
Abstract
In addition to being a critical part of host defense against pathogens, the inflammatory response can also be triggered by a number of perturbations to cellular homeostasis, including responses to protein misfolding and endoplasmic reticulum (ER) stress. Physiologically, these responses can lead to activation of tissue repair pathways, but when not properly regulated, these stress response pathways can lead to chronic inflammation. ER stress and other inflammatory pathways triggered by misfolded proteins have been implicated in the pathogenesis of several monogenic autoinflammatory diseases, and also may play a role in other conditions such as neurodegenerative diseases, where increasing evidence has accumulated about the contribution of inflammation to disease pathogenesis. Alterations in protein homeostasis can trigger autoinflammatory diseases in a number of ways, including (1) a pathogenic protein is itself misfolded, primarily activating inflammatory signaling pathways, as with the mutant tumor necrosis factor receptor 1 (TNFR1) protein in TNF receptor-associated periodic syndrome (TRAPS), or triggering an intracellular ER stress response, such as the human leukocyte antigen (HLA)-B27 protein in spondylarthropathies; (2) inflammatory responses can also be triggered by extracellular misfolded proteins, and (3) genetic defects in protein homeostasis pathways which lead to inflammatory diseases. Examples of this mechanism are proteasome mutations in chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperature (CANDLE) and related syndromes, and variants in the gene encoding ATG16L which reduce the efficiency of autophagy and related secretory pathways in inflammatory bowel disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Protein homeostasis
- Autophagy
- LC3-associated phagocytosis
- Autoinflammatory disease
- Spondyloarthropathy
- Endoplasmic reticulum (ER) stress response
- Reactive oxygen species
-
Autophagy and the proteasome control protein homeostasis, and also regulate inflammation and immunity
-
Dysregulation or disruption of these processes can contribute to pathology of a variety of diseases
-
Accumulation of misfolded proteins triggers ER stress responses and can contribute to the pathogenesis of monogenic autoinflammatory diseases, and other conditions including neurodegenerative disorders and type II diabetes
-
Genetic alterations in the efficiency of autophagy or proteosome function can contribute to autoinflammatory disease
-
Better understanding of these pathways may aid the design of therapeutic interventions in both monogenic and more complex autoinflammatory diseases
1 Cellular Mechanisms Maintaining Protein Homeostasis and Links to Inflammation Biology
Key Points
-
Damaged or ubiquitinated proteins are degraded via the proteasome, which also has essential roles in antigen presentation, the cellular stress response, and regulating cell death
-
Autophagy is primarily responsible for the degradation of long-lived proteins and cellular organelles (mitochondria, peroxisomes), and is essential for cell growth and the response to nutrient deprivation
-
The proteasome and autophagy proteins also regulate intracellular metabolism and inflammatory signaling pathways
Balancing protein synthesis, degradation and secretion is an essential part of cellular physiology, and multiple molecular mechanisms exist to maintain protein homeostasis, with over 1000 proteins estimated to participate in this process. Feedback mechanisms control the rate of protein synthesis. Separate networks of chaperones control protein folding and sensing of unfolded proteins, both in the cytoplasm and inside the vesicular network of the secretory pathway. Two major mechanisms of protein degradation, the proteasome (see also Chap. 7) and autophagy, are responsible for homeostasis of most proteins and other cellular components. Together, these systems maintain cellular viability amidst dramatic changes in protein output which occur particularly in immune cells. These cells can dramatically upregulate their biosynthetic protein flux to support rapid growth and secretion of large quantities of cytokines and antibodies. This is most evident in B lymphocytes, where in as little as 1 week, cells can differentiate from a resting B lymphocyte with little secretory capacity to plasma cells, which synthesize and secrete up to 175 million antibody molecules per day.
