
Abstract
Although experts in the genomics field now work with bioinformatics tools (software) to generate genomic diagnoses, the fact is that these solutions do not fully meet their needs. From the perspective of Information Systems (IS), the real problems lie in the lack of an approach (i.e., Software Engineering techniques) that can generate correct structures for data management. Due to the problems of dispersion, heterogeneity and the inconsistency of the data, understanding the genomic domain is a huge challenge. To demonstrate the advantages of Conceptual Modeling (CM) in complex domains -such as genomics- we propose two web-based tools for genomic diagnosis that incorporates: (i) a Conceptual Model for the direct-to-consumer genetic tests (DCGT), and (ii) our Conceptual Model of the Human Genome (CMHG), both with the aim of taking advantage of Next-Generation Sequencing (NGS) for ensuring genomic diagnostics that help to maximize the Precision Medicine (PM).
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
The study and understanding of the human genome (how life works on our planet) could probably be considered one of the great challenges of our century. Thanks to the advances in NGS (Next-Generation Sequencing) [1], there has been considerable growth in the generation of genomic and molecular information. In addition, the interactions that are available with this genomic knowledge have a direct impact on the medical environment and Precision Medicine (PM) [2].
The application of Conceptual Modeling (CM) [3] techniques to the genomic domain now provides solutions and optimizes some of the processes carried out by experts (i.e., in genetic laboratories and hospitals), and helps to solve the problems that arise in handling the large amounts of information from different sequencing methods. The use of advanced Information System (IS) engineering approaches can be useful in this domain due to the huge amount of biological information to be captured, understood and effectively managed. A considerable part of modern Bioinformatics is devoted to the management of genomic data. The existence of a large set of diverse data sources containing large amounts of data in continuous evolution makes it difficult to find convincing solutions [4]. When we addressed this problem from the IS perspective, we understood that precise CMs were required to understand the relevant information in the domain and to clearly fix and represent it to obtain an effective data management strategy.
Research and genetic diagnoses are typical examples of the work done by experts -biologists, researchers or geneticists- every day. However, some information is required to perform these tasks. Where are these data? Currently, this information is dispersed in genomic repositories including web sites, databanks, public files, etc., which are completely heterogeneous, redundant, and inconsistent (containing partial information) [5]. In addition, most of these just focus on storing specific information in order to solve a specific problem (e.g., UniProt: a catalog of information on proteins).
Due to these characteristics, we are able to estimate the difficulty of experts in finding and manipulating certain genomic information, making this goal almost impossible to achieve. Another relevant factor in the domain is the constant growth and updating of the data (i.e., biological concepts). The use of standard definitions of concepts is not mandatory, so that sometimes the same term can have different definitions, in which case the meaning of the concept depends on the interpretation given to it by the expert. After studying this situation, we decided to develop a Genomic Information System (GeIS) for facilitating the elaboration of end-user’s genetic tests. Two strategies have been followed for accomplishing an adequate data treatment and management policy:
-
1.
To provide “GenesLove.Me” (GLM) as a web application designed to generate direct-to-consumer genetic tests (DCGT) supported by BPMN [6] and CM [3] techniques to study and analyze the essential elements of the processes involved in the genomic diagnosis process and improve the development of GeIS.
The current availability of DCGT has a great number of advantages for the genomic domain, making it easier for end-users to access early genetic-origin diseases diagnosis services. Romeo-Malanda [7] defines “direct-to-consumer genetic analysisFootnote 1” as a term which is used to describe analytic services offered to detect ‘polymorphism’ and ‘health-related genetic variations’.
-
2.
To develop a prototype tool (“VarSearch”) for helping the treatment and management of genomic data. This application contrasts a set of genomic variations with the information contained in a database that follows the Conceptual Model of the Human Genome (CMHG). This model is much more general and “ambitious” with respect to the behavior of the human genome, and it consists of the following “views”: Structural, Transcription, Variation, Phenotypic, Pathways, and Bibliographic references [4, 8].
Applying GeIS to the bioinformatics domain is a fundamental requirement, since it allows us to structure our Human Genome Database (HGDB) with curated and validated data (to treat the data that will be used in the proposed application, we implemented the SILE methodology [9]).
The initial research on applying CM approaches to the human genome was reported in the works of Paton [10] and Ram [11]. The main goal in Ram’s work was to show the advantages and benefits of using CM to compare and search for the protein in 3D (see other related works in [12]). Reyes et al. describes a CMHG [4] which proposes a domain definition at the conceptual level. From this CMHG, we generated a GeIS to support VarSearch. The application of CM helps us to better understand and manage the knowledge of the human genome.
The efficient use of advances in genomic research allows the patient to be treated in a more direct way, which is reflected in results such as: “better health” and “quality of life”. The genomic domain requires methodologies and modeling techniques capable of integrating innovative ideas into: (a) data management; (b) process improvement; and (c) the inclusion of quality standards.
