
Abstract
Diabetic retinopathy (DR) is the most common microvascular complication of diabetes. Monitoring DR in diabetic patients, which can be achieved via noninvasive techniques, may be relevant for the identification and prevention of other microvascular complications of diabetes such as diabetic nephropathy. DR is hallmarked by the manifestation of progressive vascular abnormalities in the retina. These anomalies remain asymptomatic at the beginning, but may eventually advance into vision-threatening conditions. In the last half century, our understanding of the underlying pathobiology of DR has significantly increased. Although the pathogenesis of DR remains multifactorial and complex, hyperglycemia is widely recognized as the main underlying factor leading to the initial stages of DR development. Hyperglycemia induces oxidative stress, upregulation of growth factors and cytokines, inflammation, loss of pericytes, and neurodegeneration. These processes ultimately lead to vasoregression and neuroglial dysfunction, impairing visual function. Important factors in the induction of vascular anomalies are endothelial cell hypertrophy induced by vascular endothelial growth factor (VEGF) and/or the acquisition of a pro-inflammatory retinal microenvironment that is accompanied by increased leukocyte infiltration. These processes lead to luminal narrowing, capillary occlusion and the formation of acellular, non-perfused capillaries. This may cause a vicious circle of disease progression via hypoxia-induced spread of capillary non-perfusion and ischemia, mediated by VEGF and cytokines. The affected retinal vessels may have reduced barrier properties leading to fluid extravasation. Together with impaired fluid clearance by Müller cells, macular edema is formed, which is the most common cause of blindness in patients with diabetes. As a compensatory mechanism to reduce ischemia, neovascularization may develop, leading to hemorrhages, scarring, and total blindness. Despite this increased understanding of the pathobiology of DR, there are only limited therapeutic options available which still remain ineffective in some patients, including anti-VEGF therapy, corticosteroids, and laser photocoagulation. A better understanding of the basic mechanisms underlying DR development is necessary to identify new therapeutic targets and treat all patients with DR effectively in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Diabetic retinopathy
- Microvascular damage
- Nonproliferative diabetic retinopathy
- Proliferative diabetic retinopathy
- Diabetic macular edema
Introduction
Diabetic retinopathy (DR) is a major cause of vision loss and blindness among persons with diabetes mellitus. It is estimated that approximately 35% of diabetes patients develop some form of DR [1]. DR is a progressive disease that is predominantly characterized by alterations in the retinal microvasculature. It may develop from an asymptomatic nonproliferative form associated with capillary non-perfusion, microaneurysms, and retinal hemorrhages, into a vision-threating disorder such as diabetic macular edema (DME) and proliferative DR (PDR).
Although DR and diabetic nephropathy (DN) are diseases that manifest themselves in different organs, the two diseases are strongly correlated as partly overlapping mechanisms are involved in the pathobiology of DR and DN, in particular microvascular alterations [2]. Compared to other tissues, the retina is highly vulnerable for the hyperglycemic milieu induced by diabetes, which is often attributed to the fact that retinal cells are not dependent on insulin for glucose uptake and to the unique anatomy and physiology of the eye. For instance, the density of blood vessels is low in the retina to prevent absorption of light. Yet, the retina has high metabolic demands, in particular in the dark-adapted state [3], which results in physiological retinal hypoxia [4]. As such, the retina has a limited capacity to adapt to metabolic stress, which may underlie its vulnerability to diabetes [3, 5]. It is often stated that DR precedes the development of DN in diabetic patients [6]. However, not all patients with advanced DR develop DN, underscoring that the pathobiology of both diseases are also different in a number of aspects [7].
A meta-analysis of patients with type 2 diabetes and renal disease indicated that DR can be used as a predictive biomarker to distinguish DN from nondiabetic renal disease [8]. In line with this observation, the identification of DR in diabetic patients with microalbuminuria can confirm the diagnosis of DN [9]. Traditionally, renal biopsies are required for the diagnoses of DN, but the potential to indirectly monitor DN by studying the retina via noninvasive ophthalmology techniques opens up the possibility for early detection of DN and may considerably improve the outcome for patients [2].
In this chapter, we discuss the clinical manifestation and disease progression of DR, focus on the main molecular and cellular mechanisms involved, and conclude by highlighting the current treatment options for DR.
Disease Progression of DR
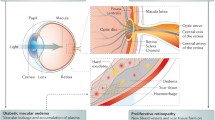
DR develops gradually in patients with diabetes. The prevalence of DR increases with the duration of diabetes. After 20 years of diabetes, almost 80% of patients have some form of DR [1]. In the clinic, DR is broadly divided into two stages: nonproliferative DR (NPDR) and proliferative DR (PDR) (Fig. 19.1). However, DR is preceded by a long preclinical phase, which is associated with the development of several microvascular and other anomalies in the retina. It is likely that diabetes affects all cell types of the retina, but the major retinal vascular changes can be easily imaged, and therefore grading of the disease is based on the severity of the vascular anomalies [10]. Recent advances in imaging techniques also enable the detection of more subtle alterations such as neurodegeneration, altered distribution of cone photoreceptors and anomalies in the thickness of the neural retina [11,12,13,14].

