
Abstract
Bladder cancer (BC) remains an aggressive disease with a poor prognosis, especially for patients with metastatic disease who have a limited median overall survival of 14 months. Urothelial carcinomas harbor frequent molecular dysregulations including recurrent mutations and copy number alteration, some of which could be potential therapeutic targets. However, no molecularly targeted agents have been approved to date for the treatment of advanced BC. Gaining new insights into the molecular landscape of BC will be critical to tailor future targeted agents for the treatment of advanced disease. The Cancer Genome Atlas (TCGA) project is cataloguing the genetic and epigenetic alterations responsible for cancer through the application of high-throughput genome analysis techniques. After the landmark paper profiling 131 patients was published in 2014, additional patients have been added with an updated TCGA analysis now including 412 patients. This chapter will review the previously described genomic alterations reported in the first manuscript and the new major highlights found in the expanded analyses recently published. The aim will be to review how this comprehensive integrated genomic analysis can inform the design of precision medicine targeted therapy for the treatment of advanced disease.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- The cancer genome atlas project
- Urothelial carcinoma
- Cancer genomics
- Whole-genome and RNA sequencing
- Molecular subtypes
- Mutational load
- Molecular biomarkers
- Molecular therapeutic targets
1 Introduction
Bladder cancer (BC) is the ninth most common tumor worldwide, with an incidence of more than 330,000 new cases each year and an annual mortality rate of 130,000 [1]. Urothelial carcinoma (UC) accounts for nearly 90% of bladder cancers. It is associated with tobacco smoking in 65–70% of cases and is three times more prevalent in men than in women. At the initial diagnosis of BC, 30% of cases present as muscle-invasive bladder cancer (MIBC), with approximately one-third of them having distant metastases at presentation. Among patients who undergo radical cystectomy, around 25% will have pathologic lymph node metastasis at the time of surgery [2]. Despite the improvements seen during the last decade in terms of both surgical quality and systemic therapy, BC remains an aggressive disease with a poor prognosis, especially for patients with locally advanced or metastatic disease who have a reduced median overall survival (OS) of around 14 months [3, 4]. Therefore, there is an important unmet need for effective anticancer treatment in order to improve outcome and survival for patients with advanced BC.
The first focused molecular analyses of patients with BC revealed that urothelial carcinomas harbor frequent molecular alterations including recurrent mutations and multiple regions of somatic copy number alteration (CNA), some of which could be potential therapeutic targets [5–8]. Moreover, it has been shown that MIBC and non-MIBC (NMIBC) each have a distinctive molecular landscape, where low-grade NMIBC is characterized by a high incidence of FGFR3 mutations and high-grade MIBC has a higher frequency of many types of mutations [9, 10]. However, no molecularly targeted agents have so far been approved for the treatment of advanced BC. Recently, pembrolizumab, an immune checkpoint inhibitor targeting the programmed cell death-1 (PD-1) receptor, showed for the first time in history an improvement in OS compared to investigator-choice standard chemotherapy (paclitaxel, docetaxel, vinflunine) as second or third line for patients with advanced UC progressing to platinum-based chemotherapy [11]. However, the population recruited for PD-1 inhibitors studies in BC have not been selected according to any specific molecular findings and programmed cell death ligand-1 (PD-L1) expression has not found to be a predictive biomarker of treatment benefit. Gaining new insights into the disease biology and the molecular landscape of BC will therefore be critical to molecularly tailor future targeted agents for the treatment of advanced disease.
The Cancer Genome Atlas (TCGA) project is a collaboration that begun in 2005, between the National Cancer Institute and the National Human Genome Research Institute with the purpose of cataloguing the genetic and epigenetic alterations responsible for cancer through the application of high-throughput genome analysis techniques, including large-scale genome sequencing and bioinformatics [12]. The final goal of TCGA is to deepen our understanding of the molecular basis of cancer in order to improve our ability to diagnose, treat, and prevent this lethal disease. Thus far, the TCGA has generated comprehensive, multidimensional maps of the key genomic alterations in 33 types of cancer, including urothelial carcinomas. The TCGA dataset, comprising more than two petabytes of genomic data of tumor tissue and matched normal tissues from more than 11,000 patients, has been made publically available and can be accessed through the TCGA Data Portal [13].