In this context, the goal of the present study, which is based on our previous work [13], is to explain the functionality of the prototype called “VarSearch”, which starts and ends its interaction in the BPMN described above in processes T10 and T11. This tool has been developed with the objective of generating the genetic diagnosis that will be provided to the end-user through the GenesLove.Me platform. The advances over our previous work [13] are:
-
The description of genetic tools based on conceptual models for the generation of genomic diagnoses, which contribute greatly to the management of the data participating in PM, and
-
The explanation of the VarSearch prototype, which is used to generate the genomic diagnosis from the HGDB. In addition, preliminary steps will be described to work with the prototype, such as loading the database, selecting different data repositories, and others.
The paper is divided into the following sections: Sect. 2 reviews the present state of the art. Section 3 describes BPMN applied to the genomic diagnosis process. Section 4 contains the two genetic tools (“GenesLove.Me” and “VarSearch”) based on conceptual models. Section 5 describes a case study with the VarSearch tool, and Sect. 6 summarizes the conclusions and outlines future work.
2 Related Work
Bioinformatics now play an important role in contributing advances to the medical and technological sector. Genetic testing reveals existing knowledge about “genes” and “variations” in the genomic domain, which is used to diagnose diseases of genetic origin in order to prevent or treat them. This brings PM closer to end-users (i.e., clients or patients).
The study of genomics (i.e., data repositories, genetic variations, diseases, treatments, etc.) is constantly growing and is increasingly seeking to ensure the application of PM. DNA sequencing began in 1977 and since then software tools have been developed for its analysis. Thanks to NGS Technologies [14], it is now possible to manipulate files (e.g., VCF: Variant Call Format) in order to generate genetic diagnoses in a more agile and efficient way [15].
2.1 Precision Medicine (PM) and Genetic Tests
PM is a way of treating patients that allows doctors to identify an illness and select the treatment most likely to help the patient according to a genetic concept of the disease in question (therefore it has also been called Personalized Medicine) [16]. The advantages of genetic tests are innumerable and allow us to identify mutations or alterations in the genes and are of great use and interest in clinical (personalized medicine) and the early diagnosis of diseases. By 2008 there were around 1,200 genetic tests available around the world [17], but they had some limitations (e.g., data management, genome sequencing, etc.) and their cost was quite high.
For this reason, companies were interested in reducing costs and providing services to end-users in the comfort of their own homes. Technological advances played a fundamental role in the genomic environment, since the introduction of the NGS for sequencing samples made it possible to obtain sequences more quickly and cheaply [18, 19].
23andMe is an American company that offers a wide range of services [20], and the type of information obtained from genetic samples is oriented to (i) genetic history (ancestors) and (ii) personal health (risk of diseases), and is presented mostly in probabilistic terms [21].
In the same way, in Spain companies of this type have emerged (e.g., TellMeGenFootnote 2 or IMEGENFootnote 3), all with the aim of providing genetic tests to end-users, simply and in the form of providing a diagnosis that allows end-users to take reactive or corrective actions (e.g., prevention and treatment) to improve their quality of life.
The genomic tools presented in this work goes one step beyond from an Information Systems and Conceptual Modeling points of view, providing a working platform strictly dependent on a precise Conceptual Model of the Human Genome (CMHG), that semantically characterizes the genomic data to be managed and interpreted.
2.2 Genetic Tools for Annotating Variations
In genomic practical settings, the annotation of variation is the most common strategy used for trying to determine which are the correct data to be considered. In this work, we consider SnpEff, Annovar, and VEP, which are three of the major tools that attempt to classify variants.
SnpEff [22] annotates and predicts the effects of genetic variants building a local database by downloading information from trusted resources. After the database is loaded, SnpEff can analyze thousands of variants per second. However, loading a database is a very expensive task from the point of view of resources, and it is even recommended to increase the default Java memory parameters.
On the other hand, although SnpEff can be run in a distributed fashion (using Amazon Cloud Services), and offers limited web interfaces, it is command-line oriented. SnpEff can also be integrated with other tools such as GATKFootnote 4 or GalaxyFootnote 5.
The Annovar software tool annotates variants [23]. The first step when using Annovar scripts is to populate its local database tables using an extensive set of external sources. It is then possible to annotate variants from a VCF file to get a separate custom tab file. wAnnovar (Web Annovar) provides an easy and intuitive web-based access to the most popular Annovar functionalities and allows users to submit their own files and wait for the results of the analysis report. Like SnpEff, Annovar is command-line oriented and does not provide a well-documented API for framework integration.
VEP (Variant Effect Predictor) [24] determines the effect of the variants by querying external databases directly, with no need to load the local database (although it is recommended for performance reasons). Like SnpEff and Annovar, it is command-line oriented and web-access is functionally limited. In order to achieve integration, basic VEP functionalities can be extended using VEP plugins. Table 1 compares VarSearch with these three tools:
As shown, VarSearch overcomes limitations by:
-
Being based on a Java EE multitier applications architecture, which is a solid approach to high-level applications in complex and heterogeneous environments. This allows VarSearch to be easily integrated with other web applications; the software is fully localized, etc.
-
Using a service oriented framework [25], which improves interoperability and integration.
-
Relying on a model-driven instead of a data-oriented paradigm. VarSearch uses a projection of the CMHG.
-
Providing a functionally complete web interface with the ability to download results in standard output file formats, which can then be post-processed by third-party tools.
