
Abstract
It has been long understood that bladder cancer harbors specific genomic and copy number alterations. More recently, increased access by way of availability and reduced costs of next-generation sequencing has resulted in an improved understanding of the molecular biology of bladder cancer. Data from genomic sequencing yield prognostic information that may aid in the decision to pursue aggressive therapies, as well as predictive information regarding response to treatments such as intravesical Bacillus Calmette-Guerin (BCG) and systemic chemotherapy. Genomic sequencing also provides data on targetable alterations that can bring novel agents from the laboratory to clinical trials. As the material for deep and broad genomic sequencing is expanded to include both plasma and urine, new avenues to screen and monitor disease are becoming available. While current guidelines recommend sequencing only for patients with advanced and metastatic disease, currently active clinical trials may expand these indications in the near future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Despite being a common cancer with a diverse and often unpredictable clinical course, bladder cancer diagnosis and management are still largely based on histologic assessment without tumor genomic profiling or routine molecular characterization. By contrast, in other malignancies, assessment for alterations known to have clinical impact on prognosis or treatment selection is guideline-recommended. For example, the National Comprehensive Cancer Network (NCCN) recommends that patients with non-small cell lung cancer undergo a panel of molecular tests to evaluate for the presence of alterations that are known to affect clinical outcomes [1]. The potential benefits of molecular characterization of malignancy include improved ability to convey prognosis to patients and their families, identify biomarkers predictive of treatment response, and identify actionable alterations for therapies, among others.
Over the last 10–15 years, there has been an influx of data that has advanced our understanding of the molecular biology of bladder cancer and has highlighted the potential utility of genomic sequencing for the diagnosis and management of patients with this disease. Bladder cancer is known to carry a significant mutational burden, akin to lung cancer and melanoma [2]. Genomic sequencing of tumors reveals a rich landscape of alterations. Some alterations are shared across grades and stages of the disease which suggest early events in tumorigenesis, while others are unique and provide insights into underlying disease biology. Further, there is significant interest in genomic sequencing to identify both prognostic and predictive biomarkers to the increasing armamentarium of local and systemic treatments for bladder cancer.
In this chapter, we review the literature supporting the role of genomic sequencing for the diagnosis and treatment of patients with bladder cancer. We first provide a primer on next-generation sequencing (NGS), including key concepts and limitations. We then review the data on sequencing for bladder cancer stratified by stage (i.e., muscle-invasive, non-muscle-invasive, and advanced/metastatic bladder cancer). We highlight the increasingly recognized importance of germline testing and address advanced approaches, such as liquid biopsy, that have the potential to radically change management in bladder cancer, especially in the adjuvant setting. Finally, we review the current guidelines and provide practical considerations in using genomic sequencing in the management of patients with bladder cancer.
Primer on Next-Generation Sequencing
Next-generation sequencing (NGS) refers to the process that reads the order of nucleic acids in DNA or RNA [3]. NGS is a significant improvement over prior methods (i.e., Sanger sequencing) in that reactions and analyses can be performed simultaneously, decreasing the time and cost of sequencing. Briefly, in DNA sequencing, the genetic material is first extracted, and a library is generated by fragmenting the DNA and adding specific adaptors. Sequencing can span from whole genome sequencing (WGS) and whole exome sequencing (WES) to more targeted panels consisting of a variable number of genes that are specific to certain pathologies or pathways. Sequencing can be performed on DNA from tumor tissue to identify somatic mutations or on normal tissues (e.g., white blood cells in a plasma sample or histologically normal tissue in a surgical pathology specimen) to identify germline mutations. The number of times a specific nucleotide is sequenced is called the coverage depth [4]. Unlike sequencing of normal tissue for germline alterations, the coverage depth required for somatic tissue sequencing is significantly greater in order to overcome contamination of the specimen by benign tissue (e.g., stromal components). Typical coverage for somatic sequencing is on the order of 1000× compared with germline sequencing coverage of around 30×. Further, mutations present in subclonal populations require higher coverage depth to detect these alterations with lower variant allele frequencies (VAF).
An important consideration in NGS is how the normal sample is derived to which the somatic tumor sequencing is compared [5]. Some assays use patient-matched normal samples, such as morphologically normal tissue like a benign lymph node in a radical cystectomy specimen or the white blood cells in a blood sample [6]. This strategy, compared with a reference genome, reduces the rates of false-positive somatic mutational calls from either germline mutations or clonal hematopoiesis of indeterminate potential.