The first comprehensive molecular landscape study of BC by the TCGA was published in 2014 and integrated data from 131 patients with chemotherapy-naïve high-grade muscle-invasive urothelial bladder carcinomas (T2–T4a, N0, M0) undergoing surgical resection with either transurethral resection or radical cystectomy [14]. This first study included data on DNA copy number, somatic mutations, gene fusions, messenger RNA expression, protein expression, DNA methylation, transcript splice variants, viral integration and pathway alterations as well as a thorough description of four expression-based molecular subtypes of BC. In 2017, a second comprehensive TCGA study was published, analyzing an expanded cohort of 412 patients with either localized or metastatic chemotherapy-naïve muscle-invasive bladder carcinoma [15]. This second analysis is derived from a threefold increase in the number of tumors, thus improving the power to detect less frequent molecular abnormalities. Moreover, this updated study provides several new analyses that were not part of the first paper including mutation burden and neoantigen load, an integrated analysis of non-coding RNA and new analytic detection of pathogens in BC. Finally, it reports for the first time a robust correlation of mutation signature clusters and expression-based subtypes in a univariate and multivariate association with survival. This chapter will review the major highlights described in these two important TCGA studies on the molecular characterization of BC and how the findings can inform the design of precision medicine targeted therapy for the treatment of advanced disease.
2 Somatic DNA Alterations
The TCGA studies confirmed that MIBC is characterized by a high overall mutation rate, similar to that of lung cancer or melanoma. This is an important result since several studies have shown that mutation load is a biomarker of response to immune checkpoint inhibitors [16, 17]. In the first TCGA analysis, there were on average 302 exonic mutations, 204 segmental alterations in genomic copy number, and 22 genomic rearrangements per sample. Whole-exome sequencing (WES) targeted 186,260 exons in 18,091 genes and identified 39,312 somatic mutations (including 38,012 point mutations and 1138 insertions or deletions) [14]. These results were internally validated by a targeted re-sequencing of all significantly mutated genes and by a comparison with whole-genome sequencing (WGS) of 18 samples, with a validation rate of 99% [14]. WES of the 412 tumors included in the second TCGA analysis increased the number of targeted exons to 193,094 and the number of genes to 18,862. It identified 131,660 somatic mutations, including 128,772 single nucleotide variants, and 2888 insertions or deletions [15]. Many of the focal mutations involved genes previously known in bladder cancer such as TP53 (49%), PIK3CA (20%), RB1 (13%), FGFR3 (12%), ERCC2 (12%), and TSC1 (8%). In addition, several other genes that have not previously been reported as significantly mutated in BC were identified, such as MLL2 (27%), CDKN1A (14%), or STAG2 (11%) [14]. Similarly, the second TCGA study identified 58 genes mutated at a frequency statistically significantly higher than background, 34 of which had not been previously identified as significantly mutated in the first study, including KMT2C (18%), ATM (14%), and FAT1 (12%) [15].