As VarSearch follows a “client/server” architecture, data loading has no impact on client performance, thus improving user experience. Data loading can be done off-line on the server so that researchers can query data on the fly with a short response time.
3 BPMN: Genomic Diagnosis Process
The GemBiosoft company is a spin-off of the Universitat Politècnica de València (UPV), founded in 2010. The main objective of this company is to define the CMHG to obtain a precise schema to manage, integrate and consolidate the large amount of genomic data in continuous growth within the genomic domain.
GemBiosoft has a web application called “GenesLove.Me” which offers DCGT to the consumer. The information provided by the genetic tests is accessible online to all users without prior registration (anonymous users). For example, non-registered users of the web application are able to consult all information related to the diagnosis of rare diseases of genetic origin, their characteristics, treatment, tutorials and videos of the way in which the process is performed.
The access security in GenesLove.Me is controlled by profiles. Users can access GLM under three profiles: (1) clients (patients), (2) provider and (3) administrator. An authenticated user with a certain access profile is authorized to carry out the operations corresponding to the access profile.
-
(1)
Clients (patients): Users with this profile are able to contract the services offered by selecting the services (DCGT) they are interested in and then paying the fee. The user is able to monitor the notifications and messages related to the diagnoses, besides consulting the histories of all the studies and treatments carried out and updating the information associated with his profile.
-
(2)
Supplier: Users with this profile are able to generate notifications about the change of status in the treatment of samples. After receiving the genetic sample, the user activates the sample by entering its code number. They can then track the sample until the sequence file is generated. They can also update their profiles and consult all the activated samples (in progress and finalized).
-
(3)
Administrator: A user with administration privileges performs administration and maintenance tasks of the web application, such as:
-
(a)
publishing online results (the administrator uploads the resulting diagnoses from the analysis performed on samples sequenced by the VarSearch tool. The application automatically notifies the user when his/her results have been published);
-
(b)
publishing advertisements;
-
(c)
publishing new diagnostic services to diagnose new diseases; and
-
(d)
consulting payment reports and the application usage report (custom time period).
-
(a)
Genetic tests are currently offered with the aim of detecting a person’s predisposition to contracting a disease of hereditary origin [26]. The bioinformatics domain seeks to provide the necessary mechanisms and means to generate genetic diagnoses that allow the end-users (patients) to obtain these results to facilitate a personalized prevention treatment.
In order to improve the understanding of the whole process using a different CM technique (business process-oriented), Fig. 1 shows a BPMN diagram describing the genetic diagnosis process (from the end-user’s service request until he/she receives his/her genetic test report).

Genetic diagnosis process [13].
Our goal is to reinforce the CM perspective of this work, in order to make CM an essential working procedure to design and implement effective and efficient genomic tools. In this process, the three actors/users specified are involved: (1) The client (patient) who requests the service to determine whether or not he/she has a disease of genetic origin; (2) The company, in this case GemBiosoft, which is in charge of managing and performing the genomic diagnosis; and (3) the Suppliers, who in this case prepare the file containing the reference of the patient involved in the genetic test.
The general process begins when the end-user (patient) enters the web application and requests the genetic analysis (t1: task 1). The company (GemBiosoft) processes this request and proceeds to send the sample container to the client (t2). When the client receives the container, he must activate it by registering its identifier in the web application (t3), then place the sample in the container and send it back to the company (t4). Upon receipt of the sample, the company confirms that it meets the necessary requirements for the study and notifies the customer of its receipt (t5). The next step is to determine the supplier who will be responsible for sequencing the samples and send him the sample (t6). The selected supplier receives the sample and notifies its reception to the company (t7). Sequence preparation is initiated through the sequencing technology used by the supplier (t8). The supplier sends the resulting sequence of the sample (file) to the company (t9). The company confirms its reception to the supplier and proceeds to analyze the sequenced sample as part of the genetic diagnosis (t10). The definitive diagnosis report (t11) is then generated. The company (in this case the administrator/user) proceeds to publish the genetic diagnosis (result) in the web application and the end-users are automatically notified of the results (t12). To end the process, the end-user accesses the web application to obtain the diagnosis and make any queries (results) (t13). In this work, we want to go deeper into tasks ‘t10’ and ‘t11’, to better understand the genomic diagnosis generation process.
The BPMN gives companies the ability to understand their internal business procedures in graphical notation and the ability to communicate these procedures in a standard way [27].
Through the model shown in Fig. 1, it facilitates the understanding of commercial collaboration and transactions between organizations. In this figure, we can see the interactions between end-users, company and suppliers [15]. The company is interested in providing a web application that allows end-users to obtain a quality genetic test in a simple way that aids the treatment and prevention of diseases of genetic origin.
3.1 Exploitation Tasks 10 and 11 (T10–T11)
In this work, we want to enhance the use of VarSearch to generate the genomic diagnostics offered through GLMFootnote 6. For this, it is important to note that GLM includes interaction with VarSearch (see tasks 10 and 11 of Fig. 1), an application developed by PROS Research CenterFootnote 7 to automatically identify the relevant information contained in the genomic databases and directly related to the genetic variations of the sequenced sample.
