
Abstract
Background
Under normal conditions, Müller cells support neuronal activity and the integrity of the blood-retinal barrier, whereas gliotic alterations of Müller cells under pathological conditions may contribute to retinal degeneration and edema formation. A major function of Müller cells is the fluid absorption from the retinal tissue, which is mediated by transcellular water transport coupled to currents through potassium channels.
Methods
Alterations of retinal Müller cells under pathological conditions were investigated by immunohistochemistry and recording their behavior under osmotic stress.
Results
In animal models of various retinopathies, e.g., retinal ischemia, ocular inflammation, retinal detachment, and diabetes, it was found that Müller cells decrease the expression of their major potassium channel (Kir4.1). This alteration is associated with an impairment of the rapid water transport across Müller cell membranes, as recognizable in the induction of cellular swelling under hypoosmolar conditions. Osmotic swelling of Müller cells is also induced by oxidative stress and by inflammatory mediators such as arachidonic acid and prostaglandins.
Conclusions
The data suggest that a disturbed fluid transport through Müller cells is (in addition to vascular leakage) a pathogenic factor contributing to the development of retinal edema. Pharmacological re-activation of the retinal water clearance by Müller cells may represent an approach to the development of new edema-resolving drugs. Triamcinolone acetonide, which is clinically used to resolve edema, prevents osmotic swelling of Müller cells as it induces the release of endogenous adenosine and subsequent A1 receptor activation which results in the opening of ion channels. Apparently, triamcinolone resolves edema by both inhibition of vascular leakage and stimulation of retinal fluid clearance by Müller cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Macular edema
Macular edema is one of the leading causes of visual impairment in patients with uveitis or diabetes, and may also occur after cataract surgery [1–3]. Retinal ischemia, oxidative stress, and inflammation are pathogenic factors implicated in the development of macular edema [4–8]. Though promising new therapeutical approaches arose from the significantly increased knowledge regarding the factors that cause retinal edema and neovascularization [8–10], the pathogenic mechanisms underlying the development of macular edema are still not fully understood. One major pathogenic event in the development of edema is an increase in the permeability of the blood retinal barrier formed by vascular endothelial and retinal pigment epithelial cells [11]. The leakage from perifoveal vessels and pigment epithelium causes fluid accumulation in the foveal tissue and subretinal space, respectively, resulting in serous macular detachment and increased thickness of the retinal tissue due to diffuse or cystoid edema. The fluid accumulation contributes to the degeneration of retinal neurons and to a decrease in visual acuity, as it results in compression of neurons, nerve fibers, and vessels (which exacerbates ischemic conditions), and in an elongation of the routes for the diffusion of metabolic substrates and oxygen. Among the various vasoactive factors that induce vascular leakage (e.g., prostaglandins, interleukin-1ß, tumor necrosis factor [12]), the vascular endothelial growth factor (VEGF) is thought to be the major cytokine causing the breakdown of the blood-retinal barrier [8, 13–15]. However, the development of chronic edema depends on two factors: the rate of fluid entry from leaky vessels into the retinal tissue, and the rate of fluid absorption from the retina back into the blood. An impairment of the fluid absorption from the retinal tissue should be considered as a major pathogenic mechanism of edema formation, for example in patients which display macular edema without angiographic vascular leakage. In the preclinical stage of diabetic retinopathy, two types of increased retinal thickness exist that are or not associated with vascular leakage [16]. It has been shown that clinically significant diabetic macular edema develops only when (in addition to vascular leakage) the active transport mechanisms of the blood-retinal barrier are dysfunctional [17]. Obviously, any anomalies in vessel permeability need to be accompanied by ineffective edema-resolving mechanisms to cause chronic edema [18].
Macular edema-a Müller cell disease?
Müller cells, the principal glial cells of the retina, constitute a functional link between neurons and vessels. They support neurons with blood-derived nutrients, remove metabolic waste, and are responsible for maintaining the homeostasis of the retinal extracellular milieu (ions, water, neurotransmitter molecules, and pH) [19]. Retinal capillaries are closely ensheathed by glial cell processes arising from astrocytes and Müller cells. Müller cells become activated upon virtually all pathogenic stimuli [19]. In the retina of diabetic animals, for example, Müller cells become reactive at an early stage owing to disruption of the blood-retinal barrier [20, 21]. Normally, Müller cells participate in the establishment of the blood-retinal barrier [22]; however, under hypoxic conditions they impair the barrier function [23]. Under hypoxic, inflammatory or glucose-deprivation conditions, Müller cells secrete factors such as VEGF that increase the vascular permeability [24–27]. Müller cells are also a source of matrix metalloproteinases [28, 29] which impair the barrier function of retinal endothelial cells, by proteolytic degradation of the tight junction protein occludin [30]. Even under normoxic conditions, Müller cells secrete factors that decrease the barrier permeability, such as the pigment epithelium-derived factor (PEDF) [31] which downregulates the expression of VEGF. Under hypoxic conditions, the expression of PEDF is reduced in the retina and Müller cells [31, 32].
In addition to the effects on the barrier state of the retinal vasculature, Müller cells may also contribute to edema development by another way. There are electron microscopic studies carried out in the early 1980s which suggest that—in addition to ischemic changes in the retinal microvasculature—swelling (i.e., intracellular edema) of Müller cells may contribute to the development of cystoid macular edema, with the cysts being formed by swollen and necrotic Müller cells [33, 34]. In contrast, other authors did not find Müller cell swelling [35]. In the brain, swelling of astrocytes usually occurs concomitantly in vascular edema, and represents a major mechanism of edema formation under ischemic and various other conditions such as hyponatremia [36]. There are further data suggesting that dysfunctional Müller cells may be a causative factor in edema development. Dominantly inherited cystoid macular edema was suggested to represent a primary disease of Müller cells since degenerated Müller cells were found to be located around a virtually intact retinal vascular endothelium [37]. In an animal model of retinal hypoxia, it has been shown that vascular leakage is associated with cellular edema of Müller cells [38]. In the present review we discuss data which support the assumption that an impairment in the fluid absorption from the retinal tissue normally carried out by Müller cells may represent one causative factor in edema development. Under distinct circumstances, Müller cells may even swell.
