
Abstract
Hepatitis C virus (HCV) is a hepatotropic virus and a major cause of chronic hepatitis and liver disease worldwide. Initial interactions between HCV virions and hepatocytes are required for productive viral infection and initiation of the viral life cycle. Furthermore, HCV entry contributes to the tissue tropism and species specificity of this virus. The elucidation of these interactions is critical, not only to understand the pathogenesis of HCV infection, but also to design efficient antiviral strategies and vaccines. This review summarizes our current knowledge of the host factors required for the HCV-host interactions during HCV binding and entry, our understanding of the molecular mechanisms underlying HCV entry into target cells, and the relevance of HCV entry for the pathogenesis of liver disease, antiviral therapy, and vaccine development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Discovery and Characterization of Hepatitis C Virus Binding and Entry Factors
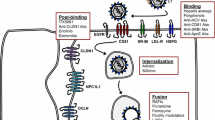
Hepatitis C virus (HCV) is a small, enveloped RNA virus that primarily targets human hepatocytes (reviewed in Lindenbach et al. 2007). The HCV envelope is composed of two virus-encoded glycoproteins, E1 and E2 (reviewed in Lavie et al. 2007). As with other enveloped viruses, the envelope glycoproteins largely define the interactions between HCV and the host cell. Moreover, HCV has been demonstrated to circulate in the blood of infected individuals in complexes with host lipoproteins and lipoprotein components (Andre et al. 2002; Miyamoto et al. 1992; Nielsen et al. 2006; Thomssen et al. 1992, 1993), which also contribute to HCV–host cell interactions. Since the discovery of HCV in 1989 (Choo et al. 1989), increasingly complex model systems have been designed to study HCV–host interactions that have allowed identification of key cell factors required for binding of HCV particles to the hepatocyte surface and entry into this cell (reviewed in Barth et al. 2006a; Bartosch and Cosset 2006; Bukh 2012; Murray and Rice 2011; von Hahn and Rice 2008). Some of these factors have been demonstrated to be involved in direct HCV envelope glycoprotein binding and/or subsequent viral entry, and are thus likely to act directly as receptors and co-receptors while others act as indirect, albeit essential, entry factors (Fig. 1).


HCV entry into hepatocytes. HCV particles circulate physically associated with lipoproteins such as VLDL, which contain protein components apolipoprotein B-100, and exchangeable apolipoproteins E and CI (shown as black circles). HCV gains access to hepatocytes by entering the space of Disse through fenestrae between hepatic endothelial cells. The first step of entry 1 is binding, when HCV envelope glycoproteins E1 and E2 (black and gray rectangles surrounding virion) and/or apoE on the lipoviral particle (LVP) binds to heparan sulfate proteoglycan (HSPG), 2 LDL receptor (LDLR) is also a high-affinity binding partner of apoE which has been reported to be important for HCV entry, 3 SR-BI may also contribute to HCV binding via lipoproteins and/or HCV E2. Moreover, SR-BI also plays a role at post-binding steps of HCV entry, 4 Receptor tyrosine kinases (RTKs) including epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2) mediate HCV entry through facilitating associations between 5 CD81 and, 6 claudin 1 (CLDN1), and through envelope glycoprotein-membrane fusion. CLDN1 is a component of tight junctions along with the entry factor, 7 occludin (OCLN) that contributes to post-binding steps of the HCV entry process. Recently identified entry factor, 8 Niemann Pick C-1 like 1 (NPC1L1) plays an important role in cholesterol reabsorption from biliary secretion of cholesterol by transporter ATP binding cassette (ABC) G5/8 heterodimeric protein, and acts as a cofactor for HCV entry during post-binding steps. Viral envelope fusion with host membranes 9 is the final step of HCV entry, as the nucleocapsid escapes the early endosome to begin translation and replication of its genome in the cytosol
The first identified entry factors were discovered by their capacity to bind directly to HCV envelope glycoprotein E2. Pileri et al. knowing that purified E2 protein could bind to human hepatocellular carcinoma cell lines, but not mouse cell lines, transduced a cDNA expression library derived from HCV-binding cells into nonbinding cells and identifíed the tetraspanin CD81 as a direct binding partner of E2 (Pileri et al. 1998). Subsequently, it has been demonstrated that CD81 contributes to post-binding steps of the HCV entry process resulting in the internalization of the viral particle (Bertaux and Dragic 2006; Farquhar et al. 2012; Koutsoudakis et al. 2006). The observation that HCV RNA in patient serum was associated with lipoproteins prompted Agnello et al. to assess the role of low-density lipoprotein receptor (LDLR) as an HCV receptor. Indeed, by using in situ hybridization of HCV RNA in HepG2 hepatoma cells that were exposed to HCV, the authors showed that LDLR is important for uptake of HCV (Agnello et al. 1999; Monazahian et al. 1999; Wunschmann et al. 2000). Recent evidence suggests that LDLR most likely is not an essential entry factor, but given its role in cholesterol metabolism it may play a role in HCV replication (Albecka et al. 2012).
Other factors that were determined to bind directly to HCV E2 were identified by observations garnered from human immunodeficiency virus (HIV) and other viruses. This strategy revealed that HCV E2 could bind the C type lectins dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin (DC-SIGN), and liver/lymph node-specific intercellular adhesion molecule 3-grabbing integrin (L-SIGN) (Gardner et al. 2003; Lozach et al. 2003; Pohlmann et al. 2003). These findings are intriguing in the role that they play in hepatocyte entry, since these molecules are not on the surface of hepatocytes, but are rather on dendritic cells and liver endothelial cells for DC-SIGN and L-SIGN, respectively. It is thought that these molecules may play a similar role as DC-SIGN plays in HIV, as an avenue of transinfection, in which the virus passes through a different type of cell to capture and localize the virus for infection of the hepatocytes that can sustain replication (Cormier et al. 2004; Lozach et al. 2004).
Soon after the identification of CD81 as a host factor playing a role in HCV entry, experimental evidence suggested that other host factors most likely contribute to HCV entry. Thus, cross-linking studies using E2 and HepG2 cells, which lack CD81 expression, lead to the identification of scavenger receptor BI (SR-BI) as an additional direct binding partner of E2 (Scarselli et al. 2002). Interestingly, since SR-BI is also a receptor for lipoproteins, the HCV virion may interact with SR-BI through associated lipoproteins and/or E2 (Barth et al. 2005a, 2008; Bartosch et al. 2003b; Catanese et al. 2007, 2010; Dreux et al. 2009). Further, by using an inter alia mutant virus in the SR-BI binding region of E2 and a lipoprotein binding-deficient SR-BI mutant as well as monoclonal SR-BI-specific antibodies, two recent studies showed that SR-BI acts at different steps during the HCV entry process (Dao Thi et al. 2012; Zahid et al. 2012). Indeed, SR-BI might first interact with the lipoprotein component of the lipoviral particle in an E2 binding independent manner (Dao Thi et al. 2012; Maillard et al. 2006). However, later during entry the interaction between SR-BI and the lipoviral particle becomes E2-dependent, for which HVR1 appears to play a major role (Dao Thi et al. 2012). These attributes point to an interesting role of SR-BI in HCV entry, as this protein acts at similar time points than CD81 forming part of the receptor complex required for HCV entry into the target cell (Kapadia et al. 2007; Zeisel et al. 2007). Interestingly, SR-BII, a splice variant of SR-BI, has also been demonstrated to promote HCV entry (Grove et al. 2007).
Subsequent to these advances, further investigations were conducted using surrogate HCV particle model systems such as HCV-like particles (HCV-LPs) isolated from insect cells (Baumert et al. 1998; Saunier et al. 2003; Triyatni et al. 2002) (see chapter “Cell Culture Systems for Hepatitis C Virus” by Steinmann and Pietschmann, this volume), and it was shown that E2 and HCV-LPs bind highly sulfated heparan sulfate proteoglycans (HSPG) either purified or on liver-derived membranes (Barth et al. 2003, 2006b). The relevance of HSPG for HCV binding was then subsequently confirmed using HCV pseudoparticles (HCVpp) and cell culture-derived HCV (HCVcc) Haberstroh et al. 2008; Jiang et al. 2012; Koutsoudakis et al. 2006; Morikawa et al. 2007) (for cell culture models see chapter “Cell Culture Systems for Hepatitis C Virus” by Steinmann and Pietschmann, this volume). Interestingly, it could be further shown that both E1 and apolipoprotein E (apoE) may also contribute to HCV attachment to HSPG (Barth et al. 2006a, b; Haberstroh et al. 2008; Jiang et al. 2012). To date, HSPG is the only known host factor that solely mediates HCV attachment to target cells without having any other described role in the viral life cycle.
