
Abstract
The term ‘epigenetic’ refers to all heritable alterations that occur in a given gene function without having any change on the DeoxyriboNucleic Acid (DNA) sequence. Epigenetic modifications play a crucial role in development and differentiation of various diseases including cancer. The specific epigenetic alteration that has garnered a great deal of attention is DNA methylation, i.e., the addition of a methyl-group to cytosine. Recent studies have shown that different tumor types have distinct methylation profiles. Identifying idiosyncratic DNA methylation profiles of different tumor types and subtypes can provide invaluable insights for accurate diagnosis, early detection, and tailoring of the related treatment for cancer. In this study, our goal is to identify the informative genes (biomarkers) whose methylation level change correlates with a specific cancer type or subtype. To achieve this goal, we propose a novel high dimensional learning framework inspired by the dynamic data driven application systems paradigm to identify the biomarkers, determine the outlier(s) and improve the quality of the resultant disease detection. The proposed framework starts with a principal component analysis (PCA) followed by hierarchical clustering (HCL) of observations and determination of informative genes based on the HCL predictions. The capabilities and performance of the proposed framework are demonstrated using a DNA methylation dataset stored in Gene Expression Omnibus (GEO) DataSets on lung cancer. The preliminary results demonstrate that our framework outperforms the conventional clustering algorithms with embedded dimension reduction methods, in its efficiency to identify informative genes and outliers, and removal of their contaminating effects at the expense of reasonable computational cost.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Dynamic Data Driven Application Systems
- Informative Genes
- Attribute Reduction Algorithm
- Informative Biomarkers
- Outlier Detection Algorithm
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The term ‘epigenetic’ refers to all heritable alterations that occur in a given gene function without having any change on the DNA sequence. Epigenetic modifications, i.e., DNA methylation and histone post-translational modifications, regulate the transcription state of a gene, and play a crucial roles in cell differentiation and proliferation [10, 22]. Accumulating evidence suggests that aberrant epigenetic changes are affiliated with various diseases such as diabetes, schizophrenia, and cancers [11, 12, 21]. Compared with genetic alterations, aberrant epigenetic modifications typically occur at an early-stage of disease. They can thus be reversed given proper interventions. The study of epigenetics is emerging but fast-growing field of science as epigenetic biomarker and therapies promise for the detection and treatment of a broad array of diseases [9, 12]. One of the most important epigenetic alteration is DNA methylation, i.e., the addition of a methyl-group to DNA. The most prevalent DNA methylation is the covalent addition of a methyl group to the 5-carbon position of the cytosine to form a 5-methylcytosine (5mC) occurring within a CpG dinucleotide (a DNA sequence in which a cytosine and guanine nucleotide appear consecutively). CpG methylation is commonly affiliated with gene silencing and most abundant in heterochromatin regions.
Recent research has shown a significant interest in understanding of the correlation between aberrant DNA methylation and cancer [12, 13, 21]. These studies revealed that cancer cells have different methylation profiles from normal cells. DNA methylation can not only be used to differentiate different tumor types, but also to distinguish tumor subtypes [17, 24, 29]. Medical understanding of DNA methylation and its implications in biology of cancer has significantly advanced in the past years enabled by high-throughput DNA-sequencing-based methylation analysis. Herein, data mining techniques for the collected high-throughput data play an increasingly important role in extracting useful information for a wide range of applications such as identifying cancer diagnosis and prognostic biomarkers.
Clustering algorithms are powerful tools for identifying idiosyncratic DNA methylation profile of different tumor types and subtypes. Basically, cluster analysis groups similar data points into same groups. Amongst the clustering algorithms presented in the literature, hierarchical clustering (HCL) is widely used for DNA methylation analysis. Variants of HCL has also been applied to the analysis of different DNA methylation patterns in different cancer types or subtypes. For instance, [29] and [4] used agglomerative hierarchical clustering algorithm for identification of aberrant DNA methylation profiles in lung cancer subtypes and lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues, respectively, while [28] performed a two-way hierarchical clustering analysis to characterize the DNA methylation profiles for clear cell sarcoma of the kidney and other pediatric renal tumors. While HCL is relatively easy to implement and comes at the expense of low computational cost when compared to other clustering algorithms such as k-means, it is still a greedy algorithm and sensitive to outliers (or influential data points). As most of the clustering algorithms, HCL assumes all information to be equally important for clustering. This assumption, however, is unlikely to be valid in most of real systems and causes HCL to mark a significant number of points as outliers, which further necessitates an implementation of a dimension reduction algorithm.