Müller cells: fluid absorption from the retina
The fluid absorption from the subretinal space and from retinal tissue into the blood is normally carried out by pigment epithelial and glial cells. This is mediated via transcellular water transport that is osmotically coupled to the fluxes of osmolytes, particularly of ions [39, 40]. Fluid absorption is carried out by both cell types under normal conditions to redistribute water which accumulates in the retinal tissue and subretinal space due to various processes (Fig. 1): the influx of water from the blood into the retinal parenchyma which is coupled to the uptake of metabolic substrates such as glucose, and the formation of water within the retinal tissue which is associated with the aerobic energy production (per glucose molecule, 42 molecules of water are formed). The water fluxes through the cells are facilitated by the polarized intramembranous expression of specialized water transporting proteins, the aquaporins. Aquaporins facilitate bidirectional water movements across membranes, in dependence on the transmembranal osmotic gradient and hydrostatic pressure, and are involved in the maintenance of the ionic and osmotic balance in the tissue [41, 42]. Pigment epithelial cells express aquaporin-1 [43] whereas Müller cells express aquaporin-4 (Fig. 2a) [44].

Water fluxes through the retina. Under normal conditions, water accumulates in the neural retina and subretinal space due to influx from the blood (coupled to the uptake of nutrients such as glucose) and the oxidative synthesis of adenosine 5′-triphosphate (ATP) that generates carbon dioxide and water. The excess water is redistributed into the blood by transcellular water transport through Müller cells (yellow) and pigment epithelial cells (RPE). The transmembranous water transport is facilitated by aquaporin (AQP) water channels. RPE cells express AQP1, whereas Müller cells express AQP4. The transcellular water transport is osmotically coupled to the transport of osmolytes, especially of potassium and chloride ions. The ion fluxes across the cellular membranes are facilitated by transporter molecules and ion channels. In Müller cells, the Kir4.1 potassium channel is co-localized with AQP4 in membranes that surround the vessels, and at both limiting membranes (compare Fig. 2)

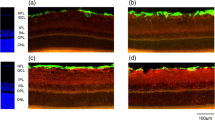
Experimental diabetes in rats alters the retinal expression of Kir4.1 potassium channels. a Localization of Kir4.1 and AQP4 proteins in slices of the neural rat retina. The Kir4.1 potassium channel is co-localized with AQP4 water channels in Müller cell membranes that surround the vessels (arrows), and at both limiting membranes (arrowheads). AQP4 is additionally expressed by Müller cells in both plexiform layers, and in the ganglion cell layer. b The prominent expression of the Kir4.1 protein around the vessels and at both limiting membranes (control retina) is absent in the retina of a diabetic animal. c View on the deep vascular plexus in the inner nuclear layer (wholemount preparation). The vessels are surrounded by AQP4 and Kir4.1 proteins in the control retina, but only by AQP4 in the retina of the diabetic animal. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer. Bars, 20 μm. With permission from ref. 52
The transcellular water transport is tightly coupled to fluxes of osmolytes, in particular of potassium and chloride ions (Fig. 1) [39, 40, 45]. Activated neurons release potassium ions. To avoid potassium-induced depolarization of neurons which may cause neuronal hyperexcitation resulting in excess release of neurotransmitters and glutamate toxicity, Müller cells take up excess potassium from the extracellular space especially in the synaptic (plexiform) layers of the retina, and release a similar amount of potassium into spaces outside of the neural retina, especially into the blood and the vitreous [46]. This “spatial buffering” of extracellular potassium is mediated predominantly by passive currents through potassium channels localized in Müller cell membranes. Müller cells express different types of potassium channels. Under normal conditions, inwardly rectifying potassium (Kir) channels of the Kir2.1 subtype are expressed in neuron-abutting membranes through which Müller cells take up excess potassium. Kir4.1 channels are expressed in membranes which are in close contact with spaces outside of the neural retina, i.e., in Müller cell processes enwrapping blood vessels and at both limiting membranes of the retina (Fig. 2a) [40, 47, 48]. Kir2.1 channels mediate only inward potassium currents into the Müller cells, whereas Kir4.1 channels mediate bidirectional currents between the extra-retinal tissues and the Müller cell interior. The co-localization of Kir4.1 potassium channels and aquaporin-4 water channels around vessels and at the limiting membranes of the retina [40] indicates a coupling of water transport to potassium currents in Müller cells. Moreover, aquaporin-4 channels are expressed by Müller cells in both plexiform layers and in the ganglion cell layer (Fig. 2a) where the cells take up excess potassium through Kir2.1 channels.
Müller cells: impairment of fluid absorption
Since the Müller cell-mediated fluid absorption from the retinal tissue predominantly relies on channel-mediated co-transport of potassium ions and water, any dysfunction of potassium channels should result in an impairment of the retinal water clearance and, therefore, will contribute to the development of chronic edema. Indeed we found in various animal models of retinopathies, e.g., after transient ischemia of the retina [49, 50], during ocular inflammation [51], in the retina of diabetic animals [52], and after retinal detachment [53, 54], that a major response of Müller cells under pathological conditions is an alteration in the expression of Kir4.1 channels. The Kir4.1 channel protein is redistributed from its prominent expression sites around the vessels and at the limiting membranes of the retina, and displays an even distribution in the retina at low level (Fig. 2b,c). This disclocation of Kir4.1 may be associated with a decrease in retinal expression of Kir4.1 protein [49], and causes a general decrease in the potassium currents flowing through Müller cell membranes [50–54]. On the other hand, the localization of Kir2.1 channels does not change after retinal ischemia [48]. Thus, Müller cells can take up potassium ions (via Kir2.1 channels), but the extrusion of excess potassium by Müller cells into extra-retinal spaces is impaired under pathological conditions. Hence, this may lead to an intracellular potassium overload and an increase in the osmotic pressure of the Müller cell interior in the diseased retina. Disruption of glial potassium homeostasis due to the downregulation of Kir4.1 channels may contribute to the death of nerve cells in the retina under pathological conditions [55], and should disturb the transport of water through Müller cells. It has been shown that Müller cells of patients with proliferative diabetic retinopathy display a strong reduction of their potassium conductance [56], suggesting that the downregulation of potassium channels may represent a feature of Müller cells in the human retina, as well as in animal models.