The development of the HCVpp system (Bartosch et al. 2003a; Hsu et al. 2003), which consists of ectopically expressed HCV envelope glycoproteins on retrovirus particles containing vector RNAs that encode reporter genes (see chapter “Cell Culture Systems for Hepatitis C Virus” by Steinmann and Pietschmann, this volume), enabled new additional screening strategies. Using such a screening strategy with a cDNA library from the highly permissive Huh7.5 cell line in human kidney cell line 293T, Evans et al. discovered claudin 1 (CLDN1) as an important entry factor for HCV (Evans et al. 2007). Interestingly, the HCV envelope glycoproteins do not directly interact with CLDN1, but CLDN1 interacts with CD81 and thereby plays an important role during post-binding steps of the HCV entry process (Evans et al. 2007; Harris et al. 2008; Krieger et al. 2010). In the human liver as well as in polarized human hepatoblastoma HepG2 cells, CLDN1 is expressed at the basal and lateral membranes as well as in tight junctions (TJs) (Mee et al. 2009; Reynolds et al. 2008). Interestingly, CD81-CLDN1 co-receptor association could only be detected at the basal membranes but not in TJ-associated pools of CLDN1 and CD81 suggesting that the nonjunctional CLDN1 plays a role in HCV entry (Mee et al. 2009). This is in line with the fact that CLDN1 is more highly expressed at the TJs in polarized cells that demonstrate reduced viral entry as compared to nonpolarized cells (Mee et al. 2009). Noteworthy, in various nonpolarized cell lines, it has been shown that the subcellular localization of CLDN1 correlates with HCV entry permissivity. Indeed, in contrast to intracellular CLDN1, only cell surface expressed CLDN1 enriched at cell–cell contact sites promotes HCV entry (Yang et al. 2008) and mutations of CLDN1 that delocalize the protein outside cell–cell contacts do not support HCV entry (Cukierman et al. 2009; Liu et al. 2009). Mutagenesis studies of CLDN1 have revealed that the intracellular domains of this entry factor, including the C-terminal domain that is important for the transport of CLDN1 into TJs, are dispensable for infection, but domains within the first extracellular loop are critical (Cukierman et al. 2009; Evans et al. 2007). The mutations that particularly disrupted HCV entry were those that interrupted cell–cell contacts but did not affect lateral interactions within the plasma membrane between either CLDN1-CLDN1 or CLDN1-CD81 (Cukierman et al. 2009). Moreover, other members of the CLDN family of proteins, e. g., CLDN6 and CLDN9, may also promote HCV entry in 293T-derived cell lines (Meertens et al. 2008; Zheng et al. 2007). In contrast to other CLDNs, CLDN6 and CLDN9 are also able to form co-receptor associations with CD81, a process that is indispensable for HCV entry (Harris et al. 2010). Since the expression of CLDN6 and 9 is very low in human hepatocytes and liver tissue (J. A. McKeating, personal communication), the relevance of these molecules for HCV entry in vivo needs to be demonstrated. Further use of similar screening strategies in mouse-derived cell lines identified occludin (OCLN) as a species-tropism defining entry factor, and it was determined that among the identified entry factors, CD81 and OCLN determine the tropism of HCV for human cells (Ploss et al. 2009). Interestingly, expression of human CD81 and human OCLN in mouse cells removes the species-specific restriction of HCV entry and allowed development of a mouse model for the early steps of HCV infection (Dorner et al. 2011). The important role of OCLN in the HCV entry process was further demonstrated by two other studies using HCVcc (Benedicto et al. 2009; Liu et al. 2009). However, whether OCLN is a true receptor that binds the HCV envelope or is an indirect, though indispensable, entry co-factor required for post-binding steps still remains an open question. Of note, HCV infection can modulate the localization and expression level of CLDN1 and OCLN in the TJ and promote superinfection exclusion (Benedicto et al. 2008; Liu et al. 2009). In this way, HCV infection appears to downregulate protein amounts in TJs of the infected cell, which may contribute to pathologic symptoms such as cholestatic disorders.
Recent functional siRNA screens that specifically focused on host cell kinases, and using HCVpp as well as HCVcc, further identified two cell surface receptor tyrosine kinases. Epidermal growth factor receptor (EGFR) and ephrin receptor A2 (EphA2) are important co-factors for HCV entry and infection (Lupberger et al. 2011). It should be noted that EGFR does not directly interact with the HCV particle, but EGFR-dependent signaling pathways lead to the formation of CD81-CLDN1 complexes required for HCV entry (Lupberger et al. 2011).
Finally, since the HCV virion is rich in cholesterol, the role of cholesterol transporter Niemann-Pick C1-like 1 (NPC1L1) was investigated recently and identified as an additional entry factor (Sainz et al. 2012), yet its exact role in HCV entry remains to be determined. Within the last years the functional in vivo relevance of most of the entry factors has been confirmed using mouse models such as the human liver chimeric upA-SCID mouse model (Lacek et al. 2012; Lupberger et al. 2011; Meuleman et al. 2008, 2012; Sainz et al. 2012) or a humanized mouse model expressing the human entry factors (Dorner et al. 2011) (see also Sect. 5).
2 The HCV Entry Pathway: From Viral Attachment to the Hepatocyte Basolateral Membrane to Fusion Within the Endosome
Viral entry plays an important role for hepatocyte tropism of HCV. Indeed, tissue tropism is at least partially defined by the interaction of the virus with a set of cell surface factors on the target cell as described above, ultimately leading to viral entry and initiation of the viral life cycle. As the virus circulates in the blood of HCV-infected patients in association with lipoproteins, both the lipoprotein components and the viral envelope glycoproteins may contribute to the first interactions between the virion and the target cell. Experimental data indicate that HCV uses a single entry pathway for initiation of productive viral infection. The entry pathway consists of three key steps: (1) viral attachment to the hepatocyte, (2) receptor-mediated endocytosis of the viral particle, and (3) endosomal fusion (Fig. 1).
Binding studies using infectious viral particles suggested that HSPG, LDLR, and SR-BI contribute to viral attachment (Albecka et al. 2012; Dao Thi et al. 2012; Catanese et al. 2010; Cormier et al. 2004; Gardner et al. 2003; Koutsoudakis et al. 2006; Lozach et al. 2003; Ludwig et al. 2004; Maillard et al. 2006; Pohlmann et al. 2003). Docking of the virus to the target cell membrane is then believed to be followed by the interaction of the virus with several (other) cell surface factors leading to molecular rearrangements at the plasma membrane and subsequently resulting in viral internalization. Among the host factors contributing to these molecular rearrangements, host cell kinases have been demonstrated to play an important role in regulating HCV entry. Indeed, phosphatidylinositol 4-kinases type III alpha (PI4KIIIα) and beta (PI4KIIIβ) have been suggested to play a role in membrane remodeling and trafficking during HCV entry in a genotype-dependent manner (Trotard et al. 2009). However, the underlying molecular mechanisms have not yet been investigated. Using protein kinase inhibitors, two studies reported that host cell kinases are able to promote co-receptor association between CD81 and CLDN1, which is mandatory for HCV entry (Farquhar et al. 2008; Harris et al. 2008, 2010; Lupberger et al. 2011). Indeed, a protein kinase A (PKA) inhibitor disrupts CD81-CLDN1 complexes and leads to intracellular localization of CLDN1, thereby reducing HCV entry (Farquhar et al. 2008). Moreover, erlotinib and dasatinib, protein kinase inhibitors of EGFR and EphA2, respectively, also disrupt CD81-CLDN1 complexes and inhibit HCV entry (Lupberger et al. 2011). These data indicate that PKA, EGFR, and EphA2 promote the CD81-CLDN1 co-receptor association that is required for HCV entry. Moreover, these data suggest that kinase signaling pathways contribute to this process (Lupberger et al. 2011). SR-BI and OCLN have also been demonstrated to participate in post-binding steps of the HCV entry process (Benedicto et al. 2009; Zeisel et al. 2007). Intriguingly, CD81, SR-BI, and CLDN1 seem to act at very closely related timepoints during HCV entry (Krieger et al. 2010; Zeisel et al. 2007). The exact role of OCLN and the interplay of the different receptors in this process are yet to be uncovered.
HCV is known to endocytose in a clathrin-dependent process (Blanchard et al. 2006; Codran et al. 2006; Coller et al. 2009; Meertens et al. 2006). Interestingly, it has been recently demonstrated that CD81-CLDN1 complexes are endocytosed in a clathrin- and dynamin-dependent manner (Farquhar et al. 2012) consistent with a previous imaging study reporting that entering HCV particles are associated with CD81 and CLDN1 (Coller et al. 2009). This mechanism involves the HCV envelope glycoproteins and the GTPase Rho, while EGFR does not appear to play a role in this process (Farquhar et al. 2012). Whether other HCV entry factors are concomitantly endocytosed along with HCV still remains to be determined. Following internalization, HCV fusion has been reported to occur in early endosomes (Coller et al. 2009; Meertens et al. 2006). This process is dependent on low pH and involves both viral and host proteins (Bartosch et al. 2003a; Blanchard et al. 2006; Haid et al. 2009; Hsu et al. 2003; Kobayashi et al. 2006; Koutsoudakis et al. 2006; Lavillette et al. 2006; Meertens et al. 2006; Tscherne et al. 2006). Indeed, the HCV envelope glycoproteins E1 and E2 contain potential fusion domains (Drummer et al. 2007; Lavillette et al. 2007) and HCV E2 has been shown to be required for liposome/HCVcc fusion in vitro (Haid et al. 2009). Moreover, the importance of the HCV envelope in viral fusion is emphasized by the fact that patient-derived anti-HCV antibodies are able to inhibit cell–cell fusion and liposome/HCVpp fusion in vitro (Haberstroh et al. 2008; Kobayashi et al. 2006). Interestingly, the CD81 and CLDN1 proteins that co-endocytose with HCV (Farquhar et al. 2012), also play a role in HCV envelope glycoprotein-dependent cell–cell fusion (Evans et al. 2007; Kobayashi et al. 2006). Moreover, the protein kinase inhibitors erlotinib and dasatinib reduced HCV envelope glycoprotein-dependent cell–cell fusion suggesting that EGFR and EphA2 may contribute to the HCV fusion process (Lupberger et al. 2011).
3 Impact of Lipoproteins and Cholesterol for HCV Entry
Soon after the structure of the viral genome of HCV was discovered, characterization of the virus in serum of infected individuals revealed that HCV RNA distributed in a wide spectrum, due to HCV forming complexes with low-density lipoproteins (LDL), and very-low density lipoproteins (VLDL) (Hijikata et al. 1993; Kanto et al. 1995; Miyamoto et al. 1992; Nielsen et al. 2006; Prince et al. 1996; Thomssen et al. 1992, 1993) (for virus structure see chapter “Virion Assembly and Release ” by Lindenbach, this volume). It was also demonstrated that HCV RNA in the higher density fractions could be immunoprecipitated with antibodies recognizing immunoglobulin G (IgG), indicating that these viruses were circulating bound with antibodies, while the lipoprotein-associated viruses had diminished antibody association (Andre et al. 2002; Thomssen et al. 1993). Virus particles that are associated with VLDL have been termed lipoviral particles (Andre et al. 2002), and their presence and composition is dependent on host factors that impact lipoprotein metabolism such as diet and lipoprotein profile (Bridge et al. 2011; Felmlee et al. 2010). Interestingly, HCV strains that were passaged through chimpanzees demonstrated a correlation between viral association with lipoproteins and infectivity: strains that had more RNA associated with lipoproteins tended to have higher infectivity (Bradley et al. 1991; Hijikata et al. 1993). This was also observed for HCVcc that were passaged in human liver transplanted mice as these HCVcc were characterized by a higher infectivity and a lower buoyant density (Lindenbach et al. 2006). These experimental findings suggested an association of infectious virions with lipoproteins in full agreement with early reports (Monazahian et al. 1999).
