
Abstract
Circadian gene expression is a pervasive feature of tissue physiology, regulating approx. 10% of transcript and protein abundance in tissues such as the liver. Technological developments have accelerated our ability to probe circadian variation of gene expression, in particular by using microarrays. Recent advances in high-throughput sequencing have similarly led to novel insights into the regulation of genes at the DNA and chromatin levels. Furthermore, tools such as RNA interference are being used to perturb gene function at a truly systems level, allowing dissection of the clockwork in increasing depth. This chapter will highlight progress in these areas, focusing on key techniques that have helped, and will continue to help, with the investigation of circadian physiology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Transcriptomics
- Genomics
- Systems biology
- Clock
- Circadian
- ChIP-chip
- ChIP-seq
- RNA-seq
- Interferomics
- Proteomics
- Metabolomics
1 Introduction
Dynamic changes in the topology of genomes and transcriptomes are not a newly recognized phenomenon—plasticity in DNA and RNA has long been recognized as a key regulatory point in most biological processes. However, on a 24-h timescale, it is only recently that the extent of changes at a systems level has begun to be appreciated (Reddy and O’Neill 2010). Progress in this arena has largely been propelled by advances in technology, which have allowed interrogation of DNA and RNA over time, at a truly global level (Akhtar et al. 2002; Panda et al. 2002; Rey et al. 2011).
The numbers of genes and transcripts regulated in a circadian fashion are not trivial. Various studies have estimated that approx. 10–15 % of mammalian transcripts undergo circadian oscillation in tissues such as the liver or heart and similar numbers of proteins oscillate over the circadian cycle (Akhtar et al. 2002; Panda et al. 2002; Storch et al. 2002; Ueda et al. 2002). Thus, when cells and tissues are viewed at the genomic, transcriptomic, and proteomic levels, they are not only in a state of flux because of their need to maintain body homeostasis but also because of the background influence of the circadian clock on the production of these macromolecules (Hastings et al. 2003; Reddy et al. 2006). In the extreme case, as occurs in cyanobacteria, the entire genome undergoes rhythmic change, which in turn sculpts the gene expression profile of thousands of genes (Johnson et al. 2008; Vijayan et al. 2009; Woelfle et al. 2007).
In this chapter, I will discuss recent advances in our understanding of the clockwork at the genomic and transcriptomic level, highlighting the importance of considering the circadian clock in analyses of cells and tissues for pharmacological experiments. I will also touch upon the types of high-throughput approaches that have been utilized to probe the clockwork at a systems level, which will have relevance to the pharmacologically minded scientist.
2 Genomic Level Analyses of the Clockwork
The genomic landscape is traditionally regarded as static and only subject to change when cells need to undergo fundamental, and often terminal, changes such as end differentiation (Kouzarides 2007). However, this line of thought has been challenged recently by observations that genome-level changes to DNA occur in disparate organisms, from bacteria to mammals. For example, in the circadian biologist’s favorite cyanobacterium, Synechococcus elongatus, its entire genome undergoes rhythmic supercoiling over a day, directing rhythmic abundance of mRNA (Kucho et al. 2005; Vijayan et al. 2009; Woelfle et al. 2007). In mammals, the changes are not thought to be as far reaching, but have been demonstrated at specific genomic loci where clock-relevant transcription factors, such as CLOCK and BMAL1, bind and modulate chromatin structure in a rhythmic manner (Ripperger and Schibler 2006). This has obvious implications for understanding RNA dynamics but also underscores the need for careful regard to sampling time in any experiment, since it cannot be assumed that the genome is “static” in terminally differentiated tissues or in cells cultured under laboratory conditions.
2.1 ChIPing Away at Chromatin
There are several techniques that can be applied to investigate the state of the genome at a given time, but one of the most versatile is chromatin immunoprecipitation (ChIP). The premise of this technique is straightforward. As transcription factors (or other proteins such as histones) bind to their native targets in the genome, they are first “frozen” in time and space by using a cross-linking reagent (usually formaldehyde). The result is that all transcription factors remain attached to the DNA they were bound to prior to fixing with the formaldehyde, and any unbound protein is cross-linked to other proteins. Cross-linked chromatin thus consists of DNA with transcription factors bound to specific regions. The cross-linked can subsequently be reversed by overnight incubation at moderate temperatures (65 °C typically), and pure DNA is extracted from this subsequently (Farnham 2009).
