
Abstract
Systemic lupus erythematosus (SLE) is a debilitating and life-threatening autoimmune disease affecting major organ systems including the kidneys, brain, joints, and skin. The etiology is unknown but microorganisms, both pathogenic and commensal, are implicated in the pathogenesis of SLE. An overview of the microbiome in human lupus and animal models is given with a summary of the most recent human microbiome association studies. Next, infectious agents with a pathogenic or protective role in the pathogenesis of SLE are summarized. The majority of the chapter reviews the contribution of infectious agents to the morbidity and mortality of SLE patients with a systematic overview organized by organ system. Invasive fungal infections and other serious infections are covered separately. Biomarkers such as C-reactive protein and procalcitonin are discussed in the context of an infectious episode versus a lupus flare. The chapter closes with a brief overview of the prevention of infections in SLE patients. In summary, this chapter encompasses the role of the microbiota and infectious agents in the pathogenesis of SLE, the infectious complications in SLE patients, and its detection and differentiation from lupus flares.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Systemic lupus erythematosus
- Microbiome
- Pathogenic microorganisms
- Pathogenesis
- Risk factors for infection
- Clinical manifestations of infection
- Morbidity and mortality
- Prevention of infection
Introduction
Systemic lupus erythematosus (SLE) is the epitome of autoimmune diseases. The pathogenesis, clinical manifestations, and management of SLE involve various aspects of microorganisms, either as potential triggers and perpetuators of disease, as infectious episodes, as complications from underlying immune dysregulation, or as adverse events from chronic immunosuppression. Despite significant progress that has been made in understanding the contributions of microorganisms to SLE, there are many questions that need to be answered.
Besides environmental factors, genetic risk factors have long been established to be important in the pathogenesis of autoimmune diseases including SLE. Several clinical case studies uncovered a family history of patients with either the same or closely related autoimmune diseases, which supports the possibility that a common genetic predisposition is at the core of autoimmune diseases [1]. Multiple genetic and genome-wide association studies have firmly established that HLA class II polymorphisms are a major genetic risk factor in many autoimmune diseases, as shown in detail for instance in rheumatoid arthritis (RA) [2, 3]. However, a genetically predisposed individual usually requires environmental exposures to initiate overt autoimmunity. With regard to potential triggers, infectious agents have long been implicated and are discussed herein.
The Role of Microorganisms in the Pathogenesis of SLE
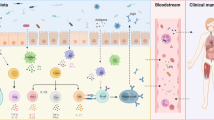
The Microbiome
Chronic inflammatory disorders such as type 1 diabetes, Crohn’s disease, or rheumatoid arthritis (RA) have been linked to gut microbial dysbiosis together with immune dysregulation. In addition, recent research suggests that early-life events and environmental factors play a significant role in the development of the adult immune system . In particular, diet and early-life exposures, e.g., infectious episodes or antibiotic exposures, influence immune cell numbers and functions. These factors are also influenced by the microbiota supporting an intricate interaction between environmental factors, the microbiota, and the host [4]. Additionally, the gut microbial communities change with increasing age, so another factor that contributes to dysbiosis is early-life perturbations [5]. Notably, several studies have shown that depletion of the gut microbiota in murine autoimmune models can affect autoantibody production as well as mortality [6]. Exact mechanisms through which the microbiota influences chronic immune-mediated diseases such as lupus remain to be established, but a multitude of effects on innate and adaptive immune cell functions are likely. In this chapter, we will focus on the effect of the microbiota on adaptive immune responses with implications for organ-specific and systemic autoimmunity and allude to potential roles in SLE.
Several groups have demonstrated in murine autoimmune models for multiple sclerosis or rheumatoid arthritis that Th17 cells, a subpopulation of CD4 T cells, are crucial in disease pathogenesis [7]. This subpopulation is characterized by expression of the chemokine receptor CCR6 as well as production of the inflammatory cytokine IL-17 [8, 9]. Th17 cells are, however, not the only cell type secreting IL-17. Voo et al. [10] have shown that there are a significant number of human CD4+ FOXP3+ regulatory T cells (Tregs) that have the capability to produce IL-17 upon activation, a subset that could not be found in the thymus. These are thought to be important in antimicrobial defense besides their regulatory function in autoimmunity and inflammation [10]. The differentiation of Tregs into an effector subset that is able to produce IL-17 may be mediated by epigenetic modifications [11]. This phenomenon may have evolved as a potent mechanism of the immune system to adapt to the vast variety of immune responses at different locations of the human body and at different stages of an immune response [12]. Th17 and Treg cells are the best-known counterparts in balancing the body’s propensity to autoimmunity versus health and homeostasis.
Interestingly, both of the above CD4 helper subsets (Th17 and Tregs) are profoundly influenced by the microbiota. Induction of Tregs by certain microbiota has been reviewed in Chap. 5. Herein, we summarize the role of gut commensals in the induction of Th17 cells. In mice, segmented filamentous bacteria (SFB), Candidatus Arthromitus or Candidatus Savagella are known as unique commensal bacteria that are able to stimulate maturation of the lymphoid cell compartments and to specifically induce Th17 cells in the small intestine [13]. Recently, a group in France succeeded for the first time in culturing SFB in vitro by mimicking their replicative niche, making it possible to study in detail its host interactions [13]. Importantly, SFB are capable of promoting pathogenic Th17 responses in mouse models of RA and multiple sclerosis. Thus, a possible influence of SFB on the pathogenesis of SLE seemed plausible, but studies using a lupus-prone mouse model suggest that SFB colonization might neither induce Th17 responses nor increase the incidence of disease [14].
Another important aspect of SFB biology is that these bacteria induce high levels of mucosal IgA and are highly IgA-coated. IgA is crucial for the homeostasis of the gut microbiota. Different levels of IgA coating of the microbiota have been exploited to identify pathogenic members of the human microbiota in inflammatory bowel disease [15]. It remains to be seen if extreme levels of IgA coating exist in non-gut diseases. An increased level of secretory IgA has been associated with intestinal dysbiosis in inflammatory bowel disease as well as psoriatic arthritis although the significance of this finding is unclear in the latter case [15, 16].
Production of high-affinity IgA is supported by a special subset of helper T cells, the T follicular helper (Tfh) cells , that have emerged as key players in the differentiation of memory B cells at several stages [17]. Interestingly, it has been suggested that Th17 cells act as progenitors for a subset of Tfh cells to promote the production of high-affinity IgA against commensal microbes [18]. This process is thought to occur via MyD88 signaling as loss of this pathway is accompanied by an inefficient IgA response as well as an altered commensal composition favoring a more inflammatory environment [19]. Several studies using lupus-prone animal models in addition to samples from human subjects demonstrated that these cells are involved in the production of pathogenic autoantibodies . A recent study showed increased Tfh-like cells (CXCR5+ICOS+PD-1+) in SLE patient blood compared to healthy controls, which did not correlate with disease activity [20]. Thus, these cells may be considered as a marker of germinal center B-cell dysregulation . As detailed above, since Tfh cells are influenced by the gut microbiota, it is plausible to assume that the microbiota plays a role in the pathogenesis of human SLE. Over the last few years, exploratory studies on the contents of the microbiota in SLE have been published. A summary of four recent studies related to the human microbiota in SLE are listed in Table 21.1.
Various mechanisms have been ascribed to microbially triggered autoimmunity including molecular mimicry. As detailed also in Chap. 6, several studies in murine models of autoimmunity have established a role for commensal bacteria as potential chronic triggers, although the exact mechanisms remain unclear [25]. As the human gut microbiota provides an enormous source of persistent antigenic variation, it is plausible that molecular mimicry via cross-reactive antigen within the microbiome could be a chronic trigger of autoreactive lymphocytes , in particular those that are recognizing the earliest autoantigen in most lupus patients, the Ro60 protein, a conserved regulatory RNA-binding protein. Interestingly, certain human gut commensals encode Ro60 orthologs, which suggests that patients that are colonized with Ro60 ortholog-containing bacteria could develop cross-reactive responses to human Ro60 protein that eventually lead to pathogenic responses based on their HLA class II-related genetic predisposition. Indeed, experimental support for such a scenario is accumPulating [26].
In summary, many facets of the microbiota could influence the pathogenesis of SLE. Besides various innate immune interactions with the microbiota, adaptive immune responses described here—Treg/Th17 balances, Tfh, and IgA levels—are influenced by the microbiota and potentially dysregulated in lupus. It is important to note that there are likely even more host-microbiota interactions impinging on innate immunity that are equally important in SLE pathogenesis, including activation of the inflammasome in lupus nephritis (LN) [27] or overstimulation of TLR signaling leading to an increased production of inflammatory cytokines and type I interferon (IFN) [28]. More research is needed to understand the multifaceted roles the microbiota may play in the pathogenesis of SLE.
The Pathogenic Microorganisms
Several studies on experimental animal models demonstrated that infectious agents are capable of breaking immunological tolerance to self-antigen inducing autoimmunity [29]. The list of microorganisms associated with human SLE includes parvovirus B19 [30], CMV [31, 32], retroviruses [31, 33], dengue virus [34], HPV vaccination or infection [35], Toxocara canis [36], and Mycobacterium tuberculosis [31], but the most compelling evidence supports a role for Epstein-Barr virus (EBV) . Many SLE patients negative for anti-dsDNA antibodies showed abnormal antibody responses to EBV: they produced IgG antibodies to EBV antigens to which healthy controls did not respond and failed to make antibodies to EBV antigens seen in healthy controls [37].