The liver is the major organ of lipid homeostasis, regulated in part by lipoproteins. VLDL particles are assembled in hepatocytes when the large 550 kDa monomeric apolipoprotein B (apoB) protein is translated in the endoplasmic reticulum (Shelness and Sellers 2001). This protein is co-translationally loaded with lipids by microsomal triglyceride transfer protein (MTP), the limiting enzyme for VLDL formation (Jamil et al. 1998). Further lipidation of the nascent VLDL may occur directly by fusion with other lipid droplets present in the lumen of the secretory pathway, where exchangeable apolipoproteins such as apoC and apoE can bind VLDL (Wang et al. 2007). Secreted VLDL undergoes significant remodeling while in circulation by docking onto lipoprotein lipase (LPL), which lines the endothelium and hydrolyzes the triglyceride (TG)-rich core for delivery of free fatty acids to skeletal muscle and adipocytes. This process will begin the conversion of a triglyceride-rich VLDL coated in exchangeable apolipoproteins, which steer the VLDL’s lipolysis and clearance rate, into a smaller, cholesterol-rich LDL particle with fewer bound exchangeable apolipoproteins (Berneis and Krauss 2002; Packard and Shepherd 1997; Zheng et al. 2008). High-density lipoproteins (HDL) are produced by another mechanism and act as a reservoir for exchangeable apolipoproteins and are key in reverse cholesterol transport from peripheral tissues to the liver (Rothblat and Phillips 2010; Rye et al. 2009; Shachter 2001).
Like for other viruses, there is a tight relation between HCV morphogenesis (see chapter “Virion Assembly and Release” by Lindenbach, this volume) and HCV entry as the composition of the viral particle largely defines virus–host cell interactions and viral entry into target cells. In human hepatoma cells, HCV has been shown to assemble in close proximity to lipid droplets and endoplasmic reticulum (Blanchard et al. 2002; Boulant et al. 2007; Miyanari et al. 2007; Op De Beeck and Dubuisson 2003; Roingeard et al. 2008). HCV NS5A and apoE play a major role in this process (Appel et al. 2008; Benga et al. 2010; Cun et al. 2010). HCV assembly and release have been linked to VLDL biogenesis (Chang et al. 2007; Gastaminza et al. 2006, 2008; Huang et al. 2007). Using mass spectrometry, a recent study reported that the lipid composition of purified HCVcc resembles that of VLDL and LDL and that the majority of E2-containing particles also contain apoE on their surface (Merz et al. 2011). Moreover, HCV virions can be precipitated with antibodies directed against apoB, apoE, and apoCI suggesting that these apolipoproteins are part of the lipoviral particles (Andre et al. 2002; Chang et al. 2007; Meunier et al. 2008). The exact morphogenesis and morphology of HCV particles still remains elusive, but as HCV E1 is able to directly interact with both apoE and apoB (Mazumdar et al. 2011), this envelope glycoprotein may contribute to the association of the viral envelope with host-derived lipoproteins. The virus may take advantage of this association with host-derived lipoproteins in order to evade the host’s immune response by masking its envelope glycoproteins and by including host-derived ligands on the virion surface for interaction with attachment factors such as HSPG, LDLR, and SR-BI (Bankwitz et al. 2010; Dreux et al. 2006; Grove et al. 2008; Prentoe et al. 2011).
While apoE on the viral particle is essential for HCV infectivity, the role of apoB for viral entry remains less well-defined (Gastaminza et al. 2008; Huang et al. 2007; Jiang and Luo 2009; Maillard et al. 2006; Merz et al. 2011; Owen et al. 2009). Indeed, apoE on HCV particles may directly interact with HSPG, LDLR, and SR-BI (Andre et al. 2002; Hishiki et al. 2010; Jiang et al. 2012; Dao Thi et al. 2012; Maillard et al. 2006; Saito et al. 2003) and antibodies directed against apoE or human apoE-derived peptides inhibit HCVcc infection by blocking virus particle binding (Chang et al. 2007; Jiang et al. 2012; Liu et al. 2012; Merz et al. 2011; Owen et al. 2009). Interestingly, it has been suggested by one study that apoE isoforms may influence HCVcc infectivity (Hishiki et al. 2010). Moreover, functional apoE gene polymorphism has been suggested as a determinant of the outcome in HCV infection: the epsilon three allele is associated with persistent HCV infection while the epsilon two allele may protect against viral persistence (Price et al. 2006). It is thus tempting to speculate that there is a link between apoE polymorphism and HCV infection. However, it has to be pointed out that another in vitro study did not report differences in HCVcc assembly and infectivity using different apoE isoforms (Cun et al. 2010).
Given the association of HCV with lipoproteins, it is not surprising that exogenous addition of various lipoproteins has been reported to influence HCV infection. Interestingly, the results of such experiments were dependent on the HCV model system used. VLDL strongly inhibited the interaction of serum-derived HCV with hepatic cells, whereas HDL and LDL did not show a significant effect (Maillard et al. 2006). In contrast, HCVcc infection and HCVpp entry were both increased by HDL while LDL had no effect (Bartosch et al. 2005; Voisset et al. 2005; von Hahn et al. 2006). Moreover, HCVcc infection was inhibited by oxidized LDL (oxLDL) and to a lesser extent by oxidized HDL (oxHDL) (Bartosch et al. 2005; Voisset et al. 2005; von Hahn et al. 2006). The physiological SR-BI ligands HDL and oxLDL/oxHDL do not appear to act as receptor agonist/antagonists, but it has been suggested that the modulation of HCV entry may involve a ternary interplay between the virus, SR-BI, and the respective lipoprotein (Bartosch et al. 2005; von Hahn et al. 2006). Furthermore, apoCI has been reported to enhance HCVcc and HCVpp infectivity and to increase fusion between viral and target membranes (Dreux et al. 2007).
Changes in the nature of the HCV-associated lipoproteins have also been demonstrated to influence HCV infectivity. Although LPL treatment of cells is able to enhance HCV attachment to target cells, at the same time it reduces HCV infectivity (Andreo et al. 2007; Maillard et al. 2011). Furthermore, treatment of HCVcc with a high dose of LPL shifts HCV to higher densities, and decreases the amount of apoE-associated HCV (Shimizu et al. 2010). In addition, the cholesterol and sphingolipid content of both the HCV virion and the target cell membrane, is important for HCV infectivity (Aizaki et al. 2008; Kapadia et al. 2007; Voisset et al. 2008; Yamamoto et al. 2011). Indeed, depletion of cholesterol from HCV particles or hydrolysis of virion-associated sphingomyelin almost completely abolished HCV infectivity without reducing viral binding (Aizaki et al. 2008). Moreover, hydrolysis of target cell plasma membrane sphingomyelin resulted in the internalization of CD81 and reduced HCV infection (Voisset et al. 2008).
In this context, it is interesting to note that the physiological roles of three identified HCV entry factors, namely SR-BI, LDLR, and NPC1L1, are to be key cholesterol transporters. SR-BI primarily functions at the basolateral surface of hepatocytes by binding esterified-cholesterol enriched HDL particles and transferring this cholesterol to the cell (Out et al. 2004). LDLR binds avidly to apoE on the surface of TG-rich lipoprotein remnants and internalizes these particles via clathrin-coated pits. The physiological role of NPC1L1 is to reabsorb unesterified cholesterol secreted into the bile by ABCG5/8 transporters. NPC1L1 is primarily located on the apical (bile canalicular) surface of hepatocytes while the other entry factors are located at the basolateral side, or are part of the TJ complex. When cellular cholesterol content is low, NPC1L1 is present at the plasma membrane to bind extracellular cholesterol. Cholesterol-bound NPC1L1, rather than acting like a cholesterol channel, is endocytosed in a caveolin-1 independent manner (Skov et al. 2011; Valasek et al. 2005) into sorting endosomes followed by progression either to the endosome recycling center or to late endosomes followed by lysosomal degradation (Jia et al. 2011). Sainz et al. showed that silencing NPC1L1, or using chemical inhibitors (ezetimibe) or antibodies to the cholesterol binding domain of NPC1L1, diminish HCVcc infection (Sainz et al. 2012). Given the physiological role for NPC1L1 and its primary location on the bile canalicular surface, NPC1L1 may act as an indirect host entry factor by modulating cholesterol levels. However, enteric cholesterol absorption has been reported to involve cooperation between SR-BI and NPC1L1, raising the possibility that these proteins may cooperate also in HCV entry. Interestingly, while ezetimibe’s primary inhibitory function is binding NPC1L1, it is intriguing that ezetimibe also binds to SR-BI (Altmann et al. 2002).
4 Viral Spread Through Cell-to-Cell Transmission
While initiation of HCV infection is dependent on cell-free infection of hepatocytes by virions entering the liver through the bloodstream, HCV dissemination within the liver and establishment of chronic HCV infection may be possible by different routes including direct viral cell-to-cell transmission between adjacent hepatocytes (Timpe et al. 2008). HCV cell-to-cell transmission appears to be more efficient than cell-free particle entry in vitro (Brimacombe et al. 2011; Timpe et al. 2008). Moreover, in contrast to cell-free HCV particle transmission, this process seems to be resistant to the majority of neutralizing antibodies thereby potentially contributing to evasion from the host humoral immune responses and establishment of chronic HCV infection in vivo (Brimacombe et al. 2011; Timpe et al. 2008). Furthermore, like cell-free particle entry, HCV cell-to-cell transmission has been described for all major genotypes, though genotype-dependent differences between the relative infection by cell-free versus cell-to-cell transmission have been reported (Brimacombe et al. 2011).
