
Abstract
The interplay of multiple biologic pathways and processes acts in synchrony to repair wounded tissue. Proangiogenic and inflammatory mediators allow enhanced delivery of oxygen and nutrients to repair wounded tissue. Fibrosis is the hallmark feature of wound healing in its final stage. In the eye, subretinal fibrosis, a nonfunctioning mass of fibrotic tissue situated in the subretinal space, is a manifestation of aberrant repair and is the characteristic end-stage subretinal lesion in exudative or neovascular age-related macular degenerative. The subretinal fibrotic lesion is preceded by gradual degeneration of the outer retinal cellular structures, the development of neovascular vessels, inflammation, and retinal tissue damage that culminates into the development of subretinal fibrosis. In this chapter, we will review the pathobiology of exudative age-related macular degeneration (AMD) as it relates to the pathogenesis of its end-stage visually devastating cicatricial lesion: subretinal fibrosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Introduction
The visual pathway is intricate and involves the concerted action of many complex intraocular structures and the central nervous system. Any pathological process along the visual pathway can lead to distorted or diminished vision. The light processing structures in the eye are analogous to capturing an image using a camera with film or a digital chip. The anterior ocular structures focus the light onto the retina which is responsible for processing the light energy and transmitting a signal to the brain.
Light from the environment is refracted principally by the cornea and the intraocular lens, and the refracted light is focused on the retina as it transverses through the vitreous in the posterior segment of the eye. The vitreous consists of a relatively transparent gelatinous structure composed mainly of serous fluid, collagen, and hyaluronic acid. The vasculature of the central retinal artery and the capillary vasculature of the underlying choroid nourish the light processing retina, which lines the inner wall of the eye. The retina absorbs photons of light via photoreceptors, generating electric impulses which are then transmitted along the optic nerve to the visual cortex in the occipital lobe.
The retina is the light processing center inside the eye. The retina is an energy-intense, highly metabolic, and oxygen-consuming intraocular tissue. It consists of a synchronized network of cellular components and axons that collectively capture light and transduce an electrical impulse to the optic nerve. A specialized region of the retina, known as the macula, has densely packed cone photoreceptors that enable high spatial visual acuity and color vision. In a highly metabolic, oxygen-intense activity, vitamin A-derived light-sensitive molecules bound to opsin within the photoreceptors process the light energy. The underlying retinal pigment epithelium (RPE) assists with recycling of photoreceptor degradative products, contributes to the blood-ocular barrier, and maintains the subneurosensory retinal space. The basement membrane of the RPE, known as Bruch’s membrane, is composed of collagen and elastic fibers. The RPE and Bruch’s membrane, therefore, serve to protect and maintain the neurosensory retina. The cellular structures of the retina cannot regenerate. Thus, once there is retinal cellular degeneration or irreparable cellular injury, retinal functional deficits often persist long-term.
In the eye, aberrant wound healing and fibrosis can affect the clarity of the visual axis depending on the ocular tissue involved. For example, fibrosis in the cornea can cause corneal opacification and thereby prohibit light from entering the eye. Retinal fibrovascular scarring and gliosis from ischemia and neovascularization on its surface can cause tractional retinal detachments and poorly functioning retinal tissue. In exudative or neovascular age-related macular degeneration (AMD), subretinal fibrosis results in damage and impaired function of the macular region of the retina resulting in severe visual impairment with difficulty discerning fine details in vision and color vision. Naturally, understanding and targeting pathways that influence the development of subretinal fibrosis and precursor lesions such as choroidal neovascularization are of interest to scientists and clinicians. In this chapter, we will review the pathobiology of exudative AMD as it relates to the pathogenesis of subretinal fibrosis.