It was reported that the positive rate of EBV-encoded latent membrane protein 1 (LMP1) in the renal tissues was significantly higher in young patients with LN compared to controls [38]. The positive rate was similar between patients of initial onset and relapse, and there was no detectable difference between the patients with and without infection. The findings support the hypothesis that EBV reactivation is associated with SLE induction with a potential role of EBV-encoded LMP1 in this process.
Rasmussen et al. [39] studied 57 SLE patients and 29 healthy controls using plasma galectin-3-binding protein as a surrogate marker of type I IFN activity . They showed that the marker’s concentrations were significantly higher in SLE patients and associated positively with EBV early antigen diffuse (EBV EA/D)-directed antibodies and the presence of antibodies against extractable nuclear antigens [39].
In addition, Draborg et al. [40] demonstrated that there was an impaired regulation of the immune response against latent and lytic cycle EBV infection in SLE, even in the absence of lymphopenia, which further supported the proposed general dysfunction of leukocytes and their cytokine regulation in SLE patients. Reviewing the numerous experimental studies on EBV establishing an association with SLE, Draborg et al. [40] theorized that the interplay between an impaired immune system and the cumulative effects of EBV and other viruses results in frequent reactivation of EBV and enhanced cell death, leading to autoreactivity and development of SLE.
Type I INF family comprises 12 IFN-α subtypes and IFN-β, IFN-ε, IFN-κ, and IFN-ω. Normally, IFN-α and IFN-β production is strictly controlled but starts rapidly when viral or bacterial nucleic acids are sensed by pattern recognition receptors (PRRs) [41]. Two cell types capable of secreting large amounts of IFN-α and IFN-β are plasmacytoid dendritic cells (PDC) and monocytes. Monocytes respond mainly to dsRNA, certain RNA viruses such as Sendai and influenza virus, whereas PDCs can be triggered to secrete type I IFN by almost all viruses and some bacteria [42].
As microbial RNA and DNA can be recognized by multiple nucleic acid sensors and thereby induce production of type I IFNs, this may be the mechanism by which several microorganisms can contribute to the development and relapse of SLE [43]. An increase in the expression of long interspersed nuclear element 1 (LINE-1) was reported in kidney biopsies from patients with LN and transcript expression correlated with the tissue expression of type I IFN [44] . This connection may suggest that endogenous retroviruses may play a role in the initiation or amplification of the autoimmune process [43].
A recent study implicated one additional specific organism, Enterococcus gallinarum, as an etiologic agent of lupus. Manfredo Vieira et al. [45] studied the (NZW × BXSB)F1 lupus model, demonstrating a role for the microbiota as evidenced by diminished disease following antibiotic therapy. Antibiotics also strengthened the gut wall barrier, preventing translocation of bacteria into the mesenteric lymph nodes, mesenteric veins, and liver. E. gallinarum specifically was able to weaken the barrier defense and induce pro-inflammatory intestinal changes in mice, and its translocation specifically upregulated lupus-associated autoantibody production, and vaccination against E. gallinarum was protective against the diseaes in mice. Finally, in humans, this organism was present in the livers of 3/3 lupus patients who underwent biopsy, compared to 0/6 healthy liver transplant donors.
The Protective Role of Infectious Agents and Parasites
Contrary to this, there are other studies that support the notion of a protective role of other organisms. Experimental animal studies showed that hepatitis B virus plays a protective role against SLE [46]. Gamma-irradiated Plasmodium chabaudi infection of lupus-prone BWF1 mice ameliorated the histopathological changes attributed to renal involvement in lupus [47]. Another study showed that infection of female BWF1 lupus mice with malaria parasites attenuated B-cell autoreactivity [48]. Chen et al. demonstrated that Toxoplasma gondii infection may prevent the progression of SLE-related nephritis in New Zealand Black/New Zealand White F1 mice and was associated with downregulated intracellular expression of IFN-γ and IL-10 [49]. Fischer et al. evaluated the seroprevalence of anti-T. gondii antibodies in European patients with rheumatoid arthritis (RA) and SLE. They found a higher prevalence of anti-T. gondii antibodies in those with RA than in SLE patients (63% vs 36%, respectively) and that the rates of seropositivity of IgG against other infectious agents were comparable between the two groups [50].
Sowalha and his group investigated the prevalence of Helicobacter pylori seropositivity in a cohort of 466 patients with SLE, finding a low rate of specific H. pylori antibodies suggesting that H. pylori might exert a protective role on the risk of developing SLE [51]. Furthermore, it has been reported that in filarial endemic areas in India, patients with SLE do not suffer from concomitant filariasis. Filarial infestation was found to be associated with a low plasma level of IL-17A, which may contribute to protection from the development of autoimmune disorders like SLE [52].
Many pathogens have developed efficient methods to overcome adaptive immune mechanisms, and there is growing evidence that they are capable of insinuating their own anti-immune strategies into a susceptible host. They can block antigen presentation interference with Toll-like receptor signaling and alter the cytokine milieu, which is crucial for an effective immune response. They may also cause antigenic competition, the tendency of strong antigens, particularly from infectious agents, to impair antibody responses to weaker ones, which would dampen immune responses against self-antigens [53, 54].
Risk Factors for Infection in SLE
SLE patients have an increased frequency of severe bacterial and viral infections, possibly due to inherited genetic and immunological defects as well as due to chronic immunosuppressive therapies [55].
Doaty et al. [31] in their review of literature cited innate immunity disturbance in SLE patients that included:
-
(a)
Breakdown of epithelial barriers in SLE patients caused by rashes, ulcers, and wounds allowing entry of infectious agents in the body.
-
(b)
Accumulation of gamma delta T cells in skin of SLE patients as compared to healthy subjects [56]. These cells are implicated in epithelial breakdown, further increasing the risk of infection [57].
-
(c)
Impaired production of IL-8 and IL-12 by PMNs with disruption of the links between the innate and adaptive systems mediated by IL-12 [58].
-
(d)
Deficiencies of the early components of the classical pathway of the complement system (C1q, C4, and C2). A higher prevalence of these genetic defects has been established in SLE, but acquired complement deficiency because of consumption due to immune complex disease may also play a role in predisposing SLE patients to infection by encapsulated organisms [57,58,61]. Furthermore, single nucleotide polymorphisms have been reported associated with mannose-binding lectin deficiency in SLE patients, further increasing their susceptibility to infection [62, 63].
-
(e)
Decreased levels of complement receptors CR1 and CR2 on B cells, PMNs, and RBCs in SLE patents [64].
-
(f)
Suppressed natural killer (NK)-cell cytotoxicity with fewer NK cells [65] and weaker NK response to IL-2 stimulation [66].
Inappropriate or dysfunctional antigen presentation by DCs might promote the breakdown of T-cell and B-cell tolerance in SLE and other autoimmune diseases. Patients with SLE show multiple DC abnormalities, including a reduced number of circulating conventional DCs but increased numbers of PDCs [67].
Neutrophils show several facets of dysregulation in SLE. Impaired phagocytosis by neutrophils in SLE has been described in multiple reports and might contribute to the increased susceptibility to infection associated with this disease [68]. In one study, neutrophils from patients with SLE showed reduced production of reactive oxygen species (ROS), which correlated with disease severity and end-organ damage [69]. Patients with chronic granulomatous disease, in which ROS production is defective, have a high incidence of SLE [70, 71]. Patients with SLE have an abnormal subset of neutrophils (termed low-density granulocytes) with an increased propensity for NETosis [72]. NETosis is a mechanism of cell death that occurs in response to various stimuli, including infectious organisms and oxidative stress. NETosis involves the extrusion of chromatin and other nuclear, cytoplasmic, and granular material from the cell. This extruded material, called neutrophil extracellular traps (NETs), contains proinflammatory cytokines, antimicrobial peptides, enzymes such as myeloperoxidase, and potentially antigenic citrullinated histones and dsDNA [73].
Other workers reported that leukopenia , in particular lymphopenia, was a common finding in SLE. In their work, however, it was not persistent [74]. SLE patients can also develop granulocytopenia due to anti-granulocyte antibodies or complications from chronic immunosuppression. If neutropenia is severe enough, the impaired function of PMNs in SLE predisposes to severe bacterial infections. It was reported [68] that regardless of infection status, medication, or disease activity, pediatric-onset SLE patients have impaired phagocytic ability against Salmonella-specific lipopolysaccharides (LPS) which is not influenced by the use of immunosuppressants. The same study [68] did not find deficiency of peroxidase production and chemotaxis activity in SLE patients; however, serum complement levels were not reported.
In the earlier review by Doaty et al. [31], they also reported adaptive immunity disturbances such as:
-
(a)
Impaired production of IFN-γ, IL-1, IL-2, and TNF-α contributing to T-cell dysfunction [75]
-
(b)
Reduced numbers and dysfunction of all B-cell lines: naïve, memory, and plasma cells
Hypogammaglobulinemia has been attributed to immunosuppressive therapy in SLE patients even in the absence of therapy with B-cell-depleting agents. Isolated IgM [76] and IgA [77] were reported in SLE patients; however their contribution to an increased risk of infection was not shown. Therefore, it has been recommended in one report to measure immunoglobulin levels during the course of SLE treatment [78].
