
Abstract
Stroke is the leading cause of death and disability worldwide (Shan and Guo, BMC Neurol 17:33, 2017). Neuroinflammation plays a significant role in the pathogenesis of stroke. In this chapter, we will first review the initial factors that trigger neuroinflammation in the ischemic brain. We will then summarize the main molecules involved in neuroinflammation after stroke as well as the major inflammatory cells derived from brain resident cells and circulating blood. In addition, we will discuss the relationship between post-ischemic inflammation and brain repairs. Lastly, anti-inflammatory therapies will be summarized. The aim of this chapter is not to meticulously review all of the abovementioned aspects, but to provide an overview of the essential components to understand neuroinflammation after stroke.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
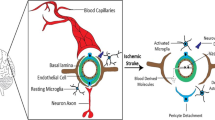
Neuroinflammation after ischemic stroke may destroy, or protect and repair brain tissues [1]. Inflammatory response after ischemic stroke involves the activation of brain-resident cells and the infiltration of peripheral blood cells into the brain parenchyma [2]. Initially, ischemic stroke results in neuronal necrosis and the production of reactive oxygen species (ROS), which triggers microglia activities. This leads to a series of inflammatory reactions, and promotes the secretion of inflammatory cytokines and chemokines cytokines, such as interleukins 1 (IL-1) and tumor necrosis factor α (TNF-α). These inflammatory mediators up-regulate the protein expressions of cerebrovascular wall attached molecules (such as P-select element and E-select element), which are also released into the peripheral blood to promote the activation of peripheral leukocytes. The expressions of adhesion molecules and chemotactic factors prompt the peripheral white blood cells to be attracted to the vascular endothelium and to infiltrate the brain parenchyma [3]. As a result, both the activated microglia/macrophages and the infiltrated leukocytes secrete cytotoxic inflammatory mediators, such as matrix metalloproteinases (MMPs), nitric oxide (NO), cytokines and ROS, enhance brain edema, promoting hemorrhagic transformation, and aggravating neuronal death.
We will discuss the major cascades of inflammatory responses involved in cerebral ischemia and brain injury. We will first introduce the factors that initiate or trigger neuroinflammation after stroke, and the associated cell signaling pathways during neuroinflammation. We will then summarize the major inflammatory products and molecules that modulate neuroinflammation. Thereafter, we will discuss the inflammatory cells which participate in brain injury after stroke, and the relationship between neuroinflammation and brain repair will be clarified. Lastly, we will discuss the current experimental evidence for anti-inflammatory therapies against stroke.
2 Neuroinflammatory Triggers, Molecules, and the Associated Cell Signaling Pathways After Stroke
The injured brain cells induce various inflammatory mediators and damage-associated molecular patterns (DAMPs), which enhance the recruitment of peripheral circulating inflammatory cells, making them more efficient participants in promoting inflammation [4]. After stroke, the lack of adequate energy delivery to the ischemic core causes a disruption of the ATP-dependent ionic gradient maintenance across the neuronal membrane, resulting in cellular swelling and organelle ruptures [5], increased intracellular calcium levels, excitotoxicity, and reactive oxygen species (ROS) production [6]. Following the initial ischemic injury, the complement system is activated, the blood-brain barrier (BBB) is interrupted, and matrix metalloproteases (MMPs) and adhesion molecules are induced [7]. These activities, along with the initial brain injury, promote DAMPs production in the ischemic brain [8], which stimulate intracellular and extracellular pattern recognition receptors (PRRs), initiating neuroinflammation. In this section, we will summarize the major DAMP and PRR proteins, and the associated cell signaling pathways.
2.1 DAMPs
DAMPsare released from stressed or damaged cells. Various substances have been found to be DAMPs, including purines (ATP and UTP), mRNA, hyaluronic acid, heat shock proteins (HSPs), high-mobility group box 1 (HMGB1) protein, and the peroxiredoxin family. For instance, purines (ATP and UTP) are released from the injured brain cells and their receptors, P2X and P2Y, function as alerting signals in the CNS. ATP also activates inflammasomes, which are large multimolecular complexes that control the activity of the proteolytic enzyme caspase-1, that cleaves pro-IL-1β to an active 17 kDa form. These DAMPs are similar to pathogen associated molecular patterns (PAMPs) expressed on the surface of gram negative bacteria [9], which are recognized by the Toll-like receptor (TLRs). DAMPS are also named as danger signals or alarmins, which induce chemotaxis and interact with the receptors on antigen-presenting cells, dendritic cells and macrophages. DAMPs also induce the activation of other PRRs, such as the receptor for advanced glycation end products (RAGE) and c-type lectin receptors [10].
HMGB1, also known as amphoterin or HMG1, is normally localized in the cell nuclei in normal brain cells, but in necrotic cells, it translocates into the cytosolic compartment, and then released into the extracellular space after stroke. It can be secreted by activated monocytes, macrophage, myeloid dendritic cells, and natural killer cells (NK cells), in response to endotoxin and other inflammatory stimuli. HMGB1 promotes necrosis and the influx of damaging inflammatory cells after ischemic stroke. The mechanism of inflammation and damage is by binding to TLR4, which mediates HMGB1-dependent activation of macrophage cytokine release. HMGB1 also activates inflammatory cells through the multiple surface receptors including TLR2, TLR4, and RAGE. RAGE expression is usually low under normal conditions, but increases to high levels upon the occurrence of inflammation [11]. TLR2 and TLR4 have been shown to be involved in infectious diseases. The expression levels of TLR4 have been found to be considerably high in brain regions that lack a tight BBB (blood-brain barrier), such as the circumventricular organs and choroid plexus [12]. The activation of NF-κB and MAP kinase usually follows the activation process of HMGB1 receptors. Recent studies indicate that a blockade of either RAGE or TLR4 contributes to the reduction of cytokine and nitric oxide production and decrease of inflammation, which indicates that HMGB1 potently participates in t inflammation induction [13].