Schematic overview of the microvascular changes occurring in retinal capillaries of diabetic patients. Hyperglycemia induces various molecular and cellular alterations ultimately leading to endothelial dysfunction, degeneration of retinal capillaries, and retinal ischemia. Before clinically relevant stages of diabetic retinopathy (DR) are manifested, thickening of the vascular lamina basalis, pericyte loss, neurodegeneration, upregulation of levels of growth factors, and increased vascular leakage occur. These processes lead to the induction of a pro-inflammatory microenvironment that is accompanied by increased leukocyte infiltration, endothelial cell hypertrophy causing narrowing of the capillary lumen and endothelial cell death, and subsequently formation of acellular, non-perfused capillaries. Nonproliferative DR is accompanied by further dysfunction of the retinal capillaries, increased leakage of capillaries and impaired fluid clearance from the retinal tissue to the circulation by Müller cells, ultimately leading to edema formation. In the more advanced stages, retinal ischemia may induce neovascularization, which can be accompanied by scarring and blindness. PCDR, preclinical DR; NPDR, nonproliferative DR; PDR, proliferative DR; Ang-2, angiopoietin-2; VEGF, vascular endothelial growth factor; CTGF, connective tissue growth factor
The first anomalies that arise in the retina during the preclinical stage are thickening of the vascular lamina basalis, pericyte loss, increased vascular permeability, and formation of acellular capillaries. Progression of the disease into NPDR is recognized by the appearance of microaneurysms and hemorrhages, both associated with areas of capillary regression, vascular leakage, and hard exudates and spreading areas of capillary non-perfusion. When the disease progresses to an even more advanced stadium, with widespread areas of retinal non-perfusion and ischemia, neovascularization can develop, which is the characteristic hallmark of PDR. PDR occurs more frequently in patients with type 1 diabetes [1]. PDR may lead to vitreous hemorrhage , fibrosis via the angio-fibrotic switch, fibrovascular membrane contraction, retinal detachment , and eventually blindness [15]. NPDR generally develops as a consequence of vascular damage caused by hyperglycemia, whereas PDR develops as a direct result of retinal ischemia induced by capillary non-perfusion and is therefore not directly affected by metabolic control [16].
An important additional manifestation of DR is DME, which can occur in combination with NPDR and PDR. Blood vessels of the retina are highly selective in regulating the entry of molecules into the retinal tissue, comparable to vessels of the blood-brain barrier. DME is caused by the breakdown of these inner blood-retinal barrier (BRB) properties, which leads to leakage of fluid and plasma proteins from the vasculature into the neural retina and ultimately to edema formation [17]. DME in the central area of the retina, the macula, often leads to severe loss of visual acuity. DME is the most prevalent disease manifestation in type 2 diabetes and therefore represents the most common cause of vision loss in patients with diabetes [1].
Early Stages of DR
Hyperglycemia is a major factor that triggers the development of DR. It elicits the activation of molecular and cellular mechanisms suggested to be involved in the disease progression such as metabolic damage, inflammation, upregulation of levels of growth factors, and neurodegeneration. All these factors may induce damage to the retinal blood vessels and may eventually lead to vessel degeneration and formation of acellular, non-perfused capillaries. This process, which is also known as vasoregression, leads to the formation of an ischemic retina, and provides the basis for the formation of NPDR, DME, and PDR.
Metabolic Damage
High plasma levels of glucose lead to vascular damage due to the induction of oxidative stress, caused by mitochondrial overproduction of reactive oxygen species (ROS) [18]. Hyperglycemia also leads to endothelial dysfunction via activation of the polyol pathway , the formation of advanced glycation end products (AGEs) , activation of protein kinase C (PKC) isoforms, and an increased flux through the hexosamine pathway [18]. These four pathways all lead to the production of ROS which are thought to be the cause of endothelial cell dysfunction and cell death. This subsequently leads to the vascular abnormalities observed during preclinical DR. For instance, increased levels of glucose activate the polyol pathway provides an alternative form of glucose metabolism in which glucose is converted to fructose [19]. However, this occurs at the expense of NADPH and NAD+, which are important cofactors involved in redox reactions, making cells more sensitive for oxidative stress , as NADPH is the major substrate for the detoxification reactions of ROS [20]. Activation of the PKC isoforms leads to endothelial cell apoptosis and the formation of acellular capillaries, whereas it also induces the expression of growth factors such as vascular endothelial growth factor (VEGF) [21, 22]. AGEs play a role in lamina basalis thickening by increasing the expression of proteins involved in synthesis of the extracellular matrix such as connective tissue growth factor (CTGF), but also induce the expression of other factors involved in the disease progression such as angiopoietin-2 (Ang-2) [23,24,25]. Activation of the hexosamine pathway, in which fructose 6-phosphate is converted into N-acetyl glucosamine, is associated with neuronal apoptosis [26]. Even when normal glucose levels are obtained in patients, progression of DR continues which suggests the existence of the phenomenon known as “metabolic memory” [27]. Oxidative stress plays an important role in the establishment of this metabolic memory, probably via modulating alterations in the epigenetic landscape [27, 28].
Inflammation
Several inflammatory mechanisms are considered to be involved in the formation of microvascular complications during the early stages of DR, which include, among others, activity of the pro-inflammatory transcription factor NF-κB , pro-inflammatory cytokines such as IL-1β and TNFα , and the intracellular adhesion molecule-1 (ICAM-1) [13, 29]. These inflammatory mediators may play a central role in the degeneration of capillaries, pericyte loss, vascular permeability, and neurodegeneration [13, 29]. Especially increased leukostasis, induced by cytokines and VEGF, has been suggested to cause the formation of early vascular lesions in the diabetic retina, via endothelial damage by FAS-FAS-L interactions, and progressive vascular occlusions and subsequent development of areas of non-perfusion [30, 31]. However, others have suggested that leukostasis is only an epiphenomenon of the retinal diabetic milieu and cannot explain by itself the pathogenesis of DR [32, 33].
Growth Factors
Several growth factors have been associated with the development of early vascular lesions in the diabetic retina, including Ang-2, VEGF, and CTGF.