More than 80 genetic loci are reported to show robust genetic associations with SLE [77,78,79,82]. More than half of these loci are connected to the type I IFN system [83]. Genome-wide association studies have revealed single nucleotide polymorphisms in the STAT4 gene , which codes for a protein involved in type I interferon receptor signal transduction, that are associated with enhanced protein production [84]. The NCF1 gene encodes the p47phox/Ncf1 protein of the NADPH oxidase (NOX2) complex, which is critical for the induction of ROS. The NCF1 gene is highly complex and has excluded SNPs in NCF1 in genome-wide association studies. It has been recently reported that an amino acid replacement in NCF1, leading to a lower capacity of inducing oxidative burst, is strongly associated with SLE [85, 86]. This observation aligns with the previously reported association of an ROS-reducing SNP in the NCF2 gene with SLE [87]. Also, some rare monogenic SLE diseases are now categorized as type I interferonopathies because of the prominent type I IFN signature [43].
In a study by Danza and Ruiz-Irastorza [88], risk factors for infection in SLE included disease activity, prednisone doses over 7.5–10 mg/day, high doses of methylprednisolone, as well as use of chemotherapeutic agents such as cyclophosphamide and multiple courses of rituximab treatment. It is difficult, however, to tease apart the therapeutics from the underlying disease severity that prompted use of these medications. Indeed, lupus patients present with multiple features that pose an increased infection risk, including hypocomplementemia, lymphopenia, and hyposplenism, and data from the 1970s prior to the introduction and widespread use of newer therapies indicated a high risk of mortality from infections early in the disease course, attesting again to the infectious risk associated with active SLE [89].
Lupus Nephritis
Renal involvement occurs in up to 60% of SLE patients [90] and remains a major determinant for morbidity and mortality among these patients [90, 91]. We will focus on this particular organ as a notorious example of the interplay between the disease and infection.
Feldman et al. [92] studied serious infections among adult Medicaid beneficiaries with SLE and LN over the years 2000–2010. They identified 33,565 patients with SLE, of whom 7113 had LN. There were 9078 serious infections reported in 7078 SLE patients, whereas in 1825 LN patients, there were 3494 reports. Infection incidence rate per 100 person-year was 10.8 in the SLE cohort and 23.9 in the LN sub-cohort.
Therapeutic modalities used in LN management, either as induction therapy or for maintenance treatment, influence the prognosis of LN. An Egyptian study retrospectively analyzed records of 928 SLE patients with biopsy-confirmed LN seen between 2006 and 2012 at Cairo University hospitals [93]. The reported complications included pneumonia requiring hospitalization in 93/575 (16.1%) patients who received intravenous cyclophosphamide as induction therapy, compared to 22 of 321 (6.9%) patients in the group that received mycophenolate. However, the difference between these two groups was not statistically significant (p = 0.270). Herpes zoster infection (HZI) was reported in 12 (1.3%) patients. The 5-year mortality was 7.4%. Sepsis was responsible for death in 68.1%, which was higher than the percentages reported in Europe (25%) [94] and China (60%) [95].
In India, Srivastava et al. [96] studied the outcome of LN in childhood-onset SLE (cSLE) retrospectively from 1989 to 2013. Among 205 children with cSLE, 134 had evidence of LN. During the follow-up period, 11 (8.2%) children died, and infections were the leading cause of death.
Lin et al. [97] conducted a nationwide cohort study of 7326 patients with newly diagnosed SLE and no history of end-stage renal disease (ESRD) . They derived their data from Taiwan’s National Health Insurance claims database from 2000 to 2011. Among all SLE patients, 316 (43%) developed ESRD . Multivariate Cox regression analysis indicated that the risk of ESRD increased with the number of infection-related hospitalizations. For patients with three or more infection-related admissions, the hazard ratio (HR) for ESRD was 5.08 (99% CI: 3.74–6.90) relative to those with no infection-related admissions. Analysis by type of infection indicated that bacteremia patients had the greatest risk for ESRD with a HR of 4.82 (95% CI: 3.40–6.85) highlighting the impact of infection on LN outcome.
In South China [98], a group of investigators studied hospital-acquired infections (HAI) in SLE patients. In a multivariate analysis, they found that a history of LN or a higher SLE disease activity index SLEDAI score correlated with HAI [OR: 3.7 (p < 0.001) and 1.1 (p < 0.001), respectively]. Moreover, treatment with high doses of glucocorticoids or cyclophosphamide was the main risk factor for HAI [OR: 2.7 (p < 0.001) and 2.9 (p < 0.001), respectively].
Murray et al. [99] studied hospitalization trends for SLE from 2000 to 2011. They identified 361,337 hospitalizations for SLE that were derived from the United States (US) Healthcare Cost and Utilization Project National Inpatient Sample (NIS). A diagnosis of SLE was associated with increased severe and opportunistic infections, including bacteremia, pneumonia, opportunistic fungal infections, herpes zoster (HZ), CMV, and Pneumocystis jirovecii pneumonia (PJP). They also found that among SLE hospitalization, rates of all these infections significantly rose between 2000 and 2011, with the exception of PJP which significantly declined. HZ was the only infection that disproportionally increased over time among SLE hospitalizations when compared with non-SLE hospitalizations. They attributed this to the increasingly widespread use of mycophenolate mofetil for induction and maintenance of LN and for severe non-renal lupus. It has been demonstrated that medications used to treat SLE, including prednisone, azathioprine, and cyclophosphamide, increase the risk of HZ [98,99,100,101,104]. In a prospective cohort study of 1485 patients with SLE, the hazard ratio for an incident diagnosis of HZ was greatest in SLE patients treated with mycophenolate mofetil (HR 5.00, 95% CI 1.40–17.60) followed by prednisone [100].
Murray et al. [99] attributed the reduction of PJP in their study to the declining use of cyclophosphamide and increasing use of mycophenolate. Even among patients receiving cyclophosphamide as induction therapy, cumulative doses may be declining over time. The Euro-Lupus Nephritis trial demonstrated equivalent efficacy and a favorable side effect profile for low-dose intravenous cyclophosphamide compared with a previously standard high-dose regime [105, 106]. Also, the American College of Rheumatology recommended mycophenolate or cyclophosphamide for induction of LN with lower cumulative doses of cyclophosphamide (“Euro-Lupus” dosing schedule), followed by mycophenolate or azathioprine for maintenance therapy [107]. Interestingly, animal studies and data from renal transplant trials suggest that mycophenolate may have antimicrobial properties against P. jirovecii, although data specific to lupus patients are lacking [108, 109].
Based on the Medicaid Analytic Extract database (2000–2010); Feldman et al. [92] found no difference in the rates of serious infection and mortality among new users of mycophenolate, azathioprine, or cyclophosphamide when examining the 29 most populated US states. Strikingly, antimalarial treatment of SLE patients was associated with an additional benefit in protection against serious infection [110]. In summary, patients with severe lupus, as evidenced by LN, have a high risk of serious infections, including death. It remains unclear the extent to which this higher risk is mediated by the underlying disease severity, versus its therapy. One favorable trend in recent years has been a lower incidence of PJP, potentially attributable to lower cumulative doses of cyclophosphamide and/or more widespread usage of mycophenolate.
The Heavy Burden of Infection on Morbidity and Mortality in SLE
It is well established that infection contributes significantly to the morbidity and mortality of SLE patients. In a South African study [111], the authors reviewed the records of hospitalized SLE patients admitted over a 79-month period. They found that infections accounted for 35.2% of admissions. Among those, pneumonia, cutaneous sepsis, urinary tract infections, and septicemia were the most common types of infection. Organisms commonly isolated were Staphylococcus aureus, Escherichia coli, and Klebsiella species, as well as Mycobacterium tuberculosis. The incidence rate ratio for infection in SLE was 1.49 compared to matched controls emphasizing its burden as a comorbidity in the disease [112]. It was found that infection-related hospitalizations are associated with an increased risk of end-stage renal disease in SLE, especially the juvenile-onset type [97]. Souza et al. reported that renal failure and infectious diseases were the most frequent causes of death in Brazilian SLE patients [113]. A Chinese study reported that over time (1986–2012) infections have increased gradually and have become the most frequent cause of death in SLE [114]. Richie et al. extensively reviewed the literature and reported a total of 17 maternal deaths in women with LN within 6 weeks postpartum. In all cases where mortality was attributed to SLE and nephritis, the patients had active disease, and infection was responsible for 41.2% of deaths [115]. In a meta-analysis that included 26,101 SLE patients with 4640 deaths, the cause-specific standardized mortality ratios (SMRs) were assessed in SLE patients. The SMR for infection was reported as 4980 with a highly significant p-value [116]. In a multicenter Southern Chinese study that included 3815 hospitalized patients, infection was the leading cause of death. Features of infection in this group included early disease onset, higher percentage of respiratory tract involvement, and predominance of Gram-negative bacteria with emergence of multidrug-resistant strains and a variety of pathogens [117].
The Clinical Presentations of Infections in SLE
Serious infections are defined as those that lead to hospitalization or death or require intravenous antibiotic treatment [118].
Shen et al. [119] studied the temporal trends among SLE patients in the intensive care unit (ICU) as part of a national population-based study in Taiwan between 1999 and 2008. The incidence of infection rose from 39.1% to 47.2%. The study reported three poor prognostic factors (i.e., older age, infection, and organ dysfunction) that were thought to potentially lessen the temporal improvement of short-term survival in ICU patients with SLE. The authors suggested that improved treatment of SLE reduces and postpones the occurrence of acute critical illness. Their study showed that the median time for SLE diagnosis to ICU admission had increased by 4 years during the 10-year study period. However, the improved survival was achieved at the costs of immune-suppression and accrual of organ damage.
