoxia, immunologic stimuli) are nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in both professional phagocytes and non-phagocytic cells (Babior 1999; Forman and Torres 2001; Novo and Parola 2008).
NADPH oxidase transfers electrons from NADPH to molecular oxygen, which results in generation of ROS. The classic phagocytic NADPH oxidase is a complex of membrane-bound cytochrome b 558, cytosolic factors p47phox, p67phox, and p40phox, and a small GTPase Rac2. The heme-binding component of cytochrome b 558 is a complex of gp91phox (91 kDa subunit of the phagocyte oxidase or NOX2) and glycosylated flavoprotein associated with p22phox (Babior 1999). NOX proteins are the catalytic subunit of NADPH oxidase. In non-phagocytic NADPH oxidase, several members of the NOX family (i.e., NOX1, NOX3, NOX4, NOX5, or DUOX1/2) have been identified (Krause 2004; Novo and Parola 2008). Different NOX proteins vary in tissue distribution in light of their varying functions (Bedard and Krause 2007). The kidney, particularly its cortical substance, expresses a high level of NOX4 (Chabrashvili et al. 2002; Geiszt et al. 2000; Shiose et al. 2001), which has been confirmed in cultured proximal TECs (Geiszt et al. 2000; Reeves et al. 2002). These studies suggest that NOX4-containing NADPH oxidase in TECs plays a pivotal role in the generation of oxidative stress or ROS in the kidney in response to inflammation and cellular stress.
Association of Oxidative Stress with Renal Pathologies
Drug/Chemical-Induced Acute Kidney Injury
Drugs are one of common factors causing damage to the kidney, which contributes to approximately 20 % of acute kidney injury (AKI) episodes (Bellomo 2006; Kaufman et al. 1991; Nash et al. 2002). Many of these drugs, such as aminoglycoside, pentamidine, cisplatin, and antiretrovirals, directly induce renal TEC necrosis, or others, such as penicillin vancomycin, lansoprazole, and thiazides, cause renal interstitial inflammation or immune-mediated tubular injury (Naughton 2008). Renal proximal TECs are a primary target of these nephrotoxic drugs because of their role in concentrating and absorbing glomerular filtrates including these nephrotoxic drugs (Perazella 2005). Oxidative stress is a common result of nephrotoxicant exposure in TECs (Baliga et al. 1997) and is induced by either mitochondrial injury or activation of NADPH oxidase. For example, the nephrotoxicity of gentamicin, cephalosporin, and aristolochic acid is associated with tubular cell death and oxidative stress (Kiyomiya et al. 2002; Pozdzik et al. 2008; Randjelovic et al. 2012), which is due to the mitochondrial injury (Pozdzik et al. 2008; Zorov 2010) or, in particular, depletion of cytochrome c in the respiratory chain of the mitochondria (Kiyomiya et al. 2002; Morales et al. 2010), whereas cisplatin-induced TEC injury is associated with oxidative stress via activation of NADPH oxidase (Kawai et al. 2006) or upregulation of NOX4 expression (Pan et al. 2009), suggesting that potential antioxidative approaches to preventing drug/chemical-induced AKI should be designed differently depending on the sources of oxidative stress, mitochondrial injury, or NADPH oxidase.
Ischemic AKI or Renal Ischemia-Reperfusion Injury
Ischemia is the leading cause of AKI, as see in many clinical settings including renal transplantation, shock, and vascular surgery (Gueler et al. 2004). Clinical and experimental studies show that renal tissue damage that occurs following ischemia-reperfusion, especially during reperfusion, is due in part to excessive production of ROS (Kaminski et al. 2002; Kim et al. 2006; McCord 1985), evidenced by the increased formation of lipid hydroperoxides and other toxic products after such an injury (Beckman et al. 1991) and by the beneficial effects of antioxidant treatment on reducing ischemia-reperfusion injury (IRI) in both in vitro and in vivo systems (Nitescu et al. 2006). The molecular pathways of inducing oxidative stress in the kidney following ischemia-reperfusion are complicated as many enzymes, including oxidases and antioxidant enzymes, are involved; early studies have shown that during ischemia the content of the antioxidants glutathione (GSH), superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GSH-Px) are decreased (Dobashi et al. 2000; Gonzalez-Flecha et al. 1993; Scaduto et al. 1988), while there is increased activation of xanthine oxidase (Sanhueza et al. 1992). The contribution of loss of antioxidant enzyme SOD to oxidative stress is further supported by the fact of worsening renal IRI in CuZnSOD (SOD1)- or extracellular SOD (SOD3)-deficient mice (Schneider et al. 2010; Yamanobe et al. 2007). The contribution of mitochondrial injury to the increase in renal oxidative stress during renal IRI has been reported in many studies (Cruthirds et al. 2003; Plotnikov et al. 2007; Sun et al. 2008) and also seen in a model of cold IRI (Saba et al. 2008), which is further supported by the beneficial effect of mitochondria-targeted antioxidants on the reduction of oxidative stress and renal IRI (Plotnikov et al. 2011; Szeto et al. 2011). On the other hand, there are not many studies in the literature showing the contribution of NADPH oxidase to oxidative stress in renal IRI. A recent study reports that NADPH oxidase (NOX4) interacts with Toll-like receptor (TLR) 4 and is required for hypoxia-induced production of ROS and cell death in renal IRI (Ben Mkaddem et al. 2010).
Septic AKI
In the intensive care unit, sepsis often leads to AKI that occurs in about 19 % of patients with moderate sepsis, 23 % with severe sepsis, and 51 % with septic shock (Rangel-Frausto et al. 1995) and is associated with oxidative stress, such as decreased total antioxidative capacity and/or increased lipid peroxidation (Roth et al. 2004; Ware et al. 2011). In animal models of sepsis-induced AKI, the tubular epithelium is the major injury target in the kidney that loses renal function as sepsis progresses (Guo et al. 2004; Wu et al. 2007; Yasuda et al. 2006). It has been proposed that ROS in combination with reactive nitrogen species (RNS) contribute to TEC injury during sepsis (Heyman et al. 2011; Wu et al. 2007; Wu and Mayeux 2007). This notion is supported by both clinical and experimental studies: the presence of nitrated plasma proteins is correlated with chronic renal failure in patients with septic shock (Fukuyama et al. 1997), nitrated proteins and oxidation products are seen in the kidney during lipopolysaccharide-induced renal injury in rats (Bian et al. 1999; Zhang et al. 2000), and, more importantly, septic AKI is prevented with antioxidant therapies, such as N-acetylcysteine (NAC) treatment (Campos et al. 2012). Fluorescence microscopic analysis analysis demonstrates that endotoxin is internalized by S1 proximal tubules through local TLR4 receptors and fluid-phase endocytosis but only induces oxidative stress in neighboring S2 tubules (Kalakeche et al. 2011). The molecular mechanisms by which sepsis induces oxidative stress in the kidney includes decreased antioxidant capacity, such as loss of SOD3 expression (Wang et al. 2003) or CAT (Perianayagam et al. 2007), mitochondrial dysfunction (Pathak et al. 2012), and/or activation of NADPH oxidase (Ben Mkaddem et al. 2010; Lanone et al. 2005; Perianayagam et al. 2007).