In DNA methylation analysis, the data is very complex with thousands of genes being collected from a single patient making the determination of an informative set of genes (biomarkers) crucial for accurate identification of cancer-related DNA methylation profiles. To this end, several dimension reduction algorithms have been proposed in the literature for the characterization of informative biomarkers. For instance, the study [29] determined seven informative genes among 24 genes using Mann-Whitney U-test, while [4] chose 500 genes with the highest variance from thousands of genes. Another study implemented a two-way hierarchical clustering to find out an informative loci [28].
In this study, we propose a dynamic data driven hierarchical clustering (3D-HCL) framework motivated by the dynamic data driven application systems (DDDAS) paradigm founded by Darema [7, 8]. The proposed 3D-HCL framework embodies an HCL algorithm with a threefold capability to detect the outliers, identify the set of informative biomarkers, and define the clusters in an efficient manner using the newly measured data from the real system. 3D-HCL initializes with a principal component analysis (PCA). Then the HCL is run as an application system and based on the results of the HCL, an outlier detection score, a cluster membership score and an informative locus score are calculated. Here, these scores steer the measurement process (in our case, informative biomarkers) based on the results of the developed clustering algorithm (HCL) for the next iteration. The bidirectional information flow between HCL and the proposed scores continues until a termination condition is satisfied. These scores are also used for real-time classification of new samples. Based on the classification results, the orchestration module can call the biomarker identification module to retrieve information from the new samples or add the samples to the cluster structure.
In this study, our proposed DDDAS based framework addresses two major issues in HCL for performing data analysis in large and complex datasets such as DNA methylation. First, the performance of all clustering algorithms is highly dependent on the performance of their embedded dimension reduction (feature selection) algorithm. To the best of our knowledge, there does not exist a single universal algorithm that promises reasonable results for all datasets. The literature presents many dimension reduction algorithms for DNA methylation analysis whose performance is dependent on the data utilized for analysis. Hence, a successful implementation of a traditional HCL would require testing with several different dimension reduction algorithms. However, our proposed 3D-HCL framework is designed as generic so that it does not depend on such high numbers of dimension reduction algorithms. While 3D-HCL is initialized with a dimension reduction algorithm, the results are minimally affected by the selected initial set of informative biomarkers. The proposed framework identifies the most informative loci at each iteration, and these loci are updated based on the informative score calculated at each iteration. Second, HCL is sensitive to outliers and noise. In the literature, outliers are also called as influential data points as they can affect the results obtained from a clustering algorithm to a significant extent. In order to mitigate this impact of outliers and make our 3D-HCL less sensitive to influential data points (i.e., outliers), we further equipped our proposed framework with an outlier detection algorithm based on a fast distance-based measure.
The proposed work is also novel in the eminent DDDAS literature. DDDAS has its power in its ability to create a symbiotic feedback loop between the real system and its application. Dynamic data obtained from the real system is incorporated into an executing application where the application in turn steers the measurement process of the real system. As such, DDDAS has been applied to a variety of areas such as supply chain systems [3], distributed electric microgrids [23, 25,26,27], smart energy management [15], data fusion analysis [2], transportation systems [14], and surveillance and crowd control [19] amongst many others. This study introduces a new dynamic data driven learning framework for identification of informative biomarkers based on the DDDAS paradigm that not only provides measures for detecting outliers but also presents an orchestration procedure for the symbiotic feedback loop between the learning mechanism and the real system application. Here, the performance of proposed 3D-HCL framework on learning mechanism is tested on the lung cancer methylation data. Our results show that the symbiotic feedback loop increases the accuracy of the learning mechanism by updating informative biomarkers based on the information obtained from clustering algorithm in the unsupervised training dataset and the real time classification results of new samples.
The proposed framework has been designed in a generic manner for wide applicability in data system with various dimensionalities. The performance of the proposed framework is demonstrated using real lung cancer DNA methylation data obtained from GEO DataSets [4] where the results reveal that our proposed framework outperforms the conventional HCL algorithm in differentiating lung cancer tissues with 3% versus 33% error margins. Last but not least, this study provides a validation for dynamic updating of massive databases for in vivo DNA methylation analysis. In such an analysis, dynamically obtained data can be processed fast in a computationally efficient way. To this end, our proposed DDDAS 3D-HCL can be considered as an online learning mechanism fed by dynamic and big data as well as archival information.
2 DNA Methylation Data
Bisulfite treatment, also known as bisulfate conversion, is used to determine a DNA methylation pattern. Bisulfite treatment converts unmethylated cytosines to uracil while methylated residues remain unaffected [30]. Samples are then subject to DNA sequencing to identify specific changes in the DNA sequence that can directly inform the methylation level of a specific CpG site.