1.1 The Proteasome
The proteasome, a multi-subunit cytoplasmic protein complex, is capable of rapid degradation of proteins marked with small molecules in the ubiquitin family, which share a structural β-grasp fold. K48-linked ubiquitin was the first identified modifier protein, which has expanded to include other related molecules including: small ubiquitin-like modifier (SUMO), neural precursor cell expressed, developmentally down-regulated 8 (NEDD8), interferon-stimulated gene 15 (ISG15), F adjacent transcript 10 (FAT10), and monoclonal non-specific suppressor factor beta (MNSFB) proteins. A special set of proteasome subunits is induced in antigen-presenting cells and forms a so-called ‘immunoproteasome’ that facilitates processing of peptides with hydrophobic C-termini that are optimized to be presented in the groove of major histocompatibility complex (MHC) class I molecules. The immunoproteasome also plays an important role in eliminating protein aggregates which can accumulate under inflammatory conditions [1]. Ubiquitination of target proteins is accomplished through three sets of enzymes termed E1, E2 and E3 ubiquitin ligases, with E1 and E2 having catalytic activity and E3 proteins serving to link the target protein to the ubiquitination machinery. Ubiquitin-like proteins are conjugated to target proteins through parallel sets of enzymes, and like ubiquitin, these proteins also have functions beyond mediating recognition of target proteins by the proteasome. Once recognized by the proteasome, target proteins are loaded into the proteolytic core of the proteasome where adenosine triphosphate (ATP)-dependent proteases digest proteins into short oligopeptides suitable for further catabolism and antigen presentation (for details on proteasomes see Chap. 7).
1.2 Autophagy
The other major proteolytic mechanism in cells is autophagy, a process in which organelles and cytoplasmic contents are enveloped in lipid membrane-enclosed vesicles which fuse with lysosomes to degrade proteins into smaller fragments. Autophagy was originally considered to be a constitutive process by which long-lived proteins and damaged organelles are degraded, but more recently, it has been found to be regulated by a series of proteins which allow environmental conditions, particularly nutrient starvation, to induce autophagy to maintain cellular protein homeostasis and function [2].
Two key nutrients sensing pathways regulate autophagy (Fig. 8.1a). Depletion of ATP or other stressors (starvation, hypoxia, oxidative stress, ER stress, infections) can activate AMP-activated protein kinase (AMPK), which phosphorylates substrates that activate autophagy. Another key nutrient sensor, the target of rapamycin complex (TORC), represses autophagy, but under conditions of amino acid starvation or removal of growth factors, TORC is repressed, which activates autophagy. Biochemically, TORC and AMPK regulate the pre-initiation complex, consisting of unc-51 like autophagy activating kinase 1/2 (ULK1/2), autophagy-related genes 13 (ATG13) and family interacting protein of 200 (FIP200) proteins (Fig. 8.1b) which regulate a class III phosphatidylinositol-3 kinase (PI-3 kinase) enzyme complex termed the initiation complex, consisting of the proteins ATG14L, beclin1, vacuolar protein sorting 34 (VPS34) and VPS15. Full activation of the initiation complex requires dissociation of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) from the beclin-1 protein, linking autophagy to regulation of apoptotic cell death. The PI-3 kinase activity of the initiation complex phosphorylates lipids on intracellular membranes, creating a substrate for binding of proteins such as WPP domain–interacting proteins 1(WIP-1 or ATG18). This converts the nascent autophagic vesicle into a crescent shaped isolation membrane (Fig. 8.1c). The completion of autophagic lipid vesicles is accomplished through a ubiquitin-like chain-reaction of protein modification termed the elongation reaction, in which a lipid modified protein LC3, takes the place of ubiquitin. The protease ATG4 cleaves LC3 to produce LC3-I, which in turn is bound by the ATG7 molecule and transferred to ATG3. A protein complex consisting of ATG5, ATG12, and ATG16L1 transfers a phosphatidylethanolamine (PE) molecule onto LC3-I, forming LC3-II, which is incorporated into the growing isolation membrane and assists in the formation of the completed double-walled autophagic vesicle (Fig. 8.1d). Fusion of autophagosomes with lysosomes to degrades the organelles and proteins within (Fig. 8.1e), providing substrates for synthesis of new macromolecules in nutrient limiting conditions.