Han et al. [120] studied the clinical presentations and outcomes of SLE patients with infection admitted to the ICU. They demonstrated that SLE patients with infections in the ICU have a higher mortality rate, and a higher APACHE II score compared to SLE patients without infections. SLE with infections also had a higher maximum temperature, higher minimum and maximum systolic blood pressure compared to SLE patients with noninfectious causes of admission.
Musculoskeletal System
Odd presentations of musculoskeletal infections at unusual anatomical sites like the sacroiliac joint [121] and uncommon organisms like Candida albicans involving the joints [122] are frequently reported in SLE patients. One rare and frequently misdiagnosed infection in SLE is tropical pyomyositis, primary muscle abscesses most frequently due to Staphylococcus aureus and frequently following trauma [123]. Also, hematogenous salmonella osteomyelitis can be encountered in immunocompromised SLE patients, a finding that can be complicated by septic arthritis if not managed promptly [124]. Infection should also be considered when dealing with cases of osteonecrosis [124, 125].
Mucocutaneous and Genital Manifestations
In a retrospective multicenter cohort study involving ten pediatric rheumatology services in São Paulo, Brazil, that included 852 childhood-onset SLE patients, the researchers reported a frequency of 14% of herpes zoster infection (HZI). Hospitalization took place in 61% of these cases, and secondary bacterial infection was reported in 13%. Postherpetic neuralgia occurred in 5%. Lymphopenia and immunosuppressive therapy seemed to be the major factors underlying this complication [126].
There is a special interest concerning human papillomavirus (HPV) in SLE patients. In a meta-analysis, the authors found that cutaneous warts (CW) were present in a higher frequency in SLE patients compared to healthy controls. It is of interest that most of the articles they cited showed that the presence of CW did not correlate with the use of immunosuppressive drugs [127].
SLE female patients have also been shown to have a higher prevalence of genital HPV infection (80.7%) compared to healthy women (35%). The odds ratio (OR) for genital HPV infection in women with SLE was 7.2 [128]. There was no evidence that the use of immunosuppressive drugs was associated with a higher prevalence of HPV infection. This finding together with the study of Silva et al. [127] suggests that the high prevalence of HPV may be due to defects in immune mechanisms that are independent of immunosuppressive drugs. Another study found that in a subset of women diagnosed with SLE in the eastern Brazilian Amazon, 75% of them were HPV positive in the 1–5 years preceding the study [129].
The Urinary Tract
Lupus patients are likely to have urinary tract infections (UTI) , with a prevalence of 36%. They usually manifest in the lower tract. They are community acquired, and the most frequently isolated uropathogen is E.coli [130].
In an Egyptian cohort of 200 SLE patients who were followed up for one year, the urinary tract was the most common site of infection (31.8%) with 74/230 infectious episodes. E.coli was the most common isolated bacterial organism (26/230), followed by Klebsiella species (11/230) and Proteus mirabilis (8/230), whereas nine cases had mixed infections [131].
Hematological Involvement
Lupus patients with episodes of bacteremia suffer from poor long-term outcome. E. coli and S. aureus are the leading pathogens reported in this setting. Community-acquired bacteremia and C-reactive protein levels lower than 8 mg/dl during bacteremic episodes are associated with lower long-term mortality [132]. A Danish study involving 5102 patient-years of follow-up [133] reported an increased incidence of arterial and venous thrombosis within 1 year after infection (2.18% and 2.56%, respectively) compared to patients who never had either a hospitalized infection or herpes simplex virus. Infections can also trigger catastrophic antiphospholipid syndrome [134] (see Chap. 22).
The Respiratory System
Respiratory tract infection is a common problem in SLE. Luijten et al. [135] reported the incidence of invasive pneumococcal infections to be 13 times higher in SLE patients than in the general Dutch population. The prevalence of latent tuberculosis infection was reported to be 26.5% in SLE patients [136]. The authors of the study cautioned that higher SLE disease activity index (SLEDAI) and increased glucocorticoid dose were associated with indeterminate results in interferon-gamma release assays. Fungal infections including aspergillosis can occur and may be associated with cavitary lesions that can lead to pneumothorax [137] (Fig. 21.1).

High-resolution chest CT scan with contrast showing a case of SLE with multiple pulmonary fungal cavitary lesions. Arrow: intracavitary mycosis. Microbiological studies diagnosed the case as aspergillosis. SLE systemic lupus erythematosus. Courtesy of Dr. Hala El-Guendy, Professor of Internal Medicine, Cairo University
Studying the incidence of diffuse alveolar hemorrhage (DAH) in SLE patients [138], researchers reported 57 episodes of DAH of 50 patients including seven recurrences. They detected infection in 22 episodes (38.6%): 8 invasive fungal infections and 16 bacterial infections, including two patients with both types. These infections were associated with treatment for SLE, requirement for mechanical ventilation, hypocomplementemia, and high CRP levels.
The Gastrointestinal System
In a large prospective cohort study of 2258 SLE patients from the Hopkins Lupus Cohort, Fangtham et al. [139] reported 53,548 cohort visits. Oral candidiasis was diagnosed at 675 visits (1.25%), in 325/2258 (14%) of SLE patients. The authors recommended inspection of the oral cavity for signs of oral candidiasis, especially in patients with active disease, proteinuria, high white blood cell count, and intake of prednisone, immunosuppressive drugs, or antibiotics.
Fawzy et al. [140] reported an increase of Giardia lamblia infection in SLE patients: 30% in SLE patients compared to 3.3% in healthy, age- and sex-matched controls from the same geographic area. This number was even higher in those who had GI symptoms (52.9%). In acute pancreatitis, the presence of concomitant infections was associated with a poor prognosis in SLE patients [141].
Opportunistic CMV colitis can lead to colonic perforation with life-threatening consequences [142, 143]. In case of significant gastrointestinal symptoms, CMV infection should be considered in SLE patients who are immunosuppressed. SLE was also found to be significantly associated with chronic hepatitis C infection [144].
The Nervous System
Infectious brain lesions (IBLs) are rare presentations in SLE patients. They can, however, be life-threatening in this context. Xu et al. [145] described 15 patients with IBLs. They reported the following characteristics: fever in 80% of cases, headache and focal neurological signs (73.3%), associated pulmonary infection (66.7%), and associated meningitis (40%). There were ring-enhancing lesions in enhanced magnetic resonance imaging in all patients (100%) (Fig. 21.2).

Cranial MRI with IV contrast, T2 weighted, showing an SLE patient whose condition was complicated with subacute bacterial endocarditis (SBE) and multiple infected emboli of the brain. Arrow heads: ring-enhancing lesions. Courtesy of Dr. Hala El-Guendy, Professor of Internal Medicine, Cairo University
Progressive multifocal leukoencephalopathy (PML) has been reported in SLE patients treated with various immunosuppressive therapies including biological drugs (e.g., rituximab or belimumab). It was suggested that severe lymphopenia may be responsible for John Cunningham virus (JCV) reactivation, the causative agent of PML. Early infection screening, antiviral therapy, and effective management of lymphopenia are important in this setting [146]. Diagnosis of PML is generally made by identification of the virus in CSF by PCR along with consistent imaging and clinical features [147]. CSF lactate is a good single indicator and a better marker, compared to other conventional markers, to distinguish bacterial meningitis from aseptic meningitis [148, 149].
Cryptococcal meningitis [150] and meningoencephalitis [151] can be fatal in SLE patients. Epidural infection , an uncommon condition, can be caused by Salmonella enteritidis, which was also reported in SLE [152]. An early diagnosis and prompt treatment is essential to prevent mortality.
Invasive and Disseminated Infections
Miliary TB with fatal consequences has been reported in juvenile SLE patients in Brazil. The authors stressed the importance of routine screening for TB in this patient population [153]. Also, the prevalence of disseminated CMV infection is rising as a complication of active treatment of SLE [154].
Invasive fungal infections (IFI) describe a group of diseases caused by cryptococcus, histoplasma, aspergillus, and candida [155]. Their frequency was 4.8% in hospitalized SLE patients in Argentina [155] and 3.9% in juvenile SLE in Brazil [156]. A Mexican study [157] found the risk of IFI in SLE to be associated with high CRP levels, high disease activity, mechanical ventilation, antibiotic treatment, hemodialysis, high dose of glucocorticoids, and treatment with mycophenolate mofetil. The mortality was four times higher in patients with IFI than in those without. Cryptococcus neoformans is the most frequent agent in Argentina and East Asia [155, 158], while Candida spp. are more common in North America [155].
IFI should be suspected in hospitalized SLE patients undergoing immunosuppressive therapy. We suggest that clinicians have a high index of suspicion for IFI in hospitalized SLE patients who have unexplained organ-specific symptoms and imaging abnormalities (e.g., neurologic, pulmonary, dermatologic, musculoskeletal, and elevated CRP with or without elevated WBC or fever). Additionally, hospitalized SLE patients who do not improve rapidly with antibiotics should undergo thorough diagnostic procedures (e.g., CSF sampling, bone marrow/tissue biopsies and/or bronchoalveolar lavage with culture, histopathology, serum antigen and antibody levels, and PCR testing) and consideration for prompt empiric antifungal treatment.