The second TCGA study tried to detect processes contributing to the high mutation rate, using a Bayesian approach. It identified five differential mutation signatures (APOBEC-a and APOBEC-b, ERCC2, C > T at CpG, POLE), which correlated with different subsets of BC with different mutation burdens and mechanisms. Clustering of four of these five mutation signatures (APOBEC-a and APOBEC-b, ERCC2 and C > T at CpG) identified four mutational signature clusters, MSig1 to MSig4. Interestingly, the activity of an endogenous mutagen, the DNA cytidine deaminase APOBEC accounted for 67% of the overall mutations. The majority of APOBEC-mediated mutations were clonal, suggesting that APOBEC activity is an early event in the carcinogenic development of BC. Moreover, the mutation signature cluster of high-APOBEC mutagenesis and high mutation burden (MSig1) was strongly associated with an improved OS (p = 1.4 × 10−4) with 75% of subjects alive 5 years after diagnosis of MIBC [15]. The authors hypothesize that the unusually good survival of this subset is due to the high mutation burden, boosting the host immune antitumor response. On the other hand, MSig2 cancers had the lowest mutation rate and the poorest 5-year survival (22%). The identification of APOBEC as the main driver of mutagenesis in BC is extremely relevant since a better understanding of its expression and activity could have a major benefit for selecting patients for clinical trials of immunotherapy.
The high mutation rate seen in BC as in lung cancer has been associated with smoking habit in several studies [18]. Moreover, several clinical trials in both BC and other advanced cancers have shown a relationship between smoking status and the relative benefit of immunotherapy and it has been postulated that this benefit is due to the high mutational load induced by smoking [11, 19]. Consequently, the TCGA has analyzed the correlation between smoking status and the presence of molecular alterations. In the first TCGA study, 72% of patients were current or past smokers. However, there was no statistically significant association between smoking status and the mutational load, frequency of mutations in any significantly mutated gene, occurrence of CNAs, or expression subtype [14]. This misbalance may reflect the fact that not all mutations might have the same potential to act as neoepitopes and consequently neoantigen load could be a more robust predictive biomarker for immunotherapy than mutational load. Hence, the second TCGA study included a neoantigen prediction analysis by enumerating peptides bearing somatic mutations and assessing their binding against the patient’s inferred HLA type. The effect of the neoantigen load was then analyzed in a univariate and multivariate analysis. Interestingly, neoantigen load did strongly correlate with mutation burden and was associated with improved survival (p = 5.2 × 10−4). On multivariate analysis, neoantigen load remained an independent predictor of survival after adjusting for age, tumor stage, histology, and node status (p = 8 × 10−4) [15].
3 MRNA Expression and Molecular Subtypes
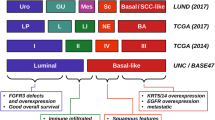
Analysis of RNA-sequencing data from the 129 tumors included in the first TCGA study identified the well-known distinction between luminal and basal subtypes of BC [20–22], and divided them into four clusters, clusters I–II being luminal and clusters III–IV being basal [14]. Luminal cluster I (or papillary-like cluster) was enriched in tumors with papillary morphology (p = 0.0002), FGFR3 mutations (p = 0.0007), FGFR3 copy number gain (p = 0.04), and elevated FGFR3 expression (p < 0.0001). Consequently, the authors hypothesized that tumors with cluster I expression and/or FGFR3 alterations could benefit from FGFR inhibitors. Moreover, luminal clusters I and II showed high protein expression of HER2, comparable to those found in TCGA HER2-positive breast cancers [23], and an elevated estrogen receptor beta signaling, flagging them as potential responders to hormone therapy and HER2 inhibitors. Luminal clusters I and II also showed characteristics similar to those of luminal A breast cancer, with high mRNA and protein expression of luminal breast differentiation markers, including GATA3 and FOXA1. On the other hand, the signature of basal cluster III (or basal/squamous-like cluster) showed molecular features similar to that of basal-like breast cancers and squamous cell cancers of the head and neck and lung [23, 24], such as high expression of KRT14, KRT5, KRT6A, and EGFR. These distinctive expression-based clusters were then externally validated using an external data set of 308 MIBC tumor samples from a prior study which confirmed the same four distinctive cluster subgroups [14, 22].