Bisulfite conversion provides methylated and unmethylated intensities at the each CpG sites to measure DNA methylation level. Beta value as defined in Eq. 12.1 is commonly used to measure DNA methylation status.
In Eq. 12.1, m i and u i represent measured ith methylated and unmethylated probe intensities, respectively. In order to avoid having negative values in probe intensities, any negative values are reset to 0. To prevent probes with very low expression levels from dominating results, an adjustment factor (α) is used. In this study, α is set to 100 as recommended by [1]. The beta value ranges between 0 and 1. A value of zero means that every copy of the CpG island in the probe is unmethylated, whereas a value of one indicates that all copies of the CpG site are completely methylated.
3 Proposed DDDAS-Based Learning Framework (3D-HCL)
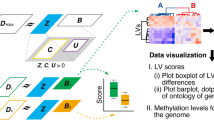
The proposed DDDAS-based learning framework first identifies: (1) candidate clusters of samples based on their DNA methylation levels and (2) informative CpG regions (biomarkers) whose methylation level change correlates with specific clusters (i.e., cancer type or subtype). The components of the proposed DDDAS-based learning framework are explained in detail in this section (see Fig. 12.1 for overview).

Overview of the proposed DDDAS 3D-HCL framework
3.1 Initialization Algorithm: Principal Component Analysis
Principal component analysis (PCA) is a widely known dimension reduction algorithm in the literature [5]. PCA performs dimensionality reduction by identifying correlations in the data while preserving as much of the variance in the high dimension data as possible. PCA converts a set of correlated variables into a set of linearly uncorrelated variables called principal components using orthogonal transformation. The steps of the principal component analysis are explained below.
Let X is an n × m matrix consisting of m dimensional observation (in our case loci) of n samples.
Step 1: Centralize the data by subtracting the mean of each variable (observation) as shown below.

Here, x ij is the data for sample i and observation j, \(\bar {X}_j\) is the mean of variable j and \(\tilde {X}\) is the centralized data matrix.
Step 2: Compute the covariance matrix C from \(\tilde {X}\) by using:
Step 3: Calculate the eigenvectors and the eigenvalues by solving the following equation for each variable
In the equation above u i represents the ith eigenvector and λ i corresponds to the ith eigenvalue. Also, each pair of eigenvector must satisfy the following conditions. These conditions ensure that the eigenvectors are orthogonal to each other.
The eigenvalues of C shows how much variance is explained by corresponding eigenvector. In dimensionality reduction, the first p eigenvectors which correspond to the largest p eigenvalues are used instead of an n × m matrix.
3.2 Clustering Algorithm: Hierarchical Clustering
Hierarchical clustering (HCL) is a widely used clustering algorithm in DNA methylation analysis to identify the DNA methylation profiles affiliated with certain cancer subtypes [4, 28, 29]. The “bottom-up” approach (agglomerative) and the “top down” (divisive) approach are the main strategies in HCL. In agglomerative, each data point starts in its own cluster, and pairs of clusters are merged until only one cluster remains, while in divisive, all data points belong to one cluster at the beginning, and they are split until each of them is in its own cluster. The agglomerative approach is more commonly applied in the literature as it is generally faster than the divisive approach in terms of computational complexity. However, it should be noted that the HCL may still perform worse in terms of solution quality as the merges in agglomerative and the splits in divisive are both determined using a greedy algorithm. Additionally, HCL provides good and understandable visualization for users, and unlike other commonly used algorithms such as k-means, HCL does not require a prior knowledge of number of clusters. Due to these reasons, hierarchical agglomerative clustering is adopted as a learning mechanism in this study. For brevity, HCL abbreviation is also used for hierarchical agglomerative clustering in the rest of the paper.
As discussed above, HCL starts with having each observation in a separate cluster, then repeatedly merges the closest pair of clusters until only one cluster is left. In this work, we base the merging operation on an average linkage where the closest pair of clusters are identified as in the following equation.
In Eq. 12.7, \(D_{avg}\left ( C_i, C_j \right )\) shows the average link (distance) between cluster i and cluster j, \(\left | C_{i} \right |\) represents the cardinality of cluster i and d xy shows the distance between data point x and point y. Since the DNA methylation levels for all probes are properly scaled using the β i values, Euclidean distance (as shown in Eq. 12.8) is used without any standardization method as a distance metric.