Age-Related Macular Degeneration
Age-related macular degeneration is a bilateral progressive retinal degenerative disorder in the geriatric population and the leading cause of irreversible visual impairment in the developed world. The burden of the disease continues to grow, as the already relatively high prevalence of AMD is expected to nearly double by the year 2050. A vision-threatening subtype of AMD characterized by choroidal neovascularization (CNV) generally responds to treatment with vascular endothelial growth factor (VEGF) inhibition, but a significant proportion of patients develop severe macular dysfunction either from inadequate treatment, persistent disease activity refractory to VEGF inhibition, or progressive atrophy and/or subretinal fibrosis despite control of CNV. Macular subretinal fibrosis results in damage to the outer retinal structures essential for light-induced signal transduction to the inner retinal cells and represents the end-stage sequelae of choroidal neovascularization in exudative macular degeneration.
Visual acuity is generally preserved in the earlier nonexudative stage of AMD. With time, accumulation of focal deposits of lipo-glyco-proteinaceous, extracellular material or “drusen,” are typically seen in Bruch’s membrane. The distribution and size of drusen varies and is found to be concordant to an individual’s risk of progression to the late stages of the disease. Along with drusen, the other hallmark features of AMD include retinal pigment epithelium (RPE) abnormalities, which signify the degeneration of the RPE and photoreceptors. In a healthy eye, the RPE is a monolayer of pigmented cells located between the neurosensory retina and the choroid that plays a critical role in the maintenance of visual function. The RPE dysfunction and the accumulation of drusen can lead to thickening of Bruch’s membrane and result in decreased diffusion of oxygen from the choriocapillaris to the photoreceptors. These changes to the outer retina stimulate CNV formation. Although the precise mechanism for the development and progression of AMD is yet to be elucidated, the development of unchecked exudative maculopathy usually results in a precipitous decline in vision. Approximately 15% of AMD patients will experience the exudative form of the disease, which is characterized by the development of CNV, retinal pigment epithelial detachments (PED), retinal pigment epithelial tears, retinal hemorrhages, and fibrovascular-induced hypertrophic scarring. Patients with exudative AMD may experience decreased vision, distortion of the vision, or central scotomata.
Fibrosis, typically in the subretinal space , is seen in the end-stage cicatricial form of exudative AMD and shares molecular mechanisms of fibrosis similar to other organs of the body. This “disciform scar,” characteristic at the end-stage of disease, refers to the round phenotypic appearance seen clinically (Fig. 17.1). Clinically, the disciform scar usually results in a central scotoma and loss of all fine details in vision with patient reliance thereafter on the rudimentary peripheral vision. Before chronic intraocular VEGF inhibition became the therapeutic mainstay, early treatments targeting choroidal neovascularization with surgical removal of choroidal neovascular membranes, laser photocoagulation therapy, and photodynamic therapy (PDT) often caused collateral damage to less affected retinal tissue and offered inadequate long-term CNV suppression.

Fundus photograph of end-stage exudative AMD with macular subretinal fibrosis (disciform scar) and atrophy. (Source: All images are from clinical patients of Dr. Martel)
Since the mid-2000s, clinical treatment of exudative AMD has transformed with the advent of intravitreal pharmacotherapy with chronic VEGF inhibition aimed at CNV suppression. These agents reduce the risk of severe vision loss; however, unsuccessful treatment outcomes have often been attributed to the progression of subretinal fibrosis or progressive retinal atrophy. Ideally, AMD therapy directed at the retinal “aging” process itself would prevent retinal degeneration. However, such a treatment has been elusive. VEGF inhibition alone remains an imperfect treatment for AMD, with clinicians and investigators interested in improving visual outcomes further. Thus, investigations into other therapeutic agents aimed at influencing other mediators of vision loss in AMD are of interest.
Antecedent Events to Subretinal Fibrosis
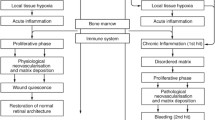
One of the cardinal features of wound healing is angiogenesis. New vessels are designed to assist in the repair of injured tissue, to increase local oxygen supply, and to recruit inflammatory cells to the damaged tissue. In exudative AMD, CNV develops in the subretinal and/or sub-RPE space, leading to hemorrhage and exudative changes, which in turn stimulates the development of subretinal fibrosis. This process is characterized by proliferation and cellular infiltration of various types. Retinal pigment epithelium, glial cells, fibroblasts, myofibroblast-like cells, and macrophages, interacting with inflammatory cytokines and growth factors, result in remodeling of the chorioretinal tissue extracellular matrix. Because of the complexity of the cellular interactions and the numerous mediators, no useful therapeutic options for subretinal fibrosis in retinal disease currently exist.