NGS identifies multiple types of alterations, such as single nucleotide variants (SNVs), as well as structural changes (insertions and deletions) and chromosomal rearrangements (translocations, duplications, and deletions). Tumor mutational burden (TMB) and microsatellite instability (MSI) , both of which may correlate with response to treatment with immunotherapy, can also be derived from NGS data. These alterations can be prognostic and/or predictive of response to treatment. Some alterations are targetable with various therapies. Collectively, alterations that are predictive, prognostic, and/or targetable are considered clinically actionable. Publicly available databases, such as OncoKB, provide curated information regarding the actionability of various alterations in many different cancer types based on guidelines, active clinical trials, and published scientific literature [7].
While NGS has led to innumerable advances in oncology and urologic oncology, several limitations are notable and important to consider . First, known intra-tumoral heterogeneity in primary bladder tumors can result in missed mutations depending on the area sequenced. There is also the issue of depth of coverage for alterations of low VAF. Second, tumor sequencing is generally performed on the primary tumor; however, differences between the primary tumor and the metastases (i.e., inter-tumoral heterogeneity) are known to exist. These differences are the result of clonal evolution and may be promoted by intervening treatments [8]. The clinical implications of these discrepancies between the primary tumor and metastases are not yet fully understood. Future studies are required to determine when and which tissues should be sequenced to best inform treatment decisions and optimize clinical outcomes.
Muscle-Invasive Bladder Cancer
Muscle-invasive bladder cancer (MIBC) is the disease state most fully characterized by genomic sequencing. Efforts have focused on the actionable genomic landscape, driver alterations in divergent differentiation, and biomarkers of treatment sensitivity.
Molecular characterization of bladder cancer was launched by the publication of two seminal manuscripts from The Cancer Genome Atlas (TCGA) [9, 10]. The first publication reported on 131 tumors, and the follow-up publication reported on the multifaceted assessment of 412 MIBC tumors. Tumors assessed in the TCGA were all high-grade muscle-invasive tumors from chemotherapy-naïve patients. Pure urothelial carcinoma not otherwise specified (NOS) comprised the majority of samples; however, 52 tumors (13%) contained some element of variant histology. WES was performed to assess mutations (i.e., SNVs) and mutational signatures, while Affymetrix Genome-Wide Human SNP Array 6.0 (Thermo Fisher Scientific) was used to determine somatic copy number alterations (SCNAs) . RNA sequencing allowed for expression-based molecular subtyping, and proteomic analysis was also performed. TCGA provided several insights into the molecular biology of bladder cancer. First, they confirmed the relatively high rate of somatic mutations, similar to melanoma and lung cancer, that has been seen in other pan-cancer studies [11]. This has important clinical implications given the United States Food and Drug Administration (FDA) approval of pembrolizumab for tumors with TMB >10 mutations per megabase, regardless of the origin of malignancy . Second, using WES and unsupervised clustering of specific mutational data, the authors were able to identify multiple mutational signatures. The first are two apolipoprotein B mRNA editing enzyme, catalytic polypeptide (APOBEC)-like signatures, which collectively account for two-thirds of all SNVs in TCGA. Patients whose tumors demonstrate APOBEC signatures were noted to have higher TMB and better overall survival. Further, these mutations were clonal, suggesting that they occurred early in bladder cancer carcinogenesis. Several more recent studies have demonstrated that even within histologically normal urothelium, chromatin-modifying alterations are common and contribute to additional mutational burden [12, 13]. The second group of mutational signatures with clinical relevance involve ERCC2. ERCC2 is a helicase involved in nucleotide excision repair and is considered a DNA-damage response (DDR) gene. More specifically, ERCC2 unwinds DNA at sites of damage to allow for other proteins and enzymes to repair the damage. These mutational signatures offer insights into the pathogenesis of bladder cancer and highlight possible avenues for therapeutic intervention.
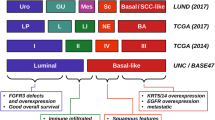
Another important finding from TCGA was the identification of RNA expression-based molecular subtypes, which have both prognostic and predictive potential. In general, these molecular subtypes paralleled those discovered in breast cancer tumors and included basal and luminal subtypes. While TCGA classified tumors into Clusters I–II (luminal) and III–IV (basal), other groups independently developed similar classification systems for muscle-invasive bladder tumors, and more recently, a consensus classification was proposed [14]. In general, luminal and basal tumors differ in appearance (papillary vs. nodular and flat), response to chemotherapy (less responsive vs. more responsive), and relative frequencies of various genomic alterations [15]. Despite the strengths and potential clinical utility of these classification systems, molecular subtype analysis has yet to be incorporated into routine clinical practice.