Nephrolithiasis
Nephrolithiasis, also known as kidney stones, is characterized by the formation of solid crystal aggregates in the kidney. Many clinical studies have reported that the incidence of nephrolithiasis is associated with many chronic inflammatory diseases (e.g., cardiovascular disease and type 2 diabetes) and systemic oxidative stress (Khan 2012; Lange et al. 2012; Tsao et al. 2007). Oxalate is a primary element most common in kidney stones. In a mouse model, the early stages of calcium oxalate crystal formation in the tubular lumen is associated with TEC injury, especially mitochondrial damage, and oxidative stress (Hirose et al. 2010). Similar data were reported in a rat model, where oxidative injury in the kidney correlated with hyperplasia of mitochondria in TECs as well as with decreased activities of SOD and GSH-Px in the mitochondria, but not with expression of NADPH oxidase subunits p47phox and NOX4 (Li et al. 2009). These studies suggest that mitochondrial injury may account for oxidative stress in the kidney with nephrolithiasis, which is further supported by an in vitro study in cultures of proximal (LLC-PK1) and distal (MDCK) tubular epithelial cells, where the intracellular O2 •− from mitochondrial sources is significantly elevated following treatment with calcium oxalate monohydrate (Khand et al. 2002). However, in cell cultures of NRK52E, a TEC line, calcium oxalate monohydrate upregulates p47phox expression and inhibition of NADPH oxidase activity reduces induction of ROS (Umekawa et al. 2009). The translocation of protein kinase C (PKC)-alpha and (PKC)-delta from the cytosol to the cell membrane, following Rac1 GTPase signaling, mediates oxalate-induced generation of ROS along with NADPH oxidase activity, lipid hydroperoxide formation, and cell death in LLC-PK1 cells (Thamilselvan et al. 2009, 2012). These studies suggest that both mitochondrial injury and NADPH oxidase may induce oxidative stress in the kidney in response to calcium oxalate crystal formation.
Diabetic Nephropathy
Diabetic nephropathy is the leading cause of end-stage renal disease in the developed world, but the mechanisms underlying hyperglycemia-induced renal injury are not fully understood. It has been suggested that the kidney normally filters ∼180g of glucose daily, most of which is reabsorbed by means of sodium-glucose cotransporter 2 (SGLT2), expressed in proximal tubules (Mitrakou 2011). However, the capacity of the SGLT1 to reabsorb glucose from renal tubules is finite, and when plasma glucose concentrations exceed a threshold, glucose appears in the urine (Mitrakou 2011). Hyperglycemia induces oxidative stress by increasing ROS generation in diabetic kidneys (Baynes and Thorpe 1999; Matsuoka et al. 2005; Nishikawa et al. 2000). In streptozotocin (STZ)-induced or db/db diabetic mice, overexpression of SOD1 attenuates diabetic nephropathy (Craven et al. 2001; DeRubertis et al. 2004), and moderate exercise suppresses the progression of early diabetic nephropathy via upregulation of SOD expression independent of hyperglycemia (Ghosh et al. 2009). In vitro high-glucose levels significantly enhance generation of ROS, reduce levels of GSH and SOD, and increase production of malondialdehyde in TECs, glomerular mesangial cells, and glomerular epithelial cells (Jiao et al. 2011; Ogawa et al. 2011), which are in line with high glucose-induced apoptosis (Allen et al. 2003; Kitamura et al. 2009; Yoo et al. 2011). These studies suggest that hyperglycemia induces oxidative stress in the kidney, which may be a key mediator of renal tubular hypertrophy in diabetic nephropathy, and protection from ROS represents a valuable therapeutic strategy to treat diabetic nephropathy. The published data also suggest that both mitochondrial injury and NADPH oxidase activity contribute to hyperglycemia-induced ROS generation in the diabetic nephropathy; normalizing mitochondrial O2 •− production prevents hyperglycemic damage (Nishikawa et al. 2000), while in both distal tubular cells and glomeruli of the kidney in STZ-induced diabetic rats, or cultured TECs treated with high glucose, the expression of NOX4 and p22phox is significantly increased (Etoh et al. 2003; Takao et al. 2011), indicating that NADPH-dependent overproduction of ROS could cause renal tissue damage in diabetes.
Pathways Activated by Oxidative Stress in the Kidney
The mitogen-activated protein kinase (MAPK) pathway is a signaling cascade of a group of serine/threonine kinases, including Ras, Raf-1, MAPK kinase (MEK)1/2, extracellular signal-regulated kinase (ERK)1/2, p38 MAPK, and c-Jun N-terminal kinase (JNK) (Kim and Choi 2010; Seger and Krebs 1995), and has been reported to be activated by oxidative stress in the kidney; direct exposure to H2O2, Tert-butyl hydroperoxide, or O2 •− production from the catabolism of hypoxanthine by xanthine oxidase induces apoptosis in cultured TECs, which is regulated by either JNK (Wang et al. 2002) or MEK1-ERK1/2 signaling pathway (di Mari et al. 1999; Lee et al. 2006; Nowak et al. 2006). Similar results were reported in TEC cultures treated with the nephrotoxic chemical arachidonic acid that stimulates ROS generation by activation of NADPH oxidase, which subsequently activates JNK (Cui and Douglas 1997), or with high glucose that induces Raf-1 and ERK1/2 (Huang et al. 2007). In renal ischemia-reperfusion, oxidative stress-induced cell death may also be mediated by the activation of this pathway because inhibition of Ras-ERK1/2 or apoptosis signal-regulated kinase (ASK)1-MAPK kinase (MKK)3-p38 MAPK signal pathway reduces renal ischemia-reperfusion injury (Sabbatini et al. 2006; Wang et al. 2009). However, the activation of ERK1/2 signaling is not sustained under conditions of severe oxidative stress (Arany et al. 2006), suggesting that the role of MAPK pathway in ROS-mediated renal injury may depend on the severity of local oxidative stress.
In addition to the MAPK pathway, renal oxidative stress also activates JAK-STAT signaling that mainly consists of a receptor, Janus kinase (JAK) and signal transducer and activator of transcription (STAT) (Aaronson and Horvath 2002). In vitro high glucose activation of the JAK2-STAT1/STAT3 pathway is associated with cell death and is blocked by cytoprotective NAC or taurine in cultured TECs (Huang et al. 2007), while inactivation of JAK2 protects TECs from H2O2 or nephrotoxic cyclosporin A (CsA)-induced cell death (Neria et al. 2009). In mouse models of renal IRI, severe oxidative stress increases tyrosine phosphorylation of STAT3 (Arany et al. 2006), and pharmacological interference of JAK2 signaling prevents renal IRI and CsA nephrotoxicity (Arany et al. 2006; Neria et al. 2009).