Although the precise mechanism of CNV development is unclear, degenerative changes in Bruch’s membrane with an accumulation of drusen and thickening of Bruch’s membrane contribute to a proangiogenic environment. Additionally, increased vascular permeability, and vasodilation accompanied by migration and proliferation of endothelial cells result in the formation of a neovascular vascular network. The interplay of proangiogenic mediators including VEGF, fibroblast growth factor (FGF), transforming growth factor (TGF), and angiopoietin with downregulation of antiangiogenic mediators result in the development of a mature choroidal neovascular membrane. The neovascularization originates in the choroid and extends through the damaged Bruch’s membrane toward the retina.
In nature’s attempt to heal Bruch’s membrane, the process of wound repair becomes aberrant. Fibroblasts accompany the abnormal choroidal neovascular vessels that spontaneously bleed and leak fluid into the retina and within the subneurosensory retinal space. Clinically, fluorescein angiography is helpful in determining the location and activity of choroidal neovascularization. On fluorescein angiography, CNV is identified by leakage of fluorescein dye into the retinal tissue represented by focal hyperfluorescence corresponding to the site of CNV (Fig. 17.3).
The spontaneous bleeding and fluid leakage from neovascular blood vessels damage the outer retinal cellular structures including the RPE and photoreceptors. Large subretinal hemorrhages are known to have a poor visual prognosis (Fig. 17.2). Animal models of subretinal hemorrhage have demonstrated the damage in the retina. For example, Glatt and Machemer’s work involving autologous blood injection into the subretinal space of rabbits showed early photoreceptor edema within 24 h, severe damage to the outer nuclear layer at 7 days, and photoreceptor degeneration. Additionally, Toth et al. demonstrated the importance of photoreceptor toxicity from fibrin degradation products in a cat model.

Fundus photograph of a patient with exudative AMD who developed a massive submacular hemorrhage from choroidal neovascularization. (Source: All images are from clinical patients of Dr. Martel)

Fluorescein angiography of a patient with exudative AMD showing fluorescein leakage at the site of choroidal neovascularization in the central macular region of the right eye. (Source: All images are from clinical patients of Dr. Martel)

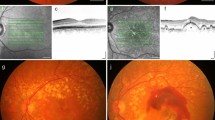
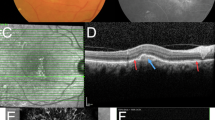
Optical coherence tomography imaging of the left eye of a patient with exudative AMD demonstrating subretinal hyper-reflective material consistent with subretinal fibrosis. Standard cross sectional analysis view, OCT on Heidelberg Spectralis. (Source: All images are from clinical patients of Dr. Martel)
Inflammation in Age-Related Macular Degeneration
The immune-privileged properties of the eye allow for transplantation of the cornea, for example, without the need for systemic immunosuppression as in other organs. Specialized immune defenses along with the blood-ocular barrier bestow the immune-privileged status of the retina. Despite the immune-privileged properties of the eye, ocular inflammation correlates with chorioretinal diseases, including AMD.
Although clinical trials of drugs targeting immune effectors have failed to alter the course of AMD, inflammation plays a critical role in the disease pathogenesis. As the macula degenerates, aberrant inflammatory mediators activate the immune system locally in the macular region. Genetic variants of immune modulators such as complement factors, cytokines, chemokines, as well as cellular mediators of innate and adaptive immunity are associated with the development and progression of AMD. Choroidal endothelial cells and invading immune cells have been shown to be responsive to complement activation products and can modulate interactions between monocytes and lymphocytes. Cytokines promoted by complement activation pathways have been shown to promote neovascularization and cell death.