There are striking similarities between the molecular mechanisms of cell-free HCV entry and HCV cell-to-cell transmission. Indeed, HCV cell-to-cell transmission appears to require numerous host factors that also play a role during cell-free entry: CD81, SR-BI, CLDN1, OCLN, EGFR, EphA2, and potentially NPC1L1 (Brimacombe et al. 2011; Lupberger et al. 2011; Sainz et al. 2012; Timpe et al. 2008). The role of LDLR in HCV cell-to-cell transmission has not been investigated to date. Interestingly, in contrast to cell-free virus entry, CD81-independent cell-to-cell transmission pathways have been described (Jones et al. 2010; Witteveldt et al. 2009). Moreover, it is worth noting that SR-BI appears to have a relevant role in cell-to-cell transmission and thus, targeting SR-BI allows to inhibit HCV spread (Brimacombe et al. 2011; Meuleman et al. 2012; Zahid et al. 2012). Furthermore, it has been shown that SR-BI may be a limiting factor in HCV cell-to-cell transmission since overexpression of SR-BI in target cells increases virus spread (Brimacombe et al. 2011).
Although HCV cell-to-cell transmission had been defined to be resistant to neutralizing antibodies, certain monoclonal antibodies directed against HCV E2 (mAb 9/27 targeting HVR1; mAb 11/20 targeting aa 436–447) have been demonstrated to partially interfere with cell-to-cell transmission, although less efficiently than with cell-free infection (Brimacombe et al. 2011). These data indicate the intriguing possibility of developing potential therapeutic neutralizing antibodies that may inhibit both cell-free HCV entry and cell-to-cell transmission, thereby limiting both initiation of HCV infection and viral spread.
5 Impact of Hepatitis C Virus Entry for Pathogenesis of Liver Disease and Antiviral Therapy
HCV entry is an important target of the host’s immune responses. Neutralizing antibodies have been reported to interfere with different steps of the HCV entry process in vitro including attachment, entry, and fusion (Fofana et al. 2012; Haberstroh et al. 2008; Jiang et al. 2012; Steinmann et al. 2004; Vieyres et al. 2011). Different anti-E2 antibodies, such as AP33 (directed against E2 aa 412–423) or 3E5-1 (directed against E2 aa 522–529), have been reported to reduce HCV attachment to target cells (Barth et al. 2005b), as well as other steps of the HCV entry process (Haberstroh et al. 2008), whereas human anti-E1 antibodies targeting aa 313–326 interfere with HCV entry during post-binding steps without blocking HCVpp or HCV-LP binding to human hepatoma cells in vitro (Haberstroh et al. 2008). Interestingly, the HCV post-binding steps targeted by neutralizing antibodies are closely related to the interaction of HCV with CD81, SR-BI, and/or CLDN1 (Haberstroh et al. 2008). Moreover a recent study demonstrated that viral escape from neutralizing antibodies also takes place during HCV post-binding steps (Fofana et al. 2012). These data highlight the impact of post-binding steps of HCV entry for neutralization and evasion of immune responses.
Viral entry has been shown to play an important role for the pathogenesis of HCV infection. Indeed, using patient-derived HCVpp and antibodies from a cohort of liver transplant patients, a recent study demonstrated that enhanced viral entry and escape from antibody-mediated neutralization play a key role for the selection of viral variants in the early phase of liver transplantation (LT) (Fafi-Kremer et al. 2010). This study suggested that the abrupt change of the host environment during LT leads to the selection of viral variants with an efficient entry phenotype (Fafi-Kremer et al. 2010). Furthermore, the analysis of HCV infection in the chimeric liver uPA-SCID mouse model corroborated this hypothesis (Fafi-Kremer et al. 2010). Using reverse genetics and chimeric HCVcc, it was then demonstrated that the enhanced entry of an escape variant isolated from a liver transplant patient is associated with an altered usage of entry factors such as CD81 and SR-BI (Fofana et al. 2012). An altered host entry factor usage associated with escape from neutralizing antibodies may thus contribute to the reinfection of the liver graft and establishment of persistent HCV infection (Fofana et al. 2012). In this context, it is interesting to point out that HCV receptor levels at the time of LT seem to modulate early HCV kinetics and that recurrence of HCV infection after LT was associated with increased levels of CLDN1 and OCLN (Mensa et al. 2011). Taken together, these data suggest a key role of virus–host interactions during HCV entry for virus spread, persistence, and liver graft infection in transplanted patients.
Given the importance of HCV entry into target cells for the initiation and maintenance of infection as well as the pathogenesis of liver disease, this multistep process offers several promising targets for antiviral strategies (see also chapter “Hepatitis C Virus-Specific Directly Acting Antiviral Drugs” by Delang et al. this volume ). Indeed, although the introduction of novel direct acting antivirals (targeting the viral protease) in combination with interferon-alfa and ribavirin, the standard treatment for chronic HCV infection, has markedly improved the outcome of patients, resistance remains an important challenge (Chevaliez and Asselah 2011; Pawlotsky 2011) (see also chapter “Treatment of Chronic Hepatitis C: Current and Future” by Pawlotsky this volume). Furthermore, important adverse effects limit the use of standard treatment or direct acting antivirals in patients with advanced liver disease, co-morbidity, co-infection, or immunosuppression (Pawlotsky 2011).
In a significant proportion of HCV infected individuals, chronic infection ultimately leads to cirrhosis and hepatocellular carcinoma, requiring LT. Unfortunately, LT is characterized by a rapid reinfection of the liver graft and an accelerated progression of infection and to date no strategy to prevent HCV reinfection of the graft is available. For this purpose, HCV entry inhibitors, by targeting cell-free virus entry, may address this unmet medical need. Furthermore, this treatment can also be efficient for established HCV infection by targeting HCV cell-to-cell transmission in combination with direct acting antivirals. A variety of entry inhibitors are in preclinical and early clinical development (reviewed in Zeisel et al. 2011). These entry inhibitors can be categorized into (1) compounds targeting the lipoviral particle, e.g., anti-envelope antibodies, heparin, and lectins (Barth et al. 2003; Davis et al. 2005; Helle et al. 2006; Koutsoudakis et al. 2006; Law et al. 2008), (2) compounds targeting essential host entry factors, e.g., antibodies blocking CD81, SR-BI, CLDN1, EGFR, EphA2, and NPC1L1 (Bartosch et al. 2003b; Catanese et al. 2007; Flint et al. 1999; Fofana et al. 2010; Lupberger et al. 2011; Sainz et al. 2012; Zeisel et al. 2007; Zahid et al. 2012) or small molecule compounds targeting CD81, SR-BI, EGFR, EphA2, and NPC1L1 (Lupberger et al. 2011; Sainz et al. 2012; Syder et al. 2011; VanCompernolle et al. 2003), and (3) compounds interfering with viral internalization and fusion, e.g., phosphorothioate oligonucleotides, arbidol, chloroquine, and silymarin (Blanchard et al. 2006; Boriskin et al. 2006, 2008; Matsumura et al. 2009; Polyak et al. 2007; Tscherne et al. 2007; Wagoner et al. 2010).
The development of liver humanized transgenic mice has allowed demonstrating proof-of-concept of entry inhibitors in vivo (for animal models see chapter “Animal Models for Hepatitis C” by Billerbeck et al. this volume). The first clinical trials with HCV entry inhibitors have been conducted (Davis et al. 2005; Hawke et al. 2010; Neumann et al. 2010; Rutter et al. 2011) or are currently being initiated. Indeed, several entry inhibitors targeting different steps of the HCV entry process have been demonstrated to prevent and/or delay HCV infection in vivo using the uPA-SCID mouse model. The first study investigated the ability of polyclonal patient-derived neutralizing antibodies to protect mice from HCV infection with a homologous viral strain (Vanwolleghem et al. 2008). This passive immunization strategy protected 5 out of 8 challenged mice. Although this study did not investigate cross-reactive neutralization of various viral strains, it provides promising evidence, which opens the perspective for potential neutralizing antibody-based immunotherapies to prevent HCV infection (Vanwolleghem et al. 2008). Recently, the ability of neutralizing antibodies to cross-neutralize different HCV genotypes in vivo was assessed (Meuleman et al. 2011b). Remarkably, protection against challenge with heterologous viral strains was achieved in a portion of the animals, though differences were observed between genotypes and depending on the viral load of the inoculum (Meuleman et al. 2011b). Importantly, although no sterilizing immunity was achieved, a delay in the establishment of HCV infection was observed and adaptive mutations in the HCV envelope glycoproteins were absent in the majority of nonprotected animals, indicating that the failure of cross-neutralization in vivo was most likely not directly linked to viral escape (Meuleman et al. 2011b). Furthermore, another study assessed the ability of human cross-neutralizing monoclonal anti-E2 antibodies derived from a chronic HCV patient and demonstrated that the majority of chimeric mice were protected from challenge with a heterologous virus (Law et al. 2008). Most recently, novel human cross-neutralizing anti-envelope antibodies targeting five novel antigenic regions (ARs) designated numerically have been described and these antibodies (particularly those that bind AR3A, AR4A, and AR5A) are also characterized by the ability to protect from heterologous virus challenge in genetically humanized mice (Giang et al. 2012). Future studies assessing the potential of potent broadly cross-neutralizing monoclonal antibodies (independently or in combination) will allow determination of whether neutralizing antibodies have the potential for clinical application in prevention of HCV infection. Interestingly, other compounds targeting the HCV envelope and preventing attachment of the virion to target cells, such as the lectin Griffithsin, also demonstrated the ability to delay HCV infection in vivo (Meuleman et al. 2011a).