Cellular autophagy pathways. Lettered steps described in the main text. AMBRA1 activating molecule in BECN1-regulated autophagy protein 1, AMPK AMP-activated kinase, ATG autophagy-related genes, FIP200 family interacting protein of 200, LC3 Microtubule-associated protein 1A/1B-light chain 3, mTORC mammalian target of rapamycin, Nox2 NAPDH oxidase 2, PI3K phosphatidylinositol-3 kinase, PI3P phosphatidylinositol 3-phosphate, ROS reactive oxygen species, Ub ubiquitin, ULK1/2 unc-51 like autophagy activating kinase 1/2, UVRAG UV radiation resistance-associated gene protein, VPS vacuolar protein sorting, WIP WPP domain–interacting proteins
In addition to being a generalized mechanism for recycling cellular components, autophagy and components of the autophagic machinery can perform targeted degradation of damaged cellular components and control exocytic and endocytic processes where lysosomes are coupled to vesicular trafficking. Even under non-starvation conditions, certain organelles, including peroxisomes and mitochondria, can be recycled through autophagy, which performs the valuable cellular function of removing sources of reactive oxygen species (ROS) and toxic lipids. In damaged mitochondria, loss of the mitochondrial electrochemical gradient causes the accumulation of the PTEN-induced putative kinase 1 (PINK1) kinase on the cytosolic-facing outer mitochondrial membrane. PINK1 activates Parkinson kinase (Parkin), an E3 ligase, which catalyzes ubiquitination of multiple substrates on the mitochondrial membrane. These substrate proteins recruit molecules such as p62/sequestrome and optineurin, which in turn recruit LC3 to the surface of these particular mitochondria and results in their selective elimination.
During exocytosis, fusion of exocytic and other intermediate vesicles to lysosomes can regulate the secretory process. Certain components of the autophagy machinery also participate in the LC3-mediated fusion of endocytic vesicles with lysosomes, which enhances the degradation of phagocytosed apoptotic cells and dampens the inflammatory response triggered by apoptotic cells, a process termed LC3-associated phagocytosis (LAP) [3] (Fig. 8.1f). Mice deficient in components of LAP have heightened inflammatory responses to apoptotic cells, and develop features of both autoinflammatory and autoimmune disease [4].
2 Protein Homeostasis in the Pathogenesis and Regulation of Monogenic Autoinflammatory Diseases
Key Points
-
Autophagy degrades activated NLRP3 inflammasomes
-
Autophagy and proteasome-mediated degradation regulate production of type I interferons via the cGAS-STING pathway
-
In TRAPS, mutant TNFR1 protein are retained in ER and are able to activate MAPK signaling via mitochondrial ROS
2.1 Degradation of Inflammasomes Through Autophagy (Fig. 8.2)
Protein homeostasis regulate many key inflammatory pathways which are disrupted in monogenic autoinflammatory diseases (Table 8.1, Fig. 8.2). For the group of syndromes caused by activating mutations in the nucleotide-binding oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3) inflammasomes, a key link was made by the observation that activated inflammasomes are degraded through autophagy [5]. This was first observed during activation of NLRP3 and absent in melanoma 2 (AIM2) and inflammasomes, where ubiquitination of activated inflammasome components led to recognition by the p62 autophagy adaptor proteins and targeted delivery of activated inflammasome components to the autophagolysosome [6]. The superfamily of tripartite motif-containing (TRIM) family proteins also specifically target NLRP3, NLRP1 and caspase-1 for autophagic degradation. Other mechanisms regulate autophagic degradation of pro-interleukin (IL)-1β [7, 8]. Multiple activators of autophagy, including amino acid starvation, have been shown to reduce production of IL-1β whereas inhibitors of autophagy or genetic deficiency of genes encoding autophagosome components enhance inflammatory responses [9, 10]. Autophagy also inhibits the release of mitochondrial DNA into the cytoplasm in cells triggered to undergo inflammatory responses, likely due to the rapid degradation of mitochondria in which the proton gradient has collapsed after triggering of inflammatory responses, short circuiting the amplification loop in inflammation [11]. Autophagy itself can also be induced by inflammasome-regulated processes, generating a feedback loop which keeps inflammasome-mediated pathology under control, while allowing appropriate activation of innate immunity for host defense.

