
Abstract
Evolutionary conserved kinase mechanistic target of rapamycin (mTOR) is the signaling hub for cellular responses to nutrients, cytokines, growth hormones, and environmental stresses in all eukaryotic cells. Increased mTOR activity has been demonstrated in numerous diseases, such as cancer and autoimmune diseases. Due to its prominent role, mTOR inhibitors are being used and tested to treat a wide variety of conditions. Recent evidence suggests that regulation of mTOR activity and function is not universal and varies between the cells. Here we summarize the latest research on the role and regulation of mTOR in osteoclasts, the unique multinucleated bone-resorbing cells, focusing on the role of mTOR as part of the mTORC1 complex. Collectively, the results suggest that mTORC1 activity plays a double role in osteoclastogenesis: at the earlier stage, it is necessary for proliferation of the precursors, and, at the later stage, it is indispensable for cytoskeletal reorganization involved in the process of bone resorption. We also present evidence that in osteoclasts, mTOR protein levels and activity are regulated differently compared to other primary cells and cell lines. Due to this prominent role of mTOR in osteoclast formation and function, mTOR inhibitors could be used to treat numerous diseases that involve overactive osteoclasts, such as osteoporosis, inflammatory arthritis, Paget’s disease, and cancer-related osteolysis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Osteoclasts
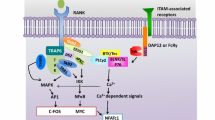
The skeleton is constantly being remodeled. New bone is deposited by osteoblasts, the bone forming cells, while old or damaged bone is removed by osteoclasts, the bone resorbing cells. These cycles of bone formation and resorption are tightly controlled, with both osteoblast and osteoclasts secreting molecules regulating each other’s activity (reviewed in [1, 2]). Osteoclasts are multinucleated cells of hematopoietic origin formed by fusion of mononuclear precursors (Fig. 5.1) [1, 3]. This precursor differentiation and fusion is initiated by two factors secreted by osteoblasts, osteocytes and stromal cells: macrophage colony stimulating factor (M-CSF) and receptor activator of nuclear factor κB ligand (RANKL). These two molecules are absolutely necessary for osteoclast differentiation, fusion, activity, and survival. Lack of either RANKL, its receptor RANK, M-CSF or M-CSF receptor CSF-1R leads to an osteoclastogenesis defect and severe osteopetrosis [4,5,6].

Osteoclastogenesis (adapted from Boyle et al. [69]). In the presence of M-CSF and RANKL, osteoclast precursors undergo differentiation and fusion. Transcription factors are listed above the cells; key functional proteins are listed below the cells. To regulate osteoclast formation and function, osteoblasts and stromal cells secrete osteoprotegerin (OPG), a decoy receptor for RANKL
To resorb bone, mature multinucleated osteoclasts attach to the bone surface and form a tight sealing zone. This sealing zone is defined by a dense cytoskeletal actin ring structure, composed primarily of F-actin. Within this sealing zone, osteoclasts form a convoluted plasma membrane called a “ruffled border,” a dynamic structure formed by continuous fusion of lysosomes and secretory vesicles delivering proteolytic enzymes for bone resorption, as well as continuous fission of transcytosing vesicles moving the degraded matrix away from the resorption site to the opposite (basolateral) side of the cell [1, 7]. The ruffled border is enriched with proton pumping vacuolar H+-ATPases (V-ATPases) and chloride proton exchangers (ClC7), the protein complexes responsible for creating an acidic environment necessary to dissolve the mineral component of bone and to allow degradation of the bone matrix proteins [1].
M-CSF is responsible for osteoclast precursor proliferation, precursor commitment, cytoskeletal organization, and survival. M-CSF binding to its tyrosine kinase receptor CSF-1R (also known as c-Fms or M-CSF receptor) activates the phosphatidyl-inositol 3-kinase (PI3K)/AKT pathway, similar to other receptor tyrosine kinases. One of the downstream targets of PI3K/AKT pathway is a serine/threonine kinase mechanistic target of rapamycin (mTOR), the main subject of this chapter (its activation and function will be discussed later in greater detail). RANKL binding to RANK initiates a number of signaling cascades and activates several transcription factors, such as nuclear factor κB (NF-κB), activator protein 1 (AP-1) and nuclear factor of activated T cells c1 (NFATc1) [1, 3, 8, 9]. These transcription factors control transcription of osteoclast-specific genes that play a role in osteoclast function (e.g., tartrate-resistant acid phosphatase (TRAP), cathepsin K (CtsK), calcitonin receptor (CTR), osteoclast-enriched V-ATPase subunits a3 and d2); attachment (e.g., integrin αvβ3); or fusion (e.g., dendritic cell-specific transmembrane protein (DC-STAMP) and osteoclast-associated receptor (OSCAR)) as outlined in Fig. 5.1 [1, 10, 11].