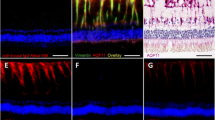
Experimental diabetes in rats affects the osmotic swelling characteristics of Müller cells, suggesting an impaired water transport across Müller cell membranes. Triamcinolone acetonide inhibits the osmotic Müller cell swelling via stimulation of endogenous adenosine signaling. a Müller cell bodies in retinal slices from control animals do not change their size upon application of a hypoosmolar solution (60% of control osmolarity). In contrast, Müller cell bodies in retinal slices from diabetic animals increase their size under osmotic stress (see the insets for an example; bar, 5 μm). b The osmotic swelling of Müller cell bodies in retinas from diabetic animals is inhibited in the presence of triamcinolone acetonide (triam; 100 μM). c The swelling-inhibitory effect of triamcinolone is prevented by inhibitors of adenosine transporters (NBTI) and adenosine A1 receptors (DPCPX), respectively. With permission from ref. 52

Putative mechanisms of Müller cell swelling and the effect of triamcinolone acetonide in the diabetic retina. a Under normal conditions, Müller cells mediate the fluid absorption from the retinal tissue into the blood by a co-transport of water (facilitated by AQP4 water channels) and osmolytes, especially potassium ions (facilitated by Kir4.1 channels). b In the diabetic retina, vascular leakage occurs due to the action of inflammatory mediators and VEGF. The Müller cells downregulate the expression of functional Kir4.1 channels. This downregulation impairs the release of potassium from Müller cells into the blood, and may result in an accumulation of potassium ions within the cells. The increase in the intracellular osmotic pressure results in an osmotic gradient across the plasma membrane that draws water into the Müller cells facilitated by AQP4 water channels. An influx of sodium ions into the cells, evoked by inflammatory mediators such as arachidonic acid (AA) and prostaglandins (PGs) which are formed in response to oxidative stress, contributes to the increase in intracellular osmotic pressure. The uptake of extravasated serum proteins by Müller cells may further increase the osmotic pressure of the cell interior. c Triamcinolone reduces edema by both inhibition of the fluid inflow from the vessels (through blocking the effects of inflammatory mediators and VEGF) and re-activation of the fluid clearance function of Müller cells. The latter action is mediated by triggering a release of endogenous adenosine and subsequent activation of adenosine A1 receptors which results in the opening of potassium channels (likely two pore domain [TASK] channels). The efflux of potassium ions is associated with a water transport out of the cells and, therefore, with a shrinkage of the cells. The triamcinolone-evoked stimulation of the potassium clearance by Müller cells facilitates the fluid absorption from the retinal tissue
An increase in intracellular osmotic pressure and an impairment of the rapid release of ionic osmolytes will favor swelling of Müller cells under distinct conditions. Indeed, we found that the swelling characteristics of Müller cells from diseased retinas were altered. Under normal conditions, Müller cells do not swell when isolated retinas are perfused with a hypoosmolar solution (Fig. 3a). However, Müller cells in pathologically altered retinas display a time-dependent swelling of their cell bodies in a hypoosmolar environment. Osmotic Müller cell swelling was observed under all circumstances which are associated with a downregulation of Kir4.1 channels, i.e., in retinas of diabetic animals (Fig. 3a) [52], after retinal ischemia [49] or detachment [54], and during endotoxin-induced ocular inflammation [51]. There is a correlation between the extent of osmotic swelling and the decrease of potassium currents in Müller cells of detached retinas [54]. The swelling of Müller cells indicates that the water transport across Müller cell membranes in response to a transmembranal osmotic gradient is impaired due to the restriction of a rapid release of potassium ions after downregulation of functional Kir4.1 channels. Moreover, a causal relationship between the downregulation of functional potassium channels and the osmotic swelling of Müller cells is suggested by the observation that a blockade of potassium channels in Müller cells of control retinas, e.g., by barium ions, induces cellular swelling under hypoosmolar conditions [49]. Both, the low potassium channel expression and the inefficient cell volume regulation, resemble properties of undifferentiated Müller cells in the young postnatal retina [57], suggesting that Müller cells dedifferentiate under pathological conditions.
In addition to the decreased expression of functional potassium channels, two further pathogenic factors are implicated in the osmotic Müller cell swelling: inflammatory mediators and oxidative stress. Inhibition of enzymes which produce arachidonic acid and prostaglandins (phospholipase A2 and cyclooxygenase) prevents the osmotic swelling of Müller cells in pathologically altered retinas [52, 54, 58]. Likewise, inhibition of oxidative stress by application of a reducing agent impedes the swelling of Müller cells under hypoosmolar conditions. Conversely, the application of arachidonic acid or prostaglandin E2, or oxidative stress evoked by H2O2, induces osmotic swelling of Müller cells in control retinas [52, 54, 58]. Local inflammation and oxidative stress are pathological factors in retinal diseases known to be associated with edema, e.g., diabetic retinopathy [6, 8, 59–65]. Under normal conditions, Müller cell bodies in the porcine retina do not express immunoreactivity for cyclooxygenase-2, whereas after experimental retinal detachment, they express this enzyme [54], suggesting that (in addition to retinal neurons) Müller cells increase the expression of enzymes that produce inflammatory mediators. A similar up-regulation of inflammatory-related proteins in Müller cells has been described during experimental diabetes [64]. Retinas of diabetic animals display an increased expression of cyclooxygenase-2, and an increased production of prostaglandin E2 [66, 67]. Arachidonic acid and its metabolites, especially prostaglandin E2, are major mediators of macular edema [5, 7]. It is known that oxidative stress evoked by acute application of H2O2 mimicks the edema-inducing effect of retinal ischemia-reperfusion [68]. Therefore, radical scavengers which inhibit vascular leakage in the ischemic retina [69] should also inhibit Müller cell swelling.