Predicting response to chemotherapy is an important clinical question and has been addressed in several studies from a genomics standpoint. Given the prevalence of ERCC2 mutational signatures from TCGA, the functional implications of ERCC2 alterations have been evaluated and demonstrate a correlation with response to cisplatin-based chemotherapy [16, 17]. In another study comparing primary MIBC and secondary MIBC that progressed from non-muscle-invasive bladder cancer (NMIBC), patients with secondary MIBC had fewer ERCC2 mutations, worse recurrence-free survival (RFS) and overall survival (OS) rates, and poorer response to neoadjuvant chemotherapy [18]. Identification of ERCC2 as a potential biomarker predictive of chemotherapy sensitivity has led to two clinical trials testing bladder preservation in genomically selected patients with specific alterations , mainly in DDR genes. The Alliance 031701 trial (NCT03609216) is evaluating bladder preservation in highly selected patients with certain DDR alterations who demonstrate a complete clinical response after dose-dense gemcitabine and cisplatin neoadjuvant chemotherapy. The RETAIN trial (NCT02710734) evaluates a similarly selected cohort based on a partially overlapping set of genes and using a different neoadjuvant chemotherapy regimen. Conversely, cisplatin chemotherapy resistance has been associated with FGFR3 alterations and clonal mutations in integrin signaling pathway genes [8, 19].
Along with prognostic and predictive biomarkers, genomic sequencing of MIBC has also revealed a rich genomic landscape of actionable alterations found at clinically relevant frequencies. In one study of nearly 100 patients with high-grade bladder cancer (85% of which were MIBC), 61% had at least one clinically actionable alteration [20]. In addition to the previously mentioned neoadjuvant trials in patients with certain DDR alterations, two trials are evaluating FGFR inhibitors given the known frequency of FGFR3 alterations in bladder cancer (NCT04197986 and NCT04294277).
Finally, the wide spectrum of histomorphologic subtypes of bladder cancer is being actively investigated to discover genomic drivers of variant histology. Some variants are enriched in specific alterations that are nearly pathognomonic. For example, plasmacytoid variant bladder cancer, which is known to present more commonly at a locally advanced stage with common positive surgical margins at cystectomy , almost always carries a deletion in CDH1, which encodes for the E-Cadherin protein [21]. Other variants, such as small cell carcinoma, resemble pure urothelial carcinoma with common TERT promoter mutations while also being enriched for TP53 and RB1 alterations [22]. Although these discoveries advance our understanding of the pathogenesis of bladder cancer with variant histology, they also expose actionable alterations that could expand treatment options in patients who are typically resistant to chemotherapy and have poor clinical outcomes.
In summary, significant sequencing data exist for patients with MIBC that aid in prognosis and treatment response prediction, although none have yet reached routine clinical practice in this disease state. Several clinical trials exist in the neoadjuvant and adjuvant space for genomically selected patients. Future work will continue to unravel the pathways that contribute to divergent differentiation and exposure of therapeutic vulnerabilities in these aggressive tumors.
Non-muscle-Invasive Bladder Cancer
Efforts in NMIBC have focused on developing molecular-subtype classifications, characterizing the genomic landscape and drivers, and attempting to correlate these findings with the diversity of clinical outcomes from this heterogeneous group of patients.
While molecular subtypes in MIBC have been independently derived by several groups and a consensus classifier has been proposed, subtypes in NMIBC are considerably less defined at this time. The most significant effort to date used RNA sequencing data to derive three molecular subtypes from a cohort of 460 patients with NMIBC and 14 patients with MIBC [23]. Class 1 represented largely luminal tumors with predictably frequent FGFR3 alterations. Class 2 was also luminal-like, when compared with other classifiers for MIBC, but expressed more epithelial to mesenchymal transition markers and had more frequent predicted mutations in TP53 and DDR genes , such as ERCC2. Class 3 tumors were more basal-like but did not represent a subtype seen in TCGA. In terms of progression, class 1 and class 3 tumors generally had favorable outcomes, while class 2 tumors were significantly more likely to progress. While these subtypes provided some biological underpinning, they largely paralleled tumor grade and stage (e.g., classes 1 and 3 consisted of mainly low-grade Ta tumors and class 2 comprised the majority of the high-grade T1 tumors among the three groups) and have yet to be adapted clinically. In another study of 140 low-grade Ta and high-grade Ta tumors, whole genome sequencing clearly demonstrated a significantly more unstable genome in subgroup 2, which consisted mainly of high-grade Ta tumors [24]. Other groups have attempted to further identify subtypes in high-grade T1 tumors, which account for the majority of progression and cancer-specific mortality in NMIBC [25, 26].