Osteoclastogenesis is usually described as a multistage process which includes proliferation of the precursors, commitment, fusion of the committed osteoclast precursors, polarization on the bone surface, formation of the sealing zone/ruffled border, and apoptosis (although the latest intravital imaging shows fission of osteoclasts at the end of the bone resorbing cycle [12]; therefore, it is possible that apoptosis is not the ultimate last stage of the osteoclast life cycle in vivo). Each stage of osteoclastogenesis is defined by the expression of key proteins—transcription factors and other proteins involved in osteoclast differentiation, fusion, and function. For example, PU.1 is the earliest hematopoietic transcription factor expressed by the osteoclast precursors, and loss of PU.1 results in the complete absence of osteoclasts and myeloid precursors [8]. To elucidate precise molecular mechanisms activated during different stages of osteoclast differentiation, two conditional gene targeting mouse models are widely used. The lysozyme M (LysM)-Cre mouse model targets osteoclast precursors, since LysM is expressed mainly by the cells of the myeloid lineage, the cells that include osteoclast progenitors, monocytes, macrophages, and dendritic cells [13]. Meanwhile, the Ctsk-Cre mouse model targets later stages of osteoclastogenesis, since CtsK is expressed primarily by mature osteoclasts and not by the precursors [14].
As mentioned earlier, M-CSF signaling through PI3K/AKT activates mTOR, an evolutionary conserved kinase responsible for cellular responses to growth factors, nutrient availability and other extracellular cues [15]. It regulates protein and lipid synthesis, lysosomal and mitochondrial biogenesis, just to name a few, in all eukaryotic cells. The purpose of this chapter is to summarize the latest research on the role of mTOR in osteoclast differentiation and function. But, first, we will briefly describe the major players involved in mTOR signaling.
5.2 Overview of mTOR Signaling
mTOR belongs to the PI3K-related family of kinases. As the name implies, TOR (target of rapamycin) was identified in yeast genetic screens as the protein target of rapamycin, a macrolide with an antifungal and immunosuppressant activity [16]. In mammalian cells, mTOR exists as part of two multiprotein complexes, complex 1 (mTORC1) and complex 2 (mTORC2) (Fig. 5.2). mTORC1 consists of mTOR, regulatory-associated protein of mTOR (Raptor), DEP-domain containing mTOR-interaction protein (DEPTOR), proline-rich AKT substrate 40 kDa (PRAS40), and mammalian lethal with Sec13 protein 8 (mLST8). The functions of the mTORC1 components are well known: Raptor assists with substrate recognition and recruitment [17, 18], mLST8 is a positive regulator of mTOR activity [19], while PRAS40 and DEPTOR are the negative regulators of mTOR activity [20, 21]. mTORC2 is made up of mTOR, rapamycin-insensitive companion of mTOR (Rictor), protein observed with Rictor (Protor-1/2), mammalian stress-activated protein kinase interacting protein (mSIN1), mLST8, and DEPTOR [22]. Since some of the components of these two complexes are the same, Raptor and Rictor are commonly used as markers to identify and distinguish mTORC1 and mTORC2, respectively. The functions of these two complexes are also different: mTORC1 is involved in regulation of cell growth, proliferation, protein and lipid biosynthesis, as well as regulation of autophagy, a lysosomal degradation pathway; while mTORC2, although less studied, is involved in cell survival, metabolism and cytoskeletal reorganization [15, 22]. Both complexes have been observed associated with various cellular compartments, such as lysosomes, mitochondria, nuclei, and the cytosol and it has been suggested that this localization is directly connected to mTOR function [23].