In Eq. 12.8, \(\beta _{i}^{g}\) is the beta value of probe g of point i and θ is the set of informative probes. HCL builds a tree-based hierarchical representation (dendrogram) using the Eqs. 12.7 and 12.8 and clusters are obtained by cutting the dendrogram at a desired level.
3.3 Orchestration Procedure: Cluster Membership Score Based Algorithm
In clustering problems, cluster assignments are made on the basis of a similarity measure, Euclidean distance in our case. Even if the clusters are formed based on the measure, the similarity measure may not answer such detailed questions related with the results of clustering algorithm, such as labeling, determining right number of clusters, detecting outliers or border points, etc. To this end, in this study, we propose a cluster membership score that shows the membership degree of a point to a cluster. The cluster membership score is developed based on the definition of an uncertainty classification measure proposed by [6, 18] for a probabilistic distance clustering algorithm. Here, we adapt the definition of uncertainty classification measure to HCL for better understanding of the clusters. The cluster membership score is explained in detail below.
Let \(d\left ( i, C_j \right )\) is the average of distances between observation i and observations assigned to cluster j.
Definition 1
Let m i is the cluster membership measure of observation i. It is the harmonic mean of the distances \(\left \{ d\left ( x, C_j \right ):j \ \in \left \{ 1, \dots , k \right \} \right \}\) divided by their geometric mean.
The proposed score is ranging from 0 to 1. A value of zero indicates that the point is certainly a member of a cluster whereas a value of one shows the current information do not explain the membership of a point.
Definition 2
Let M j denotes the cluster validation score for given cluster structure. The cluster validation score is the mean of the cluster membership scores of data points assigned to the corresponding cluster.
Based on the definition of cluster membership score, the low values of M j indicate that cluster j is explained and separated well from the other clusters with the given information (in our case, identified set of informative CpG regions). This score is designed to help orchestration of the information flow between hierarchical clustering, and outlier detection algorithm and dimension reduction algorithm. Also, it determines when the framework will be terminated.
3.4 Outlier Detection Algorithm
Outliers can arise from DNA methylation data due to measurement errors and/or the dynamic nature in epigenetic mechanisms. Identification of outliers can eliminate their contaminating effect on the methylation data and tremendously increase the performance of clustering algorithm. Because of greedy mechanism used in merging clusters, HCL is sensitive to outliers and it can result in ‘trivial’ clusters. To this end, outlier detection and removal are important tasks to improve the performance of clustering algorithm in DNA methylation analysis.
Outliers (or influential points) can be defined as data points, distant from the remainder of the data points. HCL can result in meaningless clusters due to outliers (an example is shown in Results Section). In this study, we propose fast distance measure for detecting outliers. This measure is designed by assuming that while normal data points have a dense neighborhood whereas outliers are far apart from their neighbors and thus have a less dense neighborhood.
Let o i represents the outlier score of data point i. It is number of points that are closer than distance p.
In Eq. 12.11, \(\delta \left ( d_{ij}, p \right )\) is a function such that it is 1 if d ij ≤ p and it is 0, if otherwise. The data points with small o i values are considered as outliers. Here, o i values are highly dependent on the parameter p. Small p values can find that all points are normal while large p can mark normal points as outliers. Therefore, selection of p is very important. In this study, p is determined as average of distance between data points. Other outlier detection algorithms, similar with the proposed one in this study, can be found in the literature [20].
3.5 Dimension Reduction Algorithm: Locus Information Score Based Algorithm
In conventional application of HCL on a high-dimensional space, dimension reduction algorithm (or feature selection algorithm) finds a set of informative attributes and then HCL forms clusters based on this set. However, since the results of any clustering algorithm are highly dependent on selected attributes, HCL can fail in most of the real systems especially for dynamic, complex systems such as a DNA sequence. Also, PCA has two important drawbacks. First, since PCA performs dimensionality reduction using orthogonal transformation, it complicates the identification biomarkers associated with diseases in DNA methylation analysis. Second, PCA does not take into account the contaminating effect of outliers in determining the principal components. To address these issues, we developed a locus informative score inspired by [16].
Let \(In f_{l}^{j}\) denotes the information score of locus l for the cluster j. Based on the cluster structure found by HCL, it is calculated as follows.
In Eq. 12.12, \(\beta _{i}^{l}\) is the beta value of locus l for sample (point) i, and μ and σ are the mean and standard deviation for the given set of beta values, respectively. The higher \(In f_{l}^{j}\) scores mean that the locus l is informative to differentiate the cluster j from the other clusters. In this study, if \(In f_{l}^{j}\) is greater than 1 for any cluster, the locus l is considered as an informative biomarker.