2.2 Pattern-Recognition Receptor (PRR)
The major PRRs in the ischemic brain include Toll-like receptors (TLRs) and RAGE. Toll-like receptors TLRs) play an important role in post-ischemic inflammation [14]. They are expressed in macrophages and dendritic cells that recognize structurally conserved molecules derived from microbes. TLRs play key roles in proinflammatory cytokine expressions, leading to inflammatory responses and brain tissue damage. The activation of microglial cells during ischemic stroke has a close relationship with the significant induction of several TLRs, especially TLR2 and TLR4. In addition, TLRs are widely expressed on granulocytic cells and their activation is pivotal for the recruitment of bone-borne cells to the ischemic area.
The activation of TLRs initiates signaling events that stimulate the production of proinflammatory cytokines and chemokines, leading to the recruitment of leukocytes, especially tneutrophils and macrophages. The production of ROS and nitric oxide derivatives leads to the recruitment of leukocytes to the ischemic area, which subsequently exaggerate the initial damage. Recognition of TLRs on DCs enhances antigen presentations to the adaptive immunity elements. These events induce an early innate immune response, followed by the delayed T-cell activation phase. In fact, T cells play a pivotal role in the pathogenesis of ischemic stroke, depending on their cytokine production.
RAGE is a multi-ligand receptor, consisting of three Ig-domains (V1, C1 and C2), the transmembrane domain, and the cytosolic tail, required for RAGE-dependent cell signaling. The distal Ig-domain (V1) is implicated in the binding of HMGB1 and other RAGE ligands. HMGB1/RAGE regulates the cytoskeleton and the migratory responses of cells through the small GTPases, Cdc42 and Rac. Furthermore, HMGB1 binding to RAGE regulates migration through rapid integrin activation that requires the small GTPase, Rap1 [15]. Ligand binding to RAGE broadly regulates gene expression through the transcription factors CREB, SP1 and NF-κB. RAGE ligation activates the transcription of several plasma membrane proteins, such as the VCAM-1, ICAM-1 and the adhesion molecules, AMIGO. Additionally, RAGE signaling also plays a pivotal role in inducing the expression of intracellular proteins, including Bcl-2 and chromogranin B. RAGE not only plays a key role in regulating cell motility through the modulation of the cytoskeleton, HMGB1/RAGE has also been indicated in regulating integrin functions.
3 The Production of Inflammatory Molecules
The inflammatory reaction after stroke is associated with a variety of inflammatory mediators [16], such as cytokines, chemokines, and cell adhesion molecules, which will be reviewed in this section.
3.1 Cytokines
Cytokines are small proteins secreted by specific cell types, promoting interactions between immune cells, thus modulating inflammation. There are pro-inflammatory cytokines, including IL-1β, TNFα, and IL-6, and anti-inflammatory cytokines, including IL-4, IL-10, and TGFβ. These cytokines are secreted by various cells, including T cells, monocytes, and macrophages. Their functions are to promote or inhibit activities of these inflammatory cells.
IL-1β is one of the most important cytokines that affect brain injury and neuroinflammation after stroke. It is synthesized as a precursor protein with a small molecular mass of approximately 33 kD, mainly in monocytes and macrophages. Upon stimulations, the precursor form of IL-1βhas minimal biological activity and becomes functional IL-1β by cleaving with the protease caspase-1; the activity of caspase-1 is modulated in inflammasome [17]. Thus, the mechanism of IL-1β production and caspase-1 activation regulated by inflammasome has received great attention. It is suggested that inflammasome is activated via hypoxia or ATP [17], and following the activation of caspase-1, generates active IL-1β. IL-1β is produced from monocytes and macrophages, which are activated by endogenous Toll-like receptor (TLR) ligands [18]. In contrast, gene KO of TLR2/4 results in reductions in IL-1β mRNA levels in infiltrating mononuclear cells in TLR2/4-double deficient mice after ischemic stroke.
IL-1β plays an important role in inducing the apoptosis of neuronal cells and contributes to the production of chemokine in astrocytes and microglia [19]. IL-1β is induced and expressed in the damaged brain tissue 30 min after ischemic stroke onset. IL-1β function loss is associated with infarct size reduction, which indirectly indicates that IL-1β might be a neurotoxic mediator [20].
Another important cytokine in stroke-induced inflammation is TNF-α, which is involved in the progress of acute pathological reaction of ischemic stroke, and contributes to the initiation of systemic inflammation [21]. Its expression initially increases at 1–3 h after the ischemic onset, and then reaches a second peak at 24–36 h. TNF-α protein expressions first occur in neurons, especially during the first hours after the ischemic stroke, then in microglia/macrophages, and in blood-borne inflammatory cells [22]. TNF-α also induces the expression of adhesion molecules via cerebral endothelial and glial cells; these adhesion molecules then stimulate the neutrophil accumulation and migration in the microvasculatures [23]. Additionally, TNF-α may also be implicated in BBB impairment, implement the phenotypic transformation of the endothelial cells to contribute to a pro-coagulant milieu in the cerebral microvessels, and activate glial cells to form scars as part of a repair and remodeling after ischemic tissue damage [24].
3.2 Chemokines
Chemokines are another set of cytokines. Their major function is to induce chemotaxis, i.e. they guide leukocyte migration to the inflammatory tissues. Various chemokines increase upon stroke onset. The migration of monocytes/macrophages requires the chemokine CCL2, which functions via the chemokine receptor CCR2, to attract monocytes/macrophages out of the bone marrow and into the ischemic brain. CCL2 is a small cytokine that belongs to the CC chemokine family. It is also known as monocyte chemoattractant protein 1 (MCP1) and small inducible cytokine A2 [25]. In addition to monocytes, CCL2 is also responsible for the recruitment of dendritic cells and memory T cells to the inflammation produced by ischemic insult [26]. Chemokines express in the infarct region. Intracerebroventricular injection (ICVI) of a chemokine-receptor antagonist (viral macrophage inflammatory protein-II) reduces infarct volume [27]. CCL2 expression is induced in response to ischemic insult, playing a key role in the recruitment of monocytes in ischemic tissue injury [28].