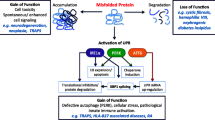
Consequences of protein misfolding and intracellular signaling complexes that play a role in the pathogenesis of specific autoinflammatory disease. (a) The effects of the misfolding of secretory proteins in the endoplasmic reticulum (ER) are depicted at the bottom of the figure. The degradation of misfolded proteins can cause a loss-of-function, whereas the accumulation of misfolded proteins can trigger abnormal intracellular signaling or, at higher levels, the induction of the unfolded-protein response (UPR), which can also lead to the induction of inflammation and programmed cell death. Different foci of abnormal cellular signaling that trigger autoinflammatory diseases are depicted in the cell. (b) In proteasome subunit beta 8 (PSMB8) deficiency, reduced degradation of misfolded proteins and peptides by the immunoproteasome leads to the accumulation of ubiquitylated proteins and cellular stress. This can lead to the production of interferon-β (IFN-β), which in turn upregulates the synthesis of immunoproteasome subunits, perpetuating the abnormalities. (c) stimulator of interferon genes (STING) senses cyclic dinucleotides generated by cyclic guanosine monophosphate-adenosine monophosphate synthetase (cGAS) from endogenous and viral DNA or directly from bacteria, and triggers induction of interferon production through TBK1 and IRF3. STING is degraded in an autophagy-dependent manner. (d) In the cryopyrin-associated periodic syndromes (CAPS), mutations in nucleotide-binding oligomerization domain (NOD) leucine-rich repeats (LRR) and pyrin domain-containing 3 (NLRP3) enhance the activation of the NLRP3 inflammasome and the processing of pro-interleukin (IL)-1β into its active form. In familial Mediterranean fever, mutant pyrin is thought to associate with the inflammasome adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and increase IL-1β processing. The NLRP3 inflammasome can also be degraded through autophagy. (e) In the spondyloarthropathies , human leukocyte antigen (HLA)-B27 is expressed at a high level (which is enhanced in inflammation), fails to fold properly and is retained in the endoplasmic reticulum (ER), triggering a partial ER stress response that leads to type I IFN and IL-23 production. (f) In tumor necrosis factor receptor-associated periodic syndrome (TRAPS), mutations in the extracellular region of the TNF receptor 1 (TNFR1) leads to accumulation of the mutant receptor in the ER, which triggers an abnormal inflammatory response that is amplified by TNF or lipopolysaccharide (LPS) signaling through cell-surface receptors. CARD caspase recruitment domain, JNK c-Jun N-terminal kinase, LRR leucine-rich repeat, PYD pyrin domain, ROS reactive oxygen species, TLR Toll-like receptor. Adapted from [42]
2.2 Regulation of Cyclic Guanosine Monophosphate-Adenosine Monophosphate Synthetase (cGAS) and Stimulator of Interferon Genes-(STING) Pathway by Autophagy
Autophagy can also regulate the activity of the cyclic guanosine monophosphate-adenosine monophosphate synthetase (cGAS)-stimulator of interferon genes (STING) pathway, another key intracellular sensor of pathogens and ectopic double stranded DNA (Fig. 8.2c). The enzyme (cGAS ) is activated by cytosolic dsDNA to synthesize the dinucleotide second messenger cyclic guanosine monophosphate–adenosine monophosphate (cGAMP). The sensor protein STING is activated by cGAMP and activates the production of type I interferons through the kinase TANK binding kinase (TBK1) and the transcription factor interferon regulatory transcription factor 3 (IRF3) [12]. Activating mutations in STING trigger the syndrome of STING-associated vasculopathy with onset in infancy (SAVI) which is marked by excess production of type I interferon in response to cGAMP and a strong in vivo interferon gene transcriptional signature [13] (see Chap. 24). Bacterial dinucleotide metabolites can activate STING directly without cGAS [14]. In parallel with activating interferon synthesis, dsDNA also activates autophagy through cGAS, and a pathway requiring activation of AMPK and the downstream kinases ULK1/2 [15]. Activated STING is also ubiquitinated, leading to recruitment of the p62/sequestrin which mediates its delivery to autophagocytic vesicles and degradation [15]. This regulatory mechanism is likely important in preventing sustained activation of STING and type I interferon and the severe clinical consequences that are seen in the SAVI syndrome.