This process can be performed on samples from different times in the circadian cycle such that a temporal map of transcription factor binding can be obtained. If a particular genomic locus is of interest, such as a promoter/enhancer region of a gene, the polymerase chain reaction (PCR) can be used to amplify the relevant target region, and its enrichment at different time points can be compared quantitatively using real-time PCR (qPCR). If, however, the target regions in the genome are unknown, or you wish to take an unbiased approach to finding transcription factor target sites, then other genome-level methods have to be used in combination with ChIP.
2.2 ChIP-chip and ChIP-seq Allow Whole-Genome Analyses of Transcription Factor Binding
Having the ability to temporally map binding sites of transcription factors (or other proteins that bind to DNA) across the genomic landscape has only recently become possible with the advent of new technologies. The first breakthrough technology was the DNA microarray. When using ChIP coupled with microarrays (also known as a DNA “chip”), the technique is termed “ChIP-chip.” An important aspect is to ensure adequate genomic coverage. This is, however, difficult given the limited capacity and packing density of probe DNA sequences on the surface of microarrays and the necessity to synthesize a plethora of probes to cover the entire genome (Buck and Lieb 2004; Scacheri et al. 2006; Wu et al. 2006). To get useful coverage, multiple microarrays have to be used, which is both expensive and experimentally time-consuming. Initially, promising studies were confined to the detailed analysis of individual chromosomes, but low-resolution studies at the “whole genome” level have been performed (Bieda et al. 2006; Horak et al. 2002).
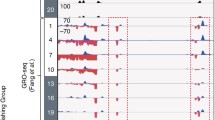
Microarrays have now been effectively usurped, as will no doubt be the case soon for transcriptomics studies, by high-throughput sequencing approaches. This technology has revolutionized genomic-scale analyses. Instead of having a defined set of probes that cover the entire genome, it is now possible to instead simply sequence all of the ChIPed DNA sequences that represent binding regions across the genome (termed “ChIP-seq”). After some relatively demanding bioinformatics to align each sequence to the reference genome (e.g., mouse or human), each sequence, and the relative number of sequences at a particular genomic locus, can be determined at a very high resolution (Farnham 2009). At binding sites, enrichment of overlapping sequences yields “peaks” where the target protein was bound (Fig. 1). Further bioinformatics can subsequently determine, with ever-increasing precision, binding motifs within the genomic DNA (Park 2009; Pepke et al. 2009).

Protein–chromatin interactions are first cross-linked in situ using, typically, formaldehyde. Specific DNA fragments are co-immunoprecipitated and sequenced to identify genome-wide sites associated with a factor or modification of interest (Adapted from Illumina Web site, http://www.illumina.com/technology/chip_seq_assay.ilmn)
Recently, some investigators have applied ChIP-seq methodology to investigate the function of the core transcription–translation feedback oscillator at a genomic scale. For example, by using an antiserum directed against BMAL1, Rey and colleagues have mapped BMAL1 binding sites across the genome of mouse liver, over the circadian cycle (Rey et al. 2011). This convincingly showed that BMAL1 rhythmically binds to over 2,000 target sites in the genome, with peak occupancy occurring in the middle of the circadian day. Functionally, these targets were diverse, but their results pointed towards carbohydrate and lipid metabolism loci as major targets for BMAL1’s action in vivo. Furthermore, using a combination of bioinformatics and modeling, these authors were able to show that E-box motifs were strongly correlated with the presence of BMAL1 binding sites and to rhythmic transcription of these loci (Rey et al. 2011). Recent comprehensive studies have further developed these principles and determined a circadian “chromatin landscape” by assaying other components of the “clock complex” (Koike et al. 2012). The true power, however, of genome-level approaches comes from the ability to relate changes at the level of DNA to transcripts and eventually to proteins, the effectors of cellular physiology.
3 Circadian Transcriptomics
Early studies in mammals implicated a “core” set of clock genes in the molecular clockwork (Buhr and Takahashi 2013; Hastings et al. 2003; Reddy and O’Neill 2009). Amongst these were the Period and Cryptochrome gene families, as well as the transcription factors driving their expression, Clock and Bmal1. It became apparent quickly that the majority of these genes were expressed rhythmically and that there were close parallels between the mammalian clockwork and what had been extensively investigated in Drosophila previously (Hastings et al. 2003). Initially, however, it was thought that relatively few genes (and their respective transcripts) were under circadian clock control. However, with the advent of microarray technology, this hypothesis became eminently testable.