Differentiating Flare from Infection in SLE Patients
SLE follows a chronic course with intermitting flares [159]. Symptoms such as fever, fatigue, and rash may be seen in an SLE flare or as a result of infection [160]. The differentiation of SLE activity and infection in febrile or otherwise acutely ill SLE patients is extremely difficult, and several biomarkers have been recognized as potential tools to differentiate between these two conditions [161].
Serum procalcitonin (PCT) and CRP are markers with strong supportive evidence. Song et al. [162] performed a meta-analysis of published studies and found that procalcitonin is more specific and has better diagnostic accuracy than PCR for bacterial infection in systemic rheumatic diseases. Bador et al. [163] conducted a study to determine predictive values of PCT and CRP for bacterial infections in SLE patients. Bacterial infection was defined as positive culture results. PCT and CRP were measured by automated immunoassays. The areas under the receiver operating characteristic curves for PCT and CRP were not significantly different (0.797 (CCI 0.614–0.979) vs 0.755 (CI 0.600–0.910)). They found that PCT but not CRP was higher in flaring lupus patients with infection (p = 0.019 vs 0.195), as compared to flaring SLE patients without infection. A PCT of <0.17 ng/ml ruled out infection with a negative predictive value (NPV) of 94%. In patients in remission, CRP but not PCT was elevated during infection (p = 0.036 vs 0.103); a CRP <0.57 ng/dl had a NPV of 96%. They concluded that PCT may be a better marker to rule out bacterial infection in lupus flares but not in remission or general screening. Serio et al. [164] conducted a systematic review on this topic and concluded that PCT levels detected during disease flares were lower than those observed during bacterial infection and that elevated PCT levels ≥0.5 μg/l strongly suggest bacterial infection. SLE patients, including patients in remission, tend to have higher CRP baseline levels when compared with controls. CRP response during flares seems to be incomplete and did not always correlate with disease activity. Values greater than 1.0 mg/dl can indicate severe flare if neither serositis nor arthritis is associated, while higher CRP levels above 5–6 mg/dl may be associated with infection [165].
Other potential biomarkers have been identified but have limited usage to date. One is the delta neutrophil index , an index which reflects the fraction of circulating immature granulocytes associated with infection [166]. The activity of adenosine triphosphate produced by CD4+ T cells was also found to be lower in patients with LN with infection compared to non-infected LN patients [167]. The ratio of erythrocyte-bound C4d to complement receptor 1 (C4d/CR1 ) was also studied. Febrile patients with disease flares had higher ratios and lower CRP levels than those with infection [168]. Ospina et al. [161] suggested that new scores, which include different biomarkers, might represent a better solution for differentiating infections from flares.
Prevention of Infections in SLE
Various strategies can be applied to reduce the risk of infections in SLE patients. These include vaccinations, antibacterial or antiviral prophylaxis, and intravenous immunoglobulins [169] (see also Chaps. 32 and 33).
Most non-live vaccines are immunogenic and safe in SLE patients, although antibody titers are frequently lower than those of healthy controls [170]. HPV vaccines can be given safely to SLE patients to avoid the increased incidence of anogenital warts and cervical epithelial dysplasia or carcinoma associated with high-risk viral genotypes [170]. Several experts [171] have recommended annual examinations of the cervical cytology in immunosuppressed patients.
Influenza vaccination is well tolerated and conveys a moderate protection against influenza infection in SLE. Considering that influenza runs a more severe course in SLE patients with a higher risk of disease exacerbation, influenza vaccination is recommended in patients with a low-to-moderate SLEDAI score or in those with stable disease. However, there were limited data and concern of the vaccine triggering a flare in severe disease [172]. Pneumococcal vaccination, however, is recommended for patients at any stage of their disease [171].
Live attenuated vaccines should generally be avoided in immunosuppressed patients. Recent studies, however, suggest that they can be considered in mildly immunosuppressed patients [171]. Serological screening for hepatitis B virus infection before starting immunosuppressive therapy is recommended for SLE patients to avoid viral reactivation [173].
Conclusion
In summary, microbial agents are involved in various aspects of lupus including the pathogenesis, treatment complications, and long-term sequelae. Further studies are needed to fully delineate the role of commensal microbiota in the pathogenesis of SLE and the entire spectrum of acute and chronic infections (bacterial, viral, parasitic) during the lifespan of lupus patients, particularly those on chronic immunosuppressive therapies.
Abbreviations
- AZA:
-
Azathioprine
- CCR6:
-
Chemokine receptor 6
- CMV:
-
Cytomegalovirus
- CRP:
-
C-reactive protein
- CSF:
-
Cerebrospinal fluid
- cSLE:
-
Childhood systemic lupus erythematosus
- CW:
-
Cutaneous warts
- ESRD:
-
End stage renal disease
- HAI:
-
Hospital-acquired infection
- HBV:
-
Hepatitis B virus
- HLA:
-
Human leukocyte antigen
- HPV:
-
Human papillomavirus
- HR:
-
Hazard ratio
- HZI:
-
Herpes zoster infection
- IBLs:
-
Infected brain lesions
- IFI:
-
Invasive fungal infection
- IFN:
-
Interferon
- JCV:
-
John Cunningham virus
- LMP1:
-
Latent membrane protein
- LN:
-
Lupus nephritis
- LPS:
-
Lipopolysaccharides
- MMF:
-
Mycophenolate mofetil
- NETs:
-
Neutrophil extracellular traps
- NKs:
-
Natural killer cells
- PCT:
-
Procalcitonin
- PDC:
-
Plasmacytoid dendritic cells
- PMNs:
-
Polymorphonuclear cells
- PRRs:
-
Pattern recognition receptors
- RA:
-
Rheumatoid arthritis
- ROS:
-
Reactive oxygen species
- SBE:
-
Subacute bacterial endocarditis
- SFB:
-
Segmented filamentous bacteria
- SLE:
-
Systemic lupus erythematosus
- SLEDAI:
-
Systemic lupus erythematosus disease activity index
- SMR:
-
Standardized mortality ratio
- SMRs:
-
Standardized mortality ratios
- TB:
-
Tuberculosis
- Tfh:
-
Follicular helper T cells
- TLR:
-
Toll-like receptors
- Tregs:
-
Regulatory T cells
- UTI:
-
Urinary tract infections
References
Mackay IR. Science, medicine, and the future: tolerance and autoimmunity. BMJ. 2000;321(7253):93–6.
Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis—insights into pathogenesis. Nat Rev Rheumatol. 2016;12(2):81–91.
van Drongelen V, Holoshitz J. Human leukocyte antigen-disease associations in rheumatoid arthritis. Rheum Dis Clin N Am. 2017;43(3):363–76.
Gollwitzer ES, Marsland BJ. Impact of early-life exposures on immune maturation and susceptibility to disease. Trends Immunol. 2015;36(11):684–96.
Arrieta MC, Stiemsma LT, Amenyogbe N, et al. The intestinal microbiome in early life: health and disease. Front Immunol. 2014;5:427.
Vieira SM, Pagovich OE, Kriegel MA. Diet, microbiota and autoimmune diseases. Lupus. 2014;23(6):518–26.
Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. 2017;87:1–15.
Veldhoen M, Hocking RJ, Flavell RA, et al. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7(11):1151–6.
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, et al. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8(9):942–9.
Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A. 2009;106(12):4793–8.
Lee YK, Mukasa R, Hatton RD, et al. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21(3):274–80.
Koenen HJ, Smeets RL, Vink PM, et al. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008;112(6):2340–52.
Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–98.
Johnson BM, Gaudreau MC, Al-Gadban MM, et al. Impact of dietary deviation on disease progression and gut microbiome composition in lupus-prone SNF1 mice. Clin Exp Immunol. 2015;181(2):323–37.
Palm NW, de Zoete MR, Cullen TW, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158(5):1000–10.
Scher JU, Ubeda C, Artacho A, et al. Decreased bacterial diversity characterizes the altered gut microbiota in patients with psoriatic arthritis, resembling dysbiosis in inflammatory bowel disease. Arthritis Rheumatol. 2015;67(1):128–39.
Wang NS, McHeyzer-Williams LJ, Okitsu SL, et al. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nat Immunol. 2012;13(6):604–11.
Hirota K, Turner JE, Villa M, et al. Plasticity of Th17 cells in Peyer's patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol. 2013;14(4):372–9.
Kubinak JL, Petersen C, Stephens WZ, et al. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe. 2015;17(2):153–63.
Choi JY, Ho JH, Pasoto SG, et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 2015;67(4):988–99.
Hevia A, Milani C, Lopez P, et al. Intestinal dysbiosis associated with systemic lupus erythematosus. MBio. 2014;5(5):e01548–14.
Rojo D, Hevia A, Bargiela R, et al. Ranking the impact of human health disorders on gut metabolism: systemic lupus erythematosus and obesity as study cases. Sci Rep. 2015;5:8310.
Lopez P, de Paz B, Rodriguez-Carrio J, et al. Th17 responses and natural IgM antibodies are related to gut microbiota composition in systemic lupus erythematosus patients. Sci Rep. 2016;6:24072.
Conti F, Ceccarelli F, Iaiani G, et al. Association between Staphylococcus aureus nasal carriage and disease phenotype in patients affected by systemic lupus erythematosus. Arthritis Res Ther. 2016;18:177.
Ruff WE, Kriegel MA. Autoimmune host-microbiota interactions at barrier sites and beyond. Trends Mol Med. 2015;21(4):233–44.