VarSearch relies heavily on a CMHG, which makes integration of external genomic databases feasible. However, due to the large amount of information available, the data loaded in VarSearch are the result of a selective loading process [9] where the selected data correspond to the relevant information on the disease to be analyzed.
4 Genetic Tools Based on Conceptual Models
It is widely accepted that applying conceptual models facilitates the understanding of complex domains (like genetics). In our case we used this approach to define two models representing:
-
(a)
the characteristics and the processes of DCGT [13], and
-
(b)
the behavior of the human genome (CMHG) [28]. One of the leading benefits of CM is that it accurately represents the relevant concepts of the analyzed domain.
After performing an initial analysis of the problem domain, the next step is to design a domain representation in the form of a conceptual model.
Our conceptual models can evolved with the new discoveries made in the field of genomics in order to improve data processing to ensure effective PM. We can thus see how CM gives positive support to the knowledge in which precision medicine plays a key role [28]. It is important to highlight that the advantage of CM for representing this domain is that it eases the integration of new knowledge into the model [5].
4.1 GenesLove.Me (GLM)
After an analysis of the requirements required for this work, important decisions were taken to arrive at an adequate representation of the basic and essential concepts in the understanding of the domain under study.
Figure 2 presents the conceptual model proposed, which can be classified into three main parts: (a) Stakeholders: represents all the participants involved in the web application; (b) Genetic diagnostics: represents everything related to diseases offered, diagnoses and samples of patients; and (c) Sales management: represents the management of services offered, purchases and payments.

GLM conceptual model [13].
The complete description of the CM can be found in the previous work [13]. Through our CM we incorporate genetic data currently used in the PM, achieving a conceptual representation that meets the needs of the bioinformatics domain. As we mentioned above, this model aims to improve the conceptual definition of the treatment related to genomic diagnosis, and thus leave a conceptual framework for further improvements.
4.2 VarSearch (Prototype)
A GeIS can be defined as a system that collects, stores, manages and distributes information related to the behavior of the human genome. As mentioned above, the GeIS described here is based on the CMHG [4, 8, 28]. This section deals with the preliminary steps and the design and implementation of the prototype.
4.2.1 First Steps
VarSearch is based upon the E-Genomic Framework (EGF), described in depth in different research papers such as [25, 29]. For the implementation of the tool, a series of steps were carried out to ensure its good performance, as explained below:
-
(a)
Human Genome Database (HGDB). The transformation of our model defined for the database schema (logical model) was almost automatic, using the Moskitt tool (https://www.prodevelop.es/es/products/moskitt). The MOSKitt project aims to provide a set of open source tools and a technological platform for supporting the execution of software development methods which are based in model driven approaches, including graphical modeling tools, model transformations, code generation and collaboration support [30]. In this task, we found two different levels of abstraction in the model. The conceptual model represents the domain from the point of view of scientific knowledge, while the database schema (Fig. 3) focuses on the efficient storage and retrieval of data.
Fig. 3. 
Database schema (Human Genome Database, HGDB).

For this reason, the details of the physical representation must be considered to improve the final implementation. It is important to emphasize the integration of the two tables “Validation” and “Curator” in the DB schema. These tables are not actually part of the knowledge representation of the domain, but are necessary for the development and implementation of the tool (Explained in detail in Sect. 4.2.2).
To load the HGDB the SILE methodology [9] was used, which was developed to improve the loading processes and guarantee the treatment of “curated data”. SILE was used to perform the “search” and “identification” of variations associated with a specific disease (a task validated by experts in the genetic domain, for example, biologists, geneticists, biomedical engineers).
When the identified and curated data have been obtained the “selective loading” is performed (through the loading module) in the HGDB. The data loaded are then “exploited” by VarSearch. Some of the diseases -of genetic origin- studied and loaded were Alcohol Sensitivity [15], Neuroblastoma (Table 2 shows a set of variations detected for Neuroblastoma) [31], and others.
-
(b)
Selection of the different data sources. For the choice of data sources, we addressed the requirements raised in this first phase of the project. After conducting studies and analysis of various genomic repositories, we selected the following databases: NCBI, dbSNP, UMD and BIC.
NCBI [32] (https://www.ncbi.nlm.nih.gov/) is a data source with curated data on structural concepts of DNA sequencing. From this repository, we extracted information related to chromosomes, genes, transcripts, exons… and everything related to the “Structural view” of our conceptual model. dbSNP [33], BIC [34] and UMD [35] are databases of variations that store curated information on genetic differences between individuals. The main reason for using dbSNP is because it not only focuses on variations of a specific gene or region, but also contains variations related to all chromosomes and updates the information immediately. BIC and UMD were selected because of the requirements of a research group that was collaborating with us in a project (Future Clinic) focused on “breast cancer”. This group helped us to test the performance of our GeIS and its associated tool. Currently, we are studying and analyzing other genomic repositories like: ClinVar, dbGaP, 1000 Genomes, ALFRED, and others [5].
-
(c)
Genetic loading module. For the loading process of the HGDB, a load module was designed to store the data from the previously measured data sources. This load module was developed using an ETL strategy [36] with three different levels: extraction, transformation, and load (see Fig. 4). Each level is completely independent of the others, facilitating and clarifying the design of the system and improving its flexibility and scalability. As can be seen in Fig. 4, all the necessary information is extracted from the source databases in the first layer (1). All this raw unstructured data goes to the second layer (2) where several transformations are made in order to format the data according to the structure of our database schema. These transformed data are sent to the third layer (3), which communicates directly with the database (following the above-mentioned SILE methodology in Task “a”, Sect. 4.2.1).