2.3 Accumulation of Misfolded Mutated Proteins
In addition to being regulated by autophagy, inflammatory signaling pathways can be activated in monogenic autoinflammatory disease by the altered protein encoded by the causal genetic mutation. The tumor necrosis factor receptor-associated periodic syndrome (TRAPS) is caused by autosomal dominant missense mutations in the extracellular domain of TNFR1, the key pro-inflammatory receptor for TNF [16] (see Chap. 18). In cells from patients with TRAPS and mice engineered to express TRAPS-associated TNFR1 mutations, the mutant protein misfolds and is retained in the endoplasmic reticulum (ER), where it signals in a ligand-independent manner to activate MAPK signaling through a pathway dependent on mitochondrial ROS [17]. Autophagy may also play a role in degrading TNFR1 [18]. Cells from patients with TRAPS have enhanced pro-inflammatory responses to innate immune stimuli and sensitivity to in vivo lipopolysaccharide (LPS) challenge in a manner dependent on the wild-type TNFR1 [19]. Accumulated TNFR1 also triggers a low but detectable activation of the unfolded protein response (UPR) [20]. Upregulation of NLRP3 and enhanced inflammasome activation and IL-1β production ensues in myeloid cells, likely explaining the clinical responsiveness of TRAPS to blockade of IL-1β [21].
3 Alteration in Protein Homeostasis Mechanisms and Triggering of Inflammatory Responses by Misfolded Proteins in Complex Diseases
Key Points
-
Intracellular misfolded proteins are key players in inducing the inflammation and ER stress responses in various diseases including ankylosing spondylitis
-
Extracellular protein aggregates can also induce ER stress and are important players in pathogenesis of Alzheimer disease and type II diabetes
In addition to genetic variants in specific proteins which can trigger ER stress responses or altered signal transduction, alterations in protein homeostasis mechanisms themselves can lead to enhanced inflammation. For diseases associated with misfolded proteins, pathogenesis can further be divided into intracellular vs. extracellular proteins, as the mechanisms by which inflammation is triggered vary depending on the location of the misfolded protein.
3.1 Defects in the Autophagy Pathway
Genetic deficiencies in components of the autophagy pathway such as ATG5 and ATG7 result in accumulation of damaged mitochondria, as do mutations in parkin, which are associated with hereditary forms of Parkinson disease linking defective mitophagy to neurodegeneration. In studying the susceptibility allele for Crohn disease linked to the gene encoding ATG16L1, it was discovered that exocytosis by intestinal Paneth cells can be regulated by autophagy, with the disease susceptibility variant reducing secretion of antimicrobial peptides [22].