mTORC1 and mTORC2 complexes composition and signaling (see text for detailed explanation)
mTORC1 is activated by several factors. Activation by growth factors is a complicated and tightly controlled multistep process (Fig. 5.2) (reviewed in detail in [15, 23,24,25]). Briefly, growth factor/cytokine signaling through, for example, receptor tyrosine kinases, activates PI3K, phosphoinositide-dependent kinase-1 (PDK1), and AKT. AKT phosphorylates TSC2 on S939 and T1462 and thus inhibits the tuberous sclerosis TSC1/TSC2/TBC1D7 complex (TSC) [26, 27]. The TSC complex is a GTPase-activating protein (GAP) for the GTPase Ras homolog enriched in brain (Rheb) that functions as a negative regulator of mTORC1. Inhibition of the TSC complex allows Rheb-GTP to bind mTORC1 and promote its kinase activity [25]. Both Rheb and active mTORC1 are localized on the lysosomal surface.
mTORC1 is also activated by amino acids (reviewed in detail in [24, 28]). The exact mechanism is still being investigated, but so far it appears to involve several multiprotein complexes that regulate cellular responses to individual amino acids. In very simplified terms, in the presence of amino acids, active mTORC1 complex is located on the lysosome where it phosphorylates its substrates: eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and ribosomal protein S6 kinase (S6K); both regulate downstream pathways necessary for protein and nucleotide synthesis. In the absence of amino acids, inactive mTORC1 has been reported to dissociate from the lysosome [29, 30]. Rheb and Rag GTPases, also located on the lysosomal surface, are necessary for mTORC1 activity. Several other multiprotein complexes, such as GATOR1 (GAP activity toward the Rag GTPases 1), GATOR2, KICSTOR (a scaffold for GATOR1) (reviewed in [15, 24]), have also been reported to regulate mTORC1 activity. Solute carrier family 38 member 9 (SLC38A9) and CASTOR1 have been described to serve as arginine sensors [31,32,33], while Sestrin1 and Sestrin2 have been identified as leucine sensors [34, 35].
In addition to all of these multiprotein complexes, active mTORC1 is tethered to the lysosome via Ragulator, a pentameric scaffolding complex that also anchors Rag GTPases to the lysosome. Furthermore, mTORC1 activation is directly linked to the V-ATPases: some of the subunits of the Ragulator complex directly interact with several V-ATPase subunits [30]. The V-ATPases are necessary for mTORC1 activation as inhibition of the V-ATPases using inhibitors or siRNA decreases mTORC1 activity; however, a precise role of the V-ATPases in mTORC1 signaling is not known [29, 30]. In the absence of amino acids, inactive mTORC1 allows initiation of autophagy, a lysosomal degradation process, to raise intracellular free amino acid levels by degrading proteins and organelles to survive this temporary “starvation” [36].
Less is known about mTORC2 signaling: mTORC2 is not activated by amino acids and is less sensitive to rapamycin treatment [22]. As described above, growth factor signaling activates AKT (S308), which, in turn phosphorylates mTOR on T2173 (in both mTORC1 and mTORC2 complexes) [37]. mTORC2 has been shown to phosphorylate AKT (S473) leading to maximal activation and stabilisation of AKT, thus connecting the mTORC1 and mTORC2 pathways (Fig. 5.2) [38].
5.3 Role of mTOR in Osteoclasts
Osteoclasts are unique cells: they are multinucleated (up to 20–30 nuclei per cell in pathological conditions); they contain numerous mitochondria; they have high levels of lysosomal membrane proteins and V-ATPases; during resorption, they secrete large amounts of proteolytic enzymes to degrade the demineralized bone matrix. All these processes require increased energy demands, as well as protein and lipid synthesis. The cellular master switch responsible for regulation of cell survival, cell proliferation, lipogenesis, protein synthesis, nucleotide synthesis, lysosome and mitochondrial biogenesis in all eukaryotic cells is, in fact, mTOR. Even though mTOR is involved in all of these cellular processes, surprisingly little is known about the precise role of mTOR in regulation of osteoclast differentiation and function. Below is the summary of what we do know so far.