Fig. 4. 
Load module.

4.2.2 Design and Implementation of VarSearch
VarSearch is a web application that allows the analysis of variations obtained from the DNA sequenciation of biological samples and which is stored in FASTA or VCF file formats [37]. Different users can access the application in private spaces in the HGDB and each user can address his own variations. The validation of variations that they consider relevant can be included. It also offers storage for the users’ variations to find similarities in the file analysis process. Another advantage is the inclusion of the information obtained from the data sources, together with the user validations in the database, which is an improvement in performance related to the search for variations. VarSearch can find variations in our database from a provided file (see Fig. 5).

VarSearch application.
The variations found are displayed to the user, and any additional information that the file lacks can be calculated and validated. Any variations of the file that have not been found in our database can also be stored. After inserting one or more variations not found in a file -because they are considered relevant to the user- and reanalyzing this file, these inserted variations will be found in our database and displayed to the user. Figure 6 shows how the functionality has been grouped into three main packages: (1) User management: a user can act as administrator and control other users, or can create new users and modify or eliminate their Information. (2) Data load management: the system allows the user to load the files to be analyzed in both VCF and FASTA format, compare the variations in these files to the variations in the HGDB used by VarSearch. (3) Data analysis: After analyzing and verifying the variations in the input files, the user can list the variations and classify them by multiple criteria (position, chromosome, etc.). There is also a series of functionalities related to the login and modification of account information that has not been grouped in any functionality package.

VarSearch’s general use case diagram (domain).
-
Confidentiality of the information. As this information is a company’s primary resource, VarSearch restricts access to it. When a user validates a variation, he can choose a privacy category:
-
(a)
Public content, if he is willing to share the knowledge with other users, or
-
(b)
Private content, allowing access only to the owner-user and hidden from other users. All the variations can only be accessed by the user who created them.
-
(a)
-
VarSearch Architecture. In order to make it accessible to all users, VarSearch was designed as a web application with HTML5 technology in a language common to all current browsers. The information is managed by the MySQL database. The VarSearch architecture consists of the following elements:
-
(a)
A distributable database based on MySQL (using software tools like: Navicat Enterprise and MySQL Workbench). For the initial validation of this database, we only loaded the information related to chromosomes 13 and 22.
-
(b)
A set of REST services [38] developed in Java using Hibernate and Jersey, which are deployed on a Tomcat server 7.
-
(c)
A web application, which uses the Bootstrap framework for general organization of the interface and files, together with jQuery to define advanced interface components and invoke REST services.
-
(d)
It also includes a “mini” REST service to manage users and roles, which is based on the same architecture and technologies as the other REST services. The data layer is based solely on MySQL (you can see the VarSearch architecture represented in Fig. 7).
Fig. 7. 
VarSearch architecture.
-
(a)

The application entry point is a file with variations detected by a sequencing machine in VCF or FASTA format. With this input the database is searched to detect any variations, additional information on the diseases they may cause and the associated bibliography. VarSearch users follow this process when working with the tool:
-
(1)
A VCF file is uploaded from the web.
-
(2)
The file is then processed and parsed. The entries are shown on an HTML table and the variants of each VCF entry can be seen.
-
(3)
The variations present in the input file can be annotated against the database and the annotated file is downloaded in *.xls, *.csv and *.pdf format or its contents viewed in another HTML table.
To parse the VCF file and annotate the variants, VarSearch relies on snpEff and snpSift [39] tools, and so well tested libraries are used instead of reinventing the wheel. This also ensures VCF standard support, using ANN files for variant annotation.
If another type of information is considered useful for annotation and not covered by the procedure described, VarSearch uses the “INFO” field to introduce the desired values. As VarSearch is based on EGF, new genome annotation files can be quickly integrated by developing the proper parser module, either by a custom development or integrating a third-party tool or library.
All the information associated with the variations found in our HGDB can be obtained. For variations in the lists, user validations can be integrated for future searches with the “Add Validation” option. Another advantage of VarSearch is the user management (new users can be created and edited using the “User Management” option).
One of the objectives of VarSearch is to continue the extension and implementation of all the knowledge defined in our CMHG, such as the treatment of pathways and metabolic routes [28]. This tool facilitates the analysis and search for variations, improving the generation of genomic diagnoses associated with diseases of genetic origin. End-users will find the web application easy to use and they are guaranteed security for their data [40].
5 Case Studies
In the previous work [13], the case study applied to GenesLove.Me was defined, explaining all the processes involved in the management of DTCG (Sect. 3). In summary, the test cases were performed with the implemented solution.
The validation scenario consisted of a group of five (5) users, who made requests for genetic testing for “lactose intolerance”. To begin the process, each user involved in the case study authorized the procedure through an “informed consent” [15, 41], which becomes a legal support that establishes the rights and obligations of the service offered and its expected scope.