Greiling TM, Dehner C, Chen X, et al. Commensal orthologs of the human autoantigen Ro60 as triggers of autoimmunity in lupus. Science Translational Medicine. 2018;10(434):eaan2306.
Kahlenberg JM, Kaplan MJ. The inflammasome and lupus: another innate immune mechanism contributing to disease pathogenesis? Curr Opin Rheumatol. 2014;26(5):475–81.
Horton CG, Farris AD. Toll-like receptors in systemic lupus erythematosus: potential targets for therapeutic intervention. Curr Allergy Asthma Rep. 2012;12(1):1–7.
Hsieh AH, Jhou YJ, Liang CT, et al. Fragment of tegument protein pp65 of human cytomegalovirus induces autoantibodies in BALB/c mice. Arthritis Res Ther. 2011;13(5):R162.
Hod T, Zandman-Goddard G, Langevitz P, et al. Does parvovirus infection have a role in systemic lupus erythematosus? Immunol Res. 2017;65(2):447–53.
Doaty S, Agrawal H, Bauer E, et al. Infection and lupus: which causes which? Curr Rheumatol Rep. 2016;18(3):13.
Rigante D, Esposito S. Infections and systemic lupus Erythematosus: binding or sparring partners? Int J Mol Sci. 2015;16(8):17331–43.
Nelson P, Rylance P, Roden D, et al. Viruses as potential pathogenic agents in systemic lupus erythematosus. Lupus. 2014;23(6):596–605.
Sane P, Amritkar V, Pooja G. Dengue viral infection triggering abnormal immune response in a case of Kikuchi disease which later evolved into SLE. J Assoc Physicians India. 2016;64(1):147.
Soldevilla HF, Briones SF, Navarra SV. Systemic lupus erythematosus following HPV immunization or infection? Lupus. 2012;21(2):158–61.
Levy M, Bourrat E, Baudouin V, et al. Toxocara canis infection: unusual trigger of systemic lupus erythematosus. Pediatr Int. 2015;57(4):785–8.
Fattal I, Shental N, Molad Y, et al. Epstein-Barr virus antibodies mark systemic lupus erythematosus and scleroderma patients negative for anti-DNA. Immunology. 2014;141(2):276–85.
Ding Y, He X, Liao W, et al. The expression of EBV-encoded LMP1 in young patients with lupus nephritis. Int J Clin Exp Med. 2015;8(4):6073–8.
Rasmussen NS, Nielsen CT, Houen G, et al. Humoral markers of active Epstein-Barr virus infection associate with anti-extractable nuclear antigen autoantibodies and plasma galectin-3 binding protein in systemic lupus erythematosus. Lupus. 2016;25(14):1567–76.
Draborg AH, Sandhu N, Larsen N, et al. Impaired cytokine responses to Epstein-Barr virus antigens in systemic lupus Erythematosus patients. J Immunol Res. 2016;2016:6473204.
Gürtler C, Bowie AG. Innate immune detection of microbial nucleic acids. Trends Microbiol. 2013;21(8):413–20.
Fitzgerald-Bocarsly P, Feng D. The role of type I interferon production by dendritic cells in host defense. Biochimie. 2007;89(6–7):843–55.
Bengtsson AA, Ronnblom L. Role of interferons in SLE. Best Pract Res Clin Rheumatol. 2017;31(3):415–28.
Mavragani CP, Sagalovskiy I, Guo Q, et al. Expression of long interspersed nuclear element 1 Retroelements and induction of type I interferon in patients with systemic autoimmune disease. Arthritis Rheumatol. 2016;68(11):2686–96.
Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, Costa FRC, Tiniakou E, Greiling T, Ruff W, Barbieri A, Kriegel C, Mehta SS, Knight JR, Jain D, Goodman AL, Kriegel MA. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359:1156–61.
Liu X, Jiao Y, Cui B, et al. The potential protective role of hepatitis B virus infection in pristane-induced lupus in mice. Lupus. 2016;25(11):1180–9.
Abdel-Maksoud MA, Abdel-Ghaffar FA, El-Amir A, et al. Infection with Plasmodium chabaudi diminishes plasma immune complexes and ameliorates the histopathological alterations in different organs of female BWF1 lupus mice. Eur Rev Med Pharmacol Sci. 2016;20(4):733–44.
Badr G, Sayed A, Abdel-Maksoud MA, et al. Infection of female BWF1 lupus mice with malaria parasite attenuates B cell Autoreactivity by modulating the CXCL12/CXCR4 Axis and its downstream signals PI3K/AKT, NFkappaB and ERK. PLoS One. 2015;10(4):e0125340.
Chen M, Aosai F, Norose K, et al. Toxoplasma gondii infection inhibits the development of lupus-like syndrome in autoimmune (New Zealand black × New Zealand white) F1 mice. Int Immunol. 2004;16(7):937–46.
Fischer S, Agmon-Levin N, Shapira Y, et al. Toxoplasma gondii: bystander or cofactor in rheumatoid arthritis. Immunol Res. 2013;56(2–3):287–92.
Sawalha AH, Schmid WR, Binder SR, et al. Association between systemic lupus erythematosus and helicobacter pylori seronegativity. J Rheumatol. 2004;31(8):1546.
Panda AK, Das BK. Diminished IL-17A levels may protect filarial-infected individuals from development of rheumatoid arthritis and systemic lupus erythematosus. Lupus. 2017;26(4):348–54.
Finlay BB, McFadden G. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell. 2006;124(4):767–82.
Bach JF. Infections and autoimmune diseases. J Autoimmun. 2005;25:74–80.
Murdaca G, Orsi A, Spano F, et al. Vaccine-preventable infections in systemic lupus Erythematosus. Hum Vaccin Immunother. 2016;12(3):632–43.
Robak E, Niewiadomska H, Robak T, et al. Lymphocytes Tgammadelta in clinically normal skin and peripheral blood of patients with systemic lupus erythematosus and their correlation with disease activity. Mediat Inflamm. 2001;10(4):179–89.
Volc-Platzer B, Anegg B, Milota S, et al. Accumulation of gamma delta T cells in chronic cutaneous lupus erythematosus. J Investig Dermatol. 1993;100(1):84s–91s.
Tsai CY, Wu TH, Yu CL, et al. Decreased IL-12 production by polymorphonuclear leukocytes in patients with active systemic lupus erythematosus. Immunol Investig. 2002;31(3–4):177–89.
Truedsson L. Classical pathway deficiencies – a short analytical review. Mol Immunol. 2015;68(1):14–9.
Rupert KL, Moulds JM, Yang Y, et al. The molecular basis of complete complement C4A and C4B deficiencies in a systemic lupus Erythematosus patient with homozygous C4A and C4B mutant genes. J Immunol. 2002;169(3):1570.
Pickering MC, Botto M, Taylor PR, et al. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv Immunol. 2000;76:227–324.
Garred P, Voss A, Madsen HO, et al. Association of mannose-binding lectin gene variation with disease severity and infections in a population-based cohort of systemic lupus erythematosus patients. Genes Immun. 2001;2(8):442–50.
Sebastiani GD, Galeazzi M. Infection—genetics relationship in systemic lupus erythematosus. Lupus. 2009;18(13):1169–75.
Marquart HV, Svendsen A, Rasmussen JM, et al. Complement receptor expression and activation of the complement cascade on B lymphocytes from patients with systemic lupus erythematosus (SLE). Clin Exp Immunol. 1995;101(1):60–5.
Park YW, Kee SJ, Cho YN, et al. Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum. 2009;60(6):1753–63.
Tanaka T, Saiki O, Negoro S, et al. Decreased expression of interleukin-2 binding molecules (p70/75) in T cells from patients with systemic lupus erythematosus. Arthritis Rheum. 1989;32(5):552–9.
Jin O, Kavikondala S, Sun L, et al. Systemic lupus erythematosus patients have increased number of circulating plasmacytoid dendritic cells, but decreased myeloid dendritic cells with deficient CD83 expression. Lupus. 2008;17(7):654–62.
Wu SA, Yeh KW, Lee WI, et al. Impaired phagocytosis and susceptibility to infection in pediatric-onset systemic lupus erythematosus. Lupus. 2013;22(3):279–88.
Bengtsson AA, Pettersson A, Wichert S, et al. Low production of reactive oxygen species in granulocytes is associated with organ damage in systemic lupus erythematosus. Arthritis Res Ther. 2014;16(3):R120.
Magnani A, Brosselin P, Beaute J, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. 2014;134(3):655–662.e8.
De Ravin SS, Naumann N, Cowen EW, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol. 2008;122(6):1097–103.
Villanueva E, Yalavarthi S, Berthier CC, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187(1):538–52.
Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol. 2015;27(5):448–53.
Lertchaisataporn K, Kasitanon N, Wangkaew S, et al. An evaluation of the association of leukopenia and severe infection in patients with systemic lupus erythematosus. J Clin Rheumatol. 2013;19(3):115–20.
Alarcon GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin N Am. 2006;20(4):849–75.
Goldstein MF, Goldstein AL, Dunsky EH, et al. Selective IgM immunodeficiency: retrospective analysis of 36 adult patients with review of the literature. Ann Allergy Asthma Immunol. 2006;97(6):717–30.
Cassidy JT, Kitson RK, Selby CL. Selective IgA deficiency in children and adults with systemic lupus erythematosus. Lupus. 2007;16(8):647–50.