4 Results and Discussion
In this work, the capabilities and performance of the proposed DDDAS-based framework are demonstrated using lung cancer DNA methylation data obtained from the Gene Expression Omnibus (GEO) repository at the National Center for Biotechnology Information archives with accession number GSE16559 [4]. In this section, we first demonstrate each step of our framework to build groups of samples with related methylation profiles on the training dataset. Then, we show how to incorporate test data into the 3D-HCL and correlate test results with 3D-HCL predictions.
4.1 Learning from Training Data
The training dataset used in our experiments is a part of larger data and consists of 33 samples and two clusters, namely Non-malignant pulmonary and lung adenocarcinoma. At the initialization step of the proposed learning framework, PCA is applied to all 1505 probes with DNA different methylation levels for all samples. When the number of variables (in our case, number of probes) is larger than the number of samples, PCA reduces the dimensionality to, the number of samples (at best). In this specific example, PCA results in 32 components without loss of information. To select the initial set of informative biomarkers, we start by looking at the cumulate variance explained by a set of principal components (Fig. 12.2).

Cumulative variance explained by a set of principal components
As shown in Fig. 12.2, the first few components have the largest variance among all components. In fact, while the first three components retain 60% of the original variance, the last 19 components are only able to explain 10% of the variation of the original data. Because some variance is expected in the original DNA methylation dataset due to the contained outliers; in this work, the components that explains 90% of the variance (13 of them) are chosen to initialize the framework.
Next, hierarchical clustering was run with the distance matrix calculated based on the components found by PCA using the Eq. 12.8. Figure 12.3 illustrates the dendrogram obtained from HCL. As shown in the Fig. 12.3, HCL could not find any meaningful clusters. Similar results were recorded when using conventional approach that incorporated PCA as a dimension reduction algorithm and HCL as a clustering algorithm. Error rate of this conventional approach was also recorded as 33%.

Dendrogram obtained from HCL (Solution 1)
In the solution, cluster 2 has only one member which is sample 28. In this example, since cluster validation score for cluster 2 was zero and the cardinality of cluster was one, an outlier score of each data point was calculated based on the current distance matrix and shown in Fig. 12.4.

Outlier score of each data point
The outlier score of point 28 is zero, meaning that there is no point closer to the point 28 than p, mean of the distance matrix. The point was marked as outlier and removed from distance matrix. Then, HCL was applied to the 32 samples. It should be noted here that after outlier removal, the points were re-indexed based on their position with respect to outlier point index. In Solution 2, HCL resulted in more meaningful clusters with error rate of 18% as shown in Fig. 12.5a. Since the cluster validation scores of both clusters were less than 0.8, the outlier removing algorithm was not executed. Next, the informative probes were determined based on the cluster formation obtained Solution 2 using the Eq. 12.12. The distance matrix was re-calculated with respect to the new set of informative probes and HCL was applied to the new distance matrix. The result of the algorithm was shown in Fig. 12.5b. Here, the error rate was down to 3.03% and the cluster validation scores of cluster 1 and cluster 2 were down to 0.36 and 0.65, respectively. The same procedure is repeated to obtain Solution 3 where the results are shown in Fig. 12.5c. While Solution 4 presents the same clustering formation as was in Solution 3, cluster validation scores are better for both clusters in Solution 4 due to an updated set of informative biomarkers. Since the same solutions were obtained in consecutive runs, the algorithm is terminated.

The results of HCL in Solution 2, Solution 3, and Solution 4
The cluster validation scores of both clusters in each run were illustrated in Fig. 12.6. It is expected that cluster validation score decreases after each run with updating the set of informative probes based on the obtained clusters. However, the validation scores of clusters in Solution 1 were smaller than the those in Solution 2. Here, in Solution 1, cluster 2 consists of only point 28 which is far from the other data points and obviously, the cluster validation score is 0 for cluster 2. Also, since the point 28 is far from the points, in Solution 1, the cluster validation score of cluster 1 was smaller than other solutions. This issue can be explained as the effect of influential points on the cluster validation score that the users should bear in mind.

Cluster validation scores
After the recursive procedure is terminated, there is one more step to finalize the solution. In this step, the outlier(s) was re-assigned to the closest cluster based on the results of HCL. The closest cluster is the one that has the minimum average distance between outlier and the points assigned to corresponding cluster. The result after the finalization step was represented in Fig. 12.7. The error rate is 3.03%, the proposed procedure mis-assigned only point 8 (non-malignant pulmonary) to the lung adenocarcinoma cluster.