Next, the following case studies performed with the prototype VarSearch are presented.
5.1 Using the VarSearch Prototype
To verify VarSearch performance, two case studies were carried out. In the first, VarSearch was used to analyze a VCF file. The second compared the time spent on searching for variations manually and using the application; it is important to highlight that this prototype has as end-users the geneticists and experts responsible for the generation of the genomic diagnostics. To access the application VarSearch users must have an account provided by Gembiosoft SME (http://gembiosoft.com/). After logging in, a file is selected for analysis. VarSearch reads all records and transforms them into variations.
These transformations depend on the file information: for example, the FASTA files contain a genetic sequence (NG), and so require the reference on which the variation is based to be to the “NG” sequence. In contrast, VCF files use positions relative to chromosomes (NC). Once the file records have been converted into variations, the next step is to search for these variations in our HGDB. After the analysis, the “variations found” and “variations not found” can be differentiated.
-
Found Variations Management. Found variations are those extracted from the file in which information has been found in the HGDB, which means that this variation has been found in at least one genomic repository. A found variation has much more information than the variation obtained from the file and allows us to calculate and submit detailed information to the user.
Having analyzed the VCF file, all the variations found are displayed to the user, in each case calculating the HGVS notationFootnote 8, its data source identifier, clinical significance, and the number of validations and databases found together with their bibliographic references. This information is calculated for VCF and FASTA; however, VCF variations are sorted by samples. Figure 8 shows the results obtained by analyzing a VCF file with a single sample. For this sample (5323-BRCAyA), a number of variations were found with the corresponding information. A variation can have validations made by users. The validation column corresponds to the number of validations that each variation has and if a validation is private, only the owner will see it. Another VarSearch feature is its support for multiple bibliographical references. A variation can be found in different databases and may contain different bibliographic references.

Analysis of VCF file using VarSearch.
-
Not found Variations (insertion and treatment). The user who is analyzing variations may find a variation in the file, which was not found in the database (see Fig. 9). Using his experience and knowledge he may consider some variations as relevant despite not being found.
Fig. 9. 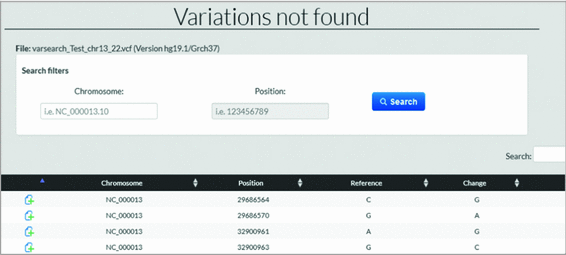
List of variations not found.

With VarSearch the user can insert the not found variations or any variation considered key to the study. If the user has inserted certain variations that had not been found, on reanalyzing the file these inserted variations are compared with the variations in the file, showing the similarities.
In order to differentiate the variations of the different repositories from user variations, the results obtained from the user’s experience and the results from years of study of different biomedical databases are differentiated.
5.2 Improved Efficiency and Time in Finding Variations with VarSearch
To validate the effectiveness and performance of the proposed software, some experiments were performed to measure efficiency and time. A study was conducted to compare the time spent searching for variations manually with an automatic search of all the repositories mentioned above using VarSearch.
A manual search of one variation involves detecting the variation in the VCF or FASTA file, a search for the variation in the different repositories, and the identification and verification of the variation. VarSearch was tested for the time it needed to search for several variations, calculating the time evolution according to the number of variations involved (2, 5 and 7). The results can be seen in Fig. 10.

Time optimization.
As can be seen in Fig. 10, the cost of performing a manual search rises to 5’32 min for 2 variations, 10’83 min for 5 variations and 18’89 min for 7 variations.
However, with VarSearch the time remains constant at between 2 and 3 s for different variations, which confirms its efficient performance. Using this prototype thus significantly reduces the time spent on the search for variations. Also, it must be remembered that the manual search process does not calculate additional information for variations. If this information were necessary, the search time would increase significantly, however, with VarSearch this time remains constant because this information has already been calculated in the search for variations.
6 Conclusions and Future Research
This paper describes a study and analysis of the implementation of two web application to facilitate DCGT, the first offer the services (genomic diagnosis) to the final user using an interface easy to use -GenesLove.Me-, and the second is a prototype for the generation of the diagnosis using our HGDB -VarSearch-. Through these applications, we can inform end-users about their predisposition to suffer certain genetically based illnesses.
Through the development of our web applications we seek to provide end-users with a genomic diagnosis in a secure and reliable way. The use of BPMN and Conceptual Modeling based approaches for this type of service aids the understanding of the participants in the processes in the genomic domain and improves the processes involved.
Bioinformatics is a domain that is constantly evolving, and with the application of conceptual models, we can extend our genomic knowledge and conceptual representation accurately and simply.
In this work, we have focused mainly on the description of the prototype “VarSearch”, which plays a fundamental role in processes 10 and 11 of the BPMN previously described, because with this prototype we generated the genomic diagnosis facilitated through GenesLove.Me.