Presumably, the osmotic swelling of Müller cells in injured retinas is caused by oxidative stress, resulting in activation of phospholipase A2 and cyclooxygenase, subsequent arachidonic acid- and prostaglandin-evoked lipid peroxidation, and intracellular sodium overload. Simultaneously, the compensatory channel-mediated efflux of potassium might be inhibited (Fig. 4b). Oxidative stress can initiate lipid peroxidation, resulting in the release of arachidonic acid from the cell membrane [70, 71]. Ischemia in the neural tissue causes formation of free polyunsaturated fatty acids, particularly arachidonic acid, from membrane phospholipids, mainly by the action of phospholipase A2 [72–75]. Free radicals and hydroperoxides produced during ischemia-reperfusion stimulate the activity of lipoxygenase and cyclooxygenase [76]. Arachidonic acid induces both vascular and cellular edema in the brain [77, 78] and evokes swelling of cultured astrocytes [79]. The increase in intracellular sodium, being an effect of arachidonic acid and prostaglandins, is mainly due to inhibition of the sodium pump activity (Fig. 4b) [80, 81]. A similar increase in intracellular sodium occurs during swelling of Müller cells [82]. However, the intracellular sodium overload leads to cellular swelling only when the cells lost their capability to release osmolytes. Under normal conditions, retinal glial cells release potassium ions via Kir4.1 channels and, thus, avoid swelling despite of their increased sodium content. However, when the channel-mediated release of potassium is disturbed, they swell. It is known that arachidonic acid blocks voltage-gated potassium channels in Müller cells [83] which are the only channels that (under normal conditions) mediate outward potassium currents across the Müller cell membrane after downregulation of Kir4.1 [84–86]. Arachidonic acid may also inhibit the swelling-activated efflux of other osmolytes such as amino acids and chloride ions [87].
The induction of osmotic Müller cell swelling, which is not observed in cells from control tissues, suggests that the water transport across Müller cell membranes in response to osmotic gradients is impaired under pathological conditions; a disturbance of this water transport should prevent the resolution of retinal edema. The data suggest that an insufficient fluid absorption by Müller cells, and even Müller cell swelling under distinct conditions associated with osmotic imbalances between retinal and extra-retinal tissues, may represent an important factor contributing to the development of edema.
Stimulation of fluid absorption
Chronic edema is caused by an imbalance between the fluid influx into the tissue and the fluid absorption from the tissue. Edema can be resolved by inhibition of vascular leakage or by stimulation of the fluid clearance. Hitherto, most of the research regarding clinically used edema-resolving therapies was focused on the inhibition of vascular leakage. Reduction in fluid extravasation can be obtained by inhibition of the release, or of the effects of the vessel-permeabilizing factors VEGF and inflammatory factors. VEGF inhibitors, e.g., bevacizumab, reduce the level of free VEGF. Ruboxistaurin blocks protein kinase-ß which is an intracellular mediator involved in VEGF-induced increase in vascular permeability [88]. The anti-inflammatory corticosteroid, triamcinolone acetonide, inhibits vascular leakage [89, 90], reduces the vitreal level of VEGF [91], the secretion of VEGF by retinal cells [92–94], and the cellular effects of VEGF, e.g., the VEGF-evoked secretion of matrix metalloproteinases (own unpublished results). However, it is not known whether the edema-resolving therapies used clinically stimulate the fluid absorption from the retinal tissue concomitantly. This is likely since anti-inflammatory steroids are also effective in resolution of retinal edema in cases which are not associated with angiographic vascular leakage.
Stimulation of the fluid clearance function of pigment epithelial and Müller cells may represent an approach to the development of novel edema-resolving drugs, as it has been shown in experimentally detached retinas. Here, activation of purinergic P2Y2 receptors stimulates the ion transport and, therefore, the fluid absorption from the subretinal space by the pigment epithelium, resulting in proper attachment of the sensory retina [95, 96]. We found that triamcinolone inhibits the osmotic swelling of Müller cells. The swelling-inhibitory effect was observed in animal models of retinal ischemia-reperfusion and detachment, and in retinas of diabetic animals (Fig. 3b) [52, 54, 58]. Moreover, triamcinolone inhibits the swelling of Müller cells in control retinas observed in the presence of the potassium channel blocker barium, the inflammatory mediators arachidonic acid or prostaglandin E2, as well as under oxidative stress [52, 54, 58]. We found that triamcinolone inhibits osmotic swelling of Müller cells by stimulation of an autocrine purinergic signaling cascade (Figs. 3c, 4c) [58]. Application of triamcinolone stimulates the nucleoside transporter-mediated release of adenosine from the cells. Adenosine activates adenosine A1 receptors which leads to the opening of barium- and arachidonic acid-insensitive potassium channels (likely, two pore-domain channels [97]), as well as of chloride channels, in the Müller cell membrane [54, 82]. The extrusion of ions draws water out of the cells thereby preventing cellular swelling under hypoosmolar conditions (Fig. 4c). The action of adenosine on ion channels does not require intracellular calcium but is mediated by cyclic adenosine monophosphate, protein kinase A, and phosphatidylinositol-3 kinase (own unpublished results) [54, 82].