Angiopoietin-2 (Ang-2)
The angiopoietin (Ang)-Tie system plays an important role in maintaining vascular stability [25]. Ang-1 is secreted by perivascular cells and activates the tyrosine kinase receptor Tie2 on endothelial cells, thereby inducing increased cell survival, endothelial barrier function and vessel stabilization. In contrast, Ang-2 inactivates Tie2, leading to endothelial cell degeneration and vessel destabilization. It is thought that Ang-2 plays a key role in initiating vasoregression in preclinical DR [25]. Hyperglycemia induces Ang-2 expression, altering the balance between the two ligands and favoring the inhibitory effects of Ang-2 on the Tie2 receptor [34]. High levels of Ang-2 lead to pericyte loss, which is one of the first morphological change observed in the diabetic eye [35]. In addition, high levels of Ang-2 lead to endothelial cell death and formation of acellular capillaries [25].
Vascular Endothelial Growth Factor (VEGF)
VEGF is an important inducer of vascular permeability and the major pro-angiogenic factor [36, 37]. These functions are mainly mediated via the VEGF family member known as VEGF-A. In this chapter, we refer to this family member when discussing the functions of VEGF . The role of VEGF in the development of the advanced stages of DR such as PDR and DME is discussed below in detail. However, VEGF also plays an important role in the preclinical stage of DR. Hypoxia is a major inducer of VEGF expression, but hyperglycemia can upregulate VEGF expression as well [3]. VEGF induces vascular permeability, increases the adhesion of leukocytes to the vasculature, and translocates the insulin-independent glucose transporter-1 (Glut-1) from intracellular stores to the membrane which may further promote hyperglycemia-induced damage in the early diabetic retina [38, 39]. VEGF over-expression in the diabetic retina may also induce endothelial cell hypertrophy, leading to narrowing of the capillary lumen and thereby eventually be the cause of capillary non-perfusion [32]. This may even incite a vicious circle of disease progression, creating areas of local ischemia, followed by more endogenous VEGF production and further luminal narrowing. This vicious circle, which is an alternative or complementary mechanism to the paradigm of VEGF -induced leukostasis as the cause of capillary occlusion, could explain why vascular lesions spread from focal points in the diabetic retina, whereas other areas remain totally unaffected [31, 32].
Connective Tissue Growth Factor (CTGF)
CTGF is a growth factor that regulates the expression of several other growth factors and extracellular matrix (ECM) proteins [40]. As a consequence, it is involved in a wide range of biological processes such as production of ECM components, angiogenesis, wound healing, and fibrosis [40]. Thickening of the lamina basalis of retinal capillaries is one of the first pathologically visible change in early diabetes, preceding the loss of pericytes [41]. CTGF appears to play a key role in this process [42, 43]. In preclinical DR, CTGF expression is upregulated by AGEs and VEGF [23, 43]. Besides the role of CTGF in lamina basalis thickening, recent findings suggest that CTGF also plays a role in inducing pericyte loss and the formation of acellular capillaries in the early stages of DR [44].
Neurodegeneration
Retinal vascular cells are closely associated with neurons and glial cells in the so-called neurovascular unit, which critically regulate their function. Before the onset of the typical microvascular lesions of NPDR, anomalies in the neuronal structure, and function of the retina can be detected [45]. For instance, in a longitudinal study it was shown that thinning of the retinal neural layers precedes the development of microvascular changes [46]. Moreover, altered neuronal function is observed in the retina, and, as consequence, contrast sensitivity and dark adaptation is reduced in the early stages of DR [45]. The causal relationship between early neurodegeneration and vasoregression or the late vascular pathologies in DR remains unclear [46].
Ischemic Retinopathy
The ischemic milieu of the retina in NPDR induces the production of VEGF and other growth factors. High VEGF levels play a central role in the pathobiology of both DME and PDR.
Diabetic Macular Edema (DME)
Breakdown of the Blood-Retinal Barrier (BRB)
Integrity of the BRB is essential for vision. Without a proper BRB, plasma proteins leak into the retinal tissue, leading to accumulation of proteins and fluid in the macula and other features of DME. In general, there are two main routes for molecules to cross the endothelial monolayer, that is, 1) via opening of the junctions between endothelial cells or 2) through the endothelial cell cytoplasm via vesicles or specific transporters. These pathways are known as the paracellular pathway and transcellular pathway, respectively. Small molecules and solutes can diffuse back and forth across the endothelium via the paracellular route, whereas molecules larger than 3 nm in radius cannot pass the BRB paracellularly and use the transcellular route. As described by the rules of Starling, the concentration of macromolecules is an important determinant of the interstitial osmotic pressure. Increased transport of macromolecules via vesicle-mediated transcytosis therefore plays an important role in the pathobiology of DME [17]. VEGF appears to be a major regulator of this process. VEGF has been shown to increase the number of caveolar vesicles in human retinal explants [47]. In addition, intraocular VEGF injections in monkey eyes shifted the distribution of vesicles from an abluminal localization to a luminal localization, without obviously altering the junctional integrity between cells [48]. This shift in distribution may reflect an altered direction of vesicular transcytosis, occurring from blood to tissue [48]. Thus, especially active transcytosis of plasma proteins via vesicles may alter the interstitial osmotic pressure, which in turn draws fluid from the leaky vessels into the retinal tissue causing DME. However, it should be noted that generally much more emphasis is put on the role of paracellular permeability in microvascular permeability and BRB breakdown in eye disease. Accordingly, disruption of the junctional interactions between retinal endothelial cells is most widely recognized as important causative factor in BRB breakdown and DME formation [17].