Complement Pathway
Complement pathway activation is known to be important in pulmonary and renal fibrosis and likely also plays a role in intraocular fibrotic changes. Genetic evidence from genome-wide association studies and genetic variant analyses suggests that the complement system, in particular the alternative complement system, is dysregulated in AMD. Components of the complement system have been found in drusen and have been shown to contribute to vascular endothelial growth factor (VEGF) expression, one of the main drivers of choroidal neovascularization. Indeed, several variants of proteins in the complement system such as complement factor H (CFH), complement factor B (CFB), and complement components 2, 3, and 5 (C2, C3, C5) are associated with the progression of or protection against AMD. The strongest association with AMD involves a polymorphism of CFH, which may be involved in up to 50% of AMD patients. Although there remains considerable enthusiasm in complement-targeted therapeutics, the historical clinical trial failures of these drug targets have simply reinforced the lack of understanding of how the complement interactions influence atrophic progression and subretinal fibrosis in AMD.
Innate Immune System
There is clear evidence from animal models demonstrating a complex role of macrophages and microglia in both preventing and encouraging exudative AMD. Microglia and macrophages are innate immune cells that function to initiate inflammatory responses , clear debris, and remodel tissue. Microglia are specialized cells that typically reside in the inner retina and the central nervous system possessing the ability to migrate into the subretinal space in response to inflammatory stimuli. Several studies have identified the presence of macrophages in AMD, in or around drusen, and at areas of retinal atrophy and choroidal neovascular membranes.
Some investigators have shown that macrophages from CNV tissue express VEGF. Macrophages can also induce proliferation and migration of vascular endothelial cells by cytokines accelerating angiogenesis and CNV formation. Macrophages can display different phenotypes as well: the M1 phenotype which is proinflammatory and more common in retinal macular atrophy or the M2 phenotype, which is anti-inflammatory, angiogenic, and is more common in the exudative AMD subtype.
Cytokines and Growth Factors
Choroidal neovascularization develops in growth factor and cytokine-rich environments, in tandem with proangiogenic molecules, most notably VEGF. While VEGF is the main proangiogenic target in clinical practice, other inflammatory mediators are thought to be contributory to CNV formation. Some of the key mediators include transforming growth factor (TGF)-β, fibroblast growth factor (FGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and tumor necrosis factor-α (TNF-α). The source of these cytokines may be RPE, infiltrating macrophages, or fibroblasts.
Chemokine receptors on various cells function to direct sites of inflammation, and their associated downstream effectors suggest that many inflammatory-related pathways contribute to the pathogenesis of AMD. In animal models expressing chemokine receptor deficiency such as CCL2, CCR2, and CX3C deficiency, phenotypic characteristics similar to AMD are present. Moreover, a chemokine-dependent immune function may have a role in photoreceptor apoptosis. In terms of CNV formation, chemokine receptors have been shown to be important. An eosinophil/mast cell chemokine receptor, CCR3, has been expressed on human CNV membranes. Both CCR3 and the chemokine CXCL8, also known as interleukin-8 (IL-8), have both been implicated in angiogenesis and are associated with AMD.
In clinical practice, intravitreal pharmacotherapy therapy targeting CNV is the mainstay of treating exudative AMD. Currently, the primary target of the intravitreal pharmacotherapy is VEGF, but other angiogenic modulator drugs targeting PDGF, FGF, and TGF are under investigation. Combined inhibition of VEGF and other growth factors or mediators of angiogenesis may be more effective than anti-VEGF alone. A phase 3 clinical trial in 2016 investigating combination therapy with anti-VEGF intravitreal pharmacotherapy combined with intravitreal pegpleranib (Fovista) anti-PDGF compared to anti-VEGF monotherapy failed to meet the primary endpoint of mean change in visual acuity at 12 months. Despite the dampened enthusiasm for targeting PDGF and other growth factors, PDGF and related growth factors are still being investigated as a potential target for reducing the incidence of subretinal fibrosis in exudative AMD.