Several other signaling pathways have also recently been reported to mediate oxidative stress-induced TEC death. H2O2 increases prostate apoptosis response factor (Par)-4 expression in human TECs, and Par-4 silencing significantly protects TECs from apoptosis via activating the PI3K/Akt signaling pathway (Sun et al. 2011). Studies of nephrotoxic drug/chemical-induced nephrotoxic pathways using whole genome DNA microarray demonstrates that the nuclear factor erythroid 2-related factor (Nrf2) pathway is the most significant signaling response for oxidative stress in a proximal TEC cell line (HK-2) following challenges with nephrotoxins (cadmium, diquat dibromide) but not with cyclosporine A (Wilmes et al. 2011). The anticancer drug carboplatin-mediated ROS stimulation of calcineurin-NFAT3 (nuclear factor of activated T-cell 3) activation that is essential for carboplatin-mediated TEC apoptosis (Lin et al. 2010).
Antioxidant Therapies for Oxidative Stress in the Kidney
Antioxidants consist of enzymes, such as SOD, CAT, and GSH-Px, and small molecular antioxidant scavengers, such as vitamin E, N-acetylcysteine (NAC), and α-lipoic acid. These antioxidants neutralize the activity of free radicals in the body by donating electrons, suggesting their therapeutic potential for mitigating oxidative stress-mediated kidney diseases. Several antioxidant therapies for reducing oxidative stress in the kidney have been recently reported (Table 114.1), but many antioxidant agents, such as NAC and vitamins, have not successfully passed the scrutiny of clinical trials for prevention and treatment of renal pathologies, while others, such as allopurinol, have varying degrees of success (Table 114.1). We discuss possible reasons for the failure or the success of these antioxidant therapies.
NAC, the N-acetyl derivative of L-cysteine, is the best studied antioxidant for preventing contrast medium-induced nephropathy (CIN) and other renal pathologies in patients, but the outcomes are not satisfactory (Table 114.1). NAC is metabolized in the gut wall and liver and is converted to L-cysteine and, ultimately, from the cysteine to the antioxidant glutathione (GSH). The antioxidant activity of NAC requires prior conversion to GSH in a study using human platelets (Gibson et al. 2009). Thus, the efficacy of antioxidant therapy using NAC is dependent on the conversion or metabolism of NAC to GSH. Recently, Nolin et al. reported that the conversion of NAC to GSH is largely limited in patients with end-stage renal disease (ESRD) as compared with that in healthy control subjects (Nolin et al. 2010), which may explain the reason why NAC is not effective in patients with renal pathologies.
Vitamin E is the term for a group of tocopherols and tocotrienols, of which α-tocopherol has the highest biological activity. As an antioxidant, vitamin E is a peroxyl radical scavenger, preventing the propagation of free radicals by lipid peroxidation by reacting with them to form a tocopherol radical that will then be reduced by a hydrogen donor (such as ascorbic acid) and thus return to its reduced state (Traber and Stevens 2011). However, in addition to antioxidant/radical scavenging, vitamin E also has other biological functions, such as inhibition of protein kinase C activity, resulting in suppressing smooth muscle growth (Schneider 2005; Zingg and Azzi 2004), upregulation of connective tissue growth factor in tumor necrosis factor-treated smooth muscle cells (Villacorta et al. 2003), and inhibition of platelet aggregation (Zingg and Azzi 2004). Therefore, the outcome of antioxidant therapy using vitamin E not only is determined by its bioavailability and antioxidant activity that can be affected by the levels of ascorbic acid or other reducing agents but also is complicated by its non-antioxidant activities in the body. This concept is supported by the clinical observations of vitamin E against kidney injury as listed in Table 114.1 and many other studies demonstrating that vitamin E is not able, at physiological concentrations, to protect against oxidant-induced damage or prevent disease allegedly caused by oxidative damage (Azzi 2007).
Ascorbic acid (vitamin C) is another antioxidant that has no benefit in the prevention and treatment of kidney injury in patients (Table 114.1). This compound does not directly reduce oxidative stress. Instead, its antioxidant activity is via the interaction with vitamins E – the vitamin E redox cycle (Halliwell and Gutteridge 1999; Liebler et al. 1989). In addition to this antioxidant activity, ascorbic acid primarily acts as an essential cofactor for a number of enzymes – α-ketoglutarate-dependent dioxygenases, particularly prolyl hydroxylases that play a role in the biosynthesis of collagen and in downregulation of hypoxia-inducible factor (HIF)-1, a transcription factor that regulates many genes responsible for tumor growth, energy metabolism, and neutrophil function and apoptosis (Traber and Stevens 2011). Therefore, like vitamin E, ascorbic acid is a nonspecific antioxidant agent.
Allopurinol is a purine-like drug that reduces uric acid production from xanthine and hypoxanthine by inhibition of xanthine oxidase (XO) activity and has been used for the clinical management of gout and conditions associated with hyperuricemia for several decades (Pacher et al. 2006). Allopurinol is metabolized by XO to an active metabolite, oxypurinol, which inhibits the production of uric acid and ROS (i.e., H2O2) from XO. Because oxypurinol is eliminated in urine via the kidneys, patients with impaired renal function are prone to prolonged excretion and elevated levels of circulating oxypurinol (Elion et al. 1968), which has been reported to be closely involved with the occurrence of allopurinol hypersensitivity syndrome (Hande et al. 1984). Therefore, although allopurinol has a protective effect on renal function in patients but not in AKI (Table 114.1), as antioxidant therapy for renal pathologies, it can be limited by its high risk for allopurinol hypersensitivity syndrome and its XO target specificity – not attenuating other sources of oxidative stress. Recently, a new generation of XO inhibitor, a non-purine-selective febuxostat, has been tested in preclinical models for preventing ischemic AKI (Tsuda et al. 2012) and unilateral ureteral obstructive nephropathy (Omori et al. 2012). This new XO inhibitor may be safe for patients with mild to moderate renal impairment without dose adjustment (Tatsuo and Iwao 2011), but its antioxidant activity in patients with renal pathologies remains under investigation.