3.2 Accumulation of Intracellular Misfolded Proteins (Fig. 8.2)
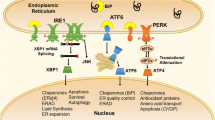
The human leukocyte antigen (HLA)-B27 protein is a well-studied example of an abundant protein where misfolding contributes to induction of an ER stress response and inflammation in spondylarthropathies. Ankylosing spondylitis (AS) is a polygenic immune mediated multisystem inflammatory chronic disorder characterized by inflammation centered on the axial spine with syndesmophyte formation resulting in the fusion of vertebral facet joints, involvement of sacroiliac and peripheral joints, enthesitis and extra-articular manifestations including inflammatory bowel disease and acute anterior uveitis. AS has a strong genetic predisposition, and presence of the HLA-B27 MHC class I allele remains the greatest genetic risk factor identified to date, conferring a relative risk of more than 80-fold in AS and somewhat lower, but significant risk in other spondylarthropathies [23]. It was initially presumed that the pathogenesis involved presentation of pathogenic peptides to class-I restricted T cells, but the failure to identify these putative peptides, and the persistence of AS-like disease in HLA-B27 transgenic rats in the absence of CD8+ T cells has refocused research into roles for HLA-B27 in triggering inflammation independent of the adaptive immune system, thus supporting the concept of AS as an autoinflammatory disease. Compared to other HLA molecules, HLA-B27 is predisposed to form homodimers in the absence of β2-microglobulin. A role in recognition of surface HLA-B27 through KIR3DL2, an activating receptor found on the surface of natural killer (NK) cells, T cells and myeloid cells has been hypothesized in AS [24, 25], and the recent development of therapeutic antibodies against KIR3DL2 may allow clinical testing of this hypothesis. Another property of HLA-B27 dimers is accumulation in the ER, likely due to protein misfolding. Accumulation of misfolded HLA-B27 can trigger the ER stress response, also known as the UPR which in turn can increase the expression of proinflammatory mediators, including IL-23 in myeloid cells. IL-23 is a powerful costimulatory signal for the development of IL-17 secreting lymphocytes [26, 27]. A possible target cell for IL-23 has emerged from studies of non-classical T cells which reside in the tendon sheath and respond to IL-23 by secreting IL-17 [28]. These findings correlate with the therapeutic efficacy of antibodies blocking the activity of IL-23 and IL-17 in AS in clinical trials [29]. Inflammatory signals including toll-like receptor (TLR) ligation can also enhance activation of the UPR, constituting a positive feedback loop [30].
3.3 Accumulation of Extracellular Misfolded Proteins
Inflammation has recently been implicated in the pathogenesis of neurodegenerative and other diseases associated with organ failure, with specific misfolded proteins triggering inflammation and cell death through shared and individual pathways. Alzheimer disease (AD) is a neurodegenerative disease that is the most common cause of dementia in the elderly. The main histopathological features of AD are intracellular deposits of neurofibrillary tangles made of hyperphosphorylated Tau proteins and extracellular aggregates of amyloid-β, which form amyloid plaques. This accumulation is associated with glial activation, increased brain inflammation in the hippocampus and the cerebral cortex and neuronal toxicity. Several studies have identified abnormal levels of ER stress in the human brain of patients with AD. Markers of dysfunctional ER proteostasis correlate with the progression of AD and are associated with an activation of the UPR machinery [31]. Amyloid-β oligomers can induce ER stress in cultured neurons via interaction with N-methyl-d-aspartate (NMDA) receptors leading to alteration of ER calcium homeostasis and neuronal dysfunction [32]. The study of brain tissue from patients with AD as well as those with Parkinson disease also revealed s-nitrosylation and inactivation of protein disulfide isomerase (PDA), a protein critical for folding in the ER. Inactivation of PDA leads to accumulation of misfolded proteins, ER stress and neuronal cell death [33]. A connection to inflammation was made through the observation that the UPR activates the production of pro-inflammatory cytokines through the nuclear factor kappa B (NF-κB) signaling pathway [34]. Some misfolded extracellular proteins, such as islet amyloid polypeptide, can directly activate the NLRP3 inflammasome, triggering IL-1β release [35]. These findings may partly explain the responsiveness of type II diabetes to therapeutic blockade of IL-1β [36], and are inspiring clinical trials of anti-inflammatory and anti-cytokine agents for the treatment of these diseases.