The first study to map the circadian transcriptome was performed in plants by Harmer and colleagues (Harmer et al. 2000). This demonstrated the pervasive nature of rhythmic transcription and the clear anticipatory advantage that clock control over plant homeostasis might have. It did not take long for similar results to emerge in other eukaryotic systems, most notably in mammals.
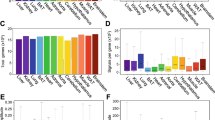
Several studies illustrated the extensive influence of the circadian clock on tissue transcriptomes, most notably in the liver, brain, and heart (Akhtar et al. 2002; Panda et al. 2002; Storch et al. 2002; Ueda et al. 2002). Subsequently, a plethora of tissue transcriptomes has been mapped over the circadian cycle, including those of adipose tissue, gut, and bone (Polidarova et al. 2011; Zvonic et al. 2006, 2007). Together, these studies and others highlight that in excess of 10 % of the transcriptome in any individual tissue could undergo robust rhythmic change over the circadian day and night (Hughes et al. 2009). The functional consequences of this widespread control over gene expression by the clock are perhaps obvious but are only beginning to be recognized more widely outside the circadian clock field (Fig. 2).

Microarray analysis of gene expression over a circadian cycle (i.e., in the absence of external time cues) in mouse liver. The top panel shows a heat map with genes that oscillate in a similar pattern clustered together. In this case, transcripts peaking in the middle of the cycle, CT12, are shown (CT circadian time; where CT0 is subjective dawn and CT12 is subjective dusk). The bottom panel shows the same data as a graphical representation for each gene. The left side of the heat map and graph assayed expression by an autocorrelation method. See Akhtar et al. (2002) for further details
As microarray technology has matured, high-throughput sequencing seems set to take over its reign over transcriptomics research, in a similar way in which ChIP-chip has given way to ChIP-seq. The power of sequencing the transcriptome (after reverse transcription and processing into DNA) is clear (Hawkins et al. 2010; Marguerat and Bahler 2010; Wang et al. 2009). RNA-seq (the term used to describe this technique) is so powerful because it cannot only interrogate messenger RNA (mRNA) but can also be used to assay small RNAs, such as microRNAs (Cheng et al. 2007; Gatfield et al. 2009; Yang et al. 2008) and other noncoding RNA species (e.g., large noncoding RNA—lncRNA). Furthermore, RNA-seq can be used to perform systems-level analyses of alternative splicing, which may add further tiers of regulation to RNA processing by the clock (Licatalosi et al. 2008; Wang et al. 2010).
4 Interferomics and Manipulating the Clockwork
Interferomics is a novel area within systems biology that aims to study the biological impact of perturbing post-transcriptional (but pre-translational) processes (Baggs and Hogenesch 2010). The main tool that is becoming used increasingly in this area is RNA interference (RNAi). Using this technique, it is possible to silence a specific gene with a cell line using a small interfering RNA molecule (siRNA). With a suitable screening platform for circadian clock function, a collection of these siRNAs could be applied onto cells and phenotypes screened for using a suitable screening assay.
The pre-eminent assay system used by circadian biologists to assay the clockwork employs bioluminescent reporter constructs (Yamaguchi et al. 2001). These consist of “clock gene” promoters driving the expression of luciferase expression (e.g., Bmal1::luciferase and mPer2::luciferase) which act as markers for circadian oscillation within the cell line. Once reporters are introduced into cells stably, siRNAs can be transfected into reporter cells and their effects on the clock determined using real-time bioluminescence monitoring (Hastings et al. 2005).
This type of approach has recently been put into practice in two major studies (Maier et al. 2009; Zhang et al. 2009), with similar paradigms also used for chemical compound screening (Hirota et al. 2010). Interestingly both kinds of approach have delineated links to canonical kinase pathways, including casein kinases (Hirota et al. 2010; Maier et al. 2009). This highlights the power of complementary approaches to dissecting components of the transcription–translation feedback loop.