Solution after finalization step
The proposed dynamic data driven high dimensional learning framework clusters the samples and identifies the biomarkers associated with these clusters. The proposed framework results in 72 probes that help classify the new samples without having to conduct a complete analysis. The original data and identified informative probes were represented in Fig. 12.8a, b, respectively.

Original data and identified informative probes
4.2 Learning from Test Data
The discriminative model obtained from learning algorithm based on the training dataset eases the classification of the test data without running the learning algorithm. This is the main motivation of the most of the learning algorithms proposed in the literature. However, especially in high dimensional data, training dataset may not reflect all of the different groups that are possible within the system (in our case, cancer types and subtypes) causing the discriminative model outputting incorrect predictions when used with the test data. For example, in unsupervised DNA methylation analysis, the biomarkers identified using a dimension reduction algorithm and a discriminative model (cluster structure in our case) are heavily dependent on each other and thus training dataset can mislead the results of a discriminative model. Here, we conduct two sets of experiments where in the first set of experiments, we demonstrate the capabilities of 3D-HCL in real-time classification of the test data, then in the second set of experiments, we show the performance of our framework in the updating of the discriminative model with respect to test data predictions in the case that training data do not reflect the true representation of the new samples. In the first set of experiments, we classify the 8 non-malignant pulmonary samples where the results of 3D-HCL are shown in Fig. 12.9.

Classification of 8 non-malignant pulmonary samples
Figure 12.9 shows the distance between each sample in the training set and the newly obtained sample. As seen in Fig. 12.9, the new samples except sample 4 have similar DNA methylation profiles based on the biomarkers identified using the discriminative model. Here, the orchestration module calculates m i and decides the class of each new sample i without running the clustering algorithm. The cluster membership of the samples except sample 4 is smaller than 0.2 which means that the identified biomarkers explain the class of these samples. However, membership score of sample 4 is quite high (approximately 0.85). This shows that this sample can be considered as an outlier or can come from a different cluster that has not been considered in training dataset. Since this data point is far from all of the samples in both the training and test datasets, it is marked as an outlier. Our outlier detection algorithm also classifies the point as an outlier with respect to its o i score.
In the second experiment, the test dataset includes 12 pleural mesothelioma samples which have not been considered in the training dataset. Similar to Figs. 12.9 and 12.10 shows the dissimilarity value between the pleural mesothelioma samples, and the non-malignant pulmonary and lung adenocarcinoma samples. Here, m i values of the samples in this test dataset are approximately 0.90. Here the orchestration module decides that all these samples belong to a different cluster since these samples have similar DNA methylation profiles based on the identified biomarkers. Then, to identify the new set of biomarkers that separates these samples from the samples in the training dataset, the locus informative score (see Eq. 12.12) is calculated for each locus assuming that the samples in the new dataset belong to a new cluster. Based on \(inf_l^j\) values, 42 additional loci are labeled as informative biomarkers. The results of updated learning model are represented in Fig. 12.11. The results show that the symbiotic feedback loop increases the accuracy of the learning mechanism by updating informative biomarkers based on the information obtained from the test data. As such, the accuracy of the updated learning model is 97.78%.

Classification of 12 pleural mesothelioma samples

Identified informative biomarkers after new test data
5 Conclusion
In this work, a dynamic data driven applications systems (DDDAS) high dimensional learning framework, namely 3D-HCL, is introduced for identifying idiosyncratic DNA methylation profile of different tumor types and/or subtypes. The proposed framework is composed of five algorithms, (1) principal component analysis that initializes the framework by determining initial set of informative biomarkers, (2) hierarchical clustering algorithm that clusters the points (samples) into groups which represent cancer types or subtypes, (3) outlier detection algorithm that finds outliers and removes their contaminating effect on the input of HCL, (4) informative probe selection procedure which identifies the biomarkers associated with clusters obtained by learning mechanism, and (5) orchestration procedure that coordinates the information flow between HCL and outlier detection algorithm and informative probe selection procedure based on the designed cluster membership score. The performance of the proposed framework was demonstrated using real lung cancer data obtained from GEO database [4]. The performance of the proposed DDDAS-based recursive procedure is noted as very promising on the case study in terms of detecting outliers and removing their contaminating effect, finding meaningful clusters, and identifying biomarkers. In particular, in the selected dataset, traditional HCL results in approximately 33% error and fails to identify meaningful clusters. Our improved iterative procedure, however successfully differentiates the non-malignant pulmonary and lung adenocarcinoma with 3% error and identifies an aberrant DNA methylation profile associated with these cancer types. Interestingly, we observed that outliers have a significant effect on dimension reduction by misleading the determination of informative genes. Our proposed procedure is also able to detect and remove the outliers from the methylation dataset to minimize their potential contamination. Collectively, the proposed framework paves the way towards analyzing complex DNA methylation data using data driven learning mechanisms.