Similar to cyclopentenone prostaglandins – endogenous activators of Nrf2 pathway and inhibitors of NF-κB (Surh et al. 2011) – bardoxolone methyl (BARD) has been demonstrated to activate the Nrf2 pathway (Liby et al. 2005; Sporn et al. 2011; Yates et al. 2007) that plays a central role in the maintenance of redox balance and protection against oxidative stress. It has been recently documented that impaired Nrf2 activity is associated with renal oxidative stress and inflammation in animals with chronic kidney disease (Kim et al. 2011; Kim and Vaziri 2010; Kim et al. 2010; Yoh et al. 2001). BARD activates the Nrf2 pathway in the kidney (Reisman et al. 2012), so that treatment with BARD ameliorates ischemic AKI in experimental studies (Wu et al. 2011) and improves eGFR in patients with diabetic nephropathy (Table 114.1). However, a phase III clinical trial to evaluate safety and efficacy of BARD (20 mg/day) in patients with CKD and type 2 diabetes has been discontinued due to safety concerns related to the many adverse side effects and high mortality rates in patients receiving BARD (October 18, 2012, www.marketwatch.com). Although the mechanism of BARD toxicity in these patients is currently unknown, it is possible that BARD may not only reverse the impaired Nrf2 pathway in diseased kidneys but could also activate Nrf2 pathways in healthy organs, whereby the expression of antioxidant enzymes (e.g., NADPH quinone oxidoreductase, glutamate-cysteine ligase, heme oxygenase) could be upregulated to increase oxidative stress instead.
Taken together, evidence from these studies indicates that the efficacy of antioxidant therapy using different agents is dependent on their bioavailability and target/disease specificity. It is likely that new avenues that are have more specific targeting as antioxidants and which possess better pharmacokinetic profiles will lead the way for future treatment strategies of patients with renal diseases. This requires a great focus on designing procedures to reduce renal mitochondrial injury and the use of specific inhibitors of Nox-4-NADPH oxidase that have better improved bioavailabilities in patients with renal pathologies.
Conclusion
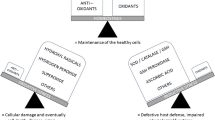
Over the past several years, both clinical and experimental studies have advanced our understanding of the pathological role of oxidative stress in the development of various renal pathologies. This review discusses the pathways that lead to the generation of ROS in the kidneys or cultured TECs following a variety of nephrotoxic insults and the pathways activated by ROS in their nephrotoxicity (Fig. 114.1). In these renal pathologies, both mitochondrial injury and NADPH oxidase are considered the main sources of ROS that activate MAPK, JAK-STAT, or other pathways. However it remains unclear whether each renal disorder has its own unique source of ROS generation. Despite the large number of studies testing antioxidant reagents in the prevention of oxidative stress-mediated renal injury, there is as yet no drug available that directly targets renal ROS activity; it is clear that this would be a valuable strategy for drug discovery to protect against drug nephrotoxicity, AKI, kidney stone or diabetes-associated kidney damage.

A simple scheme of oxidative stress in renal tubular epithelium in renal pathologies. Accumulation of superoxide (O2 •−), the precursor of ROS, results from mitochondrial dysfunction, upregulation of oxidases, and/or downregulation of antioxidant enzymes in renal tubular epithelium in the response to inflammatory stimuli, ischemia, and exposure to toxic chemicals. ROS activates or disrupts intracellular signaling pathways (e.g., MAPK), resulting in cell death. TEC tubular epithelial cells, XO xanthine oxidase, ER endoplasmic reticulum, M mitochondria
References
Aaronson DS, Horvath CM (2002) A road map for those who don't know JAK-STAT. Science 296:1653–1655
Adabag AS, Ishani A, Bloomfield HE, Ngo AK, Wilt TJ (2009) Efficacy of N-acetylcysteine in preventing renal injury after heart surgery: a systematic review of randomized trials. Eur Heart J 30:1910–1917
Allen DA, Harwood S, Varagunam M, Raftery MJ, Yaqoob MM (2003) High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J 17:908–910
Arany I, Megyesi JK, Nelkin BD, Safirstein RL (2006) STAT3 Attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int 70:669–674
Aslanger E, Uslu B, Akdeniz C, Polat N, Cizgici Y, Oflaz H (2012) Intrarenal application of N-acetylcysteine for the prevention of contrast medium-induced nephropathy in primary angioplasty. Coron Artery Dis 23:265–270
Azzi A (2007) Molecular mechanism of alpha-tocopherol action. Free Radic Biol Med 43:16–21
Babior BM (1999) NADPH oxidase: an update. Blood 93:1464–1476
Baliga R, Ueda N, Walker PD, Shah SV (1997) Oxidant mechanisms in toxic acute renal failure. Am J Kidney Dis 29:465–477
Baynes JW, Thorpe SR (1999) Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48:1–9
Beckman JK, Yoshioka T, Knobel SM, Greene HL (1991) Biphasic changes in phospholipid hydroperoxide levels during renal ischemia/reperfusion. Free Radic Biol Med 11:335–340
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Bellomo R (2006) The epidemiology of acute renal failure: 1975 versus 2005. Curr Opin Crit Care 12:557–560
Ben Mkaddem S, Pedruzzi E, Werts C, Coant N, Bens M, Cluzeaud F, Goujon JM, Ogier-Denis E, Vandewalle A (2010) Heat shock protein gp96 and NAD(P)H oxidase 4 play key roles in toll-like receptor 4-activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ 17:1474–1485
Bian K, Davis K, Kuret J, Binder L, Murad F (1999) Nitrotyrosine formation with endotoxin-induced kidney injury detected by immunohistochemistry. Am J Physiol 277:F33–40
Boscheri A, Weinbrenner C, Botzek B, Reynen K, Kuhlisch E, Strasser RH (2007) Failure of ascorbic acid to prevent contrast-media induced nephropathy in patients with renal dysfunction. Clin Nephrol 68:279–286
Campos R, Shimizu MH, Volpini RA, de Braganca AC, Andrade LC, Lopes FD, Olivo CR, Canale D, Seguro AC (2012) N-acetylcysteine prevents pulmonary edema and acute kidney injury in rats with sepsis submitted to mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 302:L604–L650
Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS (2002) Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 39:269–274