Subretinal Fibrosis
Subretinal fibrosis in exudative AMD shares common molecular and cellular components of scar formation with other organs in the body. Inflammatory and pro-fibrotic cells migrate and proliferate at the Bruch’s membrane–RPE complex producing extracellular matrix and clearing cellular damage and debris. Angiogenesis and the development of choroidal neovascularization, sometimes termed choroidal neovascular membranes (CNVM) , are associated with these events in the retina. Repeated inflammation and tissue reorganization result in irreversible scar formation.
The generation of subretinal fibrous tissue is complicated, and multiple cell types, proteins, and products are involved. As mentioned previously, the various cytokines and growth factors produced by RPE, infiltrating macrophages, or fibroblasts are thought to be contributory to the pathogenesis of subretinal fibrosis. When the fibrous tissue becomes apparent clinically, the CNV/fibrous subretinal fibrotic tissue complex may be called a disciform scar. Disciform scars may continue to grow, with new areas of neovascularization proliferating along the edges of previously unaffected regions of the retina. An essential clinical imaging tool often used in the diagnosis and surveillance of AMD patients, optical coherence tomography (OCT) imaging, allows in vivo visualization of the subretinal fibrosis by visualizing subretinal hyper-reflective material (Fig. 17.4).
Histologically , fibrous tissue not typically apparent on ophthalmoscopy clinical examination accompanies a choroidal neovascular membrane (CNVM). The CNVM consists of connective tissue components admixed with cellular components. These cellular components include damaged RPE and photoreceptors, vascular endothelial cells, macrophages, myofibroblasts, and fibroblast-like cells. This CNVM fibrovascular tissue complex may be beneath the RPE, termed type I CNV, or between the RPE and the photoreceptors, termed type II CNV. The principal location of the choroidal neovascularization either beneath the RPE or within the subneurosensory retinal space may have implications for disease progression and outcome. CNVM histology has also demonstrated that development of fibrous scar is accompanied by an increase in apoptosis and a decrease in cellularity, suggesting that subretinal fibrosis may evolve along with regression of CNV in exudative AMD. The variable responses to anti-VEGF therapy among patients may be attributable to differences in the CNV morphology, phenotype, and location of CNV growth within the chorioretinal tissue.
The subretinal fibrosis itself is composed of a matrix of collagen (mainly type I and IV with lesser amounts of types III, V, and VI collagen) and fibronectin, with alpha-smooth muscle actin (α-SMA) and cytokeratin. Macrophages can directly produce extracellular matrix including type I collagen, which is responsible for maintaining the integrity of subretinal fibrous tissue and fibronectin. Perhaps one of the critical initial steps of subretinal fibrosis is an epithelial-mesenchymal transition of RPE cells. Alpha-B crystallin is an essential regulator of epithelial-mesenchymal transition, acting as a molecular chaperone for SMAD4 and as its potential therapeutic target for preventing subretinal fibrosis development in exudative AMD. Alpha-B crystallin has also been shown to influence angiogenesis.
Various cytokines and growth factors have been shown capable of triggering the epithelial-mesenchymal transition of the RPE, resulting in the conversion to myofibroblasts, which can further promote fibrotic activities such as cell proliferation, migration, and extracellular matrix remodeling. Matricellular proteins such as thrombospondin 1 (TSP1), tenascin-C, and osteonectin are present in CNV and are thought to regulate fibrosis, binding to growth factor receptors or integrins on the cell surface. Connective tissue growth factor (CTGF) is transcriptionally activated by several factors including transforming growth factor-β (TGF-β). CTGF stimulates fibroblast proliferation, migration, adhesion, and extracellular matrix formation, and its overproduction is thought to play a role in pathways that lead to fibrosis. In the eye, CTGF is correlated with pathologic fibrosis in vitreoretinal disorders; its role in subretinal fibrosis development in exudative AMD is unclear. There remain multiple components and pathways involved in subretinal fibrosis that has yet been fully elucidated. As we better understand the mechanisms of fibrosis in AMD, new therapies and treatments can be explored.