Two pore-domain potassium channels may function as an osmolyte extrusion pathway that helps to maintain proper Müller cell volumes when Kir4.1 channels are downregulated under pathological conditions. Since the potassium currents drive the water transport through Müller cells, the activation of two pore-domain channels by adenosine should facilitate the potassium clearence and the water absorption from the edematous retinal tissue. Stimulation of two pore-domain channels, either indirectly by A1 receptor activation or directly by channel openers, may represent a method to dissolve retinal edema. It is known that activation of A1 receptors has protective effects against retinal ischemic injury [98, 99]; a stimulatory effect on the fluid clearance through Müller cells may contribute to this protective effect of A1 receptor activation.
Conclusions: retinal edema- a dysregulation at the glio-vascular interface
Retinal edema develops during ischemia/hypoxia and inflammation; under both conditions, Müller cells alter their potassium conductance and swelling characteristics [49–51]. The osmotic swelling of Müller cells indicates that the fluid transport through Müller cell membranes is impaired under conditions which are clinically associated with macular edema (ischemia, ocular inflammation, diabetes). In addition to vascular leakage, dysregulation of the water movements across the glio-vascular interface may represent a major causative factor of edema. Most likely, there are different subtypes of macular edema in individual patients, with differences in the relative contribution of vascular leakage and dysfunction of Müller cells. Additive effects of systemic diseases such as hypertension [100] may increase the variability of mechanisms for edema formation. In the presence of an osmotic gradient across the glio-vascular interface, e.g., when the blood becomes hypoosmolar due to hyponatremia or hypoalbuminemia associated with renal or hepatic failures, or when neuronal hyperexcitation results in a strong increase of the retinal osmotical pressure in comparison to the blood and vitreous, Müller cells may swell similar as described in brain astrocytes [101]. Pharmacological stimulation of the ion and water clearance function of Müller cells should aid in resolution of both vascular and cellular edema. It is suggested that triamcinolone—via stimulation of endogenous adenosine signaling in Müller cells—facilitates the fluid absorption from the edematous tissue. Adenosine A1 receptors may represent a promising target for the development of novel drugs that stimulate the absorption of excesss fluid in edema.
References
Ferris FL, Patz A (1984) Macular edema. A complication of diabetic retinopathy. Surv Ophthalmol 28:S452–S461
Rothova A, Suttorp-van Schulten MS, Frits Treffers W, Kijlstra A (1996) Causes and frequency of blindness in patients with intraocular inflammatory disease. Br J Ophthalmol 80:332–336
Ray S, D’Amico DJ (2002) Pseudophakic cystoid macular edema. Semin Ophthalmol 17:167–180
Bresnick GH (1983) Diabetic maculopathy. A critical review highlighting diffuse macular edema. Ophthalmology 90:1301–1317
Guex-Crosier Y (1999) The pathogenesis and clinical presentation of macular edema in inflammatory diseases. Doc Ophthalmol 97:297–309
van Dam PS (2002) Oxidative stress and diabetic neuropathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev 18:176–184
Miyake K, Ibaraki N (2002) Prostaglandins and cystoid macular edema. Surv Ophthalmol 47:S203–S218
Caldwell RB, Bartoli M, Behzadian MA, El-Remessy AE, Al-Shabrawey M, Platt DH, Liou GI, Caldwell RW (2005) Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets 6:511–524
Speicher MA, Danis RP, Criswell M, Pratt L (2003) Pharmacologic therapy for diabetic retinopathy. Expert Opin Emerg Drugs 8:239–250
Eichler W, Yafai Y, Wiedemann P, Fengler D (2006) Antineovascular agents in the treatment of eye diseases. Curr Pharm Des 12:2645–2660
Cunha-Vaz JG, Travassos A (1984) Breakdown of the blood-retinal barriers and cystoid macular edema. Surv Ophthalmol 28:S485–S492
Derevjanik NL, Vinores SA, Xiao WH, Mori K, Turon T, Hudish T, Dong S, Campochiaro PA (2002) Quantitative assessment of the integrity of the blood-retinal barrier in mice. Invest Ophthalmol Vis Sci 43:2462–2467
Aiello LP, Bursell SE, Clermont A, Duh E, Ishii H, Takagi C, Mori F, Ciulla TA, Ways K, Jirousek M, Smith LE, King GL (1997) Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes 46:1473–1480
Antcliff RJ, Marshall J (1999) The pathogenesis of edema in diabetic maculopathy. Semin Ophthalmol 14:223–232
Marmor MF (1999) Mechanisms of fluid accumulation in retinal edema. Doc Ophthalmol 97:239–249
Lobo CL, Bernardes RC, Cunha-Vaz JG (2000) Alterations of the blood-retinal barrier and retinal thickness in preclinical retinopathy in subjects with type 2 diabetes. Arch Ophthalmol 118:1364–1369
Mori F, Hikichi T, Takahashi J, Nagaoka T, Yoshida A (2002) Dysfunction of active transport of blood-retinal barrier in patients with clinically significant macular edema in type 2 diabetes. Diabetes Care 25:1248–1249
Bellhorn RW (1984) Analysis of animal models of macular edema. Surv Ophthalmol 28:S520–S524
Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A (2006) Müller cells in the healthy and diseased retina. Prog Retin Eye Res 25:397–424