Craven PA, Melhem MF, Phillips SL, DeRubertis FR (2001) Overexpression of Cu2+/Zn2+ superoxide dismutase protects against early diabetic glomerular injury in transgenic mice. Diabetes 50:2114–2125
Cruthirds DL, Novak L, Akhi KM, Sanders PW, Thompson JA, MacMillan-Crow LA (2003) Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Arch Biochem Biophys 412:27–33
Cui XL, Douglas JG (1997) Arachidonic acid activates c-jun N-terminal kinase through NADPH oxidase in rabbit proximal tubular epithelial cells. Proc Natl Acad Sci USA 94:3771–3776
DeRubertis FR, Craven PA, Melhem MF, Salah EM (2004) Attenuation of renal injury in db/db mice overexpressing superoxide dismutase: evidence for reduced superoxide-nitric oxide interaction. Diabetes 53:762–768
di Mari JF, Davis R, Safirstein RL (1999) MAPK activation determines renal epithelial cell survival during oxidative injury. Am J Physiol 277:F195–203
Dobashi K, Ghosh B, Orak JK, Singh I, Singh AK (2000) Kidney ischemia-reperfusion: modulation of antioxidant defenses. Mol Cell Biochem 205:1–11
Elion GB, Yu TF, Gutman AB, Hitchings GH (1968) Renal clearance of oxipurinol, the chief metabolite of allopurinol. Am J Med 45:69–77
Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H, Nawata H (2003) Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia 46:1428–1437
Evan AP, Lingeman JE, Coe FL, Shao Y, Parks JH, Bledsoe SB, Phillips CL, Bonsib S, Worcester EM, Sommer AJ, Kim SC, Tinmouth WW, Grynpas M (2005) Crystal-associated nephropathy in patients with brushite nephrolithiasis. Kidney Int 67:576–591
Farris AB, Colvin RB (2012) Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens 21:289–300
Forman HJ, Torres M (2001) Redox signaling in macrophages. Mol Aspects Med 22:189–216
Fukuyama N, Takebayashi Y, Hida M, Ishida H, Ichimori K, Nakazawa H (1997) Clinical evidence of peroxynitrite formation in chronic renal failure patients with septic shock. Free Radic Biol Med 22:771–774
Ganji MR, Harririan A (2012) Chronic allograft dysfunction: major contributing factors. Iran J Kidney Dis 6:88–93
Geiszt M, Kopp JB, Varnai P, Leto TL (2000) Identification of renox, an NAD(P)H oxidase in kidney. Proc Natl Acad Sci USA 97:8010–8014
Ghosh S, Khazaei M, Moien-Afshari F, Ang LS, Granville DJ, Verchere CB, Dunn SR, McCue P, Mizisin A, Sharma K, Laher I (2009) Moderate exercise attenuates caspase-3 activity, oxidative stress, and inhibits progression of diabetic renal disease in db/db mice. Am J Physiol Renal Physiol 296:F700–708
Gibson KR, Neilson IL, Barrett F, Winterburn TJ, Sharma S, MacRury SM, Megson IL (2009) Evaluation of the antioxidant properties of N-acetylcysteine in human platelets: prerequisite for bioconversion to glutathione for antioxidant and antiplatelet activity. J Cardiovasc Pharmacol 54:319–326
Goicoechea M, de Vinuesa SG, Verdalles U, Ruiz-Caro C, Ampuero J, Rincon A, Arroyo D, Luno J (2010) Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol 5:1388–1393
Gonzalez-Flecha B, Evelson P, Sterin-Speziale N, Boveris A (1993) Hydrogen peroxide metabolism and oxidative stress in cortical, medullary and papillary zones of rat kidney. Biochim Biophys Acta 1157:155–161
Gueler F, Gwinner W, Schwarz A, Haller H (2004) Long-term effects of acute ischemia and reperfusion injury. Kidney Int 66:523–527
Guo R, Wang Y, Minto AW, Quigg RJ, Cunningham PN (2004) Acute renal failure in endotoxemia is dependent on caspase activation. J Am Soc Nephrol 15:3093–3102
Hafiz AM, Jan MF, Mori N, Shaikh F, Wallach J, Bajwa T, Allaqaband S (2012) Prevention of contrast-induced acute kidney injury in patients with stable chronic renal disease undergoing elective percutaneous coronary and peripheral interventions: randomized comparison of two preventive strategies. Catheter Cardiovasc Interv 79:929–937
Halliwell B, Gutteridge JM (1999) Free radicals in biology and medicine. Oxford University Press, New York
Hande KR, Noone RM, Stone WJ (1984) Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med 76:47–56
Heyman SN, Rosen S, Rosenberger C (2011) A role for oxidative stress. Contrib Nephrol 174:138–148
Hilmi IA, Peng Z, Planinsic RM, Damian D, Dai F, Tyurina YY, Kagan VE, Kellum JA (2010) N-acetylcysteine does not prevent hepatorenal ischaemia-reperfusion injury in patients undergoing orthotopic liver transplantation. Nephrol Dial Transplant 25:2328–2333
Hirose M, Yasui T, Okada A, Hamamoto S, Shimizu H, Itoh Y, Tozawa K, Kohri K (2010) Renal tubular epithelial cell injury and oxidative stress induce calcium oxalate crystal formation in mouse kidney. Int J Urol 17:83–92
Huang JS, Chuang LY, Guh JY, Huang YJ, Hsu MS (2007) Antioxidants attenuate high glucose-induced hypertrophic growth in renal tubular epithelial cells. Am J Physiol Renal Physiol 293:F1072–1082
Jaffery Z, Verma A, White CJ, Grant AG, Collins TJ, Grise MA, Jenkins JS, McMullan PW, Patel RA, Reilly JP, Thornton SN, Ramee SR (2012) A randomized trial of intravenous n-acetylcysteine to prevent contrast induced nephropathy in acute coronary syndromes. Catheter Cardiovasc Interv 79:921–926
Jiao B, Wang YS, Cheng YN, Gao JJ, Zhang QZ (2011) Valsartan attenuated oxidative stress, decreased MCP-1 and TGF-beta1 expression in glomerular mesangial and epithelial cells induced by high-glucose levels. Biosci Trends 5:173–181
Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, Sandoval RM, Dagher PC (2011) Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol 22:1505–1506
Kamgar M, Zaldivar F, Vaziri ND, Pahl MV (2009) Antioxidant therapy does not ameliorate oxidative stress and inflammation in patients with end-stage renal disease. J Natl Med Assoc 101:336–344
Kaminski KA, Bonda TA, Korecki J, Musial WJ (2002) Oxidative stress and neutrophil activation-the two keystones of ischemia/reperfusion injury. Int J Cardiol 86:41–59
Kaufman J, Dhakal M, Patel B, Hamburger R (1991) Community-acquired acute renal failure. Am J Kidney Dis 17:191–198
Kawai Y, Nakao T, Kunimura N, Kohda Y, Gemba M (2006) Relationship of intracellular calcium and oxygen radicals to cisplatin-related renal cell injury. J Pharmacol Sci 100:65–72
Khan SR (2012) Is oxidative stress, a link between nephrolithiasis and obesity, hypertension, diabetes, chronic kidney disease, metabolic syndrome? Urol Res 40:95–112