5 Beyond Transcription
Proteins are of course the final effectors of cellular function; what do we know about the impact of rhythmic transcription on protein levels following translation? Intuitively, this would seem to be quite a straightforward question. However, the data in the clock field and in other domains highlights that transcriptomics datasets do not correlate well with proteomics datasets from the same samples (Hanash 2003; Reddy et al. 2006), emphasizing the importance of mapping protein abundance in addition to mRNA expression. This point is further highlighted by recent data from Selbach and colleagues, who took a systems approach to determine the “flow” of mRNA to protein quantitatively (Schwanhausser et al. 2011). More detailed descriptions of post-translational aspects (e.g., proteomics and metabolomics) are considered in other chapters within this volume (Robles and Mann 2013).
References
Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP (2002) Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol 12:540–550
Baggs JE, Hogenesch JB (2010) Genomics and systems approaches in the mammalian circadian clock. Curr Opin Genet Dev 20:581–587
Bieda M, Xu X, Singer MA, Green R, Farnham PJ (2006) Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 16:595–605
Buck MJ, Lieb JD (2004) ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics 83:349–360
Buhr ED, Takahashi JS (2013) Molecular components of the mammalian circadian clock. In: Kramer A, Merrow M (eds) Circadian clocks, vol 127, Handbook of experimental pharmacology. Springer, Heidelberg
Cheng HY, Papp JW, Varlamova O, Dziema H, Russell B, Curfman JP, Nakazawa T, Shimizu K, Okamura H, Impey S, Obrietan K (2007) microRNA modulation of circadian-clock period and entrainment. Neuron 54:813–829
Farnham PJ (2009) Insights from genomic profiling of transcription factors. Nat Rev Genet 10:605–616
Gatfield D, Le Martelot G, Vejnar CE, Gerlach D, Schaad O, Fleury-Olela F, Ruskeepaa AL, Oresic M, Esau CC, Zdobnov EM, Schibler U (2009) Integration of microRNA miR-122 in hepatic circadian gene expression. Genes Dev 23:1313–1326
Hanash S (2003) Disease proteomics. Nature 422:226–232
Harmer SL, Hogenesch JB, Straume M, Chang H-S, Han B, Zhu T, Wang X, Kreps JA, Kay SA (2000) Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290:2110–2113
Hastings MH, Reddy AB, Maywood ES (2003) A clockwork web: circadian timing in brain and periphery, in health and disease. Nat Rev Neurosci 4:649–661
Hastings MH, Reddy AB, McMahon DG, Maywood ES (2005) Analysis of circadian mechanisms in the suprachiasmatic nucleus by transgenesis and biolistic transfection. Methods Enzymol 393:579–592
Hawkins RD, Hon GC, Ren B (2010) Next-generation genomics: an integrative approach. Nat Rev Genet 11:476–486
Hirota T, Lee JW, Lewis WG, Zhang EE, Breton G, Liu X, Garcia M, Peters EC, Etchegaray JP, Traver D, Schultz PG, Kay SA (2010) High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIalpha as a clock regulatory kinase. PLoS Biol 8:e1000559
Horak CE, Mahajan MC, Luscombe NM, Gerstein M, Weissman SM, Snyder M (2002) GATA-1 binding sites mapped in the beta-globin locus by using mammalian chIp-chip analysis. Proc Natl Acad Sci USA 99:2924–2929
Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, Hogenesch JB (2009) Harmonics of circadian gene transcription in mammals. PLoS Genet 5:e1000442
Johnson CH, Mori T, Xu Y (2008) A cyanobacterial circadian clockwork. Curr Biol 18:R816–R825
Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS (2012) Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338(6105):349–354
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Kucho K, Okamoto K, Tsuchiya Y, Nomura S, Nango M, Kanehisa M, Ishiura M (2005) Global analysis of circadian expression in the cyanobacterium Synechocystis sp. strain PCC 6803. J Bacteriol 187:2190–2199
Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB (2008) HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456:464–469
Maier B, Wendt S, Vanselow JT, Wallach T, Reischl S, Oehmke S, Schlosser A, Kramer A (2009) A large-scale functional RNAi screen reveals a role for CK2 in the mammalian circadian clock. Genes Dev 23:708–718