Inflammation
The low-grade inflammatory milieu of the diabetic eye promotes breakdown of the BRB and further vascular leakage, in addition or as an alternative pathway to VEGF [49]. Pro-inflammatory mediators induce BRB breakdown primarily via the paracellular pathway. For instance, the pro-inflammatory cytokine TNFα alters the expression of several junctional proteins and increases the permeability for small molecules but not for large molecules [50]. However, the precise role of inflammation in the pathogenesis of DME remains still a manner of debate [33].
Impaired Fluid Homeostasis
Besides increased protein and fluid extravasation in edema formation, impaired fluid reabsorption from the retinal tissue to the circulation also plays a role in edema formation. A correct balance between fluid extravasation and clearance is especially important in the retina, since the retina does not have a lymphatic system to remove excess fluid from the interstitium [51]. Müller cells , which are the glial cells of the retina, play an alternative role in this process. Müller cells regulate via facilitating transcellular water transport via water channels termed aquaporins (AQPs) [52]. The transport of water by these channels is tightly coupled to the potassium current in cells. In the diabetic retina, expression of the potassium channel Kir4.1 is downregulated, resulting in the accumulation of potassium in cells, increased water influx via AQP4, and swelling of Müller cells [52]. Moreover, altered expression of the AQPs subtypes in Müller cells of the diabetic retina may also impair fluid homeostasis, leading to cellular swelling [53, 54]. Thus, altered fluid clearance in the diabetic retina may contribute to both intracellular and extracellular edema and DME.
Proliferative Diabetic Retinopathy (PDR)
There is evidence that retinal ischemia in NPDR leads to a vicious circle of disease worsening by VEGF-induced leukostasis and/or endothelial hypertrophy [31, 32], and this may eventually lead to PDR , which only occurs when widespread areas of capillary non-perfusion have developed. In a response to counteract retinal ischemia, a pro-angiogenic response is initiated leading to retinal neovascularization and the development of PDR. However, these newly forming vessels are leaky, fragile, and prone to rupture, leading to hemorrhages in the vitreous. In addition, the newly formed vessels are the visible component of a coexisting wound-healing response, which develops into fibrous tissue formation and scarring and may cause retinal detachment and eventually total blindness.
Compared to the VEGF levels detected in the eyes of patients with NPDR, the mean concentration of VEGF in the vitreous of patients with active PDR are approximately 35 times higher [55]. However, it is important to note that high levels of VEGF are not sufficient to lead to neovascularization. Altered expression of its receptors in retinal vessels is also involved. In fact, one study showed that repeated high doses of intraocular VEGF injections did not induce retinal neovascularization in monkey eyes [32, 56]. In contrast, prominent neovascularization was detected in the iris [56]. This observation was explained by the notion that retinal capillaries express only VEGFR1 in pericytes or on the abluminal side of endothelial cells, under normal physiological conditions, whereas the iris vessels constitutively express VEGFR2 [56, 57]. VEGFR1 has often been described to function only as a regulatory decoy receptor for the more important receptor VEGFR2 that mediates the angiogenic effects of VEGF. Thus, dysregulation of VEGFR signaling is necessary in the retina to facilitate neovascularization, but not in the iris. This may explain why retinal neovascularization only occurs in the advanced stages of DR, whereas increased VEGF levels are already observed in the preclinical phase of DR. In diabetic patients with established DR, altered expression of VEGFRs in the retina can be observed, showing prominent vascular expression of VEGFR1, VEGFR2, and VEGFR3 [57].
The induction of fibrosis in the fibrovascular membranes of PDR is dependent on an critical balance between VEGF and the pro-fibrotic growth factor CTGF [15, 40]. VEGF itself promotes CTGF expression . When the equilibrium between these factors reaches a certain threshold, fibrosis may overrule, initiating the angio-fibrotic switch [15].
Therapeutic Options
DR is a complex disease and many factors are involved. Hyperglycemia is the main underlying factor leading to vascular damage. Therefore, metabolic control is important for the management of DR. However, due to the phenomenon known as metabolic memory, and by the independent effects of established retinal capillary non-perfusion and ischemia, proper glycemic control alone is not effective to reduce the prevalence of DR or to treat DR when vision-threatening stages have developed [10, 58]. Other risk factors for DR include hypertension and hyperlipidemia, and management of these factors is relevant for DR, in particular for the clinical phases such as DME. Besides these general measures, there are only a few therapeutic options available for DR at the present.
Available treatments for DME are laser photocoagulation, anti-VEGF therapy , and corticosteroids . Focal and grid laser photocoagulation has been the standard care for DME for decades, but anti-VEGF agents have been shown to be superior [59]. Anti-VEGF therapy requires regular injections for 2 or more years and is associated with suboptimal responses in some patients. In addition, there are concerns that VEGF has important neuroprotective functions in the retina, suggesting the need for alternative treatment options [60]. Anti-VEGF treatment of DME has nevertheless led to a major improvement in treatment outcome in DME and has been shown not only to have a direct effect on macular edema but also a more long-term inhibitory effect on the progression of the underlying NPDR [61, 62]. The latter effect seems to underscore the presumed role of VEGF in the pathogenesis of the diabetes-induced damage leading to vasoregression and eventually NPDR.
Glucocorticoids have been shown to reduce DME, which generally is explained by their targeting of the inflammatory part of the pathogenesis and fluid clearance by Müller cells [52, 63]. However, glucocorticoids may have a direct restorative effect on the BRB [64, 65], providing an alternative explanation for their effectivity in DME. Nevertheless, corticosteroids are associated with serious side effects such as cataract formation and glaucoma due to increased intraocular pressure [66, 67]. Therefore, treatment with these agents is only advisable in patients that do not respond to anti-VEGF therapy.