During osteoclastogenesis, mTOR mRNA expression is increased at the preosteoclast stage, but returns to baseline levels in mature osteoclasts (our unpublished data and [39]). At the same time, gene expression levels of the mTORC1 and mTORC2 specific subunits Raptor and Rictor do not change over the course of osteoclastogenesis [39]. The activity of mTORC1, as measured by phosphorylation of S6K and S6, is also increased during the early/proliferation phase and then rapidly declines to almost undetectable levels in mature multinucleated osteoclasts [40, 41]. Both RANKL and M-CSF activate mTORC1 signaling, as determined by phosphorylation of mTORC1 substrates S6K, S6, and 4EBP1 [42]. mTOR has been shown to play a role in osteoclast survival: mTOR downregulates Bim (also known as BCL2-like protein 11), a proapoptotic BH3 domain only protein, and the decreased levels of Bim allow for osteoclast survival [43]. Treatment with mTORC1 inhibitor rapamycin or with mTOR siRNA inhibits osteoclast formation and induces apoptosis, confirming that mTORC1 is necessary for osteoclastogenesis and survival [42, 43]. Interestingly, it was observed that rapamycin had a more pronounced effect on osteoclast differentiation when cells were treated with the inhibitor at the earlier (days 1–2), rather than later (days 3–4) stages of osteoclastogenesis [39]. Furthermore, genetic deletion of mTOR or Raptor in vitro also significantly suppressed osteoclastogenesis in cells derived from bone marrow of mTOR fl/– or Raptor fl/fl mice [39]. Similar observations were also made in vivo: rapamycin treatment inhibited metastasis-induced osteoclastogenesis, as well as bone resorption [44]. These observations suggested that mTORC1 activity is more important at the precursor proliferation/early commitment stage rather than at the mature osteoclast stage.
Several in vivo studies have been published in the last 2 years (summarized in Table 5.1), which methodically deciphered and shed the light on the role of mTORC1 in osteoclast biology. To elucidate the role of mTORC1 in osteoclastogenesis, Wu et al. [45] created two osteoclast-specific conditional knockout mouse models by targeting the mTORC1 negative regulator TSC1 in osteoclast precursors (LysM-Cre;Tsc1 fl/fl mice) or in mature osteoclasts (Ctsk-Cre;Tsc1 fl/fl mice). Unexpectedly, hyper-activation of mTORC1 resulted in high bone density/osteopetrotic phenotype in both mouse lines; however, the underlying osteoclast defect was different [45]. LysM-Cre;Tsc1 fl/fl mice had normal weight and size, and the number of osteoclasts in vivo did not appear to be affected by the deletion; however, bone resorption parameters (lower serum C-terminal telopeptide (CTX) and urine deoxypyridinoline (DPD) levels) were decreased. In vitro, monocyte proliferation was increased, while the number of multinucleated TRAP-positive cells and bone resorption were significantly diminished. Gene expression of the differentiation markers (e.g., DC-STAMP, NFATc1, CtsK, TRAP) was decreased, mainly due to reduced NFATc1 and NF-κB activity, therefore, explaining the failure to form multinucleated cells [45]. The bone and osteoclast phenotype, as well as an inhibition of NFATc1 and NF-κB were also confirmed independently by another group [40]. At the same time, the Ctsk-Cre;Tsc1 fl/fl mice also had normal weight and size; however, the number of osteoclasts in vivo was dramatically increased [45]. Bone resorption, as measured by CTX and DPD levels, was significantly decreased in these mice, suggesting impaired osteoclast function. In vitro, the number and size of the Ctsk-Cre;Tsc1 fl/fl osteoclasts was increased; however, the bone resorbing function was decreased. The authors also reported that the number of ring-like actin structures in the bones of both mouse lines was diminished, and this defect appeared to be more pronounced in Ctsk-Cre;Tsc1 fl/fl osteoclasts, suggesting that the impaired bone resorption was due to actin ring formation defect [41, 45]. Another group, also using osteoclasts derived from Ctsk-Cre;Tsc1 fl/fl mice, showed that hyperactivation of mTORC1 in mature osteoclasts disturbed podosome belt/actin ring assembly, resulting in decreased bone resorption in vivo and in vitro [41]. Interestingly, treatment with low doses of rapamycin rescued podosome belt assembly and bone resorbing function both in vivo and in vitro, suggesting that low levels of mTORC1 activity are still required for proper osteoclast function. Xu et al. [41] further showed that this actin ring/podosome assembly defect was dependent on mTOR regulation of small GTPases Cdc42 and Rac1, the regulators of the actin cytoskeleton and the GTPases involved in osteoclast migration, formation of actin ring, podosome belt, and ruffled border [46]. The osteoclasts from Ctsk-Cre;Tsc1 fl/fl mice had lower levels of Rac1/Cdc42 activity compared to controls and the authors proposed that mTORC1 is an upstream negative regulator of Rac1/Cdc42 [41].