The future venues of this work involves itself with the testing of the proposed algorithm using datasets collected from a larger number of patients. The outcome of these studies will further validate the capability and performance of the DDDAS learning algorithm while improving its accuracy. In this study, a threshold based algorithm is developed based on designed informative locus score so that the pre-defined threshold value can affect the identification of informative biomarkers. Here, an optimal mechanism or automatic fine-tuning mechanisms for the threshold parameter can be investigated. Lastly, in its current form, the hierarchical clustering algorithm expects the number of clusters as a pre-determined parameter. The future work will focus on the exploration of a cluster membership score that will help optimize the number of clusters used in run-time.
The proposed 3D-HCL framework is tested on a real lung cancer DNA methylation dataset. As demonstrated in Sect. 12.4 the feedback loop between the clustering and identification of biomarkers provides more accurate results of aberrant DNA methylation profiles associated with cancer. The results of this study show that DDDAS based methodologies can provide invaluable insight into accurate cancer diagnosis, early detection, and treatment tailoring especially the research in the analysis of time-course data, and complex and comprehensive studies involving very large number of genes and samples such as Human Genome Project. In time-course methylation analysis, the proposed 3D-HCL framework can be further investigated to identify groups of biomarkers whose expressions are not stable over time, and then classify the new samples based on these identified biomarkers by steering the timing of data collection. In conclusion, this study highlights that DDDAS based learning methodologies offer not only more accurate results but also more efficient experimental designs for analyzing and understanding of genetic and epigenetic blueprint in the complex and comprehensive projects.
References
M. Bibikova, Z. Lin, L. Zhou, E. Chudin, E.W. Garcia, B. Wu, D. Doucet, N.J. Thomas, Y. Wang, E. Vollmer et al., High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 16(3), 383–393 (2006)
E. Blasch, Y. Al-Nashif, S. Hariri, Static versus dynamic data information fusion analysis using DDDAS for cyber security trust. Proc. Comput. Sci. 29, 1299–1313 (2014)
N. Celik, S. Lee, K. Vasudevan, Y.J. Son, Dddas-based multi-fidelity simulation framework for supply chain systems. IIE Trans. 42(5), 325–341 (2010)
B.C. Christensen, C.J. Marsit, E.A. Houseman, J.J. Godleski, J.L. Longacker, S. Zheng, R.F. Yeh, M.R. Wrensch, J.L. Wiemels, M.R. Karagas et al., Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using dna methylation profiles. Cancer Res. 69(15), 6315–6321 (2009)
J.P. Cunningham, Z. Ghahramani, Linear dimensionality reduction: survey, insights, and generalizations. J. Mach. Learn. Res. 16, 2859–2900 (2015)
H. Damgacioglu, C. Iyigun, Uncertainity and a new measure for classification uncertainity, in Uncertainty Modeling in Knowledge Engineering and Decision Making, ed. by C. Kahraman (World Scientific, Hackensack, 2012), pp. 925–930
F. Darema, Dynamic data driven application systems. Internet Process Coordination p. 149 (2002)
F. Darema, Dynamic data driven applications systems: A new paradigm for application simulations and measurements, in International Conference on Computational Science, Krakow, (Springer, 2004), pp. 662–669
S.U. Devaskar, S. Raychaudhuri, Epigenetics–a science of heritable biological adaptation. Pediatr. Res. 61, 1R–4R (2007)
A. Eccleston, N. DeWitt, C. Gunter, B. Marte, D. Nath, Epigenetics. Nature 447(7143), 395–395 (2007)
G. Egger, G. Liang, A. Aparicio, P.A. Jones, Epigenetics in human disease and prospects for epigenetic therapy. Nature 429(6990), 457–463 (2004)
M. Esteller, Epigenetics in cancer. N. Engl. J. Med. 358(11), 1148–1159 (2008)
M. Esteller, P.G. Corn, S.B. Baylin, J.G. Herman, A gene hypermethylation profile of human cancer. Cancer Res. 61(8), 3225–3229 (2001)