Patients with PDR are treated with panretinal laser photocoagulation and vitreoretinal surgery. During panretinal laser photocoagulation, focal thermal burns are used to destroy retina pigment epithelial cells and the overlying photoreceptors. Although this procedure sounds counterintuitive as a treatment option, it is significantly effective in reducing PDR, probably by downregulation of VEGF production induced by the reduction in oxygen consumption of the retina [68]. Vitreoretinal surgery may be necessary in later stages of PDR, when severe complications have been developed in the retina such as vitreous hemorrhage, tractional retinal detachment, and epimacular fibrovascular proliferations [10]. Recently, anti-VEGF therapy has also been shown to be a promising alternative treatment option for PDR targeting the pro-angiogenic phenotype of the retina [69]. However, targeting VEGF in PDR patients with advanced fibrovascular proliferations carries a risk of acceleration of the angio-fibrotic switch, inducing retinal fibrosis, contraction, and possibly retinal detachment [15, 70].
Screening
DR gradually progresses from an asymptomatic manifestation into a more advanced disease associated with vision loss. By the time clinical symptoms occur, several irrevocable cellular and molecular changes in the retina have already occurred. Although various treatment options exist for patients with DR, the currently available therapies are generally less effective in restoring vision loss beyond a certain stage of the disease. This indicates that early detection by screening of asymptomatic persons with diabetes is of uttermost importance. In addition, there is a great variability in the rate of disease progression and risk to develop clinically significant forms of DR among diabetic patients [71]. Some diabetes patients do not develop vascular anomalies and have good visual acuity after many years of diabetes, whereas others have a rapidly progressing form that does not respond to therapy [71]. An additional important aspect of screening is to identify the patients that are at risk at developing vision-threatening forms of DR. However, this remains a problem in the clinic.
Retinal Imaging
In the clinic, noninvasive imaging techniques are used to monitor the disease progression of DR. Commonly used techniques include ophthalmoscopy , fundus photography , fluorescein angiography (FA) , and optical coherence tomography (OCT) [14]. Fundus photography allows detection of microvascular abnormalities within the retina, making grading of the disease possible (Fig. 19.2). To provide additional information, FA and angio-OCT are useful. During FA, the fluorescent dye fluorescein is administrated into the systemic circulation, and the vascular filling and extravasation of fluorescein from the retinal vasculature are assessed (Fig. 19.3). FA allows the detection of vascular leakage but also characterizes areas of capillary non-perfusion. OCT generates cross-sectional, three-dimensional images of the retina, whereas angio-OCT provides a detailed image of the perfused microvasculature. OCT makes it possible to determine the thickness for each individual layer of the retina, enabling detection of fluid accumulation and monitoring of treatment effects [72, 73].

Fundus images of patients with diabetic retinopathy (DR). Color or red-free fundus images of a normal retina (a), a retina with severe nonproliferative DR and maculopathy (b), and a retina with proliferative DR (c, d). Note intraretinal hemorrhages (b, white arrow) indicating retinal ischemia, yellow intraretinal hard exudates (b, black arrow) indicating vascular leakage, and neovascularization extending from the optic disk to the surface of the retina (c, d)

Fundus image (a), fluorescein angiogram (b), optical coherence tomography (OCT) cross section (c), and OCT macular thickness map (d) of a patient with nonproliferative diabetic retinopathy and diabetic macular edema. Note hard exudates (a, white arrows), capillary non-perfusion (b, dark areas marked with white arrows) and leaky microaneurysms (b, orange arrow), retinal cysts (c, star), and thickened retina (d)
Screening for DR is very cost-effective, and fundus photography and subsequent grading is the standard approach [74]. Addition of OCT imaging to screening programs considerably improves correct detection of DME and reduces the overall costs [75]. Implementation of OCT imaging in screening programs may therefore be useful for early detection of DR. Recently, automated image analysis employing artificial intelligence software, developed with deep learning algorithms, have been presented with high sensitivity and specificity outperforming human graders [76].
Although the specific sequence of events leading to the onset of DR remains a manner of debate, it is becoming clear that neurodegeneration can be observed in the retina long before the clinical recognized vascular lesions can be detected [45, 46]. OCT imaging can detect the associated thinning of the neural layers within the retina [46].
Biomarkers
Due to the multifactorial nature of DR, it is likely that multiple biomarkers are needed in clinical practice for meaningful prognostic or predictive purposes rather than one individual biomarker [10]. Common biomarkers for DR are HbA1c, visual acuity, glucose levels, lipid levels, imaging characteristics, of which only HbA1c has a proven prognostic significance [77, 78]. Identification of novel biomarkers may have immense value for the clinic. For instance, the identification of increased VEGF levels in eyes of diabetic patients has significantly increased our understanding of the pathogenesis of DR, and the concomitant introduction of anti-VEGF agents and OCT imaging has revolutionized its treatment. The current search for novel biomarkers is mainly focused on the basic mechanisms underlying DR, which include AGEs, oxidative stress, endothelial dysfunction, inflammation, and pro-angiogenic factors [79].
Concluding Remarks
DR is the most common microvascular complication affecting diabetic patients. The threat of loss of vision from DR is one of the main concerns of persons with diabetes in relation to their disease [80]. It is becoming clear that DR should not be classified as a solely vascular complication of diabetes, as neurodegeneration and inflammation play an important role in the pathobiology of DR as well. Although significant progress has been made in the understanding of DR at a molecular and cellular level, there are still a limited number of therapies available at present. A better understanding of the basic mechanisms involved in the pathogenesis is essential for the identification of novel therapeutic targets and may explain why the currently available therapies are not effective in some patients.