Lieth E, Barber A, Xu B, Dice C, Ratz MJ, Tanase D, Strother JM (1998) Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Diabetes 47:815–820
Rungger-Brändle E, Dosso AA, Leuenberger PM (2000) Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci 4:1971–1980
Tout S, Chan-Ling T, Hollander H, Stone J (1993) The role of Müller cells in the formation of the blood-retinal barrier. Neuroscience 55:291–301
Tretiach M, Madigan MC, Wen L, Gillies MC (2005) Effect of Müller cell co-culture on in vitro permeability of bovine retinal vascular endothelium in normoxic and hypoxic conditions. Neurosci Lett 378:160–165
Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA (1995) Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol 113:1538–1544
Amin RH, Frank RN, Kennedy A, Eliott D, Puklin JE, Abrams GW (1997) Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci 38:36–47
Eichler W, Kuhrt H, Hoffmann S, Wiedemann P, Reichenbach A (2000) VEGF release by retinal glia depends on both oxygen and glucose supply. Neuroreport 11:3533–3537
Yafai Y, Iandiev I, Wiedemann P, Reichenbach A, Eichler W (2004) Retinal endothelial angiogenic activity: effects of hypoxia and glial (Müller) cells. Microcirculation 11:577–586
Behzadian MA, Wang XL, Windsor LJ, Ghaly N, Caldwell RB (2001) TGF-ß increases retinal endothelial cell permeability by increasing MMP-9: possible role of glial cells in endothelial barrier function. Invest Ophthalmol Vis Sci 42:853–859
Milenkovic I, Weick M, Wiedemann P, Reichenbach A, Bringmann A (2003) P2Y receptor-mediated stimulation of Müller glial cell DNA synthesis: dependence on EGF and PDGF receptor transactivation. Invest Ophthalmol Vis Sci 44:1211–1220
Giebel SJ, Menicucci G, McGuire PG, Das A (2005) Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest 85:597–607
Eichler W, Yafai Y, Keller T, Wiedemann P, Reichenbach A (2004) PEDF derived from glial Müller cells: a possible regulator of retinal angiogenesis. Exp Cell Res 299:68–78
Duh EJ, Yang HS, Suzuma I, Miyagi M, Youngman E, Mori K, Katai M, Yan L, Suzuma K, West K, Davarya S, Tong P, Gehlbach P, Pearlman J, Crabb JW, Aiello LP, Campochiaro PA, Zack DJ (2002) Pigment epithelium-derived factor suppresses ischemia-induced retinal neovascularization and VEGF-induced migration and growth. Invest Ophthalmol Vis Sci 43:821–829
Fine BS, Brucker AJ (1981) Macular edema and cystoid macular edema. Am J Ophthalmol 92:466–481
Yanoff M, Fine BS, Brucker AJ, Eagle RC (1984) Pathology of human cystoid macular edema. Surv Ophthalmol 28:S505–S511
Gass JD, Anderson DR, Davis EB (1985) A clinical, fluorescein angiographic, and electron microscopic correlation of cystoid macular edema. Am J Ophthalmol 100:82–86
Kimelberg HK (1995) Current concepts of brain edema. Review of laboratory investigations. J Neurosurg 83:1051–1059
Loeffler KU, Li ZL, Fishman GA, Tso MOM (1992) Dominantly inherited cystoid macular edema. A histopathologic study. Ophthalmology 99:1385–1392
Stepinac TK, Chamot SR, Rungger-Brändle E, Ferrez P, Munoz JL, van den Bergh H, Riva CE, Pournaras CJ, Wagnieres GA (2005) Light-induced retinal vascular damage by Pd-porphyrin luminescent oxygen probes. Invest Ophthalmol Vis Sci 46:956–966
Pederson JE (1994) Fluid physiology of the subretinal space. In: Wilkinson CP (ed) Retina. Mosby, St. Louis, pp 1955–1968
Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP (1999) Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Müller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26:47–54
Verkman AS (2002) Physiological importance of aquaporin water channels. Ann Med 34:192–200
Manley GT, Binder DK, Papadopoulos MC, Verkman AS (2004) New insights into water transport and edema in the central nervous system from phenotype analysis of aquaporin-4 null mice. Neuroscience 129:983–991
Stamer WD, Bok D, Hu J, Jaffe GJ, McKay BS (2003) Aquaporin-1 channels in human retinal pigment epithelium: role in transepithelial water movement. Invest Ophthalmol Vis Sci 44:2803–2808
Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP (1998) Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Müller cells and fibrous astrocytes. J Neurosci 18:2506–2519
Bialek S, Miller SS (1994) K+ and Cl− transport mechanisms in bovine pigment epithelium that could modulate subretinal space volume and composition. J Physiol (Lond) 475:401–417
Newman EA, Reichenbach A (1996) The Müller cell: a functional element of the retina. Trends Neurosci 19:307–312
Kofuji P, Biedermann B, Siddharthan V, Raap M, Iandiev I, Milenkovic I, Thomzig A, Veh RW, Bringmann A, Reichenbach A (2002) Kir potassium channel subunit expression in retinal glial cells: implications for spatial potassium buffering. Glia 39:292–303
Iandiev I, Tenckhoff S, Pannicke T, Biedermann B, Hollborn M, Wiedemann P, Reichenbach A, Bringmann A (2006) Differential regulation of Kir4.1 and Kir2.1 expression in the ischemic rat retina. Neurosci Lett 396:97–101
Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P, Wolburg H, Reichenbach A, Bringmann A (2004) A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci 26:493–502
Pannicke T, Uckermann O, Iandiev I, Biedermann B, Wiedemann P, Perlman I, Reichenbach A, Bringmann A (2005) Altered membrane physiology in Müller glial cells after transient ischemia of the rat retina. Glia 50:1–11