In contrast with CCL2, CCL5is another chemokine, which is responsible for the recruitment of T cells, basophils, and eosinophils, and plays an important role in recruiting leukocytes into ischemic lesions [29]. Aided by particular cytokines released by T cells, CCL5 also induces the proliferation and activation of certain natural-killer (NK) cells to form CHAK (CC-Chemokine-activated killer) cells [22]. CCL5 released from blood-borne cells is a pivotal mediator upon reperfusion. Recently, it is widely reported that CCL5 induced the recruitment of leukocyte and platelet adhesion in the cerebral microvasculature [30]. It has been suggested that CCL5 induces the migration of peripheral blood-borne cells across the impaired BBB by recognizing CCR1 and CCR5 receptors and adhesion molecules [31]. Additionally, CCL5 also increases cerebral damage through the secondary induction of other potent pro-inflammatory cytokines, such as IL-6.
3.3 MMPs
Another important group of molecules implicated in neuroinflammation are Matrix metalloproteinases (MMPs). MMPs, also known as matrixins, are calcium-dependent zinc-containing endopeptidases belonging to a larger family of proteases known as the metzincin superfamily [32]. Collectively, these enzymes are capable of degrading all kinds of extracellular matrix proteins, and can also process a number of bioactive molecules. They are known to be involved in the cleavage of cell surface receptors, the release of apoptotic ligands (such as the FAS ligand), and chemokine/cytokine inactivation [33]. MMPs are also thought to play a major role in cell behaviors such as cell proliferation, migration (adhesion/dispersion), differentiation, angiogenesis, apoptosis, and host defense. MMPs are essential neurotoxic mediators that promote BBB breakdown and post-ischemic inflammation. Functionally similar to IL-1β, MMPs induce apoptotic neuronal cell death by TNF-α and Fas ligands [34]. The neurotoxic function of MMP-9 is particularly established, given that infarct size is reduced in MMP-9-deficient mice compared to that in control mice.
3.4 Cell Adhesion Molecules
The adhesion, migration, and infiltration of blood leukocytes into the ischemic brain requires the involvement of cell adhesion molecules (CAMs), which are proteins that express on the inflammatory cell surface. They are involved in binding with other cells or with the extracellular matrix (ECM) in the process of cell adhesion. CAMs consist of three protein families: the integrins, the selectins, and Ig (immunoglobulin superfamily).
The integrin family includes heterodimeric membrane glycoproteins with anαand aβsubunit, which play an important role in ECM and cell-cell interactions [35]. Integrins are transmembrane receptors that stimulate cell-extracellular matrix (ECM) adhesion. Upon ligand binding, integrins are responsible for activating signal transduction pathways, which regulate cellular signals such as organization of the intracellular cytoskeleton, regulation of the cell cycle, and movement of new receptors to the cell membrane [36]. In the brain, integrins also play a pivotal role to unite the endothelial cells, astrocytes, and basal lamina that consist of the blood-brain barrier; consequently, they are extremely essential in maintaining the integrity of the cerebral microvasculature [37]. Another integrin, named α6β4 (CD104), interferes with the normal interactions between laminin-5 and astrocytes in the ECM upon ischemic insult. Above all, it is widely accepted that these integrins are involved in the progression of platelet activation [38], promoting adherence of neutrophils to endothelium and mediating cell-ECM interactions.
The selectins are a family of cell adhesion molecules that bind to sugar moieties or polymers [29], including P-, E-, and L-selectin. P-selectin expresses on platelets and endothelial cells, and its counter receptor on leukocytes consists of the oligosaccharide sialyl-Lewis X. The early re-localization of P-selectin facilitates the primary adhesion of leukocytes [39]. Nevertheless, it also plays a continuing role because its expression increases in post-ischemic cerebral vasculature.
E-selectin expresses on activated endothelium, in sequence after P-selectin under ischemic insult [40]. It has been suggested that transnasal E-selectin tolerization induces tolerance to a secondary ischemic insult in experimental stroke. Currently, a large number of studies are underway to advance these studies to clinical trials with the ultimate goal of using this form of immunomodulation for the secondary prevention of stroke [41].
L-selectin, which expresses on both endothelium and leukocytes, was originally identified as being a significant factor in homing in to the sites of infection and to peripheral lymph nodes. In addition to promoting neutrophil rolling [42], L-selectin also mediates neutrophil attachment to endothelial cells through interactions that are independent of integrin CD18 [43].
The last CAMs we will discuss are the Immunoglobulin superfamily. There are five major members of the immunoglobulin superfamily: Vascular cell adhesion molecule-1 (VCAM-1), Intercellular adhesion molecule-1(ICAM-1), Intercellular adhesion molecule-2 (ICAM-2), Mucosal vascular addressin cell adhesion molecule 1(CD146), and Platelet-endothelial cell adhesion molecule-1 (PECAM-1) [44] which are expressed on activated endothelial cells. Members of the immunoglobulin superfamily mediate a stronger binding of leukocytes to the vascular endothelium than the selectins under cerebral ischemia [45]. These cell adhesion molecules play an important role in the tight adhesion of leukocytes under ischemic insult, as well as the unique role of ICAM-2 in leukocyte-platelet interactions [46]. Therefore, treatments targeting the CAM family may interfere with the first step in the inflammatory pathway, resulting in the blocking of leukocyte transmigration into the ischemic lesions [47].
4 Inflammatory Cells and Ischemic Stroke
In the previous sections, we have discussed the initiation of post-ischemic inflammation and the molecules that modulate the inflammatory response. These inflammatory triggers and molecules modulate the central players of neuroinflammation, inflammatory cells, including both brain resident cells and peripheral blood leukocytes, which we will discuss in this section.