References
Yau JWY, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–64.
Zerbini G, Maestroni S, Turco V, Secchi A. The eye as a window to the microvascular complications of diabetes. Dev Ophthalmol. 2017;60:6–15.
Arden GB, Arden GB, Sidman RL, Sidman RL, Arap W, Arap W, et al. Spare the rod and spoil the eye. Br J Ophthalmol. 2005;89(August):764–9.
Hughes JM, Groot AJ, van der Groep P, Sersansie R, Vooijs M, van Diest PJ, et al. Active HIF-1 in the normal human retina. J Histochem Cytochem. 2010;58(3):247–54.
Antonetti DA, Barber AJ, Bronson SK, Freeman WM, Gardner TW, Jefferson LS, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–11.
Bakris GL. Overview of diabetic nephropathy: the relationship between diabetic nephropathy and diabetic retinopathy. [Internet]. [cited 2017 Oct 19]. Available from: https://www.uptodate.com/contents/overview-of-diabetic-nephropathy.
Chavers BM, Mauer SM, Ramsay RC, Steffes MW. Relationship between retinal and glomerular lesions in IDDM patients. Diabetes. 1994;43(3):441–6.
He F, Xia X, Wu XF, Yu XQ, Huang FX. Diabetic retinopathy in predicting diabetic nephropathy in patients with type 2 diabetes and renal disease: a meta-analysis. Diabetologia. 2013; 56(3):457–66.
National Kidney Foundation. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007;49(2 Suppl 2):S12–154.
Stitt AW, Curtis TM, Chen M, Medina RJ, GJ MK, Jenkins A, et al. The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res. 2016;51:156–86.
Lammer J, Prager SG, Cheney MC, Ahmed A, Radwan SH, Burns SA, et al. Cone photoreceptor irregularity on adaptive optics scanning laser ophthalmoscopy correlates with severity of diabetic retinopathy and macular edema. Investig Ophthalmol Vis Sci. 2016;57(15):6624–32.
van Dijk HW, Verbraak FD, Kok PHB, Garvin MK, Sonka M, Lee K, et al. Decreased retinal ganglion cell layer thickness in patients with type 1 diabetes. Invest Ophthalmol Vis Sci. 2010;51(7):3660–5.
Tang J, Kern TS. Inflammation in diabetic retinopathy. Prog Retin Eye Res. 2011;30:343–58.
Ramos de Carvalho JE, Verbraak FD, Aalders MC, van Noorden CJ, Schlingemann RO. Recent advances in ophthalmic molecular imaging. Surv Ophthalmol. 2014;59:393–413.
Kuiper EJ, Van Nieuwenhoven FA, de Smet MD, van Meurs JC, Tanck MW, Oliver N, et al. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS One. 2008;3(7):e2675.
Cunha-Vaz J. Characterization and relevance of different diabetic retinopathy phenotypes. Dev Ophthalmol. 2007;39:13–30.
Klaassen I, Van Noorden CJF, Schlingemann RO. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog Retin Eye Res. 2013;34:19–48.
Li C, Miao X, Li F, Wang S, Liu Q, Wang Y, et al. Oxidative stress-related mechanisms and antioxidant therapy in diabetic retinopathy. Oxidative Med Cell Longev. 2017;2017:9702820.
Lorenzi M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabesity Res. 2007;2007:61038.
Molenaar RJ, Botman D, Smits MA, Hira VV, Van Lith SA, Stap J, et al. Radioprotection of IDH1-mutated cancer cells by the IDH1-mutant inhibitor AGI-5198. Cancer Res. 2015;75(22):4790–802.
Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15(11):1298–306.
Williams B, Gallacher B, Patel H, Orme C. Glucose-induced protein kinase C activation regulates vascular permeability factor mRNA expression and peptide production by human vascular smooth muscle cells in vitro. Diabetes. 1997;46(9):1497–503.
Hughes JM, Kuiper EJ, Klaassen I, Canning P, Stitt AW, Van Bezu J, et al. Advanced glycation end products cause increased CCN family and extracellular matrix gene expression in the diabetic rodent retina. Diabetologia. 2007;50(5):1089–98.
Gardiner TA, Anderson HR, Stitt AW. Inhibition of advanced glycation end-products protects against retinal capillary basement membrane expansion during long-term diabetes. J Pathol. 2003;201(2):328–33.
Hammes HP, Feng Y, Pfister F, Brownlee M. Diabetic retinopathy: targeting vasoregression. Diabetes. 2011;60:9–16.
Nakamura M, Barber AJ, Antonetti DA, LaNoue KF, Robinson KA, Buse MG, et al. Excessive Hexosamines block the neuroprotective effect of insulin and induce apoptosis in retinal neurons. J Biol Chem. 2001;276(47):43748–55.
Kowluru RA. Diabetic retinopathy, metabolic memory and epigenetic modifications. Vis Res. 2017;139:30.
Kowluru RA, Kowluru A, Mishra M, Kumar B. Oxidative stress and epigenetic modifications in the pathogenesis of diabetic retinopathy. Prog Retin Eye Res. 2015;48:40–61.
Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–68.
Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18(12):1450–2.
Liu Y, Shen J, Fortmann SD, Wang J, Vestweber D, Campochiaro PA. Reversible retinal vessel closure from VEGF-induced leukocyte plugging. JCI insight. 2017;2(18):e95530.