Clinical Implications in AMD
AMD is the most common cause of severe visual impairment in developed countries. Since the hallmark feature of exudative AMD is choroidal neovascularization, and subfoveal CNV subretinal fibrosis is common, the CNV lesion has been the target of interest to many investigators. Although the previous clinical course of inevitable and often abrupt decline in vision is improving with the advent of anti-VEGF therapy, most patients nevertheless lose vision from exudative AMD albeit at a much slower pace. Even with treatment, only about 30% of exudative AMD patients retain their original level of vision after 5 years.
Since anti-VEGF therapy only allows short-term suppression of choroidal neovascular activity and the exudative maculopathy, there is the need for chronic administration of intravitreal anti-VEGF pharmacotherapy to slow down the disease progression. Prompt initiation of anti-VEGF therapy may be beneficial since the development of subretinal fibrosis is associated with a longer interval between diagnosis of exudative AMD and treatment with anti-VEGF drugs. However, even with prompt initiation of anti-VEGF therapy, subretinal fibrosis may nonetheless develop or progress. Once subretinal fibrosis and scarring develop, no retinal regenerative treatments for reversing the end-stage maculopathy exist.
Therapeutic strategies aimed at inhibition of subretinal fibrosis are an active area of investigation. Although chronic suppression of the neovascularization seems to reduce the risk of subretinal fibrosis and end-stage maculopathy markedly, there remains a considerable portion of the AMD population that nonetheless experience severe loss of sight. This may be due to anti-VEGF agents serving a more important role in decreasing permeability or leakage of neovascular vessels rather than inducing neovascular regression. The molecular mechanism and effect of VEGF inhibition on pro-fibrotic factors associated with subretinal fibrosis remain unclear. Moreover, there are likely other mediators of CNV formation and exudation.
With emerging therapies on the horizon, early detection of subtle maculopathy in the future will have increasing importance. Advances in ocular imaging with adaptive optics technology and optical coherence tomography imaging with optical coherence tomography angiography may allow better quantification and objective evaluation of subretinal fibrosis and neovascularization. Advancements in in-vivo ocular coherence tomography imaging such as polarization-sensitive optical coherence tomography imaging may offer a unique method of quantifying and following subretinal fibrosis in vivo over time. These retinal imaging technologies may, therefore, allow optimized disease management and evaluation of emerging therapeutic strategies.
In the eye, subretinal fibrosis is a manifestation of aberrant wound repair and is the characteristic end-stage subretinal lesion in exudative AMD. While there have been great strides in the development of anti-VEGF pharmacotherapies, further investigation is necessary to alter the course of this disease. The interplay of various proangiogenic and inflammatory mediators is influential in exudative AMD and the associated subretinal fibrosis.
Suggested Reading
Bressler SB, Silva JC, Bressler NM, Alexander J, Green WR. Clinicopathologic correlation of occult choroidal neovascularization in age-related macular degeneration. Arch Ophthalmol. 1992;110:827–32.
Daniel E, Toth CA, Grunwald JE, Jaffe GJ, Martin DF, Fine SL, Maguire MG. Risk of scar in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2014;121(3):656–66.
Diago T, Pulido JS, Molina JR, Collett LC, Link TP, Ryan EH Jr. Ranibizumab combined with low-dose sorafenib for exudative age-related macular degeneration. Mayo Clin Proc. 2008;83(2):231–4.
Rein DB. Forecasting age-related macular degeneration through the year 2050. Arch Ophthalmol. 2009;127:533.
Friedlander M. Fibrosis and diseases of the eye. J Clin Investig. 2007;117:576–86.
Friedman DS, O'Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–72.
Grossniklaus HE. Histopathologic and ultrastructural findings of surgically excised choroidal neovascularization. Arch Ophthalmol. 1998;116:745.
Grossniklaus HE, Hutchinson AK, Capone A Jr, Woolfson J, Lambert HM. Clinicopathologic features of surgically excised choroidal neovascular membranes. Ophthalmology. 1994;101:1099–111.
Ishikawa K, Kannan R, Hinton DR. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp Eye Res. 2016;142:19–25.