VarSearch is a flexible new analysis framework or web application that provides a powerful resource for exploring both “coding” and “non-coding” genetic variations. To do this, VarSearch integrates VCF format input/output with an expanding set of genome information. VarSearch (and other tools built on EGF) will therefore facilitate research into the genetic basis of human diseases. EGF can also be expected to allow the development of new tools in diverse e-genomics contexts. As genetic laboratories are now oriented to facilitating genetic procedures, web access, usability and feasibility, the definition of different profiles are therefore important goals. All this allows the user to configure the tool according to his specific needs. These necessities include inserting genetic variations and validating its own variations, thus increasing its “know-how”.
Future research work will also be aimed at:
-
The application of Data Quality (DQ) metrics to enhance our HGDB.
-
The study and treatment of new diseases of genetic origin (continue expanding the list of illnesses available in the web application).
-
Implementation of data management mechanisms to enhance the quality of personalized medicine.
-
Improving/develop the next version of VarSearch (version 2.0) for genetic diagnosis (including -haplotypes and statistical factors-).
-
We also intend to extend the model with studies on the treatment of “haplogroups”, including subjects with a similar genetic profile who share a common ancestor.
Notes
- 1.
This type of analysis is available through direct sales systems in pharmacies or other health care bodies, but the Internet has become the main selling channel for direct-to-consumer genetic analyses [7].
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
References
Buermans, H.P.J., den Dunnen, J.T.: Next generation sequencing technology: advances and applications. Biochimica et Biophysica Acta (BBA) – Mol. Basis Dis. 1842(10), 1932–1941 (2014). https://doi.org/10.1016/j.bbadis.2014.06.015
Grosso, L.A.: Precision medicine and cardiovascular diseases. Rev. Colomb. Cardiol. 23(2), 73–76 (2016). https://doi.org/10.1007/978-3-540-39390-0
Olivé, A.: Conceptual Modeling of Information Systems, pp. 1–445. Springer, Heidelberg (2007). https://doi.org/10.1007/978-3-540-39390-0
Reyes Román, J.F., Pastor, Ó., Casamayor, J.C., Valverde, F.: Applying conceptual modeling to better understand the human genome. In: Comyn-Wattiau, I., Tanaka, K., Song, I.-Y., Yamamoto, S., Saeki, M. (eds.) ER 2016. LNCS, vol. 9974, pp. 404–412. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-46397-1_31
Reyes Román, J.F., Pastor, Ó., Valverde, F., Roldán, D.: How to deal with Haplotype data: an extension to the conceptual schema of the human genome. CLEI Electron. J. 19(3) (2016). http://dx.doi.org/10.19153/cleiej.19.3.2
Object Management Group: Business Process Model and Notation (2016). http://www.bpmn.org/
Romeo-Malanda, S.: Análisis genéticos directos al consumidor: cuestiones éticas y jurídicas (2009). http://www.institutoroche.es/legalactualidad/85/analisis
Pastor López, O., Reyes Román, J.F., Valverde Giromé, F.: Conceptual Schema of the Human Genome (CSHG). Technical report (2016). http://hdl.handle.net/10251/67297
Reyes Román, J.F., Pastor, O.: Use of GeIS for early diagnosis of alcohol sensitivity. In: Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies, vol. 3, pp. 284–289 (2016). https://doi.org/10.5220/0005822902840289
Bornberg-Bauer, E., Paton, N.W.: Conceptual data modelling for bioinformatics. Briefings Bioinform. 3(2), 166–180 (2002). https://doi.org/10.1093/bib/3.2.166
Ram, S., Wei, W.: Modeling the semantics of 3D protein structures. In: Conceptual Modeling–ER 2004, Proceedings, pp. 696–708 (2004). https://doi.org/10.1007/978-3-540-30464-7_52
Pastor, M.A., Burriel, V., Pastor, O.: Conceptual modeling of human genome mutations: a dichotomy between what we have and what we should have. In: BIOSTEC Bioinformatics 2010, pp. 160–166 (2010). ISBN 978-989-674-019-1
Reyes Román, J.F., Iñiguez-Jarrín, C., Pastor, O.: GenesLove.Me: a model-based web-application for direct-to-consumer genetic tests. In: Proceedings of the 12th International Conference on Evaluation of Novel Approaches to Software Engineering, pp. 133–143, Porto, Portugal, 28–29 April (2017). ISBN 978-989-758-250-9, https://doi.org/10.5220/0006340201330143
Mardis, E.R.: The $1,000 genome, the $100,000 analysis? Genome Med. 2(11), 84 (2010)
Reyes Román, J.F.: Integración de haplotipos al modelo conceptual del genoma humano utilizando la metodología SILE. Universitat Politècnica de València (2014). http://hdl.handle.net/10251/43776
Aguilar Cartagena, A.: Medicina Personalizada, Medicina De Precisión, ¿Cuán Lejos Estamos De La Perfección? Carcinos 5, 1–2 (2015)
Grupo RETO Hermosillo, A.: El cáncer de mama (2016). http://gruporetohermosilloac.com/index.php