Hofman P, van Blijswijk BC, Gaillard PJ, Vrensen GF, Schlingemann RO. Endothelial cell hypertrophy induced by vascular endothelial growth factor in the retina: new insights into the pathogenesis of capillary nonperfusion. Arch Ophthalmol. 2001;119(6):861–6.
van der Wijk A-E, Hughes JM, Klaassen I, Van Noorden CJF, Schlingemann RO. Is leukostasis a crucial step or epiphenomenon in the pathogenesis of diabetic retinopathy? J Leukoc Biol. 2017;102(4):993–1001.
Yao D, Taguchi T, Matsumura T, Pestell R, Edelstein D, Giardino I, et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J Biol Chem. 2007;282(42):31038–45.
Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, et al. Angiopoietin-2 causes Pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes. 2004;53(4):1104–10.
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219(4587):983–5.
Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun. 1989;161(2):851–8.
Lu M, Perez VL, Ma N, Miyamoto K, Peng HB, Liao JK, et al. VEGF increases retinal vascular ICAM-1 expression in vivo. Investig Ophthalmol Vis Sci. 1999;40(8):1808–12.
Sone H, Deo BK, Kumagai AK. Enhancement of glucose transport by vascular endothelial growth factor in retinal endothelial cells. Invest Ophthalmol Vis Sci. 2000;41(7):1876–84.
Klaassen I, van Geest RJ, Kuiper EJ, van Noorden CJF, Schlingemann RO. The role of CTGF in diabetic retinopathy. Exp Eye Res. 2015;133:37–48.
Gardiner TA, Stitt AW, Anderson HR, Archer DB. Selective loss of vascular smooth muscle cells in the retinal microcirculation of diabetic dogs. Br J Ophthalmol. 1994;78:54–60.
Kuiper EJ, Witmer AN, Klaassen I, Oliver N, Goldschmeding R, Schlingemann RO. Differential expression of connective tissue growth factor in microglia and pericytes in the human diabetic retina. Br J Ophthalmol. 2004;88(8):1082–7.
Kuiper EJ, van Zijderveld R, Roestenberg P, Lyons KM, Goldschmeding R, Klaassen I, et al. Connective tissue growth factor is necessary for retinal capillary basal lamina thickening in diabetic mice. J Histochem Cytochem. 2008;56(8):785–92.
Van Geest RJ, Leeuwis JW, Dendooven A, Pfister F, Bosch K, Hoeben KA, et al. Connective tissue growth factor is involved in structural retinal vascular changes in long-term experimental diabetes. J Histochem Cytochem. 2014;62(2):109–18.
Stem MS, Gardner TW. Neurodegeneration in the pathogenesis of diabetic retinopathy: molecular mechanisms and therapeutic implications. Curr Med Chem. 2013;20(26):3241–50.
Sohn EH, van Dijk HW, Jiao C, Kok PHB, Jeong W, Demirkaya N, et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc Natl Acad Sci. 2016;113(19):E2655–64.
Wisniewska-Kruk J, Van Der Wijk AE, Van Veen HA, Gorgels TGMF, Vogels IMC, Versteeg D, et al. Plasmalemma vesicle-associated protein has a key role in blood-retinal barrier loss. Am J Pathol. 2016;186(4):1044–54.
Hofman P, Blaauwgeers HG, Tolentino MJ, Adamis AP, Nunes Cardozo BJ, Vrensen GF, et al. VEGF-A induced hyperpermeability of blood-retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Curr Eye Res. 2000;21(2):637–45.
Noma H, Mimura T, Yasuda K, Shimura M. Role of inflammation in diabetic macular edema. Ophthalmologica. 2014;232(3):127–35.
Van der Wijk AE, Vogels IMC, Van Noorden CJF, Klaassen I, Schlingemann RO. TNFα-induced disruption of the blood-retinal barrier in vitro is regulated by intracellular 30′50′-cyclic adenosine monophosphate levels. Investig Ophthalmol Vis Sci. 2017;58(9):3496–505.
Curtis TM, Gardiner TA, Stitt AW. Microvascular lesions of diabetic retinopathy: clues towards understanding pathogenesis? Eye. 2009;23(7):1496–508.
Reichenbach A, Wurm A, Pannicke T, Iandiev I, Wiedemann P, Bringmann A. Müller cells as players in retinal degeneration and edema. Graefes Arch Clin Exp Ophthalmol. 2007;245:627–36.
Fukuda M, Nakanishi Y, Fuse M, Yokoi N, Hamada Y, Fukagawa M, et al. Altered expression of aquaporins 1 and 4 coincides with neurodegenerative events in retinas of spontaneously diabetic Torii rats. Exp Eye Res. 2010;90(1):17–25.
Iandiev I, Pannicke T, Reichenbach A, Wiedemann P, Bringmann A. Diabetes alters the localization of glial aquaporins in rat retina. Neurosci Lett. 2007;421(2):132–6.
Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331(22):1480–7.
Witmer AN, van Blijswijk BC, van Noorden CJF, Vrensen GFJM, Schlingemann RO. In vivo Angiogenic phenotype of endothelial cells and Pericytes induced by vascular endothelial growth factor-A. J Histochem Cytochem. 2004;52(1):39–52
Witmer AN, Blaauwgeers HG, Weich HA, Alitalo K, Vrensen GFJM, Schlingemann RO. Altered expression patterns of VEGF receptors in human diabetic retina and in experimental VEGF-induced retinopathy in monkey. Investig Ophthalmol Vis Sci. 2002;43(3):849–57.
Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36(7):808–12.
Agarwal A, Afridi R, Hassan M, Sadiq MA, Sepah YJ, Do DV, et al. Novel therapies in development for diabetic Macular Edema. Curr Diab Rep. 2015;15:75.