Lim E, Tao Y, White AJ, et al. Hypogammaglobulinemia in pediatric systemic lupus erythematosus. Lupus. 2013;22(13):1382–7.
Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2017;29(5):423–33.
Deng Y, Tsao BP. Advances in lupus genetics and epigenetics. Curr Opin Rheumatol. 2014;26(5):482–92.
Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun. 2013;41:25–33.
Bentham J, Morris DL, Cunninghame Graham DS, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47:1457.
Bronson PG, Chaivorapol C, Ortmann W, et al. The genetics of type I interferon in systemic lupus erythematosus. Curr Opin Immunol. 2012;24(5):530–7.
Abelson AK, Delgado-Vega AM, Kozyrev SV, et al. STAT4 associates with systemic lupus erythematosus through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk. Ann Rheum Dis. 2009;68(11):1746–53.
Zhao J, Ma J, Deng Y, et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat Genet. 2017;49:433.
Olsson LM, Johansson ÅC, Gullstrand B, et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann Rheum Dis. 2017;76(9):1607–13.
Jacob CO, Eisenstein M, Dinauer MC, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A. 2012;109(2):E59–67.
Danza A, Ruiz-Irastorza G. Infection risk in systemic lupus erythematosus patients: susceptibility factors and preventive strategies. Lupus. 2013;22(12):1286–94.
Urowitz MB, Bookman AA, Koehler BE, et al. The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976;60(2):221–5.
Mok CC. Con: cyclophosphamide for the treatment of lupus nephritis. Nephrol Dial Transplant. 2016;31(7):1053–7.
Mills JA. Systemic lupus erythematosus. N Engl J Med. 1994;330(26):1871–9.
Feldman CH, Hiraki LT, Winkelmayer WC, et al. Serious infections among adult Medicaid beneficiaries with systemic lupus erythematosus and lupus nephritis. Arthritis Rheumatol. 2015;67(6):1577–85.
Momtaz M, Fayed A, Wadie M, et al. Retrospective analysis of nephritis response and renal outcome in a cohort of 928 Egyptian lupus nephritis patients: a university hospital experience. Lupus. 2017;26(14):1564–70.
Cervera R, Khamashta MA, Font J, et al. Morbidity and mortality in systemic lupus erythematosus during a 10-year period: a comparison of early and late manifestations in a cohort of 1,000 patients. Medicine (Baltimore). 2003;82(5):299–308.
Mok CC, Lau CS, Chan TM, et al. Clinical characteristics and outcome of southern Chinese males with systemic lupus erythematosus. Lupus. 1999;8(3):188–96.
Srivastava P, Abujam B, Misra R, et al. Outcome of lupus nephritis in childhood onset SLE in north and Central India: single-centre experience over 25 years. Lupus. 2016;25(5):547–57.
Lin CH, Hung PH, Hu HY, et al. Infection-related hospitalization and risk of end-stage renal disease in patients with systemic lupus erythematosus: a nationwide population-based study. Nephrol Dial Transplant. 2017;32(10):1683–90.
Zhan Z, Lao M, Su F, et al. Hospital-acquired infection in patients with systemic lupus erythematosus: a case-control study in a southern Chinese population. Clin Rheumatol. 2017;37(3):709–17.
Murray SG, Schmajuk G, Trupin L, et al. National lupus hospitalization trends reveal rising rates of herpes zoster and declines in pneumocystis pneumonia. PLoS One. 2016;11(1):e0144918.
Chakravarty EF, Michaud K, Katz R, et al. Increased incidence of herpes zoster among patients with systemic lupus erythematosus. Lupus. 2013;22(3):238–44.
Borba EF, Ribeiro AC, Martin P, et al. Incidence, risk factors, and outcome of herpes zoster in systemic lupus erythematosus. J Clin Rheumatol. 2010;16(3):119–22.
Pope JE, Krizova A, Ouimet JM, et al. Close association of herpes zoster reactivation and systemic lupus erythematosus (SLE) diagnosis: case-control study of patients with SLE or noninflammatory nusculoskeletal disorders. J Rheumatol. 2004;31(2):274–9.
Manzi S, Kuller LH, Kutzer J, et al. Herpes zoster in systemic lupus erythematosus. J Rheumatol. 1995;22(7):1254–8.
Rondaan C, de Haan A, Horst G, et al. Altered cellular and humoral immunity to varicella-zoster virus in patients with autoimmune diseases. Arthritis Rheumatol. 2014;66(11):3122–8.
Houssiau FA, Vasconcelos C, D'Cruz D, et al. Immunosuppressive therapy in lupus nephritis: the euro-lupus nephritis trial, a randomized trial of low-dose versus high-dose intravenous cyclophosphamide. Arthritis Rheum. 2002;46(8):2121–31.
Rhee C, Gohil S, Klompas M. Regulatory mandates for sepsis care--reasons for caution. N Engl J Med. 2014;370(18):1673–6.
Hahn BH, McMahon MA, Wilkinson A, et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken). 2012;64(6):797–808.
Husain S, Singh N. The impact of novel immunosuppressive agents on infections in organ transplant recipients and the interactions of these agents with antimicrobials. Clin Infect Dis. 2002;35(1):53–61.
Oz HS, Hughes WT. Novel anti-pneumocystis carinii effects of the immunosuppressant mycophenolate mofetil in contrast to provocative effects of tacrolimus, sirolimus, and dexamethasone. J Infect Dis. 1997;175(4):901–4.
Herrinton LJ, Liu L, Goldfien R, et al. Risk of serious infection for patients with systemic lupus Erythematosus starting glucocorticoids with or without Antimalarials. J Rheumatol. 2016;43(8):1503–9.
Dubula T, Mody GM. Spectrum of infections and outcome among hospitalized South Africans with systemic lupus erythematosus. Clin Rheumatol. 2015;34(3):479–88.
Rees F, Doherty M, Grainge M, et al. Burden of comorbidity in systemic lupus Erythematosus in the UK, 1999–2012. Arthritis Care Res (Hoboken). 2016;68(6):819–27.
Souza DC, Santo AH, Sato EI. Mortality profile related to systemic lupus erythematosus: a multiple cause-of-death analysis. J Rheumatol. 2012;39(3):496–503.
Fei Y, Shi X, Gan F, et al. Death causes and pathogens analysis of systemic lupus erythematosus during the past 26 years. Clin Rheumatol. 2014;33(1):57–63.
Ritchie J, Smyth A, Tower C, et al. Maternal deaths in women with lupus nephritis: a review of published evidence. Lupus. 2012;21(5):534–41.
Lee YH, Choi SJ, Ji JD, et al. Overall and cause-specific mortality in systemic lupus erythematosus: an updated meta-analysis. Lupus. 2016;25(7):727–34.
Chen D, Xie J, Chen H, et al. Infection in southern Chinese patients with systemic lupus erythematosus: spectrum, drug resistance, outcomes, and risk factors. J Rheumatol. 2016;43(9):1650–6.
Dixon WG, Watson K, Lunt M, et al. Rates of serious infection, including site-specific and bacterial intracellular infection, in rheumatoid arthritis patients receiving anti-tumor necrosis factor therapy: results from the British Society for Rheumatology biologics register. Arthritis Rheum. 2006;54(8):2368–76.
Shen HN, Yang HH, Lu CL. Temporal trends in characteristics and outcome of intensive care unit patients with systemic lupus erythematosus in Taiwan: a national population-based study. Lupus. 2013;22(6):644–52.
Han BK, Bhatia R, Traisak P, et al. Clinical presentations and outcomes of systemic lupus erythematosus patients with infection admitted to the intensive care unit. Lupus. 2013;22(7):690–6.
Pronk SM, van Ommen CH, Prince FH, et al. Venous thrombosis as a first sign of SLE. Ned Tijdschr Geneeskd. 2014;158:A7179.
Imamura H, Iwamoto T, Momohara S. Unusual case of an elbow mass caused by Candida arthritis in a patient with systemic lupus erythematosus. Hand Surg. 2014;19(3):409–11.
Meesiri S. Pyomyositis in a patient with systemic lupus erythematosus and a review of the literature. BMJ Case Rep. 2016. https://doi.org/10.1136/bcr-2016-214809.
Kim SS, Perino G, Boettner F, et al. Salmonella septic arthritis of the knees in a patient with systemic lupus erythematosus. Lupus. 2013;22(7):740–3.
Khammassi N, Kort Y. Osteonecrosis of the femoral condyles revealed by septic arthritis in systemic lupus erythematosus. Pan Afr Med J. 2015;22:94.
Ferreira JC, Marques HH, Ferriani MP, et al. Herpes zoster infection in childhood-onset systemic lupus erythematosus patients: a large multicenter study. Lupus. 2016;25(7):754–9.
Silva LM, Santos WG, Santiago MB. Prevalence of cutaneous warts in patients with systemic lupus erythematosus: a systematic review. J Infect Dev Ctries. 2016;10(9):902–6.
Lyrio LD, Grassi MF, Santana IU, et al. Prevalence of cervical human papillomavirus infection in women with systemic lupus erythematosus. Rheumatol Int. 2013;33(2):335–40.
Amaral JL, Araujo MV, Dias GA, et al. Clinical and epidemiological study of human papillomavirus infection in women with systemic lupus erythematosus in eastern Brazilian amazon. Acta Reumatol Port. 2017;42(1):47–54.
Hidalgo-Tenorio C, Jiménez-Alonso J, de Dios Luna J, et al. Urinary tract infections and lupus erythematosus. Ann Rheum Dis. 2004;63(4):431–7.