Khand FD, Gordge MP, Robertson WG, Noronha-Dutra AA, Hothersall JS (2002) Mitochondrial superoxide production during oxalate-mediated oxidative stress in renal epithelial cells. Free Radic Biol Med 32:1339–1350
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802:396–405
Kim HJ, Vaziri ND (2010) Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 298:F662–671
Kim J, Kil IS, Seok YM, Yang ES, Kim DK, Lim DG, Park JW, Bonventre JV, Park KM (2006) Orchiectomy attenuates post-ischemic oxidative stress and ischemia/reperfusion injury in mice. A role for manganese superoxide dismutase. J Biol Chem 281:20349–20356
Kim J, Cha YN, Surh YJ (2010) A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res 690:12–23
Kim HJ, Sato T, Rodriguez-Iturbe B, Vaziri ND (2011) Role of intrarenal angiotensin system activation, oxidative stress, inflammation, and impaired nuclear factor-erythroid-2-related factor 2 activity in the progression of focal glomerulosclerosis. J Pharmacol Exp Ther 337:583–590
Kitamura O, Uemura K, Kitamura H, Sugimoto H, Akaike A, Ono T (2009) Serofendic acid protects from iodinated contrast medium and high glucose probably against superoxide production in LLC-PK1 cells. Clin Exp Nephrol 13:15–24
Kitzler TM, Jaberi A, Sendlhofer G, Rehak P, Binder C, Petnehazy E, Stacher R, Kotanko P (2012) Efficacy of vitamin E and N-acetylcysteine in the prevention of contrast induced kidney injury in patients with chronic kidney disease: a double blind, randomized controlled trial. Wien Klin Wochenschr 124:312–319
Kiyomiya K, Matsushita N, Kurebe M, Nakagawa H, Matsuo S (2002) Mitochondrial cytochrome c oxidase as a target site for cephalosporin antibiotics in renal epithelial cells (LLC-PK(1)) and renal cortex. Life Sci 72:49–57
Krause KH (2004) Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn J Infect Dis 57:S28–29
Lange JN, Mufarrij PW, Wood KD, Holmes RP, Assimos DG (2012) The association of cardiovascular disease and metabolic syndrome with nephrolithiasis. Curr Opin Urol 22:154–159
Lanone S, Bloc S, Foresti R, Almolki A, Taille C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, El-Benna J, Motterlini R, Boczkowski J (2005) Bilirubin decreases NOS2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J 19:1890–1892
Lee JS, Kim SY, Kwon CH, Kim YK (2006) EGFR-dependent ERK activation triggers hydrogen peroxide-induced apoptosis in OK renal epithelial cells. Arch Toxicol 80:337–346
Li CY, Deng YL, Sun BH (2009) Taurine protected kidney from oxidative injury through mitochondrial-linked pathway in a rat model of nephrolithiasis. Urol Res 37:211–220
Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, Williams CR, Royce DB, Honda T, Honda Y, Gribble GW, Hill-Kapturczak N, Agarwal A, Sporn MB (2005) The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res 65:4789–4798
Liebler DC, Kaysen KL, Kennedy TA (1989) Redox cycles of vitamin E: hydrolysis and ascorbic acid dependent reduction of 8a-(alkyldioxy)tocopherones. Biochemistry 28:9772–9777
Lin H, Sue YM, Chou Y, Cheng CF, Chang CC, Li HF, Chen CC, Juan SH (2010) Activation of a nuclear factor of activated T-lymphocyte-3 (NFAT3) by oxidative stress in carboplatin-mediated renal apoptosis. Br J Pharmacol 161:1661–1676
Matsuoka T, Wada J, Hashimoto I, Zhang Y, Eguchi J, Ogawa N, Shikata K, Kanwar YS, Makino H (2005) Gene delivery of Tim44 reduces mitochondrial superoxide production and ameliorates neointimal proliferation of injured carotid artery in diabetic rats. Diabetes 54:2882–2890
McCord JM (1985) Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312:159–163
Mitrakou A (2011) Kidney: its impact on glucose homeostasis and hormonal regulation. Diabetes Res Clin Pract 93(Suppl 1):S66–72
Moist L, Sontrop JM, Gallo K, Mainra R, Cutler M, Freeman D, House AA (2010) Effect of N-acetylcysteine on serum creatinine and kidney function: results of a randomized controlled trial. Am J Kidney Dis 56:64–650
Momeni A, Shahidi S, Seirafian S, Taheri S, Kheiri S (2010) Effect of allopurinol in decreasing proteinuria in type 2 diabetic patients. Iran J Kidney Dis 4:128–132
Morales AI, Detaille D, Prieto M, Puente A, Briones E, Arevalo M, Leverve X, Lopez-Novoa JM, El-Mir MY (2010) Metformin prevents experimental gentamicin-induced nephropathy by a mitochondria-dependent pathway. Kidney Int 77:861–869
Najafian B, Alpers CE, Fogo AB (2011) Pathology of human diabetic nephropathy. Contrib Nephrol 170:36–47
Nash K, Hafeez A, Hou S (2002) Hospital-acquired renal insufficiency. Am J Kidney Dis 39:930–936
Naughton CA (2008) Drug-induced nephrotoxicity. Am Fam Physician 78:743–750
Neria F, Castilla MA, Sanchez RF, Gonzalez Pacheco FR, Deudero JJ, Calabia O, Tejedor A, Manzarbeitia F, Ortiz A, Caramelo C (2009) Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin a toxicity. Kidney Int 75:227–234
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Nitescu N, Ricksten SE, Marcussen N, Haraldsson B, Nilsson U, Basu S, Guron G (2006) N-acetylcysteine attenuates kidney injury in rats subjected to renal ischaemia-reperfusion. Nephrol Dial Transplant 21:1240–1247
Nolin TD, Ouseph R, Himmelfarb J, McMenamin ME, Ward RA (2010) Multiple-dose pharmacokinetics and pharmacodynamics of N-acetylcysteine in patients with end-stage renal disease. Clin J Am Soc Nephrol 5:1588–1594
Nouri-Majalan N, Ardakani EF, Forouzannia K, Moshtaghian H (2009) Effects of allopurinol and vitamin E on renal function in patients with cardiac coronary artery bypass grafts. Vasc Health Risk Manag 5:489–494
Novo E, Parola M (2008) Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 1:5
Nowak G, Clifton GL, Godwin ML, Bakajsova D (2006) Activation of ERK1/2 pathway mediates oxidant-induced decreases in mitochondrial function in renal cells. Am J Physiol Renal Physiol 291:F840–855
Ogawa D, Asanuma M, Miyazaki I, Tachibana H, Wada J, Sogawa N, Sugaya T, Kitamura S, Maeshima Y, Shikata K, Makino H (2011) High glucose increases metallothionein expression in renal proximal tubular epithelial cells. Exp Diabetes Res 2011:534872
Omori H, Kawada N, Inoue K, Ueda Y, Yamamoto R, Matsui I, Kaimori J, Takabatake Y, Moriyama T, Isaka Y, Rakugi H (2012) Use of xanthine oxidase inhibitor febuxostat inhibits renal interstitial inflammation and fibrosis in unilateral ureteral obstructive nephropathy. Clin Exp Nephrol 16:549–556
Pacher P, Nivorozhkin A, Szabo C (2006) Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev 58:87–114