The genomic landscape of NMIBC demonstrates TERT promoter and common chromatin-modifying gene alterations across all grades and stages, which are known early events in bladder cancer pathogenesis [27]. Notably, shifts in oncogenic drivers and/or targetable alterations can be observed with increasing grade and stage from low-grade Ta to high-grade Ta to high-grade T1. For example, FGFR3 mutations, a known driver in low-risk tumors, decrease in frequency from greater than 80% in low-grade Ta to less than 40% in high-grade T1. Conversely, oncogenic drivers of aggressive disease, such as TP53 and RB1 were more common with the shift from low-grade to high-grade disease, and frequencies in high-grade T1 disease approached that of a TCGA MIBC comparator cohort, correlating with the clinical experience that at least a subset of these tumors had the potential for invasion and metastases.
Ongoing efforts are focused on identifying associations with recurrence and progression, as well as predictors of response to Bacillus Calmette-Guerin (BCG), as the failure of this treatment often results in therapeutic escalation to radical cystectomy. High-grade NMIBC tumors were found to have higher TMB which correlated with more frequent mutations in DDR genes, particularly ERCC2 [27]. These findings were independently confirmed in a separate analysis consisting of 126 cases of high-grade NMIBC showing that TMB increased from low-grade NMIBC to high-grade NMIBC and that TMB and DDR alterations were positively correlated [28]. The association between TMB and response to BCG should be further explored, although theoretically, a higher mutational burden would result in a more robust response to an immunotherapy-based treatment (such as BCG) [27, 29]. On the other hand, significant associations between ARID1A alterations and BCG resistance were demonstrated in both studies, which is notable as these alterations could be a predictive biomarker of resistance to therapy and, in turn, potentially targetable.
In summary, molecular classification of NMIBC is based on comprehensive analyses of large patient cohorts but has yet to develop utility in clinical practice. NMIBC is of particular interest in terms of prognostic and predictive genomic biomarkers given the diversity of clinical outcomes that span from indolent yet recurrent low-grade tumors to quickly progressive and metastatic high-grade tumors. Finally, the lifelong invasive nature of surveillance for many patients with NMIBC provides substantial motivation for advanced approaches of sequencing cell-free tumor DNA (ctDNA) in the urine.
Advanced and Metastatic Bladder Cancer
The genomic landscape of advanced and metastatic bladder cancer is similar to muscle-invasive disease but often influenced by the selective pressures of systemic treatment. In a study of 72 chemotherapy-resistant tumors and a subgroup of matched pre- and post-chemotherapy samples, few mutations were shared between the primary and metastatic tumors [8]. However, the divergence of primary and metastatic samples on WES occurred early in the evolution of these tumors indicating that this is an early event in the natural history of the disease. In a rapid autopsy series of multiple primary and metastatic sites from seven patients with both bladder and upper-tract cancer, discordance in mutations with potentially actionable mutations occurred in 30% of samples [30]. This finding highlights the potential importance of sequencing additional sites of disease as tumors become resistant to therapy, progress, or metastasize to additional sites, which can be addressed by advanced approaches such as ctDNA.
To date, the only FDA-approved targeted therapy for bladder cancer is the pan-FGFR3 inhibitor, erdafitinib, which is approved for patients with locally advanced or metastatic disease that has progressed during or following platinum-containing chemotherapy [31]. Genetic testing for FGFR2/3 alterations is indicated to identify patients for this treatment. No guidance is provided for indicating whether primary or metastatic samples should be tested for these alterations. To optimally select patients for targeted therapies, future studies will be required to determine whether known intra- and inter-tumoral heterogeneity results in inappropriate selection of candidates for treatment and which samples are ideal for genetic testing.
Germline Alterations
Epidemiologic studies have identified that approximately 30% of urothelial cancers have a heritable component [32]. However , while germline mismatch repair (MMR) variants have been associated with Lynch syndrome and the risk of urothelial carcinoma of the upper tracts, no clear associations with bladder cancer exist. Current efforts have focused on characterizing the landscape of germline alterations, evaluating the role for germline testing, and identifying clinically relevant implications of germline alterations in patients with bladder cancer.