4.1 Brain Resident Cells in Ischemic Stroke
As we have discussed, the first immune cells activated in the ischemic lesion are the resident microglia in the brain. Microglia, the counterparts of monocytes/macrophages in the blood and other organs, are the main long-living resident immune cells in the central nervous system (CNS). Microglia have the ability to continuously sense and scan their environment for injuries and pathogens, then reacting to the damage-induced signals to protect against brain injury. Therefore, microglia maintain a stable chemical and physical microenvironment necessary for the CNS to conduct its normal functions [48]. When ischemic stroke occurs, microglia activate rapidly in response to ischemic injury [49]: their reactions reaching peaks at 2–3 days after stroke and lasting for several weeks. The role of microglia in ischemic stroke is a double-edged sword, as microglia produce inflammatory factors leading to cell damage and death. Microglia can also produce TGF-β1, which protects the central nervous system (CNS) [50].
Astrocytes are specialized and the most abundant cell type in the CNS, playing important roles in CNS disorders, including ischemic stroke [51]. Reactive astrogliosis is one of the important pathological features of ischemic stroke, which is accompanied by changes in morphology, proliferation, and gene expressions in the reactive astrocytes. Glial fibrillary acidic protein (GFAP) is overexpressed on the astrocytes after stroke [52], indicating the progress of astrocyte proliferation and differentiation. Astrocytes eventually fill up the empty space in the damaged brain to form a glial scar, replacing the CNS cells that cannot regenerate.
4.2 Peripheral Blood-Borne Cells in Ischemic Stroke
Many types of blood leukocytes participate in neuroinflammation and modulate brain injury after stroke, including neutrophils, monocytes, T cells, B cells, and NK cells. Neutrophils are the first leukocytes to infiltrate into the ischemic brain, acting from 30 min to a few hours after focal cerebral ischemia, then peaking from days 1–3, and rapidly decreasing thereafter [53]. Infiltrating neutrophils contribute to inflammation and brain injury by releasing pro-inflammatory factors, including MMPs and inducible nitric oxide (iNOS) [17]. As neutrophils are detrimental to brain injury, ischemic brain injury might be decreased by an immuno-depletion of neutrophils or an antibody blockage of neutrophil infiltration [54].
A few hours after stroke onset, monocytes are recruited into the ischemic brain, participating in neuroinflammation. There are at least two types of blood monocytes, which can be divided into “pro-inflammatory” (Ly-6Chigh/CCR2+) and “anti-inflammatory” (Ly-6Clow/CCR2−) subpopulations [55]. Monocyte/macrophage expressions can be divided into two types: inflammatory M1 and anti-inflammatory M2 macrophages [56]. M1 macrophages produce inflammatory responses by secreting inflammatory factors, such as IL-1β and TNF-α, and overexpressing iNOS, while M2 macrophages produce anti-inflammatory cytokines, such as IL-10 and TGF-β. As a result, M1 macrophages promote brain injury while M2 macrophages inhibit brain injury and promote brain recovery. Nevertheless, monocyte derived macrophages are not the only sources of M1 and M2 macrophages. Microglia are another major source of M1 and M2 macrophages, but the distinctive roles of microglia and monocytes in M1 and M2 polarization remain unknown.
Blood lymphocytes also play significant roles in neuroinflammation and brain injury [57]. Among the various types of lymphocytes, the significant role of T cells has been extensively studied [58], including CD4+, CD8+ T cells, γδT cells, and Treg cells in the pathogenesis of ischemic stroke [21]. The depletion of CD4 or CD8 T cells attenuates brain injury; γδT cells promote brain injury, while Treg activity attenuates brain injury and promotes functional recovery after stroke.
Lastly, dendritic cells (DC) play an important role in connecting innate and adaptive immunity. CD11c+ DC cells are increased in the ipsilateral hemisphere, peaking at 72 h after ischemic stroke. In addition, major histocompatibility complex (MHC)-II and co-stimulatory molecules are up-regulated in the brain resident and peripheral DC, which may be responsible for lymphocyte activation. Furthermore, the brain-resident DC might be involved in directing the early, local immune response and, later, participate in the recruitment of lymphocytes upon activation by INF-γ.
5 Post-ischemic Inflammation and Brain Repair
Neuroinflammation occurs after stroke in the ischemic brain, and is designed to remove necrotic tissues and promote lesion resolution. Generally, post-stroke neurorestoration is achieved by enhancing neurogenesis, oligodendrogenesis and angiogenesis, which collaboratively contribute to neurological recovery [59]. Neurogenesis and oligodendrogenesis produce new parenchymal cells from neural stem cells which promote plasticity, restore neuronal signal transduction, and stimulate myelination [60]. Angiogenesis and arteriogenesis are the major forms of vascular remodeling, which contribute to increased cerebral blood flow (CBF). In addition, they generate trophic factors and proteases, which are crucial for restoration by helping to construct an environment for remyelination and neurite outgrowth [61].
5.1 The Removal of Necrotic Cells
The crucial process in brain repair mainly consists of the suppression of inflammation and clearing of dead cells [62], in which neutrophils and macrophages play critical roles. Macrophages include monocytes and microglia derived macrophages, which polarize into pro-inflammatory M1 and anti-inflammatory M2. The M2 macrophages are particularly important for inflammation resolution and brain repair. Nevertheless, how microglia and monocyte derived macrophages contribute to brain repair has not been distinguished. The phygocytotic mechanisms of macrophages after stroke are not completely understood. Nevertheless, the results of cultured microglia have indicated that ATP signaling through the G protein-coupled P2Y receptors results in rapid microglial membrane ruffling and whole-cell migration. ATP, ADP and UTP might be potent agonists for P2Y G protein-coupled receptors and P2X ligand-gated ion channels [63]. The microglia/macrophages are attracted by UTP and ATP through P2Y2 receptors. In addition, UDP acts on P2Y6 receptors to stimulate microglial phagocytosis. Furthermore, phosphatidylserine (PtdSer), which translocates to the outer leaflet of the plasma membrane of apoptotic cells, enables the apoptotic cells to be recognized by the phygocytotic cells and removed [64]. Other PtdSer binding proteins include MGF-E8 on microglia and TIM4 on macrophages, which also participate in the clearance of dead cells [65].