In addition to virus-neutralizing antibodies, antivirals that target host cell surface factors are being developed (reviewed in Zeisel et al. 2011). Antibodies directed against CD81 and SR-BI have been investigated in both protection and treatment studies. While both anti-CD81 and anti-SR-BI monoclonal antibodies protect mice from challenge with HCV, only anti-SR-BI monoclonal antibodies appear also able to reduce viral spread (Lacek et al. 2012; Meuleman et al. 2008, 2012). Anti-CLDN1 antibodies are currently being evaluated in the uPA-SCID mouse model to assess their antiviral activity against HCV infection. Furthermore, the clinically available kinase inhibitor erlotinib, targeting EGFR, and the cholesterol lowering drug ezetimibe, targeting NPC1L1, significantly impaired the establishment of HCV infection in the uPA-SCID mouse model (Lupberger et al. 2011; Sainz et al. 2012). Given the relevance of host cell kinases for HCV entry (Lupberger et al. 2011) and the number of kinase inhibitors being developed to treat a wide variety of human diseases, kinase inhibitors have been suggested as a novel class of antivirals for the prevention and treatment of HCV infection.
Since HCV entry is a major target of B cell responses, the virus–host interactions during viral entry are also relevant for HCV vaccine development. Indeed, B cell vaccines inducing cross-neutralizing antibodies are in preclinical and clinical development (Frey et al. 2010; Garrone et al. 2011; Halliday et al. 2011; Houghton 2012).
6 Conclusions and Perspectives
Since the discovery of HCV more than 20 years ago, the development of model systems has allowed investigators to decipher the molecular mechanisms of the different steps of the viral life cycle. In the past decade, several host cell surface factors contributing to HCV binding and/or entry have been identified. The functional relevance of most factors has been validated in cell culture and animal model systems. The thorough characterization of the role of each of these host factors enabled researchers to gain insights into the complex process of HCV entry. Given the complexity of virus–host interactions during this step of the viral life cycle, viral entry offers numerous potential targets for antiviral therapy. Since HCV entry is a major target of B cell responses, a detailed understanding of virus–host interactions during viral entry is also relevant for HCV vaccine development. Although highly orchestrated and complex, the mechanism of viral entry is conserved for all HCV genotypes and appears to be restricted to a single pathway. In contrast to the highly variable virus, host factors are less susceptible to mutations; therefore targeting host factors may allow increase of the genetic barrier to resistance (reviewed in Zeisel et al. 2012). Furthermore, the efficient in vitro and in vivo neutralization of patient-derived HCV isolates known to escape autologous neutralizing responses, by using monoclonal antibodies directed against the HCV envelope glycoproteins or CD81, CLDN1, and SR-BI, suggests that viral entry is a viable target for prevention of HCV reinfection of the liver graft (Fafi-Kremer et al. 2010; Fofana et al. 2010; Lacek et al. 2012; Zahid et al. 2012). Given the essential role of host cell surface entry factors in HCV cell-to-cell transmission and thus for spread and maintenance of infection, it is not surprising that defined entry inhibitors have also been shown to prevent cell-to-cell transmission and viral spread in vivo (Lacek et al. 2012; Meuleman et al. 2012). Thus entry inhibitors may hold promise for the development of novel antiviral strategies for the prevention and treatment of HCV infection.
Abbreviations
- Apo:
-
Apolipoprotein
- CD81:
-
Cluster of differentiation 81
- CLDN1:
-
Claudin 1
- EGFR:
-
Epidermal growth factor receptor
- EphA2:
-
Ephrin receptor A2
- HCV:
-
Hepatitis C virus
- HCVcc:
-
Cell culture-derived HCV
- HCV-LPs:
-
HCV-like particles
- HCVpp:
-
HCV pseudoparticles
- HDL:
-
High-density lipoprotein
- IgG:
-
Immunoglobulin G
- JFH1:
-
Japanese fulminant hepatitis 1
- LDL:
-
Low-density lipoprotein
- LDLR:
-
Low-density lipoprotein receptor
- LT:
-
Liver transplantation
- mAb:
-
Monoclonal antibody
- NPC1L1:
-
Niemann-Pick C1-like 1
- OCLN:
-
Occludin
- PKA:
-
Protein kinase A
- RTKs:
-
Receptor tyrosine kinases
- SR-BI:
-
Scavenger receptor class B type I (alias SCARBI)
- TG:
-
Triglyceride
- VLDL:
-
Very-low-density lipoprotein
References
Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX (1999) Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci USA 96:12766–12771
Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, Lai MM, Miyamura T, Wakita T, Suzuki T (2008) Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol 82:5715–5724
Albecka A, Belouzard S, de Beeck AO, Descamps V, Goueslain L, Bertrand-Michel J, Terce F, Duverlie G, Rouille Y, Dubuisson J (2012) Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55:998–1007
Altmann SW, Davis HR Jr, Yao X, Laverty M, Compton DS, Zhu LJ, Crona JH, Caplen MA, Hoos LM, Tetzloff G, Priestley T, Burnett DA, Strader CD, Graziano MP (2002) The identification of intestinal scavenger receptor class B, type I (SR-BI) by expression cloning and its role in cholesterol absorption. Biochim Biophys Acta 1580:77–93
Andre P, Komurian-Pradel F, Deforges S, Perret M, Berland JL, Sodoyer M, Pol S, Brechot C, Paranhos-Baccala G, Lotteau V (2002) Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J Virol 76:6919–6928
Andreo U, Maillard P, Kalinina O, Walic M, Meurs E, Martinot M, Marcellin P, Budkowska A (2007) Lipoprotein lipase mediates hepatitis C virus (HCV) cell entry and inhibits HCV infection. Cell Microbiol 9:2445–2456
Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R (2008) Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog 4:e1000035
Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, Zeisel MB, Baumert TF, Keck ZY, Foung SK, Pecheur EI, Pietschmann T (2010) Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol 84:5751–5763
Barth H, Schäfer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E, Weizsäcker FV, Blum HE, Baumert TF (2003) Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem 278:41003–41012
Barth H, Cerino R, Arcuri M, Hoffmann M, Schurmann P, Adah MI, Gissler B, Zhao X, Ghisetti V, Lavezzo B, Blum HE, von Weizsacker F, Vitelli A, Scarselli E, Baumert TF (2005a) Scavenger receptor class B type I and hepatitis C virus infection of primary tupaia hepatocytes. J Virol 79:5774–5785
Barth H, Ulsenheimer A, Pape GR, Diepolder HM, Hoffmann M, Neumann-Haefelin C, Thimme R, Henneke P, Klein R, Paranhos-Baccala G, Depla E, Liang TJ, Blum HE, Baumert TF (2005b) Uptake and presentation of hepatitis C virus-like particles by human dendritic cells. Blood 105:3605–3614
Barth H, Liang TJ, Baumert TF (2006a) Hepatitis C virus entry: molecular biology and clinical implications. Hepatology 44:527–535
Barth H, Schnober EK, Zhang F, Linhardt RJ, Depla E, Boson B, Cosset FL, Patel AH, Blum HE, Baumert TF (2006b) Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J Virol 80:10579–10590
Barth H, Schnober EK, Neumann-Haefelin C, Thumann C, Zeisel MB, Diepolder HM, Hu Z, Liang TJ, Blum HE, Thimme R, Lambotin M, Baumert TF (2008) Scavenger receptor class B is required for hepatitis C virus uptake and cross-presentation by human dendritic cells. J Virol 82:3466–3479
Bartosch B, Dubuisson J, Cosset FL (2003a) Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197: 633-642. J Exp Med 197:633–642
Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset FL (2003b) Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem 278:41624–41630
Bartosch B, Verney G, Dreux M, Donot P, Morice Y, Penin F, Pawlotsky JM, Lavillette D, Cosset FL (2005) An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol 79:8217–8229
Bartosch B, Cosset FL (2006) Cell entry of hepatitis C virus. Virology 348:1–12
Baumert TF, Ito S, Wong DT, Liang TJ (1998) Hepatitis C virus structural proteins assemble into viruslike particles in insect cells. J Virol 72:3827–3836
Benedicto I, Molina-Jimenez F, Barreiro O, Maldonado-Rodriguez A, Prieto J, Moreno-Otero R, Aldabe R, Lopez-Cabrera M, Majano PL (2008) Hepatitis C virus envelope components alter localization of hepatocyte tight junction-associated proteins and promote occludin retention in the endoplasmic reticulum. Hepatology 48:1044–1053
Benedicto I, Molina-Jimenez F, Bartosch B, Cosset FL, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernandez A, Aldabe R, Lopez-Cabrera M, Majano PL (2009) The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J Virol 83:8012–8020
Benga WJ, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C (2010) Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles. Hepatology 51:43–53
Berneis KK, Krauss RM (2002) Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res 43:1363–1379
Bertaux C, Dragic T (2006) Different domains of CD81 mediate distinct stages of hepatitis C virus pseudoparticle entry. J Virol 80:4940–4948
Blanchard E, Brand D, Trassard S, Goudeau A, Roingeard P (2002) Hepatitis C virus-like particle morphogenesis. J. Virol. 76:4073–4079
Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y (2006) Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80:6964–6972
Boriskin YS, Pecheur EI, Polyak SJ (2006) Arbidol: a broad-spectrum antiviral that inhibits acute and chronic HCV infection. Virology J 3:56
Boriskin YS, Leneva IA, Pecheur EI, Polyak SJ (2008) Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr Medicinal Chem 15:997–1005
Boulant S, Targett-Adams P, McLauchlan J (2007) Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol 88:2204–2213