Marguerat S, Bahler J (2010) RNA-seq: from technology to biology. Cell Mol Life Sci 67:569–579
Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB (2002) Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109:307–320
Park PJ (2009) ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet 10:669–680
Pepke S, Wold B, Mortazavi A (2009) Computation for ChIP-seq and RNA-seq studies. Nat Methods 6:S22–S32
Polidarova L, Sladek M, Sotak M, Pacha J, Sumova A (2011) Hepatic, duodenal, and colonic circadian clocks differ in their persistence under conditions of constant light and in their entrainment by restricted feeding. Chronobiol Int 28:204–215
Reddy AB, O’Neill JS (2009) Healthy clocks, healthy body, healthy mind. Trends Cell Biol 20:36–44
Reddy AB, O’Neill JS (2010) Healthy clocks, healthy body, healthy mind. Trends Cell Biol 20:36–44
Reddy AB, Karp NA, Maywood ES, Sage EA, Deery M, O’Neill JS, Wong GKY, Chesham J, Odell M, Lilley KS, Kyriacou CP, Hastings MH (2006) Circadian orchestration of the hepatic proteome. Curr Biol 16:1107–1115
Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F (2011) Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol 9:e1000595
Ripperger JA, Schibler U (2006) Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet 38:369–374
Robles MS, Mann M (2013) Proteomic approaches in circadian biology. In: Kramer A, Merrow M (eds) Circadian clocks, vol 127, Handbook of experimental pharmacology. Springer, Heidelberg
Scacheri PC, Crawford GE, Davis S (2006) Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol 411:270–282
Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473:337–342
Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, Weitz CJ (2002) Extensive and divergent circadian gene expression in liver and heart. Nature 417:78–83
Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, Iino M, Shigeyoshi Y, Hashimoto S (2002) A transcription factor response element for gene expression during circadian night. Nature 418:534–539
Vijayan V, Zuzow R, O’Shea EK (2009) Oscillations in supercoiling drive circadian gene expression in cyanobacteria. Proc Natl Acad Sci USA 106:22564–22568
Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang Z, Kayikci M, Briese M, Zarnack K, Luscombe NM, Rot G, Zupan B, Curk T, Ule J (2010) iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol 8:e1000530
Woelfle MA, Xu Y, Qin X, Johnson CH (2007) Circadian rhythms of superhelical status of DNA in cyanobacteria. Proc Natl Acad Sci USA 104:18819–18824
Wu J, Smith LT, Plass C, Huang TH (2006) ChIP-chip comes of age for genome-wide functional analysis. Cancer Res 66:6899–6902
Yamaguchi S, Kobayashi M, Mitsui S, Ishida Y, van der Horst GTJ, Suzuki M, Shibata S, Okamura H (2001) View of a mouse clock gene ticking. Nature 409:684
Yang M, Lee JE, Padgett RW, Edery I (2008) Circadian regulation of a limited set of conserved microRNAs in Drosophila. BMC Genomics 9:83
Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, Liu X, Atwood A, Huss JW 3rd, Janes J, Su AI, Hogenesch JB, Kay SA (2009) A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 139:199–210
Zvonic S, Ptitsyn AA, Conrad SA, Scott LK, Floyd ZE, Kilroy G, Wu X, Goh BC, Mynatt RL, Gimble JM (2006) Characterization of peripheral circadian clocks in adipose tissues. Diabetes 55:962–970
Zvonic S, Ptitsyn AA, Kilroy G, Wu X, Conrad SA, Scott LK, Guilak F, Pelled G, Gazit D, Gimble JM (2007) Circadian oscillation of gene expression in murine calvarial bone. J Bone Miner Res 22:357–365
Acknowledgments
Supported by the Wellcome Trust (083643/Z/07/Z), the European Research Council (ERC) Grant No. 281348 (MetaCLOCK), NIHR Cambridge Biomedical Research Centre, and the MRC Centre for Obesity and Related Metabolic Disorders (MRC CORD).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Reddy, A.B. (2013). Genome-Wide Analyses of Circadian Systems. In: Kramer, A., Merrow, M. (eds) Circadian Clocks. Handbook of Experimental Pharmacology, vol 217. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-25950-0_16
Download citation
DOI: https://doi.org/10.1007/978-3-642-25950-0_16
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-25949-4
Online ISBN: 978-3-642-25950-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)