Two groups generated osteoclast-specific conditional knockout mouse models where mTORC1 was inactivated by targeting Raptor, the unique scaffolding protein in mTORC1, in osteoclast precursors and in mature osteoclasts [40, 47]. Interestingly, the mouse models had different bone phenotypes: LysM-Cre;Raptor fl/fl mice had osteopenia [40], while Ctsk-Cre;Raptor fl/fl mice were osteopetrotic [47]. The LysM-Cre;Raptor1 fl/fl mice had a reduced bone mass and a significantly higher number of osteoclasts in vivo, together with an elevated bone resorption rate (as measured by serum CTX levels). In vitro, osteoclastogenesis using the cells from the LysM-Cre;Raptor1 fl/fl mice was increased, generating higher number of larger (5+ nuclei) cells; gene expression of osteoclast-specific genes was also upregulated, suggesting an acceleration of differentiation compared to the controls. In addition, the LysM-Cre;Raptor1 fl/fl osteoclasts generated larger resorption lacunae, confirming the in vivo phenotype [40]. Since the protein levels of the transcription factors NFATc1 and NF-κB2 were increased, the authors proposed that the noncanonical NF-κB2 and NFATc1 are negatively regulated by mTORC1 during osteoclastogenesis [40]. In comparison, when mTORC1 was inactivated in mature osteoclasts, the Ctsk-Cre;Raptor fl/fl mice had lower bone mass and decreased number of osteoclasts [47]. In vitro, the number of multinucleated Ctsk-Cre;Raptor fl/fl osteoclasts was also decreased, even though osteoclast progenitor proliferation was not affected. The authors demonstrated that the expression of a constitutively active form of S6K1 rescued the osteoclast phenotype in vitro, confirming that mTORC1 activity is necessary for proper osteoclast maturation and function [47].
In summary, these latest osteoclast-specific conditional knockout models clearly demonstrate that mTORC1 signaling plays a crucial role in osteoclast formation and function. What is apparent so far, is the fact that mTORC1 has different roles during different stages of osteoclastogenesis: high mTORC1 activity is necessary for early precursor proliferation phase, while low levels of mTORC1 activity are required for the later stages—osteoclast fusion, cytoskeletal reorganization/actin ring/ruffled border formation, and bone resorption (furthermore, mTORC1 was recently shown to play a role in determining osteoclast size, both in continuous osteoclast fusion and in fusion-independent cytoplasm growth [48]). At the moment, it is hard to reconcile all the observations into a simple consistent model, but it is clear that dysregulation of mTORC1 can potentially lead to osteopetrosis or to osteopenia when activated or repressed at the wrong time.
5.4 Regulation of mTOR in Osteoclasts
Based on the studies summarized in the previous section, it is clear that mTORC1 activity levels differ at different stages of osteoclastogenesis, with higher protein levels of mTOR/mTORC1 activity at the earlier stages, and lower protein levels/activity at the later stages. The mechanisms of mTORC1 regulation in osteoclasts are still not known; however, there are potential clues suggesting that in osteoclasts mTOR is regulated differently compared to other cell types and cell lines.
The majority of published studies indicate that the following factors are involved in mTORC1 regulation: (1) nutrient/amino acid status, with mTORC1 reported to localize to the surface of the lysosomes and to dissociate during starvation; (2) autophagy, where active mTORC1 suppresses autophagy, while inactive mTORC1 induces autophagy [49]; (3) the V-ATPase function, where inhibition of the V-ATPase downregulates mTORC1 activity [29, 30]; and (4) lysosomal positioning, where peripheral vs. perinuclear localization of the lysosomes dictates mTORC1 activity levels [50]. Lysosomes appear to play a central role in mTORC1 regulation and function: it is the place for mTORC1 activity, substrate recruitment and phosphorylation; furthermore, mTORC1 activity is regulated by intraluminal amino acids via an unknown inside-out mechanism ([30] and see detailed reviews in [25, 51, 52]). Meanwhile, mTORC1 is responsible for regulation of lysosomal biogenesis: active mTORC1 phosphorylates (and inactivates) transcription factor EB (TFEB), the transcription factor considered to be a master regulator of lysosomal biogenesis [53]. This creates an interdependent relationship between mTORC1 and the lysosome: the lysosome regulates mTORC1 activity, while mTORC1 controls lysosome formation and function.