4 Targeting Protein Homeostasis for the Therapy of Autoinflammatory Diseases: Future Perspectives
Although cytokine blocking therapies have been notably successful in the treatment of some autoinflammatory diseases, preventing protein misfolding and triggering of ER stress by mutant proteins in monogenic disease or misfolding-prone protein isoforms such as HLA-B27 remains an important therapeutic goal. ‘Molecular chaperones’ which bind misfolded proteins and attenuate ER stress responses to them have been successful in some animal models of diseases such as in α-crystallin mutations which cause cataracts [37]. The discovery that autophagy and proteasome-mediated degradation control expression of inflammatory mediators and pathways involved in autoinflammatory as well as other diseases has spurred research into enhancing proteolysis of key inflammatory mediators or autophagy in general as a therapeutic strategy [38, 39]. Pharmacological agents that activate AMPK, such as methotrexate, or inactivate mTOR, such as rapamycin, can promote autophagy and degradation of aggregate-prone proteins, but what portion of their anti-inflammatory or immunosuppressive effects are due to this mode of action is not clear. Using bifunctional molecules to enhance degradation of targeted proteins through the recruitment of E3 ligases may be a more selective strategy to remove key inflammatory mediators from the cell [40]. If these strategies succeed, it will be a good example of how harnessing the powerful physiological mechanisms of protein homeostasis can lead to therapeutic benefit.
Abbreviations
- AD:
-
Alzheimer disease
- AIM:
-
Absent in melanoma
- AMPK:
-
AMP-activated protein kinase
- AS:
-
Ankylosing spondylitis
- ATG:
-
Autophagy-related genes
- Bcl-2:
-
B-cell lymphoma 2
- CANDLE:
-
Chronic atypical neutrophilic dermatitis with lipodystrophy and elevated temperature
- cGAMP:
-
cyclic guanosine monophosphate–adenosine monophosphate
- cGAS:
-
cyclic guanosine monophosphate-adenosine monophosphate synthetase
- FIP200:
-
Family interacting protein of 200
- HLA:
-
Human leukocyte antigen
- IRF:
-
Interferon regulatory transcription factor
- ISG:
-
Interferon-stimulated gene
- LAP:
-
LC3-associated phagocytosis
- LC3:
-
Microtubule-associated protein light chain 3
- MHC:
-
Major histocompatibility complex
- mTOR:
-
mammalian target of rapamycin
- NEDD:
-
Neural precursor cell expressed, developmentally down-regulated
- NF-κB:
-
Nuclear factor kappa B
- NK:
-
Natural killer
- NLRP:
-
NOD-like receptor family pyrin domain containing
- NMDA:
-
N-methyl-d-aspartate
- NOD:
-
Nucleotide-binding oligomerization domain
- PARKIN:
-
Parkinson kinase
- PDA:
-
Protein disulfide isomerase
- PE:
-
Phosphatidylethanolamine
- PI:
-
Phosphatidylinositol
- PINK:
-
PTEN-induced putative kinase 1
- ROS:
-
Reactive oxygen species
- SAVI:
-
STING-associated vasculopathy with onset in infancy
- STING:
-
Stimulator of interferon genes
- SUMO:
-
Small ubiquitin-like modifier
- TBK:
-
TANK binding kinase
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- TORC:
-
Target of rapamycin complex
- TRAPS:
-
TNF receptor-associated periodic syndrome
- TRIM:
-
The superfamily of tripartite motif-containing
- ULK:
-
unc-51 like autophagy activating kinase
- UPR:
-
Unfolded protein response
- UPS:
-
Ubiquitin–proteasome system
- VPS:
-
Vacuolar protein sorting
- WIP:
-
WPP domain–interacting proteins
References
van Deventer S, Neefjes J. The immunoproteasome cleans up after inflammation. Cell. 2010;142(4):517–8.
Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75.
Martinez J, Malireddi RK, Lu Q, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol. 2015;17(7):893–906.
Martinez J, Cunha LD, Park S, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533(7601):115–9.
Saitoh T, Akira S. Regulation of inflammasomes by autophagy. J Allergy Clin Immunol. 2016;138(1):28–36.