5.2 Oligodendrogenesis
Oligodendrocytes are quite sensitive to ischemic insult because white matter has lower blood flow than gray matter, and deep white matter has little collateral blood supply [66]. Post-ischemic neuroinflammation may have a detrimental effect on white matter cohesion by upregulating matrix MMPs, as both MMP-2 and MMP-9 exacerbate white matter lesions [67]. Oligodendrocyte progenitor cells (OPC) differentiate into mature oligodendrocytes under ischemic insult [68], and produce myelin sheaths, which wrap around axons, facilitating nerve conduction. Therefore, oligodendrogenesis plays an important role in behavioral and functional restoration after ischemic stroke [69].
Oligodendrogenesis after stroke occurs by recruiting resident OPCs from white and gray matter and generated by SVZ neural progenitor cells; these new oligodendrocytes become mature myelinating oligodendrocytes [70]. Such OPCs generated in the SVZ are observed in humans after demyelination [71]. These newly produced OPCs migrate to peri-infarct gray and white matter to participate in oligodendrogenesis. This OPCs migration is modulated by stromal-derived factor 1α (SDF-1α) and vascular endothelial growth factor (VEGF), secreted by activated cerebral endothelial cells in the ischemic boundary region [72]. In addition, glutamatergic signals resulting from damaged axons in the corpus callosum also induce the migration of OPCs from the SVZ to the peri-infarct areas.
5.3 Angiogenesis
As a crucial growth factor in post-ischemic angiogenesis, VEGF is generated by reactive astrocytes. Its action might need neutrophil MMPs, which indicates an association between inflammation and angiogenesis [73]. Angiogenesis usually occurs in the penumbra of the human ischemic brain hours after initial onset and continues to exist for several weeks [74]. The first step in angiogenesis is the nitric oxide (NO)-initiated vasodilation. Combining the vasodilation effect of NO with the increase in VEGF expression, which increases vascular permeability, allows the extravasation of plasma proteins that lay down a provisional scaffold for the migration of endothelial cells for vascular sprouting [75]. The second step starts with the dissociation of smooth muscle cells (SMCs) and loosening of the extracellular matrix that enwraps the mature vessels [76]. Angiopoietin-2 (Ang2), a Tie2 signaling inhibitor, may take part in stimulating the detachment of pericytes from endothelial cells, whereas the MMP family of proteinases decreases the matrix molecules, and then weakens vascular integrity [77]. The proliferating endothelial cells may migrate to distant sites as the sprouting path has been established. Consequently, the angiogenic process will be mediated vy a series of molecular signals such as VEGF, VEGF receptors, and placental growth factors [78]. Additionally, Ang1 will activate Tie2 receptors when the new blood vessel networks are formed [79], which will help to stabilize the networks initiated by VEGF.
6 Anti-Inflammatory Therapy in Ischemic Stroke
Since post-ischemic inflammation exacerbates brain injury after stroke, inflammation has been a target for stroke therapy. In this section, we will review some strategies targeted against pro-inflammatory responses.
6.1 Targeting Inflammasome Signaling Pathways
Inflammasome is a pivotal mediator in inflammation. This involves the activation of pro-caspase-1 into cleaved caspase-1 [80], which initiates and amplifies the generation of pro-inflammatory cytokines IL-1β and IL-18, resulting in apoptotic neuronal and glial cell death following ischemic stroke [81]. Therefore, targeting the upstream or downstream cascades of the inflammasome signaling pathways, including protein expressions in the pathway, as well as its activity and products, may provide avenues for developing therapeutics against ischemic stroke [82]. There are many potential targets involved in inflammasome signaling which can be explored: plasma membrane receptors/channels (i.e. P2X7 receptors, Pannexin 1 and K+ channels), cytokines (i.e. IL-1β and IL-18), inflammasome components (i.e. NLRPs, ASC and caspase-1), secondary messengers (i.e. ROS and PKR) [83], signaling pathways (i.e. NF-κB and MAPK) and cytokine receptors (i.e. IL-1R1and IL-18R) [84].
6.2 Targeting Neutrophil Recruitment
Clinical studies have tested three kinds of drugs or antibodies targeting neutrophil recruitment, which have potential values for ischemic stroke [85]: a humanized antibody against the CD11b/CD18, a mAb against ICAM-1, and the UK-279276. UK-279,276 is a recombinant glycoprotein, which inhibits neutrophil recruitment by selectively binding to the CD11b/CD18 integrin. Some studies have shown that these treatments are well tolerated. However, the resulting side effects, such as leukopenia and immunosuppression, make them ineffective for stroke treatment [86]. It seems that there is still a long way to go in translating neutrophil inhibitors for effective clinical use [87].
6.3 Immunosuppression Strategies
As previously discussed, T cells also participate in neuroinflammation and modulate brain injury after stroke. T cells consist of a number of subsets, including both pro-inflammatory and anti-inflammatory subsets. These subsets either promote or inhibit inflammation, leading either to brain injury exacerbation or attenuation. Nevertheless, the overall inhibition of T cell functions have been shown to attenuate brain injuries. T cell functions can be inhibited with the immunosuppressant drugs: FK-506, rapamycin, and cyclosporin A (CsA), combined with immunophilins, which are a high-affinity receptor proteins in the cytoplasm. This combination causes rotamase inhibition in T cells, thus interrupts cell activation. FK506 and CsA complexes, with their immunophilin receptors, have been indicated to impact a variety of cellular immune responses by suppressing Ca2+-dependent serine/threonine phosphatase [88], calcineurin, and by mediating Ca2+ release via the ryanodine receptor. The rapamycin-FKBP12 complex suppresses the cell cycle process by mediating cell cycle kinases, including the PI3 kinase-like molecule known as ‘target of rapamycin (TOR)’ [89]. There are many other signaling pathways shown to be modulated by immunophilins such as, the interaction with heat-shock proteins, glucocorticoid receptor (s), nitric oxide synthase activity, the transforming growth factor receptor, and protein folding [90]; which have been involved in the immunomodulatory effects of immunophilin-binding immunosuppressants.