Two large retrospective analyses have evaluated germline alterations in patients with urothelial carcinoma and identified similar rates and types of alterations [33, 34]. Using a panel of 77 cancer predisposition genes, one study found that up to 13.7% of patients had a pathogenic or likely pathogenic germline variant in a cohort of 586 patients with urothelial carcinoma, majority of whom (79%) had bladder cancer [34]. In this study, the most frequently altered gene was APC, and the most frequently altered genes specifically with moderate or high penetrance were BRCA2, MSH2, CHEK2, ERCC3, NBN, and RAD50. In total, 83% of germline variants were in DDR genes. In the subgroup with clinically annotated data, patients with any moderate-/high-penetrance variant (n = 27) were more likely to be ≤45 years old (22% vs. 6%) and of Ashkenazi Jewish ancestry (41% vs. 14%) compared with patients with no moderate-/high-penetrance variants (n = 142). Importantly, one-quarter of patients with germline variants in this study would not have been referred for germline testing based on published guidelines, suggesting that current methods to identify patients with potentially hereditary bladder cancer are inadequate. A second study comprised a larger cohort (n = 1038) tested with an assay from Invitae (San Francisco, CA), which sequenced between 1 and 130 genes (median 42) [33]. Despite the heterogenous sequencing panel, similar results were obtained. Approximately 24% of patients carried a pathogenic germline variant, of which 18.6% were in actionable genes as defined by the NCCN. This study also found that germline DDR alterations accounted for the majority (78%) of germline mutations. Combined, these studies suggest that certain high-risk cohorts would benefit from germline testing, and future studies should strive to identify how to best select these patients.
Despite their prevalence, the clinical implications of germline variants have yet to be fully realized. Current germline analyses have focused on patients with advanced and metastatic disease, thereby limiting generalizability to patients with localized muscle-invasive and non-muscle-invasive disease. Additional studies are needed to delineate the role of germline testing in select patients with bladder cancer.
Liquid Biopsy
Circulating Tumor Cells and Cell-Free Tumor DNA
There is increasing interest in genomic analysis of circulating tumor cells and cell-free tumor genomic material in patients with bladder cancer [35]. These assays, often referred to as liquid biopsies, have multiple clinical applications from screening and diagnosis to risk stratification and surveillance. Analysis of circulating tumor cells requires the identification and isolation of intact tumor cells, which can be analyzed morphologically as well as from a molecular standpoint. Circulating cell-free tumor DNA (ctDNA), on the other hand, can be isolated from a blood draw and sequenced using NGS platforms. In metastatic bladder cancer, ctDNA has been shown to reproduce the genomic landscape of MIBC based on paired tumor tissue profiling and compared with an analysis of TCGA [36]. ctDNA may potentially overcome several limitations previously discussed that apply to bulk tumor tissue sequencing of bladder cancer. First, these assays may capture alterations that are absent in bulk tumor sequencing given the known extent of intra-tumoral heterogeneity. Similarly, although many alterations in bladder cancer are thought to occur early in the development of tumors, inter-tumoral heterogeneity between primary tumors and metastatic sites may be better captured with ctDNA. This is especially possible in the setting of intervening targeted treatments (such as erdafitinib). Second, ctDNA can yield actionable genomic information in patients whose tumors are inaccessible without a high-risk invasive procedure (e.g., certain pulmonary metastases). Finally, serial ctDNA can be collected relatively simply, as this only requires a blood draw. Serial analysis of ctDNA provides the opportunity to evaluate response to treatment, guide additional therapies, and monitor resistance.
Monitoring of minimal residual disease after surgery is another potential application for liquid biopsies. In a prospective study of 68 patients with MIBC undergoing radical cystectomy, a primary tumor WES-informed customized ctDNA panel had a sensitivity and specificity of 100% and 98%, respectively, for the detection of recurrence after surgery [37]. Although this assay provides no targetable information to guide therapy selection, this sensitive assay could help guide adjuvant treatment in patients who are likely to have a recurrence. Further, ctDNA could be used to help guide treatment decisions in a variety of settings (e.g., early cystectomy, neoadjuvant chemotherapy, and consolidative surgery, among others) where the potential for over and undertreatment is substantial. Future studies are needed to better characterize the utility of ctDNA in these various disease states.