Shi CS, Shenderov K, Huang NN, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–63.
Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015;210(6):973–89.
Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–97.
Harris J. Autophagy and IL-1 family cytokines. Front Immunol. 2013;4:83.
Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8.
Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–30.
Sun L, Wu J, Du F, Chen X, Chen Z. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (New York, NY). 2013;339(6121):786–91.
Liu Y, Jesus AA, Marrero B, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–18.
Moretti J, Roy S, Bozec D, et al. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. 2017;171(4):809–23 e13.
Konno H, Konno K, Barber G. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155(3):688–98.
McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999;97(1):133–44.
Bulua AC, Simon A, Maddipati R, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J Exp Med. 2011;208(3):519–33.
Bachetti T, Chiesa S, Castagnola P, et al. Autophagy contributes to inflammation in patients with TNFR-associated periodic syndrome (TRAPS). Ann Rheum Dis. 2013;72(6):1044–52.
Simon A, Park H, Maddipati R, et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc Natl Acad Sci U S A. 2010;107(21):9801–6.
Dickie LJ, Aziz AM, Savic S, et al. Involvement of X-box binding protein 1 and reactive oxygen species pathways in the pathogenesis of tumour necrosis factor receptor-associated periodic syndrome. Ann Rheum Dis. 2012;71(12):2035–43.
De Benedetti F, Gattorno M, Anton J, et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N Engl J Med. 2018;378(20):1908–19.
Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456(7219):259–63.
Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369(9570):1379–90.
Allen RL, Trowsdale J. Recognition of classical and heavy chain forms of HLA-B27 by leukocyte receptors. Curr Mol Med. 2004;4(1):59–65.
Kollnberger S, Bowness P. The role of B27 heavy chain dimer immune receptor interactions in spondyloarthritis. Adv Exp Med Biol. 2009;649:277–85.
Goodall JC, Wu C, Zhang Y, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A. 2010;107(41):17698–703.
Colbert RA, Tran TM, Layh-Schmitt G. HLA-B27 misfolding and ankylosing spondylitis. Mol Immunol. 2014;57(1):44–51.
Sherlock JP, Joyce-Shaikh B, Turner SP, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. 2012;18(7):1069–76.
Taurog JD, Chhabra A, Colbert RA. Ankylosing spondylitis and axial spondyloarthritis. N Engl J Med. 2016;374(26):2563–74.
Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11(5):411–8.
Scheper W, Hoozemans JJ. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. 2015;130(3):315–31.
Costa RO, Lacor PN, Ferreira IL, et al. Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers. Aging Cell. 2012;11(5):823–33.
Uehara T, Nakamura T, Yao D, et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006;441(7092):513–7.
Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer’s disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. J Neuroinflammation. 2009;6:41.
Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol. 2010;11(10):897–904.
Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356(15):1517–26.
Makley LN, McMenimen KA, DeVree BT, et al. Pharmacological chaperone for alpha-crystallin partially restores transparency in cataract models. Science. 2015;350(6261):674–7.
Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16(7):487–511.
Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–30.
Cromm PM, Crews CM. Targeted protein degradation: from chemical biology to drug discovery. Cell Chem Biol. 2017;24(9):1181–90.
Agyemang AF, Harrison SR, Siegel RM, McDermott MF. Protein misfolding and dysregulated protein homeostasis in autoinflammatory diseases and beyond. Semin Immunopathol. 2015;37(4):335–47.
Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol. 2012;12(8):570–80.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cudrici, C.D., Siegel, R.M. (2019). Disruption of Protein Homeostasis and Activation of Cellular Stress Pathways in Autoinflammation. In: Hashkes, P., Laxer, R., Simon, A. (eds) Textbook of Autoinflammation. Springer, Cham. https://doi.org/10.1007/978-3-319-98605-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-98605-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-98604-3
Online ISBN: 978-3-319-98605-0
eBook Packages: MedicineMedicine (R0)