7 Summary
Neuroinflammation plays a crucial role in the pathogenesis of ischemic insult [91]. In this chapter, we have reviewed the initial trigger factors, summarized the molecules involved in post stroke neuroinflammation as well as the major inflammatory cells; the relationship between inflammation and brain repair, as well as possible anti-inflammatory therapies after stroke. Although there are many experimental results suggesting promising strategies to inhibit inflammation and attenuate brain injury, anti-inflammatory therapies for clinical translation have been unsuccessful. Therefore, there is still much to be done before pre-clinical trials can be translated for clinical use. As the role of inflammatory response upon ischemic insult is a double-edged sword, the challenge on how to curb its detrimental effects while promoting its beneficial roles for functional brain recovery remains.
Future research should synthesize each situation regarding pro- and anti-inflammatory responses under ischemic conditions, not by studying and analyzing them separately.
Many outstanding questions still remain: how to identify the exact dynamic balance between pro-and anti-inflammation generated during the different stages of ischemic stroke? Which pro-inflammatory or anti-inflammatory mediators should be targeted for therapy? How and when should the pro-inflammatory or anti-inflammatory pathways be activated or inhibited for treatment? Therefore, in order to design effective therapies, we must consider the dynamic balance between pro- and anti-inflammatory responses. Then we must identify the discrepancies between pre-clinical studies and clinical trials.
With a comprehensive understanding of the disease process, future studies will be able to identify a pipeline of new targets that control the cell signaling pathways and networks, rather than on a single mediator, and identify complementary holistic approaches to treat ischemic stroke.
References
Shan K, Guo W. Stroke caused by an inflammatory thrombus: a case report. BMC Neurol. 2017;17(1):33.
Silverman MG, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316(12):1289–97.
Bragg F, et al. Association between diabetes and cause-specific mortality in rural and urban areas of China. JAMA. 2017;317(3):280–9.
Fucikova J, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. 2016;128(26):3113–24.
Choi HW, Klessig DF. DAMPs, MAMPs, and NAMPs in plant innate immunity. BMC Plant Biol. 2016;16(1):232.
Thoudam T, et al. Role of mitochondria-associated endoplasmic reticulum membrane in inflammation-mediated metabolic diseases. Mediat Inflamm. 2016;2016:1851420.
Santos SC, et al. Immunomodulation after ischemic stroke: potential mechanisms and implications for therapy. Crit Care. 2016;20(1):391.
Nakayama T. An inflammatory response is essential for the development of adaptive immunity-immunogenicity and immunotoxicity. Vaccine. 2016;34(47):5815–8.
Versluys M, Tarkowski LP, Van den Ende W. Fructans as DAMPs or MAMPs: evolutionary prospects, cross-tolerance, and multistress resistance potential. Front Plant Sci. 2016;7:2061.
Lu L, et al. Innate immune regulations and liver ischemia-reperfusion injury. Transplantation. 2016;100(12):2601–10.
Gougeon ML, et al. HMGB1/anti-HMGB1 antibodies define a molecular signature of early stages of HIV-associated neurocognitive disorders (HAND). Heliyon. 2017;3(2):e00245.
Wang Y, et al. Cigarette smoke attenuates phagocytic ability of macrophages through down-regulating Milk fat globule-EGF factor 8 (MFG-E8) expressions. Sci Rep. 2017;7:42642.
Liu Y, et al. Blockade of HMGB1 preserves vascular homeostasis and improves blood perfusion in rats of acute limb ischemia/reperfusion. Microvasc Res. 2017;112:37–40.
Ji Y, et al. Temporal pattern of Toll-like receptor 9 upregulation in neurons and glial cells following cerebral ischemia reperfusion in mice. Int J Neurosci. 2016;126(3):269–77.
Olsson S, Jood K. Genetic variation in the receptor for advanced glycation end-products (RAGE) gene and ischaemic stroke. Eur J Neurol. 2013;20(6):991–3.
Vidale S, et al. Postischemic inflammation in acute stroke. J Clin Neurol. 2017;13(1):1–9.
Lee GA, et al. Interleukin 15 activates Akt to protect astrocytes from oxygen glucose deprivation-induced cell death. Cytokine. 2017;92:68–74.
Bronisz E, Kurkowska-Jastrzebska I. Matrix metalloproteinase 9 in epilepsy: the role of neuroinflammation in seizure development. Mediat Inflamm. 2016;2016:7369020.
Li N, et al. Bidirectional relationship of mast cells-neurovascular unit communication in neuroinflammation and its involvement in POCD. Behav Brain Res. 2017;322(Pt A):60–9.
Mijajlovic MD, et al. Post-stroke dementia – a comprehensive review. BMC Med. 2017;15(1):11.
Shukla V, et al. Cerebral ischemic damage in diabetes: an inflammatory perspective. J Neuroinflammation. 2017;14(1):21.
Zhang Y, et al. Effects of Shaoyao-Gancao decoction on infarcted cerebral cortical neurons: suppression of the inflammatory response following cerebral ischemia-reperfusion in a rat model. Biomed Res Int. 2016;2016:1859254.
Guo X, et al. miR-145 mediated the role of aspirin in resisting VSMCs proliferation and anti-inflammation through CD40. J Transl Med. 2016;14(1):211.