Metzker, M.L.: Sequencing technologies - the next generation. Nat. Rev. Genet. 11(1), 31–46 (2010)
Voelkerding, K.V., Dames, S.A., Durtschi, J.D.: Next-generation sequencing: from basic research to diagnostics. Clin. Chem. 55(4), 641–658 (2009)
23andMe: 23andMe (2016). https://www.23andme.com/
23andMe: How it works? (2016). https://www.23andme.com/howitworks/
Cingolani, P.: snpEff: variant effect prediction (2012)
Wang, K., Li, M., Hakonarson, H.: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38(16), e164–e164 (2010)
McLaren, W., Gil, L., Hunt, S.E., Riat, H.S., Ritchie, G.R., Thormann, A., Cunningham, F.: The ensembl variant effect predictor. Genome Biol. 17(1), 122 (2016)
Roldán, D., Pastor, O., Fernández, M.: An integration architecture framework for e-genomics services. In: IEEE RCIS (2014). https://doi.org/10.1109/rcis.2014.6861063
U. S. National Library of Medicine: What is genetic testing? Genetics Home Reference (2017)
Chinosi, M., Trombetta, A.: BPMN: an introduction to the standard. Comput. Stand. Interfaces 34(1), 124–134 (2012)
Reyes Román, J.F., León, A., Pastor, Ó.: Software engineering and genomics: the two sides of the same coin? In: Proceedings of the International Conference on Evaluation of Novel Approaches to Software Engineering (ENASE 2017), pp. 1–6 (2017). https://doi.org/10.5220/0006368203010307
Roldán M.D., Pastor López, Ó., Reyes Román, J.F.: E-genomic framework for delivering genomic services. An application to JABAWS. In: 9th RCIS (IEEE), pp. 516–517 (2015). https://doi.org/10.1109/RCIS.2015.7128915
Muñoz, J., Llacer, M., Bonet, B.: Configuring ATL transformations in MOSKitt. In: Proceedings of the 2nd. International Workshop on Model Transformation with ATL (MtATL 2010), CEUR Workshop Proceedings (2010)
Burriel, V., Reyes Román, J.F., Heredia C.A., Iñiguez-Jarrín, C., León, A.: GeIS based on conceptual models for the risk assessment of neuroblastoma. In: 11th RCIS (IEEE), pp. 1–2 (2017). https://doi.org/10.1109/RCIS.2017.7956581
National Center for Biotechnology Information (2017). https://www.ncbi.nlm.nih.gov/
Sherry, S.T., Ward, M.H., Kholodov, M., Baker, J., Phan, L., Smigielski, E.M., Sirotkin, K.: dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29(1), 308–311 (2001)
Szabo, C., Masiello, A., Reyes Román, J.F., Brody, L.C.: The breast cancer information core: database design, structure, and scope. Hum. Mutat. 16(2), 123 (2000)
Béroud, C., Collod-Béroud, G., Boileau, C., Soussi, T., Junien, C.: UMD (Universal mutation database): a generic software to build and analyze locus-specific databases. Hum. Mutat. 15(1), 86 (2000)
Zhou, H., Yang, D., Xu, Y.: An ETL strategy for real-time data warehouse. In: Wang, Y., Li, T. (eds.) Practical Applications of Intelligent Systems. Advances in Intelligent and Soft Computing, vol. 124, pp. 329–336. Springer, Heidelberg (2011). https://doi.org/10.1007/978-3-642-25658-5_41
Claverie, J.M., Notredame, C.: Bioinformatics for Dummies. Wiley, Hoboken (2011)
Haupt, F., Karastoyanova, D., Leymann, F., Schroth, B.: A model-driven approach for REST compliant services. In: IEEE International Conference on Web Services (ICWS), pp. 129–136 (2014)
Tolhuis, B., Wesselink, J.J.: NA12878 Platinum Genome GENALICE MAP analysis report (2015)
León, A., Reyes, J., Burriel, V., Valverde, F.: Data quality problems when integrating genomic information. In: Link, S., Trujillo, J.C. (eds.) ER 2016. LNCS, vol. 9975, pp. 173–182. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-47717-6_15
de Galicia, C.A.: Ley 3/2001, reguladora del consentimiento informado y de la historia clínica de los pacientes (2001)
Acknowledgements
This work was supported by the MESCyT of the Dominican Republic and also by the Generalitat Valenciana through project IDEO (PROMETEOII/2014/039), the Spanish Ministry of Science and Innovation through Project DataME (ref: TIN2016-80811-P).
The authors are grateful to Jorge Guerola M., David Roldán Martínez, Alberto García S., Ana León Palacio, Francisco Valverde Girome, Ainoha Martín, Verónica Burriel Coll, Mercedes Fernández A., Carlos Iñiguez-Jarrín, Lenin Javier Serrano and Ma. José Villanueva for their valuable assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this paper

Cite this paper
Reyes Román, J.F., Iñiguez-Jarrín, C., Pastor, Ó. (2018). Genomic Tools*: Web-Applications Based on Conceptual Models for the Genomic Diagnosis. In: Damiani, E., Spanoudakis, G., Maciaszek, L. (eds) Evaluation of Novel Approaches to Software Engineering. ENASE 2017. Communications in Computer and Information Science, vol 866. Springer, Cham. https://doi.org/10.1007/978-3-319-94135-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-94135-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-94134-9
Online ISBN: 978-3-319-94135-6
eBook Packages: Computer ScienceComputer Science (R0)