Saint-Geniez M, Maharaj ASR, Walshe TE, Tucker BA, Sekiyama E, Kurihara T, et al. Endogenous VEGF is required for visual function: evidence for a survival role on Müller cells and photoreceptors. Chan-Ling T, editor. PLoS One. 2008;3(11):e3554.
Bandello F, Corvi F, La Spina C, Benatti L, Querques L, Capuano V, et al. Outcomes of intravitreal anti-VEGF therapy in eyes with both neovascular age-related macular degeneration and diabetic retinopathy. Br J Ophthalmol. 2016;100(12):1611–6.
Ip MS, Domalpally A, Sun JK, Ehrlich JS. Long-term effects of therapy with ranibizumab on diabetic retinopathy severity and baseline risk factors for worsening retinopathy. Ophthalmology. 2015;122(2):367–74.
Agarwal A, Sarwar S, Sepah YJ, Nguyen QD. What have we learnt about the management of diabetic macular edema in the antivascular endothelial growth factor and corticosteroid era? Curr Opin Ophthalmol. 2015;26:177–83.
Förster C, Burek M, Romero IA, Weksler B, Couraud P-O, Drenckhahn D. Differential effects of hydrocortisone and TNFα on tight junction proteins in an in vitro model of the human blood–brain barrier. J Physiol. 2008;586(Pt 7):1937–49.
Felinski EA, Cox AE, Phillips BE, Antonetti DA. Glucocorticoids induce transactivation of tight junction genes occludin and claudin-5 in retinal endothelial cells via a novel cis-element. Exp Eye Res. 2008;86(6):867–78.
Chu YK, Chung EJ, Kwon OW, Lee JH, Koh HJ. Objective evaluation of cataract progression associated with a high dose intravitreal triamcinolone injection. Eye. 2008;22(7):895–9.
Kersey JP, Broadway DC. Corticosteroid-induced glaucoma: a review of the literature. Eye. 2006;20:407–16.
Photocoagulation treatment of proliferative diabetic retinopathy: the second report of diabetic retinopathy study findings. Ophthalmology. 1978;85(1):82–106.
Gross JG, Glassman AR, Jampol LM, Inusah S, Aiello LP, Antoszyk AN, et al. Panretinal photocoagulation vs Intravitreous Ranibizumab for proliferative diabetic retinopathy. JAMA. 2015;314(20):2137.
Van Geest RJ, Lesnik-Oberstein SY, Tan HS, Mura M, Goldschmeding R, Van Noorden CJF, et al. A shift in the balance of vascular endothelial growth factor and connective tissue growth factor by bevacizumab causes the angiofibrotic switch in proliferative diabetic retinopathy. Br J Ophthalmol. 2012 Apr;96(4):587–90.
Nunes S, Ribeiro L, Lobo C, Cunha-Vaz J. Three different phenotypes of mild nonproliferative diabetic retinopathy with different risks for development of clinically significant macular edema. Investig Ophthalmol Vis Sci. 2013;54(7):4595–604.
Garvin MK, Abràmoff MD, Abràmoff MD, Wu X, Russell SR, Burns TL, et al. Automated 3-D Intraretinal layer segmentation of macular spectral-domain optical coherence tomography images. IEEE Trans Med Imaging. 2009;28(9):1436–47.
Garvin MK, Abràmoff MD, Kardon R, Russell SR, Wu X, Sonka M. Intraretinal layer segmentation of macular optical coherence tomography images using optimal 3-D graph search. IEEE Trans Med Imaging. 2008;27(10):1495–505.
Stefánsson E, Bek T, Porta M, Larsen N, Kristinsson JK, Agardh E. Screening and prevention of diabetic blindness. Acta Ophthalmol Scand. 2000;78(4):374–85.
Olson J, Sharp P, Goatman K, Prescott G, Scotland G, Fleming A, et al. Improving the economic value of photographic screening for optical coherence tomography-detectable macular oedema: a prospective, multicentre, UK study. Health Technol Assess (Rockv). 2013;17(51):1–141.
Abràmoff MD, Lou Y, Erginay A, Clarida W, Amelon R, Folk JC, et al. Improved automated detection of diabetic retinopathy on a publicly available dataset through integration of deep learning. Investig Ophthalmol Vis Sci. 2016;57(13):5200–6.
Jenkins AJ, Joglekar MV, Hardikar AA, Keech AC, O’Neal DN, Januszewski AS. Biomarkers in diabetic retinopathy. Rev Diabet Stud. 2015;12:159–95.
Cunha-Vaz J, Ribeiro L, Lobo C. Phenotypes and biomarkers of diabetic retinopathy. Prog Retin Eye Res. 2014;41:90–111.
Simó-Servat O, Simó R, Hernández C. Circulating biomarkers of diabetic retinopathy: an overview based on physiopathology. J Diabetes Res. 2016;2016:5263798.
Quandt SA, Reynolds T, Chapman C, Bell RA, Grzywacz JG, Ip EH, et al. Older adults’ fears about diabetes: using common sense models of disease to understand fear origins and implications for self-management. J Appl Gerontol. 2013;32(7):783–803.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Bosma, E.K., van Noorden, C.J.F., Klaassen, I., Schlingemann, R.O. (2019). Microvascular Complications in the Eye: Diabetic Retinopathy. In: Roelofs, J., Vogt, L. (eds) Diabetic Nephropathy. Springer, Cham. https://doi.org/10.1007/978-3-319-93521-8_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-93521-8_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93520-1
Online ISBN: 978-3-319-93521-8
eBook Packages: MedicineMedicine (R0)