Kojima Y, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536(7614):86–90.
Elkind MS, et al. The levels of inflammatory markers in the treatment of stroke study (LIMITS): inflammatory biomarkers as risk predictors after lacunar stroke. Int J Stroke. 2010;5(2):117–25.
Reaux-Le GA, et al. Current status of chemokines in the adult CNS. Prog Neurobiol. 2013;104:67–92.
Wacker BK, Perfater JL, Gidday JM. Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J Neurochem. 2012;123(6):954–62.
Kwon MJ, Yoon HJ, Kim BG. Regeneration-associated macrophages: a novel approach to boost intrinsic regenerative capacity for axon regeneration. Neural Regen Res. 2016;11(9):1368–71.
Zemer-Wassercug N, et al. The effect of dabigatran and rivaroxaban on platelet reactivity and inflammatory markers. J Thromb Thrombolysis. 2015;40(3):340–6.
Sajedi KM, et al. Correlation of early and late ejection fractions with CCL5 and CCL18 levels in acute anterior myocardial infarction. Iran J Immunol. 2016;13(2):100–13.
Rom S, et al. miR-98 and let-7g* protect the blood-brain barrier under neuroinflammatory conditions. J Cereb Blood Flow Metab. 2015;35(12):1957–65.
Wang XH, You YP. Epigallocatechin gallate extends therapeutic window of recombinant tissue plasminogen activator treatment for brain ischemic stroke: a randomized double-blind and placebo-controlled trial. Clin Neuropharmacol. 2017;40(1):24–8.
Zhang HT, et al. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol Med Rep. 2017;15(1):57–64.
Kanazawa M, et al. Therapeutic strategies to attenuate hemorrhagic transformation after tissue plasminogen activator treatment for acute ischemic stroke. J Atheroscler Thromb. 2017;24(3):240–53.
Fujioka T, et al. Beta1 integrin signaling promotes neuronal migration along vascular scaffolds in the post-stroke brain. EBioMedicine. 2017;16:195–203.
Rom S, et al. PARP inhibition in leukocytes diminishes inflammation via effects on integrins/cytoskeleton and protects the blood-brain barrier. J Neuroinflammation. 2016;13(1):254.
Xu XR, et al. Platelets and platelet adhesion molecules: novel mechanisms of thrombosis and anti-thrombotic therapies. Thromb J. 2016;14(Suppl 1):29.
Huang H, et al. Cerebral ischemia-induced angiogenesis is dependent on tumor necrosis factor receptor 1-mediated upregulation of alpha5beta1 and alphaVbeta3 integrins. J Neuroinflammation. 2016;13(1):227.
Zhao J, et al. Cinnamaldehyde inhibits inflammation and brain damage in a mouse model of permanent cerebral ischaemia. Br J Pharmacol. 2015;172(20):5009–23.
Wu N, et al. Association of inflammatory and hemostatic markers with stroke and thromboembolic events in atrial fibrillation: a systematic review and meta-analysis. Can J Cardiol. 2015;31(3):278–86.
Kurkowska-Jastrzebska I, et al. Carotid intima media thickness and blood biomarkers of atherosclerosis in patients after stroke or myocardial infarction. Croat Med J. 2016;57(6):548–57.
Pusch G, et al. Early dynamics of P-selectin and interleukin 6 predicts outcomes in ischemic stroke. J Stroke Cerebrovasc Dis. 2015;24(8):1938–47.
Yang S, et al. Biomarkers associated with ischemic stroke in diabetes mellitus patients. Cardiovasc Toxicol. 2016;16(3):213–22.
Guo M, et al. Polymorphisms in the receptor for advanced glycation end products gene are associated with susceptibility to drug-resistant epilepsy. Neurosci Lett. 2016;619:137–41.
Zhang D, et al. Up-regulation of VCAM1 relates to neuronal apoptosis after intracerebral hemorrhage in adult rats. Neurochem Res. 2015;40(5):1042–52.
Lu W, Bromley-Coolidge S, Li J. Regulation of GABAergic synapse development by postsynaptic membrane proteins. Brain Res Bull. 2017;129:30–42.
Riehl A, et al. The receptor RAGE: bridging inflammation and cancer. Cell Commun Signal. 2009;7:12.
Schofield ZV, et al. Neutrophils—a key component of ischemia-reperfusion injury. Shock. 2013;40(6):463–70.
Tabas I. 2016 Russell Ross memorial lecture in vascular biology: molecular-cellular mechanisms in the progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37(2):183–9.
Altug CH, et al. Assessment of the relationship between serum vascular adhesion protein-1 (VAP-1) and severity of calcific aortic valve stenosis. J Heart Valve Dis. 2015;24(6):699–706.
Mandelbaum M, et al. A critical role for proinflammatory behavior of smooth muscle cells in hemodynamic initiation of intracranial aneurysm. PLoS One. 2013;8(9):e74357.
Tamma G, et al. Effect of roscovitine on intracellular calcium dynamics: differential enantioselective responses. Mol Pharm. 2013;10(12):4620–8.
Banjara M, Ghosh C. Sterile neuroinflammation and strategies for therapeutic intervention. Int J Inflamm. 2017;2017:8385961.
Courties G, Moskowitz MA, Nahrendorf M. The innate immune system after ischemic injury: lessons to be learned from the heart and brain. JAMA Neurol. 2014;71(2):233–6.
Pedersen DS, et al. Toxicological aspects of injectable gold-hyaluronan combination as a treatment for neuroinflammation. Histol Histopathol. 2014;29(4):447–56.
Zhang Y, et al. Treadmill exercise promotes neuroprotection against cerebral ischemia-reperfusion injury via downregulation of pro-inflammatory mediators. Neuropsychiatr Dis Treat. 2016;12:3161–73.
Perez-de-Puig I, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. 2015;129(2):239–57.