Pan H, Mukhopadhyay P, Rajesh M, Patel V, Mukhopadhyay B, Gao B, Hasko G, Pacher P (2009) Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. J Pharmacol Exp Ther 328:708–714
Pascual J, Perez-Saez MJ, Mir M, Crespo M (2012) Chronic renal allograft injury: early detection, accurate diagnosis and management. Transplant Rev (Orlando) 26:280–290
Pathak E, MacMillan-Crow LA, Mayeux PR (2012) Role of mitochondrial oxidants in an in vitro model of sepsis-induced renal injury. J Pharmacol Exp Ther 340:192–201
Perazella MA (2005) Drug-induced nephropathy: an update. Expert Opin Drug Saf 4:689–706
Pergola PE, Krauth M, Huff JW, Ferguson DA, Ruiz S, Meyer CJ, Warnock DG (2011a) Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b-4 CKD. Am J Nephrol 33:469–476
Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG (2011b) Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365:327–336
Perianayagam MC, Liangos O, Kolyada AY, Wald R, MacKinnon RW, Li L, Rao M, Balakrishnan VS, Bonventre JV, Pereira BJ, Jaber BL (2007) NADPH oxidase p22phox and catalase gene variants are associated with biomarkers of oxidative stress and adverse outcomes in acute renal failure. J Am Soc Nephrol 18:255–263
Plotnikov EY, Kazachenko AV, Vyssokikh MY, Vasileva AK, Tcvirkun DV, Isaev NK, Kirpatovsky VI, Zorov DB (2007) The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int 72:1493–1502
Plotnikov EY, Chupyrkina AA, Jankauskas SS, Pevzner IB, Silachev DN, Skulachev VP, Zorov DB (2011) Mechanisms of nephroprotective effect of mitochondria-targeted antioxidants under rhabdomyolysis and ischemia/reperfusion. Biochim Biophys Acta 1812:77–86
Pozdzik AA, Salmon IJ, Debelle FD, Decaestecker C, Van den Branden C, Verbeelen D, Deschodt-Lanckman MM, Vanherweghem JL, Nortier JL (2008) Aristolochic acid induces proximal tubule apoptosis and epithelial to mesenchymal transformation. Kidney Int 73:595–607
Ramos LF, Kane J, McMonagle E, Le P, Wu P, Shintani A, Ikizler TA, Himmelfarb J (2011) Effects of combination tocopherols and alpha lipoic acid therapy on oxidative stress and inflammatory biomarkers in chronic kidney disease. J Ren Nutr 21:211–218
Randjelovic P, Veljkovic S, Stojiljkovic N, Velickovic L, Sokolovic D, Stoiljkovic M, Ilic I (2012) Protective effect of selenium on gentamicin-induced oxidative stress and nephrotoxicity in rats. Drug Chem Toxicol 35:141–148
Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP (1995) The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA 273:117–123
Rasi Hashemi S, Noshad H, Tabrizi A, Mobasseri M, Tayebi Khosroshahi H, Heydarnejad M, Khalaj MR, Aghamohammadzadeh N (2012) Angiotensin receptor blocker and N-acetyl cysteine for reduction of proteinuria in patients with type 2 diabetes mellitus. Iran J Kidney Dis 6:39–43
Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, Segal AW (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416:291–297
Reisman SA, Chertow GM, Hebbar S, Vaziri ND, Ward KW, Meyer CJ (2012) Bardoxolone methyl decreases megalin and activates nrf2 in the kidney. J Am Soc Nephrol 23:1663–1673
Renke M, Tylicki L, Rutkowski P, Larczynski W, Aleksandrowicz E, Lysiak-Szydlowska W, Rutkowski B (2008) The effect of N-acetylcysteine on proteinuria and markers of tubular injury in non-diabetic patients with chronic kidney disease. A placebo-controlled, randomized, open, cross-over study. Kidney Blood Press Res 31:404–410
Rhee SG (1999) Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med 31:53–59
Rocha M, Hernandez-Mijares A, Garcia-Malpartida K, Banuls C, Bellod L, Victor VM (2010) Mitochondria-targeted antioxidant peptides. Curr Pharm Des 16:3124–3131
Roth E, Manhart N, Wessner B (2004) Assessing the antioxidative status in critically ill patients. Curr Opin Clin Nutr Metab Care 7:161–168
Saba H, Munusamy S, Macmillan-Crow LA (2008) Cold preservation mediated renal injury: involvement of mitochondrial oxidative stress. Ren Fail 30:125–133
Sabbatini M, Santillo M, Pisani A, Paterno R, Uccello F, Seru R, Matrone G, Spagnuolo G, Andreucci M, Serio V, Esposito P, Cianciaruso B, Fuiano G, Avvedimento EV (2006) Inhibition of Ras/ERK1/2 signaling protects against postischemic renal injury. Am J Physiol Renal Physiol 290:F1408–1415
Saitoh T, Satoh H, Nobuhara M, Machii M, Tanaka T, Ohtani H, Saotome M, Urushida T, Katoh H, Hayashi H (2011) Intravenous glutathione prevents renal oxidative stress after coronary angiography more effectively than oral N-acetylcysteine. Heart Vessels 26:465–472
Sanhueza J, Valdes J, Campos R, Garrido A, Valenzuela A (1992) Changes in the xanthine dehydrogenase/xanthine oxidase ratio in the rat kidney subjected to ischemia-reperfusion stress: preventive effect of some flavonoids. Res Commun Chem Pathol Pharmacol 78:211–218
Scaduto RC Jr, Gattone VH 2nd, Grotyohann LW, Wertz J, Martin LF (1988) Effect of an altered glutathione content on renal ischemic injury. Am J Physiol 255:F911–921
Schneider C (2005) Chemistry and biology of vitamin E. Mol Nutr Food Res 49:7–30
Schneider MP, Sullivan JC, Wach PF, Boesen EI, Yamamoto T, Fukai T, Harrison DG, Pollock DM, Pollock JS (2010) Protective role of extracellular superoxide dismutase in renal ischemia/reperfusion injury. Kidney Int 78:374–381
Seger R, Krebs EG (1995) The MAPK signaling cascade. FASEB J 9:726–735
Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, Hattori M, Sakaki Y, Sumimoto H (2001) A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem 276:1417–1423
Sporn MB, Liby KT, Yore MM, Fu L, Lopchuk JM, Gribble GW (2011) New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod 74:537–545
Stamp LK, O'Donnell JL, Zhang M, James J, Frampton C, Barclay ML, Chapman PT (2011) Using allopurinol above the dose based on creatinine clearance is effective and safe in patients with chronic gout, including those with renal impairment. Arthritis Rheum 63:412–421
Sun Z, Zhang X, Ito K, Li Y, Montgomery RA, Tachibana S, Williams GM (2008) Amelioration of oxidative mitochondrial DNA damage and deletion after renal ischemic injury by the KATP channel opener diazoxide. Am J Physiol Renal Physiol 294:F491–498
Sun B, Lu C, Zhou GP, Xing CY (2011) Suppression of Par-4 protects human renal proximal tubule cells from apoptosis induced by oxidative stress. Nephron Exp Nephrol 117:e53–61