Pannicke T, Uckermann O, Iandiev I, Wiedemann P, Reichenbach A, Bringmann A (2005) Ocular inflammation alters swelling and membrane characteristics of rat Müller glial cells. J Neuroimmunol 161:145–154
Pannicke T, Iandiev I, Wurm A, Uckermann O, vom Hagen F, Reichenbach A, Wiedemann P, Hammes H-P, Bringmann A (2006) Diabetes alters osmotic-swelling characteristics and membrane conductance of glial cells in rat retina. Diabetes 55:633–639
Iandiev I, Uckermann O, Pannicke T, Wurm A, Tenckhoff S, Pietsch UC, Reichenbach A, Wiedemann P, Bringmann A, Uhlmann S (2006) Glial cell reactivity in a porcine model of retinal detachment. Invest Ophthalmol Vis Sci 47:2161–2171
Wurm A, Pannicke T, Iandiev I, Bühner E, Pietsch U-C, Reichenbach A, Wiedemann P, Uhlmann S, Bringmann A (2006) Changes in membrane conductance play a pathogenic role in osmotic glial cell swelling in detached retinas. Am J Pathol 169:1990–1998
Dalloz C, Sarig R, Fort P, Yaffe D, Bordais A, Pannicke T, Grosche J, Mornet D, Reichenbach A, Sahel J, Nudel U, Rendon A (2003) Targeted inactivation of dystrophin gene product Dp71: phenotypic impact in mouse retina. Hum Mol Genet 12:1543–1554
Bringmann A, Pannicke T, Uhlmann S, Kohen L, Wiedemann P, Reichenbach A (2002) Membrane conductance of Müller glial cells in proliferative diabetic retinopathy. Can J Ophthalmol 37:221–227
Wurm A, Pannicke T, Iandiev I, Wiedemann P, Reichenbach A, Bringmann A (2006) The developmental expression of K+ channels in retinal glial cells is associated with a decrease of osmotic cell swelling. Glia 54:411–423
Uckermann O, Kutzera F, Wolf A, Pannicke T, Reichenbach A, Wiedemann P, Wolf S, Bringmann A (2005) The glucocorticoid triamcinolone acetonide inhibits osmotic swelling of retinal glial cells via stimulation of endogenous adenosine signaling. J Pharmacol Exp Ther 315:1036–1045
Kowluru RA, Kennedy A (2001) Therapeutic potential of anti-oxidants and diabetic retinopathy. Expert Opin Investig Drugs 10:1665–1676
Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, Adamis AP (2002) Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-α suppression. FASEB J 16:438–440
Santilli F, Cipollone F, Mezzetti A, Chiarelli F (2004) The role of nitric oxide in the development of diabetic angiopathy. Horm Metab Res 36:319–335
Baydas G, Tuzcu M, Yasar A, Baydas B (2004) Early changes in glial reactivity and lipid peroxidation in diabetic rat retina: effects of melatonin. Acta Diabetol 41:123–128
Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, Kern TS, Adamis AP (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18:1450–1452
Gerhardinger C, Costa MB, Coulombe MC, Toth I, Hoehn T, Grosu P (2005) Expression of acute-phase response proteins in retinal Müller cells in diabetes. Invest Ophthalmol Vis Sci 46:349–357
Leal EC, Santiago AR, Ambrosio AF (2005) Old and new drug targets in diabetic retinopathy: from biochemical changes to inflammation and neurodegeneration. Curr Drug Targets CNS Neurol Disord 4:421–434
Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158:147–152
Du Y, Sarthy VP, Kern TS (2004) Interaction between NO and COX pathways in retinal cells exposed to elevated glucose and retina of diabetic rats. Am J Physiol 287:R735–R741
Stefánsson E, Wilson CA, Lightman SL, Kuwabara T, Palestine AG, Wagner HG (1987) Quantitative measurements of retinal edema by specific gravity determinations. Invest Ophthalmol Vis Sci 28:1281–1289
Szabo ME, Droy-Lefaix MT, Doly M, Carré C, Braquet P (1991) Ischemia and reperfusion-induced histologic changes in the rat retina. Invest Ophthalmol Vis Sci 32:1471–1478
Davidge ST, Baker PN, Laughlin MK, Roberts JM (1995) Nitric oxide produced by endothelial cells increases production of eicosanoids through activation of prostaglandin H synthase. Circ Res 77:274–283
Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ (1996) Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci USA 93:15069–15074
Rehncrona S, Westerberg E, Akesson B, Siesjö BK (1982) Brain cortical fatty acid and phospholipids during and following complete and severe incomplete ischemia. J Neurochem 38:84–93
Yoshida S, Ikeda M, Busto R, Santiso M, Martinez E, Ginsberg M (1986) Cerebral phosphoinositide, triacylglycerol and energy metabolism in reversible ischemia: origin and fate of free fatty acids. J Neurochem 47:744–757
Abe K, Kogure K, Yamamoto H, Imazawa M, Miyamoto K (1987) Mechanism of arachidonic acid liberation during ischemia in gerbil cerebral cortex. J Neurochem 48:503–509
Umemura A, Mabe H, Nagai H, Sugino F (1992) Action of phospholipase A2 and C on free fatty acid release during complete ischemia in rat neocortex. Effect of phospholipase C inhibitor and N-methyl-D-aspartate antagonist. J Neurosurg 76:648–651
Asano T, Shigeno T, Johshita H, Usui M, Hanamura T (1987) A novel concept on the pathogenetic mechanism underlying ischaemic brain oedema: relevance of free radicals and eicosanoids. Acta Neurochir Suppl (Wien) 41:85–96
Chan PH, Fishman RA, Caronna J, Schmidley JW, Prioleau G, Lee J (1983) Induction of brain edema following intracerebral injection of arachidonic acid. Ann Neurol 13:625–632
Wahl M, Schilling L, Unterberg A, Baethmann A (1993) Mediators of vascular and parenchymal mechanisms in secondary brain damage. Acta Neurochir Suppl (Wien) 57:64–72