Harmon EY, et al. Anti-inflammatory immune skewing is atheroprotective: Apoe−/-FcgammaRIIb−/− mice develop fibrous carotid plaques. J Am Heart Assoc. 2014;3(6):e001232.
Yu JH, et al. Induction of neurorestoration from endogenous stem cells. Cell Transplant. 2016;25(5):863–82.
Jarosiewicz B, et al. Virtual typing by people with tetraplegia using a self-calibrating intracortical brain-computer interface. Sci Transl Med. 2015;7(313):313ra179.
Kaya AH, Erdogan H, Tasdemiroglu E. Searching evidences of stroke in animal models: a review of discrepancies a review of discrepancies. Turk Neurosurg. 2017;27(2):167–73.
Rossi PJ, et al. Proceedings of the third annual deep brain stimulation think tank: a review of emerging issues and technologies. Front Neurosci. 2016;10:119.
Duricki DA, et al. Delayed intramuscular human neurotrophin-3 improves recovery in adult and elderly rats after stroke. Brain. 2016;139(Pt 1):259–75.
ElAli A, Jean LN. The role of monocytes in ischemic stroke pathobiology: new avenues to explore. Front Aging Neurosci. 2016;8:29.
Azad TD, Veeravagu A, Steinberg GK. Neurorestoration after stroke. Neurosurg Focus. 2016;40(5):E2.
Di Cesare F, et al. Phosphodiesterase-5 inhibitor PF-03049423 effect on stroke recovery: a double-blind, placebo-controlled randomized clinical trial. J Stroke Cerebrovasc Dis. 2016;25(3):642–9.
Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016;144:103–20.
Popa-Wagner A, et al. Poststroke cell therapy of the aged brain. Neural Plast. 2015;2015:839638.
Wu X, et al. Long-term effectiveness of intensive therapy in chronic stroke. Neurorehabil Neural Repair. 2016;30(6):583–90.
Amar AP, Griffin JH, Zlokovic BV. Combined neurothrombectomy or thrombolysis with adjunctive delivery of 3K3A-activated protein C in acute ischemic stroke. Front Cell Neurosci. 2015;9:344.
Choi JC, et al. Effect of pre-stroke statin use on stroke severity and early functional recovery: a retrospective cohort study. BMC Neurol. 2015;15:120.
Wood H. Migraine: migraine is associated with increased risk of perioperative ischaemic stroke. Nat Rev Neurol. 2017;13(2):67.
Sullivan R, et al. A possible new focus for stroke treatment – migrating stem cells. Expert Opin Biol Ther. 2015;15(7):949–58.
Villapol S, et al. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain. 2015;138(Pt 11):3299–315.
Kongbunkiat K, et al. Leukoaraiosis, intracerebral hemorrhage, and functional outcome after acute stroke thrombolysis. Neurology. 2017;88(7):638–45.
Sun L, et al. L-Serine treatment may improve neurorestoration of rats after permanent focal cerebral ischemia potentially through improvement of neurorepair. PLoS One. 2014;9(3):e93405.
Algra A, Wermer MJ. Stroke in 2016: stroke is treatable, but prevention is the key. Nat Rev Neurol. 2017;13(2):78–9.
Meimounn M, et al. Intensity in the neurorehabilitation of spastic paresis. Rev Neurol (Paris). 2015;171(2):130–40.
Ruscher K, Wieloch T. The involvement of the sigma-1 receptor in neurodegeneration and neurorestoration. J Pharmacol Sci. 2015;127(1):30–5.
Abeliovich A, Gitler AD. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature. 2016;539(7628):207–16.
Jackson JL, et al. Associations of 25-hydroxyvitamin D with markers of inflammation, insulin resistance and obesity in black and white community-dwelling adults. J Clin Transl Endocrinol. 2016;5:21–5.
Gogia S, Kaiser Y, Tawakol A. Imaging high-risk atherosclerotic plaques with PET. Curr Treat Options Cardiovasc Med. 2016;18(12):76.
Liu CL, Zhang K, Chen G. Hydrogen therapy: from mechanism to cerebral diseases. Med Gas Res. 2016;6(1):48–54.
Liang LJ, Yang JM, Jin XC. Cocktail treatment, a promising strategy to treat acute cerebral ischemic stroke? Med Gas Res. 2016;6(1):33–8.
Katayama Y, et al. Neuroprotective effects of clarithromycin against neuronal damage in cerebral ischemia and in cultured neuronal cells after oxygen-glucose deprivation. Life Sci. 2017;168:7–15.
Anrather J, Iadecola C. Inflammation and stroke: an overview. Neurotherapeutics. 2016;13(4):661–70.
Satani N, Savitz SI. Is immunomodulation a principal mechanism underlying how cell-based therapies enhance stroke recovery? Neurotherapeutics. 2016;13(4):775–82.
Vafadari B, Salamian A, Kaczmarek L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem. 2016;139 Suppl 2:91–114.
Choi DH, Kang SH, Song H. Mean platelet volume: a potential biomarker of the risk and prognosis of heart disease. Korean J Intern Med. 2016;31(6):1009–17.
de Ramon L, et al. RNAi-based therapy in experimental ischemia-reperfusion injury. The new targets. Curr Pharm Des. 2016;22(30):4651–7.
Toraldo DM, et al. Statins may prevent atherosclerotic disease in OSA patients without co-morbidities? Curr Vasc Pharmacol. 2017;15(1):5–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Hao, J., Zheng, K., Zhao, H. (2018). Inflammation and Ischemic Stroke. In: Jiang, W., Yu, W., Qu, Y., Shi, Z., Luo, B., Zhang, J. (eds) Cerebral Ischemic Reperfusion Injuries (CIRI). Springer Series in Translational Stroke Research. Springer, Cham. https://doi.org/10.1007/978-3-319-90194-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-90194-7_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-90193-0
Online ISBN: 978-3-319-90194-7
eBook Packages: MedicineMedicine (R0)