Bradley D, McCaustland K, Krawczynski K, Spelbring J, Humphrey C, Cook EH (1991) Hepatitis C virus: buoyant density of the factor VIII-derived isolate in sucrose. J Med Virol 34:206–208
Bridge SH, Sheridan DA, Felmlee DJ, Nielsen SU, Thomas HC, Taylor-Robinson SD, Neely RD, Toms GL, Bassendine MF (2011) Insulin resistance and low-density apolipoprotein B-associated lipoviral particles in hepatitis C virus genotype 1 infection. Gut 60:680–687
Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, Timpe JM, Krieger SE, Baumert TF, Tellinghuisen TL, Wong-Staal F, Balfe P, McKeating JA (2011) Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J Virol 85:596–605
Bukh J (2012) Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 142(1279–1287):e1273
Catanese MT, Graziani R, von Hahn T, Moreau M, Huby T, Paonessa G, Santini C, Luzzago A, Rice CM, Cortese R, Vitelli A, Nicosia A (2007) High-avidity monoclonal antibodies against the human scavenger class B type I receptor efficiently block hepatitis C virus infection in the presence of high-density lipoprotein. J Virol 81:8063–8071
Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, Nicosia A (2010) Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol 84:34–43
Chang KS, Jiang J, Cai Z, Luo G (2007) Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J Virol 81:13783–13793
Chevaliez S, Asselah T (2011) Mechanisms of non-response to antiviral treatment in chronic hepatitis C. Clin Res Hepatol Gastroenterol 35(Suppl 1):S31–41
Choo Q-L, Kuo A, Weiner AJ, Overby LR, Bradley DW, Houghton M (1989) Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362
Codran A, Royer C, Jaeck D, Bastien-Valle M, Baumert TF, Kieny MP, Pereira CA, Martin JP (2006) Entry of hepatitis C virus pseudotypes into primary human hepatocytes by clathrin-dependent endocytosis. J Gen Virol 87:2583–2593
Coller KE, Berger KL, Heaton NS, Cooper JD, Yoon R, Randall G (2009) RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog 5:e1000702
Cormier EG, Durso RJ, Tsamis F, Boussemart L, Manix C, Olson WC, Gardner JP, Dragic T (2004) L-SIGN (CD209L) and DC-SIGN (CD209) mediate transinfection of liver cells by hepatitis C virus. Proc Natl Acad Sci USA 101:14067–14072
Cukierman L, Meertens L, Bertaux C, Kajumo F, Dragic T (2009) Residues in a highly conserved claudin-1 motif are required for hepatitis C virus entry and mediate the formation of cell-cell contacts. J Virol 83:5477–5484
Cun W, Jiang J, Luo G (2010) The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J Virol 84:11532–11541
Dao Thi VL, Granier C, Zeisel MB, Guerin M, Mancip J, Granio O, Penin F, Lavillette D, Bartenschlager R, Baumert TF, Cosset FL, Dreux M (2012) Characterization of Hepatitis C Virus Particle Subpopulations Reveals Multiple Usage of the Scavenger Receptor BI for Entry Steps. J Biol Chem 287:31242–31257
Davis GL, Nelson DR, Terrault N, Pruett TL, Schiano TD, Fletcher CV, Sapan CV, Riser LN, Li Y, Whitley RJ, Gnann JW Jr (2005) A randomized, open-label study to evaluate the safety and pharmacokinetics of human hepatitis C immune globulin (Civacir) in liver transplant recipients. Liver Transpl 11:941–949
Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, Schoggins JW, Catanese MT, Burton DR, Law M, Rice CM, Ploss A (2011) A genetically humanized mouse model for hepatitis C virus infection. Nature 474:208–211
Dreux M, Pietschmann T, Granier C, Voisset C, Ricard-Blum S, Mangeot PE, Keck Z, Foung S, Vu-Dac N, Dubuisson J, Bartenschlager R, Lavillette D, Cosset FL (2006) High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J Biol Chem 281:18285–18295
Dreux M, Boson B, Ricard-Blum S, Molle J, Lavillette D, Bartosch B, Pecheur EI, Cosset FL (2007) The Exchangeable Apolipoprotein ApoC-I Promotes Membrane Fusion of Hepatitis C Virus. J Biol Chem 282:32357–32369
Dreux M, Dao Thi VL, Fresquet J, Guerin M, Julia Z, Verney G, Durantel D, Zoulim F, Lavillette D, Cosset FL, Bartosch B (2009) Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extra-cellular domains. PLoS Pathog 5:e1000310
Drummer HE, Boo I, Poumbourios P (2007) Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J Gen Virol 88:1144–1148
Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM (2007) Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805
Fafi-Kremer S, Fofana I, Soulier E, Carolla P, Meuleman P, Leroux-Roels G, Patel AH, Cosset FL, Pessaux P, Doffoel M, Wolf P, Stoll-Keller F, Baumert TF (2010) Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J Exp Med 207:2019–2031
Farquhar MJ, Harris HJ, Diskar M, Jones S, Mee CJ, Nielsen SU, Brimacombe CL, Molina S, Toms GL, Maurel P, Howl J, Herberg FW, van Ijzendoorn SC, Balfe P, McKeating JA (2008) Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity. J Virol 82:8797–8811
Farquhar MJ, Hu K, Harris HJ, Davis C, Brimacombe CL, Fletcher SJ, Baumert TF, Rappoport JZ, Balfe P, McKeating JA (2012) Hepatitis C virus induces CD81 and claudin-1 endocytosis. J Virol 86:4305–4316
Felmlee DJ, Sheridan DA, Bridge SH, Nielsen SU, Milne RW, Packard CJ, Caslake MJ, McLauchlan J, Toms GL, Neely RD, Bassendine MF (2010) Intravascular transfer contributes to postprandial increase in numbers of very-low-density hepatitis C virus particles. Gastroenterology 139: 1774-1783, 1783 e1771-1776.
Flint M, Maidens CM, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Hittingbottom A, Levy S, McKeating J (1999) Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor CD81. J. Virol. 73:6235–6244
Fofana I, Krieger SE, Grunert F, Glauben S, Xiao F, Fafi-Kremer S, Soulier E, Royer C, Thumann C, Mee CJ, McKeating JA, Dragic T, Pessaux P, Stoll-Keller F, Schuster C, Thompson J, Baumert TF (2010) Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 39:953–964
Fofana I, Fafi-Kremer S, Carolla P, Fauvelle C, Zahid MN, Turek M, Heydmann L, Cury K, Hayer J, Combet C, Cosset FL, Pietschmann T, Hiet MS, Bartenschlager R, Habersetzer F, Doffoel M, Keck ZY, Foung SKH, Zeisel MB, Stoll-Keller F, Baumert TF (2012) Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology 143(223–233):e229
Frey SE, Houghton M, Coates S, Abrignani S, Chien D, Rosa D, Pileri P, Ray R, Di Bisceglie AM, Rinella P, Hill H, Wolff MC, Schultze V, Han JH, Scharschmidt B, Belshe RB (2010) Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 28:6367–6373
Gardner JP, Durso RJ, Arrigale RR, Donovan GP, Maddon PJ, Dragic T, Olson WC (2003) L-SIGN (CD 209L) is a liver-specific capture receptor for hepatitis C virus. Proc Natl Acad Sci USA 100:4498–4503
Garrone P, Fluckiger AC, Mangeot PE, Gauthier E, Dupeyrot-Lacas P, Mancip J, Cangialosi A, Du Chene I, LeGrand R, Mangeot I, Lavillette D, Bellier B, Cosset FL, Tangy F, Klatzmann D, Dalba C (2011) A prime-boost strategy using virus-like particles pseudotyped for HCV proteins triggers broadly neutralizing antibodies in macaques. Sci Transl Med 3: 94ra71.
Gastaminza P, Kapadia SB, Chisari FV (2006) Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074–11081
Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV (2008) Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129
Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M (2012) Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci USA 109:6205–6210
Grove J, Huby T, Stamataki Z, Vanwolleghem T, Meuleman P, Farquhar M, Schwarz A, Moreau M, Owen JS, Leroux-Roels G, Balfe P, McKeating JA (2007) Scavenger receptor BI and BII expression levels modulate Hepatitis C virus infectivity. J Virol 81:3162–3169
Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA (2008) Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J Virol 82:12020–12029
Haberstroh A, Schnober EK, Zeisel MB, Carolla P, Barth H, Blum HE, Cosset FL, Koutsoudakis G, Bartenschlager R, Union A, Depla E, Owsianka A, Patel AH, Schuster C, Stoll-Keller F, Doffoel M, Dreux M, Baumert TF (2008) Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 135(1719–1728):e1711
Haid S, Pietschmann T, Pecheur EI (2009) Low pH-dependent hepatitis C virus membrane fusion depends on E2 integrity, target lipid composition, and density of virus particles. J Biol Chem 284:17657–17667
Halliday J, Klenerman P, Barnes E (2011) Vaccination for hepatitis C virus: closing in on an evasive target. Exp Rev Vaccines 10:659–672
Harris HJ, Farquhar MJ, Mee CJ, Davis C, Reynolds GM, Jennings A, Hu K, Yuan F, Deng H, Hubscher SG, Han JH, Balfe P, McKeating JA (2008) CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol 82:5007–5020
Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, Balfe P, McKeating JA (2010) Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem 285:21092–21102
Hawke RL, Schrieber SJ, Soule TA, Wen Z, Smith PC, Reddy KR, Wahed AS, Belle SH, Afdhal NH, Navarro VJ, Berman J, Liu QY, Doo E, Fried MW (2010) Silymarin ascending multiple oral dosing phase I study in noncirrhotic patients with chronic hepatitis C. J Clin Pharmacol 50:434–449
Helle F, Wychowski C, Vu-Dac N, Gustafson KR, Voisset C, Dubuisson J (2006) Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J Biol Chem 281:25177–25183
Hijikata M, Shimizu YK, Kato H, Iwamoto A, Shih JW, Alter HJ, Purcell RH, Yoshikura H (1993) Equilibrium centrifugation studies of hepatitis C virus: evidence for circulating immune complexes. J Virol 67:1953–1958
Hishiki T, Shimizu Y, Tobita R, Sugiyama K, Ogawa K, Funami K, Ohsaki Y, Fujimoto T, Takaku H, Wakita T, Baumert TF, Miyanari Y, Shimotohno K (2010) Infectivity of hepatitis C virus is influenced by association with apolipoprotein E isoforms. J Virol 84:12048–12057
Houghton M (2012) Prospects for prophylactic and therapeutic vaccines against the hepatitis C viruses. Immunological Rev 239:99–108
Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA (2003) Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA 100:7271–7276
Huang H, Sun F, Owen DM, Li W, Chen Y, Gale M Jr, Ye J (2007) Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci USA 104:5848–5853
Jamil H, Chu CH, Dickson JK Jr, Chen Y, Yan M, Biller SA, Gregg RE, Wetterau JR, Gordon DA (1998) Evidence that microsomal triglyceride transfer protein is limiting in the production of apolipoprotein B-containing lipoproteins in hepatic cells. J Lipid Res 39:1448–1454
Jia L, Betters JL, Yu L (2011) Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annual Rev Physiol 73:239–259
Jiang J, Luo G (2009) Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 83:12680–12691
Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G (2012) Hepatitis C Virus Attachment Mediated by Apolipoprotein E Binding to Cell Surface Heparan Sulfate. J Virol 86:7256–7267
Jones CT, Catanese MT, Law LM, Khetani SR, Syder AJ, Ploss A, Oh TS, Schoggins JW, MacDonald MR, Bhatia SN, Rice CM (2010) Real-time imaging of hepatitis C virus infection using a fluorescent cell-based reporter system. Nat Biotechnol 28:167–171
Kanto T, Hayashi N, Takehara T, Hagiwara H, Mita E, Naito M, Kasahara A, Fusamoto H, Kamada T (1995) Density analysis of hepatitis C virus particle population in the circulation of infected hosts: implications for virus neutralization or persistence. J Hepatol 22:440–448
Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV (2007) Initiation of Hepatitis C Virus Infection Is Dependent on Cholesterol and Cooperativity between CD81 and Scavenger Receptor B Type I. J Virol 81:374–383
Kobayashi M, Bennett MC, Bercot T, Singh IR (2006) Functional analysis of hepatitis C virus envelope proteins, using a cell-cell fusion assay. J Virol 80:1817–1825
Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R (2006) Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320
Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, Schnober EK, Mee C, Soulier E, Royer C, Lambotin M, Grunert F, Dao Thi VL, Dreux M, Cosset FL, McKeating JA, Schuster C, Baumert TF (2010) Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 51:1144–1157
Lacek K, Vercauteren K, Grzyb K, Naddeo M, Verhoye L, Slowikowski MP, Fafi-Kremer S, Patel AH, Baumert TF, Folgori A, Leroux-Roels G, Cortese R, Meuleman P, Nicosia A (2012) Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. J Hepatol 57:17–23
Lavie M, Goffard A, Dubuisson J (2007) Assembly of a functional HCV glycoprotein heterodimer. Current issues in molecular biology 9:71–86
Lavillette D, Bartosch B, Nourrisson D, Verney G, Cosset FL, Penin F, Pecheur EI (2006) Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J Biol Chem 281:3909–3917
Lavillette D, Pecheur EI, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset FL (2007) Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol 81:8752–8765
Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR (2008) Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14:25–27
Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, Rice CM (2006) Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc Natl Acad Sci USA 103:3805–3809
Lindenbach BD, Thiel HJ, Rice CM (2007) Flaviviridae: the viruses and their replication. In “Fields Virology” (DM Knipe and PM Howley, eds.), pp. 1101-1152. Lippincott-Raven, Philadelphia
Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T (2009) Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83:2011–2014
Liu S, McCormick KD, Zhao W, Zhao T, Fan D, Wang T (2012) Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology 56:484–491
Lozach PY, Lortat-Jacob H, de Lacroix de Lavalette A, Staropoli I, Foung S, Amara A, Houles C, Fieschi F, Schwartz O, Virelizier JL, Arenzana-Seisdedos F, Altmeyer R (2003) DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J Biol Chem 278: 20358-20366
Lozach PY, Amara A, Bartosch B, Virelizier JL, Arenzana-Seisdedos F, Cosset FL, Altmeyer R (2004) C-type lectins L-SIGN and DC-SIGN capture and transmit infectious hepatitis C virus pseudotype particles. J Biol Chem 279:32035–32045
Ludwig IS, Lekkerkerker AN, Depla E, Bosman F, Musters RJ, Depraetere S, van Kooyk Y, Geijtenbeek TB (2004) Hepatitis C virus targets DC-SIGN and L-SIGN to escape lysosomal degradation. J Virol 78:8322–8332
Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF (2011) EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595
Maillard P, Huby T, Andreo U, Moreau M, Chapman J, Budkowska A (2006) The interaction of natural hepatitis C virus with human scavenger receptor SR-BI/Cla1 is mediated by ApoB-containing lipoproteins. Faseb J 20:735–737
Maillard P, Walic M, Meuleman P, Roohvand F, Huby T, Le Goff W, Leroux-Roels G, Pecheur EI, Budkowska A (2011) Lipoprotein lipase inhibits hepatitis C virus (HCV) infection by blocking virus cell entry. PLoS ONE 6:e26637
Matsumura T, Hu Z, Kato T, Dreux M, Zhang YY, Imamura M, Hiraga N, Juteau JM, Cosset FL, Chayama K, Vaillant A, Liang TJ (2009) Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology 137:673–681
Mazumdar B, Banerjee A, Meyer K, Ray R (2011) Hepatitis C virus E1 envelope glycoprotein interacts with apolipoproteins in facilitating entry into hepatocytes. Hepatology 54:1149–1156
Mee CJ, Harris HJ, Farquhar MJ, Wilson G, Reynolds G, Davis C, van ISC, Balfe P, McKeating JA (2009) Polarization restricts hepatitis C virus entry into HepG2 hepatoma cells. J Virol 83: 6211-6221
Meertens L, Bertaux C, Dragic T (2006) Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol 80:11571–11578
Meertens L, Bertaux C, Cukierman L, Cormier E, Lavillette D, Cosset FL, Dragic T (2008) The tight junction proteins claudin-1, -6, and -9 are entry cofactors for hepatitis C virus. J Virol 82:3555–3560
Mensa L, Crespo G, Gastinger MJ, Kabat J, Perez-del-Pulgar S, Miquel R, Emerson SU, Purcell RH, Forns X (2011) Hepatitis C virus receptors claudin-1 and occludin after liver transplantation and influence on early viral kinetics. Hepatology 53:1436–1445
Merz A, Long G, Hiet MS, Brugger B, Chlanda P, Andre P, Wieland F, Krijnse-Locker J, Bartenschlager R (2011) Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J Biol Chem 286:3018–3032
Meuleman P, Hesselgesser J, Paulson M, Vanwolleghem T, Desombere I, Reiser H, Leroux-Roels G (2008) Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 48:1761–1768
Meuleman P, Albecka A, Belouzard S, Vercauteren K, Verhoye L, Wychowski C, Leroux-Roels G, Palmer KE, Dubuisson J (2011a) Griffithsin has antiviral activity against hepatitis C virus. Antimicrob Agents Chemother 55:5159–5167
Meuleman P, Bukh J, Verhoye L, Farhoudi A, Vanwolleghem T, Wang RY, Desombere I, Alter H, Purcell RH, Leroux-Roels G (2011b) In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 53:755–762
Meuleman P, Catanese MT, Verhoye L, Desombere I, Farhoudi A, Jones CT, Sheahan T, Grzyb K, Cortese R, Rice CM, Leroux-Roels G, Nicosia A (2012) A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 55:364–372
Meunier JC, Russell RS, Engle RE, Faulk KN, Purcell RH, Emerson SU (2008) Apolipoprotein c1 association with hepatitis C virus. J Virol 82:9647–9656
Miyamoto H, Okamoto H, Sato K, Tanaka T, Mishiro S (1992) Extraordinarily low density of hepatitis C virus estimated by sucrose gradient centrifugation and the polymerase chain reaction. J Gen Virol 73:715–718
Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K (2007) The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097
Monazahian M, Bohme I, Bonk S, Koch A, Scholz C, Grethe S, Thomssen R (1999) Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol 57:223–229
Morikawa K, Zhao Z, Date T, Miyamoto M, Murayama A, Akazawa D, Tanabe J, Sone S, Wakita T (2007) The roles of CD81 and glycosaminoglycans in the adsorption and uptake of infectious HCV particles. J Med Virol 79:714–723
Murray CL, Rice CM (2011) Turning Hepatitis C Virus into a Real Virus. Annual Rev Microbiol 65:307–327
Neumann UP, Biermer M, Eurich D, Neuhaus P, Berg T (2010) Successful prevention of hepatitis C virus (HCV) liver graft reinfection by silibinin mono-therapy. J Hepatol 52:951–952
Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL (2006) Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J Virol 80:2418–2428
Op De Beeck A, Dubuisson J (2003) Topology of hepatitis C virus envelope glycoproteins. Rev Med Virol 13:233–241
Out R, Hoekstra M, Spijkers JA, Kruijt JK, van Eck M, Bos IS, Twisk J, Van Berkel TJ (2004) Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res 45:2088–2095
Owen DM, Huang H, Ye J, Gale M Jr (2009) Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108
Packard CJ, Shepherd J (1997) Lipoprotein heterogeneity and apolipoprotein B metabolism. Arteriosclerosis Thrombosis Vascular Biol 17:3542–3556
Pawlotsky JM (2011) Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53:1742–1751
Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S (1998) Binding of hepatitis C virus to CD81. Science 282:938–941
Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM (2009) Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886
Pohlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA (2003) Hepatitis C Virus Glycoproteins Interact with DC-SIGN and DC-SIGNR. J Virol 77:4070–4080
Polyak SJ, Morishima C, Shuhart MC, Wang CC, Liu Y, Lee DY (2007) Inhibition of T-cell inflammatory cytokines, hepatocyte NF-kappaB signaling, and HCV infection by standardized Silymarin. Gastroenterology 132:1925–1936
Prentoe J, Jensen TB, Meuleman P, Serre SB, Scheel TK, Leroux-Roels G, Gottwein JM, Bukh J (2011) Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J Virol 85:2224–2234