Mohamed DF, Habeeb RA, Hosny SM, et al. Incidence and risk of infection in Egyptian patients with systemic lupus erythematosus. Clin Med Insights Arthritis Musculoskelet Disord. 2014;7:41–8.
Marcos M, Fernandez C, Soriano A, et al. Epidemiology and clinical outcomes of bloodstream infections among lupus patients. Lupus. 2011;20(9):965–71.
Baronaite Hansen R, Jacobsen S. Infections increase risk of arterial and venous thromboses in Danish patients with systemic lupus erythematosus: 5102 patient-years of followup. J Rheumatol. 2014;41(9):1817–22.
Catoggio C, Alvarez-Uria A, Fernandez PL, et al. Catastrophic antiphospholipid syndrome triggered by fulminant disseminated herpes simplex infection in a patient with systemic lupus erythematosus. Lupus. 2012;21(12):1359–61.
Luijten RK, Cuppen BV, Bijlsma JW, et al. Serious infections in systemic lupus erythematosus with a focus on pneumococcal infections. Lupus. 2014;23(14):1512–6.
Xiao P, Dong C, Yue Y, et al. Dynamic expression of microRNAs in M2b polarized macrophages associated with systemic lupus erythematosus. Gene. 2014;547(2):300–9.
Pamuk ON, Pamuk GE, Barutcu E, et al. The development of pulmonary aspergillosis and pneumothorax in a patient with neutropenic systemic lupus erythematosus and successful treatment of the first case. BMJ Case Rep. 2014;2014:bcr2013200818. https://doi.org/10.1136/bcr-2013-200818.
Martinez-Martinez MU, Sturbaum AK, Alcocer-Varela J, et al. Factors associated with mortality and infections in patients with systemic lupus erythematosus with diffuse alveolar hemorrhage. J Rheumatol. 2014;41(8):1656–61.
Fangtham M, Magder LS, Petri MA. Oral candidiasis in systemic lupus erythematosus. Lupus. 2014;23(7):684–90.
Fawzy M, Edrees A, Okasha H, et al. Gastrointestinal manifestations in systemic lupus erythematosus. Lupus. 2016;25(13):1456–62.
Wang Q, Shen M, Leng X, et al. Prevalence, severity, and clinical features of acute and chronic pancreatitis in patients with systemic lupus erythematosus. Rheumatol Int. 2016;36(10):1413–9.
Strasser C, Wolf EM, Kornprat P, et al. Opportunistic cytomegalovirus infection causing colonic perforation in a patient with systemic lupus erythematosus. Lupus. 2012;21(4):449–51.
Tachikawa Y, Nozawa H, Tanaka J, et al. Colonic perforation in a patient with systemic lupus erythematosus accompanied by cytomegalovirus infection: a case report. Int J Surg Case Rep. 2016;23:70–3.
Mahroum N, Hejly A, Tiosano S, et al. Chronic hepatitis C viral infection among SLE patients: the significance of coexistence. Immunol Res. 2017;65(2):477–81.
Xu Y, Xu D, Zhang T, et al. The prevalence and clinical characteristics of systemic lupus erythematosus with infectious brain lesions in China. Scand J Rheumatol. 2012;41(6):466–71.
Berntsson SG, Katsarogiannis E, Lourenco F, et al. Progressive multifocal leukoencephalopathy and systemic lupus erythematosus: focus on etiology. Case Rep Neurol. 2016;8(1):59–65.
Williamson EML, Berger JR. Diagnosis and treatment of progressive multifocal Leukoencephalopathy associated with multiple sclerosis therapies. Neurotherapeutics. 2017;14(4):961–73.
Mekitarian Filho E, Horita SM, Gilio AE, et al. Cerebrospinal fluid lactate level as a diagnostic biomarker for bacterial meningitis in children. Int J Emerg Med. 2014;7(1):14.
Huy NT, Thao NT, Diep DT, et al. Cerebrospinal fluid lactate concentration to distinguish bacterial from aseptic meningitis: a systemic review and meta-analysis. Crit Care. 2010;14(6):R240.
Zhong Y, Li M, Liu J, et al. Cryptococcal meningitis in Chinese patients with systemic lupus erythematosus. Clin Neurol Neurosurg. 2015;131:59–63.
Zheng H, Li M, Wang D, et al. Gender-specific contributing risk factors and outcome of female cryptococcal meningoencephalitis patients. BMC Infect Dis. 2016;16:22.
de Araujo DB, Daolio L, Szajubok JC, et al. Epidural abscess due to Salmonella enteritidis in a patient with systemic lupus erythematosus. Lupus. 2012;21(12):1356–8.
Freire PS, Montoni JD, Ribeiro AS, et al. Miliary tuberculosis: a severe opportunistic infection in juvenile systemic lupus erythematosus patients. Rev Bras Reumatol Engl Ed. 2016;56(3):274–9.
Berman N, Belmont HM. Disseminated cytomegalovirus infection complicating active treatment of systemic lupus erythematosus: an emerging problem. Lupus. 2017;26(4):431–4.
Vinicki JP, Catalan Pellet S, Pappalardo C, et al. Invasive fungal infections in argentine patients with systemic lupus erythematosus. Lupus. 2013;22(9):892–8.
Silva MF, Ferriani MP, Terreri MT, et al. A multicenter study of invasive fungal infections in patients with childhood-onset systemic lupus erythematosus. J Rheumatol. 2015;42(12):2296–303.
Martinez-Martinez MU, Herrera-Van Oostdam D, Roman-Acosta S, et al. Invasive fungal infections in patients with systemic lupus erythematosus. J Rheumatol. 2012;39(9):1814–8.
Chen GL, Chen Y, Zhu CQ, et al. Invasive fungal infection in Chinese patients with systemic lupus erythematosus. Clin Rheumatol. 2012;31(7):1087–91.
Chung SA, Brown EE, Williams AH, et al. Lupus nephritis susceptibility loci in women with systemic lupus erythematosus. J Am Soc Nephrol. 2014;25(12):2859–70.
Firooz N, Albert DA, Wallace DJ, et al. High-sensitivity C-reactive protein and erythrocyte sedimentation rate in systemic lupus erythematosus. Lupus. 2011;20(6):588–97.
Ospina FE, Echeverri A, Zambrano D, et al. Distinguishing infections vs flares in patients with systemic lupus erythematosus. Rheumatology (Oxford). 2017;56(suppl_1):i46–54.
Song GG, Bae SC, Lee YH. Diagnostic accuracies of procalcitonin and C-reactive protein for bacterial infection in patients with systemic rheumatic diseases: a meta-analysis. Clin Exp Rheumatol. 2015;33(2):166–73.
Bador KM, Intan S, Hussin S, et al. Serum procalcitonin has negative predictive value for bacterial infection in active systemic lupus erythematosus. Lupus. 2012;21(11):1172–7.
Serio I, Arnaud L, Mathian A, et al. Can procalcitonin be used to distinguish between disease flare and infection in patients with systemic lupus erythematosus: a systematic literature review. Clin Rheumatol. 2014;33(9):1209–15.
Dima A, Opris D, Jurcut C, et al. Is there still a place for erythrocyte sedimentation rate and C-reactive protein in systemic lupus erythematosus? Lupus. 2016;25(11):1173–9.
Pyo JY, Park JS, Park YB, et al. Delta neutrophil index as a marker for differential diagnosis between flare and infection in febrile systemic lupus erythematosus patients. Lupus. 2013;22(11):1102–9.
Liu J, Pan Y, Tang LJ, et al. Low adenosine triphosphate activity in CD4+ cells predicts infection in patients with lupus nephritis. Clin Exp Rheumatol. 2014;32(3):383–9.
Chen CH, Tai SB, Chen HC, et al. Analysis of erythrocyte C4d to complement receptor 1 ratio: use in distinguishing between infection and flare-up in febrile patients with systemic lupus Erythematosus. Biomed Res Int. 2015;2015:939783.
Sciascia S, Cuadrado MJ, Karim MY. Management of infection in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2013;27(3):377–89.
Grein IH, Groot N, Lacerda MI, et al. HPV infection and vaccination in systemic lupus Erythematosus patients: what we really should know. Pediatr Rheumatol Online J. 2016;14(1):12.
Mathian A, Arnaud L, Adoue D, et al. Prevention of infections in adults and adolescents with systemic lupus erythematosus: guidelines for the clinical practice based on the literature and expert opinion. Rev Med Interne. 2016;37(5):307–20.
Liao Z, Tang H, Xu X, et al. Immunogenicity and safety of influenza vaccination in systemic lupus Erythematosus patients compared with healthy controls: a meta-analysis. PLoS One. 2016;11(2):e0147856.
Watanabe R, Ishii T, Harigae H. Pretreatment screening for hepatitis B virus infection in patients with systemic lupus erythematosus. Tohoku J Exp Med. 2015;237(1):9–15.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Ragab, G., Dehner, C., Hamza, H., Kriegel, M. (2018). Systemic Lupus Erythematosus. In: Ragab, G., Atkinson, T., Stoll, M. (eds) The Microbiome in Rheumatic Diseases and Infection. Springer, Cham. https://doi.org/10.1007/978-3-319-79026-8_21
Download citation
DOI: https://doi.org/10.1007/978-3-319-79026-8_21
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-79025-1
Online ISBN: 978-3-319-79026-8
eBook Packages: MedicineMedicine (R0)