Urinary Cell-Free Tumor DNA
Analysis of ctDNA in the urine represents a logical strategy for the detection of bladder cancer. In a large analysis of 118 patients with bladder cancer and 67 healthy adults, Dudely et al. evaluated a novel hybrid-capture target enrichment strategy to sequence ctDNA from the discarded supernatant of urine samples [38]. TERT and PLEKHS1 promoter mutations were the most commonly discovered alterations, and the concordance between mutations in tumor tissue and urinary ctDNA was between 67% and 73% and higher for clonal versus subclonal mutations. APOBEC mutational signatures were significantly more common in patients with bladder cancer compared with patients in the control group, suggesting the possibility of using this assay as a screening tool. Compared with cytology, urinary ctDNA had significantly higher sensitivity (83–93% vs. 14%) and equivalent specificity (96–97% vs. 100%). This assay was also practical in that 50 cc of urine could be stored at 4 °C for up to 7 days. Interestingly , urinary ctDNA may be more sensitive than plasma ctDNA. In one study of nearly 250 samples from 17 patients, urinary supernatant and urinary cell pellet had more frequent single nucleotide variants and higher mutant allele frequencies compared with plasma ctDNA [39]. Urinary ctDNA has many potential applications and prospective clinical trials are needed to better define its role in the management of patients with bladder cancer.
Guidelines and Practical Approach
Despite the accumulating data on the clinical applicability of genomic sequencing in bladder cancer diagnosis and management, there is no consensus approach and the major guidelines in urologic oncology do not yet uniformly recommend testing. The NCCN guidelines recommend molecular/genomic testing in patients with stages IIIB and IV disease to identify potential therapeutic targets and to screen for clinical trial eligibility [40]. The European Association of Urology (EAU) guidelines mention the potential future utility of genomic sequencing but no current indications in either the MIBC or metastatic bladder cancer guidelines. Finally, 2020 amended American Urological Association (AUA) guidelines for nonmetastatic MIBC discuss the potential of genomic prognostic and/or predictive biomarkers but do not recommend testing; the AUA NMIBC guidelines make no mention of genomic sequencing [41, 42].
At this time, genomic sequencing to guide clinical care should be limited to patients with stage IIIB or IV disease as per the NCCN guidelines. Genetic screening or testing in patients with earlier-stage disease should be limited to the clinical trial or prospective study setting. There are several practical considerations for testing in patients with advanced disease. First is the question regarding which tumor sites should be sequenced. While most targeted therapies and clinical trials will accept sequencing from any source, there are some studies that suggest genomic differences in key drivers between the primary and metastatic tumors. This would support sequencing of the metastatic material if that were available. Second, testing should be performed prior to initiation of therapy to reduce the influence of treatment on the results. Third, as previously discussed, assays that utilize matched normal samples will reduce error from germline mutations and clonal hematopoiesis of indeterminate potential. Finally, integration of genetic counselors to aid in the interpretation, education, and counseling of patients is of significant added value and likely to be more important over time as germline testing becomes more commonly indicated [43, 44].
Conclusions
There is increasing evidence to support the role of genomic sequencing in the management of patients with bladder cancer. In patients with MIBC, clinical and translational data have demonstrated that some DDR genes, specifically ERCC2, may confer cisplatin sensitivity, and current clinical trials are testing the role of genomic biomarkers to select patients for bladder preservation. Additional clinical trials are genomically selecting patients for adjuvant targeted therapies in patients at high risk of recurrence after radical cystectomy. In NMIBC, genomic analyses are helping to identify predictors of response to BCG and indications for more aggressive therapy in others. In advanced and metastatic disease, tumor genomic evolution is being investigated to understand the drivers of metastasis and how potential targeted therapies should be selected. Germline analysis may provide data to aid in risk assessment for secondary malignancies and cascade testing in patients with alterations that confer an increased risk of hereditary cancers. In nearly all disease stages, analysis of ctDNA from blood and/or urine could revolutionize how samples are collected for analysis. As NGS technology advances and the cost of deeper and more broad sequencing falls, more complete sequencing (e.g., WES, WGS) may become routine.
References
Clinical N, Guidelines P, Guidelines N. Non-small cell lung. 2021.
Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. https://doi.org/10.1038/nature12477.
Alekseyev YO, Fazeli R, Yang S, et al. A next-generation sequencing primer—how does it work and what can it do? Acad Pathol. 2018;5:1–11. https://doi.org/10.1177/2374289518766521.
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–32. https://doi.org/10.1038/nrg3642.
Ptashkin RN, Mandelker DL, Coombs CC, et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 2018. https://doi.org/10.1001/jamaoncol.2018.2297.
Parikh K, Huether R, White K, et al. Tumor mutational burden from tumor-only sequencing compared with germline subtraction from paired tumor and normal specimens. JAMA Netw Open. 2020;3(2):e200202. https://doi.org/10.1001/jamanetworkopen.2020.0202.