Surh YJ, Na HK, Park JM, Lee HN, Kim W, Yoon IS, Kim DD (2011) 15-Deoxy-delta(12,14)-prostaglandin J(2), an electrophilic lipid mediator of anti-inflammatory and pro-resolving signaling. Biochem Pharmacol 82:1335–1351
Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV (2011) Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 22:1041–1052
Takao T, Horino T, Kagawa T, Matsumoto R, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Taguchi T, Morita T, Terada Y (2011) Possible involvement of intracellular angiotensin II receptor in high-glucose-induced damage in renal proximal tubular cells. J Nephrol 24:218–224
Tatsuo H, Iwao O (2011) A repeated oral administration study of febuxostat (TMX-67), a non-purine-selective inhibitor of xanthine oxidase, in patients with impaired renal function in Japan: pharmacokinetic and pharmacodynamic study. J Clin Rheumatol 17:S27–34
Thamilselvan V, Menon M, Thamilselvan S (2009) Oxalate-induced activation of PKC-alpha and -delta regulates NADPH oxidase-mediated oxidative injury in renal tubular epithelial cells. Am J Physiol Renal Physiol 297:F1399–1410
Thamilselvan V, Menon M, Thamilselvan S (2012) Selective Rac1 inhibition protects renal tubular epithelial cells from oxalate-induced NADPH oxidase-mediated oxidative cell injury. Urol Res 40:415–423
Thiele H, Hildebrand L, Schirdewahn C, Eitel I, Adams V, Fuernau G, Erbs S, Linke A, Diederich KW, Nowak M, Desch S, Gutberlet M, Schuler G (2010) Impact of high-dose N-acetylcysteine versus placebo on contrast-induced nephropathy and myocardial reperfusion injury in unselected patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. The LIPSIA-N-ACC (prospective, single-blind, placebo-controlled, randomized Leipzig immediate PercutaneouS coronary intervention acute myocardial infarction N-ACC) trial. J Am Coll Cardiol 55:2201–2209
Traber MG, Stevens JF (2011) Vitamins C and E: beneficial effects from a mechanistic perspective. Free Radic Biol Med 51:1000–1013
Tsao KC, Wu TL, Chang PY, Sun CF, Wu LL, Wu JT (2007) Multiple risk markers for atherogenesis associated with chronic inflammation are detectable in patients with renal stones. J Clin Lab Anal 21:426–431
Tsuda H, Kawada N, Kaimori JY, Kitamura H, Moriyama T, Rakugi H, Takahara S, Isaka Y (2012) Febuxostat suppressed renal ischemia-reperfusion injury via reduced oxidative stress. Biochem Biophys Res Commun 427:266–272
Umekawa T, Tsuji H, Uemura H, Khan SR (2009) Superoxide from NADPH oxidase as second messenger for the expression of osteopontin and monocyte chemoattractant protein-1 in renal epithelial cells exposed to calcium oxalate crystals. BJU Int 104:115–120
Villacorta L, Graca-Souza AV, Ricciarelli R, Zingg JM, Azzi A (2003) Alpha-tocopherol induces expression of connective tissue growth factor and antagonizes tumor necrosis factor-alpha-mediated downregulation in human smooth muscle cells. Circ Res 92:104–110
Wang L, Matsushita K, Araki I, Takeda M (2002) Inhibition of c-Jun N-terminal kinase ameliorates apoptosis induced by hydrogen peroxide in the kidney tubule epithelial cells (NRK-52E). Nephron 91:142–147
Wang W, Jittikanont S, Falk SA, Li P, Feng L, Gengaro PE, Poole BD, Bowler RP, Day BJ, Crapo JD, Schrier RW (2003) Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. Am J Physiol Renal Physiol 284:F532–537
Wang Y, Ji HX, Zheng JN, Pei DS, Hu SQ, Qiu SL (2009) Protective effect of selenite on renal ischemia/reperfusion injury through inhibiting ASK1-MKK3-p38 signal pathway. Redox Rep 14:243–250
Ware LB, Fessel JP, May AK, Roberts LJ 2nd (2011) Plasma biomarkers of oxidant stress and development of organ failure in severe sepsis. Shock 36:12–17
Wilmes A, Crean D, Aydin S, Pfaller W, Jennings P, Leonard MO (2011) Identification and dissection of the Nrf2 mediated oxidative stress pathway in human renal proximal tubule toxicity. Toxicol In Vitro 25:613–622
Wu L, Mayeux PR (2007) Effects of the inducible nitric-oxide synthase inhibitor L-N(6)-(1-iminoethyl)-lysine on microcirculation and reactive nitrogen species generation in the kidney following lipopolysaccharide administration in mice. J Pharmacol Exp Ther 320:1061–1067
Wu L, Gokden N, Mayeux PR (2007) Evidence for the role of reactive nitrogen species in polymicrobial sepsis-induced renal peritubular capillary dysfunction and tubular injury. J Am Soc Nephrol 18:1807–1815
Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA, Grossman E, Chen J, Zhou XJ, Hartono J, Winterberg P, Chen B, Agarwal A, Lu CY (2011) Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am J Physiol Renal Physiol 300:F1180–1192
Yamanobe T, Okada F, Iuchi Y, Onuma K, Tomita Y, Fujii J (2007) Deterioration of ischemia/reperfusion-induced acute renal failure in SOD1-deficient mice. Free Radic Res 41:200–207
Yasuda H, Yuen PS, Hu X, Zhou H, Star RA (2006) Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int 69:1535–1542
Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, Guilarte TR, Yamamoto M, Sporn MB, Kensler TW (2007) Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther 6:154–162
Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S (2001) Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int 60:1343–1353
Yoo EG, Lee WJ, Kim JH, Chae HW, Hyun SE, Kim DH, Kim HS, Oh Y (2011) Insulin-like growth factor-binding protein-3 mediates high glucose-induced apoptosis by increasing oxidative stress in proximal tubular epithelial cells. Endocrinology 152:3135–3142
Zhang C, Walker LM, Mayeux PR (2000) Role of nitric oxide in lipopolysaccharide-induced oxidant stress in the rat kidney. Biochem Pharmacol 59:203–209
Zhou L, Chen H (2012) Prevention of contrast-induced nephropathy with ascorbic acid. Intern Med 51:531–535
Zingg JM, Azzi A (2004) Non-antioxidant activities of vitamin E. Curr Med Chem 11:1113–1133
Zorov DB (2010) Amelioration of aminoglycoside nephrotoxicity requires protection of renal mitochondria. Kidney Int 77:841–843
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Du, C., Wang, X., Chen, H. (2014). Oxidative Stress to Renal Tubular Epithelial Cells – A Common Pathway in Renal Pathologies. In: Laher, I. (eds) Systems Biology of Free Radicals and Antioxidants. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30018-9_187
Download citation
DOI: https://doi.org/10.1007/978-3-642-30018-9_187
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30017-2
Online ISBN: 978-3-642-30018-9
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences