
Abstract
Hyperammonaemia is a metabolic emergency and prompt treatment is paramount to optimize neurological outcome. Ammonia is extremely neurotoxic and increased levels can arise from an inherited or acquired defect in hepatic detoxification. Inborn errors of metabolism leading to hyperammonaemia mainly affect the hepatic urea cycle due to single enzyme deficiencies, transporter defects or mitochondrial dysfunction. Primary hyperammonaemia is a consequence of direct urea cycle dysfunction whereas secondary hyperammonaemia can result from disturbance of the urea cycle by toxic metabolites or substrate deficiencies. Immediate recognition and early initiation of specific treatment are of utmost importance. Prognostic factors include duration of hyperammonaemic coma and the extent of ammonia accumulation (Häberle et al. Orphanet J Rare Dis 7:32, 2012). The principles of management in the acute situation aim to rapidly remove ammonia, decrease production and replace rate limiting amino acids.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Clinical ScenarioA 2-day-old male infant presents with a 24 h history of poor feeding, not waking for feeds and increased respiratory rate. The initial blood gas shows a respiratory alkalosis (pH 7.41, pCO2 3.1, HCO3 19 and base excess −4.2) with a normal lactate and blood sugar. The plasma ammonia returns as 550 μmol/L. What are the initial differential diagnoses and initial management?
1 Ammonia Metabolism and the Urea Cycle
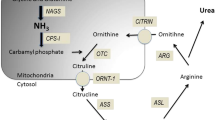
Ammonia, NH3, is an important source of nitrogen for protein synthesis, amino acid metabolism and pH homeostasis. Ammonia dissolves in water and at physiological pH ammonia exists predominantly in its ionized form, ammonium, NH4+. The normal concentration of ammonium in plasma is between 11 and 50 μmol/L. Ammonia is continuously produced and consumed during cellular metabolism and arises from breakdown of purine and pyrimidine products and deamination of several amino acids including glutamine, asparagine, serine, threonine, glycine, proline and lysine [1]. In mammals, the urea cycle is the main pathway of ammonia detoxification. The complete urea cycle is only expressed in the liver and proximal parts are expressed in the gastrointestinal tract and kidney [2]. Periportal hepatocytes receive the high nitrogen load of portal blood arriving from the intestine. The urea cycle consists of six enzymatic steps; the proximal part within the mitochondria and distal part in the cytoplasm (Fig. 27.1). The first step of the urea cycle involves the conversion of ammonia and bicarbonate into carbamoylphosphate by carbamoylphosphate synthetase 1 (CPS1). The urea cycle, i.e. CPS1 requires allosteric activation by N-acetylglutamate (NAG) which is synthesized by N-acetylglutamate synthetase (NAGS). Inherited deficiency of this enzyme is extremely rare, moreover toxic metabolites can lead to a secondary hyperammonaemia via impairment of NAGS activity.

The urea cycle (J Haberle 2013)
The second step in the urea cycle involves the condensation of carbamoylphosphate with ornithine to form citrulline, a reaction catalysed by ornithine transcarbamoyltransferase (OTC), the only X-linked urea cycle defect. The following three reactions involving argininosuccinate synthetase (ASS), argininosuccinate lyase (ASL) and arginase (AR1) take place in the cytoplasm. The final step hydrolyses arginine into ornithine and urea, and ornithine is regenerated for another rotation of the cycle. Hence transporters are required to transfer the urea cycle intermediates across the mitochondrial membrane in both directions; ornithine-citrulline antiporter (ORTN1) and aspartate-glutamate antiporter (citrin). Deficiencies of citrin, carbonic anhydrase Va (CAVA) and Δ [3]-pyrroline-5-carboxylate synthetase (P5CS) can also cause hyperammonaemia by restriction of the supply of asparate, bicarbonate and ornithine, respectively, to the urea cycle. The main focus of this chapter will be the main primary hyperammonaemic disorders, their manifestations and management. Table 27.1 shows primary and secondary causes of hyperammonaemia to be considered.
2 Clinical Manifestations of Hyperammonaemia
Clinically, hyperammonaemia presents with signs of encephalopathy. In the neonatal period, there may be a short symptom-free period and subsequent vomiting, poor feeding, increased sleepiness, irritability, muscular hypotonia, seizures, hyper- or hypoventilation and coma. In infants and older children there is commonly an intercurrent illness or other catabolic stress, e.g. fasting, post-surgery (and post-partum), rapid weight loss, treatment with steroids/chemotherapy or followed by a high-protein containing meal or load. Some children can display a history of self-selecting a low protein diet. A more chronic presentation in some may be reported with cyclical or chronic vomiting, developmental delay, faltering growth, neurocognitive and behavioural impairment. These symptoms in older children and adults can often be mistaken for encephalitis, drug or alcohol intoxication or a space occupying lesion. Hence investigations in any child or adult with unexplained reduced level of consciousness should include an ammonia measurement [4,5,6].
3 Diagnostic Tests
Accurate plasma ammonia measurement requires a free-flowing venous or arterial sample, capillary samples are not recommended to avoid spurious results as haemolysis causes positive interference. The sample should be collected into an ammonia-free specimen tube and transported immediately to the laboratory on ice, the sample needs to be separated within 15 min of collection [7]. Generally ammonia levels more than 500 μmol/L suggest an underlying IEM, however, this is not the rule. In UCDs there is often the absence of hypoglycaemia, lactic acidosis or ketosis in contrast to organic acidaemias for example, where ketosis and metabolic acidosis predominates. Ammonia is a respiratory stimulant and respiratory alkalosis is frequently present, particularly in the neonate.
Plasma amino acids can show characteristic patterns with elevation of glutamine and alanine indicating hyperammonaemia and low or high citrulline and arginine indicting specific UCDs. In OTC and ASS deficiency there is an elevation of orotic acid in the urine due to excess carbamolyphoshate, whilst this is absent in NAGS or CPS1 deficiency (Fig. 27.2).

Diagnostic algorithm (Orphanet J Rare Dis 2012)
Acylcarnitine profiles and urine organic acids can detect specific fatty acid oxidation defects or organic acidaemias.
Mutation analysis is the gold standard for diagnosis. The majority of disorders are autosomal recessive excluding OTC deficiency which is X-linked, where mutation detection has at least 80% sensitivity [8]. The use of next generation sequencing of multiple genes simultaneously is increasingly being used [9]. In deceased patients or when blood DNA is not available, some diagnoses can be made from frozen liver tissue or cultured fibroblasts. Genetic diagnosis allows appropriate counselling, prenatal diagnosis and appropriate family screening.
4 Urea Cycle Defects (UCDs)
UCDs result from loss of function of any of the enzymatic steps within the mitochondrial or cytosolic part of the urea cycle. The overall frequency is quoted as 1 in 35,000 [10].
Clinical manifestations: The classical UCDs can all lead to severe hyperammonaemia and can present at any age. The most common UCD is OTC deficiency followed by ASS deficiency. NAGS and CPS1 deficiency are extremely rare.
Classical presentation in the neonate is typically within the first few days of life with poor feeding, lethargy, vomiting and hyperammonaemic encephalopathy. Hypoglycaemia, lactic acidosis and ketosis may or may not be present. Clinical presentation outside the neonatal period is variable and often triggered by intercurrent illness, fasting and/or ingestion of a high protein containing meal or protein supplements, post-partum, treatment with steroids or chemotherapy. Older children and adults can present acutely with unexplained reduced level of consciousness and/or neurological signs (tremor, irritability, seizures) which can be mistaken for encephalitis or drug intoxication. Hence the importance of measuring plasma ammonia in patients of any age with unexplained reduced level of consciousness.
Female carriers of OTC deficiency can manifest clinical and biochemical signs which are variable due to X-inactivation (lyonization), ranging from asymptomatic to recurrent episodes of hyperammonaemia.
Children with UCDs can have ongoing episodes of hyperammonaemia triggered by intercurrent illness or fasting. Those with ASL deficiency are less prone to recurrent hyperammonaemia but can still develop neurocognitive difficulties, seizures and chronic liver disease. Hypertension is frequent in adolescent and adults with ASL deficiency and brittle hair (trichorrhexis nodosa) secondary to arginine deficiency is a pathognomonic finding. In contrast the clinical manifestations of AR1 deficiency are characterized by developmental delay, neurocognitive impairment, seizures and spastic tetra- or diplegia.
Diagnostic tests: Plasma amino acids and urine organic acids are first line tests which can distinguish between some of the UCDs. CPS1 and NAGS deficiency are biochemically indistinguishable. Enzyme assays in cultured fibroblasts or liver tissue are available in selective laboratories, however, these are recently superseded by genetic analysis. Mutation analysis can then be made available for prenatal testing.
4.1 Treatment
-
1.
Emergency management in acute hyperammonaemia must be commenced immediately. The main principles of treatment are:
-
(a)
Stop protein intake
-
(b)
Promote anabolism with adequate calorie intake from glucose (8–10 mg/kg/min) and lipid (1–2 g/kg/day)
-
(c)
Alternate pathway medications to lower ammonia (sodium benzoate and sodium phenylbutyrate) with supplementation of arginine and/or citrulline depending on the type of UCD. Carbaglutamic acid or N-carbamyl-L-glutamate, a synthetic analogue of NAG is the treatment for NAGS deficiency and CPS1 deficiency can sometimes respond to therapy.
-
(d)
Use of acute dialysis to reduce ammonia levels depending on the expertise of the centre (see CRRT section)
-
(a)
It is important to provide sufficient calories with glucose and lipid until the ammonia is lowered and protein can gradually be reintroduced. The mechanism of action of drugs is described later in the chapter and dosing is shown in Table 27.2.
-
2.
Maintenance treatment involves a carefully supervised protein-restricted diet, ammonia-lowering agents and supplementation of essential amino acids. Regular metabolic follow-up is required with monitoring of growth and development, plasma amino acid levels, vitamins, minerals and trace elements.
-
3.
Liver transplantation is curative for the proximal urea cycle defects and is a viable treatment option to prevent hyperammonaemic episodes, further neurological injury and improve quality of life. Hepatocyte transplantation has been used a ‘bridge’ to liver transplantation in the male neonate with OTC deficiency [11,12,13].
4.2 Prognosis
Urgent recognition and management of hyperammonaemia is vital. The impact on neurological outcome can be catastrophic if treatment is delayed. Mortality in neonatal onset OTCD is reported at 24% and 11% in late onset cases [14]. For UCDs collectively the mortality remains 60% for early onset OTCD in males and of the collective group 52% had developmental delay [15].
Age at onset and peak ammonia concentration at presentation (>500 μmol/L) best predict neurological outcome [16]. Prognosis is considered very poor if: hyperammonaemic coma has lasted more than 3 days, intracranial pressure is clearly increased and ammonia peaked at >1000 μmol/L (although impact of this level on prognosis depends on the duration of hyperammonaemia) [17].
5 Other IEMs Causing Hyperammonaemia
5.1 Organic Acidaemias
These are disorders of branched chain amino acid metabolism. Classical organic acidaemias associated with hyperammonaemia are propionic acidaemia, methylmalonic acidaemia, isovaleric acidaemia and maple syrup urine disease. Presentation is commonly in the neonatal period with metabolic acidosis, lactic acidosis, ketosis and hyperammonaemia. The metabolic acidosis often distinguishes this group of disorders from UCDs. The hyperammonaemia is thought to arise from a secondary inhibition of NAGS and CPS1 function and is often responsive to the addition of carbaglutamic acid in emergency management. Organic acids can cause bone marrow suppression and pancytopenia can be a feature. Diagnosis is established with urine organic acids, acylcarnitine profile and genetic confirmation. Management is aimed at restriction of natural protein, carnitine supplementation, ammonia-lowering agents as required and emergency regimens in the acute situation [18].
5.2 Fatty Acid Oxidation and Carnitine Cycle Defects
Fatty acid oxidation is a mitochondrial process and clinical presentation is variable. Medium chain acyl CoA dehydrogenase (MCAD) deficiency is the most common defect and is part of the UK newborn screening programme. Clinical presentation of other defects (e.g. Very long chain acyl CoA dehydrogenase (VLCAD) deficiency, long chain hydroxyl acyl CoA dehydrogenase (LCHAD) deficiency, carnitine-acylcarnitine translocase (CACT) deficiency) tend to exhibit hypoketotic hypoglycaemia, liver dysfunction, mild–moderate hyperammonaemia, cardiomyopathy and rhabdomyolysis. End organ effects occur due to accumulation of medium- or long-chain acylcarnitines, depending on the enzymatic deficiency. The hyperammonaemia results from lack of acetyl-CoA and subsequent urea cycle dysfunction. Urine organic acids and acylcarnitines will aid initial diagnosis and confirmatory testing is with DNA analysis and/or cultured fibroblast fatty acid oxidation studies. Treatment aims to limit fasting periods, emergency regimen in the acute situation and the use of fat restriction and medium chain triglycerides in certain defects [19].
5.3 Lysinuric Protein Intolerance (LPI)
LPI is due to a defect in the transport of dibasic amino acids (lysine, ornithine and arginine) at the basolateral intestinal membrane and renal tubular epithelium. The defect leads to impaired absorption and loss, respectively, of the dibasic amino acids. Urine and plasma amino acids can be diagnostic with genetic confirmation. This is a multisystemic disease with clinical features, including faltering growth, short stature, interstitial lung disease, chronic renal disease, osteopenia, hepatosplenomegaly and immune dysfunction [20]. Mild to moderate hyperammonaemia results from lack of substrates and subsequent impairment of the urea cycle.
5.4 Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH Syndrome)
HHH is a rare IEM of the urea cycle which has a variable phenotype ranging from mild form with learning difficulties and neurological involvement to severe form with lethargy, hepatic failure and seizures. The defect is in the protein ORC1, which is a transporter of ornithine, lysine and arginine into the mitochondrial matrix. The presence of homocitrulline is a hallmark of the disease and is detected in the urine. Hyperammonaemia is variable and often moderate [21].
5.5 Hyperinsulinism-Hyperammonaemia (HIHA) Syndrome
HIHA tends to cause mild hyperammonaemia. The disorder results from a gain of function mutation in the gene encoding glutamate dehydrogenase (GDH1). This protein is expressed in the liver, kidney, brain and pancreatic β-cells. Neonates tend to present with profound hypoketotic hypoglycaemia and mild hyperammonaemia. They have an increased risk of seizures and learning difficulties. The hypoglycaemia usually responds to diazoxide and the hyperammonaemia often does not require intervention [22].
5.6 Mitochondrial Disorders
This is a large and heterogeneous group of disorders involving energy metabolism. The resulting ATP deficiency from impaired mitochondrial oxidative phosphorylation is thought to impair urea cycle function as some of the enzymes are ATP-dependent. A specific mitochondrial condition with mutations in TMEM70 gene (which encodes a transmembrane protein involved in ATP synthase activity) tends to present in the neonatal period with hyperammonaemia and lactic acidosis [23, 24].
5.7 Carbonic Anhydrase Va Deficiency (CAVA)
This is a recently described cause of early onset hyperammonaemia with hypoglycaemia, hyperlactataemia, ketosis and metabolic acidosis. Defective hepatic bicarbonate production leads to this unique combination of biochemical findings. Diagnosis is confirmed by genetic confirmation. The hyperammonaemia responds to carbaglutamic acid and may reach levels which require haemofiltration. Outcomes in reported cases appear good in the short-term [25].
6 Intravenous Medications Used in Hyperammonaemia
Ammonia-lowering or scavenging agents commonly used are sodium benzoate and sodium phenylbutyrate, both conjugate specific amino acids within the liver and require co-enzyme A. Table 27.2 shows recommended drug doses. Loading doses followed by the maintenance infusions are used in the emergency setting, advice should always be sought from a metabolic specialist.
Sodium benzoate combines with glycine to form hippurate, which is excreted in the urine and thus removal of a nitrogen source. Adverse effects from intravenous infusion include metabolic acidosis, hypernatraemia, hyperbilirubinaemia due to displacement of bilirubin from albumin and in view of its caustic nature, protection from extravasation injury is important.
Sodium phenylbutyrate combines with glutamine to form phenylacetylglutamine, which is also excreted in the urine. Potassium can become depleted and hence should be monitored.
Arginine becomes an essential amino acid in certain defects and often requires supplementation intravenously. In large doses, resulting nitric oxide accumulation can lead to systemic hypotension.
Plasma amino acids should be monitored during treatment and doses adjusted accordingly with advice from metabolic specialists [26, 27].
7 The Role of Dialysis in Hyperammonaemia
At physiologic pH, 1–2% of plasma ammonia exists in the form of NH3, which readily permeates across cellular membranes (pK = 9.0) [28]. The process is pH dependent with a significantly higher accumulation of cerebral ammonia at alkaline pH [29].
Glutamate is converted in the astrocyte cytosol in an equimolar ratio into glutamine via glutamine synthetase. In hyperammonaemia, there is dysfunction of the astrocytes with shrinkage and secondary hyperactivation of the NMDA receptors which leads to disruption of cerebral metabolism and neuronal and glial injury [30]. Although the pathway is well described, there is no clear and consistent threshold or duration of ammonia toxicity that correlate closely with clinical signs (seizures, encephalopathy and cerebral oedema) or that is predictive of permanent neurologic injury as many confounding factors such as hypoxia or hypotension may also be present especially in the shocked neonate.
In the largest published series of neonatal hyperammonaemia over a 25-year period (56 neonates), peak blood level > 1000μmol/L had poorest survival [31]. Uchino similarly concluded raised ammonia (peak levels >350 μmol/L) had poor survival or neurodevelopmental outcome in a study of 92 neonates with urea cycle disorder [32]. In a study of 26 neonates, Msall demonstrated duration of coma was associated with peak ammonia level [33].
In contrast Westrope (n = 14) and Picca (n = 45) failed to identify peak ammonia or duration of raised ammonia as outcome markers [34, 35].
This exemplifies the complexity of modelling outcomes for rare metabolic diseases where many confounding factors are present and sample size spans many years (sometimes decades) over which historical treatment strategies have changed and been refined.
7.1 Ammonia Removal by Dialysis
Ammonia (NH3) is a small water-soluble molecule (molecular mass 17 g/mol) with low protein binding. It therefore has a favourable profile for clearance via peritoneal or extracorporeal dialysis. Using best practice as a guide, the aim of dialytic therapy is to reduce brain ammonia toxicity as fast as possible into a “safe” zone below 100–200 μmol/L within the first 12–24 hours of therapy [31, 36, 37]. With effective dialysis, ammonia blood levels should halve in 2–4 h. There are no studies addressing the speed of ammonia reduction and if there are adverse consequences to this (e.g. osmolar shifts) versus ongoing toxicity if ammonia levels are reduced more gradually.
Dialysis should be considered if ammonia levels start increasing above 300 μmol/L despite medical therapy been optimized [31]. Effective extracorporeal dialysis requires securing a large bore central venous dual lumen catheter and having a period of effective trouble-free dialysis without circuit thrombosis or downtime. In the neonatal population, it is technically challenging with even experts experiencing complications and delays [28, 38, 39].
Peritoneal dialysis has been used widely in neonatal hyperammonaemia with the advantage of being quick to initiate and relatively easy to perform. Picca demonstrated the advantages of early initiation of PD sometimes at outlying non-tertiary hospitals with similar outcome to those treated primarily by extracorporeal dialysis (HD or CVVHD) in tertiary centres [35]. In some studies, ammonia clearance rates with PD were similar to hemofiltration [35, 36]. Peritoneal dialysis (PD), when applied early, can allow safe and relatively risk-free method to reduce plasma ammonia before extracorporeal dialysis [35]. Others have demonstrated a slower initial rate of ammonia reduction with PD (time to half the plasma ammonia level) relative to CVVHD (n = 21 neonates); however, both took a similar time to reach a “safe” zone within 24 h [37]. Typically acute PD is prescribed at 10–20 mL/kg volumes. Frequent cycles (<30 min) may maximize dialytic ammonia clearance rates when ammonia levels are very high.
7.2 Extracorporeal Dialysis
At maximal efficiency, all extracorporeal dialysis modes (intermittent haemodialysis, CVVH, CVVHD and CVVHDF) may provide excellent ammonia clearance [30, 35, 39, 40]. Historically, intermittent hemodialysis has had the reputation of having the highest ammonia clearance rates using blood flow rates of 5–10 mL/kg/min and dialysate flow rates of 500 mL/min (2000–4000 mL per 1.73 m2 [38]. Ammonia extraction with these settings is above 95% [35].
McBryde demonstrated improved survival with HD as the primary therapy (n = 18) [38]. The main problems experienced are technical issues (adequate vascular access) and side effects such as hypotension which may limit the required blood flow rates. Typically, rebound hyperammonaemia may occur when the hemodialysis cycle has finished. For this reason, continuous high-dose renal replacement therapy has been proposed as an alternative [34, 37, 39, 41].
For CVVH or CVVHD, three key factors largely determine clearance of ammonia: blood flow rate, haemofilter surface area and ultrafiltration rate. For neonates, typically slightly larger filters relative to patient size are used (0.3–0.4 m2). These are designed to operate at maximal efficiency at higher blood flow rates of around 150 ml/min. Using lower blood flow rates of 20–50 mL/min (5–15 mL/kg), which is more appropriate for neonates may result in ultrafiltration on the flow dependent part of the mass solute clearance curve (Fig. 27.3, point B). Large filtration gains can be achieved by increasing blood flow rate (if tolerated by the patient) as filtration is more efficient at the same transmembrane pressure (Fig. 27.3a, point A). This concept is well described by Clarke and Huang [42, 43].

Mass transfer of solutes across a hypothetical haemofilter at three different blood flow rates (BFR). Red vertical line represents the point where maximal filter efficiency is achieved (point B). Ultrafiltration rate is higher at the same transmembrane pressure when blood flow rate is maximized (point A)
Blood flow rates of 80 mL/min are achievable in neonates with well-placed 7–8 Fr internal jugular catheters (author’s personal experience) with access pressures below 80 cmH20. Small vascular access catheters (5Fr) should be avoided as blood flow is limited and circuit thrombosis is universal (100% by 60 h) [44]. Low flow rates of 20 mL/min for a 0.3 m2 filter may be at the lower end of efficiency. Picca demonstrated ammonia extraction of just above 50% at these lower flows [35].
The second factor to consider in hemofiltration is dialysate flow rate. Troyanov compared CVVH versus CVVHD for ammonia clearance in adult patients and identified maximal efficiency with a dialysate flow rate of 4.5 L per hour (blood blow rate of 150 mL/min or 9 L/h) [45].
This is a ratio of dialysate to blood flow of 50%. In contrast, neonates may use a similar size filter (0.3–0.4 m2) yet blood flow rates are as much as 10-fold lower (20–50 mL/min). If dialysate flow rates are greater than the blood flow rate, no benefit is gained as only a fixed concentration of ammonia is presented to the filter over time. Figure 27.4 shows curves for dialysate flow rate (mls/h) in a 3.5 kg patient at different blood flow rates. It demonstrates the wide variations and dialysate flow rates for some published cases. For points B and C, ammonia extraction was just above 50%. This demonstrates that ammonia clearance can be maximized at higher blood flow rates (greater clearance at the same ratio of dialysate to blood flow rate). It also highlights that it is important to be aware of the relationship of blood flow rate and dialysis rate in neonates who are 3–5 kg in size. With CVVH, predilution affords a greater flow rate (30–60 mL/kg/h) but this is partially offset by a dilution effect and a loss of 40% efficiency especially if low blood flow rates are used [45].

Absolute dialysate flow rate (mL/h) for a 3.5 kg neonate at three different blood flow rates. The x-axis is the ratio of dialysate to blood flow. The vertical red line is the point where dialysate flow equals blood flow rate. The horizontal line is the dialysate flow rate suggested by Spinale [11]. Point A represents a neonate on 40 mL/min blood flow rate [11], Point B and C are neonates from series by Picca et al (112). Point C, the neonate received 33.3 mL/min dialysate at 20 mL/min blood flow rate
7.3 Recirculation
In adult patients, femoral site vascular access demonstrates the highest recirculation rates using saline dilution technique of 26% compared to jugular venous access [46]. This is because there is less of blood reservoir around the femoral vein compared to the right atrium. Recirculation rate also increase significantly at higher flow. Unfortunately, recirculation rates have not been measured in paediatric dialysis, but are likely to be much higher around 50% as the distance between the proximal and distal lumen in paediatric dual lumen lines is very close, as little as 5–10 mm apart. This is partly why efficiency of CVVH or CVVHD is only about 50% in neonates. The femoral vein is also significantly smaller in diameter in neonates and is more prone to catheter related complications and thrombosis.
The ideal site for vascular access is therefore the internal jugular vein, preferable the right as it has a more direct course to the right atrium.
7.4 Maximizing Efficiency of Dialysis
In some circumstances the ammonia levels fail to decline or even increase despite apparently adequate dialysis. Occasionally ammonia production will exceed even the most effective dialytic clearance. Important points to consider in this scenario are:
-
1.
Minimize recirculation through the vascular access. Use the proximal and distal catheter drainage ports as the manufacturer intended. “Swapping” the lumens around, even if flow appears better only increases recirculation. Try using internal jugular access.
-
2.
Optimize blood flow rate(BFR). BFR >40 mL/min enhances filtration but this is dependent on good vascular access. Recirculation will increase with higher blood flow rate if the catheter position is problematic.
-
3.
Ensure adequate delivery of intravenous sodium benzoate and sodium phenylacetate. Both are small molecular weight molecules that are not protein bound and are freely diffusible [47]. Giving excessive doses may also precipitate neurologic deterioration [31].
-
4.
Consider dual mode dialysis (peritoneal dialysis and hemofiltration or HD)
-
5.
In hyperammonaemia due to organic acid disorders, larger organic acids may be more effectively cleared with convective hemofiltration as opposed to counter current haemodialysis or haemodiafiltration.
-
6.
Alkalosis promotes ammonia entry into the brain. If pH >7.45, aim for normal pCO2. If the patient is haemodynamically stable, mild to moderate acidosis could be tolerated to reduce cerebral ammonia uptake.
Conclusion
Hyperammonaemia secondary to IEM is a common neonatal/paediatric emergency. Measurement of ammonia levels should form a part of investigations for any collapsed newborn or in fact any child who presents with sudden onset of loss of consciousness. A very close liaison should always be sought between the treating intensive care team and metabolic specialist. Peritoneal dialysis is a good modality to lower serum ammonia levels awaiting transfer to tertiary centres for extracorporeal dialysis or even as the primary form of renal replacement therapy. There are no head to head studies comparing convective versus dialysis mode of renal replacement therapy. All efforts should be made to optimize CRRT including minimizing circuit downtimes. Prognosis depends on the type of inborn error of metabolism, peak ammonia levels and the duration of hyperammonaemic coma. Despite optimizing dialytic therapy, one might not be able to control ammonia levels if the rate of production of ammonia is higher than what the extracorporeal therapy can remove.
Key Learning Points
-
Hyperammonaemia secondary to IEM can occur at any age
-
Measure ammonia in any patient with unexplained reduced level of consciousness
-
Age of onset and peak ammonia levels at presentation impact neurological outcome
-
Consider dialysis to acutely lower blood ammonia levels above 300 μmol/L. Ideal target is to half ammonia in 2–4 h with a level below 150–200 μmol/L within 24 h.
-
At maximal efficiency, all modes of dialysis including perineal dialysis can acutely lower blood ammonia levels. The mode and type of dialysis should be chosen according to the expertise of the clinical team or institution.
-
For neonatal haemofiltration (CVVH or CVVHD), a large bore right internal jugular vascular access catheter (e.g. 7Fr) is preferable for optimal blood flow. Femoral catheters may have significant recirculation.
References
Adeva MM, Souto G, Blanco N, Donapetry C. Ammonium metabolism in humans. Metabolism. 2012;61(11):1495–511.
Jackson MJ, Beaudet AL, O’Brien WE. Mammalian urea cycle enzymes. Annu Rev Genet. 1986;20:431–64.
Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32.
Bowker R, Green A, Bonham JR. Guidelines for the investigation and management of a reduced level of consciousness in children: implications for clinical biochemistry laboratories. Ann Clin Biochem. 2007;44:506–11.
Smith W, Kishnani PS, Lee B, Singh RH, Rhead WJ, Sniderman King L, Smith M, Summar M. Urea cycle disorders: clinical presentation outside the newborn period. Crit Care Clin. 2005;21(4 Suppl):S9–17.
Saudubray JM, Nassogne MC, de Lonlay P, Touati G. Clinical approach to inherited metabolic disorders in neonates: an overview. Semin Neonatol. 2002;7(1):3–15.
Barsotti RJ. Measurement of ammonia in blood. J Pediatr. 2001;138(1 Suppl):S11–9.
Wertheim-Tysarowska K, Gos M, Sykut-Cegielska J. Bal J; genetic analysis in inherited metabolic disorders--from diagnosis to treatment. Own experience, current state of knowledge and perspectives. Dev Period Med. 2015;19(4):413–31.
Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum Mutat. 2006;27(7):626–32.
Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, Kirmse B. The incidence of urea cycle disorders. European registry and network for intoxication type metabolic diseases (E-IMD). Mol Genet Metab. 2013;110(1–2):179–80.
Legido-Quigley C, Cloarec O, Parker DA, Murphy GM, Holmes E, Lindon JC, Nicholson JK, Mitry RR, Vilca-Melendez H, Rela M, Dhawan A, Heaton N. First example of hepatocyte transplantation to alleviate ornithine transcarbamylase deficiency, monitored by NMR-based metabonomics. Bioanalysis. 2009;1(9):1527–35.
Kido J, Matsumoto S, Momosaki K, Sakamoto R, Mitsubuchi H, Endo F, Nakamura K. Liver transplantation may prevent neurodevelopmental deterioration in high-risk patients with urea cycle disorders. Pediatr Transplant. 2017;21(6)
Yu L, Rayhill SC, Hsu EK, Landis CS. Liver transplantation for urea cycle disorders: analysis of the united network for organ sharing database. Transplant Proc. 2015;47(8):2413–8.
Bachmann C. Outcome and survival of 88 patients with urea cycle disorders: a retrospective evaluation. Eur J Pediatr. 2003;162(6):410–6.
Batshaw ML, Tuchman M, Summar M, Seminara J, Members of the Urea Cycle Disorders Consortium. A longitudinal study of urea cycle disorders. Mol Genet Metab. 2014;113(1–2):127–3.
Burgard P, Kölker S, Haege G, Lindner M, Hoffmann GF. Neonatal mortality and outcome at the end of the first year of life in early onset urea cycle disorders—review and meta-analysis of observational studies published over more than 35 years. J Inherit Metab Dis. 2016;39(2):219–2.
Posset R, Garcia-Cazorla A, Valayannopoulos V, Teles EL, Dionisi-Vici C, Brassier A, Burlina AB, Burgard P, Cortès-Saladelafont E, Dobbelaere D, Couce ML, Sykut-Cegielska J, Häberle J, Lund AM, Chakrapani A, Schiff M, Walter JH, Zeman J, Vara R, Kölker S. Additional individual contributors of the E-IMD consortium; age at disease onset and peak ammonium level rather than interventional variables predict the neurological outcome in urea cycle disorders. J Inherit Metab Dis. 2016;39(5):661–72.
Fraser JL, Venditti CP. Methylmalonic and propionic acidemias: clinical management update. Curr Opin Pediatr. 2016;28(6):682–93.
Oplin SE. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J Inherit Metab Dis. 2013;36(4):645–58.
Ogier de Baulny H, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106(1):12–7.
Martinelli D, Diodato D, Ponzi E, Monné M, Boenzi S, Bertini E, Fiermonte G, Dionisi-Vici C. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J Rare Dis. 2015;10:29.
Palladino AA, Stanley CA. The hyperinsulinism/hyperammonemia syndrome. Rev Endocr Metab Disord. 2010;11(3):171–8.
Häberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch Biochem Biophys. 2013;536(2):101–8.
Magner M, Dvorakova V, Tesarova M, Mazurova S, Hansikova H, Zahorec M, Brennerova K, Bzduch V, Spiegel R, Horovitz Y, Mandel H, Eminoğlu FT, Mayr JA, Koch J, Martinelli D, Bertini E, Konstantopoulou V, Smet J, Rahman S, Broomfield A, Stojanović V, Dionisi-Vici C, van Coster R, Morava E, Sperl W, Zeman J, Honzik T. TMEM70 deficiency: long-term outcome of 48 patients. J Inherit Metab Dis. 2015;38(3):417–26.
Diez-Fernandez C, Rüfenacht V, Santra S, Lund AM, Santer R, Lindner M, Tangeraas T, Unsinn C, de Lonlay P, Burlina A, van Karnebeek CD, Häberle J. Defective hepatic bicarbonate production due to carbonic anhydrase VA deficiency leads to early-onset life-threatening metabolic crisis. Genet Med. 2016;18(10):991–1000.
Auron A, Brophy PD. Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr Nephrol. 2012;27(2):207–22.
Häberle J. Clinical practice: the management of hyperammonemia. Eur J Pediatr. 2011;170(1):21–34.
Lang W, Blöck TM, Zander R. Solubility of NH3 and apparent pK of NH4+ in human plasma, isotonic salt solutions and water at 37 degrees C. Clin Chim Acta. 1998;273(1):43–58.
da Fonseca-Wollheim F, Heinze KG. Solubility of NH3 and apparent pK of NH4+ in human plasma, isotonic salt solutions and water at 37 degrees C. Eur J Clin Chem Clin Biochem. 1992;30(12):867–9.
Thrane V, Thrane A, Wang W, et al. Solubility of NH3 and apparent pK of NH4+ in human plasma, isotonic salt solutions and water at 37 degrees C. Nat Med. 2013;19(12):1643–8.
Berry E, Rhead S, Brusilow W, Hamosh S. Survival after treatment with phenyl acetate and benzoate for urea cycle disorders. NEJM. 2007;356:2282–92.
Uchino T, Endo F, Matsuda I. Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis. 1998;21(Suppl 1):151–9.
Msall M, Batshaw ML, Suus R, Brusilow SW, Mellitis ED. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea-cycle enzymopathies. N Engl J Med. 1984;310(23):1500–5.
Westrope C, Morris K, Burford D, Morrison G. Continuous hemofiltration in the control of neonatal hyperammonemia: a 10-year experience. Pediatr Nephrol. 2010;25(9):1725–30.
Picca S, Dionisi-Vici C, Bartuli A, De Palo T, Papadia F, Montini G, Materassi M, Donati MA, Verrina E, Schiaffino MC, Pecoraro C, Iaccarino E, Vidal E, Burlina A, Emma F. Short-term survival of hyperammonemic neonates treated with dialysis. Pediatr Nephrol. 2015;30:839–47.
Schaefer F, Straube E, Oh J, Mayatepeck E. Dialysis in neonates with inborn errors of metabolism. Nephrol Dial Transplant. 1999;14:910–8.
Arbeiter AK, Kranz B, Wingen AM, Bonzel KE, Dohna-Schwake C, Hanssler L, Neudorf U, Hoyer PF, Büscher R. Continuous venovenous haemodialysis (CVVHD) and continuous peritoneal dialysis (CPD) in the acute management of 21 children with inborn errors of metabolism. Nephrol Dial Transplant. 2010;25(4):1257–65.
McBryde KD, Kershaw DB, Bunchman TE, Maxvold NJ, Mottes TA, Kudelka TL, Brophy PD. Renal replacement therapy in the treatment of confirmed or suspected inborn errors of metabolism. J Pediatr. 2006;148:770–8.
Spinale JM, Laskin BL, Sondheimer N, Swartz SJ, Goldstein SL. High-dose continuous renal replacement therapy for neonatal hyperammonemia. Pediatr Nephrol. 2013;28:983–6.
Hackbarth R, Bunchman TE, Chua AN, et al. The effect of vascular access location and size on circuit survival in pediatric continuous renal replacement therapy: a report from the PPCRRT registry. Int J Artif Organs. 2007;30:1116–21.
Picca S, Dionisi-Vici C, Abeni D, Pastore A, Rizzo C, Orzalesi M, Sabetta G, Rizzoni G, Bartuli A. Extracorporeal dialysis in neonatal hyperammonemia: modalities and prognostic indicators. Pediatr Nephrol. 2001;16(11):862–7.
Lai Y-C, Huang H-P, Tsai I-J, Tsau Y-K. High-volume continuous venovenous hemofiltration as an effective therapy for acute management of inborn errors of metabolism in young children. Blood Purif. 2007;25:303–8.
Clark WR, Turk JE, Kraus MA, Gao D. Dose determinants in continuous renal replacement therapy. Int J Artif Organs. 2003;27:815–20.
Huang Z, Letteri J, Clark WJ, Ronco C, Gao D. Operational characteristics of continuous renal replacement modalities used for critically ill patients with acute kidney injury. Int J Artif Organs. 2008;31(6):525–34.
Troyanov S, Cardinal J, Geadah D, Parent D, Courteau S, et al. Solute clearances during continuous venovenous haemofiltration at various ultrafiltration flow rates using Multiflow-100 and HF1000 filters. Nephrol Dial Transplant. 2003;18:961–6.
Little MA, Conlon PJ, Walshe JJ. Access recirculation in temporary hemodialysis catheters as measured by the saline dilution technique. Am J Kidney Dis. 2000;6:1135–9.
Bunchman TE, Barletta GM, Winters JW, Gardner JJ, Crumb TL, McBryde KD. Phenylacetate and benzoate clearance in a hyperammonemic infant on sequential hemodialysis and haemofiltration. Pediatr Nephrol. 2007;22(7):1062–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Vara, R., Durward, A. (2018). Hyperammonaemia and IEM. In: Deep, A., Goldstein, S. (eds) Critical Care Nephrology and Renal Replacement Therapy in Children. Springer, Cham. https://doi.org/10.1007/978-3-319-90281-4_27
Download citation
DOI: https://doi.org/10.1007/978-3-319-90281-4_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-90280-7
Online ISBN: 978-3-319-90281-4
eBook Packages: MedicineMedicine (R0)