Chakravarty D, Gao J, Phillips S, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017. https://doi.org/10.1200/po.17.00011.
Faltas BM, Prandi D, Tagawa ST, et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat Genet. 2016. https://doi.org/10.1038/ng.3692.
Cancer T, Atlas G. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22. https://doi.org/10.1038/nature12965.
Robertson AG, Kim J, Al-Ahmadie H, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171(3):540–556.e25. https://doi.org/10.1016/j.cell.2017.09.007.
Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. https://doi.org/10.1038/nature12213.
Li R, Du Y, Chen Z, et al. Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science (80- ). 2020;370(6512):82–9.
Lawson ARJ, Abascal F, Coorens THH, et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science (80- ). 2020;370(6512):75–82.
Kamoun A, de Reyniès A, Allory Y, et al. A consensus molecular classification of muscle-invasive bladder cancer [Formula presented]. Eur Urol. 2020;77(4):420–33. https://doi.org/10.1016/j.eururo.2019.09.006.
Seiler R, Ashab HAD, Erho N, et al. Impact of molecular subtypes in muscle-invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur Urol. 2017;72(4):544–54.
Van Allen EM, Mouw KW, Kim P, et al. Somatic ERCC2 mutations correlate with cisplatin sensitivity in muscle-invasive urothelial carcinoma. Cancer Discov. 2014;4(10):1140–53. https://doi.org/10.1158/2159-8290.CD-14-0623.
Li Q, Damish AW, Frazier Z, et al. ERCC2 helicase domain mutations confer nucleotide excision repair deficiency and drive cisplatin sensitivity in muscle-invasive bladder cancer. Clin Cancer Res. 2019;25(3):977–88. https://doi.org/10.1158/1078-0432.CCR-18-1001.
Pietzak EJ, Zabor EC, Bagrodia A, et al. Genomic differences between “primary” and “secondary” muscle-invasive bladder cancer as a basis for disparate outcomes to cisplatin-based neoadjuvant chemotherapy. Eur Urol. 2019;75(2):231–9. https://doi.org/10.1016/j.eururo.2018.09.002.
Teo MY, Mota JM, Whiting KA, et al. Fibroblast growth factor receptor 3 alteration status is associated with differential sensitivity to platinum-based chemotherapy in locally advanced and metastatic urothelial carcinoma. Eur Urol. 2020;78(6):907–15. https://doi.org/10.1016/j.eururo.2020.07.018.
Iyer G, Al-Ahmadie H, Schultz N, et al. Prevalence and co-occurrence of actionable genomic alterations in high-grade bladder cancer. J Clin Oncol. 2013;31(25):3133–40. https://doi.org/10.1200/JCO.2012.46.5740.
Al-Ahmadie HA, Iyer G, Lee BH, et al. Frequent somatic CDH1 loss-of-function mutations in plasmacytoid variant bladder cancer. Nat Genet. 2016;48(4):356–8. https://doi.org/10.1038/ng.3503.
Chang MT, Penson A, Desai NB, et al. Small-cell carcinomas of the bladder and lung are characterized by a convergent but distinct pathogenesis. Clin Cancer Res. 2018;24(8):1965–73. https://doi.org/10.1158/1078-0432.CCR-17-2655.
Hedegaard J, Lamy P, Nordentoft I, et al. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell. 2016;30(1):27–42. https://doi.org/10.1016/J.CCELL.2016.05.004.
Hurst CD, Alder O, Platt FM, et al. Genomic subtypes of non-invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell. 2017;32(5):701–715.e7. https://doi.org/10.1016/j.ccell.2017.08.005.
Patschan O, Sjödahl G, Chebil G, et al. A molecular pathologic framework for risk stratification of stage T1 urothelial carcinoma. Eur Urol. 2015; https://doi.org/10.1016/j.eururo.2015.02.021.
Robertson AG, Groeneveld CS, Jordan B, et al. Identification of differential tumor subtypes of T1 bladder cancer. Eur Urol. 2020. https://doi.org/10.1016/j.eururo.2020.06.048.
Pietzak EJ, Bagrodia A, Cha EK, et al. Next-generation sequencing of nonmuscle invasive bladder cancer reveals potential biomarkers and rational therapeutic targets. Eur Urol. 2017;72(6):952–9. https://doi.org/10.1016/j.eururo.2017.05.032.