Our laboratory is interested in investigating the role of the lysosomal pH in osteoclast differentiation and signaling. One of the model systems we use is a mouse model with the R740S mutation in the V-ATPase a3 subunit, where an evolutionary conserved arginine involved in proton translocation across the membrane is replaced with serine [54]. The a3 R740S mutation does not affect protein expression, and the V-ATPase multisubunit complexes are assembled and targeted to the lysosome; however, the proton pumping is impaired [55, 56]. The a3 containing V-ATPases are preferentially expressed in osteoclasts and are localized to the lysosomes and to the ruffled border membrane [57, 58]. Due to this high expression level, the a3 R740S mutation significantly affects osteoclast bone resorption: homozygous (R740S/R740S) mice have severe osteopetrorickets, and heterozygous (+/R740S) animals have mild osteopetrosis [56, 59]. Lysosomal pH in osteoclasts with the R740S mutation is higher compared to the wild type (+/+) controls: pH ~6.3 in the R740S/R740S cells vs. pH ~5.7 in +/R740S cells vs. pH ~4.7 in +/+ controls [55, 60]. This gene-dosage effect makes the R740S cells a perfect model to study the role of lysosomal pH in osteoclast signaling.
During characterization of the +/R740S osteoclasts, we demonstrated that the these cells had decreased osteoclastogenesis due to accumulation of regulator of calcineurin 1 (RCAN1), an endogenous inhibitor of NFATc1, resulting in impaired NFATc1 nuclear translocation [55]. As RCAN1 protein levels in the cells are controlled by lysosomal degradation [61], we investigated autophagy, a lysosomal degradation process dependent on proper lysosomal function. We made three interesting observations: (1) osteoclastogenesis was severely impaired in R740S/R740S cells (Fig. 5.3); (2) autophagic flux was blocked in cells with the R740S mutation; and (3) mTOR protein levels and mTORC1 activity was increased in cells with the R740S mutation [62, 63]. The last observation appeared to contradict the current model of mTORC1 signaling, a model stating that active mTORC1 inhibits autophagy and, therefore, cannot coexist with active autophagic degradation. To verify our findings, we treated +/+ cells with the lysosomal inhibitors ammonium chloride (NH4Cl) or chloroquine (CHQ) and confirmed that increased lysosomal pH resulted in higher mTOR protein levels and mTORC1 activity [62]. Based on our results, we hypothesized that in osteoclasts mTOR is regulated by lysosomal degradation. Treatment of +/+ and +/R740S osteoclasts with CHQ and proteasomal inhibitor MG132 increased mTOR protein levels in +/+ cells, but not in +/R740S osteoclasts, confirming our hypothesis. Cycloheximide blockade (inhibition of new protein synthesis) showed a decrease of mTOR protein levels in +/+ cells; however, the rate of the decrease in +/R740S cells was significantly slower, further supporting our lysosomal degradation hypothesis [62]. Our finding contradicting the current model of mTOR regulation is not unique and have been also observed in at least two other cell types: in primary mouse chondrocytes [64, 65] and in primary mouse hippocampal neurons [66]. Bartolomeo et al. reported that this increased mTORC1 activity was only observed in chondrocytes from the mouse model for mucopolysaccharidosis type VII (MPSVII), a lysosomal storage disease, but not in fibroblasts from MSPVII mice or HeLa cells lacking the same gene [64]. Furthermore, Hwang et al. showed that in ischemia-induced hippocampal neurons, mTOR was preferentially degraded via the autophagy/lysosomal pathway [66]. These results collectively suggest that mTOR/mTORC1 regulation by lysosomal degradation could be a special property of highly specialized cells, such as neurons, chondrocytes, and osteoclasts.

Osteoclastogenesis in cells with R740S mutation. Spleen-derived osteoclasts were differentiated in the presence of M-CSF and RANKL for 4 days. The cells were then fixed and stained for TRAP, an osteoclast marker. R740S/R740S cells had almost no cells with more than four nuclei (unpublished observations)
Lysosomal positioning is another factor reported to be involved in regulation of mTORC1 activity [50]. Using HeLa cells, Korolchuk et al. showed that during “starvation” (corresponding to inactive mTORC1), the lysosomes are located in the perinuclear region of the cells, while in the presence of the nutrients/amino acids (corresponding to active mTORC1), the lysosomes are dispersed in the cytosol and move toward cell periphery [50]. Furthermore, overexpression of factors that redistributed lysosomes to the periphery, e.g., kinesins KIF1Bβ and KIF2 and the small GTPase ARL8B, increased mTORC1 activity. Contrary to HeLa cells [50], in osteoclasts, lysosomes were primarily perinuclear during “fed” conditions, while “starvation” caused the lysosomes to move to the periphery [63]. Similar observations were reported for human adipose microvascular endothelial cells, primary human macrophages, and dendritic cells [67, 68], suggesting that different cell types have different pattern of lysosomal distribution. In addition to lysosomal distribution, we also observed that in osteoclasts mTOR does not disassociate from the lysosome during “starvation”. Using an ultrapure lysosomal purification method, we demonstrated that absence of mTORC1/lysosome dissociation in the absence of nutrients was only observed in differentiated mature osteoclasts, but not in undifferentiated mouse monocyte cell line RAW264.7 [63].
In summary, we believe that mTOR regulation in osteoclasts (and possibly in other highly specialized cells, such as neurons) is different compared to other cell types and cell lines: (1) mTOR protein levels and mTORC1 activity appears to be regulated by lysosomal/autophagic degradation; (2) mTORC1 activity does not depend on lysosomal distribution; and (3) mTORC1 does not dissociate from the lysosome and remains associated with the lysosome even during “starvation.” However, the exact mechanisms involved in mTORC1 regulation in osteoclasts are not known and still need to be elucidated.
5.5 Conclusion
Osteoclasts are unique bone-resorbing cells involved in maintaining bone homeostasis; however, increased osteoclast activity is responsible for pathological bone loss in numerous conditions, such as osteoporosis, osteoarthritis, rheumatoid arthritis, Paget’s disease, and cancer-related osteolysis. As mTOR plays a key role in regulating osteoclast formation, activity, and function, mTORC1 signaling pathway could become a therapeutic target to treat diseases that involve overactive osteoclasts [38]. Since regulation of mTOR can be different in very specialized cells as we and others have shown, caution is necessary in extrapolating treatment paradigms from one cell/organ type to another.
References
Crockett JC, et al. Bone remodelling at a glance. J Cell Sci. 2011;124(Pt 7):991–8.
Teti A. Mechanisms of osteoclast-dependent bone formation. Bonekey Rep. 2013;2:449.
Ono T, Nakashima T. Recent advances in osteoclast biology. Histochem Cell Biol. 2018;149:325.
Kong YY, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–23.
Hsu H, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96(7):3540–5.
Wiktor-Jedrzejczak W, et al. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc Natl Acad Sci U S A. 1990;87(12):4828–32.
Salo J, et al. Removal of osteoclast bone resorption products by transcytosis. Science. 1997;276(5310):270–3.
Tondravi MM, et al. Osteopetrosis in mice lacking haematopoietic transcription factor PU.1. Nature. 1997;386(6620):81–4.
Carey HA, et al. Enhancer variants reveal a conserved transcription factor network governed by PU.1 during osteoclast differentiation. Bone Res. 2018;6:8.
Yagi M, et al. DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med. 2005;202(3):345–51.
Miyamoto H, et al. OC-STAMP and DC-STAMP cooperatively modulate cell-cell fusion to form osteoclasts and foreign body giant cells. J Bone Miner Res. 2012;27:1289.
McDonald M, et al. Intravital imaging of osteoclasts in vivo reveals cellular recycling as a novel cell fate mechanism. J Bone Miner Res. 2017;32(Suppl 1):Abstrac #1109.
Clausen BE, et al. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–77.
Chiu WS, et al. Transgenic mice that express Cre recombinase in osteoclasts. Genesis. 2004;39(3):178–85.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–76.
Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–9.
Hara K, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–89.
Nojima H, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278(18):15461–4.
Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904.
Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–86.
Sancak Y, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903–15.
Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126(Pt 8):1713–9.
Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203(4):563–74.
Meng D, Frank AR, Jewell JL. mTOR signaling in stem and progenitor cells. Development. 2018;145(1):pii: dev152595.
Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med. 2012;18(9):524–33.
Manning BD, et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–62.
Han S, et al. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal. 2008;20(6):1084–91.
Wolfson RL, Sabatini DM. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab. 2017;26(2):301–9.
Sancak Y, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303.
Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–83.
Wang S, et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science. 2015;347(6218):188–94.
Rebsamen M, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519(7544):477–81.
Chantranupong L, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165(1):153–64.
Saxton RA, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science. 2016;351(6268):53–8.
Wolfson RL, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–8.
Shen HM, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. 2014;39(2):61–71.
Halova L, et al. Phosphorylation of the TOR ATP binding domain by AGC kinase constitutes a novel mode of TOR inhibition. J Cell Biol. 2013;203(4):595–604.
Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. 2016;12(3):169–82.
Indo Y, et al. Metabolic regulation of osteoclast differentiation and function. J Bone Miner Res. 2013;28(11):2392–9.
Zhang Y, et al. mTORC1 inhibits NF-kappaB/NFATc1 signaling and prevents osteoclast precursor differentiation, in vitro and in mice. J Bone Miner Res. 2017;32(9):1829–40.
Xu S, et al. TSC1 regulates osteoclast podosome organization and bone resorption through mTORC1 and Rac1/Cdc42. Cell Death Differ. 2018;
Glantschnig H, et al. M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003;10(10):1165–77.
Sugatani T, Hruska KA. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J Biol Chem. 2005;280(5):3583–9.
Hussein O, et al. Rapamycin inhibits osteolysis and improves survival in a model of experimental bone metastases. Cancer Lett. 2012;314(2):176–84.
Wu H, et al. Bone size and quality regulation: concerted actions of mTOR in mesenchymal stromal cells and osteoclasts. Stem Cell Rep. 2017;8(6):1600–16.
Touaitahuata H, Blangy A, Vives V. Modulation of osteoclast differentiation and bone resorption by Rho GTPases. Small GTPases. 2014;5:e28119.
Dai Q, et al. Inactivation of regulatory-associated protein of mTOR (raptor)/mammalian target of rapamycin complex 1 (mTORC1) signaling in osteoclasts increases bone mass by inhibiting osteoclast differentiation in mice. J Biol Chem. 2017;292(1):196–204.
Tiedemann K, et al. Regulation of osteoclast growth and fusion by mTOR/raptor and mTOR/rictor/Akt. Front Cell Dev Biol. 2017;5:54.
Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998;273(7):3963–6.
Korolchuk VI, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13(4):453–60.
Puertollano R. mTOR and lysosome regulation. F1000Prime Rep. 2014;6:52.
Rabanal-Ruiz Y, Korolchuk VI. mTORC1 and Nutrient homeostasis: the central role of the lysosome. Int J Mol Sci. 2018;19(3):pii: E818.
Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–7.
Kawasaki-Nishi S, Nishi T, Forgac M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc Natl Acad Sci U S A. 2001;98(22):12397–402.
Voronov I, et al. The R740S mutation in the V-ATPase a3 subunit increases lysosomal pH, impairs NFATc1 translocation, and decreases in vitro osteoclastogenesis. J Bone Miner Res. 2013;28(1):108–18.
Ochotny N, et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J Bone Miner Res. 2011;26(7):1484–93.
Toyomura T, et al. Three subunit a isoforms of mouse vacuolar H(+)-ATPase. Preferential expression of the a3 isoform during osteoclast differentiation. J Biol Chem. 2000;275(12):8760–5.
Manolson MF, et al. The a3 isoform of the 100-kDa V-ATPase subunit is highly but differentially expressed in large (>or=10 nuclei) and small (<or= nuclei) osteoclasts. J Biol Chem. 2003;278(49):49271–8.
Ochotny N, et al. The R740S mutation in the V-ATPase a3 subunit results in osteoclast apoptosis and defective early-stage autophagy. J Cell Biochem. 2013;114(12):2823–33.
Johnson L, et al. V-ATPases containing a3 subunit play a direct role in enamel development in mice. J Cell Biochem. 2017;118(10):3328–40.
Liu H, et al. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009;23(10):3383–92.
Hu Y, et al. Lysosomal pH plays a key role in regulation of mTOR activity in osteoclasts. J Cell Biochem. 2016;117(2):413–25.
Wang A, et al. Activity-independent targeting of mTOR to lysosomes in primary osteoclasts. Sci Rep. 2017;7(1):3005.
Bartolomeo R, et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J Clin Invest. 2017;127(10):3717–29.
Newton PT, et al. Pharmacological inhibition of lysosomes activates the MTORC1 signaling pathway in chondrocytes in an autophagy-independent manner. Autophagy. 2015;11(9):1594–607.
Hwang JY, et al. Global ischemia induces lysosomal-mediated degradation of mTOR and activation of autophagy in hippocampal neurons destined to die. Cell Death Differ. 2017;24:317.
Johnson DE, et al. The position of lysosomes within the cell determines their luminal pH. J Cell Biol. 2016;212(6):677–92.
Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108(3):855–64.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–42.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zirngibl, R.A., Voronov, I. (2018). The Role of mTOR in Osteoclasts. In: Turksen, K. (eds) Autophagy in Health and Disease. Stem Cell Biology and Regenerative Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-98146-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-98146-8_5
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-98145-1
Online ISBN: 978-3-319-98146-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)