A similar RNA-sequencing analysis was undertaken on the second TCGA study in the expanded cohort of 412 patients, recapitulating the same two major luminal and basal transcriptional subtypes identified in the first study [15]. Moreover, this expanded analysis provided further discrimination within these subgroups which led to a re-cataloguing of the different subtypes into five entities: luminal-papillary (n = 142), luminal-infiltrated (n = 78), luminal (n = 26), basal-squamous (n = 142), and to the identification of a novel neuronal subtype (n = 20). As described before, the luminal-papillary cluster was enriched with papillary tumors (58% vs. 20% in the other subtypes; p < 10−13) and with lower-stage T1 or T2 tumors (55% vs. 23%, p < 10−8). Similarly, it was characterized by frequent FGFR3 alterations (44%), either mutations, amplification, overexpression, or fusions, which indicates that many tumors of the luminal-papillary cluster tumors might have developed from a precursor non-MIBC. This subtype was also characterized by a major loss of DNA methylation and included cases that were almost all node-negative, from younger patients (median age 61 vs. 69; p < 4 × 10−3), and had better survival (p < 0.05) [15].
The luminal-infiltrated subtype, on the other hand, was distinguished from other luminal subtypes by a strong expression of smooth muscle and myofibroblast gene signatures and a p53-like expression which has previously been associated with chemoresistance [21]. The luminal-infiltrated subtype correlated with the prior luminal cluster II, which has been reported to benefit from anti-PDL1 inhibitor atezolizumab [16, 17]. The basal-squamous subtype was characterized by high expression of basal and stem-like markers (KRT5, KRT6A, KRT14) and squamous differentiation markers (TGM1, DSC3, PI3) and included 82% of tumors containing squamous cell features (p < 10−11). This subtype was enriched in TP53 mutations (p = 5 × 10−3) and had a high carcinoma-in situ (CIS) expression signature score, indicating that they may have originated from bladder basal cells through CIS lesions. The basal-squamous subtype also showed the strongest immune expression signature, including T cell markers and inflammation genes, indicating the presence of lymphocytic infiltrates. Interestingly, the basal-squamous subtype correlated with the prior clusters III and IV, which were the clusters showing greatest benefit from anti-PDL1 inhibitor atezolizumab after cluster II [16, 17].
The neuronal subtype, finally, included three of four histologic small cell neuroendocrine tumors found in the whole cohort, but showed no apparent histologic distinction from other types of MIBC in the majority of cases (85%). This subtype was characterized by a high expression of many neuronal differentiation genes, as well as neuroendocrine markers. Half of the samples had mutations in both TP53 and RB1, which is a genetic hallmark of small cell neuroendocrine cancer, regardless of the primary origin. This subtype was the most infrequent cluster (5%) and had the highest CIS expression signature score, also indicating it may have originated from CIS lesions. Importantly, the neuronal subtype had the poorest survival compared to the other four subgroups (p = 1.4 × 10−3) in keeping with the known aggressive behavior of neuroendocrine BCs [15].
The identification of these mRNA expression subtypes as five distinctive molecular entities is a critical discovery that will promote gaining new insights into the specific biology of each subtype, a critical requisite to using molecular correlates to tailor future tumor-personalized targeted therapies.
4 Pathway Analysis and Therapeutic Targeting
The information obtained from the somatic mutation analysis and copy number data was integrated in order to identify the more frequently altered pathways and potential targets amenable for therapeutic intervention. Importantly, most of the canonical signaling pathways that were consistently altered in both TCGA studies provide significant opportunities for a molecular-targeted therapeutic blockade [14, 15]. Integrated analysis revealed three frequently dysregulated pathways: p53/cell cycle regulation (89%), RTK/RAS/PI3 K signaling (71%), and chromatin remodeling pathways with alterations in the histone-modifying genes in 52% of cases, and in the nucleosome remodeling complex in 26% [15]. p53/cell cycle alterations included TP53 mutations in 48% of cases, MDM2 amplification (copy number >4) in 6%, and MDM2 overexpression (>twofold above the median) in 19%, with strong mutual exclusivity between these events (p < 10−16) [15]. Mutations in chromatin-modifying and regulatory genes were common, with 10 such genes having a mutation frequency greater than 5%, and with 66% of samples showing a mutation in one or more genes. Of note, 10 of the 39 significantly mutated genes with mutation frequency >5% were either chromatin-modifying or chromatin-regulatory genes, such as KDM6A (a histone de-methylase), histone methyltransferases (KMT2A, KMT2C, KMT2D), or histone acetylases (CREBBP, EP300, KANSL1) [15]. Mutations in these ten genes were predominantly inactivating, which suggests that they are functionally relevant. Taken together, this data indicate that dysregulation of gene expression mediated by alterations in chromatin-regulatory genes is a driver of BC development [15]. Moreover, the presence of abnormalities in chromatin-modifying enzymes identifies a subset of BC patients who could benefit from drugs targeting chromatin modifications, such as agents that bind acetyl-lysine binding motifs (bromodomains) [25, 26].