Nassar AH, Umeton R, Kim J, et al. Mutational analysis of 472 urothelial carcinoma across grades and anatomic sites. Clin Cancer Res. 2019;25(8):2458–70. https://doi.org/10.1158/1078-0432.CCR-18-3147.
Bellmunt J, Kim J, Reardon B, et al. Genomic predictors of good outcome, recurrence, or progression in high-grade T1 non–muscle-invasive bladder cancer. Cancer Res. 2020;80(20):4476–86. https://doi.org/10.1158/0008-5472.CAN-20-0977.
Winters BR, De Sarkar N, Arora S, et al. Genomic distinctions between metastatic lower and upper tract urothelial carcinoma revealed through rapid autopsy. JCI Insight. 2019;4(13). https://doi.org/10.1172/jci.insight.128728.
Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381(4):338–48. https://doi.org/10.1056/nejmoa1817323.
Mucci LA, Hjelmborg JB, Harris JR, et al. Familial risk and heritability of cancer among twins in nordic countries. JAMA. 2016;315(1):68–76. https://doi.org/10.1001/jama.2015.17703.
Nassar AH, Abou Alaiwi S, AlDubayan SH, et al. Prevalence of pathogenic germline cancer risk variants in high-risk urothelial carcinoma. Genet Med. 2019;0(0):1–10. https://doi.org/10.1038/s41436-019-0720-x.
Carlo MI, Ravichandran V, Srinavasan P, et al. Cancer susceptibility mutations in patients with urothelial malignancies. J Clin Oncol. 2020;38(5):406–14. https://doi.org/10.1200/JCO.19.01395.
Kouba E, Lopez-Beltran A, Montironi R, et al. Liquid biopsy in the clinical management of bladder cancer: current status and future developments. Expert Rev Mol Diagn. 2019;00(00):1–10. https://doi.org/10.1080/14737159.2019.1680284.
Vandekerkhove G, Lavoie J, Annala M, et al. Plasma ctDNA is a tumor tissue surrogate and enables clinical-genomic stratification of metastatic bladder cancer. Nat Commun. 2021:1–12. https://doi.org/10.1038/s41467-020-20493-6.
Christensen E, Birkenkamp-Demtröder K, Sethi H, et al. Early detection of metastatic relapse and monitoring of therapeutic efficacy by ultra-deep sequencing of plasma cell-free DNA in patients with urothelial bladder carcinoma. J Clin Oncol. 2019;37(18):1547–57. https://doi.org/10.1200/JCO.18.02052.
Dudley JC, Schroers-Martin J, Lazzareschi DV, et al. Detection and surveillance of bladder cancer using urine tumor DNA. Cancer Discov. 2019;9(4):500–9. https://doi.org/10.1158/2159-8290.CD-18-0825.
Patel KM, van der Vos KE, Smith CG, et al. Association of plasma and urinary mutant DNA with clinical outcomes in muscle invasive bladder cancer. Sci Rep. 2017;7(1):5554. https://doi.org/10.1038/s41598-017-05623-3.
Flaig TW. NCCN guidelines updates: management of muscle-invasive bladder cancer. J Natl Compr Cancer Netw. 2019;17(5.5):591–3.
Chang Sam S, Boorjian SA, Chou R, et al. Diagnosis and treatment of non-muscle invasive bladder cancer: AUA/SUO guideline. J Urol. 2016;196(4):1021–9.
Chang SS, Bochner BH, Chou R, et al. Treatment of non-metastatic muscle-invasive bladder cancer: AUA/ASCO/ASTRO/SUO guideline (Amended 2020). 2020.
Wicklund CAL, Duquette DA, Swanson AL. Clinical genetic counselors: an asset in the era of precision medicine. Am J Med Genet Part C Semin Med Genet. 2018;178(1):63–7. https://doi.org/10.1002/ajmg.c.31605.
Jacobs MF, Milliron KJ. Genetic counseling and previvorship in patients with urologic malignancies. Curr Opin Urol. 2019;29(4):371–7. https://doi.org/10.1097/MOU.0000000000000638.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lenis, A.T., Pietzak, E.J. (2021). Bladder Cancer Genomics: Indications for Sequencing and Diagnostic Implications. In: Bjurlin, M.A., Matulewicz, R.S. (eds) Comprehensive Diagnostic Approach to Bladder Cancer. Springer, Cham. https://doi.org/10.1007/978-3-030-82048-0_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-82048-0_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-82047-3
Online ISBN: 978-3-030-82048-0
eBook Packages: MedicineMedicine (R0)