R. Fujimoto, R. Guensler, M. Hunter, H.K. Kim, J. Lee, J. Leonard II, M. Palekar, K. Schwan, B. Seshasayee, Dynamic data driven application simulation of surface transportation systems, in International Conference on Computational Science, the University of Reading, UK (Springer, 2006), pp. 425–443
R.M. Fujimoto, N. Celik, H. Damgacioglu, M. Hunter, D. Jin, Y.J. Son, J. Xu, Dynamic data driven application systems for smart cities and urban infrastructures, in Winter Simulation (WSC), Washington, D.C. (IEEE, 2016), pp. 1143–1157
T.R. Golub, D.K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J.P. Mesirov, H. Coller, M.L. Loh, J.R. Downing, M.A. Caligiuri et al., Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286(5439), 531–537 (1999)
K. Holm, C. Hegardt, J. Staaf, J. Vallon-Christersson, G. Jönsson, H. Olsson, Å. Borg, M. Ringnér, Molecular subtypes of breast cancer are associated with characteristic dna methylation patterns. Breast Cancer Res. 12(3), 1 (2010)
C. Iyigun, A. Ben-Israel, Semi-supervised probabilistic distance clustering and the uncertainty of classification, in Advances in Data Analysis, Data Handling and Business Intelligence ed. by A. Fink (Springer, Berlin/Heidelberg, 2009), pp. 3–20
A.M. Khaleghi, D. Xu, Z. Wang, M. Li, A. Lobos, J. Liu, Y.J. Son, A DDDAMS-based planning and control framework for surveillance and crowd control via UAVs and UGVs. Expert Systems with Applications 40(18), 7168–7183 (2013)
E.M. Knox, R.T. Ng, Algorithms for mining distance based outliers in large datasets, in Proceedings of the International Conference on Very Large Data Bases, New York City, NY (Citeseer, 1998) pp. 392–403
P.W. Laird, R. Jaenisch, The role of DNA methylation in cancer genetics and epigenetics. Annu. Rev. Genet. 30(1), 441–464 (1996)
E. Li, C. Beard, R. Jaenisch, Role for dna methylation in genomic imprinting. Nature 366(6453), 362–365 (1993)
X. Shi, H. Damgacioglu, N. Celik, A dynamic data-driven approach for operation planning of microgrids. Proc. Comput. Sci. 51, 2543–2552 (2015)
K.D. Siegmund, P.W. Laird, I.A. Laird-Offringa, A comparison of cluster analysis methods using dna methylation data. Bioinformatics 20(12), 1896–1904 (2004)
A.E. Thanos, X. Shi, Sáenz, J.P., N. Celik, A DDDAMS framework for real-time load dispatching in power networks, in Proceedings of the 2013 Winter Simulation Conference: Simulation: Making Decisions in a Complex World, Washington, D.C. (IEEE Press, 2013), pp. 1893–1904
A.E. Thanos, D.E. Moore, X. Shi, N. Celik, System of systems modeling and simulation for microgrids using DDDAMS, in Modeling and Simulation Support for System of Systems Engineering Applications (Wiley, Hoboken, 2015), p. 337
A.E. Thanos, M. Bastani, N. Celik, C.H. Chen, Dynamic data driven adaptive simulation framework for automated control in microgrids. IEEE Trans. Smart Grid 8(1), 209–218 (2017)
H. Ueno, H. Okita, S. Akimoto, K. Kobayashi, K. Nakabayashi, K. Hata, J. Fujimoto, J.I. Hata, M. Fukuzawa, N. Kiyokawa, DNA methylation profile distinguishes clear cell sarcoma of the kidney from other pediatric renal tumors. PLoS One 8(4), e62233 (2013)
A.K. Virmani, J.A. Tsou, K.D. Siegmund, L.Y. Shen, T.I. Long, P.W. Laird, A.F. Gazdar, I.A. Laird-Offringa, Hierarchical clustering of lung cancer cell lines using DNA methylation markers. Cancer Epidemiol. Biomark. Prev. 11(3), 291–297 (2002)
R.Y.H. Wang, C.W. Gehrke, M. Ehrlich, Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res. 8(20), 4777–4790 (1980)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Damgacioglu, H., Celik, E., Yuan, C., Celik, N. (2018). Dynamic Data Driven Application Systems for Identification of Biomarkers in DNA Methylation. In: Blasch, E., Ravela, S., Aved, A. (eds) Handbook of Dynamic Data Driven Applications Systems. Springer, Cham. https://doi.org/10.1007/978-3-319-95504-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-95504-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95503-2
Online ISBN: 978-3-319-95504-9
eBook Packages: Computer ScienceComputer Science (R0)