PI3K signaling alterations included activating point mutations in PIK3CA (22%), which could potentially benefit from PI3K inhibitors; mutations or deletions of TSC1 (8%), which could potentially benefit from mTOR inhibitors [15] and overexpression of AKT3 (10%), potentially responsive to AKT inhibitors [14]. As mentioned earlier, FGFR3 pathway was also frequently altered, including mutations (14%) and fusions (2%), all potentially responsive to FGFR inhibitors [15]. FGFR3 mutations were more frequent in lower-stage tumors (21% in T1–T2 vs. 10% in T3–T4; p = 0.003) and correlated with better survival (p = 0.04) [15]. Other frequent altered pathways with therapeutic implications included amplification of EGFR (9%, potentially responsive to EGFR inhibitors), mutations of HER3 (6%, potentially sensitive to HER inhibitors), and mutation or amplification of HER2 (9%, potentially sensitive to HER2 inhibitors) [14]. Of note, the frequency of HER2 alterations was comparable to that of the TCGA HER2-positive breast cancers, albeit with less amplifications and more mutations [23]. DNA repair pathways also showed frequent genomic alterations (16%) including mutations in ATM (14%) and ERCC2 (9%), and deletions in RAD51B (2%) [15], and could indicate responsiveness to platinum agents or PARP inhibitors.
5 Other Significant Findings
-
Viral DNA integration: RNA-sequencing and WGS data were used to identify evidence of viral DNA genomic integration due to infection by several virus, such as cytomegalovirus (CMV), BK polyomavirus, human papilloma virus (HPV), or human herpes virus (HHV) [14, 15]. The first analysis identified viral DNAs in 7 of 122 tumors (6%), and viral transcripts in 5 of 122 (4%) [14]. Taking both studies together, there was evidence of infection by CMV (n = 3), HPV (n = 11), HHV4 (n = 6), HHV5 (n = 6), and polyomavirus (n = 1), indicating that viral infection might have a role in the development of a small subset of BC [14, 15].
-
Non-coding RNAs (lncRNAs and miRNAs) subdivide mRNA expression subtypes: The second TCGA study provided for the first time an integrated analysis of non-coding RNA, including long non-coding RNA (lncRNA) and microRNA (miRNA). Clustering by lncRNA and miRNA expression was concordant with the mRNA subtypes while providing further discrimination within them, with differential epithelial-mesenchymal transition (EMT), CIS scores, histologic features, and survival [15]. For example, lncRNA cluster 3 was a subset of the luminal-papillary subtype with a better survival. It was characterized by a low frequency of TP53 mutations and high frequency of FGRF3 mutations/fusions and was associated with papillary histology, node-negative disease, or low T-stage/node-positive cases. Similarly, the four miRNA clusters were concordant with subtypes for mRNA (p = 2.4 × 10−52), lncRNA (p = 1.5 × 10−45), hypomethylation (p = 4.5 × 10−30) and were associated with histological subtype (papillary vs. non-papillary), combined T-stage/node+, node positive/negative, and CIS gene sets [15]. miRNA subtype 3 was enriched in lncRNA 3, and had better survival, consistent with low EMT scores. On the other hand, miRNA subtype 4 and 2 contained most of the basal/squamous mRNA subtype, and had relatively poor survival, consistent with relatively high EMT scores.
-
Proteomic data subtypes: The second TCGA study also included for the first time an unsupervised clustering using reverse phase protein array (RPPA) proteomic data analysis. This analysis identified five robust clusters with differential protein expression profiles, pathway activities, and overall survival (p = 0.019), several of them displaying alterations suitable for therapeutic intervention [15]. Proteomic cluster C1 (epithelial/papillary) was associated with low EMT scores, papillary differentiation, and improved survival. Cluster C2 (epithelial/intermediate) had a more intermediate outcome profile. Both clusters C1 and C2 are enriched in HER2 expression levels, indicating they might benefit from HER2 inhibitors [15]. Cluster C3 (proliferative/low signaling) had a high cell cycle pathway score, low PI3K and mTOR pathway signaling, but high EGFR expression levels, making it a potential candidate for EGFR-directed therapies. Clusters C4 and C5 had higher EMT pathway scores, of which cluster C4 (EMT/hormone signaling) had the worst outcome and was associated with non-papillary samples and pathologic advanced stage 3 and 4.
6 Survival Univariate and Multivariate Analysis
The second TCGA analysis assessed the correlation of 101 clinical and molecular variables with overall survival in a univariate and multivariate log-rank test. Of the 101 covariates analyzed by univariate log-rank tests, 13 were selected for multivariate Cox regression analysis. LASSO regression analysis was chosen to fit a multivariate model. The best-survival subtype was set as the reference variable for each of mRNA, lncRNA, miRNA, and MSig.
The variables with largest coefficients were AJCC stages III and IV, the mRNA neuronal and luminal subtypes, the low mutation rate MSig 2, and miRNA subtype 4, which is a subset of basal-squamous cases, and KLF4 regulon activity, all of which were associated with poorer survival. The mRNA luminal-infiltrated subtype, age, GATA3 regulon activity, and MSig4 were retained with smaller coefficients. The ranking order from poorer to better survival were: mRNA neuronal subtype, AJCC stage IV, MSig2, miRNA subtype 4, AJCC stage III, mRNA luminal subtype, KLF4 regulon, mRNA luminal-infiltrated, age, GATA3 regulon, and MSig 4.15. Tumor stages III and IV correlated with worse survival and were associated with a 45% and 112% higher risk of death, respectively, than stage I and II tumors combined. Mutational signature cluster MSig1 showed a 47% lower risk of death than MSig4, while cluster MSig2 had a 38% higher risk of death. The mRNA neuronal subtype had the worse survival outcome and had a 63% higher risk of death than the basal/squamous subtype. This latter showed no significant risk differences with the three luminal subtypes. Finally, miRNA cluster 4, a poorer-survival subset of the basal/squamous mRNA subtype as mentioned earlier, had a 36% higher risk of death than miRNA subtype 1 [5].
7 Subtype-Stratified Potential Treatments
By integrating mRNA subtype classification, altered pathways data, EMT and CIS signatures, and immune infiltrate analyses, the second study of the TCGA proposed potential specific therapeutic recommendations for each subtype of MIBC, depending on their specific molecular landscape, that can be tested in prospective clinical trials:
-
Luminal-papillary subtype: It is characterized by FGFR3 mutation, fusions and/or amplification, papillary histology and a very low CIS score. This subtype can be assessed as having relatively low risk for progression, and when diagnosed as localized disease, it might not need to be treated with neoadjuvant chemotherapy (NAC). On the other hand, FGFR3 tyrosine kinase inhibitors should be tested in patients with metastatic disease.