Knickelbein JE, Chan C-C, Sen HN, Ferris FL, Nussenblatt RB. Inflammatory mechanisms of age-related macular degeneration. Int Ophthalmol Clin. 2015;55(3):63–78.
Leibowitz HM, Krueger DE, Maunder LR, et al. The Framingham eye study monograph: an ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol. 1980;24:335–610.
Nussenblatt RB, Ferris F. Perspectives: age related macular degeneration and the immune response – implications for therapy. Am J Ophthalmol. 2007;144(4):618–26.
Patel M, Chan C-C. Immunopathological aspects of age-related macular degeneration. Semin Immunopathol. 2008;30:97–110.
Schachat AP. Ryans retina. Edinburgh: Elsevier; 2018.
Schlingemann RO. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2003;242:91–101.
Seddon JM, Willett WC, Speizer FE, et al. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276:1141–6.
Siedlecki J, Wertheimer C, Wolf A, Liegl R, Priglinger C, Priglinger S, Eibl-Lindner K. Combined VEGF and PDGF inhibition for neovascular AMD: anti-angiogenic properties of axitinib on human endothelial cells and pericytes in vitro. Graefes Arch Clin Exp Ophthalmol. 2017;255:963–72.
Wang Y, Wang VM, Chan C-C. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye. 2010;25:127–39.
Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Investig. 2007;117:524–9.
Subfoveal neovascular lesions in age-related macular degeneration. Arch Ophthalmol. 1991;109:1242.
Glatt H, Machemer R. Experimental subretinal hemorrhage in rabbits. Am J Ophthalmol. 1982;94(6):762–73.
Toth CA, Morse LS, Hjelmeland LM, Landers MBIII. Fibrin directs early retinal damage after experimental subretinal hemorrhage. Arch Ophthalmol. 1991;109(5):723–9.
Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006;103:2328–33.
Sivaprasad S, Adewoyin T, Bailey TA, Dandekar SS, Jenkins S, Webster AR, et al. Estimation of systemic complement C3 activity in age-related macular degeneration. Arch Ophthalmol. 2007;125:515–9.
Tuo J, Smith B, Bojanowski CM, et al. The involvement of sequence variation and expression of CX3CR1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:1297–9.
Kent D, Sheridan C. Choroidal neovascularization: a wound healing perspective. Mol Vis. 2003;9:747–55.
Nakama T, Yishida S, Ishikawa K, Kobayashi Y, Zhou Y, Nakao S, Sassa Y, Oshima Y, Takao K, Shimahara A, Yoshikawa K, Hamasaki T, Ohgi T, Hayashi H, Matsuda A, Kudo A, Nozaki M, Ogura Y, Kuroda M, Ishibashi T. Inhibition of choroidal fibrovascular membrane formation by a new class of RNA interference therapeutic agent targeting periostin. Gene Ther. 2015;22:127–37.
Nicolò M, Piccolino FC, Zardi L, Giovannini A, Mariotti C. Detection of tenascin-C in surgically excised choroidal neovascular membranes. Graefes Arch Clin Exp Ophthalmol. 2000;238:107–11. https://doi.org/10.1007/s004170050018.
Kobayashi Y, Yoshida S, Zhou Y, Nakama T, Ishikawa K, Kubo Y, et al. Tenascin-C secreted by transdifferentiated retinal pigment epithelial cells promotes choroidal neovascularization via integrin αV. Lab Investig. 2016b;96:1178–88. https://doi.org/10.1038/labinvest.2016.99.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Martel, J.N., Nguyen, V.Q., Eller, A.W. (2019). Fibrotic Remodeling in Exudative (Wet) Macular Degeneration. In: Willis, M., Yates, C., Schisler, J. (eds) Fibrosis in Disease . Molecular and Translational Medicine. Humana Press, Cham. https://doi.org/10.1007/978-3-319-98143-7_17
Download citation
DOI: https://doi.org/10.1007/978-3-319-98143-7_17
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-98142-0
Online ISBN: 978-3-319-98143-7
eBook Packages: MedicineMedicine (R0)