Price DA, Bassendine MF, Norris SM, Golding C, Toms GL, Schmid ML, Morris CM, Burt AD, Donaldson PT (2006) Apolipoprotein epsilon3 allele is associated with persistent hepatitis C virus infection. Gut 55:715–718
Prince AM, Huima-Byron T, Parker TS, Levine DM (1996) Visualization of hepatitis C virions and putative defective interfering particles isolated from low-density lipoproteins. J Viral Hepat 3:11–17
Reynolds GM, Harris HJ, Jennings A, Hu K, Grove J, Lalor PF, Adams DH, Balfe P, Hubscher SG, McKeating JA (2008) Hepatitis C virus receptor expression in normal and diseased liver tissue. Hepatology 47:418–427
Roingeard P, Hourioux C, Blanchard E, Prensier G (2008) Hepatitis C virus budding at lipid droplet-associated ER membrane visualized by 3D electron microscopy. Histochem Cell Biol 130:561–566
Rothblat GH, Phillips MC (2010) High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol 21:229–238
Rutter K, Scherzer TM, Beinhardt S, Kerschner H, Stattermayer AF, Hofer H, Popow-Kraupp T, Steindl-Munda P, Ferenci P (2011) Intravenous silibinin as ‘rescue treatment’ for on-treatment non-responders to pegylated interferon/ribavirin combination therapy. Antiviral Therapy 16:1327–1333
Rye KA, Bursill CA, Lambert G, Tabet F, Barter PJ (2009) The metabolism and anti-atherogenic properties of HDL. J Lipid Res 50(Suppl):S195–200
Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL (2012) Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285
Saito H, Dhanasekaran P, Nguyen D, Baldwin F, Weisgraber KH, Wehrli S, Phillips MC, Lund-Katz S (2003) Characterization of the heparin binding sites in human apolipoprotein E. J Biol Chem 278:14782–14787
Saunier B, Triyatni M, Ulianich L, Maruvada P, Yen P, Kohn LD (2003) Role of the asialoglycoprotein receptor in binding and entry of hepatitis C virus structural proteins in cultured human hepatocytes. J Virol 77:546–559
Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A (2002) The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025
Shachter NS (2001) Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism. Curr Opin Lipidol 12:297–304
Shelness GS, Sellers JA (2001) Very-low-density lipoprotein assembly and secretion. Curr Opin Lipidol 12:151–157
Shimizu Y, Hishiki T, Sugiyama K, Ogawa K, Funami K, Kato A, Ohsaki Y, Fujimoto T, Takaku H, Shimotohno K (2010) Lipoprotein lipase and hepatic triglyceride lipase reduce the infectivity of hepatitis C virus (HCV) through their catalytic activities on HCV-associated lipoproteins. Virology 407:152–159
Skov M, Tonnesen CK, Hansen GH, Danielsen EM (2011) Dietary cholesterol induces trafficking of intestinal Niemann-Pick Type C1 Like 1 from the brush border to endosomes. Am J Physiol 300:G33–40
Steinmann D, Barth H, Gissler B, Schürmann P, Adah MI, Gerlach JT, Pape GR, Depla E, Jacobs D, Maertens G, Patel AH, Inchauspé G, Liang TJ, Blum HE, Baumert TF (2004) Inhibition of hepatitis C virus-like particle binding to target cells by antiviral antibodies in acute and chronic hepatitis C. J Virol 78:9030–9040
Syder AJ, Lee H, Zeisel MB, Grove J, Soulier E, Macdonald J, Chow S, Chang J, Baumert TF, McKeating JA, McKelvy J, Wong-Staal F (2011) Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J Hepatol 54:48–55
Thomssen R, Bonk S, Propfe C, Heermann K-H, Koechel HG, Uy A (1992) Association of hepatitis C virus in human sera with beta-lipoprotein. Med Microbiol Immunol 181:293–300
Thomssen R, Bonk S, Thiele A (1993) Density heterogeneities of hepatitis C virus in human sera due to the binding of beta-lipoproteins and immunoglobulins. Med Microbiol Immunol 182:329–334
Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA (2008) Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24
Triyatni M, Saunier B, Maruvada P, Davis AR, Ulianich L, Heller T, Patel A, Kohn LD, Liang TJ (2002) Interaction of hepatitis C virus-like particles and cells: a model system for studying viral binding and entry. J Virol 76:9335–9344
Trotard M, Lepere-Douard C, Regeard M, Piquet-Pellorce C, Lavillette D, Cosset FL, Gripon P, Le Seyec J (2009) Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. Faseb J 23:3780–3789
Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM (2006) Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741
Tscherne DM, Evans MJ, von Hahn T, Jones CT, Stamataki Z, McKeating JA, Lindenbach BD, Rice CM (2007) Superinfection exclusion in cells infected with hepatitis C virus. J Virol 81:3693–3703
Valasek MA, Weng J, Shaul PW, Anderson RG, Repa JJ (2005) Caveolin-1 is not required for murine intestinal cholesterol transport. J Biol Chem 280:28103–28109
VanCompernolle SE, Wiznycia AV, Rush JR, Dhanasekaran M, Baures PW, Todd SC (2003) Small molecule inhibition of hepatitis C virus E2 binding to CD81. Virology 314:371–380
Vanwolleghem T, Bukh J, Meuleman P, Desombere I, Meunier JC, Alter H, Purcell RH, Leroux-Roels G (2008) Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 47:1846–1855
Vieyres G, Dubuisson J, Patel AH (2011) Characterization of antibody-mediated neutralization directed against the hypervariable region 1 of hepatitis C virus E2 glycoprotein. J Gen Virol 92:494–506
Voisset C, Callens N, Blanchard E, Op De Beeck A, Dubuisson J, Vu-Dac N (2005) High density lipoproteins facilitate hepatitis C virus entry through the scavenger receptor class B type I. J Biol Chem 280:7793–7799
Voisset C, Lavie M, Helle F, Op De Beeck A, Bilheu A, Bertrand-Michel J, Terce F, Cocquerel L, Wychowski C, Vu-Dac N, Dubuisson J (2008) Ceramide enrichment of the plasma membrane induces CD81 internalization and inhibits hepatitis C virus entry. Cell Microbiol 10:606–617
von Hahn T, Lindenbach BD, Boullier A, Quehenberger O, Paulson M, Rice CM, McKeating JA (2006) Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 43:932–942
von Hahn T, Rice CM (2008) Hepatitis C virus entry. J Biol Chem 283:3689–3693
Wagoner J, Negash A, Kane OJ, Martinez LE, Nahmias Y, Bourne N, Owen DM, Grove J, Brimacombe C, McKeating JA, Pecheur EI, Graf TN, Oberlies NH, Lohmann V, Cao F, Tavis JE, Polyak SJ (2010) Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology 51:1912–1921
Wang H, Gilham D, Lehner R (2007) Proteomic and lipid characterization of apolipoprotein B-free luminal lipid droplets from mouse liver microsomes: implications for very low density lipoprotein assembly. J Biol Chem 282:33218–33226
Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, Keck ZY, Foung SK, Pietschmann T, Rice CM, Patel AH (2009) CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol 90:48–58
Wunschmann S, Medh JD, Klinzmann D, Schmidt WN, Stapleton JT (2000) Characterization of hepatitis C virus (HCV) and HCV E2 interactions with CD81 and the low-density lipoprotein receptor. J Virol 74:10055–10062
Yamamoto M, Aizaki H, Fukasawa M, Teraoka T, Miyamura T, Wakita T, Suzuki T (2011) Structural requirements of virion-associated cholesterol for infectivity, buoyant density and apolipoprotein association of hepatitis C virus. J Gen Virol 92:2082–2087
Yang W, Qiu C, Biswas N, Jin J, Watkins SC, Montelaro RC, Coyne CB, Wang T (2008) Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J Biol Chem 283:8643–8653
Zahid MN, Turek M, Xiao F, Thi VL, Guerin M, Fofana I, Bachellier P, Thompson J, Delang L, Neyts J, Bankwitz D, Pietschmann T, Dreux M, Cosset FL, Grunert F, Baumert TF, Zeisel MB (2012) The post-binding activity of scavenger receptor BI mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology. Oct 18. doi: 10.1002/hep.26097. [Epub ahead of print]
Zeisel MB, Koutsoudakis G, Schnober EK, Haberstroh A, Blum HE, Cosset F-L, Wakita T, Jaeck D, Doffoel M, Royer C, Soulier E, Schvoerer E, Schuster C, Stoll-Keller F, Bartenschlager R, Pietschmann T, Barth H, Baumert TF (2007) Scavenger receptor BI is a key host factor for Hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 46:1722–1731
Zeisel MB, Fofana I, Fafi-Kremer S, Baumert TF (2011) Hepatitis C virus entry into hepatocytes: Molecular mechanisms and targets for antiviral therapies. J Hepatol 54:566–576
Zeisel MB, Lupberger J, Fofana I, Baumert TF (2012) Host-targeting agents for prevention and treatment of viral hepatitis C - perspectives and challenges. J Hepatol Oct 4. pii: S0168–8278(12)00753-2. doi: 10.1016/j.jhep.2012.09.022. [Epub ahead of print]
Zheng A, Yuan F, Li Y, Zhu F, Hou P, Li J, Song X, Ding M, Deng H (2007) Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J Virol 81:12465–12471
Zheng C, Khoo C, Furtado J, Ikewaki K, Sacks FM (2008) Dietary monounsaturated fat activates metabolic pathways for triglyceride-rich lipoproteins that involve apolipoproteins E and C-III. Am J Clin Nutri 88:272–281
Acknowledgments
The authors acknowledge financial support of their work by the European Union (ERC-2008-AdG-233130-HEPCENT and INTERREG-IV-FEDER-Hepato-Regio-Net 2009 and 2012), Laboratoire d’Excellence HEPSYS (Investissement d’Avenir; ANR-10-LAB-28), ANRS (2008/354, 2009/183, 2011/132, 2012/239), Inserm, University of Strasbourg and the Strasbourg University Hospitals, France. DJF is a recipient of an EASL fellowship. We also would like to thank Isabel Fofana (Inserm U748, Strasbourg, France) for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Zeisel, M.B., Felmlee, D.J., Baumert, T.F. (2013). Hepatitis C Virus Entry. In: Bartenschlager, R. (eds) Hepatitis C Virus: From Molecular Virology to Antiviral Therapy. Current Topics in Microbiology and Immunology, vol 369. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-27340-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-642-27340-7_4
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-27339-1
Online ISBN: 978-3-642-27340-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)