-
Luminal-infiltrated subtype: It has high expression of both EMT and myofibroblast markers. It is enriched on immune markers such as PD-L1 and CTLA4, which is in keeping with the fact this subtype has been reported to benefit from anti-PDL1 inhibitor atezolizumab [16, 17], as mentioned earlier. Thus, patients presenting with this molecular subtype could not only benefit from PD-L1 inhibitors on the metastatic setting but also in both the neoadjuvant and postoperative adjuvant settings. Neoadjuvant cisplatin-based chemotherapy could also be used but is expected to produce infrequent tumor responses, as this subtype has been associated with chemoresistance.
-
Luminal subtype: It is characterized by a very high expression of luminal markers (uroplakins). Because this subtype had not been previously described as a separate entity, the potential therapies are not well defined. Consequently, it could benefit from NAC for localized disease and/or therapies targeting each specific molecular alteration.
-
Basal-squamous subtype: It is characterized by female enrichment, squamous differentiation, and basal keratin expression. This subtype has the strongest immune expression signature (indicating the presence of lymphocytic infiltrates) and is enriched on immune markers such as PD-L1 and CTLA4. This is illustrated by the fact that this subtype also showed benefit in the atezolizumab trials [16, 17]. This subtype could benefit from NAC for localized disease and from immune checkpoint inhibitors for the metastatic setting.
-
Neuronal subtype: It is characterized by expression of both neuroendocrine and neuronal markers and a high cell cycle signature indicating a high proliferative status. Similarly to small cell neuroendocrine tumors originating from other organs, etoposide-platinum combination chemotherapy should be the preferred option, in both the neoadjuvant and metastatic setting.
Finally, the authors suggest that this molecularly driven therapeutic sub-classification should be prospectively validated in future clinical trials, as well as tested retrospectively in ongoing or completed clinical trials that assessed similar treatment strategies.
8 Conclusions
In the past 30 years, the treatment of advanced bladder cancer has barely moved beyond platinum-based combination chemotherapy and surgery. The recent approval of the immune checkpoint inhibitor pembrolizumab as second or third line in the metastatic setting, after showing an improved overall survival, has been the major breakthrough revolution in bladder cancer therapy of the last decades [11]. However, this immune therapy, in the same way as classical platinum chemotherapy, is administered to all patients in an unselected manner and no robust predictive biomarkers of response have been identified. Consequently, a significant proportion of patients will never benefit from these therapies but we are unable to predict that in advance given the lack of clinical or molecular biomarkers. That is why the main objective of the TCGA studies is to provide a comprehensive molecular characterization of the genetic landscape of MIBC in order to improve our ability to personalize the therapy of this lethal disease. The two TCGA studies on urothelial cancer have shown that the molecular landscape of this disease is rich in several genetic and epigenetic alterations and that up to two-thirds of patients have potentially actionable mutations. The first TCGA analysis integrated genomic data from 131 MIBC samples and showed several relevant findings: a high somatic mutation rate, similar to that of lung cancer and melanoma; statistically significant recurrent mutations in 32 genes; four mRNA expression subtypes showing a distinctive molecular landscape; and potential therapeutic targets in 69% of the samples [14]. The second TCGA study expanded the cohort to 412 samples and demonstrated several other relevant findings: The high mutational load in BC is mainly driven by the APOBEC-mediated mutagenesis; tumors with high-APOBEC and high mutation load had an extraordinary improved survival; mRNA clustering identified a novel neuronal subtype with small cell neuroendocrine features and poor survival. Finally, the integration of mRNA subtype classification, altered pathways data, EMT and CIS signatures, and immune infiltrate analyses provided one the most important findings of this second TCGA study: the identification of five expression-based distinctive molecular subtypes with different developmental mechanisms and distinct therapeutic potential [15]. Although this molecular sub-classification still needs to be prospectively validated in future clinical trials, it opens a massive window of opportunities into personalized treatment of bladder cancer.