Staub F, Winkler A, Peters J, Kempski O, Kachel V, Baethmann A (1994) Swelling, acidosis, and irreversible damage of glial cells from exposure to arachidonic acid in vitro. J Cereb Blood Flow Metab 14:1030–1039
Lees GJ (1991) Inhibition of sodium-potassium-ATPase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res Rev 16:283–380
Owada S, Larsson O, Arkhammar P, Katz AI, Chibalin AV, Berggren PO, Bertorello AM (1999) Glucose decreases Na,K-ATPase activity in pancreatic ß-cells: an effect mediated via Ca2+-independent phospholipase A2 and protein kinase C-dependent phosphorylation of the α-subunit. J Biol Chem 274:2000–2008
Uckermann O, Wolf A, Kutzera F, Kalisch F, Beck-Sickinger AG, Wiedemann P, Reichenbach A, Bringmann A (2006) Glutamate release by neurons evokes a purinergic inhibitory mechanism of osmotic glial cell swelling in the rat retina: activation by neuropeptide Y. J Neurosci Res 83:538–550
Bringmann A, Skatchkov SN, Biedermann B, Faude F, Reichenbach A (1998) Alterations of potassium channel activity in retinal Müller glial cells induced by arachidonic acid. Neuroscience 86:1291–1306
Chao TI, Henke A, Reichelt W, Eberhardt W, Reinhardt-Maelicke S, Reichenbach A (1994) Three distinct types of voltage-dependent K+ channels are expressed by Müller (glial) cells of the rabbit retina. Pflügers Arch 426:51–60
Francke M, Pannicke T, Biedermann B, Faude F, Wiedemann P, Reichenbach A, Reichelt W (1997) Loss of inwardly rectifying potassium currents by human retinal glial cells in diseases of the eye. Glia 20:210–218
Bringmann A, Francke M, Pannicke T, Biedermann B, Faude F, Enzmann V, Wiedemann P, Reichelt W, Reichenbach A (1999) Human Müller glial cells: altered potassium channel activity in proliferative vitreoretinopathy. Invest Ophthalmol Vis Sci 40:3316–3323
Sanchez-Olea R, Morales-Mulia M, Moran J, Pasantes-Morales H (1995) Inhibition by polyunsaturated fatty acids of cell volume regulation and osmolyte fluxes in astrocytes. Am J Physiol 269:C96–C102
Aiello LP (2002) The potential role of PKC beta in diabetic retinopathy and macular edema. Surv Ophthalmol 47:S263–S269
Sakamoto T, Miyazaki M, Hisatomi T, Nakamura T, Ueno A, Itaya K, Ishibashi T (2002) Triamcinolone-assisted pars plana vitrectomy improves the surgical procedures and decreases the postoperative blood-ocular barrier breakdown. Graefes Arch Clin Exp Ophthalmol 240:423–429
Edelman JL, Lutz D, Castro MR (2005) Corticosteroids inhibit VEGF-induced vascular leakage in a rabbit model of blood-retinal and blood-aqueous barrier breakdown. Exp Eye Res 80:249–258
Brooks HL Jr, Caballero S Jr, Newell CK, Steinmetz RL, Watson D, Segal MS, Harrison JK, Scott EW, Grant MB (2004) Vitreous levels of vascular endothelial growth factor and stromal-derived factor 1 in patients with diabetic retinopathy and cystoid macular edema before and after intraocular injection of triamcinolone. Arch Ophthalmol 122:1801–1807
Sears JE, Hoppe G (2005) Triamcinolone acetonide destabilizes VEGF mRNA in Müller cells under continuous cobalt stimulation. Invest Ophthalmol Vis Sci 46:4336–4341
Matsuda S, Gomi F, Oshima Y, Tohyama M, Tano Y (2005) Vascular endothelial growth factor reduced and connective tissue growth factor induced by triamcinolone in ARPE19 cells under oxidative stress. Invest Ophthalmol Vis Sci 46:1062–1068
Itakura H, Akiyama H, Hagimura N, Doi H, Tanaka T, Kishi S, Kurabayashi M (2006) Triamcinolone acetonide suppresses interleukin-1 beta-mediated increase in vascular endothelial growth factor expression in cultured rat Müller cells. Graefes Arch Clin Exp Ophthalmol 244:226–231
Maminishkis A, Jalickee S, Blaug SA, Rymer J, Yerxa BR, Peterson WM, Miller SS (2002) The P2Y2 receptor agonist INS37217 stimulates RPE fluid transport in vitro and retinal reattachment in rat. Invest Ophthalmol Vis Sci 43:3555–3566
Meyer CH, Hotta K, Peterson WM, Toth CA, Jaffe GJ (2002) The effects of INS37217, a P2Y2 receptor agonist, on experimental retinal detachment and electroretinogram in adult rabbits. Invest Ophthalmol Vis Sci 43:3567–3574
Skatchkov SN, Eaton MJ, Shuba YM, Kucheryavykh YV, Derst C, Veh RW, Wurm A, Iandiev I, Pannicke T, Bringmann A, Reichenbach A (2006) Tandem-pore domain potassium channels are functionally expressed in retinal (Müller) glial cells. Glia 53:266–276
Larsen AK, Osborne NN (1996) Involvement of adenosine in retinal ischemia. Studies on the rat. Invest Ophthalmol Vis Sci 37:2603–2611
Ghiardi GJ, Gidday JM, Roth S (1999) The purine nucleoside adenosine in retinal ischemia-reperfusion injury. Vision Res 39:2519–2535
Gardner TW, Antonetti DA, Barber AJ, LaNoue KF, Levison SW (2002) Diabetic retinopathy: more than meets the eye. Surv Ophthalmol 47:S253–S262
Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6:159–163
Acknowledgements
This work was supported by grants from the Deutsche Forschungsgemeinschaft (GRK 1097/1) and from the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) at the Faculty of Medicine of the University of Leipzig (C21, Z10).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reichenbach, A., Wurm, A., Pannicke, T. et al. Müller cells as players in retinal degeneration and edema. Graefe's Arch Clin Exp Ophthalmol 245, 627–636 (2007). https://doi.org/10.1007/s00417-006-0516-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00417-006-0516-y