References
Sobin LH, Gospodariwicz M, Wittekind C (eds) (2009) TNM classification of malignant tumors. UICC International Union Against Cancer, 7th edn. Wiley-Blackwell, pp 262–265
Witjes JA, Compérat E, Cowan NC et al (2014) EAU guidelines on muscle-invasive and metastatic bladder cancer: summary of the 2013 guidelines. Eur Urol 65(4):778–792
Loehrer PJ, Einhorn LH, Elson PJ et al (1992) A randomized comparison of cisplatin alone or in combination with methotrexate, vinblastine, and doxorubicin in patients with metastatic urothelial carcinoma: a cooperative group study. J Clin Oncol 10(7):1066–1073
von der Maase H, Sengelov L, Roberts JT et al (2005) Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol 23(21):4602–4608
Goebell PJ, Knowles MA (2010) Bladder cancer or bladder cancers? Genetically distinct malignant conditions of the urothelium. Urol Oncol 28(4):409–428
Forbes SA, Bindal N, Bamford S et al (2011) COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in cancer. Nucleic Acids Res 39(Database issue):D945–950
Lindgren D, Sjödahl G, Lauss M et al (2012) Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma. PLoS ONE 7(6):e38863
Hurst CD, Platt FM, Taylor CF, Knowles MA (2012) Novel tumor subgroups of urothelial carcinoma of the bladder defined by integrated genomic analysis. Clin Cancer Res 18(21):5865–5877
van Rhijn BW, Lurkin I, Radvanyi F, Kirkels WJ, van der Kwast TH, Zwarthoff EC (2001) The fibroblast growth factor receptor 3 (FGFR3) mutation is a strong indicator of superficial bladder cancer with low recurrence rate. Cancer Res 61(4):1265–1268
Knowles MA, Hurst CD (2015) Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer 15(1):25–41
Bellmunt J, de Wit R, Vaughn DJ et al (2017) Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med
https://cancergenome.nih.gov/. Last accessed June 2017
https://tcga-data.nci.nih.gov/docs/publications/tcga/. Last accessed June 2017
Network CGAR (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507(7492):315–322
Robertson G, Kim J, Al-Ahmadie H et al (2017) Comprehensive molecular characterization of muscle-invasive urothelial carcinoma. Cell
Rosenberg JE, Hoffman-Censits J, Powles T et al (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–1920
Balar AV, Galsky MD, Rosenberg JE et al (2016) Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet
Lawrence MS, Stojanov P, Polak P et al (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499(7457):214–218
Yang Y, Pang Z, Ding N et al (2016) The efficacy and potential predictive factors of PD-1/PD-L1 blockades in epithelial carcinoma patients: a systematic review and meta analysis. Oncotarget 7(45):74350–74361
Choi W, Czerniak B, Ochoa A et al (2014) Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat Rev Urol 11(7):400–410
Choi W, Porten S, Kim S et al (2014) Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25(2):152–165
Sjödahl G, Lauss M, Lövgren K et al (2012) A molecular taxonomy for urothelial carcinoma. Clin Cancer Res 18(12):3377–3386
Network CGA (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Network CGAR (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489(7417):519–525
Filippakopoulos P, Qi J, Picaud S et al (2010) Selective inhibition of BET bromodomains. Nature 468(7327):1067–1073
Wu X, Liu D, Tao D et al (2016) BRD4 regulates EZH2 transcription through upregulation of C-MYC and represents a novel therapeutic target in bladder cancer. Mol Cancer Ther 15(5):1029–1042
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Rodriguez-Vida, A., Lerner, S.P., Bellmunt, J. (2018). The Cancer Genome Atlas Project in Bladder Cancer. In: Daneshmand, S., Chan, K. (eds) Genitourinary Cancers . Cancer Treatment and Research, vol 175. Springer, Cham. https://doi.org/10.1007/978-3-319-93339-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-93339-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-93338-2
Online ISBN: 978-3-319-93339-9
eBook Packages: MedicineMedicine (R0)